
Abstract
Autism spectrum disorder (ASD) is linked to ion channel dysfunction, including chloride voltage-gated channel-4 (CLCN4). We generated Clcn4 knockout (KO) mice by deleting exon 5 of chromosome 7 in the C57BL/6 mice. Clcn4 KO exhibited reduced social interaction and increased repetitive behaviors assessed using three-chamber and marble burying tests. Surprisingly, these symptoms were improved by Risperidone treatment, a drug commonly used to treat ASD. RNA sequencing data from mouse neural progenitor cells (mNPCs) showed that the genes regulating trans-synaptic signaling, transmembrane transport, and neuronal projection development were significantly decreased in Clcn4 knockdown (KD) cells compared to wild type (WT). Moreover, Risperidone treatment increased the genes related to the ion transmembrane transport, membrane potential, and neuron projection development in Clcn4 KD. Abnormalities in synaptic plasticity and dendritic spine formation were also observed in Clcn4 KO compared to WT. We observed that phosphorylation of SYNAPSIN, PSD95, ERK and CREB, as well as the expression of CDK5, were reduced in the brains of Clcn4 KO mice. In Clcn4 KO cortical neurons, the phosphorylation of SYNAPSIN and PSD95 expressions also decreased compared to WT, indicating disrupted synaptic function. Additionally, Sholl analysis revealed a reduction in dendritic branching and neuronal projection length in both mouse and human CLCN4 KD neurons. Finally, the decreased phosphorylation of SYNAPSIN and expression of PSD95 along with dendrite abnormalities were restored after Risperidone treatment. These data suggest that dendritic outgrowth and synapse remodeling may serve as endophenotypic targets for drug efficacy in ASD.
Similar content being viewed by others


Introduction
Autism spectrum disorder (ASD) is a complex neurodevelopmental disorder [1] characterized by deficits in social interaction, communication difficulties, and restricted, repetitive behaviors [2]. The etiology of ASD is multifactorial, involving genetics, environmental, and neurobiological factors [3]. Among the genetic factors, the dysfunction of ion channels, particularly chloride ion channels, has emerged as a significant contributor of ASD pathology [4, 5]. These disruptions in ion channels can impair key neurodevelopmental processes such as neurogenesis, neuronal migration, and synaptic plasticity, which are crucial for brain development [6, 7].
Chloride voltage-gated channel-4 (CLCN4), a member of the chloride channel family, is abundantly expressed in the brain and is primarily localized to the endoplasmic reticulum and endosomes, where it plays a key role in maintaining ion homeostasis and intracellular trafficking [8,9,10,11,12]. Chloride ions regulate important functions such as synaptic vesicle acidification and glutamate uptake, with the coupling of glutamate influx and chloride efflux maintain osmotic and electrical neutrality, ensuring proper synaptic vesicle filling and transmission [13,14,15]. This process is vital for controlling the rate of vesicle acidification and efficient neurotransmitter loading, which is central to maintaining neuronal communication and synaptic plasticity.
Moreover, CLCN4 expression is closely linked to neuronal differentiation, influencing the number of dendritic branches in neurons [15]. In human studies have shown that CLCN4 dysfunction is associated with neurodevelopmental disorders such as X-linked intellectual disability and seizure disorders [16, 17]. Despite these findings, previous studies have reported that Clcn4 knockout (KO) mice do not exhibit neurological alterations in the brain, creating a paradox between the clinical phenotype observed in humans and the absence of such findings in animal models [17, 18]. In contrast, our study identified clear phenotypic changes in Clcn4 KO, further supporting the role of Clcn4 in neurodevelopmental processes.
The relevance of CLCN4 to ASD can be understood through its role in maintaining intracellular ion balance, particularly chloride and proton exchange [6, 7]. This balance is crucial for synaptic transmission and plasticity, both of which are disrupted in ASD [19]. CLCN4 dysfunction lead to impaired chloride/proton exchange, resulting in abnormal intracellular pH and ion homeostasis, which disrupt synaptic signaling and contribute to the excitatory-inhibitory imbalance commonly observed in ASD [19, 20].
In addition to its role in maintaining chloride homeostasis, CLCN4 dysfunction has been linked to disruptions in key signaling pathways such as MAPK/ERK and PI3K/AKT, which regulate synaptic plasticity and neuronal survival [21, 22]. These pathways are essential for processes like dendritic spine formation, protein clustering at synapses, and neurotransmitter release. Dysregulation of MAPK/ERK leads to impaired protein synthesis and synaptic transmission [21, 23], while disruptions in PI3K/AKT signaling affect neuronal growth and connectivity [24, 25].
Risperidone, a second-generation antipsychotic, was approved by the US Food and Drug Administration for the treatment of irritability and aggression in children with ASD from the age of 5 years [26, 27]. Risperidone improves behavioral symptoms of aggression and irritability in children with autism regardless of the genetic origin of their neurological disturbance. Its potential as a therapeutic intervention stems partially from its ability to modulate critical signaling pathways, specifically by upregulating the MAPK/ERK and PI3K/AKT pathways. This modulation promotes synaptic plasticity and has been shown to reverse cognitive deficits in animal models. By restoring synaptic protein activity through the normalization of MAPK/ERK signaling, risperidone may help correct the synaptic deficits associated with Clcn4 dysfunction, making it a promising candidate for addressing behavioral and cognitive impairments in ASD.
This study aims to further elucidate the mechanisms underlying the connection between CLCN4 deficiency and neurodevelopment. Risperidone was utilized to examine the predictive validity of the CLCN4 KD as a model for human ASD and to provide insights into the therapeutic potential of targeting ion channel and synaptic plasticity related signaling pathways in ASD.
Materials and methods
Generation of Clcn4 Knockout (KO) mice
Cas-9 protein tagged with a nuclear localization signal and gRNAs targeting exon 5 of the Clcn4 gene were purchased from Toolgen, Inc. (Seoul, Korea). Clcn4 KO were generated by Toolgen, Inc. Male mice with a Clcn4 KO on the C57BL/6 background were mated with female Clcn4 KO. All animal protocols were approved by the NCMH (No. NCMH-1711-003-003-05, NCMH-1808-003-001-02) and Dongguk University (No. IACUC-2020-001-1) and were performed according to the guidelines of the National Institute of Health. The animals were housed at a constant temperature of 22 ± 1 °C and relative humidity of 55 ± 1% on a 12 h light/12 h dark cycle (lights on at 9:00 p.m.) and given free access to food and water.
Real-time PCR
RNA was extracted from cortical tissues of wild type (WT) and Clcn4 KO using TRIzol reagent (Invitrogen), and reverse transcription was performed with RNA to cDNA EcoDry Premix (Takara Bio Company, Japan). cDNA was amplified by PCR with 0.2 ng of gene-specific primers. Real-time PCR was performed using SYBR Green (Bio-Rad, Hercules, CA, USA), and the following primer pairs were used: mClcn4F, GGATTGGATGACGGACCTCA; mClcn4R, ACACTTGTCCCTGTCCTCAA; mClcn-3F, GGATTTGCCCTCAGAAGAGACC; mClcn-3R, GGACTTTCTGCTGGAAGAGATGG; mClcn-5F, GCACCTTATGCCTGTGGTTCTG; mClcn-5R, CAGGCTCAAGCCAGATGACACC; GAPDHF, AACTTTGGCATTGRGGAAGG; GAPDHR, CACATTGGGGGTAGGAACAC for mice and CLCN4F, GCTGGAGTCTCTGTTGCCTT; CLCN4R, TCCACAAGGTCTTCAGGGGA; ACTBF, GCCAACCGCGAGAAGATGAC; ACTBR, GAGGCGTACAGGGATAGCACAG for human.
Golgi staining
Golgi-Cox staining is one of the most commonly used techniques to study the normal and abnormal morphology of neurons. We used the FD Rapid GolgiStain Kit (FD NeuroTechnologies, Columbia, MD, USA). The fixed brain tissues were sectioned (100 μm) with a vibratome (VT1000S, Leica). The sections were stained with solution D/E and visualized using a confocal microscope (Olympus, Shinjuku, Japan).
Behavioral tests
Seven- to eight-week-old second-generation male mice were used. A camera was placed 2.0 m above the apparatus, and the camera was connected to a computer to record video and data via the SMART video tracking system, version 3.0.05 (Panlab Harvard Apparatus, Barcelona, Spain).
In this study, no blinding was performed during animal experiments. Detailed protocols on behavioral testing are described in the Supplementary Information (SI). Comprehensive statistical details, including specifics of the ANOVA analysis, can be found in the Supplementary Statistics file.
Drug administration
Mice were randomly assigned to the treatment groups. Risperidone (Sigma‒Aldrich, St. Louis, MO, USA) was dissolved in 25% hydroxypropyl-β-cyclodextrin (TCI, Sigma‒Aldrich) and diluted to a concentration of 1 mg/ml in saline. The mice were randomly divided into 3 groups: the control group (WT, 0.2% hydroxypropyl-β-cyclodextrin), KO group (KO, 0.2% hydroxypropyl-β-cyclodextrin) and KO-risperidone group (KO, 0.2 mg/kg risperidone in 0.2% hydroxypropyl-β-cyclodextrin). The mice were injected intraperitoneally with vehicle (25% hydroxypropyl-β-cyclodextrin diluted in Saline to 0.2%) or risperidone for 2 weeks (5 days/week). Then, after a 1 week washout period, behavior tests (Three chamber test, Marble burying test, Y-maze test, Passive avoidance test) were performed. Three chamber and Marble burying test were performed as described above.
Culture of neural progenitor cells (NPCs) and neurons derived from embryonic stem cells (ESCs)
C57BL/6 mouse embryonic stem cells (mESCs) and MEF cells (from CF-1) purchased from ATCC (SCRC-1002, SCRC-1040) were maintained on mitomycin C-treated MEFs. mESC-derived mNPCs were maintained on NPC media with FGF2, mEGF, and heparin and differentiated on NPC media with BDNF, GDNF, cAMP, retinoic acid, L-ascorbic acid, and laminin.
HUES6 human embryonic stem cells (hESCs) obtained from the HSCI iPS Core were maintained on mitomycin C-treated MEFs. MEFs were isolated from E13.5 pregnant CF-1 mouse embryos [28]. NPCs were produced as described in Kim et al. [29]. The hESC protocols were performed in accordance with the ethical requirements and regulations of the Institutional Review Board of KAIST (KH2017-109). The protocol for MEF preparation was approved by the Committee on Animal Research at KAIST (KA2020-37).
Mouse Clcn4 knockdown (KD)
A short hairpin RNA (shRNA) targeting CLCN4 was designed using the Broad Institute GPP web portal. The target sequence in Clcn4 was 5’-CGGGAGCTAATCTTGGCTATA-3’, which is in chromosome 7. The shRNA oligos were annealed and inserted into the pLKO.1 plasmid (Addgene #8453). shRNA-pLKO.1 plasmids were transfected into mNPCs using Lipofectamine. (Invitrogen, Carlsbad CA, USA) To test the effects of risperidone, 10 µM risperidone in DMSO was added to the neuronal medium every other day.
Human CLCN4 KD
We used shRNA or CRISPR-Cas9 method to knock down CLCN4 in human NPCs. An shRNA targeting CLCN4 was designed through the Broad Institute GPP Web Portal (Fig. S2a), the shRNA oligos were then annealed, and they were subsequently inserted into the pLKO.1 plasmid (Addgene #8453). The obtained lentiviral DNA plasmids were used for lentivirus production. Lentiviruses were produced and used to infect into the human NPCs [29]. Following a five-day period of viral transduction, the NPCs were split at the desired ratio and subsequently differentiated into neurons 24 h later. In CRISPR-Cas9 method, the cells were transfected with 25 nM CRISPR-Cas9 sgRNA targeting human CLCN4 (5’-GCTAATCAAGACAGTCACGC-3’) or a negative control sgRNA (Integrated DNA Technologies, San Diego, CA, USA), using the CRISPRMAX transfection reagent. Subsequently, the cells were employed in experimental procedures for 48 h post-transfection. Following a seven-day differentiation period, western blot analysis and immunohistochemistry were conducted.
RNA sequencing of mNPC
Total RNA was extracted using TRIzol reagent (Invitrogen). The RNA samples were sequenced using DNBSEQ by BGI techsolutions Co. (Hongkong). Following sequencing read filtering by BGISEQ, genome mapping was performed using HISAT [30]. Clean reads were mapped to a reference (Mus_musculus, GCF_000001635.26_GRCm38.p6) using Bowtie2 [31], and then the gene expression level was calculated with DESeq2 within R-3.5.1. Significant up and downregulated genes based on p-value < 0.01 and Fold Change > 2. A volcano plot was generated with the enhanced Volcano package in R using significant up and downregulated genes. A heatmap was visualized using all the altered genes on a bioinformatics website (http://bioinformatics.sdstate.edu/idep/). Network plots were created by selecting a subset of enriched terms for upregulated or downregulated gene lists that are connected by edges if their Kappa similarity is >0.3. To ensure clarity, we chose the terms with the best p-values from each of the 20 clusters, with a maximum of 15 terms per cluster and a total of 250terms in the plot. Each node represents an enriched term and is colored by its cluster ID.
Primary neuronal cell culture
Neuronal cultures were prepared from the cortices of WT and Clcn4 KO on embryonic day E17. The culture protocol was produced as described in Hilgenberg & Smith [32]. Experiments were performed on the day in vitro 7 or 14 (DIV7 or DIV14). To measure cell viability and morphology, WT and KO groups were treated with 0.1% DMSO, while the KO+Ris group was treated with 5 µM or 10 µM risperidone (dissolved in 0.1% DMSO) starting from DIV1. For analysis of cell viability, cells were seeded in a 24-well plate, and at DIV7, MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl 2H-tetrazolium bromide) solution was added for 1 h at 37 °C. Then, formazan crystals were dissolved by the addition of DMSO. Following incubation for 30 min, the optical density of the solubilized formazan crystals was examined by using a spectrophotometer (Molecular Devices, USA) at 580 nm.
Western blotting
Cortical and hippocampal tissues or cortical neurons were isolated from Clcn4 KO and WT mice. Western blotting is described in detail in the SI.
Immunocytochemistry
Immunocytochemistry was conducted by primary cortical neurons from mice at DIV 7 and human NPCs and neurons. An Immunocytochemistry is described in detail in the SI.
Sholl analysis
ImageJ software was used to quantify branching. Fluorescent signals were segmented using the threshold tool, and fluorescence images were converted to gray images to trace complete cells. The end of the longest branch from the soma was identified, a 10 μm diameter circle from the soma to the end of the dendrite was made, and the contact point was calculated. Branches that contacted the circle were counted. The degree of dendritic branching was determined by the number of intersections per circle.
Statistical analysis
For sample size calculation, we referred to previous papers with similar research design, then calculated the sample sizes using the G*Power program. All data were analyzed using Prism 8 software (GraphPad Software, La Jolla, CA, USA). Data normality was determined using the Shapiro-Wilk normality test. Normally distributed data were analyzed using Student’s t-test and analysis of variance (ANOVA), followed by post hoc tests, while data that failed the normality test were analyzed using the Mann-Whitney U test. Results are presented as the mean ± standard error of the mean (SEM). P-values < 0.05 were considered statistically significant. For western analysis, the first sample of the three control samples was set to 100% and the other samples were analyzed. For the gene expression data, nbinomial Wald test was used to assess difference between WT and KO, and p-value < 0.01 were considered statistically significant.
Results
Clcn4 KO showed decreased number of dendritic spines in hippocampus
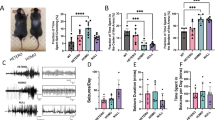
To generate Clcn4 knockout (KO) mice, exon 5 of the Clcn4 gene on chromosome 7 was deleted and we measured the mRNA expression of Clcn3, Clcn4, and Clcn5 by using real-time PCR with specific probes (Fig. 1a). There was no difference in the mRNA expression of Clcn3 (WT, 1.02 ± 0.08; KO, 1.03 ± 0.12, n = 6) and Clcn5 (WT, 1.06 ± 0.14; KO, 1.02 ± 0.09, n = 6) between wild-type (WT) and Clcn4 KO mice, but Clcn4 mRNA expression (WT, 1.03 ± 0.12; KO, 0 ± 0, p < 0.01, n = 6) was completely suppressed in Clcn4 KO mice. The distribution of pyramidal neurons and the number of spines in the hippocampus were examined through Golgi staining (Fig. 1b) at postnatal day (PND) 7. The number of dendritic spines per 10μm was decreased in Clcn4 KO compared to WT (WT, 20.2 ± 0.80; KO, 11.4 ± 0.33, n = 3, p < 0.001; Fig. 1b).

a To generate Clcn4 KO, exon 5 of the Clcn4 gene on chromosome 7 was deleted. Clcn4 mRNA expression mice were completely suppressed in Clcn4 KO when measured by qPCR. Clcn4 KO showed downregulation of Clcn4 but not Clcn3 or Clcn5. b The distribution of pyramidal neurons and the number of spines in the hippocampus were examined through Golgi staining. The number of dendritic spines per 10μm decreased in Clcn4 KO compared to WT. c The three-chamber test was performed at 8 weeks of age. In the social approach, WT mice spent significantly more time in the novel mouse (NM) -containing chamber than in the novel object (NO)-containing chamber (left panel), but there was no difference in the Clcn4 KO group. In social novelty, the WT spent a significantly longer time in the novel mouse (NM)-containing chamber than in the familiar mouse (FM)-containing chamber (right panel), but there was no difference in the Clcn4 KO group. d The marble burying test was performed at 8 weeks of age. Clcn4 KO showed a significant increase in the number of marbles buried, suggesting increased stereotypic behavior. All data were analyzed by t-test and are presented as the mean ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001).
Clcn4 KO showed ASD associated behaviors
At very early time points, we did not observe a developmental delay in motor ability in Clcn4 KO compared to WT except for a decreasing in score in the surface righting test on PND 7 (Fig. S3). To explore the effects of Clcn4 deficiency on ASD associated behaviors, the three-chamber test (WT, n = 14; KO, n = 14) and the marble burying test (WT, n = 7; KO, heterozygote n = 7, homozygote n = 7) was performed when the mice were 8 weeks of age. The three-chamber test is a method for evaluating social behavior in mouse models of ASD (Fig. 1c). In the social approach phase, WT spent significantly more time in the novel mouse (NM)- containing chamber than in the novel object (NO)-containing chamber (WT, NM 241 ± 24 s, NO 157 ± 14 s; n = 14, p < 0.01), but there was no difference in the Clcn4 KO group (KO, NM 227 ± 22 s, NO 179 ± 20 s; n = 14; Fig. 1c; left panel). Regarding social novelty, the WT mice spent a significantly longer time in the novel mouse (NM)-containing chamber than in the familiar mouse (FM)-containing chamber (WT, NM 233 ± 21 s, FM 162 ± 26 s; n = 14, p = 0.043), but there was no difference in the Clcn4 KO group (KO, NM 201 ± 22 s, FM 168 ± 27 s; n = 14, Fig. 1c; right panel). The results of this experiment indicate that there is a reduction in social interaction in Clcn4 KO compared to WT. The marble burying test is a behavioral method for repetitive behavior. We conducted the marble burying test with 7 animals but decided to present results from only 6 animals in the final data set, as one animal was identified as an outlier and excluded from the analysis. Clcn4 KO showed a significant increase in the number of marbles buried (WT, 0.83 ± 0.3; KO, heterozygote, 2.16 ± 0.83, n = 6, homozygote 3.5 ± 0.84, n = 6, WT vs Homo, p < 0.05), suggesting increased stereotypic behavior (Fig. 1d) [33].
Risperidone reversed behavioral deficits in Clcn4 KO mice
Risperidone was utilized to examine the predictive validity of the Clcn4 KO as a model for human ASD. We tested whether risperidone could reverse ASD associated behavior in Clcn4 KO. Risperidone was administered according to a predetermined schedule for 2 weeks at 5 weeks of age and validated by behavioral experiments (Fig. 2a). In Y maze test, the spontaneous alternation was significantly decreased in Clcn4 KO compared with WT, and risperidone treated Clcn4 KO showed a significant improvement in the spontaneous alternation (WT, 60.7 ± 1.8; KO, 47.4 ± 2.4; KO+Ris, 66.1 ± 2.7; n = 17; WT vs KO, p < 0.001; KO vs KO+Ris, p < 0.0001, Fig. 2b). In passive avoidance test, the latency time for dark room avoidance was significantly decreased in Clcn4 KO compared with WT, and risperidone treated Clcn4 KO showed a significant increase in the latency time for dark room avoidance (WT, 138.3 ± 32.6; KO, 15.8 ± 8.7; KO+Ris, 66.2 ± 17.2; n = 5; WT vs KO, p < 0.01; KO vs KO+Ris, p < 0.05, Fig. 2c). In the social approach phase of three chamber test, all three groups (WT, KO, KO+Ris) of mice spent significantly more time in the NM chamber than in the NO chamber (Fig. 2d, right panel). In social novelty, WT tended to spend more time in the NM-containing chamber than in the FM-containing chamber (WT, NM 274 ± 16 s, FM 191 ± 13 s; n = 8, p < 0.01), but there was no difference in the Clcn4 KO group (KO, NM 249 ± 21 s, FM 229 ± 25 s; n = 9). After risperidone treatment, Clcn4 KO spent significantly more time in the NM-containing chamber than in the FM-containing chamber. After risperidone treatment, Clcn4 KO spent significantly more time in the NM-containing chamber than in the FM-containing chamber (KO+Ris, NM 284 ± 21 s, FM 196 ± 17 s; n = 8, p < 0.01, Fig. 2d, left panel). The Clcn4 KO group also showed a significant increase in marble burying behavior, which was decreased after risperidone treatment (WT, 5.8 ± 1.0, n = 10; KO, 9.1 ± 1.2, n = 8; KO+Ris, 6.3 ± 0.5; n = 9; WT vs KO, p < 0.05; KO vs KO+Ris, p < 0.05, Fig. 2e). These results suggest that the effects of Clcn4 KO can be ameliorated by risperidone treatment, adding predictive validity to Clcn4 KO as model for human ASD.

a Risperidone was administered according to a predetermined schedule for 2 weeks. Next, we tested whether the antipsychotic drug risperidone reversed cognitive impairment and ASD associated behavior in Clcn4 KO. b In Y-maze test, the spontaneous alternation was significantly decreased in Clcn4 KO compared with WT, and risperidone treated Clcn4 KO showed a significant improvement in the spontaneous alternation. c In passive avoidance test, the latency time for dark room avoidance was significantly decreased in Clcn4 KO compared with WT, and risperidone treated Clcn4 KO showed a significant improvement in the latency time for dark room avoidance. d In the social approach phase of three chamber test, all three group (WT, KO, KO+Ris) spent significantly more time in the NM chamber than in the NO chamber (left panel). After risperidone treatment, Clcn4 KO spent significantly more time in the NM-containing chamber than in the NO-containing chamber. In social novelty, WT mice tended to spend more time in the NM-containing chamber than in the FM-containing chamber (right panel). After risperidone treatment, Clcn4 KO spent significantly more time in the NM-containing chamber than in the FM-containing chamber. e The Clcn4 KO group showed a significant increase in marble burying behavior, which decreased after risperidone treatment. All data were analyzed by t-test or one-way ANOVA are presented as the mean ± SEM. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.001).
Clcn4 KD in mNPCs showed downregulation of the neuronal development genes
To explore the effects of Clcn4 deficiency, RNA sequencing was performed on mNPCs. Among 14,208 expressed genes (FPKM > 1) in either WT or Clcn4 KD mNPC, 2446 and 2661 genes were significantly increased and decreased in Clcn4 KD mNPC compared to WT, respectively (Fig. 3a). We then analyzed the gene ontology (GO) terms and KEGG pathways of the differentially expressed genes (DEGs). The GO terms related to trans-synaptic signaling (p = 8.13E-34), the regulation of transmembrane transport (p = 6.31E-17), modulation of chemical synaptic transmission (p = 4.24E-27), neuron projection development (p = 2.99E-31), regulation of neuron projection development (p = 1.16E-21), and regulation of membrane potential (p = 1.34E-19) were significantly decreased expression in Clcn4 KD compared to WT (Fig. 3b). The GO terms related to skeletal muscle development, vascular system development, and cell adhesion were significantly enriched in genes that were increased in Clcn4 KD compared to WT (Fig. 3c).

a To explore the effects of Clcn4 deficiency, RNA sequencing was performed on mNPCs. Hierarchical clustering of three WT and KD lines based on differentially expressed RNA transcripts (2.0 FC, nbinomWaldTest test p < 0.01) from DESeq2. The expression level of each gene in a single sample is depicted according to the color scale. Volcano plot of DEGs in KD. The Y-axis displays the -log10 p-value for each gene, while the X-axis displays the log2 fold change for that gene relative to KD. b–f Top 10 enriched GO terms for DEGs. b GO term enrichment analysis for downregulated genes in WT and KD, (c) upregulated genes. d Hierarchical clustering of three KD and KD+Ris lines based on differentially expressed RNA transcripts (2.0 FC, nbinomWaldTest test p < 0.01) from DESeq2. The expression level of each gene in a single sample is depicted according to the color scale. Volcano plot of DEGs in KD. The Y-axis displays the -log10 p-value for each gene, while the X-axis displays the log2 fold change for that gene relative to KD. e GO term enrichment analysis for downregulated genes in KD and KD+Ris, f upregulated genes. Node size represents gen ratio; node color represents -log10 p-value. GO, gene ontology.
In Clcn4 KD versus Clcn4 KD+Ris comparison, 212 and 266 genes significantly increased and decreased in Clcn4 KD+Ris compared to Clcn4 KD (Fig. 3d). The GO terms related to the regulation of vascular permeability (p = 1.35E-04) and the skeletal system development (p = 1.29E-07) were decreased in Clcn4 KD+Ris compared to Clcn4 KD (Fig. 3e). The expression of genes associated with the regulation of potassium ion transmembrane transport (p = 8.71E06), the regulation of membrane potential (p = 3.31E-05), post-translational protein phosphorylation (p = 1.05E-07), and the development of neuronal projections (p = 0.0024) significantly increased in Clcn4 KD+Ris compared to Clcn4 KD (Fig. 3f).
From the gene expression data, we formulated schematic hypothesis of CLCN4 function in the synapse (Fig. S4). We postulated that CLCN4 dysfunction causes aberration in synaptic plasticity through signaling pathways such as MAPK/ERK, which are essential for processes like dendritic spine formation, protein clustering at synapses, and neurotransmitter release.
Expression and phosphorylation of synaptic proteins decreased in Clcn4 KO
We examined the protein expression and phosphorylation of SYNAPSIN 1, PSD95, CREB, ERK in Clcn4 KO. The phosphorylation of SYNAPSIN at Ser9, Ser549, and Ser609 was significantly reduced in the cortex (Ser9: WT, 100 ± 1.5; KO, 61.0 ± 6.1; Ser549: WT, 100 ± 1.2; KO, 68.1 ± 1.2; Ser609: WT, 100 ± 3.0; KO, 60.8 ± 3.0; p < 0.01, p < 0.001) and hippocampus (Ser9: WT, 100 ± 2.8; KO, 40.0 ± 6.7; Ser549: WT, 100 ± 2.8; KO, 51.3 ± 5.5; Ser609: WT, 100 ± 0.6, KO, 54.0 ± 4.9; p < 0.01, p < 0.001) of CLCN4 KO (Fig. 4a).

To investigate the expression changes of SYNAPSIN 1, PSD95, CREB and ERK along with the phosphorylation of these proteins in Clcn4 KO, the brain tissues were evaluated by immunoblotting. a There was no difference in the expression of SYNAPSIN 1 between Clcn4 KO and WT. The phosphorylation of SYNAPSIN 1 at Ser9, Ser549, and Ser609 was significantly reduced in the cortex and hippocampus in Clcn4 KO compared to WT. b In the cortex of Clcn4 KO, the expression level of PSD95 was decreased, but its phosphorylation level was not changed. However, both the phosphorylation and the expression levels of PSD95 were significantly decreased in the hippocampus of Clcn4 KO. CREB activation was markedly reduced and ERK activity decreased in the cortex and hippocampus in Clcn4 KO compared to WT. c After treatment with risperidone, the levels of p-SYNAPSIN 1 (Ser9), SYNAPSIN 1, p-PSD95, PSD95, CDK5 and β-ACTIN in the cortex of the Clcn4 KO and WT were assessed. The phosphorylation of SYNAPSIN 1 at Ser9 in the cortex was also restored in Clcn4 KO by risperidone administration, and PSD95 phosphorylation was increased in risperidone treated Clcn4 KO compared to WT and untreated Clcn4 KO. Risperidone reversed the reduction in expression of CDK5 and PSD95. All data were analyzed by t-test or one-way ANOVA are presented as the mean ± SEM. (*p < 0.05, **p < 0.01, ***p < 0.001).
In the cortices of Clcn4 KO, the protein expression level of PSD95 was decreased (WT, 100 ± 0.6; KO, 63.7 ± 4.1, p < 0.001), but its phosphorylation level was not changed (WT, 100 ± 1.5; KO, 76.2 ± 9.0). Both the phosphorylation level (WT, 100 ± 2.7; KO, 50.8 ± 2.1; p < 0.001) and the expression level of PSD95 (WT, 100 ± 4.1; KO, 68.0 ± 5.8; p < 0.05) were significantly decreased in the hippocampus of Clcn4 KO. Clcn4 KO exhibited a marked reduction in ERK activity (cortex: WT, 100 ± 1.2; KO, 33.0 ± 4.6; hippocampus: WT, 100 ± 2.9; KO, 56.1 ± 2.6; p < 0.001) and CREB (cortex: WT, 100 ± 6.8; KO, 36.9 ± 4.3, p < 0.01; hippocampus: WT, 100 ± 11.5; KO, 55.7 ± 7.3, p < 0.05) (Fig. 4b).
After treatment with risperidone, the levels of p-SYNAPSIN 1 (Ser9), SYNAPSIN 1, p-PSD95, PSD95, CDK5 and β-ACTIN in the cortex of the Clcn4 KO and WT were assessed (Fig. 4c). The phosphorylation of SYNAPSIN 1 at Ser9 in the cortex was also restored in Clcn4 KO by risperidone administration (WT, 100 ± 0.88; KO, 68.1 ± 5.8; KO+Ris, 110.0 ± 10.0; p < 0.01), and PSD95 phosphorylation was increased in risperidone treated Clcn4 KO compared to WT and untreated Clcn4 KO (WT, 100 ± 5.4; KO, 112 ± 2.5; KO+Ris, 131.6 ± 5.4; p < 0.05). Risperidone reversed the reduction in expression of CDK5 (WT, 100 ± 1.9; KO, 75.8 ± 4.3; KO+Ris, 110.7 ± 6.9; WT vs KO, p < 0.01, KO vs KO+Ris, p < 0.05) and PSD95 (WT, 100 ± 2.2; KO, 77.4 ± 1.8; KO+Ris, 107.5 ± 3.3; p < 0.01).
Clcn4 KO neurons show decreased expression of neuronal cell markers and phosphorylation of synaptic proteins
To examine the involvement of Clcn4 in neuronal cell viability and morphology, primary cortical neurons from Clcn4 KO pups were harvested and immune-stained with MAP2 (Fig. 5a). In Clcn4 deleted neurons, cell viability decreased by 50% (KO, 51.21 ± 4.54% vs WT, p < 0.001, n = 3) (Fig. 5b). Risperidone (5 and 10 μM) restored the viability of neurons from Clcn4 KO in a dose-dependent manner (KO+Ris 5 μM, 64.25 ± 5.85% vs WT, p < 0.001, KO+Ris 10 μM, 72.07 ± 2.44% vs WT, p < 0.01, n = 3; Fig. 5b). The number of MAP2-positive neurons were decreased in Clcn4 KO cortical neurons but restored after risperidone treatment to WT level (WT, 28.2 ± 1.3; KO, 23.2 ± 1.7; KO+Ris, 27.6 ± 1.6; WT vs. KO p < 0.05; KO vs KO+Ris p < 0.05, n = 14; Fig. 5c). In Sholl analysis, the distance from soma of Clcn4 deleted neurons, was significantly shorter compared to WT, but the length of the dendrites after risperidone treatment was restored to WT level (WT, 120.7 ± 12.47; KO, 57.14 ± 7.944; KO+Ris, 87.86 ± 12.54; WT vs. KO, p < 0.001, KO vs KO+Ris, p < 0.05, n = 14; Fig. 5d, e).

a Primary cortical neurons from Clcn4 KO pups were harvested and immune-stained with MAP2. b In Clcn4 deleted neurons, cell viability decreased by 50% compared to WT. Five and 10 µM risperidone restored the viability of neurons from Clcn4 KO in a dose-dependent manner. c The number of MAP2-positive neurons decreased in KO cortical neurons compared to WT but restored after risperidone treatment. d In Sholl analysis, the length of the dendrites of Clcn4 KO neurons, was significantly shorter compared to WT, but the length of the dendrites after risperidone treatment was restored to WT level. e Sholl analysis showed that the neurite complexity of Clcn4 KO neurons was decreased. Risperidone treatment restored the neurite complexity of Clcn4 KO neurons to normal levels. f SYNAPSIN 1 expression was not different between neuronal lysates from Clcn4 KO and those from WT mice at DIV7 and DIV14, but the phosphorylation of SYNAPSIN 1 at Ser9 and Ser549 was significantly reduced in neurons from Clcn4 KO at DIV14. g At DIV7, risperidone treatment increased SYNAPSIN 1 Ser9 phosphorylation in Clcn4 KO compared to untreated KO. Additionally, PSD95 expression, but not phosphorylation, was elevated in risperidone-treated KO compared to WT and untreated KO. All data were analyzed by t-test or one-way ANOVA are presented as the mean ± SEM. (*p < 0.05, **p < 0.01, ***p < 0.001).
We examined the protein expression of SYNAPSIN 1 and PSD95 in DIV7 and DIV14 cells (Fig. 5f). The phosphorylation of SYNAPSIN 1 at Ser9 and Ser549 was significantly reduced in neurons from Clcn4 KO at DIV14 (Ser9: WT, 100 ± 7.5; KO, 47.8 ± 8.5, n = 6, p < 0.001; Ser549: WT, 100 ± 9.5; KO, 69.3 ± 8.1, n = 6, p < 0.05; Fig. 5f). PSD95 expression was decreased at DIV7 but restored at DIV14. The phosphorylation level of PSD95 was not changed at DIV7 (WT, 100 ± 11.3; KO, 99.5 ± 8.2, n = 3), but it was dramatically reduced at DIV14 (WT, 100 ± 19.2; KO, 25.2 ± 7.1, n = 6, p < 0.01; Fig. 5f). Next, to confirm the recovery of SYNAPSIN 1 activity after risperidone treatment, the phosphorylation levels of SYNAPSIN 1 were examined by immunoblotting. Synapsin1 phosphorylation at Ser9 (WT, 100 ± 6.1; KO, 75.8 ± 6.3; KO+Ris, 141.2 ± 4.86; n = 3; KO vs KO+Ris, p < 0.01) was increased in risperidone treated Clcn4 KO compared with untreated Clcn4 KO. Furthermore, the expression level but not the phosphorylation level of PSD95 (WT, 97.3 ± 2.2; KO, 75.3 ± 1.8; KO+Ris, 104.7 ± 3.2; n = 3; WT vs KO, p < 0.01; KO vs KO+Ris, p < 0.01) was markedly increased in risperidone treated Clcn4 KO compared to WT and untreated Clcn4 KO (Fig. 5g).
Risperidone reversed morphological changes in human neurons resulting from CLCN4 KD
To investigate the effects of CLCN4 gene deficiency in human cells, experiments were conducted using human NPCs (hNPCs) from hESCs. In both NPCs and differentiated neurons, CLCN4 mRNA was expressed, but the expression level of CLCN4 mRNA gradually increased upon neuronal differentiation (neuron vs. NPCs at Day 3, 2.34 ± 0.34 p < 0.05; Day 6, 3.13 ± 0.33, p < 0.01; Day13, 4.04 ± 0.33, p < 0.001), indicating that the biological roles of CLCN4 are more important in neurons than in NPCs (Fig. 6a). hNPCs expressed SOX2 and NESTIN, and differentiated neurons expressed TUJ1 and MAP2 (Fig. 6b). Real-time PCR and immunocytochemistry showed that CLCN4 expression levels were decreased by CLCN4 KD (Ctrl, 1.00 ± 0.00, CLCN4 KD, 0.75 ± 0.03, p < 0.01, Fig. 6c; Ctrl, 100 ± 14.9, CLCN4 KD, 0 ± 0, p < 0.01, n = 3, Fig. 6d).

a CLCN4 mRNA expression was increased upon neuronal differentiation. b Representative images of hESC-derived NPCs and neurons. SOX2 and NESTIN were used as NPC markers, and TUJ1 and MAP2 were used as neuron markers for immunocytochemistry. c Real-time PCR showed that CLCN4 mRNA levels were decreased by CLCN4 KD. d Representative images of control (Ctrl) and CLCN4 KD. The number of CLCN4-positive neurons decreased in CLCN4 KD compared to Ctrl (e) CLCN4 KD were immunostained with MAP2 (f) Sholl analysis showed that the neurite complexity of CLCN4 KD neurons was decreased. Risperidone treatment restored the neurite complexity of CLCN4 KD to normal levels. All data were analyzed by t-test or one-way ANOVA are presented as the mean ± SEM. (Ctrl vs. shCLCN4; *p < 0.05, **p < 0.01, ***p < 0.001).
CLCN4 KD neurons showed less complex neurite structures than Ctrl (Ctrl, n = 16, CLCN4 KD, n = 24, Fig. 6f). Next, we treated CLCN4 KD with risperidone and Sholl analysis showed that risperidone treatment rescued the decrease in neurite complexity in CLCN4 KD (CLCN4 KD, n = 24, CLCN4 KD+Ris, n = 12, Fig. 6f). We also treated Ctrl neurons with risperidone and conducted Sholl analysis for Ctrl+vehicle and the Ctrl+Ris groups, and no significant differences were observed between the two groups (Fig. S5). This finding suggests that the involvement of the CLCN4 gene in neurite morphology may extend beyond animal models to human neurons. This is evidenced by the observed neurite defects in human neurons resulting from CLCN4 dysfunction.
Discussion
For the first time, we report that Clcn4 KO showed impairment of social interaction and an increase in stereotypic behavior thereby establishing face validity for ASD. In the three-chamber test, 8-week-old WT spent significantly more time in the NM-containing chamber than 8 week-old Clcn4 KO. Moreover, Clcn4 KO showed a significant increase in marble burying behavior, suggesting stereotypic behavior [33]. Previous studies have reported that Clcn4 KO do not exhibit neurological alterations in the brain [17, 18]. These findings have been known to directly conflict with reports that CLCN4 mutations cause a wide range of neurodevelopmental disorders in humans [16, 17, 34,35,36]. Mutations in CLCN4 in humans have been reported to cause neurodevelopmental disorders such as ASD, X-linked intellectual disability, and seizure disorders [17, 34,35,36,37]. It was postulated that Clcn4 KO exhibit no neurodevelopmental phenotype because Clcn3 plays the main role in the brain [11]. However, we found that Clcn4 KO on the C57BL/6 background also shows neurodevelopmental endophenotypes consistent with ASD in humans.
It should be noted that in C57BL/6 mice, Clcn4 is located on autosomal chromosome 7. In humans, Clcn4 is usually in the pseudoautosomal area, a small region on the X chromosome susceptible to rearrangement. Previous reports showed that in C57BL/6 mice complex rearrangements transferred the Clcn4 gene to the autosome on chromosome 7 [38,39,40]. We observed similar abnormalities resulting from knocking down the same Clcn4 genes on two different chromosomes in heterogeneous species, supporting the previous finding that Clcn4 was indeed transferred from its original location on the X chromosome in C57BL/6 J mice through evolutionary rearrangement [38,39,40].
ASD children have shown disruption in multiple prenatal stages, including proliferation, maturation, synaptogenesis, and neural activity [41,42,43], suggesting that the pathogenetic mechanisms that contribute to ASD are in effect during the embryonic and fetal periods [44]. To identify the molecular target of Clcn4 during the initiation stage of neuronal differentiation from NPCs, we acquired gene expression data via KD in mNPCs. The results show that KD of Clcn4 in mNPCs affected neuronal projection development and synapse organization. PSD95, SYNAPSIN 1, CDK5, and ERK, all of which are associated with synapse formation, dendritic outgrowth and neuronal development [38, 39, 45,46,47,48,49,50] were found to be significantly decreased in KD mNPCs compared to WT mNPCs. On a protein level, we were able to confirm that the protein expressions of CDK5, PSD95, ERK, CREB and SYNAPSIN 1 along with the phosphorylation levels of SYNAPSIN 1 at Ser9, Ser549, and Ser609 were significantly reduced in the cortex and hippocampus in Clcn4 KO. SYNAPSIN 1 are a family of synaptic vesicle-associated phosphoproteins that directly contribute to the fine tuning of synapse formation, maturation, and plasticity [51,52,53]. CDK5 plays regulatory roles in synaptogenesis, synaptic transmission, and plasticity [49, 54, 55]. By phosphorylating key substrates at both the presynapse and postsynapse, CDK5 acts as a key regulator of synaptic transmission in a concerted manner with the phosphatase calcineurin [56,57,58,59]. The expression level of CDK5 was reduced in the brains of Clcn4 KO, suggesting that the Clcn4 KO-induced reduction in CDK5 expression may be involved in the dephosphorylation of SYNAPSIN 1, resulting in poor synaptic transmission and spine formation.
Next, we explored the possibility of dendritic outgrowth and synapse remodeling being endophenotypic targets for drug efficacy in ASD. The gene expression findings in Clcn4 KD mNPCs are consistent with our observation in Clcn4 KO that the number of cortical dendritic spines per 10 μm and the number of dendritic branches decreased in cortical neurons from Clcn4 KO compared to WT. In individuals with ASD, there is a noticeable reduction in dendritic branching observed in the hippocampus compared to controls [60,61,62] and fewer dendrites as detected by MAP2 in the prefrontal cortex [63]. Synaptic formation and dendritic outgrowth are regulated by various extracellular factors, adhesion molecules, and cytoskeleton regulating molecules [64].
We observed that risperidone improved early development deficits along with synaptic dendritic abnormalities. Risperidone decreased stereotypic behavior in the marble burying test and improved social interaction in the three-chamber test, not only demonstrating the predictive validity of Clcn4 KO as an ASD model, but also suggesting the potential efficacy of risperidone in alleviating core symptoms of ASD when administered at a very early age. Similar findings were reported in other ASD animal models; postnatal administration of risperidone reduced anxiety, social impairment, and aggravation of stereotyped grooming in adolescent rats exposed prenatally to valproic acid, an ASD model [65]. Risperidone ameliorated striatal dopaminergic dysfunction in a Cntnap2 KO autism mouse model [66] and rescued synaptic abnormalities and repetitive behavior through attenuation of neuronal excitability and excitatory synaptic functions in the prefrontal cortex of salt-inducible kinase 1(SIK1)-truncated mice another possible ASD model, showing increased repetitive behavior, social novelty preference deficits, and an imbalance between excitatory and inhibitory synaptic functions [67]. The NMDAR antagonist MK801 has been used as a behavioral animal model of ASD [68]. Risperidone alleviates MK801-induced cognitive impairment in rats via upregulation of BDNF signaling in the hippocampus [69].
We also observed that risperidone restored the number of MAP2-positive neuronal cells and the number of dendritic branches in primary cortical neurons from Clcn4 KO. The expression level of PSD95 and CDK5 and the phosphorylation level of Synapsin were restored after 2-week treatment of risperidone in Clcn4 KO brains. Furthermore, risperidone reversed morphological changes in human neurons resulting from CLCN4 KD, in accordance with observations from the Clcn4 KO mouse brain, suggesting that our findings can be extrapolated to humans. These findings are consistent with previous reports showing that antipsychotics promote neurite outgrowth [47] and neural plasticity [48] through phosphorylation of PSD95 by CDK5-mediated TrkB phosphorylation. TrkB activity stimulates the ERK and PI3K/AKT pathways, representing a potential therapeutic target for psychiatric conditions [70,71,72]. The activity of SYNAPSIN is tightly regulated by several proteins and phosphatases, such as CDK5, which modulates the association of SYNAPSIN with synaptic vesicles, actin filaments, and other synaptic proteins [53]. Deletion of SYNAPSIN 1 leads to severely dispersed synaptic vesicles and a considerably reduced total number of synaptic vesicles within the presynapse [73].
Our results suggest that CLCN4 dysfunction contributes to synaptic and dendritic abnormalities associated with ASD. Clcn4 KO exhibited reduced social interaction and increased repetitive behaviors, both improved by risperidone. RNA-seq in Clcn4 KD mNPCs revealed decreased expression of genes related to synaptic signaling and neuron projection development, with risperidone reversing these effects. Synaptic protein phosphorylation and dendritic complexity were also reduced in Clcn4 KO brains and neurons but restored following risperidone treatment. These data suggest that dendritic outgrowth and synapse remodeling may serve as endophenotypic targets for drug efficacy in ASD.
Data availability
Raw data can be provided for investigations from JH and YK.
References
CDC, P. C. Autism and developmental disabilities monitoring (ADDM) network data & statistics on autism spectrum disorder. https://www.cdc.gov/autism/addm-network/index.html. Accessed 16 May 2024.
Edition, F. Diagnostic and statistical manual of mental disorders. Am Psychiatric Assoc. 2013;21:591–643.
Almandil NB, Alkuroud DN, AbdulAzeez S, AlSulaiman A, Elaissari A, Borgio JF. Environmental and genetic factors in autism spectrum disorders: special emphasis on data from Arabian studies. Int J Environl Res Public Health. 2019;16:658.
Daghsni M, Rima M, Fajloun Z, Ronjat M, Brusés JL, M’rad R, et al. Autism throughout genetics: perusal of the implication of ion channels. Brain Behavior. 2018;8:e00978.
Lee J, Ha S, Ahn J, Lee S-T, Choi JR & Cheon K-A. The role of ion channel related genes in autism spectrum disorder: a study using next-generation sequencing. Front Gene. 2021;12:595934.
Gascoigne DA, Drobyshevsky A, Aksenov DP. The contribution of dysfunctional chloride channels to neurovascular deficiency and neurodegeneration. Front Pharmacol. 2021;12:754743.
Huang Y, Wang Q, Peng Y, Du W, Wang Q, Qi J, et al. Spatiotemporal expression patterns of genes coding for plasmalemmal chloride transporters and channels in neurological diseases. Mol Brain. 2023;16:30.
Weinert S, Gimber N, Deuschel D, Stuhlmann T, Puchkov D, Farsi Z, et al. Uncoupling endosomal CLC chloride/proton exchange causes severe neurodegeneration. EMBO J. 2020;39:e103358.
Mohammad-Panah R, Ackerley C, Rommens J, Choudhury M, Wang Y, Bear CE. The chloride channel ClC-4 co-localizes with cystic fibrosis transmembrane conductance regulator and may mediate chloride flux across the apical membrane of intestinal epithelia. J Biol Chem. 2002;277:566–74.
Mohammad-Panah R, Harrison R, Dhani S, Ackerley C, Huan L-J, Wang Y, et al. The chloride channel ClC-4 contributes to endosomal acidification and trafficking. J Biol Chem. 2003;278:29267–77.
Guzman RE, Bungert-Plümke S, Franzen A, Fahlke C. Preferential association with ClC-3 permits sorting of ClC-4 into endosomal compartments. J Biol Chem. 2017;292:19055–65.
Jentsch TJ. CLC chloride channels and transporters: from genes to protein structure, pathology and physiology. Critical Rev Biochem Mol Biol. 2008;43:3–36.
Martineau M, Guzman RE, Fahlke C, Klingauf J. VGLUT1 functions as a glutamate/proton exchanger with chloride channel activity in hippocampal glutamatergic synapses. Nat Commun. 2017;8:1–13.
Yu Y, Tsai M-F, Yu W-P, Chen T-Y. Modulation of the slow/common gating of CLC channels by intracellular cadmium. J General Physiol. 2015;146:495–508.
Hur J, Jeong H, Park J, Jeon S. Chloride channel 4 is required for nerve growth factor-induced TrkA signaling and neurite outgrowth in PC12 cells and cortical neurons. Neuroscience. 2013;253:389–97.
Palmer E, Stuhlmann T, Weinert S, Haan E, Van Esch H, Holvoet M, et al. De novo and inherited mutations in the X-linked gene CLCN4 are associated with syndromic intellectual disability and behavior and seizure disorders in males and females. Mol Psychiatry. 2018;23:222–30.
Hu H, Haas SA, Chelly J, Van Esch H, Raynaud M, de Brouwer AP, et al. X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol Psychiatry. 2016;21:133–48.
Rickheit G, Wartosch L, Schaffer S, Stobrawa SM, Novarino G, Weinert S, et al. Role of CIC-5 in renal endocytosis is unique among CIC exchangers and does not require PY-motif-dependent ubiquitylation. J Biol Chem. 2010;285:17595–603.
Xiong Y, Chen J, Li Y. Microglia and astrocytes underlie neuroinflammation and synaptic susceptibility in autism spectrum disorder. Front Neurosci. 2023;17:1125428.
Picollo A. Vesicular CLC chloride/proton exchangers in health and diseases. Front Pharmacol. 2023;14:1295068.
Vithayathil J, Pucilowska J, Landreth GE. ERK/MAPK signaling and autism spectrum disorders. Prog Brain Res. 2018;241:63–112.
Rai SN, Dilnashin H, Birla H, Singh SS, Zahra W, Rathore AS, et al. The role of PI3K/Akt and ERK in neurodegenerative disorders. Neurotox Res. 2019;35:775–95.
Meyza KZ, Defensor EB, Jensen AL, Corley MJ, Pearson BL, Pobbe RL, et al. The BTBR T+ tf/J mouse model for autism spectrum disorders–in search of biomarkers. Behav Brain Res. 2013;251:25–34.
Khanal P, Hotulainen P. Dendritic spine initiation in brain development, learning and diseases and impact of BAR-domain proteins. Cells. 2021;10:2392.
Zhong H, Xing C, Zhou M, Jia Z, Liu S, Zhu S, et al. Alternating current stimulation promotes neurite outgrowth and plasticity in neurons through activation of the PI3K/AKT signaling pathway: neurite growth enhanced by alternating current stimulation via PI3K/AKT pathway. Acta Biochim Biophysica Sinica. 2023;55:1718.
Food and Drug Administration approval for risperdal (Risperidone) in treatment of the irritability associated with autistic disorder. 2006; https://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/020272Orig1s036,s041,020588Orig1s024,s028,s029,21444Orig1s008,s015.pdf.
Kent JM, Kushner S, Ning X, Karcher K, Ness S, Aman M, et al. Risperidone dosing in children and adolescents with autistic disorder: a double-blind, placebo-controlled study. J Autism Dev Dis. 2013;43:1773–83.
Son G, Na Y, Kim Y, Son J-H, Clemenson GD, Schafer ST, et al. miR-124 coordinates metabolic regulators acting at early stages of human neurogenesis. Commun Biol. 2024;7:1393.
Kim S-G, Lee S, Kim Y, Park J, Woo D, Kim D, et al. Tanc2-mediated mTOR inhibition balances mTORC1/2 signaling in the developing mouse brain and human neurons. Nat Commun. 2021;12:1–16.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60.
Langdon WB. Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. BioData Mining. 2015;8:1–7.
Hilgenberg LG & Smith MA. Preparation of dissociated mouse cortical neuron cultures. J Vis Exp. 2007;10:562.
Pogorelov VM, Rodriguiz RM, Insco ML, Caron MG, Wetsel WC. Novelty seeking and stereotypic activation of behavior in mice with disruption of the Dat1 gene. Neuropsychopharmacology. 2005;30:1818–31.
Scheel O, Zdebik AA, Lourdel S, Jentsch TJ. Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature. 2005;436:424–7.
Picollo A, Pusch M. Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature. 2005;436:420–3.
Veeramah KR, Johnstone L, Karafet TM, Wolf D, Sprissler R, Salogiannis J, et al. Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia. 2013;54:1270–81.
Xu X, Lu F, Zhang L, Li H, Du S, Tang J. Novel CLCN4 variant associated with syndromic X-linked intellectual disability in a Chinese girl: a case report. BMC Pediatrics. 2021;21:1–5.
Guzman RE, Sierra-Marquez J, Bungert-Plümke S, Franzen A & Fahlke C. Functional characterization of CLCN4 variants associated with X-linked intellectual disability and epilepsy. Front Mol Neurosci. 2022;15:872407.
Perry J, Palmer S, Gabriel A, Ashworth A. A short pseudoautosomal region in laboratory mice. Genome Res. 2001;11:1826–32.
Rugarli EI, Adler DA, Borsani G, Tsuchiya K, Franco B, Hauge X, et al. Different chromosomal localization of the Clcn4 gene in Mus spretus and C57BL/6J mice. Nat Genet. 1995;10:466–71.
Courchesne E, Gazestani VH, Lewis NE. Prenatal origins of ASD: the when, what, and how of ASD development. Trend Neurosci. 2020;43:326–42.
DeRosa BA, El Hokayem J, Artimovich E, Garcia-Serje C, Phillips AW, Van Booven D, et al. Convergent pathways in idiopathic autism revealed by time course transcriptomic analysis of patient-derived neurons. Sci Rep. 2018;8:8423.
Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell. 2015;162:375–90.
Prem S, Millonig JH & DiCicco-Bloom E. Dysregulation of neurite outgrowth and cell migration in autism and other neurodevelopmental disorders. Adv Neurobiol. 2020;25:109–53.
Pao P-C, Tsai L-H. Three decades of Cdk5. J Biomed Sci. 2021;28:1–17.
Yang F, Kaul R, Alkan C, Antonellis A, Friery KF, Zhu B, et al. Clcn4-2 genomic structure differs between the X locus in Mus spretus and the autosomal locus in Mus musculus: AT motif enrichment on the X. Genome Res. 2011;21:402–9.
Johnstone AL, Reierson GW, Smith RP, Goldberg JL, Lemmon VP, Bixby JL. A chemical genetic approach identifies piperazine antipsychotics as promoters of CNS neurite growth on inhibitory substrates. Mol Cell Neurosci. 2012;50:125–35.
Tendilla-Beltrán H, Meneses-Prado S, Vázquez-Roque RA, Tapia-Rodríguez M, Vázquez-Hernández AJ, Coatl-Cuaya H, et al. Risperidone ameliorates prefrontal cortex neural atrophy and oxidative/nitrosative stress in brain and peripheral blood of rats with neonatal ventral hippocampus lesion. J Neurosci. 2019;39:8584–99.
Hawasli AH, Bibb JA. Alternative roles for Cdk5 in learning and synaptic plasticity. Biotechnol J Healthcare Nutrition Technol. 2007;2:941–8.
Shah K, Rossie S. Tale of the good and the bad Cdk5: remodeling of the actin cytoskeleton in the brain. Mol Neurobiol. 2018;55:3426–38.
Jovanovic JN, Sihra TS, Nairn AC, Hemmings HC, Greengard P, Czernik AJ. Opposing changes in phosphorylation of specific sites in synapsin I during Ca2+-dependent glutamate release in isolated nerve terminals. J Neurosci. 2001;21:7944–53.
Onofri F, Giovedı̀ S, Kao H-T, Valtorta F, Borbone LB, De Camilli P, et al. Specificity of the binding of synapsin I to Src homology 3 domains. J Biol Chem. 2000;275:29857–67.
Cesca F, Baldelli P, Valtorta F, Benfenati F. The synapsins: key actors of synapse function and plasticity. Prog Neurobiol. 2010;91:313–48.
Guan J-S, Su SC, Gao J, Joseph N, Xie Z, Zhou Y, et al. Cdk5 is required for memory function and hippocampal plasticity via the cAMP signaling pathway. PloS One. 2011;6:e25735.
Barnett DG, Bibb JA. The role of Cdk5 in cognition and neuropsychiatric and neurological pathology. Brain Res Bull. 2011;85:9–13.
Cousin MA, Robinson PJ. The dephosphins: dephosphorylation by calcineurin triggers synaptic vesicle endocytosis. Trend Neurosci. 2001;24:659–65.
Tan TC, Valova VA, Malladi CS, Graham ME, Berven LA, Jupp OJ, et al. Cdk5 is essential for synaptic vesicle endocytosis. Nat Cell Biol. 2003;5:701–10.
Tomizawa K, Sunada S, Lu Y-F, Oda Y, Kinuta M, Ohshima T, et al. Cophosphorylation of amphiphysin I and dynamin I by Cdk5 regulates clathrin-mediated endocytosis of synaptic vesicles. J Cell Biology. 2003;163:813–24.
Evergren E, Benfenati F, Shupliakov O. The synapsin cycle: a view from the synaptic endocytic zone. J Neurosci Res. 2007;85:2648–56.
Phillips M, Pozzo-Miller L. Dendritic spine dysgenesis in autism related disorders. Neurosci Lett. 2015;601:30–40.
Herms J, Dorostkar MM. Dendritic spine pathology in neurodegenerative diseases. Annual Rev Pathol Mech Dis. 2016;11:221–50.
Martínez‐Cerdeño V. Dendrite and spine modifications in autism and related neurodevelopmental disorders in patients and animal models. Dev Neurobiol. 2017;77:393–404.
Mukaetova‐Ladinska E, Arnold H, Jaros E, Perry R, Perry E. Depletion of MAP2 expression and laminar cytoarchitectonic changes in dorsolateral prefrontal cortex in adult autistic individuals. Neuropathol Appl Neurobiol. 2004;30:615–23.
Reiner O, Karzbrun E, Kshirsagar A, Kaibuchi K. Regulation of neuronal migration, an emerging topic in autism spectrum disorders. J Neurochem. 2016;136:440–56.
Atia AA, Ashour RH, Zaki MM, Rahman KM & Ramadan NM. The comparative effectiveness of metformin and risperidone in a rat model of valproic acid-induced autism, potential role for enhanced autophagy. Psychopharmacology 2023;240(6):1313–32.
Peñagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. 2011;147:235–46.
Badawi M, Mori T, Kurihara T, Yoshizawa T, Nohara K, Kouyama-Suzuki E, et al. Risperidone mitigates enhanced excitatory neuronal function and repetitive behavior caused by an ASD-Associated Mutation of SIK1. Front Mol Neurosci. 2021;14:706494.
Saunders JA, Gandal MJ, Roberts TP, Siegel SJ. NMDA antagonist MK801 recreates auditory electrophysiology disruption present in autism and other neurodevelopmental disorders. Behav Brain Res. 2012;234:233–7.
Yu W, Zhu M, Fang H, Zhou J, Ye L, Bian W, et al. Risperidone reverses the downregulation of BDNF in hippocampal neurons and MK801-induced cognitive impairment in rats. Front Behav Neurosc. 2019;13:163.
Alonso M, Medina JH, Pozzo-Miller L. ERK1/2 activation is necessary for BDNF to increase dendritic spine density in hippocampal CA1 pyramidal neurons. Learn Mem. 2004;11:172–8.
Yuan P, Zhou R, Wang Y, Li X, Li J, Chen G, et al. Altered levels of extracellular signal-regulated kinase signaling proteins in postmortem frontal cortex of individuals with mood disorders and schizophrenia. J Affect Dis. 2010;124:164–9.
Enriquez-Barreto L, Morales M. The PI3K signaling pathway as a pharmacological target in Autism related disorders and Schizophrenia. Mol Cell Therap. 2016;4:1–12.
Baldelli P, Fassio A, Valtorta F, Benfenati F. Lack of synapsin I reduces the readily releasable pool of synaptic vesicles at central inhibitory synapses. J Neurosci. 2007;27:13520–31.
Acknowledgements
This research was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI18C1077), and a research grant (NRF-2018R1A2B6003640, 2022R1A2C3010655) from the National Research Foundation of Korea.
Author information
Authors and Affiliations
Contributions
YK, SJ, and JH jointly conceived and designed the study. SML, YC, HJJ, SJ, HJC, YHD, and BL performed animal behavior analysis, histological analysis and primary culture and analyzed the data. SML, YC, DK, HJJ, and SJ performed real-time reverse transcriptase polymerase chain reaction (RT‒PCR) and Western blotting. DK and YC designed KD hNPC, performed the human neuron experiment and analyzed the data. SML, YC, DK, JH, SJ, JMO, and YK wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All methods in this study were conducted in accordance with relevant guidelines and regulations. The protocol for MEF preparation was approved by the Institutional Animal Care and Use Committees of KAIST (KA2020-37). All animal experiments were approved by the National Center for Mental Health (NCMH-1711-003-003-05, NCMH-1808-003-001-02) and Dongguk University (IACUC-2020-001-1). The hESC protocols and waiver of informed consent were approved by the Institutional Review Board of KAIST (KH2017-109) and Dongguk University International Hospital (2020-04-016).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lee, S.M., Choi, Y., Kim, D. et al. Developmental deficits, synapse and dendritic abnormalities in a Clcn4 KO autism mice model: endophenotypic target for ASD. Transl Psychiatry 15, 28 (2025). https://doi.org/10.1038/s41398-024-03201-6
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41398-024-03201-6
This article is cited by
-
FOXG1 Hierarchically Shapes Synaptic Functions in Striatal iSPNs and Contributes to ASD Etiology
Neuroscience Bulletin (2026)
-
Prenatal Exposure To Valproic Acid Induces Increased Autism-Like Behaviors and Impairment of Learning and Memory Functions in Rat Offspring by Upregulating ADAM10 Expression
Neurochemical Research (2025)
-
Targeting synaptic plasticity and acetylcholine dysregulation in the medial prefrontal cortex: Rosmarinic acid attenuates Autism-like phenotypes in Shank3B−/− mice via the CREB/BDNF pathway
Psychopharmacology (2025)
