
Abstract
The chromosome 15q11.2 locus is deleted in 1.5% of patients with genetic epilepsy and confers a risk for intellectual disability and schizophrenia. Individuals with this deletion demonstrate increased cortical thickness, decreased cortical surface area and white matter abnormalities. Human induced pluripotent stem cell (iPSC)-derived neural progenitor cells from 15q11.2 deletion individuals exhibit early adhesion junction and migration abnormalities, but later neuronal development and function have not been fully assessed. Imaging studies indicating altered structure and network connectivity in the anterior brain regions and the cingulum suggest that in addition to alterations in neural progenitor dynamics, there may also be structural and functional changes within discrete networks of neurons. To explore this, we generated human forebrain cortical neurons from iPSCs derived from individuals with or without 15q11.2 deletion and used longitudinal imaging and multielectrode array analysis to evaluate neuronal development over time. 15q11.2 deleted neurons exhibited fewer connections and an increase in inhibitory neurons. Individual neurons had decreased neurite complexity and overall decreased neurite length. These structural changes were associated with a reduction in multiunit action potential generation, bursting and synchronization. The 15q11.2 deleted neurons also demonstrated specific functional deficits in glutamate- and GABA-mediated neuronal network activity and synchronization with a delay in the maturation of the inhibitory response to GABA. These data indicate that deletion of the 15q11.2 region is sufficient to impair the structural and functional maturation of cortical neuron networks, and suggest an in vitro correlate to the pathologic changes in humans with the 15q11.2 deletion.
Similar content being viewed by others

Introduction
Neurodevelopmental and neuropsychiatric disorders such as epilepsy, schizophrenia, autism and intellectual disability can be acquired but are most commonly caused by genetic changes that may be inherited or occur de novo. Among inherited forms, copy number variations (CNVs) are common [1,2,3,4]. These can involve large deletions or duplications affecting many genes and regulatory sequences, making mechanistic study and predictions difficult using standard models. However, the use of induced pluripotent stem cell derived cells (iPSCs) from individuals bearing these changes provides a mechanism to uncover changes responsible for altered neuronal development and excitability. A CNV in the 15q11.2 region in humans is implicated in both neurodevelopmental and neuropsychiatric disorders [5,6,7,8,9]. Microdeletion between breakpoints 1 and 2 results in haploinsufficiency of 4 non-imprinted genes adjacent to the imprinted regions involved in Prader Willi and Angelman syndromes, nonimprinted in Prader-Willi/Angelman syndrome-1 (NIPA1), nonimprinted in Prader-Willi/Angelman syndrome-2 (NIPA2), tubulin gamma complex-associated protein-5 (TUBGCP5) and Cytoplasmic fragile X mental retardation-interacting protein-1 (CYFIP1) [10]. This microdeletion is found in up to 1.4% of patients with generalized epilepsy, making it one of the most common CNVs identified in epilepsy [3, 8, 11]. 15q11.2 haploinsufficiency also confers risk for schizophrenia and intellectual disability [1, 5, 6, 12]. Confounding the characterization of the effects of the deletion on development is the fact that in families which have multiple family members with the deletion, there is significant heterogeneity in the presentation, with some severely affected children inheriting the deletion from more mildly affected or unaffected parents [5]. However, independent of neuropsychiatric diagnoses, the 15q11.2 microdeletion has been shown to affect overall IQ [6] as well as the presence of dyscalculia and dyslexia [7].
Structural analyses of brain magnetic resonance imaging studies (MRIs) of individuals with the 15q11.2 microdeletion have revealed reduced surface area and increased cortical thickness predominately in the frontal lobe, anterior cingulate and the precentral and postcentral gyri, as well as decreased volume of the nucleus accumbens compared to controls, even in the absence of clinically apparent disease [13]. Analysis of white matter structure using diffusion tensor imaging (DTI) studies demonstrates that 15q11.2 deletion carriers also have increased fractional anisotropy (FA) in the internal capsule and the hippocampal portion of the cingulum and decreased FA in the posterior thalamic radiation compared to controls, with the latter being associated with worse cognitive function [14]. Alterations in white matter tract microstructure detected by DTI can reflect changes in myelination or in the structure, packing density or branching of axons [15]. It is unknown which process is represented by the DTI changes in individuals with 15q11.2 deletions and the developmental and cellular mechanisms that lead to them are unknown.
Human induced pluripotent stem cells (hiPSCs) harboring the 15q11.2 microdeletion exhibit dysregulation of adherens junctions in neural progenitor cells (NPCs) in early neuronal development [16]. This has been attributed specifically to haploinsufficiency of CYFIP1, one of the four genes on this locus. Notably, deletion of Cyfip1 in mice results in ectopic embryonic NPCs [16], as well as disruption of the adult neurogenic niche in the subventricular zone and altered proliferation of NPCs in adult mice [17]. CYFIP1 interacts with the WAVE regulatory complex that functions in actin nucleation and polymerization required for cell motility, cell polarity, neurite outgrowth and synaptic remodeling [18,19,20,21,22,23]. It also binds the Fragile X mental retardation protein (FMRP) and is required for FMRP-mediated activity-dependent translation repression at synapses [24, 25]. Loss of Cyfip1 in rodents leads to multiple behavioral changes that are consistent with the positive and negative symptoms of schizophrenia [25,26,27,28]. Further, loss of Cyfip1 leads to alterations in white matter structure in rodents [27, 28]. This phenotypic overlap between CYFIP1-deficient rodents and 15q11.2 deletion harboring humans, as well as impaired neurogenesis in 15q11.2 deletion iPSCs, suggests that loss of CYFIP1 may be a primary contributor to the alterations in neuronal activity in these patients. However, the structure and function of 15q11.2 deletion neurons in later stages of development have not been determined.
Given the variable phenotypes and incomplete penetrance observed in individuals with 15q11.2 deletions, a fundamental question is whether this deletion is sufficient to bring about the altered neuronal activity and connectivity present in patients with intellectual disability, schizophrenia and epilepsy. To address this question, we used confocal microscopy and multielectrode array (MEA) analysis to examine the structural and functional development of human iPSCs derived from three 15q11.2 deletion harboring donors as well as six healthy controls without 15q11.2 deletion over extended periods of time in culture. The results indicate that 15q11.2 deletion neurons have structural changes including decreased neurite arborization, an increase in the proportion of inhibitory neurons within cultures and deficits in electrophysiologic maturation. We hypothesize that such changes predispose 15q11.2 deletion individuals to the macroscopic structural changes and the neuropsychiatric phenotypes of 15q11.2 deletion individuals.
Methods
Differentiating hiPSCs into cortical NPCs
Human induced pluripotent stem cells (hiPSCs) were maintained in Essential 8 media (ThermoFisher Scientific, Waltham, MA, USA) as described previously [29]. hiPSC derived cortical neural progenitor cells (NPCs) were generated from either control or 15q11.2 deletion bearing patient lines (Supplementary Table 1) using the human forebrain cortical differentiation protocol as adapted from Wen et al. 2014 [30]. On day in culture (DIC) 1, hiPSC plates were cleaned and were lifted from the plate with 1 mg/mL collagenase (ThermoFisher) in DMEM/F12 media (Corning, Corning, NY, USA) for 1 h at 37 °C. The colonies were transferred to a 15 mL conical tube, allowed to settle to the bottom of the tube by gravity and the media was aspirated. Colonies were then gently resuspended in DA media (DMEMF12 (-) L – Glutamine (Corning), 20% Knockout Serum Replacement, 1:100 Glutamax, 1:100 Pen-Strep, 1:100 Non Essential Amino Acids (NEAA), 1:1000 β-mercapthoethanol (ThermoFisher), 2 µM Dorsomorphin, 2 µM A83-01 (StemCell Technologies, Cambridge, MA, USA)) supplemented with 20 µM Rho-associated coiled-coil forming protein serine/threonine kinase inhibitor (ROCK-I, compound Y-27632) (ThermoFisher) and transferred to a 6-well ultra-low attachment plate (Corning) to form embryoid bodies (EBs) in suspension. On DIC 2–4, EBs were washed and media was fully changed with DA media. On DIC 5, media was changed to NPC media (DMEMF12 (-) L - Glutamine, 1:100 Glutamax, 1:100 N2 supplement (ThermoFisher), 2 µM Cyclopamine (StemCell Technologies), 2 µg/mL Heparin) for 48 h. On Day 7, the EBs were transferred in fresh NPC media to plates coated with 2% Matrigel (Corning) for 24 h at 37 °C. Partial NPC media exchanges were performed every other day until DIC 21. On DIC 22, the resulting neural rosettes were washed with phosphate buffered saline (PBS) and incubated in StemDiff dissociation media (ThermoFisher) for 1 h at 37 °C. Rosettes were lifted using pipet pressure application of pre-warmed DMEM-F12 directed at the center of the rosette and transferred to a six-well ultra-low adhesion plate and incubated overnight (16–20 h) at 37 °C in NPC media supplemented 1:100 with B27 Supplement (ThermoFisher). On DIC 23, rosettes were transferred to StemPro Accutase media (ThermoFisher) for 10 min at 37 °C before trituration to dissociate to single cells followed by centrifugation at 350 x g for 5 min. NPCs were pelleted and resuspended in neuronal media (Neurobasal media (GIBCO-ThermoFisher Scientific), Glutamax, nonessential amino acids, B27 supplement, 10 ng / mL BDNF and 10 ng / mL GDNF (Preprotech – ThermoFisher) and transferred to cryovials containing DMSO (Millipore Sigma, Darmstadt, Germany) for a final DMSO concentration of 10% DMSO. Cryovials were transferred to −80 °C in sealed Styrofoam for a minimum of 24 h prior to storage in liquid nitrogen.
Neuronal differentiation
Liquid nitrogen stocks of DIC 23 NPCs were diluted in 10 mL prewarmed Neurobasal media after brief incubation in a 37 °C water bath, centrifuged at 350 x g for 5 min and resuspended in 10 mLs of neuronal media supplemented with 20 μM of ROCK-I (1:500 DMSO) and plated on a 10 cm plate coated with poly-L-ornithine (PLO) and laminin as previously described [29]. After 48 h, ROCK-I was removed, and Neuronal media supplemented with 125 nM of the notch inhibitor, Compound E (StemCell Technologies, Vancouver, Canada) was exchanged every 48–72 h with every other media change containing 1 ug/mL laminin (ThermoFisher). After 7–10 days differentiation, neuronal plates were treated for 48 h with 0.2 uM cytosine arabinoside (ARA-C) in neuronal media to reduce actively proliferating cells.
Neuron and astrocyte co-culture
DIC 37–45 neurons and cortical rat astrocytes at passage 7–8 [31] were washed once in PBS and then lifted with 0.05% trypsin (ThermoFisher), centrifuged at 350 x g for 5 min and resuspended in 1 mL neuronal medium supplemented with compound E, 1:100 antibacterial – antifungal (Anti-Anti) (ThermoFisher) and 5% fetal bovine serum (FBS) (MEA-FBS media) with 20 µM ROCK-I. Neurons were counted and resuspended at a density of 10,000 cells / µL and astrocytes resuspended at 5000 cells / µL. For multielectrode array experiments, neurons and astrocytes were combined at a volume ratio of 1:1 (cell count ratio of 2:1) and plated as a 5 µl droplet to the center of a PLO and laminin-coated electrode array in an Axion Lumos 24-well MEA plate (Axion Biosystems, Atlanta, GA, USA). For imaging experiments, cells were plated at 50,000 neurons and 25,000 astrocytes per coverslip onto washed 30 mm #1 German borosilicate coverslips coated with PLO and laminin per well of a 24-well plate. For each coverslip, 10 µL of the cell suspension was mixed with 40 µL of MEA media with 20 µM ROCK – I and placed on the center of the coverslip. After a 30-min incubation at 37 °C 5% CO2, the wells of both the MEA plates and the 24-well coverslip plates were flooded with 500 µL MEA-FBS media added in 2 – 250 µL parts to each side of the well.
Cell transfection and viral infection
Plasmid DNA from the pAAV-hSyn-EGFP construct (Addgene, Watertown, MA, USA) was diluted into 50 µL of Opti-MEM Reduced Serum Media (ThermoFisher) and mixed gently. Lipofectamine 2000 (ThermoFisher) was mixed into another 50 µL of Opti-MEM medium and incubated for 5 min at room temperature. The diluted DNA and diluted Lipofectamine 2000 were mixed gently and incubated at room temperature for 20 min. 100 µL of the mixture was added to each well and incubated in a 37 °C incubator for 24 h before a complete media change was performed. Neurons were transfected 3–7 days after co-culture.
For viral infection, pAAV-CaMKIIa-hChR2(H134R)-EYFP was a gift from Karl Deisseroth [32] (Addgene viral prep # 26969-AAV9; http://n2t.net/addgene:26969; RRID:Addgene_26969). pAAV-mDlx-GFP-Fishell-1 was a gift from Gordon Fishell [33] (Addgene viral prep # 83900-AAV9; http://n2t.net/addgene:83900 ; RRID:Addgene_83900). Viral preps were generated by Addgene, thawed on ice prior to use, and added directly to cell culture media of DIC 90 cultures at 5 × 1013 GC / 50,000 neurons. Virus was left in media for 48 h prior to exchanging 3 times to wash out virus.
Multi-electrode array (MEA) recording and drug treatment
MEA recordings were obtained on a Maestro Edge multi-well MEA system (Axion Biosystems) with environmental control set to 37 °C, 5% CO2. On the day of recording plates allowed to equilibrate for 5 min prior to recording. For longitudinal recordings of spontaneous activity, plates were recorded for 5 min. The entire 5-min record was used for analysis. Raw voltage was sampled at 12.5 Hz and filtered with a 200 Hz high pass and 3 kHz low pass Butterworth filter. Spikes were defined as adaptive threshold crossings of greater than six times the standard deviation. Bursts were defined by a minimum of 5 spikes on a single electrode with a maximum inter spike interval (ISI) of 100 ms and network bursts were defined by an envelope threshold with a threshold factor of 1.25 with a minimum inter-burst interval of 100 ms, minimum of 35% of electrodes included and 75% of bursts [34]. Weighted mean firing rate (WMFR) (the mean firing rate averaged across active electrodes), the burst percentage (the number of spikes occurring in bursts divided by the total number of spikes multiplied by 100) and synchronization (area under normalized cross correlation - AUNCC) [35] were analyzed. For drug application, a minimum of 2 min baseline recording was obtained prior to manually pipetting 20 µL of either warmed media or warmed media with DMSO vehicle while continuous recording continued for 3 min prior to adding 20 µL of warmed media containing sufficient drug to achieve the final desired concentration of each drug. Recording continued for another 3 min prior to removing plates and washing out drugs. The drug-dependent effects were determined by the percent change in activity compared to the vehicle application. Bicuculline (Millipore-Sigma) was diluted in DMSO to a stock concentration of 20 mM and used at a final concentration of 10 µM (1:2000 DMSO dilution). CNQX (6-cyano-7-nitroquinoxaline-2,3-dione) (ThermoFisher) was diluted in DMSO to a concentration of 50 mM and used at a working concentration of 50 µM (1:1000 DMSO dilution). Both reagents were stored at – 80 °C. For analysis of percent change in response to drug application, 2 min of spontaneous activity after drug application was compared to the spontaneous activity after media or vehicle application.
Quantitative reverse transcription PCR of RNA transcripts
Co-cultures of human iPSC-derived neurons and rat primary astrocytes were maintained in 24-well plates under identical conditions, densities, and time points as those used for immunostaining and in parallel experiments. At DIC 141, cells were lysed and homogenized using TRIzol™ reagent (Invitrogen). Total nucleic acid was extracted with the RNeasy Mini Kit (Qiagen, 74104), and its quality and quantity were assessed using a NanoDrop spectrophotometer (Thermo Fisher Scientific). Contaminating DNA was removed via DNase I digestion, following the manufacturer’s instructions (Invitrogen).
Total RNA was then reverse-transcribed into complementary DNA (cDNA) using the iScript™ cDNA synthesis kit (Bio-Rad) per the manufacturer’s protocol. Human-specific primers (Supplementary Table 2) were purchased from OriGene, verified for human specificity via BLAST analysis, and tested prior to use. Quantitative PCR was performed using the SYBR Green KiCqStart™ qPCR Master Mix (Millipore Sigma) in four technical replicates per sample. Four control conditions and three disease conditions were analyzed. Gene expression levels were normalized to the housekeeping gene (Homo sapiens ribosomal protein S18 (RPS18)) and the mean of control conditions using the comparative Ct method, as previously described [36].
Immunocytochemistry
Cultures grown on coverslips were washed once with PBS, fixed with 4% paraformaldehyde for 10 min at room temperature, permeabilized with 0.1% Triton X-100 (Sigma) in a 3% bovine serum albumin (BSA) in PBS solution for 20 min at room temperature and washed three times with PBS before the addition of 3% BSA in PBS blocking solution for 60 min at room temperature. Primary (Supplementary Table 3) and secondary (Supplementary Table 4) antibodies were diluted in a 3% BSA in PBS and either 3% normal goat serum or 3% normal donkey serum (Millipore-Sigma) depending on the secondary antibody host species. The coverslips were incubated in primary antibodies for 16 h at 4 °C and washed three times with PBS before incubating in the secondary antibody solution for 1 h at room temperature. Nuclear Stain Hoechst 33342 (Millipore Sigma) was added to the secondary solution at a dilution of 1:1000. Stained coverslips were mounted onto Superfrost Plus glass slides (ThermoFisher) using ProLong Antifade mounting media (ThermoFisher).
Image acquisition, processing, and quantification
Low power images to quantify cell numbers across networks were performed on a Carl Zeiss Axio Imager.Z2 upright microscope equipped with a Carl Zeiss ApoTome.2 apotome using a 20X air objective. In cases where cultures on coverslips were analyzed quantitatively, the average of five frames was determined for each coverslip. Cell numbers were quantified using ImageJ software [37].
3D confocal images were obtained on a Carl Zeiss LSM 880 Airyscan.2 microscope (Carl Zeiss, Oslo, Norway) with either a 63X or 25X oil immersion objective and analyzed using Imaris software (Oxford Instruments, Oxon, UK). Equal settings for laser intensity, step distance and pinhole aperture were used within individual experiments as much as possible. Modifications to the brightness or contrast of images were performed equally to all images within an experiment. For Sholl analysis using the pAAV-hSyn-EGFP construct, to ensure that excitatory neurons were being sampled, bipolar neurons were excluded and half of the neurons analyzed were counterstained with vesicular glutamate transporter 1 (VGLUT) and the statistics were not different between this group and the rest.
For synaptic quantification, Imaris software was used to generate 3D reconstructions of intracellular GFP and create a mask of proximal neurites. The pre-synaptic puncta were defined by vesicular glutamate transporter 1 (VGLUT1) or glutamic acid decarboxylase 65 (GAD65) immunoreactive puncta of 0.5 µm in diameter located outside of the neurite mask and within 1 µm of the external surface of the neurite. Post synaptic puncta were defined by post synaptic density 95 (PSD95) or GEPHRYN immunoreactive puncta of 0.3 µm within the neurite. Synapses were defined as pre- and post-synaptic puncta with 1 µm of each other. These were quantified and normalized to the area of the neurite determined by the GFP mask. The individual performing imaging and analysis was blinded to the genotype of the neuronal cultures.
Quantification and statistical analyses
Data are presented as the mean ± standard error of the mean (SEM). For single comparisons, two-way unpaired t tests were performed to determine significance at the level of P < 0.05. For multiple comparisons and grouped analysis, one way or two-way ANOVA with Tukey’s or Sidak’s multiple comparison’s correction was performed. Sample sizes were not predetermined using statistical methods. Statistical analysis was performed using Prism 7 (GraphPad Software, Boston, MA, USA).
Results
15q11.2 deletion alters network structure and cellular composition
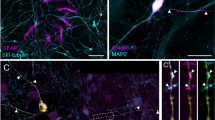
HiPSC-derived cortical neurons were co-cultured with rat astrocytes beginning at day in culture (DIC) 37 and examined by florescence microscopy for markers of differentiation and maturation. By DIC 80, 6 weeks after co-cultures were established, the neurons derived from both control and 15q11.2 deletion bearing hiPSCs exhibited features consistent with upper layer cortical neuron fate, as shown by the predominance of the post-mitotic, upper cortical layer transcription factor SATB2 (special AT-rich sequence-binding protein 2) [38] expressing neurons (87.4 ± 4.3% of neuron specific beta-tubulin III (TUJ1)+ neurons, n = 3 control donor lines vs 89.44 ± 0.7%, n = 3 deletion donor lines; P = 0.67) (Supplementary Fig. 1). There were also a small subset of neurons immunoreactive for SATB2 and the deep layer transcription factor CTIP2 (chicken ovalbumin upstream promoter transcription factor (COUP-TF)-interacting protein 2) [38, 39] with no significant differences between the two groups (10.5 ± 1.04% TUJ1+ neurons, n = 3 control donor lines vs 14.3 ± 11.5% n = 3 15q11.2 deletion donor lines; P = 0.76). At DIC 80, cultures consisted of dense TUJ1+ neurites connecting clusters of neuronal cell bodies (Fig. 1A). However, there appeared to be fewer and more disorganized interconnecting neurites in the 15q11.2 deletion cultures. This difference was more pronounced further in development at DIC 146 (Fig. 1B), despite similar numbers of TUJ1+ neurons (26.9 ± 4.95 cells per high powered field, n = 4 control donor lines with 3 independent cultures per line vs 29.5 ± 4.64 cells per high powered field, n = 3 deletion donor lines with 2–3 independent cultures per line; P = 0.71). When neurons were stained for alpha - calcium calmodulin dependent kinase II (CAMKIIα) as an indicator of glutamatergic neuron fate [40,41,42], most neurons were immunoreactive and there were similar proportions of CAMKIIα+ cells between the control and 15q11.2 deletion cultures at both DIC 80 (91.2 ± 1.2%, n = 4 control donor lines vs 93.1 ± 1.7%, n = 3 donor lines; P = 0.48) and DIC 146 (95.8 ± 0.9 vs 95.1 ± 1.4; P = 0.92) (Fig. 1A–C). Immunofluorescent staining for glutamic acid decarboxylase 67 (GAD67), as an indicator of the amino acid gamma-aminobutyric acid (GABA) production [43, 44], demonstrated a minority of cells staining positive in both control and 15q11.2 deletion lines. However, there was a doubling of GAD67+ cells in the 15q11.2 deletion lines compared to controls at DIC 146 (7.1 ± 0.94% N = 12 cultures from n = 4 control donor lines vs 18.0 ± 3.41% N = 9 cultures from n = 3 15q11.2 deletion lines, P < 0.0001) (Fig. 1A, B, D) suggesting that there is an increase in the proportion of GABAergic neurons in 15q11.2 deletion bearing neurons. Further analysis of the GAD67+ immunoreactive neurons in both control and deletion cultures demonstrated that the most of these cells were also immunoreactive for CAMKIIα (Fig. 1A, B, E), prompting additional characterization of these neuronal populations.

A, B Representative images of TUJ1+ (green, left merged panel) neurons stained for CAMKII (blue) and GAD67 (red) in control and deletion neurons to identify glutamatergic and GABAergic neurons, respectively, demonstrate deficits in network organization. Images taken on day in culture (DIC) 80 A and DIC 146 B. Red arrows indicate GAD67+ neurons. Scale bar 50 µm. C Quantification of the percent of TUJ1+ neurons expressing CAMKIIα at DIC 80 and 146 presented by donor line with each column representing a donor and each symbol representing an independent culture of that line. D Quantification of the percent of TUJ1+ neurons expressing GAD67 at DIC 80 and 146 presented by donor line as in C. Nested one-way ANOVA P = 0.01 with Sidak’s multiple comparison’s test. E Quantification of the percent of TUJ1+ neurons expressing GAD67 as well as GAD67+ CAMKIIα+ and GAD67+ CAMKIIα- neurons. Each symbol represents the mean of a minimum of three cultures from an individual donor line. Bars are the mean ± standard error of the mean (SEM) of four control and three 15q11.2 deletion lines. Two-way ANOVA with Tukey’s multiple comparison correction. F Reverse transcription quantitative PCR (qRT-PCR) data from DIC 141 cultures using human specific primers for interneuron transcripts and the pan-neuronal NKCC1 and KCC2 transcripts. All transcripts were normalized to the expression of the housekeeping gene, human RPS18 (Ribosomal protein S18), and to the mean of the control conditions. Each symbol represents 4 technical replicates for an individual donor’s line. Each bar represents the mean of 4 control and 3 deletion donor lines. Two-way ANOVA with Tukey’s multiple comparison test. * = P < 0.05, ** = P < 0.01, *** = P < 0.001 and **** = P < 0.0001.
To independently confirm the observation of increased GABAergic cells, we probed the cultures for a set of human interneuron specific transcripts using reverse transcription quantitative PCR (qRT-PCR) on co-cultures at DIC 141 (Fig. 1F). We confirmed elevated GAD67 mRNA expression in the deletion lines with a significant 4-fold increase compared to controls (4.01 ± 0.68 times control; P < 0.0001). There was also an increase in the interneuron fate determining transcription factor DLX1(Distal-Less Homeobox 1) which is active in early interneuron differentiation [45,46,47] (2.85 ± 1.0 times control; P < 0.01). Transcription of the interneuron subtype specific genes, parvalbumin (PV) and cholecystokinin (CCK), which are frequently expressed together [48] and are expressed in interneurons that make perisomatic synapses were both increased (15q11.2 deletion PV transcripts = 2.67 ± 0.45 x control, P < 0.05; 15q11.2 deletion CCK transcripts = 3.33 ± 0.58 x control, P < 0.001). In contrast, there was a non- significant decrease (0.134 ± 0.06, P = 0.1) in expression of somatostatin (SST), a GABAergic subtype specific gene that is present in cells that typically form synapses onto distal dendrites and mature later, in vivo [49, 50], suggesting that altered GABAergic neuron populations may be subtype specific with or without maturational delays. The latter hypothesis is supported by a relative increase in the expression of NKCC1 (Na-K-2Cl cotransporter isoform 1), the cation chloride cotransporter, responsible for chloride accumulation in immature cells, in the deletion lines (3.27 ± 0.17 x control, P < 0.001). Together, these data suggest alterations in cell fate in 15q11.2 deletion cultures.
Decreased neurite outgrowth and complexity in 15q11.2 deleted neurons
The 15q11.2 region contains at least one gene, CYFIP1, that encodes a protein involved in both cytoskeletal and synaptic development. The observation that cultures are less interconnected in the 15q11.2 deletion cultures (Fig. 1A) suggests that there may be primary deficits in neurite growth and expansion. To examine structural changes in individual cells, hiPSC-derived cortical forebrain neurons in co-culture with rat astrocytes were sparsely transfected with a plasmid expressing the green fluorescent protein (GFP) under the promotor for SYNAPSIN (pAAV-hSYN-EGFP) (SYNAPSIN-GFP) and matured until DIC 80 and 146 for single cell neurite analysis.
The morphology of the neurons from 15q11.2 deletion lines was less complex with fewer neurites and branches when compared to control lines at both DIC 80 and 146 (Fig. 2A). Sholl analysis of DIC 80 neurons demonstrated a significant reduction in the number of intersections at 20–160 μm from the soma between deletion and control lines (Fig. 2A, B). At DIC 146 there was also a significant difference in the number of intersections between control and deletion lines at 30 µm and from 60 µm to 180 µm (Fig. 2A, C). As expected for expanding neuronal arbors, the distance to the peak number of intersections increased in both populations over time. This distance to peak and the value of the peak number of intersections was greater in control compared to 15q11.2 deletion neurons at both timepoints (Fig. 2B, C). At 80 µm, the distance at which the peak number of intersections occurred in control neurons at day 146, there were significantly fewer intersections in the 15q11.2 deletion cells at DIC 80 compared to controls (control = 5.17 ± 0.119 intersections, N = 29 neurons form n = 3 donor lines vs 15q11.2 deletion = 2.73 ± 0.253 intersections, N = 25 neurons from n = 3 donor lines; P = 0.039). At DIC 146 there continued to be fewer intersections in 15q11.2 deletion neurons (5.9 ± 0.8, N = 45 neurons from n = 3 donor lines) compared to the control neurons (8.4 ± 0.51, N = 49 neurons from n = 4 donor lines; P = 0.024) (Fig. 2D, E). While the number of intersections increased from DIC 80 to 146, the number of intersections in the deletion bearing cells at dDIC 146 was only equal to the number in controls at DIC 80 (P = 0.758), suggesting a delay in structural maturation.

A 3D reconstructions of 25X oil immersion confocal images of pAAV-hSyn-EGFP (SYNAPSYN-GFP) expressing representative neurons with corresponding Sholl intersections in control and 15q11.2 deletion neurons at DIC 80 and DIC 146. Panels on the left demonstrate reconstruction of GFP (green) with Sholl intersection overlay (multicolored spheres). Panels on the right demonstrate inverted images with Sholl intersection and neurite skeleton only. Scale bar 50 µm. B Quantification of Sholl analysis of N = 29 control cells from 4 lines and N = 26 deletion cells from 3 lines at DIC 80. Two-way ANOVA P < 0.0001. C Sholl analysis of N = 49 control cells from 4 lines and N = 45 deletion cells from 3 lines at DIC 146. Two-way ANOVA P < 0.0001. D Average Scholl intersections by line at 80 µm from data in B and C. Individual data points are means of 6–12 cells per donor line. Columns represent means ± SEM of n = three to four donor lines. Individual symbols are the mean of a single donor line. Two-way ANOVA P < 0.001. E Data from D presented by donor line. Columns represent mean per line and symbols are individual neurons. F Average total neurite length per cell is decreased in deletion compared to control donor lines. Symbols are average of cells from each donor line. Columns are means of N = 3 or 4 donor lines per condition and timepoint. G Data from F presented per donor line. Column is the mean per line and each symbol is a neuron. Two-way ANOVA P < 0.001. Tukey’s multiple comparisons * = P < 0.05, ** = P < 0.01, *** = P < 0.001, **** = P < 0.0001. H Representative 3D reconstructions of 63X confocal images of control (top) and 15q11.2 deletion (bottom) proximal neurites of SYNAPSIN-GFP (green) expressing neurons at DIC146 immunostained for either glutamatergic (left two columns) or GABAergic (right two columns) synapses. Glutamatergic synapses are identified based on immunostaining for vesicular glutamate transporter 1 (VGLUT1) (red) outside of the neurite and intracellular post synaptic density protein 95 (PSD95) (blue) (left panels). Panels on the right show the neurite mask generated by SYNAPSIN-GFP staining (green). The presynaptic puncta from glutamatergic synapses on to the masked neurons are generated from VGLUT1 staining outside of the mask (red spheres) that are within 1 µm of the PSD95 puncta within the neurite (blue spheres, masked by neurite). GABAergic synapses are identified by immunostaining for presynaptic glutamic acid decarboxylase 65 (GAD65) (red) and post synaptic scaffolding protein GEPHRYN (blue). Panels on the right show the mask generated by GFP staining (green). The presynaptic puncta from GABAergic synapses on to the masked neurons are generated from GAD65 staining outside of the mask and within 1 µm of the GEPHRYN puncta within the neurite. Post synaptic puncta are generated from GEPHRYN staining within the masked region. I Quantification of glutamatergic synapses, defined as pre and post synaptic puncta colocalizing within 1 µm of each other, normalized to the surface area of the masked region to determine density. J Quantification of glutamatergic synapses, defined as pre and post synaptic puncta colocalizing within 1 µm of each other, normalized to the surface area of the masked region to determine density. For I and J, each symbol type represents a different donor line with each symbol representing a neurite from a unique cell. Scale bar 3 µm. Not significant by two-tailed t test.
In addition to fewer intersections, the total dendritic length per cell was decreased in the 15q11.2 deletion neurons at both DIC 80 (Mean difference 453 µm; 95% CI 68.97–837.1; P = 0.0216) and DIC 146 (513.5 µm; 68.97–837.1; P = 0.007), with the deletion neurons achieving the same length at DIC 146 as the controls at DIC 80 (−102.3 µm; −486.4–281.8; P = 0.8384) (Fig. 2F, G). Together with the differences in complexity of the arbors, the difference in total dendritic length indicates a delay in structural maturation in the 15q11.2 deletion lines.
Proximal synaptic density is not affected by 15q11.2 deletion
The simplicity of the neurite tree revealed by the experiments in Fig. 2 could be due to a primary cytoskeletal, adhesion or synaptic defect. Early changes in neurite outgrowth argue for a primary cytoskeletal or adhesion defect, while the increased magnitude of differences later in development argue that synaptic defects contribute to these changes. We examined the density of both glutamatergic and GABAergic synapses onto the proximal neurites of SYNAPSIN-GFP transfected cells at DIC 146 (Fig. 2H). Excitatory synapses were assessed by 3D confocal microscopy of co-cultures immunolabeled for presynaptic VGLUT and post synaptic PSD95. The resultant synapse density was similar between control and 15q11.2 deletion cultures (Two-tailed unpaired t-test, P = 0.19) (Fig. 2I).
Inhibitory synapses were labeled with antibodies targeting presynaptic GAD65 and the inhibitory post synaptic scaffolding protein, GEPHRYN (Fig. 2H and J). There was no significant difference between the control and the 15q11.2 deletion cultures (Two-tailed unpaired t test, P = 0.992) (Fig. 2J). These findings suggest that the 15q11.2 deletion does not determine the density of glutamatergic or GABAergic synapses, but do not speak to functional or structural changes within individual synapses. Additionally, with the same density of synapses, the absolute number of synapses would be decreased with a smaller neurite arbor in the 15q11.2 deletion neurons and does not preclude a functional change at the network level.
15q11.2 deletion decreases neurite complexity and length in both glutamatergic and GABAergic neurons
Given the results in Fig. 1 that suggest an increase in the proportion of GABAergic neurons in the 15q11.2 deletion cultures, the lack of an increase GABAergic synapse density is somewhat surprising. However, this could be explained by a simplification of the morphology of presynaptic GABAergic neurons resulting in a decrease in the overall number of synaptic connections per cell. To address this possibility, co-cultures were infected with either pAAV9-CaMKIIa-hChR2(H134R)-EYFP (CAMKIIα-YFP) for specific labeling of excitatory glutamatergic neurons [32] or pAAV9-mDlx-GFP-Fishell-1 (DLX-GFP) for specific labeling of GABAergic interneurons [33] at DIC 90 and matured until DIC 141. Confocal microscopy demonstrated robust YFP labeling of the CMAKIIα-YFP transduced glutamatergic neurons (Fig. 3A). Sholl analysis confirmed the qualitative observation that that the 15q11.2 deletion donor neurons have less complex morphology than the control neurons with significant differences in the number of Sholl intersections from 30–180 µm (Fig. 3C). Cultures infected with DLX-GFP had fewer GFP expressing cells than CAMKIIα-YFP infected cultures as expected for GABAergic neurons being a smaller proportion of the cultures, but there were qualitatively more DLX-GFP+ cells per field and each neuron had a more simplified neurite arbor in the 15q11.2 deletion cultures (Fig. 3B). Sholl analysis demonstrated significantly decreased Sholl intersections from 10–150 µm from the soma in the 15q11.2 deletion neurons (Fig. 3D). Similar to the SYNAPSIN-GFP expressing neurons in Fig. 2, there was a significant decrease in Sholl intersections at 80 µm (Fig. 3E) and cumulative neurite length per cell (Fig. 3F) in 15q11.2 deletion neurons from both CAMKIIα-YFP infected glutamatergic and DLX-GFP infected GABAergic neurons. These results suggest that 15q11.2 deletion results in structural immaturity or defects in both glutamatergic and GABAergic neurons.

A, B 3D reconstructions of 25X oil immersion confocal images of pAAV9-CaMKIIa-hChR2(H134R)-EYFP (CAMKIIα-YFP) and pAAV9-mDlx-GFP-Fishell-1 (DLX-GFP) expressing representative neurons with corresponding Sholl intersections in control and 15q11.2 deletion neurons at DIC 141. Panels on the left in A and B demonstrate reconstruction of anti-GFP immunostaining (green) with Sholl intersection overlay (multicolored spheres). Panels on the right demonstrate inverted images with Sholl intersection and neurite skeleton only. Scale bar 50 µm. C Sholl analysis of CAMKIIα-YFP+ neurites from N = 33 control cells from 3 lines and N = 33 deletion cells from 3 lines ad DIC 141. Two-way ANOVA P < 0.0001. D Sholl analysis of DLX-GFP+ neurites from N = 41 control cells from 4 donor lines and N = 30 deletion cells from 3 donor lines at DIC 141. Two-way ANOVA P < 0.0001. Tukey’s multiple comparisons test * = P < 0.05, ** = P < 0.01, *** = P < 0.001, **** = P < 0.0001. E Scholl intersections by line at 80 µm from data in C and D. Columns represent mean per line and symbols are individual neurons. Nested one-way ANOVA P < 0.0001. F Average total neurite length per cell is decreased in deletion compared to control donor lines. Column is the mean per line and each symbol is total neurite length for a single neuron. Nested one-way ANOVA P < 0.0001. Sidak’s multiple comparisons test * = P < 0.05 and *** = P < 0.001.
15q11.2 deletion neurons display delayed functional maturation
The apparent deficits in neurite development and altered network morphological interconnectivity suggests that functional neuronal development may be affected by the 15q11.2 deletion. To assess the effects of 15q11.2 deletion on functional maturation of neurons, co-cultures of hiPSC derived cortical neurons and rat astrocytes were plated onto 24-well MEA plates at DIC 37. Voltage recordings were conducted beginning at DIC 39 and subsequently every one to two weeks until DIC 202. Discrete spikes recorded from each electrode over time (Fig. 4A) were used to determine weighted mean firing rate (WMFR), burst firing and eventually synchronized network bursts. Spikes appeared in some cultures as early as DIC 39, and consistent spiking across multiple electrodes were recorded after day 50 (Fig. 4A, B), with consistent bursting after DIC 60 (Fig. 4A,C) and synchronized network bursting after DIC 70 (Fig. 4A,D).

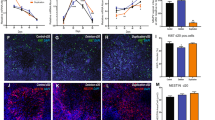
A Raster plots of spikes determined from voltage recordings from 16 electrodes from representative single wells of control (top) and 15q11.2 deletion (bottom) cultures at DIC 65, 106 and 154. Each row is data from a single electrode. Single spikes for a given electrode are represented by black vertical marks. Blue vertical lines are spikes on a single electrode meeting criterion for an electrode burst (5 spikes occurring within 100 ms). Black tracing at the top of each panel represents relative spikes per time per second across the entire well. Time scale bar is 50 s. Analysis of four control and three 15q11.2 deletion lines over at least 3 independent differentiations for B weighted mean firing rate (Two-way ANOVA P < 0.0001), C percent of spikes that occur in bursts (burst percentage) (Two-way ANOVA P < 0.0001), Tukey’s multiple comparison test and D area under normalized cross correlogram (AUNCC) as a quantification of synchronization (TwoWay ANOVA P < 0.0001). Mean values ± SEM are indicated by symbols. Linear regressions (solid lines) with 95% confidence intervals (dashed lines) are in grey and blue. Two-way ANOVA P < 0.0001. Tukey’s multiple comparisons * = P < 0.05. For B and C, Tukey’s multiple comparison’s testing indicates significance from P < 0.01 to P < 0.0001 for all data points to right of the vertical dashed line.
In 15q11.2 deletion cultures, while spikes emerged at similar time points as controls, the weighted mean firing rates (WMFR) showed delayed maturity with significantly different slopes (Control slope = 0.057, 95% CI 0.047–0.067 vs Deleted slope = 0.025, 95% CI 0.020–0.030; P < 0.0001) and significant differences between the two populations at DIC 106 (P < 0.05) and at all time points From DIC 117 to DIC 202 (P < 0.05 to P < 0.001) (Fig. 4B). Bursting emerged consistently in control neurons at DIC 58 and there was a progressive increase in the percentage of spikes that occurred in bursts until DIC 130 when this plateaued between 50 and 75%. In contrast, the 15q11.2 deletion cultures exhibited less bursting at DIC 58, and the increase in burst percentage was lower and continued to increase at the point the control cultures had leveled off, with a significant difference between control and deletion at DIC 86 and at every timepoint afterwards (P < 0.05 to P < 0.0001) (Fig. 4C).
As cultures mature, neurons begin to exhibit synchronized periods of high activity alternating with low activity across most electrodes (Fig. 4A). These spontaneous slow network oscillations started by DIC 70 in control cultures, consistent with the development of functional synaptic connectivity between neurons and synchronization of the network [51]. To assess for the development of synaptic synchronization of the network across time, the area under normalized cross correlation (AUNCC) from each culture and at each time point was determined. The AUNCC is a measure of the area under interelectrode cross-correlation normalized to the autocorrelation, and is a measure of synchronicity of the network with higher values indicating greater synchronicity [35]. In controls, synchronized activity occurred later than bursting, with a significant rise between DIC 65 and 106 (Fig. 4A, D). However, the development of synchronized activity occurred over a more protracted time course in the 15q11.2 deletion neurons (Control slope = 0.057, 95% CI 0.0014–0.0020 vs Deleted slope = 0.0012, 95% CI 0.00096–0.0015; P < 0.05) (Fig. 4D). This suggests altered functional development of synaptic connectivity as well as basic functional neuronal properties.
15q11.2 hiPSC-neurons demonstrate a delay in AMPA / kainite receptor-mediated signaling
To determine whether differences in synaptic activity contribute to differences in the functional maturation of these nascent neuronal networks, we examined glutamatergic network connectivity by blocking the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainate glutamate receptors with CNQX. We examined three timepoints at which robust spiking and bursting had already been established in control cultures (Fig. 5). As might be expected for early network development when there is less or variable synaptic activity, the effects of 50 µM CNQX application at the earliest timepoint, DIC 100, were equivalent in both the control and deletion cultures with similar numbers of cultures demonstrating an increase or a decrease in spiking, bursting or synchronized activity (Fig. 5A–D). There was no significant difference in the change in WMFR between control and 15q11.2 deletion cultures at any timepoint even though CNQX had a progressively larger effect on firing over time (Fig. 5B). However, at DIC 138, most control cultures responded to CNQX with a marked reduction in burst percentage and there was a significant difference between control and deletion cultures with less of a decrease in deletion cultures (control = −46.64 ± 7.7%, n = 25 cultures; vs. 1511.2 deletion = −7.7 ± 18.7%, n = 20 cultures; P = 0.012). By DIC 210, the deletion cultures had a similar reduction in burst percentage; suggesting a developmental delay that resolves (control = −61.261 ± 4.049%, n = 15; vs. 1511.2 deletion = −60.7 ± 9.2%, n = 12; P = 0.98) (Fig. 5C).

A Raster plots of spikes determined from voltage recordings from 16 electrodes from representative single wells of control (left) and 15q11.2 deletion (right) cultures. Each row is data from a single electrode. Single spikes for a given electrode are represented by black vertical marks. Blue vertical lines are spikes on a single electrode, meeting criteria for an electrode burst - 5 spikes occurring within 100 ms. Black tracing at the top of each panel represents spikes per time per second across the entire well. Pink boxes are network bursts. Time scale bar is 40 s. Black vertical line demarcates time of acute CNQX application. Quantification of CNQX mediated effects on B weighted mean firing rate (WMFR), C burst percentage and D synchronization in control and deletion cultures at DIC 100, 138 and 210. Each data point is the mean activity across 16 electrodes for a single well. Individual symbols are independent cultures with the type of symbol indicating a unique donor. Columns are the mean ± SEM of all cultures with 4 control and 3 deletion cultures represented. Two-way ANOVA P < 0.0001. Tukey’s multiple comparisons * = P < 0.05, ** = P < 0.01, *** = P < 0.001.
Like developmental changes in burst percentage, CNQX-dependent changes in synchronization were slower to emerge in the 15q11.2 deletion cultures, with a significantly smaller decrease in synchronicity in response to CNQX at DIC 138 (Control = −69.3 ± 5.3%, n = 25; vs. 1511.2 deletion = −34.316 ± 12.107%, n = 17; P = 0.01). By DIC 210, the deletion cultures have similar reductions in synchronicity in response to CNQX as the control cultures (Control = −73.494 ± 6.084%, n = 14; vs. 1511.2 deletion = −67.0 ± 6.6%, n = 11, P = 0.71) (Fig. 5D). Taken together, these data suggest that 15q11.2 iPSC-neurons have a delay in the development of AMPA / Kainate receptor-mediated synaptic signaling.
Development of an inhibitory GABAergic response is altered in 15q11.2 deletion neurons
We evaluated the effects of GABAergic influence on network development using the GABA-A receptor antagonist bicuculline. At DIC 79 and DIC 124, application of bicuculline decreased the WMFR in both control and 15q11.2 deletion cultures (Fig. 6A and B). A decrease in firing in response to blocking GABA-A receptors suggests that the overall effect of baseline endogenous GABAergic transmission on the network is increased neuronal firing, consistent with the presence of excitatory GABAergic signaling in immature neurons [52]. There was a non-significant reduction in the amount of bicuculline-dependent inhibition between DIC 79 and 124 in both deletion and control cultures. However, by DIC 148 in the control cultures, there was a switch in the response to bicuculline to one that reflects an increase in firing rate (127 ± 26.2%; n = 11) while the response in 15q11.2 deletion cultures is still a decrease in firing (−15.9 ± 12.02%; n = 12) (P < 0.0001) (Fig. 6B). Analysis of burst percentage in response to bicuculline also revealed no significant difference in the mean percentage change between control and 15q11.2 deletion cultures and either a reduction in firing or an equivocal response at DIC 79 and 124. However, at DIC 148 all control cultures demonstrated an increase in burst percentage in response to bicuculline (56.5 ± 25.6%, n = 10) while15q11.2 deletion cultures continued to demonstrate a reduction in burst percentage in response to bicuculline (−51.9 ± 10.3%, n = 11) (P = 0.012) (Fig. 6A and C). Similarly, the bicuculline dependent change in the AUNCC was either equivocal or negative in both the control and deletion cultures at DIC 79 and 124, but the two genotypes diverged at DIC148 with an increase in synchronicity based on a positive percent change in control cultures (93.4 ± 20.4% n = 11) and an equivocal response in the 15q11.2 deletion cultures (−2.3 ± 19.0%, n = 11) (P = 0.006) (Fig. 6D). These findings suggest either a delay in the maturation of a chloride gradient, supported by increased NKCC1 expression demonstrated in Fig. 1F, or a delay in synaptic GABAergic activity in the 15q11.2 deletion cultures, and are consistent with overall structural or functional delays or a combination of both.

A Example raster plots demonstrating acute GABA receptor blockade with application of bicuculline. Raster plots of spikes determined from voltage recordings from 16 electrodes from representative single wells of control (left) and 15q11.2 deletion (right) cultures. Each row is data from a single electrode. Single spikes for a given electrode are represented by black vertical marks. Blue vertical lines are spikes on a single electrode meeting criterion for an electrode burst - 5 spikes occurring within 100 ms. Black tracing at the top of each panel represents spikes per time per second across the entire well. Pink boxes are network bursts. Time scale bar is 40 s. Black vertical line demarcates time of acute bicuculline application. Quantification of bicuculline mediated effects on B weighted mean firing rate (WMFR), C burst percentage and D synchronization in control and deletion cultures. Each data point is the mean activity across 16 electrodes for a single well. Individual symbols are independent cultures with the type of symbol indicating a unique donor. Columns are the mean ± SEM of all cultures with 4 control and 2–3 deletion cultures represented. Two-way ANOVA. Tukey’s multiple comparisons * = P < 0.05, ** = P < 0.01, and **** = P < 0.0001.
Discussion
While 15q11.2 deletion has been shown to alter NPC biology [16], this is the first detailed assessment of later neuronal development and function in a human cell model. This study demonstrates that deletion of the 15q11.2 region is sufficient to cause structural changes at the cellular level including decreased neurite length and complexity in both glutamatergic and GABAergic neurons. This is associated with early altered structural connectivity and neuronal composition at the network level and functional delays in spontaneous activity and later deficits in synaptic mediated firing, bursting and synchronization. This developmental dysregulation in vitro suggests a possible cellular mechanism for the phenotypes of individuals affected by the 15q11.2 deletion, namely that deficits in neurite development alter maturation of both glutamatergic and GABAergic connectivity.
The overall decrease in spiking, spike bursting and synchronized activity in 15q11.2 deletion neurons that is shown in this study to be due in part to a functional delay in AMPA / Kainate receptor mediated connectivity is consistent with postmortem studies of patients with schizophrenia having decreased dendritic spines and other iPSC models of schizophrenia indicating impaired glutamatergic transmission [53]. However, at least in the proximal portions of the neurite arbor this was not due to a significant change in the density of excitatory synapses. There may be structural and functional changes within the synapses that cannot be identified at the resolution used here or changes in synaptic density more distally. Regardless of any additional functional or structural synaptic changes, the overall decreased neurite length and arborization results in overall decreased excitatory input despite similar synaptic densities.
Given the predominance of epilepsy phenotypes in 15q11.2 deletion bearing individuals, the decreased activity and glutamatergic influence of neuronal networks, in vitro, is somewhat counterintuitive to the prevailing hypothesis that the underlying cause of epilepsy is hyperexcitability, in part due to increased excitatory activity [54]. However, loss of appropriate inhibition can also lead to pathologic hyperexcitable states. In this study, there is a delayed maturation of the inhibitory response to GABA, demonstrated by application of bicuculline as well as a relative increase in the expression of the NKCC1 transporter responsible for accumulation of chloride intracellularly. The activity of this transporter is responsible for the phenomenon of higher intracellular chloride concentrations in immature neurons than in mature neurons in brain development. The resultant chloride efflux and neuronal depolarization in response to the activation of GABA receptors makes GABA an excitatory neurotransmitter early in development. This is required for primitive network activity, regulates proliferation and migration and strongly influences synaptic plasticity [55]. A prolonged window of excitatory GABAergic activity or failure to establish the appropriate chloride gradient can pathologically alter the excitation inhibition balance and synaptic plasticity through numerous mechanisms [56].
Cultures of 15q11.2 deletion neurons not only demonstrate a continued excitatory response to GABA at time points where the control neurons are inhibited, but there is an increase in the percentage of neurons in the culture that express the GABAergic protein GAD67. Increased numbers of neurons expressing GAD67 in the 15q11.2 deletion cultures could indicate a primary difference in developmental fate choice or a reactive increase in response to dysfunctional GABAergic signaling. The decreased neurite complexity of the GABAergic neurons supports both hypotheses and may explain why there are not more GABAergic synapses despite a greater proportion of cells. The additional presence of CAMKIIα antibody immunoreactivity in the GAD67+ neurons from both control and 15q11.2 deletion cultures is somewhat unexpected. However, CAMKIIα has been reported in both postmitotic neural progenitor cells [57] as well as different subtypes of interneurons [58, 59] and these findings may suggest early stages of GABAergic development or specific GABAergic neuron subtypes. The expression of other GABAergic neuron transcripts and the presence of GFP driven by the mDLX promotor, which is specific for GABAergic neurons [33], confirms the presence of GABAergic neurons in these cultures.
Altered dendritic development and synaptic function predispose individuals to neurodevelopmental disorders such as schizophrenia, epilepsy and ID. In schizophrenia, changes in dendritic spine density and morphology and decreased dendritic arborization, length and number have been shown in the prefrontal cortex [60, 61]. Decreased dendritic arborization has not only been shown in postmortem studies but also in living patients with neurite density imaging modeling of MRI data [62, 63] and is consistent with the changes demonstrated in this study, in vitro, using 15q11.2 deletion neurons.
Variable changes in dendrite and dendritic spine density have also been described in multiple types of epilepsy. In type II focal cortical dysplasia, synaptic spine density is decreased on the dendrites of dysplastic neurons and dendritic branching is altered with increased and disorganized dendrites in dysplastic neurons and decreased dendritic branch points in the relatively normal adjacent neurons [64]. Dendritic branch points, dendritic length and dendritic spines are also decreased in surgical samples from humans with temporal lobe epilepsy [65,66,67,68]. Similar findings of decreased branch points and dendritic lengths have been described in syndromic forms of intellectual disability with an increased risk of epilepsy in Down syndrome and Rhett syndrome [69]. However, particularly when looking at post-surgical or postmortem cases of epilepsy and schizophrenia, the question of whether the changes are causative or reactive to years of abnormal excitation and medication effects lingers. The results of the current study suggest that in the case of 15q11.2 deletion, a primary defect in dendrite development that coincides with a functional change in the activity of neuronal networks is a common mechanism for the different phenotypes.
Disease causing copy number variants involving multiple genes are difficult to study because manipulation of one gene may not be sufficient to recapitulate a phenotype. hiPSC derived cells offer the ability to study human cells with the genetic change directly but what is known about single genes in these regions can also provide significant insight. There are four genes that located at the 15q11.2 BP1-BP2 locus. NIPA1 is expressed in neuronal tissue and mediates Mg++ transport as well as transport of other divalent cations. NIPA1 pathogenic variants, but not deletions result in autosomal dominant hereditary spastic paraplegia (HSP) [70]. Interestingly, although initially described as a pure phenotype of spastic paraplegia, families with HSP due to certain pathogenic variants in NIPA1 have also been reported to have epilepsy and ALS possibly related to reduced protein expression [71, 72]. NIPA2 is a selective magnesium transporter that mediates renal magnesium conservation [73]. TUBGCP5 is a member of a family of 5 similar proteins that bind gamma tubulin and function in spindle assembly [74, 75]. While NIPA1 and TUBGCP5 have the potential to be important in brain physiology, CYFIP1 is proposed to be a critical regulator of neural development and synaptic plasticity [21, 25].
CYFIP1 is highly conserved with expression in both excitatory and inhibitory neurons and complete knockout in mice is embryonic lethal [20, 25]. Down regulation of the CYFIP1 protein leads to disruption of the organization of NPC’s in embryonic mice in vivo [16] as well as altered proliferation of pre- and postnatal neural progenitor cells [17], suggesting that it plays an important role in early neural development. Haploinsufficiency in mice leads to decreased dendritic complexity [76] similar to what is demonstrated here in 15q11.2 deletion neurons. The most likely mechanism for CYFIP1 mediated changes in dendritic structure is dysregulation of the WAVE regulatory complex (WRC) which is required for actin nucleation in dendritic branching and elongation [77]. Studies in mice with reduced CYFIP1as well as in human cells with 15q11.2 deletion cells have demonstrated that a reduction of CYFIP1 results in a reduction of multiple other WRC proteins [78]. Haploinsufficiency of Cyfip1 in rodents also results in immature synaptic spine structure and function due to its dual interactions with FMRP and the WAVE regulatory complex [21, 76, 79]. It is present at both glutamatergic and GABAergic synapses and decreased expression has been shown to stabilize GABAergic synapses in mature neurons [23]. While this study did not determine a difference in gross density of synapses as a result of 15q11.2 deletion, electrophysiology demonstrated delays in both synaptic systems. This could be a result of overall reduced connectivity due to decreased numbers and length of dendrites or CYFIP1 mediated molecular changes at the synapses themselves.
Conclusions
15q11.2 deletion results in decreased neurite complexity at the single cell level. At the network level, there is altered structural interconnectedness, proportions of glutamatergic and GABAergic neurons, and delays in the maturation of spike rates, busting and synchronization. This is associated with a decrease in the influence of AMPA / Kainate receptors on the network (glutamatergic synaptic excitation) as well as a prolonged excitatory response to GABA. These findings suggest several mechanisms by which neuronal network deficits can lead to pathological changes in human development and result in the complex phenotypes manifesting in 15q11.2 deletion bearing individuals, but interpretation is limited by the simplified networks present in vitro. Some, but not all the neuronal changes in 15q11.2 deletion neurons are like what has been described in animal models of Cyfip1 deletion and further exploration of the intersection between in vivo animal models and in vitro human models is likely to shed light on pathologic development in this disorder as well as potential therapeutic strategies.
Data availability
All relevant data are included in the main text and supplementary material and available upon request.
References
Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43:838–46. https://doi.org/10.1038/ng.909
Caseras X, Kirov G, Kendall KM, Rees E, Legge SE, Bracher-Smith M, et al. Effects of genomic copy number variants penetrant for schizophrenia on cortical thickness and surface area in healthy individuals: analysis of the UK biobank. Br J Psychiatry. 2021;218:104–11. https://doi.org/10.1192/bjp.2020.139
Mefford HC, Yendle SC, Hsu C, Cook J, Geraghty E, McMahon JM, et al. Rare copy number variants are an important cause of epileptic encephalopathies. Ann Neurol. 2011;70:974–85. https://doi.org/10.1002/ana.22645
Coe BP, Witherspoon K, Rosenfeld JA, van Bon BWM, Vulto-van Silfhout AT, Bosco P, et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet. 2014;46:1063–71. https://doi.org/10.1038/ng.3092
Cox D, Butler M. The 15q11.2 BP1-BP2 microdeletion syndrome: a review. Int J Mol Sci. 2015;16:4068–82. https://doi.org/10.3390/ijms16024068
Jønch AE, Douard E, Moreau C, Van Dijck A, Passeggeri M, Kooy F, et al. Estimating the effect size of the 15Q11.2 BP1-BP2 deletion and its contribution to neurodevelopmental symptoms: recommendations for practice. J Med Genet. 2019;56:701–10. https://doi.org/10.1136/jmedgenet-2018-105879
Ulfarsson MO, Walters GB, Gustafsson O, Steinberg S, Silva A, Doyle OM, et al. 15q11.2 CNV affects cognitive, structural and functional correlates of dyslexia and dyscalculia. Transl Psychiatry. 2017;7:e1109 https://doi.org/10.1038/tp.2017.77
de Kovel CGF, Trucks H, Helbig I, Mefford HC, Baker C, Leu C, et al. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain. 2010;133:23–32. https://doi.org/10.1093/brain/awp262
Jähn JA, von Spiczak S, Muhle H, Obermeier T, Franke A, Mefford HC, et al. Iterative phenotyping of 15q11.2, 15q13.3 and 16p13.11 microdeletion carriers in pediatric epilepsies. Epilepsy Res. 2014;108:109–16. https://doi.org/10.1016/j.eplepsyres.2013.10.001
Bittel DC, Kibiryeva N, Butler MG. Expression of 4 genes between chromosome 15 breakpoints 1 and 2 and behavioral outcomes in Prader-Willi syndrome. Pediatrics. 2006;118:e1276–e1283. https://doi.org/10.1542/peds.2006-0424
Mullen SA, Carvill GL, Bellows S, Bayly MA, Berkovic SF, Dibbens LM, et al. Copy number variants are frequent in genetic generalized epilepsy with intellectual disability. Neurology. 2013;81:1507–14. https://doi.org/10.1212/WNL.0b013e3182a95829
Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet. 2017;49:27–35. https://doi.org/10.1038/ng.3725
Writing Committee for the ENIGMA-CNV Working Group, Sønderby IE, Kaufmann T, Walters GB, Abdellaoui A, Ames D, et al. Association of copy number variation of the 15q11.2 BP1-BP2 region with cortical and subcortical morphology and cognition. JAMA Psychiatry. 2020;77:420–30. https://doi.org/10.1001/jamapsychiatry.2019.3779
Silva AI, Kirov G, Kendall KM, Bracher-Smith M, Wilkinson LS, Hall J, et al. Analysis of diffusion tensor imaging data from the UK biobank confirms dosage effect of 15q11.2 copy number variation on white matter and shows association with cognition. Biol Psychiatry. 2021;90:307–16. https://doi.org/10.1016/j.biopsych.2021.02.969
Winklewski PJ, Sabisz A, Naumczyk P, Jodzio K, Szurowska E, Szarmach A. Understanding the physiopathology behind axial and radial diffusivity changes-what do we know? Front Neurol. 2018;9:92 https://doi.org/10.3389/fneur.2018.00092
Yoon KJ, Nguyen HN, Ursini G, Zhang F, Kim NS, Wen Z, et al. Modeling a genetic risk for schizophrenia in iPSCs and mice reveals neural stem cell deficits associated with adherens junctions and polarity. Cell Stem Cell. 2014;15:79–91. https://doi.org/10.1016/j.stem.2014.05.003
Habela CW, Yoon KJ, Kim NS, Taga A, Bell K, Bergles DE, et al. Persistent Cyfip1 expression is required to maintain the adult subventricular zone neurogenic niche. J Neurosci. 2020;40:2015–24. https://doi.org/10.1523/JNEUROSCI.2249-19.2020
Steffen A, Rottner K, Ehinger J, Innocenti M, Scita G, Wehland J, et al. Sra-1 and Nap1 link Rac to actin assembly driving lamellipodia formation. EMBO J. 2004;23:749–59. https://doi.org/10.1038/sj.emboj.7600084
Schenck A, Qurashi A, Carrera P, Bardoni B, Diebold C, Schejter E, et al. WAVE/SCAR, a multifunctional complex coordinating different aspects of neuronal connectivity. Dev Biol. 2004;274:260–70. https://doi.org/10.1016/j.ydbio.2004.07.009
Schenck A, Bardoni B, Langmann C, Harden N, Mandel JL, Giangrande A. CYFIP/Sra-1 controls neuronal connectivity in Drosophila and links the Rac1 GTPase pathway to the fragile X protein. Neuron. 2003;38:887–98. https://doi.org/10.1016/s0896-6273(03)00354-4
De Rubeis S, Pasciuto E, Li KW, Fernández E, Di Marino D, Buzzi A, et al. CYFIP1 coordinates mRNA translation and cytoskeleton remodeling to ensure proper dendritic spine formation. Neuron. 2013;79:1169–82. https://doi.org/10.1016/j.neuron.2013.06.039
Hsiao K, Harony-Nicolas H, Buxbaum JD, Bozdagi-Gunal O, Benson DL. Cyfip1 regulates presynaptic activity during development. J Neurosci. 2016;36:1564–76. https://doi.org/10.1523/JNEUROSCI.0511-15.2016
Davenport EC, Szulc BR, Drew J, Taylor J, Morgan T, Higgs NF, et al. Autism and schizophrenia-associated CYFIP1 regulates the balance of synaptic excitation and inhibition. Cell Rep. 2019;26:2037–51. https://doi.org/10.1016/j.celrep.2019.01.092. e6
Napoli I, Mercaldo V, Boyl PP, Eleuteri B, Zalfa F, De Rubeis S, et al. The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell. 2008;134:1042–54. https://doi.org/10.1016/j.cell.2008.07.031
Bozdagi O, Sakurai T, Dorr N, Pilorge M, Takahashi N, Buxbaum JD. Haploinsufficiency of Cyfip1 produces fragile X-like phenotypes in mice. PLoS ONE. 2012;7:e42422. https://doi.org/10.1371/journal.pone.0042422
Kim NS, Ringeling FR, Zhou Y, Nguyen HN, Temme SJ, Lin YT, et al. CYFIP1 dosages exhibit divergent behavioral impact via diametric regulation of NMDA receptor complex translation in mouse models of psychiatric disorders. Biol Psychiatry. 2022;92:815–26. https://doi.org/10.1016/j.biopsych.2021.04.023
Domínguez-Iturza N, Lo AC, Shah D, Armendáriz M, Vannelli A, Mercaldo V, et al. The autism- and schizophrenia-associated protein CYFIP1 regulates bilateral brain connectivity and behaviour. Nat Commun. 2019;10:3454. https://doi.org/10.1038/s41467-019-11203-y
Silva AI, Haddon JE, Ahmed Syed Y, Trent S, Lin TCE, Patel Y, et al. Cyfip1 haploinsufficient rats show white matter changes, myelin thinning, abnormal oligodendrocytes and behavioural inflexibility. Nat Commun. 2019;10:3455. https://doi.org/10.1038/s41467-019-11119-7
Taga A, Habela CW, Johns A, Liu S, O'Brien M and Maragakis NJ Establishment of an electrophysiological platform for modeling ALS with regionally-specific human pluripotent stem cell-derived astrocytes and neurons. J Vis Exp 2021. https://doi.org/10.3791/62726.
Wen Z, Nguyen HN, Guo Z, Lalli MA, Wang X, Su Y, et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature. 2014;515:414–8. https://doi.org/10.1038/nature13716
Datta Chaudhuri A, Dasgheyb RM, DeVine LR, Bi H, Cole RN, Haughey NJ. Stimulus-dependent modifications in astrocyte-derived extracellular vesicle cargo regulate neuronal excitability. Glia. 2020;68:128–44. https://doi.org/10.1002/glia.23708
Lee JH, Durand R, Gradinaru V, Zhang F, Goshen I, Kim DS, et al. Global and local fMRI signals driven by neurons defined optogenetically by type and wiring. Nature. 2010;465:788–92. https://doi.org/10.1038/nature09108
Dimidschstein J, Chen Q, Tremblay R, Rogers SL, Saldi GA, Guo L, et al. A viral strategy for targeting and manipulating interneurons across vertebrate species. Nat Neurosci. 2016;19:1743–9. https://doi.org/10.1038/nn.4430
Chiappalone M, Novellino A, Vajda I, Vato A, Martinoia S, van Pelt J. Burst detection algorithms for the analysis of spatio-temporal patterns in cortical networks of neurons. Neurocomputing. 2005;65-66:653–62. https://doi.org/10.1016/j.neucom.2004.10.094
Halliday DM, Rosenberg JR, Breeze P, Conway BA. Neural spike train synchronization indices: definitions, interpretations, and applications. IEEE Trans Biomed Eng. 2006;53:1056–66. https://doi.org/10.1109/TBME.2006.873392
Taga A, Dastgheyb R, Habela C, Joseph J, Richard JP, Gross SK, et al. Role of human-induced pluripotent stem cell-derived spinal cord astrocytes in the functional maturation of motor neurons in a multielectrode array system. Stem Cells Transl Med. 2019;8:1272–85. https://doi.org/10.1002/sctm.19-0147
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82. https://doi.org/10.1038/nmeth.2019
Britanova O, de Juan Romero C, Cheung A, Kwan KY, Schwark M, Gyorgy A, et al. Satb2 is a postmitotic determinant for upper-layer neuron specification in the neocortex. Neuron. 2008;57:378–92. https://doi.org/10.1016/j.neuron.2007.12.028
Alcamo EA, Chirivella L, Dautzenberg M, Dobreva G, Fariñas I, Grosschedl R, et al. Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron. 2008;57:364–77. https://doi.org/10.1016/j.neuron.2007.12.012
Liu XB, Murray KD. Neuronal excitability and calcium/calmodulin-dependent protein kinase type II: location, location, location. Epilepsia. 2012;53:45–52. https://doi.org/10.1111/j.1528-1167.2012.03474.x
Jones E, Huntley G, Benson D. Alpha calcium/calmodulin-dependent protein kinase II selectively expressed in a subpopulation of excitatory neurons in monkey sensory-motor cortex: comparison with GAD-67 expression. J Neurosci. 1994;14:611–29. https://doi.org/10.1523/JNEUROSCI.14-02-00611.1994
Benson DL, Isackson PJ, Gall CM, Jones EG. Contrasting patterns in the localization of glutamic acid decarboxylase and Ca2 + /calmodulin protein kinase gene expression in the rat central nervous system. Neuroscience. 1992;46:825–49. https://doi.org/10.1016/0306-4522(92)90188-8
Kaufman DL, Houser CR, Tobin AJ. Two forms of the gamma-aminobutyric acid synthetic enzyme glutamate decarboxylase have distinct intraneuronal distributions and cofactor interactions. J Neurochem. 1991;56:720–3. https://doi.org/10.1111/j.1471-4159.1991.tb08211.x
Erlander MG, Tillakaratne NJK, Feldblum S, Patel N, Tobin AJ. Two genes encode distinct glutamate decarboxylases. Neuron. 1991;7:91–100. https://doi.org/10.1016/0896-6273(91)90077-d
Panganiban G, Rubenstein JLR. Developmental functions of the Distal-less/Dlx homeobox genes. Development. 2002;129:4371–86. https://doi.org/10.1242/dev.129.19.4371
Cobos I, Long JE, Thwin MT, Rubenstein JL. Cellular patterns of transcription factor expression in developing cortical interneurons. Cereb Cortex. 2006;16:i82–i88. https://doi.org/10.1093/cercor/bhk003
Le TN, Zhou QP, Cobos I, Zhang S, Zagozewski J, Japoni S, et al. GABAergic interneuron differentiation in the basal forebrain is mediated through direct regulation of glutamic acid decarboxylase isoforms by Dlx homeobox transcription factors. J Neurosci. 2017;37:8816–29. https://doi.org/10.1523/JNEUROSCI.2125-16.2017
Grieco SF, Johnston KG, Gao P, Garduño BM, Tang B, Yi E, et al. Anatomical and molecular characterization of parvalbumin-cholecystokinin co-expressing inhibitory interneurons: implications for neuropsychiatric conditions. Mol Psychiatry. 2023;28:5293–308. https://doi.org/10.1038/s41380-023-02153-5
Urban-Ciecko J, Barth AL. Somatostatin-expressing neurons in cortical networks. Nat Rev Neurosci. 2016;17:401–9. https://doi.org/10.1038/nrn.2016.53
Tanaka DH, Maekawa K, Yanagawa Y, Obata K, Murakami F. Multidirectional and multizonal tangential migration of GABAergic interneurons in the developing cerebral cortex. Development. 2006;133:2167–76. https://doi.org/10.1242/dev.02382
Neske GT. The slow oscillation in cortical and thalamic networks: mechanisms and functions. Front Neural Circuits. 2016;9:88. https://doi.org/10.3389/fncir.2015.00088
Ben-Ari Y. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci. 2002;3:728–39. https://doi.org/10.1038/nrn920
Habela CW, Song H, Ming G. Modeling synaptogenesis in schizophrenia and autism using human iPSC derived neurons. Mol Cell Neurosci. 2016;73:52–62. https://doi.org/10.1016/j.mcn.2015.12.002
Shao LR, Habela CW, Stafstrom CE. Pediatric epilepsy mechanisms: expanding the paradigm of excitation/inhibition imbalance. Children. 2019;6:23. https://doi.org/10.3390/children6020023
Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007;87:1215–84. https://doi.org/10.1152/physrev.00017.2006
Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–38. https://doi.org/10.1016/j.neuron.2009.03.003
Arruda-Carvalho M, Restivo L, Guskjolen A, Epp JR, Elgersma Y, Josselyn SA, et al. Conditional deletion of α-CaMKII impairs integration of adult-generated granule cells into dentate gyrus circuits and hippocampus-dependent learning. J Neurosci. 2014;34:11919–28. https://doi.org/10.1523/JNEUROSCI.0652-14.2014
Keaveney MK, Rahsepar B, Tseng H, Fernandez FR, Mount RA, Ta T, et al. CaMKIIα-positive interneurons identified via a microRNA-based viral gene targeting strategy. J Neurosci. 2020;40:9576–88. https://doi.org/10.1523/JNEUROSCI.2570-19.2020
Larsson M Non-canonical heterogeneous cellular distribution and co-localization of CaMKIIα and CaMKIIβ in the spinal superficial dorsal horn. Brain Struct Funct. 2017. https://doi.org/10.1007/s00429-017-1566-0.
Kalus P, Muller TJ, Zuschratter W, Senitz D. The dendritic architecture of prefrontal pyramidal neurons in schizophrenic patients. Neuroreport. 2000;11:3621–5. https://doi.org/10.1097/00001756-200011090-00044
Moyer CE, Shelton MA, Sweet RA. Dendritic spine alterations in schizophrenia. Neurosci Lett. 2015;601:46–53. https://doi.org/10.1016/j.neulet.2014.11.042
Nazeri A, Mulsant BH, Rajji TK, Levesque ML, Pipitone J, Stefanik L, et al. Gray matter neuritic microstructure deficits in schizophrenia and bipolar disorder. Biol Psychiatry. 2017;82:726–36. https://doi.org/10.1016/j.biopsych.2016.12.005
Stauffer EM, Bethlehem RAI, Warrier V, Murray GK, Romero-Garcia R, Seidlitz J, et al. Grey and white matter microstructure is associated with polygenic risk for schizophrenia. Mol Psychiatry. 2021;26:7709–18. https://doi.org/10.1038/s41380-021-01260-5
Rossini L, De Santis D, Mauceri RR, Tesoriero C, Bentivoglio M, Maderna E, et al. Dendritic pathology, spine loss and synaptic reorganization in human cortex from epilepsy patients. Brain. 2021;144:251–65. https://doi.org/10.1093/brain/awaa387
Aliashkevich AF, Yilmazer-Hanke D, Van Roost D, Mundhenk B, Schramm J, Blümcke I. Cellular pathology of amygdala neurons in human temporal lobe epilepsy. Acta Neuropathol. 2003;106:99–106. https://doi.org/10.1007/s00401-003-0707-0
Swann JW, Al-Noori S, Jiang M, Lee CL. Spine loss and other dendritic abnormalities in epilepsy. Hippocampus. 2000;10:617–25. https://doi.org/10.1002/1098-1063(2000)10:5
Bothwell S, Meredith GE, Phillips J, Staunton H, Doherty C, Grigorenko E, et al. Neuronal hypertrophy in the neocortex of patients with temporal lobe epilepsy. J Neurosci. 2001;21:4789–4800. https://doi.org/10.1523/JNEUROSCI.21-13-04789.2001
Kumari S, Brewster AL. Exploring dendritic and spine structural profiles in epilepsy: insights from human studies and experimental animal models. Epilepsy Curr. 2023;24:40–46. https://doi.org/10.1177/15357597231218603
Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex. 2000;10:981–91. https://doi.org/10.1093/cercor/10.10.981
Rainier S, Chai JH, Tokarz D, Nicholls RD, Fink JK. NIPA1 gene mutations cause autosomal dominant hereditary spastic paraplegia (SPG6). Am J Hum Genet. 2003;73:967–71. https://doi.org/10.1086/378817
Tanti M, Cairns D, Mirza N, McCann E, Young C. Is NIPA1-associated hereditary spastic paraplegia always ‘pure’? Further evidence of motor neurone disease and epilepsy as rare manifestations. Neurogenetics. 2020;21:305–8. https://doi.org/10.1007/s10048-020-00619-0
Fang SY, Chou YT, Hsu KC, Hsu SL, Yu KW, Tsai YS, et al. Clinical and genetic characterization of NIPA1 mutations in a Taiwanese cohort with hereditary spastic paraplegia. Ann Clin Transl Neurol. 2023;10:353–62. https://doi.org/10.1002/acn3.51724
Goytain A, Hines RM, Quamme GA. Functional characterization of NIPA2, a selective Mg2+ transporter. Am J Physiol Cell Physiol. 2008;295:C944–C953. https://doi.org/10.1152/ajpcell.00091.2008
Haren L, Farache D, Emorine L and Merdes A A stable sub-complex between GCP4, GCP5 and GCP6 promotes the assembly of gamma-tubulin ring complexes. J Cell Sci. 2020, 133. https://doi.org/10.1242/jcs.244368.
Farache D, Jauneau A, Chemin C, Chartrain M, Rémy MH, Merdes A, et al. Functional analysis of gamma-tubulin complex proteins indicates specific lateral association via their N-terminal domains. J Biol Chem. 2016;291:23112–25. https://doi.org/10.1074/jbc.M116.744862
Pathania M, Davenport EC, Muir J, Sheehan DF, López-Doménech G, Kittler JT. The autism and schizophrenia associated gene CYFIP1 is critical for the maintenance of dendritic complexity and the stabilization of mature spines. Transl Psychiatry. 2014;4:e374. https://doi.org/10.1038/tp.2014.16
Rottner K, Stradal TEB, Chen B. WAVE regulatory complex. Curr Biol. 2021;31:R512–R517. https://doi.org/10.1016/j.cub.2021.01.086
Abekhoukh S, Sahin HB, Grossi M, Zongaro S, Maurin T, Madrigal I, et al. New insights into the regulatory function of CYFIP1 in the context of WAVE- and FMRP-containing complexes. Dis Model Mech. 2017. https://doi.org/10.1242/dmm.025809.
Di Marino D, Chillemi G, De Rubeis S, Tramontano A, Achsel T, Bagni C. MD and docking studies reveal that the functional switch of CYFIP1 is mediated by a butterfly-like motion. J Chem Theory Comput. 2015;11:3401–10. https://doi.org/10.1021/ct500431h
Acknowledgements
This work was supported in part by CNCDPK12, NIH K08NS102526, Doris Duke Foundation Clinical Scientist Award to CWH; NIH R25N065729 to A.T.; NIH 5R01NS117604 and Department of Defense ALSRP W81XWH1810175 to N.J.M.; NIH R35NS116843 to H.S. and NIH R35NS137480 and R35NS097370, and Dr. Miriam and Sheldon G. Adelson Medical Research Foundation to G.-l.M. We also thank Department of Department of Neuroscience Imaging Center, Johns Hopkins University and Haiwen Chen, MD PhD for logistical support.
Author information
Authors and Affiliations
Contributions
CWH conceptualized experiments, performed experiments, performed analysis, contributed materials, wrote first draft, wrote final draft of the manuscript, SL: performed experiments, contributed to the writing of first draft. AT: performed experiments, contributed to design and analysis, contributed to writing; SO: performed experiments. RD contributed to analysis, experimental design and writing. NH provided materials and contributed to experimental design and final draft; DB: contributed to experimental design and analysis, contributed to writing first and final draft; G-LM, HS and NM: contributed materials, experimental protocols, design and analysis and writing of first and final draft and over saw the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics and consent to participate
De-identified fibroblast samples and iPSCs were obtained from commercial sources. Informed consent for sample donation was obtained at the Coriell Institute Biorepositories (Camden, New Jersey) and Cedar Sinai (West Hollywood, California) prior to deidentification and material transfer. All methods were performed in accordance with institutional safety and internal review board guidelines.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Habela, C.W., Liu, S., Taga, A. et al. Altered development and network connectivity in a human neuronal model of 15q11.2 deletion-related neurodevelopmental disorders. Transl Psychiatry 15, 329 (2025). https://doi.org/10.1038/s41398-025-03453-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-025-03453-w
