
Abstract
Attention deficit hyperactivity disorder (ADHD) is a neurodevelopmental disorder characterized by genetic predisposition and alterations in brain structural connectivity. While existing studies have established associations between genetic variants and neuroanatomical features, the specific relationships in ADHD remained poorly understood. To address this gap, we developed adversarial deep canonical correlation analysis models (A-DCCA) to disentangle ADHD-specific and non-specific “gene-white matter” association patterns. Utilizing diffusion tensor imaging and genotype data from six-hundred ADHD and typically developed children in a Chiese cohort, the current study revealed ADHD-specific correlations between the right cerebral peduncle, right posterior limb of the internal capsule, and genes regulating neural apoptotic processes (CAMK1D, METTL15, and MAP2K4). In contrast, associations involving the left cerebral peduncle, left posterior limb of the internal capsule, right superior longitudinal fasciculus, and right posterior thalamic radiation with genes related to early neural development (FYN, PHF2, ZSCAN31, and CD82) presented associations shared by ADHD and non-ADHD groups. Incorporating interpretable deep learning models, the current study unveiled white matter regions vulnerable to genetic influences in ADHD-specific and non-specific ways, shedding light on the understanding of biological substrates of ADHD.
Similar content being viewed by others
Introduction
Attention deficit hyperactivity disorder (ADHD) is a highly prevalent and debilitating condition that affects individuals in multiple aspects across the lifespan, which often causes significant burdens to individuals and families [1, 2]. Similar to other psychiatric disorders, the specific mechanisms underlying ADHD remain unclear. Currently, the genetic susceptibility and the succedent alterations in brain structure and function were suggested as core biological substrates in ADHD [3,4,5]. Genetic factors represent the most significant contributor to ADHD, with heritability estimates as high as 74% [3, 6, 7].
Advances in neuroimaging have enabled analysis of intricate brain connectivity networks, highlighting white matter (WM) as a critical structural framework for information exchange between grey matter (GM) regions [8,9,10]. Diffusion tensor imaging (DTI) is the most commonly used approach to identify in-vivo white matter (WM) properties, which helped researchers to identify altered microstructural WM integrity in psychiatric disorders. Studies on ADHD have indicated widespread WM compromises, including the Corpus Callosum (CC), the cerebellum, and the projection tracts [11,12,13,14,15,16].
Previous evidence found a robust genetic foundation underlying WM structure among typically developed (TD) population, with heritability estimates for fractional anisotropy (FA) metrics reaching up to 90% [17,18,19]. In ADHD, concordant WM changes have been observed between affected individuals and their unaffected siblings, providing evidence for strong genetic basis on WM development in this population [20, 21]. Subsequent studies further highlighted that genetic risks might act through WM alterations to act on ADHD symptoms [22, 23]. Incorporating 1314 participants collected among eight European sites, Albaugh et al. reported significant associations between ADHD symptoms and left inferior fronto-occipital, inferior longitudinal, uncinate fasciculi [22]. Based on 544 individuals, Sudre et al. identified that ADHD-PRSs were significantly associated with reduced FA in the anterior corona radiata, which mediated 29% of the genetic effects on hyperactive/impulsive symptoms, suggesting WM integrity as a critical endophenotype [23]. However, the scope remains constrained, GWAS studies predominantly reported genomic background of WM in TD population, leaving unanswered questions about potential differences in the interactive patterns between ADHD diagnosis and genes on WM microstructure.
Canonical correlation analysis (CCA) is a useful tool for identifying co-varying patterns in high-dimensional multimodal datasets [24]. Presently, it has been employed to elucidate interactions between neuroimaging and genetic data [25]. Using CCA, Lin et al., identified multivariate association patterns in schizophrenia (SCZ) between genes and brain, linking glutaminergic and GABAergic genes with the frontal lobe and the thalamus [26]. In Alzheimer’s disease (AD), single nucleotide polymorphisms (SNPs) locating within the AD-related gene APOE were detected as associated with both brain structural and functional endophenotypes [27,28,29,30,31].
Andrew extended CCA by developing Deep CCA (DCCA), an unsupervised deep learning model that maximizes correlations between datasets through nonlinear transformations [32]. Compared to CCA, DCCA learns from gradient-based iterations, which could better capture the real-world nonlinear transformations. Previous studies have demonstrated that DCCA could prominently improve the correlation coefficients between genetic and MRI latent features [30]. Meanwhile, DCCA and its variations present good performance in prediction tasks [26, 33,34,35,36]. Li et al. reported an accuracy up to 95.65% in diagnosing SCZ [26]. Hu et al., reported excellent performance in predicting cognitive abilities, with an accuracy of 96.64% [33].
Given the intricate involvement of gene and brain in ADHD, integrating DCCA holds potential in providing crucial insights into the etiological mechanisms of ADHD. However, no such work has been undertaken in this context thus far.
Based on the aforementioned background in ADHD and DCCA, the current study utilized novel techniques and firstly developed a set of adversarial DCCA (A-DCCA) models: 1) An ADHD-A-DCCA model (AA-DCCA) aimed to learn association patterns between white matter imaging measures and genetic variants which are unique to ADHD; 2) Shared-A-DCCA model (SA-DCCA) integrated a gradient reverse layer (GRL) to disregard diagnostic distinctions, to identify shared patterns between ADHD and TD individuals. We hypothesized that the gene-WM patterns contributing to the SA-DCCA model constitute a background common to both ADHD and TD participants, while genes observed within the AA-DCCA pattern exert different effects on WM in ADHD group.
Methods
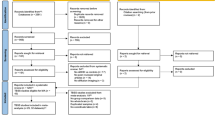
The main workflow of the study is illustrated in Fig. 1, which consists of two key components: 1) training a set of adversarial DL models and 2) interpreting the models to extract ADHD-specific and non-specific “gene-brain” association patterns (Fig. 1).

Flow diagram of the current study.
Study participants
All ADHD individuals met the diagnostic criteria of the Statistical Manual of Mental Disorders-IV or 5 (DSM-IV/DSM-5 for participants recruited in different time period) for ADHD. Diagnosis procedures involved initial assessment by senior child psychiatrists, followed by confirmation through semi-structured interviews utilizing the Chinese version of the Schedule for Affective Disorders and Schizophrenia for School-Age Children-Present and Lifetime version (KSADS-PL) [37, 38] For the controls, any evidence of current or past major psychiatric disorders in the K-SADS-PL interview and/or present or lifetime neurological disorders led to exclusion. The protocol was reviewed and approved by the Ethics Committee of Peking University Health Science Center. Written informed consent was obtained from the parents and from both the participants themselves and their parents if they were over 8 years old.
MRI acquisition and DTI preprocessing
To ensure data harmonization across cohorts, we adopted ENIGMA’s DTI preprocessing pipeline (https://enigma.ini.usc.edu/protocols/dti-protocols/). Details of the DTI parameters for each cohort can be found in the Supplementary Methods section. The FSL software (http://www.fmrib.ox.ac.uk/fsl) was employed for preprocessing in following steps: 1). Motion correction, eddy current distortion correction, brain extraction, and computation of fractional anisotropy (FA), mean diffusivity (MD), radial diffusivity (RD), and axial diffusivity (AD) maps were performed for individual participants; 2). FA images were registered to the ENIGMA FA template in the MNI-ICBM-152 standard space; 3). the registered FA maps were masked with the ENIGMA brain skeleton mask; 4). Other metric maps were registered using the FA transformations, and then masked by the skeletonized template. Post-preprocessing, skeletonized images in four modalities were resized into (N x (64, 64, 64)) and segmented into 64 slices (N x 64 x (64x64x1)) for input into the 2D autoencoder (AE) in DCCA models.
Genotyping and feature selection
One hundred sixteen samples were genotyped using Affymetrix6.0 array and four hundred eighty-four samples were genotyped using the InfiniumPsychArray-24 array by CapitalBio Ltd. (Beijing).
Quality control (QC) procedures for genetic data encompassed the following steps: 1). Exclusion of individuals with per-individual autosomal heterozygosity >3 SD larger than the mean, missing age or sex information, or a per-individual call rate <98%; 2). Exclusion of SNPs with low call rates (<95%), significant deviation from Hardy–Weinberg equilibrium (P < 1e-04), or a minor allele frequency (MAF) lower than 5%. QC procedures were performed for each genotyping dataset separately, followed by pre-phasing and imputation implemented using SHAPEIT and IMPUTE2 [39, 40]. The merged genotyping data comprised 600 samples with 2,268,177 variants for subsequent analysis.
For the A-DCCA models construction, the feature selection of Single Nucleotide Polymorphisms (SNPs) was conducted in reference to white matter-based [17] and ADHD-based GWAS results [3]. SNPs demonstrating a GWAS-P < 1E-05 were incorporated, resulting in 6874 SNPs remained for further analysis in this study. Each SNP was encoded to reflect the major alleles (A) it harbored. This encoding scheme yielded a (Nsamples × 4) one-hot vector representation, wherein the genotypes “aa,” “Aa,” “AA,” and “NA” were represented as (1, 0, 0, 0), (0, 1, 0, 0), (0, 0, 1, 0), and (0, 0, 0, 1), respectively.
DL model development and training
Based on the DCCA model, the current study developed a set of data-fusion models (A-DCCAs): AA-DCCA and SA-DCCA. Both DCCA models employed two convolutional autoencoders (CAEs) to extract features from DTI (64 × 64 x 1) and genetic inputs (6874 × 4), respectively. Each convolutional autoencoder (CAE) was architecturally composed of three sequential convolutional blocks, each incorporating a convolutional layer followed by batch normalization and max-pooling operations to progressively refine spatial feature hierarchies. The hierarchical representations were subsequently flattened into a 1D feature vector, enabling dimensional reduction while preserving discriminative patterns. This transformation facilitated the projection of input data into a 10-dimensional latent space (output dimension: N×10), where N denotes the batch size.
To optimize cross-modal feature alignment, a canonical correlation analysis (CCA) layer was integrated into the pipeline. This layer dynamically quantified the statistical correlation between the latent embeddings of paired modalities (DTI and gene) through eigen-decomposition of their covariance structures [32]. The CCA formulation is:
Then, to build AA-DCCA model, a classifier module with “Sigmoid” activation was appended to the final network layer, utilizing the binary classification of ADHD diagnosis as supervised labels. A joint optimization strategy was implemented combining Binary Cross-Entropy (BCE) and Canonical Correlation Analysis loss (CCA Loss).
In SA-DCCA architecture, a Gradient Reversal Layer (GRL) was integrated prior to the classifier. During backpropagation, this module applied negative weighting to diagnosis-related gradients, establishing an adversarial training mechanism [41]. This design significantly reduced the model’s sensitivity to diagnostic state variations.
The dataset was randomly divided into a training set (80%), and a validation set (20%). Model training initiated using the Xavier method for initialization [42]. Additional specifications included a batch size of 128, optimization with the Adam method [43], dropout layers with a dropout rate set to 0.4, a learning rate of 5E-3, early stopping after 30 epochs of unchanged validation loss, and a kernel size of 3.
Interpretation of the DL model
The DeepExplain package [44] facilitated the model’s interpretation. The “Saliency” method was employed to compute the contributions of individual voxels and SNPs to the trained models. We extracted WM clusters with saliency scores within the top 0.1% and consisting of over 100 voxels. Manual verification was conducted on gene variants with the top 10 saliency scores, and subsequent mapping of the top 200 SNPs to potential genes was performed for enrichment analyses. Regression and interaction analyses between the top SNPs and the entire AA/SA clusters were performed to identify potential associations and interactive effects.
Mapping risk genes and enrichment analyses
To link variants to potential genes, three methods for gene mapping were leveraged: 1) positional mapping, 2) eQTL mapping, and 3) chromatin interaction mapping. For positional mapping, we used a maximum distance of 10 kb [45]. The eQTL data and chromatin interaction data were utilized to identify genes through the implementation of FUMA [45] (details of the datasets can be found in the Supplementary materials).
Additionally, enrichment analyses were conducted through over-representation analysis in FUMA [45] and Metascape [46] to identify significant associations with gene sets or functional categories related to the identified variants.
Various databases encompassing both phenotypic and biological function information were utilized, comprising GWAScatalog, Gene Ontology (GO), Reactome, and the WikiPath database [47,48,49,50].
Pathway-based polygenic risk scores (p-PRSs)
The polygenic risk score (PRS) was utilized to calculate the effect of a set of SNPs in a test data set based on the GWAS summary statistics of a discovery dataset. In the current study, we selected four Gene ontology (GO) terms closely associated with neural system development and were suggested by the enrichment analyses (GO:0051402 neuron apoptotic process; GO:0016358 dendrite development; GO:0001764 neuron migration; GO:0045202 synapse) and calculate the pathway PRSs using PRSice-2 [51] based on ADHD GWAS summary statistics. Before generating the scores, clumping was used to obtain SNPs in linkage equilibrium (r2 < 0.1, 250 bp window). The P-value threshold for significance was set from 0 to 0.5, increasing by 0.00005. The associations between the polygenic profile and the target phenotypes were examined in linear regression models with age and sex as covariates for each DTI metric, P-values were adjusted by using 10,000 label-swapping permutations. The GWAS P-value threshold with the largest Nagelkerke’s r2 was considered as best-fitting.
Results
Clinal characteristics of the recruited subjects
The current study encompassed a dataset comprising 600 samples, consisting of 430 individuals diagnosed with ADHD and 170 TD individuals, all processed through the same rigorous quality control procedure. Mean age of the included samples was 121.49 ( ± 25.21) months and the male/female ratio is 4.7:1. Brain scans were performed at four different sites. No significant difference in age between ADHD and TD groups. A notable difference in sex ratio was observed, though still aligning with the recognized epidemiological features in ADHD (Further details were showed in the Supplementary Materials).
ADHD-specific and non-specific non-linear multivariate “Gene-DTI” interaction pattern
We identified WM regions and genetic variants contributed most to the A-DCCA models through saliency scores. The AA-DCCA model highlighted a cluster extending from the right cerebral peduncle (CP) to the right posterior limb of the internal capsule (PLIC) (Fig. 2). Conversely, the top contributors to the SA-DCCA model encompassed six clusters across diverse WM tracts, including the right superior longitudinal fasciculus (SLF), the right posterior thalamic radiation (PTR), the left CP, and the left PLIC.

Key white matter clusters to the AA-DCCA (A) and SA-DCCA (B), and the grey matter areas projected (C, D); DTI metrics in the whiter matter clusters in ADHD and TD groups (E & F); In the AA-DCCA cluster, AD values were significantly increased in ADHD group (E); in SA-DCCA regions, the FA values were decreased in ADHD, compared with TD.
Deterministic fiber tracking mapped the GM regions connected by these identified WM regions (Fig. 2). The AA-DCCA WM cluster connects the bilateral supplementary motor area, right dorsal lateral superior frontal gyrus, and bilateral cerebellum. The six SA-DCCA clusters displayed widespread connections across frontal, parietal, occipital, temporal lobes, and the cerebellum.
Top 10 SNPs and subsequent genes in each model were listed in the Table 1. Five of 10 SNPs contributing to the AA-DCCA model located at genes involved in apoptotic processes (METTL15, MAP2K4, and CAMK1D), with CAMK1D additionally regulating dendrite development. In contrast, the top gene in the SA-DCCA model, FYN, plays critical roles in neural developmental processes, including neuron migration and projection.
Enrichment analyses based on phenotypes suggested that the AA genes were most significantly enriched in a mental disorder, major depressive disorder (MDD); while the SA genes were mostly enriched in brain morphology (Supplementary Figure 3). These findings might suggest that the AA genes exhibit more disorder-specific roles. Results based on the biological function terms highlighted a distinct role of top genes in SA-DCCA model in the exocytosis, which is essential for synaptic function (Supplementary figure 4).
Validating ADHD-specific and non-specific associations by post-hoc analyses
Comparative analyses between ADHD and TD cohorts were conducted in the previously identified WM regions (AA, SA regions) and top 25 SNPs from each model. Results demonstrated significantly higher AD values in the AA-model cluster for the ADHD group compared to the TD group (P = 0.00427, Pbon = 0.017). Conversely, FA values within the SA regions were notably reduced in individuals with ADHD (P = 0.00195, Pbon = 0.0078). Moreover, the majority of SNPs from the AA-model contributors (16 out of 20, 80%) exhibiting significant differences between diagnostic groups (Fig. 3A). Exploration of interaction effects revealed differential associations between the AA regions and specific SNPs. For instance, the variant rs7330238 (chr13:113703404), located within MCF2L, exhibited distinct associations with MD values in ADHD and TD groups. Similarly, an interaction effect was observed between RD values and rs1051861 (chr14:58838701), located within ARID4A—an ADHD-associated gene. Conversely, such interaction effects were not evident in SA associations.

A SNPs features that differed in ADHD and TD individuals (red denotes the variant is contributor to the AA-DCCA model; blue denotes contributor of SA-DCCA); B Association patterns between top 5 SNPs and DTI metrics in the key white matter clusters (left: AA-DCCA; right: SA-DCCA); C Interaction effects in rs7330238 and rs1051861 with the AA-DCCA cluster.
Calculation of pathway-based Polygenic Risk Scores (p-PRSs) highlighted significant associations between WM properties and distinct neurodevelopmental processes. Specifically, the AA cluster showed a significant link to dendrite development (Padj = 0.023), while processes related to neuron migration and synapse were associated with the SC clusters (P = 0.0023, 0.0062, 0.0067; Padj = 0.010, 0.027, 0.027) (Fig. 4).

A, B Correlations between p-PRS and DTI metrics in two models. A Dendrite development P-PRS scores were associated with FA values in the AA-DCCA region; B In the SA regions, synapse p-PRSs were associated with RD and AD values (right and left), neuron migration p-PRSs were related to MD values (middle). C the correlations between dendrite p-PRS and AA-DCCA region differed among ADHD and TD groups. D best-fit R2 in each pathway in two models (asterisk * denotes significant adjust P-values).
Further examination of interaction effects between p-PRSs and WM regions revealed differential associations between the p-PRS of dendrite development and FA values in the AA region (P = 0.049). However, no such interactions were identified in the SA associations.
Discussion
Through interpretation of a set of A-DCCA models, we delineated shared and distinct gene-White Matter (WM) patterns in ADHD and TD cohorts. Specifically, our findings indicated differential effects of gene variants associated with apoptosis and dendrite development on WM regions, on the right CP and the right PLIC between ADHD individuals and TDs. Meanwhile, genes involved in the processes related to neuron migration and synapse had effects on widespread WM regions, involving the left CP, the left PLIC, the right SLF, and the right PTR.
Discerning the intricate interactions between genes and the brain remains a significant challenge, especially regarding potential differences in these associations across psychiatric disorders. The complexities inherent in individual variations, alongside the high-dimensional and nonlinear nature of the data, make direct association assessment challenging. Our study addressed this issue by employing DL models to conduct non-hypothesis-driven analyses on multimodal data. This approach represents a crucial step toward unraveling the etiological mechanisms of ADHD.
Despite their informative nature, DL models are often not sufficiently interpreted. In the field of psychiatry, in the absence of stable biomarkers, the essentialities of DL models have also been foregrounded by the ability of recognizing the latent mechanistic patterns under phenotypes. For instance, Gao et al. achieved a prominent accuracy when predicting three ADHD subtypes, pointing out that the connections between the right inferior occipital gyrus (IOG) and the right superior temporal gyrus (STG), and between the left supplementary motor area (SMA) and the right Heschl gyrus (HES) were the discriminative features [52]. Based on functional connectivity, Feng et al. informed ADHD biotypes, identified two subtypes response differently to pharmacological treatment [53]. As mentioned above, GWAS is an effective way to detect relevant signals in genome-wide, however, it also has limitations in detecting the real-world non-linear relationships. Liu’s work demonstrated that the convolutional neural network (CNN)-based model could capture ADHD-related SNPs which tend to be neglected by GWAS, serve as an important supplementary method [54]. The current study also holds expectations for using DL models to identify the underlying complex mechanisms.
Our results in adversarial models demonstrated two types of multivariate correlational patterns between gene and WM structure. The ADHD specific one raised between the right CP, right PLIC and genes related to apoptosis and dendrite. The pattern shared between ADHD and TD presented to be more widespread, including left projection and right association tracts associated with genes involved in basic neural system development. Interestingly, while the left CP and PLIC exhibited shared transdiagnostic association patterns, their right homologs were uniquely mapped to ADHD-specific genetic loading profiles. This hemispheric asymmetry aligns with emerging evidence of disrupted structural lateralization in ADHD neurodevelopment. A recent meta-analysis identified increased asymmetry in the frontal, subcortical, and cerebellum areas in ADHD individuals in grey matter volume (GMV), most of the GM regions exhibited “left-ward” shifts except the superior frontal lobe [55]. Same changes were observed in the Surface Area (SA) of the frontal lobe [56]. WM asymmetry alterations have been consistently reported across multiple diffusion metrics, showing leftward deviations in the frontal-striatal circuit and the corticospinal tract [57,58,59]. The current results implied the potential genetic basis underlying the “leftward” alterations in ADHD, provided further insight regarding this issue.
In this study, we found different association pattern between WM and genetic variants in ADHD cases and general population. On one hand, by interpreting AA-DCCA, we found an interaction pattern, in which the associations between gene and WM differed among ADHD and TD groups. The brain features were mostly contributed by a cluster located within the right CP and the right PLIC. This cluster mainly located in the projection tract, communicating between the right cerebellum and the bilateral supplementary motor area and the right superior frontal gyrus, which are both ADHD-related regions according to previous literature [60, 61], especially the prefrontal regions, providing “top-down” regulation of attention, inhibition/cognitive control, motivation through key neural networks includes dopaminergic circuits, reward processing networks, and default-mode network (DMN) [62, 63]. Showing consistencies with previous evidence, our findings also highlight the frontal-striatum-cerebellum circuit susceptible to genetic changes.
The top genes suggested a trend of being involved in the apoptotic processes (CAMK1D, METTL15, and MAP2K4). Enrichment analyses based on GO terms also detect such inclinations (R-HAS-140432: apoptosis induced DNA fragmentation, supplementary materials), and further suggested certain roles in neurogenesis and glia cell development. Moreover, p-PRS results also emphasized that the genetic loadings of dendrite development are associated with the AA cluster in different ways between ADHD and TD groups, which were also implied by the top contributed gene CAMK1D (Table 1). Briefly, our results suggested that possibly both apoptotic and dendrite-related processes impact the white matter in the right CP and PLIC in ADHD specific ways.
On the other hand, we identified patterns shared between individuals with and without ADHD using the SA-DCCA model. Unsurprisingly, wide-range WM tracts were identified as contributors. We identified six clusters bilaterally located. Four clusters locating in the left hemisphere were involved in the left CP, extended to the left PLIC. Other two clusters locating within the right cerebellum, within the right SLF, and the right PTR to the right SS, respectively. Tractography suggested that the clusters connected wide-range grey matter areas, including the five lobes and the bilateral cerebellum. Genetic analyses highlighted genes involved in more basic neurodevelopmental processes and synaptic function. For instance, the gene FYN, which is responsible for several important neural system developmental processes and the synaptic plasticity [64, 65]. Three zinc-finger protein family members, including the PHF2, ZSCAN31, and CD82 were also suggested. Zinc-finger proteins served as the DNA-binding transcriptional factors, regulate the development of neural system [66]. Based on previous evidence, PHF2 and ZSCAN31 both regulate the prenatal development [67, 68], including the proliferation of progenitors; CD82 is involved in the oligodendrocyte precursor differentiation [69, 70]. Enrichment analyses suggested the involvement of exocytosis, which is a synaptic process which regulate impulse conduction. P-PRS further suggested the regulation of genetic loadings in synapse-related pathways to the SA WM regions. Distinct from AA regions, no interaction effects were found here. To summarize, the interpretations of SA-DCCA model presented a shared pattern among individuals with and without ADHD, specifically, interactions between widespread WM tracts, involved in both projection and association tracts, and genes associated with more general and basic neurodevelopmental processes, including the prenatal brain development and synaptic plasticity.
Conclusion
The current study introduced a set of adversarial DCCA models, which identified ADHD-specific and non-specific gene-brain non-linear multivariate association patterns. Correlations between the right CP, PLIC and the genes responsible for apoptotic and dendrite development process were found to be different in ADHD and TD individuals; the interactions between widespread WM regions, involving bilateral projection and association tracts, and the genes involved in early neural development shared between two diagnostic groups. To conclude, the current results unveiled the etiological pathways of white matter development in ADHD and identified ADHD-specific genetic-brain associations.
Data availability
The data supporting this study are not publicly available. However, access may be granted upon reasonable request to the corresponding author, Li Yang.
Code availability
The code for developing the deep learning models was implemented in Python 3.9 using Keras. Detailed code can be provided upon request.
References
Posner J, Polanczyk GV, Sonuga-Barke E. Attention-deficit hyperactivity disorder. Lancet. 2020;395:450–62. https://doi.org/10.1016/S0140-6736(19)33004-1.
Faraone SV, Asherson P, Banaschewski T, Biederman J, Buitelaar JK, Ramos-Quiroga JA, et al. Attention-deficit/hyperactivity disorder. Nat Rev Dis Primers. 2015;1:15020 https://doi.org/10.1038/nrdp.2015.20.
Demontis D, Walters GB, Athanasiadis G, Walters R, Therrien K, Nielsen TT, et al. Genome-wide analyses of ADHD identify 27 risk loci, refine the genetic architecture and implicate several cognitive domains. Nat Genet. 2023;55:198–208. https://doi.org/10.1038/s41588-022-01285-8.
Wu Z, Yang L, Wang Y. Applying imaging genetics to ADHD: the promises and the challenges. Mol Neurobiol. 2014;50:449–62. https://doi.org/10.1007/s12035-014-8683-z.
Klein M, Onnink M, van Donkelaar M, Wolfers T, Harich B, Shi Y, et al. Brain imaging genetics in ADHD and beyond - Mapping pathways from gene to disorder at different levels of complexity. Neurosci Biobehav Rev. 2017;80:115–55. https://doi.org/10.1016/j.neubiorev.2017.01.013.
Faraone SV, Larsson H. Genetics of attention deficit hyperactivity disorder. Mol Psychiatry. 2019;24:562–75. https://doi.org/10.1038/s41380-018-0070-0.
Demontis D, Walters RK, Martin J, Mattheisen M, Als TD, Agerbo E, et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat Genet. 2019;51:63–75. https://doi.org/10.1038/s41588-018-0269-7.
Thomason ME, Thompson PM. Diffusion imaging, white matter, and psychopathology. Annu Rev Clin Psychol. 2011;7:63–85. https://doi.org/10.1146/annurev-clinpsy-032210-104507.
Wang Y, Olson IR. The original social network: white matter and social cognition. Trends Cogn Sci. 2018;22:504–16. https://doi.org/10.1016/j.tics.2018.03.005.
Wandell BA. Clarifying human white matter. Annu Rev Neurosci. 2016;39:103–28. https://doi.org/10.1146/annurev-neuro-070815-013815.
Sudre G, Norman L, Bouyssi-Kobar M, Price J, Shastri GG, Shaw P. A mega-analytic study of white matter microstructural differences across 5 cohorts of youths with attention-deficit/hyperactivity disorder. Biol Psychiatry. 2023;94:18–28. https://doi.org/10.1016/j.biopsych.2022.09.021.
Wu ZM, Bralten J, Cao QJ, Hoogman M, Zwiers MP, An L, et al. White matter microstructural alterations in children with ADHD: categorical and dimensional perspectives. Neuropsychopharmacology. 2017;42:572–80. https://doi.org/10.1038/npp.2016.223.
Connaughton M, Whelan R, O’Hanlon E, McGrath J. White matter microstructure in children and adolescents with ADHD. Neuroimage Clin. 2022;33:102957 https://doi.org/10.1016/j.nicl.2022.102957.
Chen L, Hu X, Ouyang L, He N, Liao Y, Liu Q, et al. A systematic review and meta-analysis of tract-based spatial statistics studies regarding attention-deficit/hyperactivity disorder. Neurosci Biobehav Rev. 2016;68:838–47. https://doi.org/10.1016/j.neubiorev.2016.07.022.
Zhao Y, Yang L, Gong G, Cao Q, Liu J. Identify aberrant white matter microstructure in ASD, ADHD and other neurodevelopmental disorders: a meta-analysis of diffusion tensor imaging studies. Prog Neuropsychopharmacol Biol Psychiatry. 2022;113:110477 https://doi.org/10.1016/j.pnpbp.2021.110477.
Parlatini V, Itahashi T, Lee Y, Liu S, Nguyen TT, Aoki YY, et al. White matter alterations in Attention-Deficit/Hyperactivity Disorder (ADHD): a systematic review of 129 diffusion imaging studies with meta-analysis. Mol Psychiatry. 2023;28:4098–123. https://doi.org/10.1038/s41380-023-02173-1.
Zhao B, Li T, Yang Y, Wang X, Luo T, Shan Y, et al. Common genetic variation influencing human white matter microstructure. Science (New York, NY). 2021;372:eabf3736 https://doi.org/10.1126/science.abf3736.
Kochunov P, Jahanshad N, Marcus D, Winkler A, Sprooten E, Nichols TE, et al. Heritability of fractional anisotropy in human white matter: a comparison of Human Connectome Project and ENIGMA-DTI data. Neuroimage. 2015;111:300–11. https://doi.org/10.1016/j.neuroimage.2015.02.050.
Vuoksimaa E, Panizzon MS, Hagler DJ, Hatton SN, Fennema-Notestine C, Rinker D, et al. Heritability of white matter microstructure in late middle age: A twin study of tract-based fractional anisotropy and absolute diffusivity indices. Hum Brain Mapp. 2017;38:2026–36. https://doi.org/10.1002/hbm.23502.
Tung YH, Lin HY, Chen CL, Shang CY, Yang LY, Hsu YC, et al. Whole brain white matter tract deviation and idiosyncrasy from normative development in autism and ADHD and unaffected siblings link with dimensions of psychopathology and cognition. AJP. 2021;178:730–43. https://doi.org/10.1176/appi.ajp.2020.20070999.
Chiang HL, Hsu YC, Shang CY, Tseng WYI, Gau SSF. White matter endophenotype candidates for ADHD: a diffusion imaging tractography study with sibling design. Psychol Med. 2020;50:1203–13. https://doi.org/10.1017/S0033291719001120.
Albaugh MD, Hudziak JJ, Ing A, Chaarani B, Barker E, Jia T, et al. White matter microstructure is associated with hyperactive/inattentive symptomatology and polygenic risk for attention-deficit/hyperactivity disorder in a population-based sample of adolescents. Neuropsychopharmacology. 2019;44:1597–603. https://doi.org/10.1038/s41386-019-0383-y.
Sudre G, Frederick J, Sharp W, Ishii-Takahashi A, Mangalmurti A, Choudhury S, et al. Mapping associations between polygenic risks for childhood neuropsychiatric disorders, symptoms of attention deficit hyperactivity disorder, cognition, and the brain. Mol Psychiatry. 2020;25:2482–92. https://doi.org/10.1038/s41380-019-0350-3.
Hotelling H. Relations between two sets of variates. Biometrika. 1936;28:321–77. https://doi.org/10.2307/2333955.
Yan W, Qu G, Hu W, Abrol A, Cai B, Qiao C, et al. Deep learning in neuroimaging: promises and challenges. IEEE Signal Processing Magazine. 2022;39:87–98. https://doi.org/10.1109/MSP.2021.3128348.
Li G, Han D, Wang C, Hu W, Calhoun VD, Wang YP. Application of deep canonically correlated sparse autoencoder for the classification of schizophrenia. Comput Methods Programs Biomed. 2020;183:105073 https://doi.org/10.1016/j.cmpb.2019.105073.
Du L, Zhang J, Liu F, Wang H, Guo L, Han J, et al. Identifying associations among genomic, proteomic and imaging biomarkers via adaptive sparse multi-view canonical correlation analysis. Med Image Anal. 2021;70:102003 https://doi.org/10.1016/j.media.2021.102003.
Du L, Zhang J, Zhao Y, Shang M, Guo L, Han J, et al. inMTSCCA: an integrated multi-task sparse canonical correlation analysis for multi-omic brain imaging genetics. Genomics Proteomics Bioinformatics. 2023;21:396–413. https://doi.org/10.1016/j.gpb.2023.03.005.
Yan J, Du L, Kim S, Risacher SL, Huang H, Moore JH, et al. Transcriptome-guided amyloid imaging genetic analysis via a novel structured sparse learning algorithm. Bioinformatics. 2014;30:i564–571. https://doi.org/10.1093/bioinformatics/btu465.
Wang M, Shao W, Hao X, Huang S, Zhang D. Identify connectome between genotypes and brain network phenotypes via deep self-reconstruction sparse canonical correlation analysis. Bioinformatics. 2022;38:2323–32. https://doi.org/10.1093/bioinformatics/btac074.
Hao X, Li C, Yan J, Yao X, Risacher SL, Saykin AJ, et al. Identification of associations between genotypes and longitudinal phenotypes via temporally-constrained group sparse canonical correlation analysis. Bioinformatics. 2017;33:i341–i349. https://doi.org/10.1093/bioinformatics/btx245.
Andrew G, Arora R, Bilmes J, Livescu K Deep Canonical Correlation Analysis. In: ; 2013:9.
Hu W, Cai B, Zhang A, Calhoun VD, Wang YP. Deep collaborative learning with application to the study of multimodal brain development. IEEE Trans Biomed Eng. 2019;66:3346–59. https://doi.org/10.1109/TBME.2019.2904301.
Jia W, Muhammad K, Wang SH, Zhang YD. Five-category classification of pathological brain images based on deep stacked sparse autoencoder. Multimed Tools Appl. 2019;78:4045–64. https://doi.org/10.1007/s11042-017-5174-z.
Venugopalan J, Tong L, Hassanzadeh HR, Wang MD. Multimodal deep learning models for early detection of Alzheimer’s disease stage. Sci Rep. 2021;11:3254 https://doi.org/10.1038/s41598-020-74399-w.
Zhang YD, Zhang Y, Hou XX, Chen H, Wang SH. Seven-layer deep neural network based on sparse autoencoder for voxelwise detection of cerebral microbleed. Multimed Tools Appl. 2018;77:10521–38. https://doi.org/10.1007/s11042-017-4554-8.
Yang L, Wang YF, Qian QJ, Biederman J, Faraone SV. DSM-IV subtypes of ADHD in a Chinese outpatient sample. J Am Acad Child Adolesc Psychiatry. 2004;43:248–50. https://doi.org/10.1097/00004583-200403000-00004.
Dun Y, Li QR, Yu H, Bai Y, Song Z, Lei C, et al. Reliability and validity of the Chinese version of the kiddie-schedule for affective disorders and schizophrenia-present and lifetime version DSM-5 (K-SADS-PL-C DSM-5). J Affect Disord. 2022;317:72–78. https://doi.org/10.1016/j.jad.2022.08.062.
Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat Methods. 2011;9:179–81. https://doi.org/10.1038/nmeth.1785.
Howie B, Marchini J, Stephens M. Genotype imputation with thousands of genomes. G3 (Bethesda). 2011;1:457–70. https://doi.org/10.1534/g3.111.001198.
Kamath A, Gupta S, Carvalho V Reversing Gradients in Adversarial Domain Adaptation for Question Deduplication and Textual Entailment Tasks. In: Proceedings of the 57th Annual Meeting of the Association for Computational Linguistics. Association for Computational Linguistics; 2019:5545-50. https://doi.org/10.18653/v1/P19-1556.
Glorot X, Bengio Y Understanding the difficulty of training deep feedforward neural networks. In: Proceedings of the Thirteenth International Conference on Artificial Intelligence and Statistics. JMLR Workshop and Conference Proceedings; 2010:249-56. Accessed July 13, 2023. https://proceedings.mlr.press/v9/glorot10a.html.
Kingma DP, Ba J Adam: A Method for Stochastic Optimization. Published online January 29, 2017. https://doi.org/10.48550/arXiv.1412.6980.
Ancona M, Ceolini E, Öztireli C, Gross M Towards better understanding of gradient-based attribution methods for Deep Neural Networks. Published online March 7, 2018. https://doi.org/10.48550/arXiv.1711.06104.
Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8:1826 https://doi.org/10.1038/s41467-017-01261-5.
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10:1523 https://doi.org/10.1038/s41467-019-09234-6.
Aleksander SA, Balhoff J, Carbon S, Cherry JM, Drabkin HJ, Ebert D, et al. The Gene Ontology knowledgebase in 2023. Genetics. 2023;224:iyad031 https://doi.org/10.1093/genetics/iyad031.
Gillespie M, Jassal B, Stephan R, Milacic M, Rothfels K, Senff-Ribeiro A, et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022;50:D687–D692. https://doi.org/10.1093/nar/gkab1028.
Agrawal A, Balcı H, Hanspers K, Coort SL, Martens M, Slenter DN, et al. WikiPathways 2024: next generation pathway database. Nucleic Acids Res. Published online November 6, 2023:gkad960. https://doi.org/10.1093/nar/gkad960.
MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017;45:D896–D901. https://doi.org/10.1093/nar/gkw1133.
Choi SW, O’Reilly PF. PRSice-2: polygenic risk score software for biobank-scale data. GigaScience. 2019;8:giz082 https://doi.org/10.1093/gigascience/giz082.
Gao Y, Ni H, Chen Y, Tang Y, Liu X. Subtype classification of attention deficit hyperactivity disorder with hierarchical binary hypothesis testing framework. J Neural Eng. 2023;20:056015 https://doi.org/10.1088/1741-2552/acf523.
Feng A, Feng Y, Zhi D, Jiang R, Fu Z, Xu M, et al. Functional imaging derived ADHD biotypes based on deep clustering may guide personalized medication therapy. Res Sq. Published online September 14, 2023:rs.3.rs-3272441. https://doi.org/10.21203/rs.3.rs-3272441/v1.
Liu L, Feng X, Li H, Cheng Li S, Qian Q, Wang Y. Deep learning model reveals potential risk genes for ADHD, especially Ephrin receptor gene EPHA5. Brief Bioinform. 2021;22:bbab207 https://doi.org/10.1093/bib/bbab207.
He N, Palaniyappan L, Linli Z, Guo S. Abnormal hemispheric asymmetry of both brain function and structure in attention deficit/hyperactivity disorder: a meta-analysis of individual participant data. Brain Imaging Behav. 2022;16:54–68. https://doi.org/10.1007/s11682-021-00476-x.
Postema MC, Hoogman M, Ambrosino S, Asherson P, Banaschewski T, Bandeira CE, et al. Analysis of structural brain asymmetries in attention-deficit/hyperactivity disorder in 39 datasets. J Child Psychol Psychiatry. 2021;62:1202–19. https://doi.org/10.1111/jcpp.13396.
Wu ZM, Wang P, Yang L, Liu L, Sun L, An L, et al. Altered brain white matter microstructural asymmetry in children with ADHD. Psychiatry Res. 2020;285:112817 https://doi.org/10.1016/j.psychres.2020.112817.
Silk TJ, Vilgis V, Adamson C, Chen J, Smit L, Vance A, et al. Abnormal asymmetry in frontostriatal white matter in children with attention deficit hyperactivity disorder. Brain Imaging Behav. 2016;10:1080–9. https://doi.org/10.1007/s11682-015-9470-9.
Li D, Cui X, Yan T, Liu B, Zhang H, Xiang J, et al. Abnormal rich club organization in hemispheric white matter networks of ADHD. J Atten Disord. 2021;25:1215–29. https://doi.org/10.1177/1087054719892887.
Faraone SV, Bellgrove MA, Brikell I, Cortese S, Hartman CA, Hollis C, et al. Attention-deficit/hyperactivity disorder. Nat Rev Dis Primers. 2024;10:11 https://doi.org/10.1038/s41572-024-00495-0.
Hoogman M, Muetzel R, Guimaraes JP, Shumskaya E, Mennes M, Zwiers MP, et al. Brain imaging of the cortex in ADHD: a coordinated analysis of large-scale clinical and population-based samples. Am J Psychiatry. 2019;176:531–42. https://doi.org/10.1176/appi.ajp.2019.18091033.
Arnsten AFT, Rubia K. Neurobiological circuits regulating attention, cognitive control, motivation, and emotion: disruptions in neurodevelopmental psychiatric disorders. J Am Acad Child Adolesc Psychiatry. 2012;51:356–67. https://doi.org/10.1016/j.jaac.2012.01.008.
Cubillo A, Halari R, Smith A, Taylor E, Rubia K. A review of fronto-striatal and fronto-cortical brain abnormalities in children and adults with Attention Deficit Hyperactivity Disorder (ADHD) and new evidence for dysfunction in adults with ADHD during motivation and attention. Cortex. 2012;48:194–215. https://doi.org/10.1016/j.cortex.2011.04.007.
Rajani V, Sengar AS, Salter MW. Src and Fyn regulation of NMDA receptors in health and disease. Neuropharmacology. 2021;193:108615 https://doi.org/10.1016/j.neuropharm.2021.108615.
Fão L, Coelho P, Rodrigues RJ, Rego AC. Restored Fyn levels in Huntington’s disease contributes to enhanced synaptic GluN2B-composed NMDA receptors and CREB activity. Cells. 2022;11:3063 https://doi.org/10.3390/cells11193063.
Bu S, Lv Y, Liu Y, Qiao S, Wang H. Zinc finger proteins in neuro-related diseases progression. Front Neurosci. 2021;15:760567 https://www.frontiersin.org/articles/10.3389/fnins.2021.760567
Pappa S, Padilla N, Iacobucci S, Vicioso M, Álvarez de la Campa E, Navarro C, et al. PHF2 histone demethylase prevents DNA damage and genome instability by controlling cell cycle progression of neural progenitors. Proc Natl Acad Sci. 2019;116:19464–73. https://doi.org/10.1073/pnas.1903188116.
Pi H, Li Y, Zhu C, Zhou L, Luo K, Yuan W, et al. A novel human SCAN/(Cys)2(His)2 zinc-finger transcription factor ZNF323 in early human embryonic development. Biochem Biophys Res Commun. 2002;296:206–13. https://doi.org/10.1016/s0006-291x(02)00772-6.
Mela A, Goldman JE. CD82 blocks cMet activation and overcomes hepatocyte growth factor effects on oligodendrocyte precursor differentiation. J Neurosci. 2013;33:7952–60. https://doi.org/10.1523/JNEUROSCI.5836-12.2013.
Mela A, Goldman JE. The tetraspanin KAI1/CD82 is expressed by late-lineage oligodendrocyte precursors and may function to restrict precursor migration and promote oligodendrocyte differentiation and myelination. J Neurosci. 2009;29:11172–81. https://doi.org/10.1523/JNEUROSCI.3075-09.2009.
Acknowledgements
We would like to express sincere gratitude to all the participants and their families.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81671358), the Major State Basic Research Development Program of China (973 Program, 2014CB846100), Beijing Municipal Science and Technology Commission (Z181100001518005), National Key R&D Program of China (2016YFC1306103), and the National Natural Science Foundation of China (31871259 to Suhua Chang). The National Natural Science Foundation of China (Grant No. 81761128035).
Author information
Authors and Affiliations
Contributions
Li Yang conceptualized the research and supervised the data collection, analysis and the manuscript composing. Yilu Zhao conducted data cleaning, performed the analyses, and drafted the manuscript. Xiangyu Zheng, Xuping Gao, Ning Wang, Zhao Fu, Junbin Tian, Kangfuxi Zhang, and Peng Wang contributed to sample recruitment and assisted in refining the writing. Shaoxian Li, Jichang Zhang, Xuetong Ding, and Hui Zhang were involved in data analysis. Li Sun, Binrang Yang, Shuyu Li, Suhua Chang, Qingjiu Cao, and Yufeng Wang provided valuable input and guidance for the research. This study was financially supported by grants from the National Natural Science Foundation of China (81671358 to Li Yang, 31871259 to Suhua Chang), the Major State Basic Research Development Program of China (973 Program, 2014CB846100), the Beijing Municipal Science and Technology Commission (Z181100001518005), and the National Key R&D Program of China (2016YFC1306103), which funded sample recruitment and the development of the computing environment for data analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics
The protocol was reviewed and approved by the Ethics Committee of Peking University Health Science Center. Written informed consent was obtained from the parents and from both the participants themselves and their parents if they were over 8 years old. All the methods were performed in accordance with the relevant guidelines and regulations.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, Y., Zheng, X., Gao, X. et al. Deep adversarial learning identifies ADHD-specific associations between apoptotic genes and white matter microstructure in frontal-striatum-cerebellum circuit. Transl Psychiatry 15, 320 (2025). https://doi.org/10.1038/s41398-025-03493-2
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41398-025-03493-2
