
Abstract
This study aimed to explore the neurobiological mechanism underlying intellectual disability (ID) in patients with Waardenburg syndrome (WS) identified in a Chinese family. The proband was initially diagnosed with severe ID and then 10 of the 11 extended family members underwent further medical examinations (I-1 declined to be examined). Whole-exome sequencing (WES) revealed that 6 (II-1, II-3, III-1, III-4, III-5, and III-6) of the members share the pathogenic variant of c.230 C > G of SNAI2 (also known as SLUG), a causing gene of WS. All of the mutation carriers in the third generation presented moderate to severe ID, along with severe anxiety, mild level of depression, and serious social dysfunction. But they did not show any signs of hearing loss and heterochromia iris, which are considered features of WS. Animal experiments with Snai2−/− (also known as Slugh−/−) mice were used to model the WS patients. All the Snai2−/− mice exhibited cognitive impairment, depigmented hair, and lower neural activity in the brain. The bulk RNA-seq revealed transcriptional alterations related to energy metabolism, cell growth and differentiation. The snRNA-seq and spatial transcriptomics further showed a developmental arrest of astrocyte lineage cells in the Snai2−/− mice. Moreover, Snai2−/− mice presented higher expression of genes related to IFN-α, IFN-γ, and IL-6, reduced Fos+ and GFAP+ cells, as well as low expression of EAAT1 in the hippocampus and frontal cortex. These data demonstrate that Snai2 deletion leads to developmental arrest of astrocyte lineage cells thereby impairing neuron-astrocyte interactions, ultimately resulting in cognitive impairment as seen in the WS patients.
Similar content being viewed by others

Introduction
Waardenburg syndrome (WS) is a rare genetic disorder characterized by sensorineural hearing loss, dystopia canthorum, depigmentation, and other developmental defects [1, 2]. Based on the clinical manifestations, WS is classified into four subtypes (WS1-4). WS1 and WS2 present the common features of sensorineural hearing loss and depigmentation but are distinguishable by the presence or absence of dystopia canthorum. WS3 is featured with musculoskeletal abnormalities, and the typical manifestation of WS1 [3]. WS4 is characterized by its association with intestinal aganglionosis, in addition to deafness and depigmentation [4].
Relevant to the phenotypic heterogeneity of WS, a variety of genetic mutations have been reported to be associated with specific subtypes of WS. So far, it is known that six pathogenic mutations in several genes including PAX3, MITF, SOX10, EDN3, EDNRB, and SNAI2 are associated with different subtypes of WS [5]. Because of the technological advances, new mutations in these genes have been reported in recent years [4, 6,7,8,9]. A recent study on the genotype spectrum of WS in Chinese populations identified 14 mutations that have never been reported, including c.230 C > G and c.365 C > T in SNAI2 [4]. The heterogeneity in the phenotype and genotype of WS suggests the complexity of the genotype-phenotype correlation in WS. Indeed, WS1 and WS3 were related to mutations in PAX3 and EDNRB genes [3, 6, 10]. WS2 was associated with mutations in MITF, SOX10, and SNAI2 genes [11,12,13,14]. WS4 was attributed to mutations in SOX10, EDNRB, EDN3, as well as SNAI2 [2, 4, 12, 14, 15].
SNAI2 (SLUG), a member of the Snail family, is a zinc-finger neural crest transcription factor encoded by the Snai2 (Slugh) gene and is critical for the development of hematopoietic stem cells, germ cells, and melanoblasts [16, 17]. Moreover, Snai2 plays a pivotal role in embryonic development, particularly in the differentiation and migration of neural crest-derived tissues [18]. Previous research has demonstrated that Snai2 not only regulates the fate determination of neural crest cells, such as the differentiation of melanocytes and peripheral neurons, but also contributes to the maturation and function of astrocytes in the central nervous system [19,20,21]. In humans, mutations in SNAI2 are closely associated with WS, a condition characterized by hearing loss, pigmentary abnormalities, and neural crest developmental defects, with a small proportion of patients exhibiting intellectual disability (ID) as reviewed above [4].
Here we report four adolescents with WS in a relatively large Chinese family. All of them have ID and the same pathogenic variant of SNAI2 (c.230 C > G). They also share the facial features of broad forehead, microphthalmia, and dystopia canthorum. In addition, the proband has supernumerary teeth, and a conspicuous cavum septum pellucidum (CSP) in his brain. But they did not show any signs of hearing loss and heterochromia iris, which are considered features of WS.
In animal experiments, Snai2−/− mice showed microcephaly and decreased neural activity in cerebral cortex, cerebellum, hippocampus, caudate putamen (CPu), and septum, in addition to cognitive impairment. The single-nucleus RNA-sequencing (snRNA-seq) revealed a developmental arrest of astrocyte lineage cells in Snai2−/− mouse brain. Immunohistochemistry (IHC) staining showed decreased number of Fos+ cells in cerebral cortex and hippocampus of the mouse brain. Furthermore, GFAP+ cells and excitatory amino acid transporter (EAAT1)-IHC staining intensity dramatically decreased in the foregoing brain regions. These data from Snai2−/− mice demonstrate a correlation between astrocytopathy and cognitive impairment thereby revealing a neurobiological mechanism for the ID in WS patients.
Materials and methods
Ethics approval and consent to participate
The clinical study was approved by the Ethics Committee of Shantou Central Hospital (No. SCH2019-062), and all procedures followed the Declaration of Helsinki. Written informed consent was obtained from the healthy adults or the parents of patients with ID for the use of their photographs in this publication. In addition, specific consent was obtained for the display of patient images. A record of the informed consent has been retained in accordance with ethical guidelines. The animal experiments were approved by the Ethics Committee of the Animal Experimental Center of Shantou University Medical College (No. SUMC2021-013).
Clinical phenotype analysis
The clinical manifestations of the proband and his family members were assessed by two psychiatrists and a psychometrist, with special attention to neurological, morphological, ocular, cutaneous, skeletal and facial features. The proband (III-6) was subjected to magnetic resonance imaging (MRI) and computed tomography (CT) analyses. All six affected family members (II-1, II-3, III-1, III-4, III-5, III-6) were subjected to hearing screening by means of AccuScreen (Natus, Middleton, WI, USA). The Chinese Wechsler Intelligence Scale for Children (C-WISC) was used to assess the intelligence quotient (IQ). The Hamilton Anxiety Rating Scale (HAMA, 14 items) and Hamilton Depression Rating Scale (HAMD, 24 items) were employed to evaluate the emotional states, and the Social Disability Screening Schedule (SDSS, 10 items) was applied to analyze the social functioning of all examinees.
Karyotype analysis and gene sequencing
Peripheral blood samples from II-3, II-4, III-5, and III-6 were sent to Da’an Clinical Laboratory Center (Guangzhou, China) for chromosome karyotype analysis by means of the G-banding technique. Whole exome sequencing (WES) was performed with the blood samples from nine of the ten members in generations 2 and 3 (III-3 declined to the test) by taking advantage of the MGISEQ-2000 (PE100) sequencing platform of Novogene (Beijing, China). The BGI MGIEasy V4 chip was used for the sequencing, which covered the exons of all human genes and their adjacent intronic regions of 20 base pairs. The pathogenic variant of SNAI2 (c.230 C > G) was further validated by Sanger sequencing with blood samples of the family members II-1, II-3, III-1, III-4, III-5, and III-6, but was not found in II-2, II-4, and III-2.
Western Blot analysis of SNAI2 protein expression in peripheral blood mononuclear cells
Human blood samples were collected from participants or their parents (II-1, II-2, II-3, II-4, III-1, III-2, III-4, III-5, and III-6) after obtaining written informed consent. Peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll-Paque PLUS density gradient centrifugation. The collected cells were washed with phosphate-buffered saline (PBS) and lysed in RIPA buffer supplemented with protease and phosphatase inhibitors to extract total protein. Protein concentrations in the cell lysates were determined using a BCA protein assay kit (Beyotime Biotechnology) according to the manufacturer’s instructions. A standard curve was generated using bovine serum albumin (BSA) for accurate quantification.
Western blot procedure: Protein samples (20 µg) from each person were mixed with 4× loading buffer, denatured by heating, and separated in 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Following electrophoresis, proteins were transferred onto polyvinylidene difluoride (PVDF) membranes at 100 V for 1 h. The membranes were blocked at room temperature for 1 h in Tris-buffered saline with Tween 20 (TBST) containing 5% non-fat milk. Subsequently, the membranes were incubated with a rabbit anti-SNAI2 primary antibody (Abcam, ab180714, diluted 1:1000) overnight at 4 °C. After washing three times with TBST (5 min each), the membranes were incubated with a horseradish peroxidase (HRP)-conjugated anti-rabbit IgG secondary antibody (Thermo Fisher Scientific, 31460, diluted 1:2000) at room temperature for 1 h. Finally, protein bands were visualized using an enhanced chemiluminescence (ECL) detection system for 1–5 min, and images were captured and analyzed using ImageJ software.
Animals
The breeding and animal experiments were conducted in accordance with the regulations for the management and use of laboratory animals issued by the National Center for Health Guidance. Knockout C57BL/6N-Snai2tm1cyagen mice were designed by Cyagen Biosciences (Suzhou, China). The Snai2 knockout mice (Snai2−/−) were generated by employing the CRISPR/Cas9 technology. A total of 3 male and 6 female heterozygous (Snai2+/−) mice at 4 weeks of age were obtained from Cyagen Biosciences. The nine Snai2+/− mice were raised and bred in the experimental animal center of Shantou University Medical College. All the mice were housed in a temperature- and humidity-controlled environment with free access to standard chow and water and on a 12/12 h light/dark cycle. When the mice grew up to 8 weeks of age, two females were paired with one male, and pregnancy would be considered if a copulatory plug was found in the vagina of an adult female. When an offspring grew up to one month old, genotyping was done by PCR using a segment (3–5 mm) of mouse tail. In the animal experiment, littermate mice were divided into three groups based on the mouse genotype (Snai2+/+ mice, Snai2+/− mice, and Snai2−/− mice). Throughout the entire course of the experiment and during the evaluation of outcomes, the investigator remained in a state of blindness regarding the group allocation. Anesthesia was induced via intraperitoneal injection of 1% pentobarbital sodium (1.0 ml/100 g body weight).
Phenotypic assessments for Snai2 −/− mice
The Snai2−/− mice showed global developmental delay compared with Snai2+/+ and Snai2+/− mice. At twelve weeks of age, the body length, body weight, and brain weight of each of the three groups (n = 28 per group, 11 males and 17 females) were measured. Their brain size, eyes, hair, and skin were also photographed using a Sony A6000 camera (Sony Corporation, Tokyo, Japan) and analyzed. At eight weeks of age, three groups (n = 3 per group, 2 males and 1 female) were transported to Guangdong Zhichen Biotechnology Co., Ltd for CT scanning and X-ray imaging. After anesthesia, cranial and craniofacial imaging was performed using a CT scanner (Siemens Healthineers, Erlangen, Germany) to assess skull morphology, craniofacial bone structure, and dental anatomy. Additionally, X-ray imaging was performed using an X-ray device (Siemens Healthineers, Erlangen, Germany) to capture both anteroposterior and lateral views of the skull, as well as images of the cervical spine. Over the course of the study, the lifespans of three groups (n = 50 per group, with an equal number of males and females) were monitored and documented from week 0 to week 25.
Behavioral tests
Behavioral tests, including the open-field (OF) test, barnes maze (BM) test, novel object recognition (NOR) test, and nest building (NB) test, were performed on 8-week-old mice. Except for the NB test, all other behavioral tests were performed between 8:00 am and noon. Before starting a behavioral test, mice were acclimatized to the behavioral test room environment for 30 min. During the interval between two tests, the apparatus was cleaned thoroughly with 75% ethanol to avoid smell cues. The experimenter was blind to the genotype of the tested mice. The SMART video tracking system (Panlab, Barcelona, Spain) was employed to record the performances of mice during behavioral tests.
OF test
The OF test was performed on Snai2+/+ (n = 32), Snai2+/− (n = 32), and Snai2−/− (n = 18) mice. The open field arena (40 × 40 × 30 cm, white background) was placed in the test room with an indirect artificial light source (100 lux). The mouse was placed on the center of the arena and his/her spontaneous behavior was recorded for 5 min by the SMART system for later analysis [22].
BM test
The BM test was performed on Snai2+/+, Snai2+/−, Snai2−/− mice (n = 10 per group). The BM apparatus consists of a circular, dry, open platform (with a diameter of 90 cm) with 20 holes around the perimeter of the platform. Under one of the holes is a small dark recessed chamber. The test consists of spatial acquisition trial done in the first 4 days, followed by the assessment of reference short-term memory on day 5. On the first day, an additional adaptation period was given preceded a trial. During the adaptation period, a mouse was individually placed in a cylindrical black start chamber for 10 s. Then the mouse was released from the start chamber when the light and fan were turned on, and the animal was gently guided to the escape box and remained there for 2 min. In the spatial acquisition phase, each mouse was subjected to four trials daily with a 15-min break between two adjacent trials. During each trial, a mouse underwent the same procedure as that in the adaptation period, but was allowed to explore the maze for 180 s. If a mouse did not find the escape box in 180 s, it was gently guided towards it. On day 5, the escape box was removed and replaced by a closed area (plastic plate) to assess the reference short-term memory of the mouse. After its removal from the start chamber, the mouse was allowed to explore the maze for 90 s. Primary errors, total errors, and latency time to reach the former target (i.e., the escape box) location were recorded and analyzed [23].
NOR test
The NOR test was performed on Snai2+/+ (n = 32), Snai2+/− (n = 32), and Snai2−/− (n = 18) mice. On the experiment day, a mouse was placed on the arena (familiarization phase) with two identical objects placed at the opposite corners and allowed to explore for 5 min. The mouse then returned to the home cage. After an interval of 3 h, the mouse returned to the arena (testing phase) for 5 min. During the recognition test, one of the original objects was replaced with a novel object that was the same size as the original one but different in color and shape. The SMART system was used to record and quantify exploration behavior during the familiarization and testing phases. Novel object preference was determined as the percentage of time spent on the novel object relative to the familiar one. Discrimination index was calculated according to the formula: (time with novel object minus time with familiar object)/total object exploration time [24].
NB test
The NB test was performed on Snai2+/+ (n = 32), Snai2+/− (n = 32), and Snai2−/− (n = 18) mice. Before the experiment, each mouse was raised individually in a cage filled with 1 cm thick shavings for the mouse to adapt to a single-cage environment. On the first day of the test, 5 × 5 cm paper towels (divided into 2 stacks of 8 sheets each) were placed in the cage 2 h before the commencement of the night cycle, and the performance of the tested mouse was scored at 2, 12, 24, and 48 h, following the scoring criteria established in previous studies [25, 26].
Micro PET/CT imaging of the mouse brain
PET/CT imaging of the mouse brain was conducted on twelve-week-old mice (n = 6 per group, with equal number of males and females). A mouse was deeply anesthetized and then 18F-FDG (140 uci) was injected through the mouse tail vein. Standard uptake values (SUVs) in major brain regions of mice were measured using Micro PET/CT (Siemens Medical Devices, Erlangen, Germany). Analysis was conducted using PMOD version 4.2 software. The averaged mouse brain atlas-based volume of interest (VOI) set was obtained from Guangdong ZhiChen biotechnology co., LTD., laboratory. The images were co-registered with the mouse brain atlas [27]. Then the neural activity of a brain region was normalized using standard uptake value [SUV = 18F-FDG activity in each VOI (kBq/mL)/injected dose in kBq]/body weight of each animal] as described in previous studies [28,29,30]. Analysis was conducted using PMOD version 4.2 software, and the mean mouse brain atlas-based volume of interest (VOI) set was obtained.
Auditory brainstem response testing
Twelve-week-old mice from three groups (n = 6 per group, with equal number of males and females) were used for ABR testing using the multi-channel auditory brainstem response recorder (GAT-ABR365, Ju’an Technology, Shenzhen, China), provided by Shenzhen Juyan Technology Co., Ltd. Mice were anesthetized to ensure proper sedation during the experiment. The procedures are as follows: (1) an active electrode was placed on the vertex of the skull; (2) a reference electrode was positioned behind the ear; (3) a ground electrode was attached to the lateral thigh; (4) ABR testing: Mice were placed in a soundproof chamber, and acoustic stimuli were delivered via a speaker. The stimuli consisted of 3 ms tone bursts at intensities ranging from 90–15 dB sound pressure level (SPL) and frequencies from 4–32 kHz. Each stimulus was repeated 500 times at a rate of 21 Hz. Electrophysiological signals were recorded within a 15 ms window following each stimulus. The 500 repetitions of each tone burst were averaged to obtain the ABR waveform.
Bulk RNA-seq
Twelve-week-old mice were divided into three groups (n = 12 per group, with equal number of males and females). After anesthesia, the mice were decapitated, and their brains were removed and divided into three regions: cerebral cortex (Region_1), hippocampus (Region_2), and remaining tissues (Region_3). A total of 108 samples were frozen in liquid nitrogen in RNase free tubes and sent to Novogene (Beijing, China) for bulk RNA-seq according to the following procedures. (1) RNA libraries were prepared using the NEBNext Ultra II RNA Library Prep Kit for Illumina. The procedure included mRNA isolation with poly-T oligo-attached magnetic beads, fragmentation, cDNA synthesis, end-repair, A-tailing, adapter ligation, and PCR amplification. The RNA libraries were quantified using a Qubit 2.0 Fluorometer and assessed for quality with the Agilent 2100 Bioanalyzer. (2) RNA sequencing was performed on a NovaSeq 6000 platform, generating 150 bp paired-end reads. (3) Raw sequence data were processed using Trimmomatic to remove adapters and low-quality bases. Reads were aligned to the GRCm38 mouse reference genome using STAR aligner. (4) BAM files were sorted and duplicates were marked with Picard tools. And gene expression was quantified using feature counts from the Subread package to produce a raw count matrix. (5) The principal component analysis (PCA) was performed to explore the major sources of variation in gene expression data. The raw count matrix was normalized using DESeq2, and log2-transformed counts were used for PCA. The first two principal components were visualized in a 2D scatter plot to reveal sample relationships and potential outliers. (6) Gene expression analysis was conducted with DESeq2 to compare gene expression profiles of the mice in three groups. Results were visualized using a volcano plot, plotting log2 fold changes against -log10 adjusted p-values. Significantly expressed genes (adjusted p-value < 0.05 and absolute log2 fold change > 1) were highlighted to show up-regulated and down-regulated genes. (7) A clustering heatmap was created to display gene expression patterns of the mice. The top 500 most variable genes were selected based on variance-stabilized counts from DESeq2. Hierarchical clustering was created with these genes using Euclidean distance and complete linkage. The heatmap was generated using the ComplexHeatmap package in R (R Foundation for Statistical Computing, Vienna, Austria). (8) Pairwise correlations between samples were computed using the Pearson correlation coefficient. The correlation matrix was visualized as a heatmap to show sample relationships based on gene expression profiles, applying hierarchical clustering to rows and columns to identify patterns. (9) DESeq2 results were used to identify up-regulated and down-regulated genes, and the Venn Diagram package in R was used to create diagrams showing gene overlap and unique gene sets between the animal groups. (10) The gene ontology (GO) and pathway enrichment analysis: Functional enrichment analysis was performed using the cluster profiler package in R to interpret the biological significance of DEGs. GO terms and pathways were analyzed for enrichment with adjusted p-values < 0.05. Results were visualized with dot plots and bar plots showing the top enriched GO terms and pathways.
snRNA-seq
At twelve weeks of age, one male Snai2+/+ mouse and one male Snai2−/− mouse were anesthetized, followed by decapitation and immediate brain extraction. The mouse brain samples were frozen in liquid nitrogen in RNase free tubes and sent to Novogene (Beijing, China) for snRNA-seq following the procedures of library preparation and sequencing. 10 X Genomics snRNA-seq was sued to perform single-nucleus transcriptome analysis with mouse brain samples [31]. Quality control of the snRNA-seq data was performed to filter out low-quality nuclei and to remove potential doublets. Nuclei with fewer than 200 genes detected or with more than 10% mitochondrial gene content were excluded from further analysis. Then, cell clustering was performed using the Seurat package (v4.0.0). Differentially expressed genes (DEGs) were identified using the FindAllMarkers function in Seurat with a minimum log2 fold change of 0.25 and adjusted p-value < 0.05. Marker genes were used to characterize and label cell types, and the results were visualized using uniform manifold approximation and projection (UMAP) plots and heatmaps. Gene Set Variation Analysis (GSVA) was conducted to evaluate pathway activities across different cell types. Normalized expression data from Seurat were used to perform GSVA with the GSVA R package. Gene sets from the MSigDB database were used to assess variations in pathway activity across cell clusters. GSVA scores were visualized using heatmaps to show pathway activity distributions. Pseudotime analysis was performed to infer the temporal progression of cellular states using the Monocle3 package. The expression matrix from Seurat was used to construct a cell-by-gene matrix, which was then analyzed using Monocle3 to perform dimensionality reduction, trajectory inference, and pseudotime ordering. Pseudotime trajectories were visualized with trajectory plots, and gene expression changes along pseudotime were analyzed to identify dynamic processes. Finally, stemness characteristics were assessed by evaluating the expression of stemness-related gene signatures. A set of stemness-related gene signatures were obtained from the literature and used for scoring with the Cellular (Cyto) Trajectory Reconstruction Analysis using gene Counts and Expression (CytoTRACE) package. The stemness scores for different cell types were visualized using bar plots to compare the stemness potential across cell types.
Scissor algorithm
To identify astrocyte subpopulations robustly associated with the clinical phenotypes, we employed the Scissor algorithm [32], integrating bulk RNA-seq data with snRNA-seq transcriptomic profiles. Initially, a correlation matrix was constructed between bulk samples and individual single nuclei based on transcriptomic features to quantify their associations. Subsequently, a cell-cell similarity network was generated from snRNA-seq data using a graph-based approach to capture transcriptional homogeneity among astrocytes. This network was incorporated as a regularization constraint into a sparse regression framework (L1-regularized regression) to model phenotypic variation. The regression model was optimized to select astrocyte subpopulations exhibiting statistically significant correlations with the target clinical phenotype. Cells demonstrating positive (Scissor+) or negative (Scissor−) associations with the phenotype were identified based on regression coefficients and adjusted statistical significance thresholds (p < 0.05). Following this, differential expression analysis and pathway enrichment analysis were performed on Scissor⁺ and Scissor⁻ subpopulations to delineate their molecular signatures and functional roles in biological processes. Regularization parameters, including the strength (λ), were tuned via cross-validation to ensure model robustness. All computational workflows were executed using the Scissor package in R, adhering to default parameters unless explicitly modified.
CytAssist spatial transcriptome sequencing
At twelve weeks of age, one male Snai2+/+ mouse and one male Snai2−/− mouse were anesthetized, followed by decapitation and immediate brain extraction. Spatial transcriptome sequencing of mouse brain tissue was performed using the CytAssist platform of CapitalBio Technology (Beijing, China). The protocol was optimized to create high-resolution spatial gene expression profiles of the mouse brain tissue. The size of gene expression chip was 6.5 × 6.5 mm. Each chip contained two capture regions with 4992 spots per region, and each spot contained a unique spatial barcode and millions of nucleotide primers. After incubating the sections with specific probe pairs on glass slides, the probe captures mRNA molecules, and an RNA template-mediated probe ligation reaction occurs. The Poly A structures carried by the probe molecules released by the cells can be combined with primers with spatial barcode, the probe is extended and the library is constructed. Each barcode corresponds to 1–10 cells, depending on the tissue type. Spatially barcoded mRNA was captured on the Visium slides, followed by reverse transcription and cDNA synthesis. Then, high-quality spots (cells) were selected based on UMI counts and mitochondrial gene expression, followed by PCA to reduce the dimensionality of the data. The top principal components (PCs) were used as input for clustering. Differential gene expression analysis was done to define cell clusters using the Wilcoxon rank-sum test implemented in Seurat. Marker genes specific to each cluster were identified, and clusters were annotated based on known cell-type-specific markers from the literature and publicly available Single-cell RNA sequencing (scRNA-seq) datasets, including Nefl, Nefm, Camk2n1, Gfap, Ptn, S100b, Mbp, Mobp, Cnp, and so on. Finally, GSVA was conducted to assess pathway activity across spatial locations. Differential pathway activity was assessed using the Limma package (v3.46.0). To identify spatial patterns of gene expression, the gene spatial function in the Seurat package was employed. This function leverages spatial coordinates to highlight genes with significant spatial variation. Spatially variable genes were identified using a Moran’s I test, and the top spatially variable genes were visualized using spatial feature plots.
Immunohistochemical (IHC) staining
Twelve-week-old mice in three groups (n = 6 per group, with equal number of males and females) were used for IHC staining. Under deep anesthesia, transcardial perfusion was performed with a saline solution followed by 4% paraformaldehyde. After removed from the skull, the brain samples of mice were post-fixed in 4% paraformaldehyde. After dehydration in ascending ethanol and xylene, the brain blocks were embedded in paraffin. Then, brain sections were cut at 10 μm of thickness with a microtome (Leica RM2235, Leica Biosystems, Wetzlar, Germany). IHC staining of the brain sections was performed using the following primary antibodies: mouse anti-c-Fos (1:1000, Abcam; Cat#ab208942), goat anti-GFAP (1:500, ABclonal; Cat#A0237), and rabbit anti-EAAT1/SLC1A3 (1:500, Abcam; Cat#ab181036). The secondary antibodies and relevant chromogenic agents were the HRP-Polymer anti-Mouse IHC Kit (1:1000, MaxVision; Cat#KIT-5001) and the HRP-Polymer anti-Rabbit IHC Kit (1:1000, MaxVision; Cat#KIT-5004). Bright-field images were acquired using an A2 upright confocal microscope (Carl Zeiss, Oberkochen, Germany) and an LSM800 laser confocal microscope (Carl Zeiss, Oberkochen, Germany). Case Viewer software was used to obtain randomly selected images at high power field (40×), and the labeling in cerebral cortex, hippocampus and hippocampal dentate gyrus was scored to obtain the IHC score, which were semi-quantitatively evaluated by a pathologist (Guangping Zhang) who was unaware of the animal groups. The IHC staining was scored based on the percentage of stained cells (0–100%) and staining intensity (0 = negative, 1 = weak positive, 2 = moderate positive, 3 = strong positive). The semi-quantitative IHC score was calculated using the formula: (1 × percentage of weakly positive cells) + (2 × percentage of moderately positive cells) + (3 × percentage of strongly positive cells) [33]. The total score ranged from 0–300.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 8. Data are presented as mean ± SEM. One-way ANOVA was done followed by post-hoc Tukey’s multiple comparisons. Student’s t-test (two-tailed) was done for two-group comparisons. Kaplan-Meier survival curves were analyzed using the log-rank (Mantel-Cox) test. Statistical significance was defined as ns = not significant, *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
Clinical manifestations of the proband and his family members
A 13-year-old boy, accompanied by his parents, visited our outpatient department for ID on September 15, 2015. The patient exhibited significant communication deficits, characterized by non-verbal behavior and difficulty engaging with peers, leading to social withdrawal and a preference to remain at home rather than attending school. Neurocognitive assessment by the C-WISC [34] reported his IQ of 39, indicating a moderate ID. Moreover, the four boys in the third generation presented higher scores of HAMA, HAMD, and SDSS, but their mothers (II-1 and II-3) had normal values in these assessments (Table 1). Notably, the proband appeared distinctive facial features of broad forehead, wide eyebrow distance, microphthalmia and dystopia canthorum. In addition, he showed a flat nasal bridge with a rounded nasal tip, and low-set and anteriorly rotated pinnas, but did not have any pigmentation abnormality. Interestingly, the boy was found to have supernumerary teeth in both maxilla and mandible, along with excessive salivation. Moreover, a conspicuous CSP between the lateral ventricles of the proband was seen in the T2-weighted MRI (Fig. 1A).

A The headshot and clinical images of the proband. The headshot on the top panel shows the impressive facial features of the proband. The left CT image on the bottom panel shows supernumerary teeth. The right MRI image on the bottom panel shows a conspicuous CSP (see the red arrow) between the two lateral ventricles in his brain. B Pedigree analysis of a six WS family members with SNAI2 mutations. Affected individuals are represented by filled symbols, with color coding indicating phenotypic severity: purple denotes WS-affected individuals presenting characteristic facies and dental abnormalities without ID, while red represents individuals exhibiting the complete phenotype including characteristic facies, dental abnormalities, and ID. Unaffected family members are indicated by empty symbols, and deceased individuals are denoted by slashed symbols. Individuals with undetermined WS status are marked with a question mark (?). Asterisks indicate family members selected for whole-exome sequencing analysis. C A missense substitution of c.230 C > G in SNAI2 of 8q11.21 in six members (II-1, II-3, III-1, III-4, III-5 and III-6) of this family. D Conservation analysis of SNAI2 protein sequence across species and structural characterization. The upper panel illustrates the evolutionary conservation of a SNAI2 domain among five vertebrate species: Human (Homo sapiens), Rhesus monkey (Macaca mulatta), Mouse (Mus musculus), Dog (Canis lupus familiaris), and Zebrafish (Danio rerio). Within the highlighted grey region, the amino acid residue at position 77 demonstrates species-specific variation: while human and other species maintain serine (Ser) at this position, mouse uniquely possesses threonine (Thr). The middle panel is a schematic representation of the SNAI2 protein with the S77C mutation indicated by a red dot. The bottom panel applied Swiss-Model to present the predicted structural model of the SNAI2 protein in which Ser77 is labeled.
The unique phenotype of this proband led us to interview all members of his extended family (Fig. 1B), except for the grandmother (I-1) who disagreed with the interview. II-1 and II-3 are sibling sisters who married two unrelated adult males (II-2 and II-4), respectively. The two subfamilies have four (III-1, III-2, III-3, and III-4) and two (III-5 and III-6) kids, respectively. Of all the kids in the third generation, III-1, III-4, III-5, and III-6 (the proband) share the facial features of broad forehead, wide eyebrow distance, microphthalmia and dystopia canthorum. But they did not show obvious heterochromia, skin hypo-pigmentation, and white forelock, except III-1 who showed premature graying of the hair (Fig. 1B). Importantly, all of these four kids in third generation had moderate to severe ID, while III-2 (male) and III-3 (female) had their IDs within the normal range. In addition, we assessed the emotional states and social behavior ability of all the affected family members and their mothers using Chinese versions of Hamilton Anxiety Rating Scale (HAMA, 14 items), Hamilton Depression Rating Scale (HAMD, 24 items), and Social Disability Screening Schedule (SDSS, 10 items), respectively. The data from these assessments are shown in Table 1. Notably, all the affected members in the third generation showed severe anxiety, mild level of depression, and serious social dysfunction. However, their mothers (II-1 and II-3) had these assessment scores within the normal ranges.
SNAI2 c.230C > G mutation associated with reduced SNAI2 protein expression in peripheral blood mononuclear cells of WS patients
To identify the potential genetic variations associated with the phenotype, the whole-exome sequencing (WES) was performed with blood samples from the six family members (II-3, II-4, III-1, III-4, III-5, and III-6). After filtering (rare, segregating with affected individuals and predicted to be deleterious), four missense substitutions of variants in CHD7, OR2AG2, SNAI2 and SMTN were found in the blood samples of II-3, III-1, III-4, III-5, and III-6, but not II-4. These variants are minor allele frequencies (MAF) lower than 0.001 in public and proprietary databases. Ultimately, the heterozygous variant in SNAI2 gene [c. C230G; p.S77C, 33/282,548 alleles in Genome Aggregation Database (gnomAD)] were detected. The high conservation of the homologous genes, indicated by the Genomic Evolution Rate Analysis (GERP) score of 5.59, supports their pathogenicity. Indeed, this gene is the only one closely related to neural development among the four detected genes. Furthermore, the missense substitution of c.230 C > G at SNAI2 in 8q11.21 was confirmed in the subsequent Sanger sequencing with blood samples of nine members of this family (all members in the second and third generations, except III-3 who refused to provide her blood sample). Specifically, six blood samples (II-1, II-3, III-1, III-4, III-5, and III-6) showed the missense substitution, while II-2, II-4, and III-2 did not show this single nucleotide variant (SNV) (Fig. 1C). According to the Swiss-Model, this SNV of SNAI2 had the substitution of 77th amino acid serine with cysteine (p.S77C) in the SNAI2 protein as shown in Fig. 1D. Taken together, these data demonstrate that the SNAI2 (c.230 C > G) mutation was responsible for the phenotype.
It is worth noting that the affected members (III-1, III-4, III-5, and III-6) and their mothers (II-1 and II-3) did not exhibit heterochromia irides (Fig. S1A) and hearing loss (Fig. S1B). Furthermore, III-5 and III-6, like their parents (II-3 and II-4), had normal karyotypes without chromosomal rearrangements (Fig. S2).
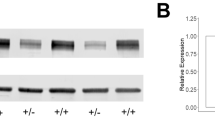
The results of Western blot showed that SNAI2 protein levels in the affected members (II-1, II-3, III-1, III-4, III-5, III-6) were significantly lower than those in the unaffected members (II-2, II-4, III-2) (p < 0.01) (Fig. S1C), suggesting that the SNAI2 c.230 C > G mutation was associated with the reduced SNAI2 protein expression and contributed to the pathogenesis of WS.
The Snai2 −/− mice recapitulated the phenotype seen in humans with SNAI2 c.230C > G
To understand how the SNAI2 c.230 C > G variant led to the phenotype seen in affected members of this family, heterozygous C57BL/6N-Snai2tm1cyagen (Snai2+/−) mice were generated by CRISPR/Cas9 technology (Fig. S3A). Compared to Snai2+/+ and Snai2+/− mice, homozygous Snai2−/− mice exhibited global developmental delay as indicated by shorter body length, lower body weight, smaller brain volume, and lower brain weight, although the brain/body weight ratios of the three groups were comparable (Fig. 2A). In addition, the Snai2−/− mice exhibited depigmented hair along the midline of abdominal wall and around the eye fissures (Fig. 2B). Furthermore, auditory brainstem response (ABR) testing reported significantly elevated hearing thresholds in Snai2−/− mice compared to both Snai2+/+ and Snai2+/− mice (p < 0.001 or p < 0.01, respectively) across all tested frequencies (4–32 kHz) (Fig. S3C), indicating the hearing loss in Snai2−/− mice.

A The left panel consists of photographs of Snai2+/+, Snai2+/−, and Snai2−/− mice, along with those of the brains from each of the groups. The bar charts on the right panel compare the body weight (g), body length (cm), brain weight (g), and the brain weight/body weight ratios of mice across the three groups (n = 28 per group, 11 males and 17 females). Data are presented as mean ± SEM. Statistical analysis was done with one-way ANOVA followed by post-hoc Tukey’s multiple comparisons. ns = not significant, ***p < 0.001. B The depigmented hair along the midline of abdominal wall and around the eye fissures of the Snai2−/− mouse, whereas Snai2+/+ and Snai2+/− mice have no depigmentation of skin and hair. C The radiographic analysis comprises two imaging modalities: the first column displays X-ray images of the skull, cervical vertebrae, lumbar vertebrae, and rib cage, while the second and third columns present three-dimensional CT reconstructions of skull morphology, craniofacial bone architecture, and dental anatomy, respectively. Distinctive morphological abnormalities in Snai2−/− mice are indicated by red arrows. These marked regions in Snai2−/− mice exhibit significant developmental retardation and structural anomalies when compared with both Snai2+/+ and Snai2+/− mice. D Kaplan-Meier survival curves comparing the lifespan of Snai2+/+, Snai2+/−, and Snai2−/− mice (n = 50 per group, with an equal number of males and females).
The CT imaging on the Snai2−/− mice showed smaller craniums with poorly calcified sutures, as well as disrupted maxilla and mandible. X-ray imaging showed disrupted cervical and thoracic segments of spinal column in Snai2−/− mice (Fig. 2C). Last but not least, the Snai2−/− mice had much shorter lifespan as indicated by significantly lower survival rates at indicated postnatal time points compared to Snai2+/+ and Snai2+/− mice (Fig. 2D).
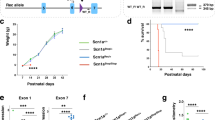
The cognitive function and emotional state of all mice in the three groups were assessed with the behavioral tests including the OF, BM, NOR, and NB tests. In the OF test, Snai2−/− mice moved a shorter total distance (TD) on the whole arena compared to the Snai2+/+ and Snai2+/− mice (p < 0.001 in both comparisons), indicating a deficit in exploratory behavior. And the Snai2−/− mice showed a lower CD/TD ratio compared with Snai2+/+ and Snai2+/− mice (p < 0.05 in both comparisons), indicating a higher level of anxiety (Fig. 3A). In the BM test, the Snai2−/− mice showed an increased primary latency before finding the escape box compared to the Snai2+/+ and Snai2+/− mice (p < 0.05 at trial 4 and p < 0.01 at trials 8, 9 and 10) (Fig. 3B). These data indicated that Snai2−/− mice had deficits in spatial working memory and reference memory. In the NOR test, Snai2−/− mice exhibited a significantly lower discrimination index compared with Snai2+/+ (p < 0.01) but not with Snai2+/− mice (Fig. 3C), indicating a moderate recognition memory deficit. In NB test, Snai2−/− mice presented significantly lower nest scores at each bout of nesting compared with Snai2+/+ and Snai2+/− mice (p < 0.001 at hours 2, 12 and 20 and p < 0.05 at hour 48) (Fig. 3D), indicating a significantly lower nesting ability. In 18F-fluorodeoxyglucose positron emission tomography (18F-FDG PET) analyses assessing regional brain glucose metabolism, Snai2−/− mice exhibited significantly reduced FDG uptake in multiple brain regions, including the cerebral cortex, cerebellum, striatum, hippocampus, and septum, compared to both Snai2+/+ and Snai2+/− mice, indicating decreased neural activity in these brain areas (Fig. 3E & F).

A The bar charts showing results from the OF test done with Snai2+/+, Snai2+/−, and Snai2−/− mice (Snai2+/+ n = 32, Snai2+/− n = 32, Snai2−/− n = 18). B The line chart showing results from BM test done with Snai2+/+, Snai2+/−, and Snai2−/− mice (Snai2+/+ n = 10, Snai2+/− n = 10, Snai2−/− n = 10). C The bar charts showing results from the NOR test done with Snai2+/+, Snai2+/−, and Snai2−/− mice (Snai2 +/+ n = 32, Snai2+/− n = 32, Snai2−/− n = 18). D The line chart showing the results from the NB test done with Snai2+/+, Snai2+/−, and Snai2−/− mice (Snai2+/+ n = 32, Snai2+/− n = 32, Snai2−/− n = 20). E Representative images of regional brain glucose metabolism measured by 18F-FDG PET in Snai2+/+, Snai2+/−, and Snai2−/− mice. The heat maps show differential glucose uptake in various brain regions of mice. F Standardized uptake value (SUV) of 18F-FDG in various brain regions of Snai2+/+, Snai2+/−, and Snai2−/− mice (n = 6 per group). Data are presented as mean ± SEM in (A–D), and (F). Each group has an equal number of males and females. Statistical analysis was done with one-way ANOVA followed by post-hoc Tukey’s multiple comparisons. ns=not significant, *p < 0.05, **p < 0.01, ***p < 0.001.
The Snai2 −/− mice showed transcriptional changes related to energy metabolism, cell growth and differentiation in the brain tissues
To explore possible changes in the downstream genes following Snai2 deletion, we performed bulk RNA sequencing (bulk RNA-seq) with brain samples of Snai2+/+, Snai2+/−, and Snai2−/− mice. The principal component analysis (PCA) of the data revealed a distinctive gene expression profile in the Snai2−/− mice brain, which had no overlap with those of Snai2+/+ and Snai2+/− mice (Fig. 4A), indicating substantial alterations in global gene expression profiles. Subsequent analysis identified numerous differentially expressed genes (DEGs) across multiple brain regions, including the cerebral cortex and hippocampus. These DEGs were visualized in volcano plots (Fig. 4B), which specifically highlighted the significant downregulation of both Snai2 and c-fos expression. Of all DEGs in Snai2−/− mice, up-regulated genes are A930036l15Rik, Ccl28, Gm24270, Lars2, Lcn2, Lrg1, and Plin4, and down-regulated ones are Arl4d, c-fos, Depp1, Hspa1a, Hspa1b, and mt-Nd4l compared to Snai2+/+ mice (Fig. 4C). These identified genes are also presented in the Venn diagram utilizing cross-tabulation analysis (Fig. 4D). The overlaps of these up- and down-regulated genes in the cerebral cortex, hippocampus, and the other regions of the Snai2−/− mice are shown in Fig. 4E. In the GO enrichment, both the cerebral cortex and the other regions of the Snai2−/− mouse brain had the same up-regulated genes related to leukocyte cell-cell adhesion, while the hippocampus had no overlap with cerebral cortex and the other brain regions in up-regulated genes. Interestingly, cerebral cortex and hippocampus presented the same down-regulated genes related to skeletal muscle cell differentiation and positive regulation of programmed cell death, while cerebral cortex and the other regions shared the same lower expression of the genes related to regulation of precursor metabolites and energy production, fat cell differentiation, and regulation of microtubule cytoskeleton organization (Fig. 4F). Of the down-regulated genes, c-fos involves in cell proliferation and differentiation following extracellular stimuli [35]. Rapid and transiently, c-fos can be expressed by almost every cell as an important piece in cell signal transduction derived from growth, maturation, intracellular calcium release, or physical stress (wounds, heat, etc.) [36]. Hspa1a and Hspa1b play a direct role in protecting cells against a variety of stresses by inhibiting stress-induced apoptosis, exemplified by the reports that Hspa1a/1b inhibited caspase-dependent apoptosis via decreasing the translocation of Bax (a Bcl-2 family member) [37]. In the other studies, Hspa1a/1b decreased cytochrome release from the mitochondria thus preventing the activation of caspase-3 through modulation of the apoptosome [38,39,40,41]. The mt-Nd4l is a subunit of NADH dehydrogenase, a component of the electron transport chain (ETC) responsible for the oxidative phosphorylation process. The dysfunction of mt-Nd4l may cause energy deficiency in cells thus resulting in metabolic disorders such as obesity and diabetes [42,43,44]. Among the upregulated genes, Ccl28-encoded protein (CCL28) is a functional ligand for CC chemokine receptor (CCR)10, which belongs to the G protein-coupled receptor superfamily and is normally expressed by melanocytes, plasma cells and skin-homing T cells [45]. Bi-allellic mutations in LARS2 were reported in the case of a young girl with bilateral severe hearing loss [46]. Lcn2 is involved in the control of cell differentiation, energy expenditure, cell death, chemotaxis, cell migration, and many other biological processes [47].

A PCA of RNA-seq data from brain tissue of Snai2+/+, Snai2+/−, and Snai2−/− mice. The black dashed-line contours delineate the approximate distribution boundaries of sample clusters according to their respective genotypes. Notably, the PCA reveals distinct transcriptional profiles, with Snai2−/− mice forming a separate cluster that is clearly segregated from the overlapping distribution of Snai2+/+ and Snai2+/− mice, indicating substantial transcriptomic differences between Snai2−/− mice and the other two groups. B Volcano plots showing DEGs in the cerebral cortex (top), hippocampus (bottom left), and the other brain regions (bottom right) in Snai2−/− mice relative to Snai2+/+ mice. The down-regulated expression of c-fos is labeled. C Heatmap of selected DEGs in the cerebral cortex, hippocampus, and the other brain regions of Snai2+/+ and Snai2−/− mice. Gene expression levels are represented as z-scores, with red indicating up-regulated expression and blue indicating down-regulated expression. Genes of interest are labeled on the right side of the heatmap. The expressions of Snai2 and c-fos decrease significantly in the Snai2−/− mice. D Correlation heatmap of the gene expression in Snai2−/− mice. The heatmap shows the Pearson correlation coefficients between gene pairs, with red indicating a positive correlation and blue indicating a negative correlation. Robust positive correlations were observed between c-fos and Arl4d, Depp1, Hspa1b, Hspa1a, as well as mt − Nd4l. E Venn diagrams showing the overlap of up-regulated (top) and down-regulated (bottom) genes in the cerebral cortex, hippocampus, and other brain regions of Snai2−/− mice. F GO enrichment analysis of up-regulated (top) and down-regulated (bottom) genes in the cerebral cortex, hippocampus, and other brain regions of Snai2−/− mice. Snai2+/+, Snai2+/−, and Snai2−/− (n = 12 per group). Each group has an equal number of males and females.
Developmental arrest of astrocyte lineage cells in the Snai2 −/− mouse brain detected by snRNA-seq
Following bulk RNA-seq, we did snRNA-seq with the brain samples of Snai2+/+ and Snai2−/− mice. This technique focuses on nascent transcripts and captures actively ongoing nuclear transcription better than single-cell RNA sequencing (scRNA-seq) due to the depletion of cytosolic and mature RNAs [48]. A total of 13174 individual brain cells were obtained from the brain samples of one Snai2+/+ mouse and one Snai2−/− mouse. With these cells, six cell clusters (astrocyte, endothelial cell, neuron, oligodendrocyte, progenitor cell, and others) were identified based on their transcriptomic signatures (Fig. 5A, Table S1). Notably, the Snai2+/+ and Snai2−/− mice were different in the proportion of each cell cluster as shown in the stacked bar charts. Compared to the Snai2+/+ mouse, the Snai2−/− mouse presented lower proportions of astrocyte, endothelial cell, oligodendrocyte, and progenitor cell, but higher proportions of neuron and others (Fig. 5A). Moreover, five astrocyte clusters were identified in brains of Snai2+/+ and Snai2−/− mice according to their transcriptomic signatures (Table S2).

A UMAP plots and cell type composition in brain tissue of Snai2+/+ and Snai2−/− mice. The UMAP plots on the left panel show six cell clusters in brain tissue of Snai2+/+(13174 cells) and Snai2−/− (13614 cells) mice. Each colored region represents a distinct cell type, including oligodendrocyte, neuron, others, astrocyte, progenitor cell, and endothelial cell. Black dashed-line boxes delineate significant differences in both the spatial distribution and morphological characteristics of astrocytes between Snai2+/+ and Snai2−/− mice. The stacked bar plots on the right panel show the proportion of each of the six cell clusters in brain tissue of Snai2+/+ and Snai2−/− mice. B Pseudotime analysis of astrocyte lineage in brain tissue of Snai2+/+ and Snai2−/− mice. The UMAP plots on the left panel show the developmental trajectory of astrocyte lineage in brain tissue of Snai2+/+ and Snai2−/− mice. Distinct spatial patterns were observed between genotypes: in Snai2+/+ mice, C1 astrocytes demonstrate a more concentrated distribution pattern with broader tissue coverage compared to the relatively dispersed and restricted distribution observed in Snai2−/− mice, as demarcated by black dashed lines. The stacked bar plots on the right panel show the proportion of each of the five astrocyte clusters in brain tissue of Snai2+/+ and Snai2−/− mice. The red rectangular frame highlights a significantly higher proportion of C1 astrocytes in Snai2+/+ mice compared to Snai2−/− mice, indicating genotype-dependent differences in astrocyte subpopulation distribution. C GSVA of metabolic processes in the five astrocyte clusters (C1-C5). The GSVA plot illustrates the metabolic processes in the five astrocyte clusters, with C1 exhibiting significantly higher enrichment scores for genes involved in acetylcholine metabolism compared to other metabolic pathways. D Pseudotime trajectory of astrocyte clusters in brain tissue of Snai2+/+ and Snai2−/− mice. Astrocytes in Snai2+/+ mouse predominantly differentiate into C1 and C5 clusters, while the differentiation process almost stops at C2 thereby leading to significant decreases in C1 and C5 clusters in the Snai2−/− mouse brain. E Box plots show the predicted ordering of cell clusters within astrocyte lineage (C1-C5) based on cellular (Cyto) trajectory reconstruction analysis using gene counts and expression (CytoTRACE) scores. Higher CytoTRACE scores indicate less differentiated cells, while the comparatively lower scores observed in C1 astrocytes indicate a more terminally differentiated cellular state.
The Snai2+/+ and Snai2−/− mice showed significant differences in the five astrocyte clusters, which were orthogonally generated on the UMAP and labeled by cell type annotation. Specifically, the Snai2−/− mouse showed much lower proportions of C1 and C5 clusters, but significantly higher proportion of C2 cluster, compared to the Snai2+/+ mice (Fig. 5B). Each of these clusters had its characteristic RNA expression profile as seen in the gene set variation analysis (GSVA). Specifically, C1 cluster showed the highest levels of RNA relating to acetate ester transport and acetylcholine metabolic process. C2 cluster presented the highest levels of RNA relating to 2-oxoglutarate metabolic process, 10-formyl tetrahydrofolate metabolic process, acetyl coenzyme A biosynthetic process from pyruvate, and acidic amino acid transport. C3 had the highest levels of RNA relating to 3-phosphoadenosine 5-phosphosulfate biosynthetic process, 3-utr (untranslated regions) mediated mRNA stabilization, 5-phosphoribose 1-diphosphate metabolic process, acrosomal vesicle exocytosis, and acrosome assembly. C4 had the highest levels of RNA relating to 3-phosphoadenosine 5-phosphosulfate metabolic process, 3-utr mediated mRNA destabilization, 5-phosphoribose 1-diphosphate metabolic process, and 5S rRNA transcription by RNA polymerase III. C5 showed middle levels of RNA relating to abscission, acetate ester transport, acetyl coenzyme A biosynthetic process, acetyl coenzyme A metabolic process, acid secretion, and acrosome reaction (Fig. 5C). These clusters are at different developmental stages from early to late, in the order of C4-C3-C2-C1-C5 as shown in Fig. 5D. Significantly, the Snai2−/− mouse presented many more C2 cells but fewer C1 and C5 cells compared to the Snai2+/+ mouse (Fig. 5E). Taken together, these data demonstrate that Snai2 deletion may lead to developmental arrest of astrocyte lineage cells in the mouse brain.
Scissor analysis identifies astrocyte subpopulations linked to Snai2-dependent phenotypic changes
To identify if astrocyte subpopulations link to phenotypic alterations in Snai2−/− mice, we employed the Scissor algorithm to integrate bulk RNA-seq-derived phenotypic data with snRNA-seq profiles. Scissor constructs a correlation matrix between bulk and single-cell transcriptomes, followed by a regularized regression that incorporates a cell-cell similarity network to pinpoint individual cells significantly associated with the phenotype. This approach enables the classification of cells as Scissor⁺ (positively associated) or Scissor⁻ (negatively associated) with the phenotype of interest. As shown in Fig. S5A & B, Scissor⁺ cells were predominantly enriched within the astrocyte two (C2) cluster in Snai2+/+ mice, but this enrichment was markedly diminished in Snai2−/− mice. In contrast, Scissor⁻ cells were more abundant in the astrocyte one (C1) cluster in the Snai2+/+ mice and substantially decreased in Snai2−/− mice. These observations suggest that C1 and C2 populations may contribute to the Snai2-dependent phenotypic differences. To further characterize the biological roles of these subpopulations, we performed differential gene expression and GO enrichment analyses on Scissor⁺ and Scissor⁻ cells (Fig. S5C). Scissor⁺ cells exhibited significant up-regulation of genes involved in the regulation of glutamatergic synaptic transmission, protein complex oligomerization, cell-cell adhesion via plasma-membrane adhesion molecules, and blood vessel morphogenesis. Conversely, genes up-regulated in Scissor⁻ cells were primarily enriched in pathways related to the cellular process involved in reproduction and regulation of growth in multicellular organism. Together, these results reveal distinct molecular and functional signatures of astrocyte subtypes that are associated with Snai2-mediated phenotypic modulation.
Spatial transcriptome changes in the Snai2 −/− mouse brain
After the bulk RNA-seq and snRNA-seq analyses, we conducted spatial transcriptomics analysis with the brain tissue of a Snai2+/+ mouse and a Snai2−/− mouse to identify specific brain regions with the changes detected by bulk RNA-seq and snRNA-seq. As shown in the UMAPs, a total of 18 cell clusters, including 2 clusters of astrocytes, one endothelial cell cluster, 10 clusters of neurons, 3 clusters of oligodendrocytes, and one progenitor cell cluster, as well as one cluster of others, were identified in brains of the Snai2+/+ and Snai2−/− mice according to their transcriptomic signatures (Table S3). The Snai2+/+ and Snai2−/− mice are different in terms of proportions of these cell clusters (Fig. 6A). In the two representative sagittal brain sections (lateral 0.24 mm) of Snai2+/+ and Snai2−/− mice, all cell clusters are mapped at specific brain region/regions of the forebrain between 2.0 mm (anterior to bregma) and −4.0 mm (posterior to bregma) (Fig. 6B). Comparing the location and number of each cell cluster in brains of Snai2+/+ and Snai2−/− mice, it was revealed that the Snai2−/− mouse had fewer C1 (Neuron 2) in cerebral cortex, C4 (Oligodendrocyte 2) in striatum, C8 (Neuron 4) in the hippocampus, C10 (Neuron 5) in cerebellum, C12 (Neuron 6) in midbrain, C13 (Others) in cerebellum, and C16 (Neuron 9) in septum, but many more C2 (Oligodendrocyte 1) in amygdala, C3 (Endothelial cell) in thalamus, and C5 (Neuron 3) in the cerebral cortex (Fig. 6C). The heatmap showing pathway activities scored per cluster by GSVA is shown in Fig. 6D. Notably, the Snai2−/− mouse brain tissue showed increased expression of genes relating to IFN-α response, IFN-γ response, IL-6/JAK/STAT3 signaling, and allograft rejection, but decreased expression of genes relating to IL-2/STAT5 signaling, TNF-α signaling via NF-κB, and Kras signaling dominant negative (dn). Of all these cell clusters, C1 (Neuron 2) showed the lowest expression of genes in IL-2/STAT5 signaling, which is consistent with the notion that IL-2 is also a modulator of the central nervous system (CNS) [49], in addition to multiple regulatory functions within the immune system. C2 (Oligodendrocyte 1), C11 (Progenitor cell), and C17 (Neuron 10) presented the lowest expression in genes relating to TNF-α signaling via NF-κB, which is in line with the regulation of brain cells, including neurons and non-neuronal cells of astrocytes, oligodendrocytes and progenitor cells, by TNF-α signaling [50]. In contrast, C6 (astrocyte 1) and C7 (astrocyte 2) showed the highest expression of genes relating to IFN-α and IFN-γ responses, as well as IL-6/JAK/STAT3 signaling, strongly suggesting that these two clusters play crucial homeostatic roles in the healthy brain, and undergo transcriptomic and functional changes in the chronic disease condition due to Snai2 deletion in this study. Strikingly, the Snai2−/− mouse showed apparently fewer c-fos cells and lower c-fos expression in the forebrain compared with Snai2+/+ mouse (Fig. 6E).

A UMAP plots of cell clusters in brain tissue of Snai2+/+ and Snai2−/− mice. The UMAP plots on the left panel show 18 cell clusters in brain tissue of Snai2+/+(3833 cells) and Snai2−/− (3899 cells) mice. The stacked bar plots on the right panel show proportion of each cell cluster in brain tissue of Snai2+/+ and Snai2−/− mice. Cell clusters are color-coded as indicated in the legend. B Spatial transcriptomic maps projected onto the same sagittal brain sections from each Snai2+/+ and Snai2−/− mouse. The regions of interest shown here are cerebral cortex, septum, hippocampus, amygdala, thalamus, hypothalamus, and parts of cerebellum and striatum. The mRNA expression profiles relate to 18 cell clusters from C0 to C17, including subtypes of astrocyte, endothelial cell, oligodendrocyte, progenitor cell, and others, in addition to those of neuron. The black dashed lines demarcate the anatomical boundaries of the cerebral cortex and hippocampus, corresponding to clusters C1, C5, C6, and C8 in panel A. C The stacked bar plots show different proportions of certain cell clusters in certain brain regions of Snai2+/+ and Snai2−/− mice. D GSVA of signaling pathways in all (C0-C17) cell clusters of the Snai2−/− mouse brain. E Levels and spatial distribution of c-fos expression in the same sagittal brain sections from each Snai2+/+ and Snai2−/− mouse. The white dashed lines delineate the cerebral cortex and hippocampus, which are known to play pivotal roles in cognitive processes.
Functional defects of astrocytes in cerebral cortex and hippocampus of the Snai2 −/− mouse
The foregoing transcriptomics data led us to focus on astrocytes in Snai2−/− mice by performing IHC staining to assess the expression of Fos, GFAP, and EAAT1 in the cerebral cortex and hippocampus, which are the regions of interest in the spatial transcriptomics experiment. GFAP is a molecular marker of mature and reactive astrocytes [51, 52]. EAAT1 is one of the abundant transporters in astrocytes and plays an important role in removing extracellular glutamate [53, 54]. As shown in Fig. S4A, Snai2−/− mice showed lower Fos IHC scores in both cerebral cortex and hippocampus compared to Snai2+/+ and Snai2−/− mice (p < 0.001), whereas the latter two groups were comparable in this measure. This data set indicates that Snai2−/− mice had fewer Fos positive cells and/or lower expression of Fos protein in the positively labeled cells in cerebral cortex and hippocampus. The Snai2−/− mice also showed lower GFAP IHC scores in both cerebral cortex and hippocampus compared to Snai2+/+ and Snai2−/− mice (p < 0.001), the latter two were comparable in this measure (Fig. S4B). Furthermore, Snai2−/− mice showed lower EAAT1 IHC score in cerebral cortex compared to Snai2+/+ and Snai2+/− mice (p < 0.001), the latter two groups significantly differed too in this measure, i.e., Snai2+/− mice presented a lower EAAT1 IHC score than the Snai2+/+ mice (p < 0.01). In the hippocampus, Snai2−/− mice showed lower EAAT1 IHC score compared to Snai2+/+ and Snai2+/− mice (p < 0.001), the latter two groups were comparable in this measure (Fig. S4C). It is worth noting that the IHC scores of Fos in the cerebral cortex and hippocampus appear higher than those of GFAP and EAAT1 in the same brain regions, indicating that some of Fos positive cells are not astrocytes in the Fos IHC staining. This is understandable given that c-fos can be expressed by almost all brain cells as biomarker in cell signal transduction related to cell growth, maturation, intracellular calcium release, or physical stress (wounds, heat, etc.) as reviewed above.
Discussion
To our knowledge, the phenotype of patients with the SNAI2 c.230 C > G mutation reported here differs from all previously reported WS subtypes. Firstly, the proband and the other three affected members in this family had no hearing loss and heterochromia irides. Secondly, all four affected members in the third generation of this family presented moderate to severe ID, plus supernumerary teeth and CSP in the proband. None of the previous studies reported these features in a same WS patient. Thirdly, none of the members in this family reported intestinal obstruction associated with Hirschsprung’s disease (WS4). These clinical features suggest that the phenotype reported here is a novel subtype of WS.
The four boys in the third generation of this family presented both lower IQ and higher scores of HAMA, HAMD, and SDSS, but their mothers (II-1 and II-3) had normal values in these assessments while carrying the SNAI2 mutation, indicating the presence of recessive heredity. The data suggest a possible correlation between ID and emotional anomalies in the WS patients of this study. Another new finding is the presence of CSP seen in the proband’s brain. The concurrent presence of CSP and ID in the proband suggests a possible contribution of CSP to ID in the patient. Unfortunately, the other affected family members with ID did not undergo brain MRI scans (they were not willing to be scanned). And CSP was regarded a normal structural brain variation in a population-based sample of school-aged children as reported in a prospective cohort in Rotterdam, the Netherlands, with 4.6% of school-aged children having CSP [55]. Interestingly, the prevalence rates of CSP in Chinese Mainland and Taiwan are 1.24 and 0.93%, respectively [56], far lower than that in the Netherlands.
Although the limited research on CSP in children mentioned above (previous and current studies) is unlikely to lead to any relationship between CSP and human ID, the animal experimental results of this study do provide new insights into this issue. First, in multiple brain regions, including the septum where CSP is located, the FDG uptake of Snai2−/− mice was significantly lower than that of Snai2+/+ and Snai2+/− mice (Fig. 3E & F). Second, spatial transcriptomics analysis revealed that the Snai2−/− mouse had fewer C16 (Neuron 9) in the septum compared to the Snai2+/+ mouse (Fig. 6C). Third, the Snai2−/− mice presented cognitive impairment compared to Snai2+/+ and Snai2+/− mice. Taken together, it is plausible to conclude that abnormal cell development and decreased neural activity in the septum of Snai2−/− mice may contribute to cognitive impairment in the mice. This interpretation helps us understand the ID phenotype seen in the affected family members in the present study.
As early as 2002, Sanchez-Martın et al. reported SNAI2 deletions in two unrelated WS2 individuals who showed heterochromia irides and profound hearing loss but no dysmorphic features [13]. None of the individuals had a family history of hearing impairment or pigmentation anomaly, suggesting the presence of recessive heredity. Similar to this previous study, the WS individuals in the present study are of recessive heredity and have SNAI2 mutation. However, none of the individuals with SNAI2 mutation in the present study showed heterochromia irides and hearing loss.
A recent study with ninety WS patients and 24 additional family members reported the SNAI2 c.230 C > G mutation in one person [4]. According to this study, 14 of all 90 WS patients (15.6%) had the white forelock and all reported hair color in WS patients was prematurely graying and turning white. In the present study, none of family members with SNAI2 mutation had the white forelock although III-1 showed the gray hair in his forelock. There were another two earlier case-reports mentioning mental retardation in WS patients, but they did not provide genetic information of the patients while describing heterochromia irides and sensorineural deafness [57, 58], which were not seen in anyone of the family members in the present study.
As depicted in Fig. 1D, the amino acid residue at position 77 of SNAI2 exhibits species-specific variation, with serine (S) in humans and threonine (T) in mice. This evolutionary divergence in key functional domains results in fundamental limitations in developing precise Snai2 knock-in (KI) mouse models with human-equivalent point mutations, rendering such genetic engineering approaches biologically unfeasible. Nevertheless, the Snai2−/− mice was shown to have a white forehead blaze and areas of depigmentation on the ventral body, tail and feet in a previous animal study [59]. Because of the phenotypic similarity of these mice to patients with WS2, the Snai2−/− (Slugh−/−) mouse was used as an animal model of WS2 to investigate a possible role of the human SNAI2 gene in WS [60]. In support of this inference, the Snai2−/− mice in the present study showed impaired auditory function, depigmented hair along the midline of abdominal wall, and around the eye fissures. In addition, Snai2−/− mice in the present study showed cognitive impairment in various behavioral tests, which were not described in the previous study [60]. Compared to the Snai2+/+ and Snai2+/−, Snai2−/− mice showed lower FDG-PET signal in their cerebral cortex, cerebellum, striatum, hippocampus, and septum, suggesting lower neural activity in these brain regions (Fig. 3E & F). This discovery helps us understand the cognitive impairment exhibited by Snai2−/− mice. Moreover, Snai2−/− mice showed overall developmental delay as indicated by shorter body length, lower body weight, and smaller brain volume, as well as shorter lifespans (Fig. 2), suggesting the importance of Snai2 gene in the growth, development, and survival of the mice.
Bulk RNA-seq analysis uncovered significant transcriptional changes associated with two key biological processes: (1) energy metabolism, including alterations in oxidative phosphorylation pathways and lipid metabolism; (2) cell growth and differentiation regulation, particularly in proliferation-related and cell cycle progression genes (Fig. 4). Of the down-regulated genes, c-fos is involved in cell proliferation and differentiation in response to extracellular stimuli [35]. Hspa1a and Hspa1b play a direct role in protecting cells against a variety of stresses by inhibiting stress-induced apoptosis [37,38,39,40,41]. And lower expression of mt-Nd4l may cause energy deficiency in cells [42,43,44]. Relevantly, the concurrent up-regulated gene expression in Snai2−/− mice represents molecular responses to the Snai2 deletion-induced cell damage/death in the brain. As examples supporting this inference, Lcn2 has been shown as an important player in neuroinflammation and neuronal death in various animal brain injury models [61, 62]. Indeed, Lcn2 is currently employed as a biomarker of brain injury [63, 64]. And CCL28 has been demonstrated as a potent growth promoting factor with the ability to support the human hematopoietic cells in vitro and in vivo [65].
c-fos exhibits ubiquitous expression across all cell types, including both neuronal and non-neuronal populations in the nervous system, where it was used as a transynaptic marker for neuronal activity [66]. It has been reported that immobilization stress can induce intense expression of c-fos in several brain areas such as hypothalamus [67]. Therefore, the observed reduction in c-fos expression in the Snai2−/− mouse brain suggests that Snai2 deletion may impair cellular responsiveness to both intrinsic and extrinsic stimuli, potentially compromising the brain’s adaptive capacity to dynamic physiological changes. As mentioned above, c-fos is also involved in cell proliferation and differentiation [35]. Furthermore, in the context of memory formation and consolidation, c-fos plays a pivotal regulatory role by promoting memory generalization through the organization of Fos-dependent neuronal ensembles. These specialized ensembles exhibit heightened excitatory synaptic inputs originating from the medial entorhinal cortex, a key hub in the memory circuitry. This enhanced connectivity facilitates the integration and processing of memory-related information, thereby significantly contributing to the dynamic modulation of cognitive functions associated with memory encoding, storage, and retrieval [68]. Overall, the down-regulation of c-fos in brain tissue found in this study may partially account for the observed reduction in astrocyte populations and cognitive impairments in Snai2−/− mice. This correlation suggests that c-fos-mediated transcriptional regulation plays a critical role in maintaining astrocyte homeostasis and supporting cognitive functions, potentially through its involvement in neural circuit plasticity and cellular stress responses. Pseudotime trajectory analysis further confirmed that C1 and C5 represent terminally differentiated astrocyte populations at late developmental stages, highlighting the critical role of Snai2 in promoting astrocyte maturation.
Consistent with the findings from bulk RNA-seq analysis, snRNA-seq revealed a profound disruption in astrocyte lineage development in Snai2−/− mice. This was characterized by a marked expansion of the C2 progenitor-like cell population coupled with significant reductions in the mature C1 and C5 astrocyte populations, suggesting an arrest in astrocyte differentiation and maturation processes (Fig. 5B). Notably, Scissor+ cells are enriched in C1 and C2 astrocyte cluster, which may play a crucial role in the phenotypic differences determined by the Snai2 gene (Fig. S5A). Particularly, GSVA revealed distinct RNA expression profiles for each cluster, with the C1 astrocyte having the highest RNA levels related to acetylcholine metabolic process (Fig. 5C). Acetylcholine may enhance memory encoding via strengthening afferent input, promoting theta rhythm, activating spiking, and modifying synapses. These effects vary across cortical structures, with those in entorhinal, perirhinal cortex, and hippocampus being vital for new episodic memory encoding [69]. Therefore, decreases in cells of these two clusters would significantly affect the neuron-astrocyte interaction and impair cognitive function, which were reflected by decreased c-fos expression in the mouse brain as indicated by the spatial transcriptomics experiment. Indeed, spatial transcriptomics data showed c-fos signal intensity decreases in extensive forebrain regions of the Snai2−/− mouse compared to those of Snai2+/+ mouse, in addition to decreased number of c-fos expressing cells (Fig. 6).
Decreased c-fos expressing cells in Snai2−/− mouse brain may be partially attributed to cell reduction/death, and/or functional defects. Indeed, the Snai2−/− mouse showed decreased cells in C1 (Neuron 2), C4 (Oligodendrocyte 2), C8 (Neuron 4), C10 (Neuron 5), C12 (Neuron 6), C13 (Others), and C16 (Neuron 9), compared to their counterparts in the Snai2+/+ mouse. Strikingly, C6 (Astrocyte 1) and C7 (Astrocyte 2) presented the highest expression of genes relating to IFN-α and IFN-γ responses, as well as an elevated level of IL-6/JAK/STAT3 signaling (Fig. 6D). It has been demonstrated that IFN-α exerts an inhibitory effect on human astrocytes on cell proliferation, cell signaling and glucose utilization, which are potentially mediated through the negative regulation of the GSK-3β/TSC2/mTOR signaling axis [70]. IFN-γ has been shown to activate astrocytes to acquire immune functions, while down- regulation of IFN-γ was confined to pathways involved in growth regulation, cell differentiation and cell adhesion [71]. Similarly, the physiological function of interleukin-6 (IL-6) within the central nervous system (CNS) is complex. It exerts neurotrophic and neuroprotective effects, and can also function as a mediator of inflammation and demyelination [72]. Notably, IL-6 has been implicated in triggering astrocyte apoptosis in the hippocampus and mediating depression-like behaviors [73]. These findings suggest that IFN-α, IFN-γ, and IL-6 signaling pathways may collectively contribute to the Snai2−/− phenotype by disrupting astrocytic energy metabolism and differentiation processes. Moreover, the IHC scores of Fos protein, GFAP, and EAAT1 in both the cerebral cortex and hippocampus of the Snai2−/− mouse brain decreased relative to those of Snai2+/− and Snai2+/+ mice (Fig. S4). And the GO enrichment analysis of up-regulated genes in Scissor+ cells, enriched biological processes include regulation of glutamatergic synaptic transmission (Fig. S5C).
Mature astrocytes perform multiple functions within the CNS. For example, astrocytes contribute to synaptic signaling as the third partner of the tripartite synapse [74], in which astrocytes sense and facilitate intake and removal of glutamate from the regions surrounding the synaptic cleft via the actions of EAAT1 and EAAT2, and mGlur5 receptors located on their processes [75, 76]. In this way, astrocytes protect synaptic transmission and prevent excitotoxicity of glutamate to neurons. In addition, astrocytes facilitate neuronal calcium signaling, which in turn leads to the release of ATP, gamma aminobutyric acid (GABA), and glutamate [77,78,79,80]. SNAI2 dysfunction disrupts the regulation of key extracellular matrix proteins, including collagens, laminins, and thrombospondins, impairing neuronal guidance and astrocyte-mediated synaptic maturation processes [21]. The experimental data presented in this study provide evidence that Snai2 deletion leads to the down-regulation of c-fos and abnormal activation of IFN-α, IFN-γ, and IL-6 signaling pathways in astrocytes. This disruption impairs the energy metabolism and differentiation processes of astrocytes, resulting in the developmental arrest of the astrocyte lineage. Consequently, it causes astrocyte dysfunction, disrupts neuron-astrocyte interactions, and ultimately impairs advanced brain functions such as cognition and emotional regulation.
Individuals with the mutation of SNAI2 c.230 C > G show a novel phenotype of WS featured with moderate to severe ID, but have no hearing loss and heterochromia irides. Snai2−/− mice, which was used as an animal model of WS2, recapitulated cognitive impairment and emotional anomalies relevant to those seen in the WS individuals with the SNAI2 mutation. Neuroimaging, morphological, and transcriptomics data from Snai2−/− mice provided concordant evidence that Snai2 deletion leads to developmental arrest of the astrocyte lineage, thereby disrupting neuron-astrocyte interactions and ultimately impairing cognition and emotional regulation. ID is a neurological developmental disorder stemming from diverse etiologies, presenting as a clinical syndrome marked by low learning ability, poor social adaptation, and limited self-care ability that becomes evident before the age of 18, with a global prevalence of around 1 to 3% [81]. Although extensive research has well-established the pivotal roles of sensorineural hearing impairment and pigmentation abnormalities in WS [11, 12], it is not evident in this study. This study demonstrates that SNAI2 mutation makes a substantial contribution to cognitive impairment. This research expands the phenotypic spectrum of WS to incorporate cognitive impairment resulting from the disruption of neuron-astrocyte interactions, highlighting the genetic heterogeneity that has not been previously recognized [82]. These findings highlight the remarkable genetic complexity of WS and provide crucial evidence underlying mechanisms of SNAI2-mediated pathways associated with the development of ID.
Data availability
The sequence data have been deposited in the Genome Sequence Archive [83] in National Genomics Data Center [84], China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA-Human: HRA008988 and GSA: CRA018396). This study did not generate any new code. The use of these data was approved by the National Committee for Data Protection. Any additional information required to reanalyze the data reported in this paper is available from the lead contact, g_ghzhang@stu.edu.cn (Guohong Zhang).
References
Pingault V, Ente D, Dastot-Le Moal F, Goossens M, Marlin S, et al. Review and update of mutations causing Waardenburg syndrome. Hum. Mutat. 2010;31:391–406.
Bondurand N, Dastot-Le Moal F, Stanchina L, Collot N, Baral V, Marlin S, et al. Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4. Am. J. Hum. Genet. 2007;81:1169–85.
Hoth CF, Milunsky A, Lipsky N, Sheffer R, Clarren SK, Baldwin CT. Mutations in the paired domain of the human PAX3 gene cause Klein-Waardenburg syndrome (WS-III) as well as Waardenburg syndrome type I (WS-I). Am. J. Hum. Genet. 1993;52:455–62.
Li W, Mei L, Chen H, Cai X, Liu Y, Men M, et al. New genotypes and phenotypes in patients with 3 subtypes of Waardenburg Syndrome identified by diagnostic next-generation sequencing. Neural Plast. 2019;2019:7143458.
Huang S, Song J, He C, Cai X, Yuan K, Mei L, et al. Genetic insights, disease mechanisms, and biological therapeutics for Waardenburg syndrome. Gene Ther. 2022;29:479–97.
Guo M, Li Q, Jiang C, Li S, Ruan B. A novel PAX3 mutation in a Chinese Han family with Waardenburg syndrome type 1. Int. J. Pediatr. Otorhinolaryngol. 2021;147:110758.
Ma J, Zhang TS, Lin K, Sun H, Jiang HC, Yang YL, et al. Waardenburg syndrome type II in a Chinese patient caused by a novel nonsense mutation in the SOX10 gene. Int. J. Pediatr. Otorhinolaryngol. 2016;85:56–61.
Ma J, Zhang Z, Jiang HC, Sun H, Ming C, Zhao LP, et al. A novel dominant mutation in the SOX10 gene in a Chinese family with Waardenburg syndrome type II. Mol. Med. Rep. 2019;19:1775–80.
Thongpradit S, Jinawath N, Javed A, Jensen LT, Chunsuwan I, Rojnueangnit K, et al. Novel SOX10 mutations in Waardenburg syndrome: functional characterization and genotype-phenotype analysis. Front. Genet. 2020;11:589784.
Tassabehji M, Read AP, Newton VE, Patton M, Gruss P, Harris R, et al. Mutations in the PAX3 gene causing Waardenburg syndrome type 1 and type 2. Nat. Genet. 1993;3:26–30.
Tachibana M. Evidence to suggest that expression of MITF induces melanocyte differentiation and haploinsufficiency of MITF causes Waardenburg syndrome type 2A. Pigment. Cell Res. 1997;10:25–33.
Zhang H, Chen H, Luo H, An J, Sun L, Mei L, et al. Functional analysis of Waardenburg syndrome-associated PAX3 and SOX10 mutations: report of a dominant-negative SOX10 mutation in Waardenburg syndrome type II. Hum. Genet. 2012;131:491–503.
Sánchez-Martín M, Rodríguez-García A, Pérez-Losada J, Sagrera A, Read AP, Sánchez-García I. SLUG (SNAI2) deletions in patients with Waardenburg disease. Hum. Mol. Genet. 2002;11:3231–6.
Chaoui A, Watanabe Y, Touraine R, Baral V, Goossens M, Pingault V, et al. Identification and functional analysis of SOX10 missense mutations in different subtypes of Waardenburg syndrome. Hum. Mutat. 2011;32:1436–49.
Song J, Feng Y, Acke FR, Coucke P, Vleminckx K, Dhooge IJ. Hearing loss in Waardenburg syndrome: a systematic review. Clin. Genet. 2016;89:416–25.
Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000;2:76–83.
Assani G, Zhou Y. Effect of modulation of epithelial-mesenchymal transition regulators Snail1 and Snail2 on cancer cell radiosensitivity by targeting of the cell cycle, cell apoptosis and cell migration/invasion. Oncol. Lett. 2019;17:23–30.
Nieto MA, Sargent MG, Wilkinson DG, Cooke J. Control of cell behavior during vertebrate development by Slug, a zinc finger gene. Science. 1994;264:835–9.
Li J, Perfetto M, Materna C, Li R, Thi Tran H, Vleminckx K, et al. A new transgenic reporter line reveals Wnt-dependent Snai2 re-expression and cranial neural crest differentiation in Xenopus. Sci. Rep. 2019;9:11191.
Dong L, Gao L. JMJD3 and SNAI2 synergistically protect against Parkinson’s disease by mediating the YAP/HIF1α signaling pathway in a mouse model. Hum. Mol. Genet. 2023;32:3040–52.
Krencik R, Hokanson KC, Narayan AR, Dvornik J, Rooney GE, Rauen KA, et al. Dysregulation of astrocyte extracellular signaling in Costello syndrome. Sci. Transl. Med. 2015;7:286ra66.
Antiorio AT, Alemán-Laporte J, Zanatto DA, Pereira MAA, Gomes MS, Wadt D, et al. Mouse behavior in the open-field test after meloxicam administration. J. Am. Assoc. Lab. Anim. Sci. 2022;61:270–4.
Malikowska-Racia N, Podkowa A, Sałat K. Phencyclidine and scopolamine for modeling amnesia in rodents: direct comparison with the use of barnes maze test and contextual fear conditioning test in mice. Neurotox. Res. 2018;34:431–41.
Joshi S, Williams CL, Kapur J. Limbic progesterone receptors regulate spatial memory. Sci. Rep. 2023;13:2164.
Neely CLC, Pedemonte KA, Boggs KN, Flinn JM. Nest building behavior as an early indicator of behavioral deficits in mice. J. Vis. Exp. 2019. https://doi.org/10.3791/60139
Deacon RM. Assessing nest building in mice. Nat. Protoc. 2006;1:1117–9.
Paxinos, G & Franklin, KBJ (eds) The Mouse Brain in Stereotaxic Coordinates. 2nd edn. San Diego: Academic Press; 2001.
Minoshima S, Cross D, Thientunyakit T, Foster NL, Drzezga A. 18F-FDG PET imaging in neurodegenerative dementing disorders: Insights into subtype classification, emerging disease categories, and mixed dementia with copathologies. J. Nucl. Med. 2022;63:2S–12S.
Coleman RA, Liang C, Patel R, Ali S, Mukherjee J. Brain and brown adipose tissue metabolism in transgenic Tg2576 mice models of Alzheimer disease assessed using 18F-FDG PET imaging. Mol. Imaging. 2017;16:1536012117704557.
Mirbolooki MR, Upadhyay SK, Constantinescu CC, Pan ML, Mukherjee J. Adrenergic pathway activation enhances brown adipose tissue metabolism: a 18F-FDG PET/CT study in mice. Nucl. Med. Biol. 2014;41:10–16.
Wu H, Kirita Y, Donnelly EL, Humphreys BD. Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: rare cell types and novel cell states revealed in fibrosis. J. Am. Soc. Nephrol. 2019;30:23–32.
Sun D, Guan X, Moran AE, Wu LY, Qian DZ, Schedin P, et al. Identifying phenotype-associated subpopulations by integrating bulk and single-cell sequencing data. Nat. Biotechnol. 2022;40:527–38.
Pirker R, Pereira JR, von Pawel J, Krzakowski M, Ramlau R, Park K, et al. EGFR expression as a predictor of survival for first-line chemotherapy plus cetuximab in patients with advanced non-small-cell lung cancer: analysis of data from the phase 3 FLEX study. Lancet Oncol. 2012;13:33–42.
Gong YX, Ye SQ, Zhang JP. The revised Chinese Webster’s intelligence scale for children. Chin. J. Clin. Psychol. 1994;2:1–6.
Velazquez FN, Caputto BL, Boussin FD. c-Fos importance for brain development. Aging. 2015;7:1028–9.
Cruz-Mendoza F, Jauregui-Huerta F, Aguilar-Delgadillo A, García-Estrada J, Luquin S. Immediate early gene c-fos in the brain: focus on glial cells. Brain Sci. 2022;12:687.
Stankiewicz AR, Lachapelle G, Foo CP, Radicioni SM, Mosser DD. Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J. Biol. Chem. 2005;280:38729–39.
Li CY, Lee JS, Ko YG, Kim JI, Seo JS. Heat shock protein 70 inhibits apoptosis downstream of cytochrome c release and upstream of caspase-3 activation. J. Biol. Chem. 2000;275:25665–71.
Matsumori Y, Northington FJ, Hong SM, Kayama T, Sheldon RA, Vexler ZS, et al. Reduction of caspase-8 and -9 cleavage is associated with increased c-FLIP and increased binding of Apaf-1 and Hsp70 after neonatal hypoxic/ischemic injury in mice overexpressing Hsp70. Stroke. 2006;37:507–12.
Steel R, Doherty JP, Buzzard K, Clemons N, Hawkins CJ, Anderson RL. Hsp72 inhibits apoptosis upstream of the mitochondria and not through interactions with Apaf-1. J. Biol. Chem. 2004;279:51490–9.
Tsuchiya D, Hong S, Matsumori Y, Shiina H, Kayama T, Swanson RA, et al. Overexpression of rat heat shock protein 70 is associated with reduction of early mitochondrial cytochrome C release and subsequent DNA fragmentation after permanent focal ischemia. J. Cereb. Blood Flow. Metab. 2003;23:718–27.
Wang BD, Rennert OM, Su YA. Mitochondrial oxidative phosphorylation, obesity and diabetes. Cell Sci. Rev. 2008;4:4.
Takamura T, Misu H, Matsuzawa-Nagata N, Sakurai M, Ota T, Shimizu A, et al. Obesity upregulates genes involved in oxidative phosphorylation in livers of diabetic patients. Obesity. 2008;16:2601–9.
Hunter CA, Kartal F, Koc ZC, Murphy T, Kim JH, Denvir J, et al. Mitochondrial oxidative phosphorylation is impaired in TALLYHO mice, a new obesity and type 2 diabetes animal model. Int. J. Biochem. Cell Biol. 2019;116:105616.
Moed H, Boorsma DM, Tensen CP, Flier J, Jonker MJ, Stoof TJ, et al. Increased CCL27-CCR10 expression in allergic contact dermatitis: implications for local skin memory. J. Pathol. 2004;204:39–46.
Kosaki R, Horikawa R, Fujii E, Kosaki K. Biallelic mutations in LARS2 can cause Perrault syndrome type 2 with neurologic symptoms. Am. J. Med. Genet. A. 2018;176:404–8.
Schröder SK, Gasterich N, Weiskirchen S, Weiskirchen R. Lipocalin 2 receptors: facts, fictions, and myths. Front. Immunol. 2023;14:1229885.
Waag R, Bohacek J. Single-nucleus RNA-sequencing in brain tissue. Curr. Protoc. 2023;3:e919.
Shen Y, Liu SS, Zhan MY, Luo JH, Zhu LJ. Interleukin-2 enhances dendritic development and spinogenesis in cultured hippocampal neurons. Anat. Rec. 2010;293:1017–23.
Pozniak PD, White MK, Khalili K. TNF-α/NF-κB signaling in the CNS: possible connection to EPHB2. J. Neuroimmune Pharmacol. 2014;9:133–41.
Roybon L, Lamas NJ, Garcia AD, Yang EJ, Sattler R, Lewis VJ, et al. Human stem cell-derived spinal cord astrocytes with defined mature or reactive phenotypes. Cell Rep. 2013;4:1035–48.
Battistella I, Cutarelli A, Zasso J, Clerici M, Sala C, Marcatili M, et al. Cortical astrocyte progenitors and astrocytes from human pluripotent stem cells. J. Pers. Med. 2023;13:538.
Rose CR, Ziemens D, Untiet V, Fahlke C. Molecular and cellular physiology of sodium-dependent glutamate transporters. Brain Res. Bull. 2018;136:3–16.
Chierzi S, Kacerovsky JB, Fok AHK, Lahaie S, Shibi Rosen A, Farmer WT, et al. Astrocytes transplanted during early postnatal development integrate, mature, and survive long term in mouse cortex. J. Neurosci. 2023;43:1509–29.
Dremmen MHG, Bouhuis RH, Blanken LME, Muetzel RL, Vernooij MW, Marroun HE, et al. Cavum septum pellucidum in the general pediatric population and its relation to surrounding brain structure volumes, cognitive function, and emotional or behavioral problems. AJNR Am. J. Neuroradiol. 2019;40:340–6.
Chen JJ, Chen CJ, Chang HF, Chen DL, Hsu YC, Chang TP. Prevalence of cavum septum pellucidum and/or cavum Vergae in brain computed tomographies of Taiwanese. Acta Neurol. Taiwan. 2014;23:49–54.
Cantani A, Bamonte G, Tacconi ML. Mental retardation and EEG abnormalities in Waardenburg’s syndrome: two case reports (EEG anomalies in Waardenburg’s syndrome). Padiatr. Padol. 1989;24:137–40.
Kawabata E, Ohba N, Nakamura A, Izumo S, Osame M. Waardenburg syndrome: a variant with neurological involvement. Ophthalmic Paediatr. Genet. 1987;8:165–70.
Pérez-Losada J, Sánchez-Martín M, Rodríguez-García A, Sánchez ML, Orfao A, Flores T, et al. Zinc-finger transcription factor Slug contributes to the function of the stem cell factor c-kit signaling pathway. Blood. 2002;100:1274–86.
Sánchez-Martín M, Pérez-Losada J, Rodríguez-García A, González-Sánchez B, Korf BR, Kuster W, et al. Deletion of the SLUG (SNAI2) gene results in human piebaldism. Am. J. Med. Genet. A. 2003;122:125–32.
Jin M, Jang E, Suk K. Lipocalin-2 acts as a neuroinflammatogen in lipopolysaccharide-injected mice. Exp. Neurobiol. 2014;23:155–62.
Wang G, Weng YC, Han X, Whaley JD, McCrae KR, Chou WH. Lipocalin-2 released in response to cerebral ischaemia mediates reperfusion injury in mice. J. Cell Mol. Med. 2015;19:1637–45.
Jha MK, Suk K. Glia-based biomarkers and their functional role in the CNS. Expert. Rev. Proteom. 2013;10:43–63.
Chan CP, Jiang HL, Leung LY, Wan WM, Cheng NM, Ip WS, et al. Multiple atherosclerosis-related biomarkers associated with short- and long-term mortality after stroke. Clin. Biochem. 2012;45:1308–15.
Karlsson C, Baudet A, Miharada N, Soneji S, Gupta R, Magnusson M, et al. Identification of the chemokine CCL28 as a growth and survival factor for human hematopoietic stem and progenitor cells. Blood. 2013;121:3838–42.
Bullitt E. Expression of c-fos-like protein as a marker for neuronal activity following noxious stimulation in the rat. J. Comp. Neurol. 1990;296:517–30.
Cullinan WE, Herman JP, Battaglia DF, Akil H, Watson SJ. Pattern and time course of immediate early gene expression in rat brain following acute stress. Neuroscience. 1995;64:477–505.
Sun X, Bernstein MJ, Meng M, Rao S, Sørensen AT, Yao L, et al. Functionally distinct neuronal ensembles within the memory engram. Cell. 2020;181:410–23.
Hasselmo ME. The role of acetylcholine in learning and memory. Curr. Opin. Neurobiol. 2006;16:710–5.
Wang T, Takikawa Y, Sawara K, Yoshida Y, Suzuki K. Negative regulation of human astrocytes by interferon (IFN) α in relation to growth inhibition and impaired glucose utilization. Neurochem. Res. 2012;37:1898–905.
Halonen SK, Woods T, McInnerney K, Weiss LM. Microarray analysis of IFN-gamma response genes in astrocytes. J. Neuroimmunol. 2006;175:19–30.
Van Wagoner NJ, Benveniste EN. Interleukin-6 expression and regulation in astrocytes. J. Neuroimmunol. 1999;100:124–39.
Shen SY, Liang LF, Shi TL, Shen ZQ, Yin SY, Zhang JR, et al. Microglia-derived interleukin-6 triggers astrocyte apoptosis in the hippocampus and mediates depression-like behavior. Adv. Sci. 2025;12:e2412556.
Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–15.
Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–7.
Hasan U, Singh SK. The astrocyte-neuron interface: an overview on molecular and cellular dynamics controlling formation and maintenance of the tripartite synapse. Methods Mol. Biol. 2019;1938:3–18.
Kruyer A, Kalivas PW, Scofield MD. Astrocyte regulation of synaptic signaling in psychiatric disorders. Neuropsychopharmacology. 2023;48:21–36.
Hong SI, Kang S, Baker M, Choi DS. Astrocyte-neuron interaction in the dorsal striatum-pallidal circuits and alcohol-seeking behaviors. Neuropharmacology. 2021;198:108759.
Soto JS, Jami-Alahmadi Y, Chacon J, Moye SL, Diaz-Castro B, Wohlschlegel JA, et al. Astrocyte-neuron subproteomes and obsessive-compulsive disorder mechanisms. Nature. 2023;616:764–73.
Nam MH, Ko HY, Kim D, Lee S, Park YM, Hyeon SJ, et al. Visualizing reactive astrocyte-neuron interaction in Alzheimer’s disease using 11C-acetate and 18F-FDG. Brain. 2023;146:2957–74.
Maulik PK, Mascarenhas MN, Mathers CD, Dua T, Saxena S. Prevalence of intellectual disability: a meta-analysis of population-based studies. Res. Dev. Disabil. 2011;32:419–36.
Zhang S, Xu H, Tian Y, Liu D, Hou X, Zeng B, et al. High genetic heterogeneity in Chinese patients with Waardenburg syndrome revealed by next-generation sequencing. Front. Genet. 2021;12:643546.
National Genomics Data Center Members and Partners. The genome sequence archive family: toward explosive data growth and diverse data types. Genomics Proteom. Bioinforma. 2021;19:578–83.
National Genomics Data Center Members and Partners. Database resources of the National Genomics Data Center, China National Center for bioinformation in 2022. Nucleic Acids Res. 2022;50:D27–D38.
Acknowledgements
We greatly appreciate the willingness of the proband and his family member to participate in this study. We also thank the Central Laboratory of Shantou University Medical College for providing technical supports.
Funding
This work was financially supported by National Natural Science Foundation of China (NSFC) (No. 31671183, No. 81972923, No. 81573510, No. 81971256), Guangdong Basic and Applied Basic Research Foundation (No. 2020A1515011174, No. 2024A1515010890), Shantou Medical Science and Technology Program (No. 200408165266290), and Clinical Research Cultivation Project of Shantou Central Hospital (No. 20190126).
Author information
Authors and Affiliations
Contributions
CX, XH, MS, LH, HL and GZ performed experiments. XH, SC, ZD, TC, CC and YL did neuroimaging and clinical assessments for the family members. CX, HX, ZL, GZ and WL analyzed the data and wrote the paper. HX reviewed and edited the manuscript. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xue, C., Xu, H., Huang, X. et al. Developmental arrest of astrocyte lineage in Snai2 deletion mice: implication for the intellectual disability in patients with Waardenburg syndrome. Transl Psychiatry 15, 391 (2025). https://doi.org/10.1038/s41398-025-03616-9
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41398-025-03616-9
