
Abstract
A personalized prognostic model that takes into account the unique molecular features of primary myelodysplastic neoplasm (MDS) in Asia patients is lacking. Diagnostic clinicopathologic features, cytogenetic changes, and gene mutations of ethnic Asian patients with primary MDS were analyzed. Variables were evaluated for associations with overall survival (OS), leukemia-free survival (LFS), and time to progression to secondary AML (TTP-sAML). Prognostic scores were built as a weighted sum of prognostic variables for each patient. The cohort comprised 1225 patients, with at least one gene mutation identified in 1177 patients (96%). Genomic factors associated with inferior outcomes included monosomy 7, del(5q), and GNAS and TP53 mutations for OS; trisomy 19, del(5q), monosomy 7, and GNAS, PTPN11 and TP53 mutations for LFS; and i(17q), del(5q), and NPM1, NRAS, GNAS, IDH2, SF3B1 and RUNX1 mutations for TTP-sAML. The Asian Prognostic Scoring System (APSS) was determined, stratifying patients into six prognostic risk categories. The APSS, compared with the International Prognostic Scoring System molecular (IPSS-M), showed superior concordance indices (C-indices) for OS (0.73 versus 0.57), LFS (0.72 versus 0.59), and TTP-sAML (0.75 versus 0.65) for this Asian cohort. In conclusion, the APSS enhanced prognostication of primary MDS in Asia.
Similar content being viewed by others


Introduction
In Asia, treatment decisions in myelodysplastic neoplasms (MDS) are conventionally based on the Revised International Prognostic Scoring system (IPSS-R) [1]. The IPSS-R was developed from 7012 patients with untreated primary MDS derived from Spanish, French, Italian, and Brazilian registries. More recently, somatic mutations have been incorporated into the prognostic assessment of MDS [2]. A personalized prediction model was developed based on American and German cohorts, which, by including genomic information, showed enhanced prognostic prediction [3]. A unique clinical-molecular prognostic model, the IPSS-molecular (IPSS-M) [4], was developed from 2957 patients with de-novo MDS, secondary/therapy-related MDS, and MDS/myeloproliferative neoplasm (MPN) overlap syndrome by the International Working Group for Prognosis in MDS (IWG-PM). In the IWG-PM cohort, only 102 patients from Asia were included [4]. The IPSS-M was externally validated in 754 Japanese patients with MDS [4]. In this Japanese cohort, the prognostic power in terms of concordance index (C-index) was lower for the prediction of leukemic transformation than that obtained from the IWG-PM cohort [4]. In a recent Chinese cohort comprising 852 patients, the C-index (<0.70) for overall survival (OS) was significantly lower compared with that in the original IWG-PM cohort in the IPSS-M [5].
Differences in presenting clinicopathologic and genomic characteristics were observed between Western and Asian Cohorts [4, 5]. Less than 30% of Asian patients presented with IPSS-R or IPSS-M very low/low risk disease [4,5,6]. In comparison, ~40–60% of Western patients presented with IPSS-R or IPSS-M very low/low risk disease [4, 7, 8]. In addition, higher frequencies of ASXL1, RUNX1, and TP53 mutations and lower frequencies of SF3B1 mutations were observed in Asian patients compared with Western cohorts [3,4,5,6, 9].
To address the under-representation of Asian patients in existing prognostic models and the differences in the clinicopathologic and genomic characteristics between Asian and Western patients, an Asian MDS Genomics project was conducted under the Asian Myeloid Working Group (AMWG), in order to define the hematologic, cytogenetic, and molecular landscape of Asian patients with primary MDS. Based on clinical and genomic features, a personalized prognostic model for MDS in Asia was established.
Subjects and methods
Study population
The study was conducted by the AMWG (ClinicalTrials.gov Identifier: NCT03169296) and was approved by the Institutional Review Boards and Ethics committees of the University of Hong Kong, the National University Singapore, and the National Taiwan University Hospital in accordance with the Declaration of Helsinki, and all patients provided informed consent. Inclusion criteria included age ≥18 years, a diagnosis of primary MDS according to the World Health Organization (WHO) 2016 criteria, and the availability of bone marrow aspirate at diagnosis for pathologic review and next-generation sequencing for mutation analysis. Patients with therapy-related myeloid neoplasm (t-MN) and myelodysplastic syndrome/myeloproliferative neoplasm (MDS/MPN) were excluded.
Genomic analyses
At diagnosis, cytogenetic analysis was performed using standard G-banding, and karyotypes were classified using the International System for Cytogenetic Nomenclature Criteria. Mutation screening was performed on the diagnostic bone marrow mononuclear cells using next-generation sequencing (NGS) for a 54-gene myeloid panel (Supplementary File 1). These genes comprised ABL1, JAK3, ASXL1, KDM6A, ATRX, KIT, BCOR, KRAS, BCORL1, KMT2A, BRAF, MPL, CALR, MYD88, CBL, NOTCH1, CBLB, NPM1, CBLC, NRAS, CDKN2A,PDGFRA, CEBPA, PHF6, CSF3R, PTEN, CUX1, PTPN11, DNMT3A, RAD21, ETV6, RUNX1, EZH2, SETBP1, FBXW7, SF3B1, FLT3, SMC1A, GATA1, SMC3, GATA2, SRSF2, GNAS, STAG2, HRAS, TET2, IDH1, TP53, IDH2, U2AF1, IKZF1, WT1, JAK2, and ZRSR2. Mutect2 from GATK was used for somatic variant calling [10]. SNV and INDEL were also called independently by VarScan2 (version 2.4.0) [11] generated by Samtools (version 1.17) [12] using default parameters. Variants called from both software were concatenated for annotations. Detection of FLT3 internal tandem duplication (FLT3-ITD) and KMT2A partial tandem duplication (KMT2A-PTD) was also performed by Pindel [13] in addition to Mutect2 and VarScan2. Snpeff (version 5.1) and ANNOVAR were used for annotation for predicted effects of the variants, while SnpSift (version 5.1) filtering further removed single-nucleotide polymorphisms (SNP) when allele frequencies were ≥ 0.01 based on population databases [14, 15]. These databases included Exome Sequencing Project (ESP) with 6500 exomes, 1the 000 Genome Project, TCGA ExAC Cancer database, International Cancer Genome Consortium (ICGC), ClinVar database, database of functional predicters (dbnsfp, version 42), gnomAD whole-genome version 3.1.2, and COSMIC (version 92). A list of variants with known diagnostic or prognostic significance in myeloid malignancies was constructed to assist variant filtration of the current dataset [16,17,18,19,20]. In addition, Varsome Clinical [21] was used to refine and exclude benign variants based on American College of Medical Genetics and Genomics (ACMG)/Association of Molecular Pathology (AMP) classifications [22]. Variants occurring in more than 1% in any one of population databases, intronic, non-coding, synonymous, or variants reported benign or likely benign by ClinVar were removed by SnpSift [23]. Variants were retained if one of the predicted variants was pathogenic based on ClinVar and COSMIC, or predicted pathogenic based on at least one function prediction algorithm (including but not limited to POLYPHEN-2, LRT, MutationTaster, FATHMM, PROVEAN, VEST4, MetaSVM, MetaRNN, MutPred, and SIFT). The genomic data generated during the current study are available in the NCBI repository, BioProject ID PRJNA1187253, http://www.ncbi.nlm.nih.gov/bioproject/1187253.
Statistical analyses
Overall survival (OS) was defined as the time from diagnosis to death (event) or latest follow-up (censor). Leukemia-free survival (LFS) was defined as the time from diagnosis to progression to secondary acute myeloid leukemia (sAML) (event), death (event), or latest follow-up (censor). Time to progression to sAML (TTP-sAML) was defined as the time from diagnosis to the development of sAML (event). Additional censoring was performed at the time of allogeneic hematopoietic stem cell transplantation (allo-HSCT). In the assessment of survivals following allo-HSCT, OS post-HSCT was defined as the time from allo-HSCT to death; LFS post-HSCT was defined as time from allo-HSCT to progression to sAML or death; and TTP-sAML post-HSCT was defined as time from allo-HSCT to the development of sAML. All data were censored on December 31, 2022. A model comprising clinicopathologic, cytogenetic, and molecular features was constructed to predict OS, LFS, and TTP-sAML. Variable selection was performed to determine the prognostic significance of age, sex, and classification of MDS, included as confounders. (Supplementary file 2). The relative weights of the selected variables were estimated using a Cox multivariate model adjusted for confounders. According to the weights of each prognostic variable, individual patient-specific prognostic scores were calculated (Supplementary file 2). Sex, classification of MDS, gene mutations, and karyotypic abnormalities were encoded as binary variables. Age, hemoglobin, total white blood cell (WBC) count, platelet count, absolute neutrophil count, and peripheral blood (PB) and bone marrow (BM) blast percentages were encoded linearly as continuous variables. Owing to sample size, variant allele frequencies and subtypes of gene mutations and gene-by-gene interactions were not included in the multivariable analysis. Three separate models were developed to predict OS, LFS, and TTP-sAML. The statistical predictive power of these prognostic models was assessed by the Harrell’s concordance index (C-index). All analyses were performed using R version 4.3.1 (R Foundation for Statistical Computing, Vienna, Austria). Details on the statistical methodology were shown in Supplementary file 2.
Results
Clinicopathologic characteristics
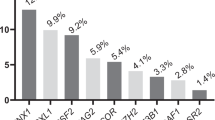
The AMWG cohort comprised 1225 patients with primary MDS enrolled from three tertiary referral centers in Asia between 1 January 2006 and 30 May 2021 (Supplementary file 3 and Table 1). There were 778 men (63.5%) and 447 women (36.5%) with a median age of 68 years (interquartile range, IQR: 57.1–77.4 years) at diagnosis. The median duration of follow-up was 2.9 years (IQR: 1–7.1 years). Cytogenetic risks grouped by IPSS-R were very good (N = 20, 1.6%), good (N = 619, 50.5%), intermediate (N = 241, 19.7%), poor (N = 70, 5.7%), and very poor (N = 193, 15.8%). The IPSS-R risk scores were very low (N = 49, 4%), low (N = 281, 22.9%), intermediate (N = 279, 22.8%), high (N = 258, 21.1%), and very high (N = 269, 20%). The IPSS-M risk scores were very low (N = 1, 0.1%), low (N = 36, 2.9%), moderately low (N = 55, 4.5%), moderately high (N = 81, 6.6%), high (N = 245, 20%), and very high (N = 670, 54.7%). Mutations were identified in one or more cases for each of the 54 genes in the panel (Fig. 1 and Supplementary files 4–6). Three hundred and ninety-seven patients (32.4%) received hypomethylating agents (HMA) (azacitidine, N = 370; decitabine, N = 27). One hundred and fifty-eight patients (12.9%) underwent allo-HSCT. Two hundred and eighty-four patients developed sAML.

A Overall survival of primary MDS in Asia; B Adverse prognostic impact of monosomy 7 (-7) on overall survival in primary MDS in Asia; C Adverse prognostic impact of del(5q) on overall survival in primary MDS in Asia; D Adverse prognostic impact of GNAS mutations on overall survival in primary MDS in Asia; E Adverse prognostic impact of TP53 mutations on overall survival in primary MDS in Asia; F Leukemia-free survival of primary MDS in Asia; G Adverse prognostic impact of trisomy 19 (+19) on leukemia-free survival in primary MDS in Asia; H Adverse prognostic impact of 5q deletion [del(5q)] on leukemia-free survival in primary MDS in Asia; I Adverse prognostic impact of monosomy 7 (-7) on leukemia-free survival in primary MDS in Asia; J Adverse prognostic impact of GNAS mutations on leukemia-free survival in primary MDS in Asia; K Adverse prognostic impact of PTPN11 mutations on leukemia-free survival in primary MDS in Asia; L Adverse prognostic impact of TP53 mutations on leukemia-free survival in primary MDS in Asia. All P-values were obtained using log-rank test. The genomic factors shown were significant on multivariable analysis (see Table 2).
Genomic landscape
Gene mutations were identified in 1177 patients (96%), with 1059 patients (86%) harboring ≥2 mutations (Supplementary files 4–6). The most commonly mutated genes were TET2, DNMT3A, and ASXL1 (Supplementary files 4–6). Mutation enrichment analysis for MDS subgroups based on the WHO 2016 classification showed significant enrichment of mutations of SF3B1, KMT2A, TET2, NOTCH1 and MYD88 in MDS with ring sideroblasts and single lineage dysplasia; CDKN2A, CUX1, STAG2, KDM6A, RUNX1 and ASXL1 in MDS with excess blasts-1 (MDS-EB-1); and TP53, GATA1, BRAF, CSF3R, IDH2, EZH2 and PHF6 in MDS-EB-2 (Supplementary files 5 and 7). Mutation enrichment analysis in MDS subgroups based on the WHO 2022 classification showed significant enrichment of mutations of SF3B1, BCOR, BCORL1, GNAS, KMT2A, CSF3R, ABL1 and IDH1 in MDS with low blasts and SF3B1 mutation; and TP53, ASXL1, IKZF1, NOTCH1, TET2, SMC1A, DMNT3A, IDH1, ATRX, PDGFRA, CUX1 and CEBPA in MDS with biallelic TP53 inactivation (Supplementary files 5 and 7). TET2 mutations significantly co-occurred with IKZF1, ATRX, ASXL1 and DMNT3A mutations (P < 0.05) (Supplementary file 8). Mutations of DNMT3A and EZH2 and mutations of RUNX1, GATA2, and ZRSR2 were mutually exclusive (Supplementary file 8). Characteristics of variants for each of the sequenced genes were shown in Supplementary file 9.
Impact of ASXL, SF3B1 and TP53 mutations
ASXL1 variant allele frequency of ≥0.2 was independently associated with inferior OS (P < 0.001), LFS (P < 0.001), and TTP-sAML (P = 0.005) (Supplementary file 10). ASXL1 frameshift variants were associated with significantly inferior OS (P < 0.001) and LFS (P < 0.001) (Supplementary file 11). SF3B1 co-mutations did not significantly impact on OS, LFS, and TTP-sAML (Supplementary file 11). A TP53 variant allele frequency of ≥0.2 was independently associated with inferior OS, LFS, and TTP-sAML (P < 0.001) (Supplementary file 12). There was no significant difference in OS, LFS, and TTPs-AML between monoallelic and multiallelic/multihit TP53 mutations (Supplementary file 12). TP53 frameshift and splicing co-mutations were associated with a significantly inferior OS (P = 0.02), LFS (P = 0.03), and TTP-sAML (P = 0.002) (Supplemental file 13).
Validation of the IPSS-R and the IPSS-M in the current Asian cohort
The IPSS-R and the IPSS-M were validated using the current Asian cohort (Supplementary files 14 and 15). Separation of prognostic categories was observed (Supplementary file 15). The C-indices obtained for IPSS-R were 0.68 for OS, 0.68 for LFS, and 0.71 for TTP-sAML; whereas those obtained for IPSS-M were 0.57 for OS, 0.59 for LFS, and 0.65 for TTP-sAML (Supplementary file 14).
Outcome
The median OS of the cohort was 3.7 years (95% confidence interval, C.I.: 2.90–4.54). The 5-year and 10-year OS were 46.7% and 41% respectively (Fig. 1A). Genomic factors associated with inferior OS on multivariable analysis included monosomy 7, del(5q), and mutations in GNAS and TP53 (Table 2 and Fig. 1B–E). The median LFS of the cohort was 2.5 years (95% CI: 2.19–3.19). The 5-year and 10-year LFS were 42.4% and 37.2% respectively (Fig. 1F). Genomic factors associated with inferior LFS on multivariate analysis included trisomy 19, del(5q), monosomy 7, and mutations in GNAS, PTPN11, and TP53 (Table 2 and Fig. 1G–L). The 5-year and 10-year cumulative risk of progression to sAML was 28.8% and 29.5% respectively (Fig. 2A). Genomic factors associated with inferior TTP-sAML on multivariate analysis include i(17q), del(5q), and mutations in NPM1, NRAS, GNAS, IDH2, SF3B1, and RUNX1 (Table 2 and Fig. 2B–I). Increasing age, as a continuous variable, was associated with worse OS and LFS (Table 2). Male sex was associated with worse OS, LFS, and TTP-sAML (Table 2). Increasing peripheral blood blast percent, as a continuous variable was associated with worse LFS and TTP-sAML (Table 2).

A Time to progression to secondary AML in primary MDS in Asia; B Adverse prognostic impact of isochromosome 17q [i(17q)] on time to progression to secondary AML in primary MDS in Asia; C Adverse prognostic impact of 5q deletion [del(5q)] on time to progression to secondary AML in primary MDS in Asia; D Adverse prognostic impact of NPM1 mutations on time to progression to secondary AML in primary MDS in Asia; E Adverse prognostic impact of NRAS mutations on time to progression to secondary AML in primary MDS in Asia; F Adverse prognostic impact of GNAS mutations on time to progression to secondary AML in primary MDS in Asia; G Adverse prognostic impact of IDH2 mutations on time to progression to secondary AML in primary MDS in Asia; H Adverse prognostic impact of SF3B1 mutations on time to progression to secondary AML in primary MDS in Asia; I Adverse prognostic impact of RUNX1 mutations on time to progression to secondary AML in primary MDS in Asia. All P values were obtained using log-rank test. The genomic factors shown were significant on multivariable analysis (see Table 2).
Personalized prognostic model
Variables impacting on OS, LFS, and TTP-sAML on multivariate analysis had their C-indices determined (Supplementary files 16-18). Variables with the highest C-indices were selected for inclusion in the prognostic models for OS, LFS, and TTP-sAML (Table 2). A six-category risk stratification (Asian Prognostic Scoring System, APSS) was defined: very good, good, intermediate-1, intermediate-2, poor, and very poor. The risk categories allowed prognostic risk separation for all three clinical end-points (Table 3 and Fig. 3A–F). To facilitate clinical utility, an APSS Web calculator has been constructed (https://medicine-intranet.hku.hk/mds/index_v3.html). With patient-specific clinicopathologic, hematological, karyotypic, and molecular parameters, a specific APSS score, the corresponding risk category, and the median survival could be calculated.

A Prognostic risk categories for overall survival according to the APSS; B Distribution of prognostic risk categories for overall survival according to the APSS (X-axis represents the prognostic score); C Prognostic risk categories for leukemia-free survival according to the APSS; D Distribution of prognostic risk categories for leukemia-free survival according to the APSS (X-axis represents the prognostic score); E Prognostic risk categories for time-to-progression to secondary acute myeloid leukemia according to the APSS; F Distribution of prognostic risk categories for time-to-progression to secondary acute myeloid leukemia according to the APSS (X-axis represents the prognostic score); G Comparison of concordance (C) indices obtained using the APSS, molecular international prognostic scoring system (IPSS-M) and the Revised International Prognostic Scoring System (IPSS-R) in the current Asian primary myelodysplastic neoplasm cohort; H Validation of the APSS risk categories on overall survival (OS) in the external IPSS-M dataset; I Validation of the APSS risk categories on leukemia-free survival (LFS) in the external IPSS-M dataset; J Validation of the APSS risk categories on the time-to-progression (TTP) to secondary acute myeloid leukemia in the external IPSS-M dataset.
In this Asian primary MDS cohort, the C-indices for OS, LFS, and TTP-sAML obtained using IPSS-R (OS: 0.68; LFS: 0.68; TTP-sAML: 0.71) and IPSS-M (OS: 0.57; LFS: 0.59; TTP-sAML: 0.65) were lower than those obtained using the APSS (OS: 0.73; LFS: 0.72; TTP-sAML: 0.75) (Fig. 3G and Supplementary file 14).
Validation of the APSS
The APSS was externally validated using the publicly available IPSS-M dataset (https://github.com/papaemmelab/ipssm) from the International Working Group for Prognosis in MDS (IWG-PM) cohort [4]. The cohort comprised 2957 patients with MDS, with 102 patients from Taiwan [4]. The number of patients of Asian descent in the entire cohort was not specified [4]. It showed clear separation of prognostic subgroups for OS (C-index 0.70), LFS (C-index 0.71), and TTP-sAML (C-index 0.74) that were statistically significant (Fig. 3H–J and Supplementary file 19).
Restratification of patients from IPSS-M to APSS
Mapping was performed between the IPSS-M to APSS risk categories for overall survival (Supplementary files 20). In 1098 patients where IPSS-M was available, restratification occurred in 917 (83.5%) of patients. Of these, 775 patients (84.5%) were downstaged and 142 patients (15.5%) were upstaged.
Prognostic impact of APSS in untreated and treated patients
The prognostic impact of the APSS was assessed in 3 subgroups of the current Asian cohort: primary untreated patients (N = 727); patients treated with HMA with data censored at the time of HSCT (N = 388); and patients receiving allogeneic HSCT (N = 122) (Supplementary files 21–23). The APSS was predictive of outcomes in all 3 groups of patients (Untreated patients: C-index for OS 0.74, C-index for LFS 0.74, C-index for TTP-sAML 0.79; patients treated with HMA: C-index for OS 0.69, C-index for LFS 0.64, C-index for TTP-sAML 0.66; patients receiving allo-HSCT: C-index for OS post-HSCT 0.61, C-index for LFS post-HSCT 0.63, C-index for TTP-sAML post-HSCT 0.62) (Supplementary files 21–23). The prediction was the best in primary untreated patients with clear separation of prognostic risk groups for OS, LFS, and TTP-sAML (P < 0.0001 for all 3) (Supplementary file 21). In patients treated with HMA, the APSS was significant for the prediction OS, LFS, and TTP-sAML (P < 0.001 for all) (Supplementary file 22). Nevertheless, in patients receiving HMA, there was overlap between the very good, good, and intermediate-1 risk categories for OS; between the very good, good, and intermediate-1 risk categories and between intermediate-2 and poor risk categories for LFS; and between the very good, good, and intermediate-1 risk categories for TTP-sAML. In patients receiving allo-HSCT, APSS was significantly predictive of post-HSCT OS (P = 0.01) and post-HSCT LFS (P = 0.001), but not TTP-sAML post HSCT (P = 0.24) (Supplementary file 23). In patients receiving allo-HSCT, there was overlap between the good, intermediate-1, intermediate-2, and poor risk categories for post-HSCT OS and post-HSCT LFS.
Discussion
We analyzed the largest cohort to date of Asian patients with primary MDS and defined their clinicopathologic features and genomic landscape. A significant proportion of patients presented with high-risk disease. At presentation, 508 patients (41.5%) and 118 patients (9.6%) had MDS with increased blasts and MDS with biallelic TP53 inactivation. With respect to IPSS-R and IPSS-M, 43% and 74.7% of them had high/very high-risk disease. Interesting differences existed between our patients and other reported cohorts in Western populations [4, 8, 9], including a lower proportion of patients with lower-risk MDS at presentation, and a lower frequency of SF3B1 but higher frequency of TET2, DNMT3A, ASXL1, and RUNX1 variants. Such differences might be expected because our cohort was enriched with patients having higher risks. Furthermore, with the use of multiple bioinformatic tools, we included variants previously reported as pathogenic and likely pathogenic, together with other novel variants predicted likely to alter protein function, which may partly explain the differences observed between our and other cohorts. Similar to our study, reports from other Asian primary MDS cohorts showed higher frequency of ASXL1, DNMT3A, TET2, and RUNX1 mutations and lower frequency of SF3B1 mutations [5, 24].
Unique genomic factors associated with inferior survival were defined in our cohort. These factors included monosomy 7, del(5q), and GNAS and TP53 mutations with OS; trisomy 19, del(5q), monosomy 7, and GNAS, PTPN11 and TP53 mutations with LFS; and i(17q), del(5q), and NPM1, NRAS, GNAS, IDH2, SF3B1 and RUNX1 mutations with TTP-sAML. The adverse prognostic impact of monosomy 7 is widely reported and validated in MDS [1, 25]. MDS with isolated del(5q) was uncommon in our cohort (accounting for 1% of patients). Co-occurrence of other aberrations, especially TP53 mutations and additional karyotypic abnormalities, could account for the adverse prognostic impact of del(5q) [26]. Isochromosome 17q results in deletion of TP53, portending an adverse outcome. Trisomy 19 is associated with myeloid malignancies, and acquisition of trisomy 19 is associated with progression to secondary AML [27]. The adverse prognostic impact of GNAS variants has not been previously reported. GNAS is an important oncogene, and activating mutations have been reported in various malignancies, including myeloid neoplasm [28, 29]. Alternative splicing of GNAS has been shown to be an important driver of MDS, together with splicing factor SFSF2 and U2AF1 mutations [30]. Mutations in RAS pathway genes, including NRAS and PTPN11, are associated with increased rates of leukemic transformation [31,32,33]. NPM1 mutations in our cohort occurred exclusively in MDS with less than 10% bone marrow blasts. Consistent with reports of other non-acute myeloid neoplasms with NPM1 mutations, higher rates of leukemia transformations were observed [34]. Although the formal diagnosis of AML with NPM1 mutation requires bone marrow blast percent of 10% or more, the threshold may be arbitrary. Whether this group of patients represented an early stage in the continuum of progression to NPM1-mutated AML remains to be determined [34]. Pathogenic mutations in IDH2 in MDS are associated with progression to secondary AML [35, 36]. RUNX1 mutations in MDS were associated with shorter latency to progression to sAML [37]. RUNX1 mutations were also reported to portend worse survival in patients with higher-risk MDS treated with venetoclax and hypomethylating agents in a large Chinese cohort [38]. ASXL1 frameshift variants were associated with adverse outcomes, consistent with previous reports [39]. However, the negative prognostic impact of ASXL1 mutation with VAF ≥ 0.2 has not been previously reported. SF3B1 mutations were not associated with a favorable outcome in our cohort, a finding consistent with that in previous reports [4].
Significant differences in outcome were not observed between monoallelic and biallelic TP53 mutations, consistent with previous reported results [40]. However, TP53 mutations with VAF ≥ 0.2 were independently associated with inferior outcome. This observation concurred with previous reports, where TP53 mutations with VAF ≥ 0.2 were associated with outcomes similar to multi-hit TP53 mutations, especially in MDS with del(5q) [41]. An adverse prognostic impact of TP53 mutations with higher VAF was also reported in other subgroups of MDS [42]. In particular, co-occurrence of frameshift and splicing mutations was associated with adverse outcome, which was also consistent with previous observations [43]. Our findings corroborate with the diverse characteristics and prognostic impact of TP53 mutations, including the mutation characteristics, VAF, allelic state, and co-occurring mutations [44].
MDS prognostication has conventionally been based on IPSS and IPSS-R. The recent advent of the IPSS-M has enabled a personalized risk assessment incorporating hematologic, cytogenetic, and molecular features to be made. In the original tested cohorts, IPSS-M successfully stratified patients with respect to OS, LFS, and transformation to AML [4], which was confirmed to be reproducible by subsequent studies of different populations [7, 8, 45]. In Asian populations, however, IPSS-M was observed to separate prognostic groups with C-indices lower than those in Western populations [4, 5]. The pathogenesis of MDS is complicated, involving putatively interactions of exposures to genotoxic agents (environmental or therapeutic) and genetic constitutions and susceptibility, which may be different for various populations. Hence, population-specific prognostication may be more clinically relevant. This is supported by Asian reports unraveling unique gene signatures associated with treatment outcome [38, 46].
With the current Asian cohort, we developed a prognostic model, APSS, based on clinicopathologic, karyotypic, and genomic data. The APSS is similar to IPSS-M in separating patients into six prognostic categories, with a continuous score that provides a personalized unique risk assessment. The APSS provides separate sets of prognostic indicators for OS, LFS, and transformation to sAML, taking into account the differential effects of clinical and genomic parameters in predicting OS, LFS, and transformation to sAML. The APSS also improved prognostication in the current Asian MDS cohort, as evidenced by the higher C-indices obtained using the APSS compared with the IPSS-M. The APSS also accounts for missing information similar to the IPSS-M by providing the best, mean, and worst score on the web calculator. A high percentage of patients were restratified using the APSS. This was likely due to the relatively higher proportion of patients in the current Asian cohort presenting with higher risk IPSS-M categories. The APSS was predictive of outcomes in primary untreated MDS, MDS treated with HMA, and MDS receiving allo-HSCT. The prediction was the best with primary untreated MDS. In patients receiving HMA or allo-HSCT, the prediction was more apparent for the very good and very poor risk categories, with significant overlap between other risk categories. The relatively small sample size may account for this observation, and further validation of the APSS in largely cohorts of patients treated with HMA and patients receiving allo-HSCT is warranted. Previous studies with larger real-world datasets on the impact of IPSS-M in patients receiving HMA and allo-HSCT showed significant separation of risk categories [8].
An important caveat in the currently available prognostic models and the APSS is the possibility of having populations with wide range of genomic backgrounds and ancestries. Further validation of the APSS in different Asian populations is warranted. Furthermore, the prognostic performance of the APSS vis-à-vis IPSS-M in Asian populations residing inside and outside of Asia may allow the impact of environmental factors on the pathogenesis and outcome of MDS to be delineated.
Data availability
The de-identified data are available from the corresponding author on reasonable request. The genomic data generated during the current study are available in the NCBI repository, BioProject ID PRJNA1187253, http://www.ncbi.nlm.nih.gov/bioproject/1187253.
References
Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Sole F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454–65.
DeZern AE, Greenberg PL. The trajectory of prognostication and risk stratification for patients with myelodysplastic syndromes. Blood. 2023;142:2258–67.
Nazha A, Komrokji R, Meggendorfer M, Jia X, Radakovich N, Shreve J, et al. Personalized prediction model to risk stratify patients with myelodysplastic syndromes. J Clin Oncol. 2021;39:3737–46.
Bernard E, Tuechler H, Greenberg PL, Hasserjian RP, Arango Ossa JE, Nannya Y, et al. Molecular international prognostic scoring system for myelodysplastic syndromes. NEJM Evid. 2022;1:EVIDoa2200008.
Wu J, Zhang Y, Qin T, Xu Z, Qu S, Pan L, et al. IPSS-M has greater survival predictive accuracy compared with IPSS-R in persons≥ 60 years with myelodysplastic syndromes. Exp Hematol Oncol. 2022;11:73.
Lee W-H, Tsai M-T, Tsai C-H, Tien F-M, Lo M-Y, Tseng M-H, et al. Validation of the molecular international prognostic scoring system in patients with myelodysplastic syndromes defined by international consensus classification. Blood Cancer J. 2023;13:120.
Kewan T, Bahaj W, Durmaz A, Aly M, Ogbue OD, Carraway HE, et al. Validation of the molecular international prognostic scoring system in patients with myelodysplastic syndromes. Blood. 2023;141:1768–72.
Sauta E, Robin M, Bersanelli M, Travaglino E, Meggendorfer M, Zhao L-P, et al. Real-world validation of molecular international prognostic scoring system for myelodysplastic syndromes. J Clin Oncol. 2023;41:2827–42.
Bersanelli M, Travaglino E, Meggendorfer M, Matteuzzi T, Sala C, Mosca E, et al. Classification and personalized prognostic assessment on the basis of clinical and genomic features in myelodysplastic syndromes. J Clin Oncol. 2021;39:1223–33.
Benjamin D, Sato T, Cibulskis K, Getz G, Stewart C, Lichtenstein L. Calling somatic SNVs and indels with mutect2. bioRxiv. 2019. https://doi.org/10.1101/861054.
Tavtigian SV, Greenblatt MS, Harrison SM, Nussbaum RL, Prabhu SA, Boucher KM, et al. Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med. 2018;20:1054–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–71.
Cingolani P, Platts A, Wang Le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Bernard E, Tuechler H, Greenberg PL, Hasserjian RP, Ossa JEA, Nannya Y, et al. Molecular international prognostic scoring system for myelodysplastic syndromes. NEJM Evid. 2022;1:EVIDoa2200008.
Duncavage EJ, Schroeder MC, O’Laughlin M, Wilson R, MacMillan S, Bohannon A, et al. Genome sequencing as an alternative to cytogenetic analysis in myeloid cancers. New Engl J Med. 2021;384:924–35.
Hinai A, Pratcorona M, Grob T, Kavelaars FG, Bussaglia E, Sanders MA, et al. The landscape of KMT2A-PTD AML: concurrent mutations, gene expression signatures, and clinical outcome. Hemasphere. 2019;3:e181.
Dai B, Yu H, Ma T, Lei Y, Wang J, Zhang Y, et al. The application of targeted RNA sequencing for KMT2A-partial tandem duplication identification and integrated analysis of molecular characterization in acute myeloid leukemia. J Mol Diagn. 2021;23:1478–90.
Awada H, Durmaz A, Gurnari C, Kishtagari A, Meggendorfer M, Kerr CM, et al. Machine learning integrates genomic signatures for subclassification beyond primary and secondary acute myeloid leukemia. Blood. 2021;138:1885–95.
Sorrentino E, Cristofoli F, Modena C, Paolacci S, Bertelli M, Marceddu G. Integration of VarSome API in an existing bioinformatic pipeline for automated ACMG interpretation of clinical variants. Eur Rev Med Pharm Sci. 2021;25:1–6.
Gallo M, Adinolfi V, Morviducci L, Acquati S, Tuveri E, Ferrari P, et al. Early prediction of pancreatic cancer from new-onset diabetes: an Associazione Italiana Oncologia Medica (AIOM)/Associazione Medici Diabetologi (AMD)/Societa Italiana Endocrinologia (SIE)/Societa Italiana Farmacologia (SIF) multidisciplinary consensus position paper. ESMO Open. 2021;6:100155.
Cingolani P. Variant annotation and functional prediction: SnpEff. Methods Mol Biol. 2022;2493:289–314.
Wang W, Zhang Y, Yang W, Liu X, Jiang L, Lang W, et al. The molecular prognostic scoring system for normal karyotype myelodysplastic syndromes. J Transl Med. 2025;23:76.
Della Porta MG, Tuechler H, Malcovati L, Schanz J, Sanz G, Garcia-Manero G, et al. Validation of WHO classification-based Prognostic Scoring System (WPSS) for myelodysplastic syndromes and comparison with the revised International Prognostic Scoring System (IPSS-R). A study of the International Working Group for Prognosis in Myelodysplasia (IWG-PM). Leukemia. 2015;29:1502–13.
Ogawa S. Genetics of MDS. Blood, J Am Soc Hematol. 2019;133:1049–59.
Johansson B, Billström R, Mauritzson N, Mitelman F. Trisomy 19 as the sole chromosomal anomaly in hematologic neoplasms. Cancer Genet Cytogenet. 1994;74:62–5.
Tirosh A, Jin DX, De Marco L, Laitman Y, Friedman E. Activating genomic alterations in the Gs alpha gene (GNAS) in 274 694 tumors. Genes Chromosomes Cancer. 2020;59:503–16.
He S, Li Y, Wang L, Li Y, Xu L, Cai D, et al. DNA methylation landscape reveals GNAS as a decitabine-responsive marker in patients with acute myeloid leukemia. Neoplasia. 2024;49:100965.
Wheeler EC, Vora S, Mayer D, Kotini AG, Olszewska M, Park SS, et al. Integrative RNA-omics discovers GNAS alternative splicing as a phenotypic driver of splicing factor–mutant neoplasms. Cancer Discov. 2022;12:836–55.
Buradkar A, Bezerra E, Coltro G, Lasho TL, Finke CM, Gangat N, et al. Landscape of RAS pathway mutations in patients with myelodysplastic syndrome/myeloproliferative neoplasm overlap syndromes: a study of 461 molecularly annotated patients. Leukemia. 2021;35:644–9.
Murphy DM, Bejar R, Stevenson K, Neuberg D, Shi Y, Cubrich C, et al. NRAS mutations with low allele burden have independent prognostic significance for patients with lower risk myelodysplastic syndromes. Leukemia. 2013;27:2077–81.
Alfayez M, Issa GC, Patel KP, Wang F, Wang X, Short NJ, et al. The clinical impact of PTPN11 mutations in adults with acute myeloid leukemia. Leukemia. 2021;35:691–700.
Patel SS, Ho C, Ptashkin RN, Sadigh S, Bagg A, Geyer JT, et al. Clinicopathologic and genetic characterization of nonacute NPM1-mutated myeloid neoplasms. Blood Adv. 2019;3:1540–5.
DiNardo CD, Jabbour E, Ravandi F, Takahashi K, Daver N, Routbort M, et al. IDH1 and IDH2 mutations in myelodysplastic syndromes and role in disease progression. Leukemia. 2016;30:980–4.
Kosmider O, Gelsi-Boyer V, Slama L, Dreyfus F, Beyne-Rauzy O, Quesnel B, et al. Mutations of IDH1 and IDH2 genes in early and accelerated phases of myelodysplastic syndromes and MDS/myeloproliferative neoplasms. Leukemia. 2010;24:1094–6.
Tsai S-C, Shih L-Y, Liang S-T, Huang Y-J, Kuo M-C, Huang C-F, et al. Biological activities of RUNX1 mutants predict secondary acute leukemia transformation from chronic myelomonocytic leukemia and myelodysplastic syndromes. Clin Cancer Res. 2015;21:3541–51.
Wang J, Fang Z, Yang S, Yan K, Zhang J, Yu Y, et al. Venetoclax and hypomethylating agent in previously untreated higher-risk myelodysplastic syndromes and genotype signatures for response and prognosis: a real-world study. Am J Hematol. 2025;100:314–9.
Thol F, Friesen I, Damm F, Yun H, Weissinger EM, Krauter J, et al. Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. J Clin Oncol. 2011;29:2499–506.
Grob T, Al Hinai AS, Sanders MA, Kavelaars FG, Rijken M, Gradowska PL, et al. Molecular characterization of mutant TP53 acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood J Am Soc Hematol. 2022;139:2347–54.
Montoro MJ, Palomo L, Haferlach C, Acha P, Chan O, Navarro V, et al. Influence of TP53 gene mutations and their allelic status in myelodysplastic syndromes with isolated 5q deletion. Blood. 2024;144:1722–31.
Deng J, Wu X, Ling Y, Liu X, Zheng X, Ye W, et al. The prognostic impact of variant allele frequency (VAF) in TP53 mutant patients with MDS: a systematic review and meta-analysis. Eur J Haematol. 2020;105:524–39.
Bernard E, Nannya Y, Hasserjian RP, Devlin SM, Tuechler H, Medina-Martinez JS, et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat Med. 2020;26:1549–56.
Zhao Y, Chen W, Yu J, Pei S, Zhang Q, Shi J, et al. TP53 in MDS and AML: biological and clinical advances. Cancer Lett. 2024;588:216767.
Aguirre LE, Al Ali N, Sallman DA, Ball S, Jain AG, Chan O, et al. Assessment and validation of the molecular international prognostic scoring system for myelodysplastic syndromes. Leukemia. 2023;37:1530–9.
Tang Y, Wang H, Zhang Z, Yao Y, Han Y, Wu D. DIAPH1 mutations predict a favorable outcome for de novo MDS. Cancer Lett. 2024;598:217125.
Acknowledgements
The study was supported by the General research fund, Research Grants Council, Hong Kong (HKU project code: 17118914); the Hong Kong Anti-Cancer Society Research Grant (HKU project code: AR190027); Celgene International II SARL (HKU project code: AR180044); the Ministry of Science and Technology, Taiwan (MOST 108-2628-B-002-015 and 111-2314-B-002-279), and the Ministry of Health and Welfare, Taiwan (MOHW 107-TDU-B-211-114009 and 111-TDU-B-221-114001).
Author information
Authors and Affiliations
Contributions
H.G., H.A.H. and Y.L.K. conceived the study. H.G., X.C.H.T., M.O., G.M.K.L., T.K.Y.W., C.Y.Y.L., W.J.C., H.F.T., H.A.H. and Y.L.K. treated the patients. H.G., R.Y., P.L., X.C.H.T., V.W.K.L., G.M.K.L., M.O., R.R., L.C., L.A., Q.Z., T.K.Y.W., C.Y.Y.L. and H.A.H. collected and assembled the data. H.G., R.Y., P.L., X.C.H.T., V.W.K.L., G.M.K.L., T.S.H. and H.A.H analyzed and interpreted the data. All authors wrote and approved the final manuscript. W.J.C., H.F.T., H.A.H. and Y.L.K. contributed equally as senior authors.
Corresponding authors
Ethics declarations
Competing interests
The study was supported by the General research fund, Research Grants Council, Hong Kong (HKU project code: 17118914); the Hong Kong Anti-Cancer Society Research Grant (HKU project code: AR190027); Celgene International II SARL (HKU project code: AR180044); the Ministry of Science and Technology, Taiwan (MOST 108-2628-B-002-015 and 111-2314-B-002-279), and the Ministry of Health and Welfare, Taiwan (MOHW 107-TDU-B-211-114009 and 111-TDU-B-221-114001). There are no other relevant competing interests to disclosed.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gill, H., Yim, R., Lee, P. et al. A clinico-genomic prognostic model for primary myelodysplastic neoplasm in Asia. Blood Cancer J. 15, 128 (2025). https://doi.org/10.1038/s41408-025-01339-0
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41408-025-01339-0
