
Abstract
Background
The molecular characterisation of pediatric acute lymphoblastic leukemia (pALL) is essential for accurate diagnosis and risk stratification. However, standard-of-care (SoC) methods have limited sensitivity and resolution.
Methods
This study evaluates the clinical utility of emerging genomic technologies—including optical genome mapping (OGM), digital multiplex ligation-dependent probe amplification (dMLPA), RNA sequencing (RNA-seq), and targeted next-generation sequencing (t-NGS)—in the largest cohort of pALL patients analysed to date, with 60 cases using OGM.
Results
Considering clinically relevant alterations identified with at least two different methods, OGM as a standalone test demonstrated superior resolution, detecting chromosomal gains and losses (51.7% vs. 35%, p = 0.0973) and gene fusions (56.7% vs. 30%, p = 0.0057), while resolving 15% of non-informative cases. Combining dMLPA and RNA-seq was the most effective approach, achieving precise classification of complex subtypes and uniquely identifying IGH rearrangements undetected by other techniques. OGM identified clinically relevant alterations in 90% of cases, and the dMLPA-RNAseq combination reached 95%, compared to 46.7% with SoC techniques.
Conclusions
Integrating these technologies into diagnostic workflows overcomes SoC limitations. OGM and the dMLPA-RNAseq combination emerge as front-line strategies, addressing pALL heterogeneity, streamlining molecular testing, and informing treatment decisions to improve outcomes.
Similar content being viewed by others
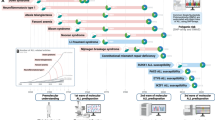


Introduction
Acute lymphoblastic leukemia (ALL) is the most common pediatric malignancy, characterised by the uncontrolled proliferation of lymphoid progenitor cells. The genetic landscape of ALL is highly heterogeneous, encompassing a wide range of structural variants (SVs) and single-nucleotide variants (SNVs) that play a critical role in leukemogenesis. Notably, SVs, including chromosomal rearrangements, deletions, and copy number alterations (CNAs), are enriched in pediatric ALL (pALL) compared to adult cases [1]. These genetic abnormalities define distinct molecular subtypes and are crucial for predicting prognosis and response to therapy [2].
The latest update of the World Health Organization (WHO) [3] and International Consensus Classification (ICC) [4] increasingly emphasise the role of molecular alterations in defining leukemia subtypes. This is exemplified by the inclusion, for the first time, of entities defined by SNVs (IKZF1 N159Y, PAX5 P80R). In parallel, modern treatment protocols, such as the ALLTogether (NCT04307576) consortium guidelines, have implemented risk stratification strategies based on fusions and CNA profiling, as well as targeted inhibitors for ABL-class patients and immunotherapy into frontline therapy. These advancements highlight the heterogeneity of diagnostic entities, and thus the need to combine genomic approaches for a precise molecular characterisation that exceeds the capabilities of current standard-of-care (SoC) methods.
Traditionally, the SoC methods for genetic diagnosis in ALL relied on conventional cytogenetics, including chromosome banding analysis (CBA) and fluorescence in situ hybridisation (FISH). While these methods have been instrumental in detecting recurrent chromosomal abnormalities, they exhibit significant limitations, such as low resolution, limited detection capacity for cryptic alterations, and reliance on viable metaphases [5]. To overcome these constraints, emerging methodologies such as multiplex ligation-dependent probe amplification (MLPA), targeted next-generation sequencing (t-NGS), digital multiplex ligation-dependent probe amplification (dMLPA), RNA sequencing (RNAseq), or optical genome mapping (OGM) have been progressively implemented into the diagnostic workup, offering enhanced sensitivity and broader detection capabilities [6,7,8].
Despite the improvement potential of these novel technologies, each method has inherent strengths and weaknesses in detecting specific types of genetic alterations. To date, no single approach has demonstrated a comprehensive coverage of the entire mutational spectrum of ALL, leaving the optimal diagnostic strategy for clinical practice unresolved [9]. A systematic evaluation of these methodologies is required to determine the most effective approach for an accurate and efficient diagnosis in the clinical setting.
In this study, we analysed a cohort of 60 pALL patients using the standard-of-care, OGM, t-NGS, MLPA, dMLPA, and RNAseq (when sufficient genetic material was available). The primary aim was to assess the diagnostic yield of each method, both individually and in combination, to identify the most robust strategy for the comprehensive genomic characterisation of pALL in the clinical setting. To date, this cohort represents the largest pALL series characterised by OGM in a clinical setting within a single institution, providing valuable insights into the optimal diagnostic workflow for this disease.
Material and methods
Patients and samples
A total of 60 bone marrow (BM) or peripheral blood (PB) samples (55 diagnoses and 5 relapses) were obtained from pALL patients (49 B-ALL, 11 T-ALL) referred to our institution between August 2021 and August 2024. The cohort included 36 males (60%) and 24 females (40%), with a median age of 5 years (range 1–16). The median blast percentage was 90% (range 20–100). Patients were selected solely based on the availability of high-quality samples to ensure that the cohort accurately reflected real-world clinical testing conditions.
Written consents were obtained from parents or legal guardians of all patients according to the recommendations of the Human Rights Declaration and the Helsinki Conference. This study was approved by the institutional ethics committee for clinical research.
Standard-of-care baseline
Immunophenotyping was performed by flow cytometry following standardised procedures. The antibody panel included anti-CD45, CD34, CD123, CD10, CD19, CD20, CD22, CD9, CD24, CD25, CD15, NG2, CD66c (KORSA), CD33, CD13, cytoplasmic MPO, nuclear TdT, and cytoplasmic CD3 (Beckman Coulter, CA, USA), as well as CD38, cytoplasmic IgM, and kappa/lambda light chains (Dako, CA, USA). Cytogenetic analysis was conducted using G-banding on metaphase chromosomes, with karyotypes interpreted according to the International System for Human Cytogenomic Nomenclature [10]. FISH were performed on interphase nuclei using commercial probes for BCR::ABL1, KMT2A, ETV6::RUNX1, TCF3, CRLF2, ABL2, EPOR, PDGFRB, and JAK2, following the manufacturer’s instructions. Additionally, ETV6::RUNX1 and BCR::ABL1 rearrangements were also assessed by RT-qPCR as described by Gabert et al. [11].
DNA and RNA isolation
Genomic DNA (gDNA) and total RNA were extracted using the QIAsymphony SP/AS instrument (Qiagen, Valencia, CA) automated platform. DNA extraction was performed with the QIAamp DNA Mini Kit (Qiagen), while RNA was isolated using the RNeasy Midi Kit (Qiagen), following the manufacturer’s instructions. The extracted nucleic acids were quantified using the Qubit Fluorometer (Thermo Fisher Scientific, San Francisco, CA, USA) with the Qubit dsDNA High Sensitivity Assay Kit for DNA and the Qubit RNA HS Assay Kit for RNA.
Emerging methods
Multiplex ligation-dependent probe amplification
The gDNA was isolated from BM or PB samples as described above. MLPA was performed in 100 ng of gDNA using the SALSA MLPA P335 (BTG1, CDKN2A/B, EBF1, ETV6, IKZF1, PAR1 region, PAX5 and RB1) (MRC-Holland, Amsterdam, The Netherlands) following the manufacturer’s instructions. Capillary electrophoresis was carried out on a SeqStudio Genetic Analyzer (Applied Biosystems, Foster City, CA, USA), and data were analysed using Coffalyser.Net software (MRC-Holland) according to established guidelines.
Digital multiplex ligation-dependent probe amplification
dMLPA was performed on 50 ng of gDNA using SALSA digitalMLPA D007 Acute Lymphoblastic Leukemia probemix (MRC-Holland) according manufacturer’s recommendations. The probemix includes target probes to identify recurrent microdeletions or amplifications, and karyotyping probes to detect gross chromosomal abnormalities along all chromosomes.
Reactions were pooled and sequenced on a MiSeq sequencer with 150 bp single-read chemistry (Illumina, San Diego, CA, USA). Coffalyser digitalMLPA software (MRC-Holland) was used to analyse the copy number status. Regions with a probe ratio value around 1.0 (±0.15) were considered unaffected, while an increased or decreased value indicated the presence of a gain or loss, respectively. Leukemic cell burden (LCB) was considered to interpret the results. Subclonal CNAs were only reported if consecutive probes had dosage values unambiguously falling outside the range but not reaching the expected ratio for a loss/gain based on the LCB, and also compared with other affected regions within the same sample.
Optical genome mapping
OGM was conducted on fresh (less than 24 h after sample collection) or frozen PB or BM samples according to the standard protocol (Bionano Genomics, San Diego, CA, USA). Briefly, ultra-high molecular weight genomic DNA (UHMW-DNA) was isolated and labelled using DLE-1 enzyme and the Bionano Prep direct labelling and staining (DLS) protocol. A total of 750 ng of labelled UHMW-DNA was loaded on a Saphyr G2.3 chip and run on Bionano’s Saphyr for imaging. Quality criteria were as follows: map rates greater than 60%, molecule N50 values >250 kb (for molecules >150 kb), and effective genome coverage >300×. Genome analysis was performed using the human genome GRCh38 as a reference, and Bionano Access 1.6 and Bionano Solve 3.6 software. Variant calling was performed with Rare Variant Pipeline and Guided assembly with standard filter settings.
Next-generation sequencing
t-NGS was performed using the ALLseq panel (Gil et al. [12]), designed to detect SNVs, insertions/deletions (indels), CNAs, gene fusions, and gene expression. The full list of targeted genes is provided in Table S1. Briefly, 10 ng of gDNA and RNA were used for library preparation, which was automated on the Ion Chef™ System (Thermo Fisher Scientific). Sequencing was conducted on the Ion S5 sequencer (Thermo Fisher Scientific). Variant calling was performed using the Ion Reporter software (Thermo Fisher Scientific), and variants were considered relevant when their allelic frequency exceeded 3%.
RNAseq
Total RNA was extracted from BM or PB as described above. Due to limited sample availability, RNAseq was performed in 20 patients. Quantification and integrity were assessed using a Qubit fluorometer (Thermo Fisher Scientific) and an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), respectively. RNA libraries were prepared using the TruSeq Stranded Total RNA Library Prep Kit (Illumina), followed by sequencing using paired-end 150 nt reads on the NovaSeq 6000 platform (Illumina), with approximately 20 million reads per sample. Data processing and bioinformatic analysis were conducted using HISAT2 for alignment and STARfusion for fusion detection. As STARfusion often reported multiple fusion genes per sample, including false positives, only high-confidence fusions supported by at least 2 junction reads and 1 spanning fragment read (at least 10,000 nucleotides apart if both genes were on the same chromosome) were considered. Additionally, specific parameters were used to identify IG rearrangements as described by Thomson et al. [13]. Differential gene expression levels were quantified by DESeq2, and diagnostic entities were predicted using the ALLCatchR classifier, with >0.5 score considered relevant.
Diagnosis yield assessment across different techniques versus standard-of-care testing
The diagnostic performance of each technique was evaluated both individually and in combination with the other. The diagnostic yield was assessed based on two levels of clinical relevance: (1) Capability to identify entities recognised by the WHO 2022 and/or ICC 2022 classifications, as well as risk stratification markers according to the ALLTogether guidelines; and (2) Any other pathogenic alterations affecting known drivers in pALL or associated with prognosis but not currently incorporated into clinical risk stratification. Diagnostic entities were assigned according to the WHO 2022/ICC classification if a defining driver or a compatible gene expression profile (GEP) was identified by any of the methods used.
Results were classified at the patient level into three categories based on concordance: (1) Completely concordant: all detected alterations were identified by the compared techniques; (2) Partially concordant: some but not all abnormalities were detected by both techniques; and (3) Discordant: entirely different abnormalities were identified by the techniques in comparison. Clinically relevant alterations identified exclusively by a single technique were further validated using orthogonal methods.
Statistical analysis
Categorical variables were compared using Fisher’s exact test or the chi-square test, as appropriate, while continuous variables were analysed using the Mann–Whitney U test or Student’s t-test. The sensitivity and specificity values of each method were calculated against the standard of care. Co-segregation of genetic alterations was analysed using the “somaticInteraction” function from the maftools package. All statistical analyses were performed using R software (version 4.4.2), with a significance threshold set at p < 0.05 or p < 0.01. Plots were generated with R software (version 4.4.2) using the ggplot2 package and BioRender.
Results
A total of 60 pALL patients underwent initial testing using SoC techniques. CBA identified high hyperdiploidy (HeH) in 11 cases (11/60, 18.3%), gene rearrangements in 6/60 (10%) cases, monosomy/trisomy in 7/60 (11.7%) cases, and was non-informative in 9 cases (9/60, 15%). FISH detected ETV6::RUNX1 fusion in 7/60 (11.7%) patients, all of which were confirmed by RT-qPCR. Additionally, CRLF2-r were identified in 3/60 (5%) patients, while KMT2A-r, JAK2-r, and TCF3-r were each detected in one patient. Main characteristics of the cohort are shown in Table S2. Patients with sufficient genetic material were further analysed as outlined in Fig. S1.
Structural variants
Chromosomal gains and losses
dMLPA
dMLPA identified a total of 191 whole chromosomal gains and 14 losses across the genome, with an average of 2.55 alterations per patient. Notably, 76.6% of these whole-chromosome gains and losses were found in 16 patients with a hyperdiploid karyotype, involving 5–9 chromosomes. The most frequently gained chromosomes were 21, 14, 10, X, 4, 17 and 6, with recurrent multiple copies of chromosomes 21, X, 4, and 8 (Fig. 1). Overall, dMLPA detected five additional cases of hyperdiploidy, with no non-informative results observed.

Bars represent the number of alterations detected per chromosome, categorised as losses (red), single gains (blue), or multiple gains (purple).
OGM
OGM identified the same chromosomal gains and losses as dMLPA. No false positives or false negatives were observed. Notably, both techniques identified five additional cases of hyperdiploidy compared to standard-of-care methods, with complete concordance between them.
Copy number alterations
MLPA
MLPA analysis for CNA-UKALL (BTG1, CDKN2A/B, EBF1, ETV6, IKZF1, JAK2, PAX5, RB1, and PAR1 region) revealed a total of 79 subchromosomal CNAs across the cohort, with an average of 1.3 alterations per patient. The most frequently altered genes were CDKN2B (27/60, 45%) and CDKN2A (25/60, 41.7%).
dMLPA
dMLPA demonstrated complete concordance with MLPA for CNA-UKALL and identified 207 additional subchromosomal alterations in 37/60 (61.7%) patients. The most frequent lesions were MTAP (11/60, 18.3%) and MLLT3 deletions (18/60, 30%), both occurring as part of 9p21 deletions, which also harbour CDKN2A/B. In fact, 83.8% of all biallelic losses and 64.3% of subclonal alterations were located in 9p (MTAP-MLLT3-CDKN2A/B). Other commonly observed deletions included VPREB1 (8/60, 13.3%), IGHM (7/60, 11.7%), and ERG (7/60, 11.7%). In contrast, RUNX1 showed the highest frequency of multiple gains, associated with iAMP21 in two patients. The deletion pattern at the 1p region was suggestive of STIL::TAL1 fusion in one patient, further confirmed by t-NGS and OGM.
OGM
OGM demonstrated full agreement with MLPA and dMLPA in 55/60 (91.7%) patients for CNA-UKALL. However, discrepancies were observed in five cases, mainly affecting CDKN2A/B and PAR1. In two patients, OGM detected a heterozygous CDKN2A/B deletion, whereas MLPA reported a biallelic loss in one case and no alteration in another. In another patient, OGM identified a CDKN2A/B loss with a low variant allele frequency (VAF = 10%) that was undetected by MLPA and dMLPA. Further discrepancies included an ETV6 homozygous deletion with a PAR1 duplication detected by MLPA but classified as wild-type by OGM. Similarly, MLPA identified heterozygous CDKN2A loss and PAR1 duplication in two cases, which were not confirmed by OGM (Fig. S2).
Aside from these discrepancies, OGM detected 16 losses that dMLPA missed due to its targeted genomic coverage. Among them, four deletions in 11q involved key genes such as KMT2A and ATM, while two deletions in 13q12.2 affected FLT3. Conversely, dMLPA identified 16 alterations that OGM did not detect, including six deletions in IGHM. Of these, 13/16 were too small to be detected by OGM, one was identified as a subclonal event by dMLPA, and two alterations in CTCF were incorrectly classified by OGM (Table S3). The complete distribution of CNAs is depicted in Fig. 2a.

a Chromosomal ideogram illustrating subchromosomal gains (green circles) and losses (orange triangles) detected across the cohort. The colour gradient represents the frequency of alterations, ranging from none (blue) to high (red). b Heatmap displaying significant co-occurrence and mutual exclusivity relationships among genomic alterations. The intensity of the colour represents the –log10(p-value), with darker shades indicating stronger statistical significance. Significant interactions are marked with * (p < 0.01) and • (p < 0.05).
Co-occurrence of CNAs was analysed to identify potential association or exclusion patterns (Fig. 2b). This analysis revealed 41 significant positive correlations, with the strongest associations observed in the 9p region (JAK2, MLLT3, MTAP, CDKN2A/B), as well as between IGHM and ERG, and TBL1XR1 and FHIT losses. Additionally, a trend towards mutual exclusivity with other alterations was noted in hyperdiploid cases; however, no significant negative correlations were identified.
Fusions
t-NGS
t-NGS targeting recurrent fusions and deregulated expression in ALL (t-NGS-RNA) (Table S1) identified a total of 18 fusions across 18/60 (30%) patients. The most frequent fusion was ETV6::RUNX1 (7/18, 38.9%), followed by P2RY8::CRLF2 (4/18, 22.2%). Additionally, two cases harboured PICALM::MLLT10 (2/18, 11.1%) and, the least common fusions were STIL::TAL1, MEF2D::BCL9, PAX5::JAK2, TCF3::ZNF384, and ZMIZ1::ABL1, each detected in one patient (Fig. 3a). CRLF2 overexpression was found in 3/4 patients carrying CRLF2-r, and TAL1 in a case harbouring STIL::TAL1, and TLX3 overexpression was detected in two patients despite the absence of detectable fusions, suggesting the presence of a potential non-productive rearrangement (data not shown).

a t-NGS-RNA (n = 60). b OGM (n = 60). c RNAseq (n = 20). Each arc represents a fusion, with gene name colours indicating different entities. The thickness of each arc is proportional to the number of detected events.
OGM
OGM detected 37 fusions across 34/60 (56.7%) patients, identifying a significantly higher number of fusions than t-NGS-RNA and confirming all fusions previously identified by SoC methods and t-NGS-RNA. Notably, OGM provided a broader characterisation of PAX5-r, detecting 6 vs. 1 fusions compared with t-NGS-RNA. Of note, 2 of these 6 cases carried additional concomitant fusions. In one of them, t-NGS-RNA only identified the subclonal P2RY8::CRLF2 secondary fusion, whereas OGM also uncovered the primary event (PAX5::PML) (Fig. 3b).
Beyond clinically relevant fusions, OGM revealed the novel transcripts RUNX1::GATA3, CDKN2A::KVS1, and ETV6::HNRNPH1, as well as additional fusions in four patients where t-NGS-RNA had already identified one rearrangement (Fig. 3b). Furthermore, OGM showed cryptic or complex structural alterations, such as t(5;14)(q35.1;q32.2) and multiple translocations, where the precise fusion partner could not be determined in 5/60 (8.3%) patients.
RNAseq
RNAseq was performed in 20/60 (33.3%) patients, providing a more detailed characterisation of fusion events compared to those identified by t-NGS-RNA or OGM. A total of 21 fusions were detected across 16 patients, yielding a higher detection rate than t-NGS-RNA, validating OGM findings and offering additional insights into cases with IGH translocations. Notably, RNAseq was the only technique that identified the IGH::DUX4 (n = 2), IGH::CEBPE, and IGH::EPOR fusions, underscoring the complexity of their detection (Fig. 3c). Furthermore, RNAseq not only confirmed previously detected fusions but also allowed the refinement of fusion breakpoints and validated some alterations through expression levels (Table S4).
SNV/Indels
t-NGS
A total of 80 SNV/Indels were identified by t-NGS in 52/60 (86.7%) patients, with a median of 1.5 alterations per patient. The majority of these variants were missense mutations, representing 62 out of 80 (77.5%) of the detected alterations (Fig. 4a). Genes involved in the RAS signalling pathway were the most frequently altered, with NRAS (23%), KRAS (21%), and FLT3 (13%) being the most commonly affected (Fig. 4b). Notably, a substantial proportion of these mutations were detected at subclonal levels, suggesting the presence of multiple leukemic subpopulations (Fig. 4c).

a Classification of variant types, showing a predominance of missense mutations. b Top 10 most frequently mutated genes. c Distribution of variant allele frequencies (VAF) for recurrently mutated genes. d Oncoprint depicting individual mutations across samples, with immunophenotypic and molecular classifications annotated at the bottom. Tumour mutational burden (TMB) is shown at the top, and the frequency of gene alterations across samples is summarised on the right. Colour codes indicate mutation types and sample characteristics. Red asterisk indicates IGHM deletion with a common B-cell phenotype. Entities were assigned if a defining driver was found by any techniques as per the WHO/ICC 2022 diagnostic classification criteria.
Association of genetic lesions with leukemia subtypes
The complete genetic landscape (detailed in Table S5) was analysed collectively with demographic data, and the most frequent events were summarised in Fig. 4d. A significant association was observed between the co-mutation of IGHM and ERG deletions, and DUX4 rearrangements (p = 0.012) (Fig. 2b), suggesting a potential cooperative role in this leukemic subtype. Interestingly, IGHM deletions as a single event showed a strong correlation with Pre-B phenotype, whereas this alteration was detected in only one patient with common B-cell phenotype (red asterisk in Fig. 4d). JAK2 deletions were significantly enriched within the PAX5-alterations subgroup (p = 0.016), while IKZF1 deletions and NRAS mutations were predominantly associated with iAMP21 (p = 0.03) and BCR-ABL-like/JAK-STAT-activated cases (p = 0.04), respectively (Fig. 4).
Diagnostic yield of emerging techniques
The diagnostic yield of each technique varied depending on the type of genetic alteration analysed (Table 1). For chromosomal gains and losses, OGM and dMLPA identified the highest number of events (31/60, 51.7%), and the clinical utility was superior to the standard-of-care approach (61.3% vs. 66.7%). The assessment of CNA-UKALL showed full concordance between MLPA and dMLPA, detecting alterations in nearly two-thirds of the cohort (38/60, 63.3%) with 100% clinical relevance, while OGM was slightly less sensitive, detecting alterations in 36/60 (60%) patients. In contrast, CNAs outside the UKALL-CNA risk panel were detected with similar sensitivity by dMLPA and OGM (37-38/60), but only a small fraction were clinically relevant (~5%) (Fig. 5a).

a Proportion of patients with genetic alterations detected by each technique, categorised by clinical relevance. b Concordance analysis between techniques for different types of genetic alterations. c Number of genetic entities identified by each technique in the entire cohort (n = 60). d Number of genetic entities identified by each technique in the subset of 20 patients with available RNAseq data. Statistically significant differences are indicated by asterisks (*p < 0.01, **p < 0.001).
The most pronounced differences were observed in fusion detection. Both the SoC methods and t-NGS identified fusions in 30% of patients (18/60), with clinical relevance rates of 72.2% and 100%, respectively. In contrast, OGM and RNAseq detected fusions in a significantly higher proportion of patients (56.7% and 80%, p < 0.01), with clinical relevance rates of 76.5% and 81.3%. SNVs and indels were exclusively detected by t-NGS (52/60), but none of these were classified as clinically relevant under the criteria applied (Fig. 5a).
The concordance analysis revealed notable differences among some techniques. Of note, none of the described discrepancies among methods could be attributed to a low blast percentage. For chromosomal gains and losses, soc methods showed discrepancies in 9/60 (15%) of patients, primarily due to non-informative cases, whereas OGM and dMLPA exhibited complete concordance. For the CNA-UKALL risk assessment, MLPA and dMLPA were fully concordant, while OGM showed partial concordance in 1/60 (1.7%) of cases and discordance in 4/60 (6.7%) patients. Regarding other CNAs, complete concordance was achieved in 60% of patients and partial concordance in 38.3%, mostly due to non-covered regions by dMLPA. Discrepancies were observed in only one case (1.7%), where dMLPA detected an ERG deletion missed by OGM. For gene fusions, the highest concordance (complete and partial) was observed between OGM and RNAseq, with only 10% of patients showing discrepancies, mainly due to missed IGH rearrangements by OGM. In contrast, the greatest discordance was found between t-NGS-RNA and RNAseq, with 65% of patients being discordant, due to the differences in the detection capabilities of these techniques (Fig. 5b).
The potential of emerging technologies to improve molecular diagnosis and risk stratification was assessed against SoC methods and various technical combinations commonly used in the clinical setting. To minimise bias, cases with available RNAseq data were analysed separately, ensuring a balanced evaluation of each method’s contribution. OGM as a standalone test demonstrated significantly higher performance in refining B-NOS and T-NOS cases compared to SoC methods (25% vs. 40%, and 3.3% vs. 13.3%, p < 0.01) and tNGS (25% vs. 55%, p < 0.001) (Fig. 5c). OGM also outperformed SoC methods in detecting PAX5 alterations (0% vs. 10%, p < 0.01), and provided a more accurate diagnosis than combined approaches, including SoC and t-NGS or dMLPA and t-NGS (Fig. 6a). Additionally, RNAseq evaluation in 20/60 patients showed a superior refinement of the B-NOS group compared to OGM due to the identification of IGH rearrangements (Fig. 5d and Fig. S2).

a Sankey diagram showing the distribution and reclassification of patients across four diagnostic strategies: standard-of-care, standard-of-care with t-NGS, dMLPA combined with t-NGS, and OGM. Each vertical axis represents one diagnostic approach, while the coloured flows depict how individual patient classification shifts among categories with each successive approach. b Risk stratification comparison across methodologies, showing the proportion of patients classified into different genetic risk groups. The width of each node is proportional to the number of patients in each category.
In terms of risk stratification, OGM and dMLPA with t-NGS outperformed SoC methods with MLPA and SoC with MLPA and t-NGS combinations, especially in the good-risk genetic subgroup (58% vs. 50%, p < 0.05) (Fig. 6b).
Overall, OGM and RNAseq outperformed SoC methods and commonly used combinatorial approaches in identifying clinically relevant biomarkers. Standard-of-care methods exhibited the lowest detection rate, identifying biomarkers in only 28/60 patients (46.7%), which increased to 36/60 (60%) when combined with t-NGS. In contrast, RNAseq and OGM, as standalone methods, detected biomarkers in 16/20 (80%) and 54/60 (90%) patients, respectively. The combination of dMLPA and RNAseq demonstrated the highest performance, identifying clinically relevant biomarkers in 95% of cases. These findings underscore OGM as a powerful standalone tool and highlight dMLPA combined with RNAseq or OGM as the most promising approaches for clinical practice.
Discussion
This study represents the largest cohort of pALL patients analysed by OGM, compared with standard-of-care methods and emerging techniques, performed at a single institution in a clinical laboratory setting. While the analytical performance of these techniques has been reported by different groups, this is the first study to assess their diagnostic yield as individual methods and in combination within the same cohort [14,15,16]. Our results support the incorporation of OGM or the combination of dMLPA with RNAseq into clinical practice, as these approaches significantly increase the proportion of patients with clinically relevant markers to 90–95%, compared to only 46.7% using standard-of-care methods (Table 1). This improvement has direct clinical implications, as the refinement in diagnosis and/or risk stratification may influence treatment decisions, including eligibility for targeted therapies or enrolment in clinical trials, which could significantly impact survival in these patients.
Cytogenetic analysis based on CBA has long been the gold standard for detecting chromosomal gains and losses in pALL. These alterations appear in up to 35–40% of cases, and are critical for prognosis and treatment decisions [17]. Recent studies have demonstrated the high resolution and sensitivity of novel approaches like OGM and dMLPA, and our findings further reinforce these observations, revealing a significant reduction in non-informative cases and the reclassification of 8.3% of patients only considering chromosomal gains and losses [18, 19].
Subchromosomal alterations have gained increasing recognition in recent years, particularly those included in the UKALL-CNA risk profile, which has demonstrated prognostic significance and is now mandatory for detection in clinical laboratories [20]. As CBA has a limited resolution, MLPA is prompted as the gold standard to identify these alterations. In agreement with Bédics et al. [21], our data also support dMLPA as a feasible alternative for detecting these alterations, demonstrating comparable results to MLPA, which is expected given their shared methodological principles. In contrast, OGM showed discordant results in 8.3% of cases, primarily affecting the CDKN2A/B and PAR1 regions. A rational explanation for these discrepancies could be the intrinsic limitations of OGM in certain genomic regions [22]. Like sequencing-based techniques, OGM exhibits reduced sensitivity in low-coverage regions and highly repetitive sequences, such as pseudoautosomal regions (PAR), which may compromise the detection of specific CNAs [23]. Additionally, the limit of detection for CNAs with OGM is another critical factor. Levy et al. reported that a minimum coverage of 300× ensures a reliable limit of detection of 10%–15% VAF; however, this threshold declines sharply in samples with lower coverage, further impacting sensitivity. CNAs beyond the UKALL-CNA profile remain of limited clinical significance. However, certain alterations like ERG deletions are already integrated into the IKZF1plus risk profile and are used by some groups for risk stratification [24, 25]. Additionally, we identified recurrent VPREB1 deletions, an essential gene for lymphoid development, whose loss may contribute to maturation arrest at early stages (Fig. 2). Similarly, IGHM deletions may impair the formation of a functional BCR, also leading to a developmental block at the pre-B stage and potentially contributing to leukemic transformation [26]. Interestingly, IGHM deletions as a single event showed a strong correlation with Pre-B phenotype, while co-mutation of IGHM and ERG correlated with IGH::DUX4 and common B phenotype. These findings support the hypothesis that IGHM deletions may, in some cases, act as surrogate markers of IGH fusions and contribute to leukemogenesis by disrupting early B-cell receptor formation and cooperating with transcriptional programmes such as those driven by DUX4 [27]. Thus, although most CNAs beyond the UKALL-CNA profile are not currently considered clinically relevant, their identification may be useful, as they could have future implications in pALL classification and risk assessment.
Structural variants giving rise to gene fusions are a hallmark in pALL, and current diagnostic guidelines mandate the identification of more than 10 recurrent rearrangements in clinical settings. t-NGS emerged a decade ago as a more comprehensive alternative to karyotype and FISH, and is now widely implemented in clinical laboratories. However, only ~30% of fusions in pALL are recurrent, posing a major challenge for t-NGS-RNA panels, which are inherently limited to detecting known targets [28, 29]. This limitation is evident in our results and those reported by others, while all fusions detected by t-NGS-RNA in our cohort were clinically relevant; the number of rearrangements identified by OGM or RNAseq was significantly higher, underscoring the value of unbiased genome-wide approaches in the molecular characterisation of pALL [2, 30]. Moreover, a substantial number of pALL subtypes are now defined by GEP rather than a single genetic alteration. Given the pivotal role of these entities in patient management and the increasing affordability of RNAseq, this technology is emerging as the logical transition from t-NGS-RNA in clinical practice. Another important challenge that could be addressed is the identification of intricate fusions located in highly repetitive or variable regions, such as IGH::DUX4 or IGH::CEBPE, where most techniques often fail [31]. The ability to integrate fusion calling with gene expression profiling enhances the robustness for detecting these alterations. In fact, RNAseq was the sole technique to identify four patients carrying IGH rearrangements that were missed by any other method, further supporting its role as a powerful tool in pALL molecular diagnostics. Regarding OGM, it enables the detection of complex structural variants that may not result in the formation of fusion transcripts. This is particularly relevant in T-ALL, where non-canonical or transcriptionally silent rearrangements could be found in up to 60% of cases.
In contrast to adult ALL and other hematological neoplasms, where diagnostic classifications, therapeutic protocols and clinical trials increasingly incorporate the SNVs into clinical decision-making, current treatment guidelines for pALL, such as ALLTogether protocol, do not consider these alterations for therapeutic stratification. Although some SNVs remain relevant for diagnostic classification—particularly under the ICC 2022 framework—all of these entities are also characterised by distinct transcriptional signatures. Hence, the identification of the underlying point mutation may not be essential when transcriptomic profiling is available. Moreover, RNA-based approaches can also detect SNVs/indels when sufficient read depth is achieved, further reducing the need for separate targeted sequencing [32, 33]. In line with this, our results show that while over 85% of patients harbour at least one SNV/indel, the vast majority lack current clinical relevance in the pediatric setting. Therefore, genomic approaches that focus primarily on the detection of SNVs/indels, such as targeted NGS panels, may have limited utility for the pALL clinical setting.
Despite the strengths of our study, including the comprehensive genetic characterisation of pALL using multiple methodologies, some limitations should be acknowledged. As the study was conducted at a single institution, the applicability of these findings may vary across centres depending on local infrastructure and expertise. Moreover, the cohort size is relatively modest, which may limit the statistical power to detect associations involving rare alterations or specific subtypes. Samples with low blast % may be assumed to be misdiagnosed regardless of the method used. However, a low blast count in pALL at diagnosis or relapse is unusual. Regarding the potential misclassification of rare entities, GEP has been demonstrated to accurately detect up to 21 B-ALL subtypes [34] and 10 T-ALL classes [35], which grants rare entity identification, especially in B-ALL. Intergenic fusions are frequent in T-ALL, and these alterations have been broadly demonstrated to be correctly detected by OGM. Therefore, the identification of rare entities not included in our work should be correctly identified by these technologies. Nonetheless, additional studies are needed to validate our results and assess the feasibility of implementing these strategies more broadly.
Conclusion
In summary, this study demonstrates the value of emerging technologies to improve the molecular characterisation of pediatric ALL. Large-scale multicenter studies have paved the way for defining the molecular basis of this disease, providing critical insights into its genetic landscape. However, it is essential to translate this growing knowledge into accessible and feasible diagnostic workflows that can be implemented in routine clinical settings. This study validates novel techniques such as OGM, evaluates their individual and combined performance, and ultimately proposes a diagnostic strategy based either on OGM as a standalone approach or on the combination of RNAseq with dMLPA. Both strategies significantly increase the detection of clinically relevant alterations and could streamline current diagnostic algorithms by reducing the need for cascade testing, which often leads to increased costs and prolonged turnaround times that are not aligned with the demands of real-world diagnostic laboratories. This technological adaptation would ensure a more comprehensive diagnosis, facilitating treatment decisions and, ultimately, improving patient outcomes.
Data availability
All data is available upon reasonable request to llop_margar@gva.es or jose_gil@iislafe.es.
References
Roberts KG. Genetics and prognosis of ALL in children vs adults. Hematology. 2018;2018:137–45. https://doi.org/10.1182/asheducation-2018.1.137.
Brady SW, Roberts KG, Gu Z, Shi L, Pounds S, Pei D, et al. The genomic landscape of pediatric acute lymphoblastic leukemia. Nat Genet. 2022;54:1376–89. https://doi.org/10.1038/s41588-022-01159-z.
Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBdO, Berti E, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia. 2022;36:1720–48. https://doi.org/10.1038/s41375-022-01620-2.
Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka H-M, et al. International consensus classification of myeloid neoplasms and acute leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140:1200–28. https://doi.org/10.1182/blood.2022015850.
Medeiros BC, Othus M, Estey EH, Fang M, Appelbaum FR. Unsuccessful diagnostic cytogenetic analysis is a poor prognostic feature in acute myeloid leukaemia. Br J Haematol. 2013;164:245–50. https://doi.org/10.1111/bjh.12625.
Rack KA, Van Den Berg E, Haferlach C, Beverloo HB, Costa D, Espinet B, et al. European recommendations and quality assurance for cytogenomic analysis of haematological neoplasms. Leukemia. 2019;33:1851–67. https://doi.org/10.1038/s41375-019-0378-z.
Janic D, Peric J, Karan-Djurasevic T, Kostic T, Marjanovic I, Stanic B, et al. Application of targeted next generation sequencing for the mutational profiling of patients with acute lymphoblastic leukemia. J Med Biochem. 2019. https://doi.org/10.2478/jomb-2019-0017.
Levy B, Kanagal-Shamanna R, Sahajpal NS, Neveling K, Rack K, Dewaele B, et al. A framework for the clinical implementation of optical genome mapping in hematologic malignancies. Am J Hematol. 2024;99:642–61. https://doi.org/10.1002/ajh.27175.
Walter W, Iacobucci I, Meggendorfer M. Diagnosis of acute lymphoblastic leukaemia: an overview of the current genomic classification, diagnostic approaches, and future directions. Histopathology. 2024. https://doi.org/10.1111/his.15338
ISCN 2020. 2020. https://doi.org/10.1159/isbn.978-3-318-06867-2.
Gabert J, Beillard E, Van Der Velden VHJ, Bi W, Grimwade D, Pallisgaard N, et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia – a Europe Against Cancer Program. Leukemia. 2003;17:2318–57. https://doi.org/10.1038/sj.leu.2403135.
Gil JV, Such E, Sargas C, Simarro J, Miralles A, Pérez G, et al. Design and Validation of a Custom Next-Generation Sequencing Panel in Pediatric Acute Lymphoblastic Leukemia. Int J Mol Sci. 2023;24:4440. https://doi.org/10.3390/ijms24054440.
Thomson AJ, Rehn JA, Heatley SL, Eadie LN, Page EC, Schutz C, et al. Reproducible bioinformatics analysis workflows for detecting IGH gene fusions in B-Cell acute lymphoblastic leukaemia patients. Cancers. 2023;15:4731. https://doi.org/10.3390/cancers15194731.
Heyer EE, Deveson IW, Wooi D, Selinger CI, Lyons RJ, Hayes VM, et al. Diagnosis of fusion genes using targeted RNA sequencing. Nat Commun. 2019. https://doi.org/10.1038/s41467-019-09374-9.
Thakral D, Kaur G, Gupta R, Bernard-SlagterA, Savola S, Kumar I, et al. Rapid identification of key copy number alterations in B- and T-Cell acute lymphoblastic leukemia by digital multiplex Ligation-Dependent probe amplification. Front Oncol. 2019. https://doi.org/10.3389/fonc.2019.00871.
Rack K, De Bie J, Ameye G, Gielen O, Demeyer S, Cools J, et al. Optimizing the diagnostic workflow for acute lymphoblastic leukemia by optical genome mapping. Am J Hematol. 2022;97:548–61. https://doi.org/10.1002/ajh.26487.
Mrózek K, Harper DP, Aplan PD. Cytogenetics and molecular genetics of acute lymphoblastic leukemia. Hematol Oncol Clin North Am. 2009;23:991–1010. https://doi.org/10.1016/j.hoc.2009.07.001.
Loghavi S, Wei Q, Ravandi F, Quesada AE, Routbort MJ, Hu S, et al. Optical genome mapping improves the accuracy of classification, risk stratification, and personalized treatment strategies for patients with acute myeloid leukemia. Am J Hematol. 2024;99:1959–68. https://doi.org/10.1002/ajh.27435.
Levy B, Baughn LB, Akkari Y, Chartrand S, LaBarge B, Claxton D, et al. Optical genome mapping in acute myeloid leukemia: a multicenter evaluation. Blood Adv. 2022;7:1297–307. https://doi.org/10.1182/bloodadvances.2022007583.
Moorman AV, Barretta E, Butler ER, Ward EJ, Twentyman K, Kirkwood AA, et al. Prognostic impact of chromosomal abnormalities and copy number alterations in adult B-cell precursor acute lymphoblastic leukaemia: a UKALL14 study. Leukemia. 2021;36:625–36. https://doi.org/10.1038/s41375-021-01448-2.
Bedics G, Egyed B, Kotmayer L, Bernard-Slagter A, de Groot K, Beko A, et al. PersonALL: a genetic scoring guide for personalized risk assessment in pediatric B-cell precursor acute lymphoblastic leukemia. Br J Cancer. 2023;129:455–65. https://doi.org/10.1038/s41416-023-02309-8.
Vieler LM, Nilius-Eliliwi V, Schroers R, Vangala DB, Nguyen HP, Gerding WM. Optical genome mapping reveals and characterizes recurrent aberrations and new fusion genes in adult ALL. Genes. 2023;14:686 https://doi.org/10.3390/genes14030686.
Rehn JA, O’Connor MJ, White DL, Yeung DT. DUX hunting—clinical features and diagnostic challenges associated with DUX4-rearranged leukaemia. Cancers. 2020;12:2815 https://doi.org/10.3390/cancers12102815.
Stanulla M, Dagdan E, Zaliova M, Moricke A, Palmi C, Cazzaniga G, et al. IKZF1Plus defines a new minimal residual disease–dependent very-poor prognostic profile in pediatric B-Cell precursor acute lymphoblastic leukemia. J Clin Oncol. 2018;36:1240–9. https://doi.org/10.1200/jco.2017.74.3617.
Palmi C, Bresolin S, Junk S, Fazio G, Silvestri D, Zaliova M, et al. Definition and Prognostic value of PH-like and IKZF1Plus status in children with down syndrome and B-cell precursor acute lymphoblastic leukemia. HemaSphere. 2023;7:e892 https://doi.org/10.1097/hs9.0000000000000892.
Naji P, Khatami M, Heidari MM, Hashemi A, Jenabzadeh A, Chamani R, et al. Mutational analysis of the VPREB1 gene of Pre-BCR complex in a cohort of sporadic pediatric patients with B-Cell acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2022;44:210–9. https://doi.org/10.1097/mph.0000000000002456.
Chen D, Zheng J, Gerasimcik N, Lagerstedt K, Sjorgen H, Abrahamsson J, et al. The expression pattern of the pre-B cell receptor components correlates with cellular stage and clinical outcome in acute lymphoblastic leukemia. PLoS ONE. 2016;11:e0162638 https://doi.org/10.1371/journal.pone.0162638.
Vega-Garcia N, Benito R, Esperanza-Cebollada E, Llop M, Robledo C, Vicente-Garcés C, et al. Helpful criteria when implementing NGS panels in childhood lymphoblastic leukemia. J Pers Med. 2020;10:244 https://doi.org/10.3390/jpm10040244.
Llop M, Sargas C, Barragán E. The role of next-generation sequencing in acute myeloid leukemia. Curr Opin Oncol. 2022. https://doi.org/10.1097/cco.0000000000000899.
Umeda M, Ma J, Westover T, Ni Y, Song G, Maciaszek JL, et al. A new genomic framework to categorize pediatric acute myeloid leukemia. Nat Genet. 2024;56:281–93. https://doi.org/10.1038/s41588-023-01640-3.
Gu Z, Churchman ML, Roberts KG, Moore I, Zhou X, Nakitandwe J, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet. 2019;51:296–307. https://doi.org/10.1038/s41588-018-0315-5.
Atak ZK, Gianfelici V, Hulselmans G, De Keersmaecker K, Devasia AG, Geerdens E, et al. Comprehensive analysis of transcriptome variation uncovers known and novel driver events in T-Cell acute lymphoblastic leukemia. PLoS Genet. 2013;9:e1003997 https://doi.org/10.1371/journal.pgen.1003997.
Hu Z, Jia Z, Liu J, Mao A, Han H, Gu Z. MD-ALL: an integrative platform for molecular diagnosis of B-acute lymphoblastic leukemia. Haematologica. 2023. https://doi.org/10.3324/haematol.2023.283706. Published online November 16.
Beder T, Hansen B-T, Hartmann AM, Zimmermann J, Amelunxen E, Wolgast N, et al. The gene expression classifier ALLCatchR identifies B-cell precursor ALL subtypes and underlying developmental trajectories across age. HemaSphere. 2023;7:e939 https://doi.org/10.1097/hs9.0000000000000939.
Gu A, Schmidt B, Lonsdale A, Jalaldeen R, Kosasih HJ, Brown LM, et al. TALLSorts: a T-cell acute lymphoblastic leukemia subtype classifier using RNA-seq expression data. Blood Adv. 2023;7:7402–6. https://doi.org/10.1182/bloodadvances.2023010385.
Acknowledgements
We wish to thank the patients, family members, and staff from all the clinical units involved in this study.
Funding
This work was funded by the Spanish Ministry of Science and Innovation (PID2021-126138OB-I00) and supported by a grant from the Scientific Foundation of the Spanish Association Against Cancer to JVG (PRDVA246268GIL).
Author information
Authors and Affiliations
Contributions
Conceptualisation, ES and ML; original draft, JVG; laboratory work, JVG, GA, SdlH, AM, AD-G and CL-B; clinical data recollection, MdC, AV, RA, CF, JMF and EB; results discussion, JVG, EB, ES, CF, JMF and ML. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was approved by the institutional ethics committee of Instituto de Investigación Sanitaria La Fe for studies involving humans on December 22, 2021 (2021-045-1).
Consent for publication
Written informed consents were obtained from parents or legal guardians of all patients according to the recommendations of the Human Rights Declaration and the Helsinki Conference.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gil, J.V., Avetisyan, G., Miralles, A. et al. Benchmarking standard-of-care and emerging genomic approaches to enhance diagnosis in pediatric acute lymphoblastic leukemia. Br J Cancer 133, 1744–1754 (2025). https://doi.org/10.1038/s41416-025-03204-0
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41416-025-03204-0
