
Abstract
Cancer stem cells (CSCs) are a type of stem cell that possesses not only the intrinsic abilities of stem cells but also the properties of cancer cells. Therefore, CSCs are known to have self-renewal and outstanding proliferation capacity, along with the potential to differentiate into specific types of tumor cells. Cancers typically originate from CSCs, making them a significant target for tumor treatment. Among the related cascades of the CSCs, mammalian target of rapamycin (mTOR) pathway is regarded as one of the most important signaling pathways because of its association with significant upstream signaling: phosphatidylinositol 3‑kinase/protein kinase B (PI3K/AKT) pathway and mitogen‑activated protein kinase (MAPK) cascade, which influence various activities of stem cells, including CSCs. Recent studies have shown that the mTOR pathway not only affects generation of CSCs but also the maintenance of their pluripotency. Furthermore, the maintenance of pluripotency or differentiation into specific types of cancer cells depends on the regulation of the mTOR signal in CSCs. Consequently, the clinical potential and importance of mTOR in effective cancer therapy are increasing. In this review, we demonstrate the association between the mTOR pathway and cancer, including CSCs. Additionally, we discuss a new concept for anti-cancer drug development aimed at overcoming existing drawbacks, such as drug resistance, by targeting CSCs through mTOR inhibition.
Similar content being viewed by others

Facts
-
Cancer treatment faces challenges due to its complexity, with chemotherapy limited by drug resistance, prompting the exploration of advanced therapeutic approaches.
-
Cancer Stem Cells (CSCs) are a key focus, sharing characteristics with normal stem cells and being targeted to prevent differentiation into tumor cells and promote self-renewal.
-
The mTOR pathway plays a pivotal role in activating stem and immune cells within cancer tissues, leading to the development and utilization of mTOR inhibitors in FDA-approved cancer treatments.
-
Clinical trials categorize mTOR inhibitors for various cancer types, demonstrating their efficacy in suppressing CSC differentiation, intrinsic stem cell properties, and reducing metastasis.
-
Despite promising results, caution is warranted in mTOR inhibition due to its involvement in normal cellular activities, necessitating careful strategies to avoid potential side effects and the development of advanced, targeted tumor therapies.
Open questions
-
How can CSC-targeting therapy effectively prevent tumor recurrence, considering the pivotal role of CSCs in tumor relapse, metastasis, and resistance to radiotherapy and chemotherapy?
-
What intrinsic factors contribute to the development of resistance in CSCs, including their longer cell cycle, DNA repair system, and involvement of oxidative modulators and metabolic plasticity regulators?
-
In what ways does the PI3K/AKT/mTOR axis contribute to CSC resistance, and what are the outcomes and implications of clinical trials involving mTOR pathway inhibitors for various cancers?
-
What challenges are associated with delivering anti-cancer drugs to CSCs, and how effective are viral delivery systems like adenovirus and nanoparticle therapeutics in reaching and targeting CSCs, considering existing challenges in these delivery methods?
Introduction
Stem cells are known for their three unique properties: self-renewal ability [1], differentiation capability [2], and exceptional proliferation potential [3, 4]. Due to their intrinsic ability to self-renew and differentiate into multiple lineages [2, 3], stem cells have been applied to generate complex tissues and organoids [5,6,7,8,9]. However, stem cells also have the capacity to develop tumors through cellular growth, recurrence, and metastasis [10]. Therefore, some of stem cells, which possess not only stem ability but also cancer ability, are classified as cancer stem cells (CSCs) [11]. CSCs can self-renew and differentiate into tumors, contributing to the growth, metastasis, and drug resistance of cancers. To understand the framework of these stem cells, including CSCs, several signaling pathways have been studied: Wnt signaling [12], Hedgehog signaling [13], Janus kinase/signal transducer and activator of transcription protein (JAK/STAT) signaling [14], Notch signaling [15], and the mammalian target of rapamycin (mTOR) signaling [16].
During the cellular proliferation of stem cells, the expression of Wnt protein and Hedgehog protein is upregulated [17]. Therefore, Wnt and Hedgehog signaling actively progress in stem cells, including CSCs, for self-renewal and proliferation [13]. In CSCs, JAK/STAT signaling pathway has been known to be associated with maintenance of self-renewal property, hematopoiesis, and neurogenesis [14, 18]. After phosphorylation, both JAK and STAT proteins are dimerize, leading to the activation of downstream transcription. As the Notch protein acts as a promoter or suppressor during transcriptional process, Notch signaling is considered significant in the regulation of CSCs’ properties, including differentiation into various types of tumor cells [15]. The Notch pathway’s effect depends on the types of substances binding to the receptors.
In addition to those signaling pathways, mTOR signaling has been considered the most important for understanding and regulating CSCs [19,20,21,22,23]. mTOR, also referred to as FK506-binding protein 12-rapamycin-associated protein 1 (FRAP1), belongs to the phosphatidylinositol 3-kinase (PI3K)-related protein kinase family and is a component of both mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [24,25,26,27]. mTOR is a serine/threonine kinase that regulates the cell cycle [28], glycolysis [28], the immune response [29], and many other activities in mammalian cells. Particularly, mTOR signaling is known to be essential for regulating the self-renewal ability, differentiation capacity, and proliferation property of the stem cells [11, 30,31,32]. It has been revealed that mTOR maintains pluripotency in human embryonic stem cells (hESCs) by preserving the expression of pluripotency-related genes such as octamer-binding transcription factor 4 (OCT4), NANOG, and sex determining region Y-box 2 (SOX2) [33, 34]. This cascade is related to upstream signaling pathway such as the PI3K/protein kinase B (AKT) pathway in pluripotency maintenance [35]. However, in mesenchymal stem cells (MSCs), in contrast to hESCs, mTOR signaling induces differentiation into various types of cells, including myoblasts, osteoblasts, adipocytes, and neurons [36]. In hematopoietic stem cells (HSCs), hyperproliferation and loss of quiescence occur due to inhibition of the mTOR pathway [37]. Therefore, the mTOR pathway is concluded to have different cellular effects depending on the types of stem cells. The mTOR signaling pathway not only in stem cells but also in various types of cells such as normal cells, cancer cells, and CSCs is described in Fig. 1.

Effect of PI3K/AKT/mTOR axis is described depending upon types of cells. Normal cells can synthesize proteins, grow, and proliferate through activation of mTOR pathway [38, 41]. Stem cells can proliferate and self-renew through mTOR signaling [11, 30,31,32]. Also, pluripotency of the stem cells can be maintained by mTOR axis [33,34,35]. In cancer cells, cell division resulting in cell growth and angiogenesis are affected by mTOR signaling pathway [39, 40]. In CSCs, which have both characteristics of stem cells and cancer cells, unlimited tumorigenesis and metastasis are enhanced due to improved cell proliferation and angiogenesis [19,20,21,22,23].
Since cancers originate from CSCs, and mTOR signaling plays an important role in CSC regulation, the clinical potential and importance of mTOR in effective therapy are increasing [38]. Through anti-cancer therapy targeting CSCs by precisely regulating the mTOR pathway, the effectiveness of chemotherapy could be enhanced, potentially eliminating cancer metastasis and recurrence [39, 40]. In this review, we demonstrate the association between the mTOR pathway and cancers, including CSCs. Furthermore, we discuss how to develop new anti-cancer drugs and overcome the drug resistance, which is known to be the most challenging hurdle in CSC-targeting treatments.
Cancer and mTOR pathway
Since mTOR signaling is strongly linked with CSCs, it is also associated with cancer, which typically originates from CSCs [41]. mTOR in cancer is related to tumorigenesis [42, 43], metastasis [44, 45], tumor development [46, 47] and angiogenesis [48, 49]. The mTORC1/ribosomal S6 kinase (S6K) pathway contributes to growth signal-mediated genome instability and tumorigenesis by inhibiting of the function of ring finger protein 168 (RNF168) [50]. Additionally, mTORC2/AKT signaling activates proliferative cell cycles in cancer cells, leading to tumor development, through the binding of mutated rat sarcoma virus (RAS) proteins to mTOR components of mTORC2 and mitogen-activated protein kinase-associated protein 1 (MAPKAP1) [51].
Abnormal activation of mTOR pathway leads to anabolism and energy storage, supplying a plethora of nutrients to the tumor, thus promoting tumor proliferation. Furthermore, mTOR regulates the expression of survival factors in cancer cells, such as cellular myelocytomatosis (C-MYC), and hypoxia induced gene 1 (HIG1), as well as angiogenic factors, including vascular endothelial growth factor (VEGF) [52,53,54,55]. Consequently, activation of the mTOR pathway improves and accelerates the generation, metastasis, proliferation, and angiogenesis of tumor cells [56, 57]. In Fig. 2, molecules contributing to mTOR pathway-mediated tumorigenesis and tumor development are described according to the types of cancer.

The PI3K/AKT/mTOR axis and related molecules, classified as oncogenes (green and yellow) and tumor suppressor (red), are described depending upon types of cancer. PTEN and p53 are the representative tumor suppressor [60,61,62,63,64,65,66, 112, 137, 138], whereas 4EBP-1 and SREBP1 are major downstream oncogenes. Breast and colorectal cancer are affected by 4EBP-1 and eIF4E [59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76], and gynecologic and lung cancer are induced by 4EBP-1 and S6K [77,78,79,80,81,82,83, 102,103,104]. Both LDLRAD2 and MiT/TFE are significant and direct oncogenes causing pancreatic cancer [193,194,195,196]. Liver and urothelial cancer are associated with expression of SREBP1 [180,181,182,183,184].
Breast cancer
Breast cancer, a malignant tumor that originates in mammary glands, has been a significant cause of cancer-associated death, not only in women but also in men [58]. In breast cancer cells, the PI3K/AKT/mTORC1/sterol regulation element-binding protein (SREBP) pathway is recognized as the primary cascade inducing lipid synthesis and increasing the proliferation of tumor cells [59]. The tumor suppressor gene, phosphatase and tensin homolog (PTEN), plays an important role in breast cancer [60, 61]. Since PTEN regulates the PI3K/AKT/mTOR signaling pathway, it is involved in cell growth, survival, migration, and progression [62, 63]. Mutation in the phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) gene and alterations in the breast cancer type 1 and 2 susceptibility protein (BRCA 1 and 2) genes lead to PTEN inactivation, resulting in the overexpression of the human epidermal growth factor receptor 2 (HER2) gene and the activation of the PI3K pathway [64,65,66]. It is known that 18 ~ 20% of breast cancers are caused by the overexpression of HER2 oncogenes, which activate the PI3K/AKT/mTOR pathway, promoting tumor development [65].
Colorectal cancer
Colorectal cancer, including colon cancer, consists of malignant tumor cells in the large intestine tissues [67, 68]. In colorectal cancer, downstream molecules of the mTOR pathway such as eukaryotic translation initiation factor 4E (eIF4E) and eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1), play a crucial role in cancer development [69,70,71,72]. In colon cancer cell lines with overexpressed eIF4E, uncontrollable cellular growth and transformation into malignant forms were observed [73, 74]. In a transgenic in vivo mouse model induced to overexpress eIF4E, ubiquitination of the β-actin promoter was induced, and therefore, the development of tumor was also investigated [75]. Additionally, activated 4E-BP1, which was highly expressed in colon carcinoma, was found to be important for lymph node metastasis in colorectal cancer patients [76].
Gynecologic cancer
Gynecologic cancer occurs in women’s reproductive organs and includes endometrial cancer (EC), cervical cancer (CC), ovarian cancer (OC), and others [77, 78]. In Phase II clinical trial, mTOR inhibition showed a significant effect on EC treatment; however, severe side effects such as diarrhea were also observed [79]. Other clinical trials have been conducted targeting the mTOR signal to explore its correlation with EC development [80, 81]. In the case of CC, blocking the phosphorylation of 4E-BP1 in human papillomaviruses (HPVs)-infected cells led to an increased number of cells in the G1 phase, resulting in apoptosis of the CC cells [82]. When mTOR inhibitor was applied solely to OC cells, it exhibited a moderate response, whereas dual inhibitor showed effective inhibition of the PI3K signaling pathway, deactivating the proliferation of the OC cell line [83].
Liver cancer
When hepatocytes are damaged due to various external factors such as viruses, alcohol, and obesity, diseases are caused and then maintained chronically. The repetition of cell death and regeneration during disease symptoms eventually leads to the generation of hepatocellular carcinoma (HCC) [84]. The major causes of failure in liver cancer treatment are the invasion and metastasis of hepatocellular carcinoma (HCC) [85]. Since the PI3K/AKT/mTOR pathway is activated in HCC, activated mTOR serves as a representative marker for detecting the recovery or recurrence of liver cancer, including cholangiocarcinoma [86, 87] and hepatoblastoma [88, 89]. Therefore, expression of phosphorylated mTOR (p-mTOR) signifies and increased tumor grade, and elevated alpha-fetoprotein (AFP) indicates enhanced metastasis [90, 91], invasion [92, 93], and proliferation [94] of liver cancer cells.
Tumor cells invaded and metastasized into other tissues by degrading the extracellular matrix (ECM) and the basement membrane. Matrix metalloproteinase (MMP), the ECM-degrading enzyme, has been considered crucial for tumor invasion and metastasis. Among MMPs, MMP-2 and -9 have been highlighted as key factors in this function [95, 96]. The PI3K/AKT/mTOR pathway upregulates the expression of both MMP-2 and -9, thus promoting the invasion and metastasis of HCC [97, 98]. mTORC2 is also associated with liver cancer, as it induces the synthesis of fatty acids and lipids. The mTORC2 pathway has been a significant target for eliminating lipid-related liver tumors [99, 100]. According to the results from in vivo liver cancer mouse model, hepatic steatosis and the development of HCC were observed through the activation of the mTOR signal [101].
Lung cancer
Lung cancer has two subtypes: non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC). Most cases of lung cancer are NSCLC, with only about 15% being SCLC [102]. Recently, it has been revealed that the PI3K/AKT/mTOR signaling pathway influences the aggressiveness of lung cancer. It stimulates transcription factors, cytokines, and receptor tyrosine kinases (RTKs), activating epithelial-mesenchymal transition (EMT) markers, which in turn cause invasion and migration in lung cancer [103]. Since the mTOR pathway is upregulated in NSCLC, the concentration of p-mTOR increases by up to 90% in NSCLC patients with adenocarcinoma, and up to 60% and 40% in NSCLC patients with large cell carcinoma and squamous cell carcinoma (SCC), respectively [103]. Additionally, the concentration of representative downstream molecules of the mTOR signal, such as ribosomal protein S6 (rpS6) and 4E-BP1, increased by up to 58% in NSCLC patients [103, 104].
Pancreatic cancer
Pancreatic cancer is one of the leading causes of cancer-related deaths worldwide [105, 106]. It is known that low-density lipoprotein receptor class A domain-containing 2 (LDLRAD2) plays a significant role in the progression of pancreatic cancer [107,108,109]. LDLRAD2 is a gene involved in various human diseases, particularly common in pancreatic cancer cell lines and tissues. While LDLRAD2 is primarily regulated by the Wnt/β-catenin pathway, it has been also found that phosphorylation of AKT and mTOR molecules is significantly reduced in an LDLRAD2 knockout model [110]. Knocking out LDLRAD2 successfully decreases the proliferation, invasion, and metastasis of pancreatic cancer cells [110]. Therefore, it has been confirmed that LDLRAD2 is involved in the progression of pancreatic cancer by regulating the AKT/mTOR pathway. Additionally, RAS-related guanosine triphosphate (GTP)-binding protein D (RRAGD)-mediated activation of mTORC1 has been observed in pancreatic ductal adenocarcinoma patients, and an increase in proliferation and cancer growth has also been investigated [111].
Prostate cancer
Prostate cancer is the second leading cause of cancer-related death in men, following lung cancer [112]. In prostate cancer, mTOR downregulates the expression of glycogen synthase kinase 3 (GSK-3) [113]. The decrease in GSK-3 inhibits the caspase-3 signaling pathway, reducing ROS production. Consequently, the apoptosis of tumor cells is inhibited [113]. Thus, mTOR activation induces the development of prostate cancer by inhibiting cellular apoptosis.
Skin cancer
Skin cancer is divided into various types, represented as melanoma and keratinocyte carcinoma, depending on the area it occurs and its relationship to the mTOR pathway [114, 115]. In melanoma patients, the level of phosphorylated AKT (p-AKT) level is high, and the PI3K/AKT/mTOR pathway is activated due to PTEN loss [116]. It has already been found that PTEN is reduced by 10 ~ 30% in melanoma cell lines [117]. Serine/threonine protein kinase (BRAF), a member of the RAF kinase family, plays a vital role in regulating the MAPK pathway, influencing cell division, differentiation, and secretion [118,119,120]. Mutation of BRAF is known to induce melanoma genesis and metastasis [117]. The BRAF mutation is found in 60% of melanoma cells [121] and 90% of melanoma patients [122].
In keratinocyte carcinoma, there are three major types of cells: basal cell carcinoma (BCC), SCC, and Merkel cell carcinoma (MCC). The generation of BCC is strongly linked to the PI3K/AKT/mTOR pathway [123]. Treatment of an mTOR inhibitor showed significant disease recession in BCC patients [124]. SCC exhibits a higher mTOR level than BCC [125, 126]. Furthermore, enhanced expression of cyclic-dependent kinase 2 (CDK2) induces the malignant transition of cancer cells through AKT/mTOR signal [127]. MCC, a nonmelanoma skin cancer derived from neuroendocrine cells, is associated with the activation of 4E-BP1 and rpS6, downstream molecules of the mTOR pathway [128]. Administering a dual drug that inhibits both mTORC1 and mTORC2 in a xenograft mouse model effectively suppressed the growth of MCC [129, 130].
Urothelial cancer
Urothelial cancer is a type of tumor that can occur in the urethra, bladder, ureters, renal pelvis, and other urinary organs [131]. Bladder cancer is the fifth most common cancer in the world and the most common cancer among urothelial cancers [132]. In bladder cancer, the AKT/mTOR signaling pathway is activated through the upregulation of pyruvate kinase M2 (PKM2), leading to increased expression of SREBP-1c and the genes related to fatty acid synthesis [133, 134]. As fatty acid production increases, tumor growth is also enhanced [51]. PTEN, a suppressor of the mTOR signal, is related to various types of urothelial cancers and their mechanisms [135, 136]. In particular, the p53 pathway, one of the PTEN downstream pathways, has the greatest influence on bladder cancer, inducing apoptosis in tumor cells [132, 137]. Therefore, inactivation of PTEN and p53 together increases mTOR signaling and promotes tumor growth [132, 138]. Phosphorylated-rpS6 (p-rpS6), as a marker of mTOR activity, is frequently expressed in muscle-invasive urothelial cancer cells [139,140,141]. Thus, p-rpS6 is associated with lymph node metastasis. According to related studies, it was demonstrated that cell proliferation and migration decreased, and the tumor volume was also reduced by 55% in the T24-xenograft in vivo model through the inhibition of rpS6 phosphorylation and subsequent inactivation of the mTOR pathway [142].
Targeting mTOR signaling In Cscs
Cancer cells are not composed of only one type of cell but are a group of multiple cells [143, 144]. This characteristic is called heterogeneity, and research on various types of cancer has been ongoing since the 1800s [76, 145]. Therefore, tumor cells exhibit diverse characteristics and functions that support cancer development [146].
Initially, CSCs were identified in leukemia in 1994 as a type of cancer cell with the ability to continuously grow and self-renew [147]. It was revealed that a small subset of cancer cells could (re)generate tumor cells, leading to investigations into the capacity of self-renewal and differentiation into heterogeneous tumor cells [148]. There have been various opinions and contentions regarding the crucial role of CSCs, which represent only a small portion of the total cancer cells within a tumor that contains various cell types.
CSCs are believed to be generated within a niche, which includes fibroblasts, blood vessels, and secreted factors that surround and influence CSCs [149]. This process is similar to normal stem cells, and the accumulation of many mutations in stem cells has been considered an important factor in their conversion into CSCs [150, 151]. Another hypothesis suggests that cancer cells can mutate into CSCs during the chemotherapy process of metastasis [152]. Metastatic cancer cells can acquire the ability to differentiate into other types of cancer cells, resulting in the generation of CSCs [150, 153]. According to another viewpoint, when a small subset of cancer cells is stimulated and activated by chemotherapy, they can differentiate into cancer cells with the ability to induce cancer recurrence [154, 155].
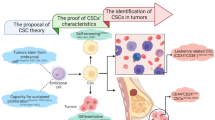
Figure 3 depicts the hypothesis on CSC generation, followed by the results of CSC-targeting anti-cancer therapeutics through mTOR inhibition. Additionally, various types of mTOR inhibitors and their outcomes are presented, depending on the types of CSCs, in Table 1.

There have been several hypotheses on how the CSCs, which possess a crucial role in cancer development, are generated (purple box in left). Accumulation of mutations in normal stem cells are considered as an important cause of CSC generation [150, 151]. Also, cancer cells can be mutated into CSCs by chemotherapy during the process of metastasis [152]. The metastatic cancer cells can acquire the ability to differentiate into other types of cancer cells, resulting in generation of CSCs [150, 153]. In addition, when a small part of the cancer cells is stimulated and activated by radiotherapy and chemotherapy, it can be differentiated into cancer cells, which have ability to induce cancer recurrence [154]. And these cancer cells with recurrence ability can be activated into CSCs [155]. Through differentiation of the CSCs, CSC-derived cancer tissues are generated (middle of the scheme). Through treatment of mTOR inhibitor to the CSC-derived cancer tissues, CSC-targeting anti-cancer therapy is possible (pink box in right). By inhibition of mTOR signaling pathway, viability and ability of CSC decrease [89, 95, 97,98,99, 102, 109], radio/chemo-sensitivity of the CSCs increase [100], and number or size of tumor sphere decrease [87, 88, 90,91,92, 108, 111], resulting in apoptosis of the cancer tissues [86, 96, 112, 115].
Breast CSCs
Rapamycin (ABI-009), a representative mTORC1 inhibitor, has been approved by the U.S. Food and Drug Administration (FDA) as an immunosuppressant for renal transplantation and for the treatment of lymphangioleiomyomatosis (LAM) [156, 157]. Furthermore, rapamycin (ABI-009) is being considered for use as an anti-cancer drug targeting the mTOR signal in breast cancer stem cells [158]. Since rapamycin (ABI-009) inhibits the mTOR pathway, resulting in the downregulation of manganese superoxide dismutase (MnSOD) expression, it has been revealed that rapamycin (ABI-009) reduces the number of mammospheres, which are the markers of breast cancer development, and makes breast CSCs sensitive to radiation therapy [159].
Metformin is a drug that inhibits the mTOR pathway by activating adenosine monophosphate-activated protein kinase (AMPK) [160]. When metformin was treated to breast CSCs, the size and number of the CSCs significantly decreased, and their radiosensitivity increased [161, 162]. In addition, metformin reduced the recurrence ability of breast CSCs when co-treated with fluorouracil (5-FU), epirubicin, and cyclophosphamide (FEC) [163].
Investigations have also been conducted to assess the effects of another mTOR inhibitor, everolimus (RAD001) [99, 107, 111, 164, 165]. According to the results, everolimus (RAD001) was found to increase the expression of caspase-3 and -8 in breast cancer cells, inducing apoptosis of the tumors by upregulating apoptotic gene expression [99, 107]. Furthermore, it was confirmed that the growth of breast cancer cells decreased with everolimus (RAD001) treatment in an MCF-7 bearing in vivo mouse model [107]. Additionally, co-treatment of everolimus (RAD001) with the anti-estrogen drug exemestane (FCE-24304) for hormone receptor-positive breast cancer therapy showed alleviation of breast cancer by inactivating the mTOR signal in patients [111, 164, 165].
As a dual inhibitor targeting both PI3K and mTOR, VS-5584 (SB2343) efficiently decreased the number of tumor spheres and induced apoptosis of CSCs when co-treated with paclitaxel (PTX), which regulates the cell cycle to stay at G2, inhibiting the cell division [166, 167].
Central nervous system CSCs
Glioblastoma multiforme (GBM) is one of the most aggressive cancers that occur in the central nervous system (CNS) and is known to be difficult to treat due to strong resistance to chemotherapy [168]. FC85, a drug targeting AKT/mTOR signal, reactivates the functionality of p53 by blocking its endogenous inhibitor, murine double minute 2 homologue (MDM2) [169]. Since p53 is a tumor suppressor that induces apoptosis in cancer cells, co-treatment of FC85 with ISA27 influences the promotion of differentiation and the inhibition of proliferation in GBM CSCs [169, 170].
All-trans retinoic acid (ATRA), derived from retinol, is known to induce the differentiation of neuro-related progenitor cells and stem cells [171]. Therefore, co-treatment of ATRA with rapamycin (ABI-009) reduces the size of tumor and decreases the motility of cancer cells, inducing differentiation of the CSCs in GBM [171].
In addition to GBM, neuroblastoma is another type of CNS cancer derived from neural crest tissue that frequently occurs in young people [172]. To investigate the anti-cancer efficiency of triciribine, an AKT inhibitor, co-treatment with rapamycin (ABI-009) was applied to both GBM cell line (U251) and the neuroblastoma cell line (SH-SY5Y) [173]. Inhibition of proliferation and reduction in migration were observed in both cell lines. Furthermore, rapamycin (ABI-009) was applied to CSCs derived from GBM and neuroblastoma, resulting in successful suppression of cell growth and downregulation of CD133, indicating the inhibition of the neural stem cell surface marker expression [174].
Colon CSCs
In human metastatic colon cancers, including colon CSCs, mTORC2 is strongly expressed, and thus, the mTORC2 pathway activates serum/glucocorticoid-regulated kinase 1 (SGK1), enhancing the cancer properties [175]. Therefore, one of the most popular mTORC2 inhibitors, torin-1, has been used to reduce the growth, survival, and invasion of colon CSC populations [175]. Accordingly, torin-1 decreased the stem cell markers related to pluripotency in colon CSCs in an in vivo model.
Another mTOR inhibitor, torkinib (PP242), was also treated with rapamycin (ABI-009), resulting in reduced tumor formation ability and aldehyde dehydrogenase 1 (ALDH1) activity, inducing autophagy [176]. Additionally, co-treatment of rapamycin (ABI-009) with dactolisib (NVP-BEZ235) caused the colon CSCs to lose their differentiation capability due to the dual inhibition of PI3K/mTOR [177]. It was confirmed that treatment of dactolisib (NVP-BEZ235) alone inhibited the proliferation of colon CSCs and reduced the expression of stem cell markers such as CD133 and leucine-rich repeat-containing G-protein coupled receptor 5 (LGR5) [178].
As an ATP-competitive PI3K/mTOR dual inhibitor, gedatolisib (PF-04691502) has been utilized to inhibit tumor growth and CSC proliferation in both in vitro and in vivo colon cancer models [179].
Liver CSCs
Since HCC causes mutations in hepatic progenitor cells, liver CSCs are generated and activated, resulting in chemoresistance, tumor relapse, and metastasis [180]. Sorafenib and regorafenib (BAY 73-4506), the representative multi-kinase inhibitors used in patients with advanced HCC, lose their effectiveness due to drug resistance with frequent administration [181]. Therefore, to overcome resistance issues in liver CSCs, rapamycin (ABI-009) was administered before the sorafenib. This resulted in the suppression of mTOR by rapamycin (ABI-009) and a reduction in the liver CSC population by sorafenib. Additionally, treatment with sorafenib followed by rapamycin (ABI-009) decreased the proportion of liver CSCs and reduced their ability to form tumor spheres [182].
Baicalein, a drug used to regulate inflammatory factors and treat cancer [183], was co-administered with temsirolimus (CCI-779) to an in vivo animal model. This combination prevented HCC growth and also suppressed the ability of autophagy [184].
Lung CSCs
Recently, studies have shown that CSCs are initiated during tumor development and affect tumor progression and metastasis in lung cancer [185, 186]. In lung CSCs, overexpression of CD164 and activation of AKT/mTOR signaling pathway have been observed [187, 188]. Therefore, rapamycin (ABI-009) has shown significant effects in pre-clinical investigations by suppressing sphere formation and tumor growth. This is achieved through the upregulation of C-X-C motif chemokine receptor 4 (CXCR4), which is involved in the growth and metastasis of lung cancer cells [187].
Cisplatin (CDDP), an anti-cancer drug most effective against tumor cells in the resting period, and β- elemene, which inhibits the PI3K/AKT/mTOR axis, were co-treated to lung cancer cell lines such as A549 and NCI-H1650 [189]. According to the results, the combination treatment reduced resistance to chemotherapy and stem-like behaviors. In clinical trials, rapamycin (ABI-009) was also investigated in a phase I clinical trial through co-treatment with sunitinib (SU11248), which targets receptors for NSCLC cell growth [190]. Additionally, temsirolimus (CCI-779), an mTORC1 inhibitor, was studied in a phase II clinical trial for patients with stage III NSCLC or stage IV NSCLC [191]. Furthermore, the dual mTOR inhibitor, sapanisertib (MLN0128), was tested in a phase II clinical trial for patients with lung cancer, which is at stage IV or recurrent due to mutation in multiple genes related to cancer growth [192].
Pancreatic CSCs
In the case of pancreatic cancer, CSCs frequently contribute to cancer relapse and induce resistance to typical therapeutics [193]. Co-administration of sonidegib (LDE225), a PI3K/mTOR inhibitor, with dactolisib (NVP-BEZ235), a smoothened inhibitor, in an in vivo mouse model, resulted in the downregulation of pluripotency markers such as OCT4, NANOG, SOX2, and c-MYC in pancreatic CSCs. This led to suppression of cancer growth and the formation of tumor spheres [193]. Additionally, the combination treatment of sonidegib (LDE225) with dactolisib (NVP-BEZ235) regulated genetic expression related to cellular proliferation and apoptosis [193].
Rapamycin (ABI-009) is known to inhibit formation of tumor spheres and anchorage-independent growth in pancreatic cancer cells [21]. Therefore, when rapamycin (ABI-009) was co-administered with glioma-associated inhibitor 61 (GANT-61), a Hedgehog signal inhibitor targeting Zinc finger protein GLI1, a reduction in cell viability and sphere growth was observed [194].
BMS-777607, a small-molecule met kinase inhibitor, also inhibits the tyrosine kinase of the MET receptor and is known to have therapeutic effects in tumor xenograft in vivo model [195]. Thus, BMS-777607 was co-treated with an mTOR inhibitor AZD8055, to pancreatic CSCs. The combination treatment not only reduced cell viability but also enhanced therapeutic synergy effects [196].
Other CSCs
Experiments involving various drugs have also been conducted on several other CSCs. For example, dactolisib (NVP-BEZ235), a dual inhibitor targeting both PI3K and mTOR, was co-treated with cisplatin (CDDP) to ovarian CSCs, leading to a reduction in stem cell colony formation and downregulation of stem cell markers [197]. Additionally, treatment with dactolisib (NVP-BEZ235) reduced stem cell properties in smooth muscle CSCs [198].
Another trial-targeting drug that inhibits three molecules (PI3K, AKT, and mTOR), S14161, has been found to significantly reduce the proportion of side population cells (SP cells) with stem cell-like characteristics [199].
To inhibit mTOR of mTORC1, rapamycin (ABI-009) was co-treated with temsirolimus (CCI-779) to salivary grand CSCs, resulting in promotion of apoptosis in the cells [200]. Rapamycin (ABI-009), being one of the most popular and effective mTOR inhibitors, was also applied to nasopharyngeal CSCs, which occur in the throat, nose, and back of the mouth [201].
Consequently, when drugs inhibiting mTOR signaling were used in diverse combinations on various types of CSCs, mostly the mTOR inhibitor showed a significant effect on reducing stem cell markers and cellular proliferation, leading to a decrease in tumor development.
Efficient targeting Of Cscs
CSCs have been considered as ‘cancer-initiating cells’, performing a pivotal role in tumor relapse, metastasis, and resistance to radiotherapy and chemotherapy [202, 203]. Tumors often regrow after treatments because the regrowth process is accelerated, and the self-renewal ability of CSCs remains unrestricted. Hence, there is a need for CSC-targeting therapy to prevent tumor recurrence [204,205,206,207].
Targeting CSC biomarkers
There have been some representative biomarkers which have been used to identify and efficiently target the CSCs [208, 209]: CD44, CD133, epithelial cell adhesion molecule (EpCAM), and ALDH. CD44, a receptor for hyaluronic acid (HA) in ECM, promotes anti-apoptosis and chemo-resistance in CSCs by activating signaling pathways such as STAT3 and c-Src kinase [210,211,212]. CD133, another well-characterized biomarker, is associated with tumorigenicity, spheroid formation, and EMT [213,214,215]. Since EpCAM regulates cell adhesion, proliferation, migration, and EMT, overexpression of EpCAM is directly linked to tumor formation and poor differentiation grade [216,217,218]. ALDHs, crucial in aldehyde metabolism, are upregulated in highly malignant tumors and contribute to CSC maintenance through interactions with retinoic acid, reactive oxygen species (ROS), and chemotherapeutic drugs [219,220,221,222]. SOX2/OCT4 and Wnt/β-catenin signaling pathways interact with ALDH to maintain CSC stemness and therapy resistance [223,224,225]. Although those identified CSC markers have been used to target the CSCs with high selectivity, not all of the CSC biomarkers are equally effective. Thus, profound research on efficacy comparison in vivo and multiple targeting is required for enhanced CSC-targeting.
Based on those studies, CSC biomarker-targeting agents have been investigated in some clinical trials [226]. Rituximab, targeting CD20, has shown efficacy in treating follicular lymphoma and mantle-cell lymphoma, although adverse reactions have been noted [227, 228]. Alemtuzumab, directed against CD52, has received approval for chronic lymphocytic leukemia (CLL) treatment, and combining CD20 and CD52 antibodies for refractory CLL has shown positive results [229]. For head and neck squamous cell carcinoma (SCC) treatment, bivatuzumab, targeting CD44v6, demonstrated safety [230, 231]. Further, various CD123 antibodies, such as XmAb14045, MGD006, and talacotuzumab, have been developed with improved effects, showing promise in clinical trials [232,233,234]. Additionally, research is also ongoing using EpCAM antibodies, like adecatumumab and catumaxomab, which have shown potential in treating hormone-resistant prostate cancer and ovarian cancer [235,236,237,238]. Also, combinational approaches involving EpCAM antibodies and CAR-T technology have been explored in phase II clinical trials for various cancers (NCT02729493, and NCT02725125).
Given the diversity in CSC surface markers of various types of cancers, developing effective treatment strategies requires a tailored approach. Each type of cancer may exhibit unique characteristics in terms of CSC markers, influencing how therapies are designed and conducted. Consequently, clinical trials must adopt combined approaches that account for these variations to maximize treatment efficacy. This approach ensures that treatments are specifically targeted to the CSCs present in each cancer type, thereby improving the chances of successful outcomes for patients undergoing experimental therapies.
Targeting CSC pathways
Another CSC-targeting technology is based on CSC-associated key signaling pathways [226]: Notch [239, 240], Hedgehog [241], Wnt [242], TGF-β [243, 244], JAK-STAT [245, 246], PI3K [20], and NFκB [247]. Abnormal function of those signaling pathways is crucial in CSCs [226]. And these pathways often interact with each other through crosstalk during tumor development. Although Notch signaling, vital for CSC maintenance and differentiation, is dysregulated in various cancers, trials with Notch inhibitors showed mixed results across different cancers [248, 249]. For instance, a representative γ-secretase inhibitor (GSI) MK-0752, despite initial promise in leukemia, yielded poor outcomes in solid tumors [250, 251]. In case of Hedgehog signaling, which contributes to CSC stemness and chemoresistance [252,253,254], approved inhibitors like vismodegib, sonidegib, and glasdegib demonstrated efficacy in basal cell carcinoma and acute myeloid leukemia (AML) [255,256,257]. Trials combining these kinds of inhibitors with chemotherapy are ongoing for treatment of various cancers [257]. By targeting these CSC-associated pathways, CSC-driven tumor growth and treatment resistance could be relieved.
Thus, the integration of CSC-targeting strategies with mTOR-targeted therapeutics as outlined above holds promise for developing a novel therapeutic paradigm aimed specifically at mTOR inhibition in CSCs. This approach is designed to enhance the eradication of CSCs, potentially decreasing drug resistance mechanisms and preventing tumor recurrence. By selectively targeting mTOR in CSCs, this therapeutic strategy offers an advanced approach to anti-cancer treatment, aiming to achieve efficacy while minimizing off-target effects on non-CSC populations. Therefore, such a precise approach in targeting CSCs through mTOR inhibition emphasizes its potential as a safer and more effective treatment method. And further investigations in clinical settings to validate its therapeutic utility in cancer management are required.
Recommendations for following research
Combinational approaches with mTOR inhibition
Unlike other stem cells, CSCs have a considerably longer cell cycle due to the activation of anti-apoptotic signals, such as B cell lymphoma 2 (BCL-2) [258], and the induction of myeloid leukemia cell differentiation protein 1 (MCL-1) [259]. CSCs also possess a skillful DNA repair system, which significantly contributes to therapy resistance [260,261,262]. Furthermore, there are intrinsic factors involved in the development of resistance, such as oxidative modulators, metabolic plasticity regulators, and drug efflux-regulating pumps [263]. Among a variety of signaling pathways associated with the CSCs, PI3K/AKT/mTOR axis is deeply involved in cascades such as anti-oxidative mechanisms [264] and anti-quiescence signaling [265], resulting in cancer resistance and survival in the end. Consequently, numerous clinical trials have been conducted, involving various types of cancers treated with drugs that inhibit the mTOR pathway (Table 2). These trials have addressed breast cancer, colon cancer, lung cancer, ovarian cancer, pancreatic cancer, prostate cancer, and urothelial cancer, utilizing various mTOR inhibitors such as rapamycin, everolimus, metformin, and sirolimus. Since these sole treatments utilizing mTOR pathway inhibitors only have been faced with limitations especially in efficacy, there have been combinational approaches for enhanced anti-cancer therapy.
First, DNA/RNA-targeting anti-cancer drugs have been co-treated with mTOR pathway inhibitors (Fig. 4A). Azacitidine priorly targets RNA and induces cytotoxicity in G1 phase cells [266], carboplatin targets replicating DNA inhibiting transcription [267], and capecitabine for oral administration disrupts DNA and RNA synthesis inducing cellular apoptosis especially in metastatic cancer [268, 269]. Second, drugs targeting mTOR partners which crosstalk with mTOR signaling pathway in diverse cancers have been used combined with mTOR inhibitors (Fig. 4B). Cetuximab, monoclonal antibody of epidermal growth factor receptor (EGFR), blocks activation of EGFR cascades through ligand binding inhibition, hindering cell proliferation [270]. Enzalutamide which targets androgen pathway [271], crenolanib, the representative inhibitor targeting platelet-derived growth factor receptor (PDGFR) signaling pathway [272], and numerous vascular endothelial growth factor receptor (VEGFR) inhibitors [273] have been co-treated with mTOR inhibitors.

For enhanced anti-cancer treatment, mTOR-targeting strategy can be combined with DNA/RNA-targeting anti-cancer therapies (A), mTOR partner inhibition (B), TME-targeting immunotherapy (C), anti-tumor immunity activation through M1 macrophage polarization or T cell activation (D), and advanced drug delivery system using virus, nanoparticles, or nanovesicles (E).
Despite the ongoing efforts in combinational therapies targeting DNA/RNA or mTOR partners, it is crucial to acknowledge that the clinical outcomes derived from these approaches are currently limited to phases 1 and 2 trials. These early stages primarily focus on assessing initial safety profiles, optimal dosing, and preliminary effectiveness. Therefore, it remains imperative to conduct subsequent phase 3 trials and long-term studies to establish the definitive efficacy and safety of these treatments. Only through rigorous evaluation across diverse patient populations, we can verify their potential impact and usefulness as standard therapies in the future.
Tumor immune modulation through mTOR inhibition
In addition to the intrinsic factors, one of the most well-known key factors contributing to resistance capabilities of the CSCs is tumor microenvironment (TME) [274,275,276]. The TME determines the fate of tumor by interacting with molecules in the surrounding cancer tissue [277, 278]. In TME conditions, there is an abundance of M2 macrophages, which mediate immunosuppression. Consequently, the anti-inflammatory environment induced by these M2 macrophages contributes to therapeutics resistance [263, 279,280,281]. Hence, to develop improved anti-cancer therapy, there have been recent approaches target the TME developed by tumor-associated immune systems. These approaches involve removing nutrients or signals essential to the TME, creating different environments that induce cancer quiescence [282, 283]. Additionally, the TME itself can be eliminated using enzymes that degrade the components of the ECM within the TME [284, 285]. In recent studies on immunotherapy, tumor-associated macrophages (TAM), specifically M2 macrophages with pro-tumor activity, have been induced to polarize into anti-tumor macrophages, known as inflammatory M1 macrophages. This immune modulation aims to alter the immune composition and properties of cancer circumstances [286, 287].
Since mTOR has been known to modulate not only the cancer tissue itself but also those tumor immune systems, the role and importance of the mTOR in innate and adaptive immune responses is recently spotlighted [288]. First, the mTOR is known to regulate antigen-presenting cells (APCs), such as dendritic cells (DCs) [289]. As a representative mTOR inhibitor, rapamycin induces differentiation of the APC, and inhibits antigen uptake by DCs, modulating their antigen presentation ability [290]. Second, mTOR is also known to activate effector T cells [291], and to induce proliferation of regulatory T cells (Tregs) [292, 293]. In detail, rapamycin-based mTOR inhibition affects the T cell activation process, disrupting some part of the cell cycle, and also separates the activated T cells, which are ready to fight [294]. Simultaneously, rapamycin induces the Tregs to proliferate, and increases the population of forkhead box P3-positive (FOXP3 + ) T cells, which will be changed into major immune modulators [295, 296].
Therefore, based on the understanding of the relationship between cancer and the immune system, it is anticipated that anti-cancer immunotherapies could maximize the efficacy of cancer treatments. By applying anti-cancer immunotherapies targeting the TME, it is possible to efficiently disrupt the interaction between TME and CSCs (Fig. 4C). Furthermore, by polarizing the abundant pro-tumor M2 macrophages in the TME into anti-tumor M1 macrophages, persistent and effective anti-cancer treatment efficacy is anticipated (Fig. 4D). In addition to these approaches, simultaneously inhibiting mTOR, which exerts immunosuppressive effects on both innate and adaptive immunity, is expected not only to effectively eliminate cancer but also to potentially provide long-term benefits by reducing resistance to cancer treatment and preventing cancer recurrence.
Advanced drug delivery systems
Regarding the effective delivery of anti-cancer drugs to CSCs, which often reside in the inner parts of tumor tissues, various drug delivery platforms are under development [297]. Since cancer cells typically originate from CSCs, primarily located at the center of the tumor mass, many drugs struggle to reach these CSCs, resulting in the failure of CSC eradication [298].
Recently, the development of drug delivery systems has involved the use of viruses, nanoparticles, and nanovesicles. Among viral delivery systems, adenovirus is a well-known carrier [299, 300]. However, adenovirus-based delivery faces challenges related to low efficacy and high toxicity that need to be addressed [282]. Viruses can also be employed to deliver microRNAs that target and block genes involved in CSC resistance and relapse [301]. Notably, microRNAs such as miR-15a and miR-16-1 are known for their tumor-suppressive properties [302, 303].
Nanoparticle therapeutics have demonstrated enhanced efficacy while simultaneously reducing side effects, making nanoparticle-based anti-cancer drug delivery systems promising for cancer treatment [304]. Although there are a number of nanoparticle types depending upon their components and ingredients, generally using the nanoparticles allows for targeted drug localization in tumors, resulting in increased cellular uptake of anti-cancer drugs [305, 306]. Consequently, not only pre-clinical studies but also clinical investigations have been conducted to advance cancer treatment using various types of nanoparticles [307, 308].
Nanovesicles include both extracellular vesicles (EVs) which are spontaneously generated by cells, and cell-derived nanovesicles (CDNVs) which are generally produced using serial extrusion methods [309]. Since the nanovesicles are not only easy to prepare at a large scale but also suitable for drug loading, they have emerged as promising carriers for anti-cancer therapy [310, 311]. Furthermore, leveraging the intrinsic characteristics of the source cell, nanovesicles can be applied to various types of cancer cells, with potential for expansion into immunotherapy, gene therapy, and cell therapy when derived from suitable source cell types [312].
The development of advanced drug delivery systems such as viruses, nanoparticles, and nanovesicles represents a promising frontier in overcoming the challenges of targeting CSCs within tumor tissues (Fig. 4E). These systems not only enhance drug delivery efficiency but also alleviate the toxicity associated with traditional treatments. Considering the unique properties of each delivery platform, researchers are advancing towards more effective and targeted anti-cancer treatments, potentially transforming the landscape of cancer treatment by improving outcomes and patient quality of life.
Conclusions
Cancer is a highly complex disease to treat, given its various causes and symptoms that depend on the type of cancer and its location of occurrence [313]. Chemotherapy, a common method of tumor treatment, faces limitations due to drug resistance, necessitating the development of improved cancer therapy approaches [314, 315]. A range of hypotheses related to cancer development and regeneration have emerged, with one prominent strategy focusing on eliminating CSCs by targeting crucial pathways associated with them [202]. CSCs, akin to normal stem cells, possess the ability to differentiate into tumor cells and exhibit self-renewal ability [148]. In CSCs, several representative signaling pathways are involved in maintaining pluripotency and resistance to therapeutics. Consequently, numerous studies have been conducted, encompassing in vitro, in vivo and clinical trials. Among these pathways, the mTOR pathway is particularly noteworthy for its significant role in activating not only stem cells but also immune cells within cancer tissues [28, 29]. In 1999, the representative mTOR inhibitor, rapamycin, received FDA approval as an immune suppressor that reduces the activation of T helper 17 cells (Th17 cells), which are implicated in inflammation and immune homeostasis [316]. Subsequently, with the discovery of the association between mTOR signaling and cancer, FDA-approved mTOR inhibitors have been employed in clinical trials for cancer treatment, while various other mTOR inhibiting molecules are currently under development [28]. Table 2 presents clinical trials for anti-cancer therapy using mTOR inhibitors categorized by cancer types.
As described in this manuscript and the tables, research involving the treatment of mTOR inhibitors alone or in combination with other anticancer drugs on various types of CSCs has demonstrated that inhibiting the mTOR pathway suppresses not only CSC differentiation into tumor cells but also intrinsic stem cell properties, such as self-renewal and uncontrolled proliferation [161,162,163, 178, 182, 197, 198]. Additionally, mTOR-inhibited CSCs have shown increased apoptosis, resulting in decreased metastasis [167, 169, 189, 200]. The CSC-targeting therapy via mTOR inhibition has shown enhanced anti-cancer efficacy, offering hope for addressing existing limitations in cancer treatment, such as drug resistance caused by the active CSCs (Fig. 5) [159, 184, 317,318,319]. However, it is important to note that the mTOR pathway is involved in various cellular activities, not only CSCs but also in normal cells. Therefore, strategies involving mTOR inhibition require careful consideration to avoid potential side effects [320, 321]. Nonetheless, the elimination of CSCs using mTOR inhibition remains a potent approach in anti-tumor therapy, and advanced tumor-targeting techniques with specificity are required for enhanced efficiency.

The mTOR pathway-mediated cellular mechanisms in CSCs are summarized (purple box in left), and the results of mTOR inhibition is also described (pink box in right). In CSCs, mTOR signal induce unlimited tumorigenesis, metastasis [19,20,21,22,23], tumor development, and radio/chemo-resistance [39, 40], resulting in relapse of cancer and failure in anti-cancer therapy. However, by inhibiting mTOR signaling pathway, CSC viability and ability decrease [89, 95, 97,98,99, 102, 109], radio/chemo-sensitivity increase, alleviating resistance [100], tumor suppression is induced, resulting in cancer cell apoptosis [87, 88, 90,91,92, 108, 111]. Therefore, elimination of the CSCs using mTOR inhibition is a powerful strategy with potentials in anti-cancer therapy.
Data availability
The published article includes all data sets generated/analyzed for this study.
References
De Los Angeles A, Ferrari F, Xi R, Fujiwara Y, Benvenisty N, Deng H, et al. Hallmarks of pluripotency. Nature. 2015;525:469–78.
Weissman IL. Stem cells: units of development, units of regeneration, and units in evolution. Cell. 2000;100:157–68.
Fuchs E, Segre JA. Stem cells: a new lease on life. Cell. 2000;100:143–55.
Jordan CT, Guzman ML, Noble M. Cancer stem cells. N. Engl J Med. 2006;355:1253–61.
Dutta D, Heo I, Clevers H. Disease modeling in stem cell-derived 3D organoid systems. Trends Mol Med. 2017;23:393–410.
Lancaster MA, Knoblich JA. Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc. 2014;9:2329–40.
Sato T, Clevers H. SnapShot: growing organoids from stem cells. Cell. 2015;161:1700.e1.
Wang X. Stem cells in tissues, organoids, and cancers. Cell Mol Life Sci. 2019;76:4043–70.
Yin X, Mead BE, Safaee H, Langer R, Karp JM, Levy O. Engineering stem cell organoids. Cell Stem Cell. 2016;18:25–38.
Chang JC. Cancer stem cells: Role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine. 2016;95:S20–S25.
Prasad S, Ramachandran S, Gupta N, Kaushik I, Srivastava SK. Cancer cells stemness: A doorstep to targeted therapy. Biochim Biophys Acta Mol Basis Dis Bba-mol Basis Dis. 2020;1866:165424
Kanwar SS, Yu Y, Nautiyal J, Patel BB, Majumdar AP. The Wnt/β-catenin pathway regulates growth and maintenance of colonospheres. Mol Cancer. 2010;9:1–13.
Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–7.
Herrera SC, Bach EA JAK/STAT signaling in stem cells and regeneration: from Drosophila to vertebrates. Development. 2019;146.
Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 2011;11:338–51.
Bajaj J, Diaz E, Reya T. Stem cells in cancer initiation and progression. J Cell Biol. 2019;219:e201911053.
Staal FJ, M. Sen J. The canonical Wnt signaling pathway plays an important role in lymphopoiesis and hematopoiesis. Eur J Immunol. 2008;38:1788–94.
Matsui WH Cancer stem cell signaling pathways. Medicine. 2016;95:S8–S19.
Dogan F, Avci CB. Correlation between telomerase and mTOR pathway in cancer stem cells. Gene. 2018;641:235–9.
Fath MK, Ebrahimi M, Nourbakhsh E, Hazara AZ, Mirzaei A, Shafieyari S, et al. PI3K/Akt/mTOR signaling pathway in cancer stem cells. Pathol Res Pr. 2022;237:154010.
Matsubara S, Ding Q, Miyazaki Y, Kuwahata T, Tsukasa K, Takao S. mTOR plays critical roles in pancreatic cancer stem cells through specific and stemness-related functions. Sci Rep. 2013;3:1–10.
Mohammed A, Janakiram NB, Brewer M, Ritchie RL, Marya A, Lightfoot S, et al. Antidiabetic drug metformin prevents progression of pancreatic cancer by targeting in part cancer stem cells and mTOR signaling. Transl Oncol. 2013;6:649–59.
Xia P, Xu X-Y. PI3K/Akt/mTOR signaling pathway in cancer stem cells: from basic research to clinical application. Am J Cancer Res. 2015;5:1602–9.
Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, et al. A mammalian protein targeted by G1-arresting rapamycin–receptor complex. Nature. 1994;369:756–8.
Mitra A, Luna JI, Marusina AI, Merleev A, Kundu-Raychaudhuri S, Fiorentino D, et al. Dual mTOR inhibition is required to prevent TGF-β-mediated fibrosis: implications for scleroderma. J Invest Dermatol. 2015;135:2873–6.
Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78:35–43.
Sabers C, Martin MM, Brunn GJ, Williams JM, Dumont FJ, Wiederrecht G, et al. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem. 1995;270:815–22.
Mossmann D, Park S, Hall MN. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat Rev Cancer. 2018;18:744–57.
Crino PB. The mTOR signalling cascade: paving new roads to cure neurological disease. Nat Rev Neurol. 2016;12:379–92.
Chan Y-S, Göke J, Ng J-H, Lu X, Gonzales KA, Tan C-P, et al. Induction of a human pluripotent state with distinct regulatory circuitry that resembles preimplantation epiblast. Cell Stem Cell. 2013;13:663–75.
Gangloff Y-G, Mueller M, Dann SG, Svoboda P, Sticker M, Spetz J-F, et al. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol Cell Biol. 2004;24:9508–16.
Murakami M, Ichisaka T, Maeda M, Oshiro N, Hara K, Edenhofer F, et al. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol Cell Biol. 2004;24:6710–8.
Alhasan BA, Gordeev SA, Knyazeva AR, Aleksandrova KV, Margulis BA, Guzhova IV, et al. The mTOR Pathway in Pluripotent Stem Cells: Lessons for Understanding Cancer Cell Dormancy. Membranes. 2021;11:858.
Zhou J, Su P, Wang L, Chen J, Zimmermann M, Genbacev O, et al. mTOR supports long-term self-renewal and suppresses mesoderm and endoderm activities of human embryonic stem cells. Proc Natl Acad Sci USA. 2009;106:7840–5.
Fan C, Zhao C, Zhang F, Kesarwani M, Tu Z, Cai X, et al. Adaptive responses to mTOR gene targeting in hematopoietic stem cells reveal a proliferative mechanism evasive to mTOR inhibition. Proc Natl Acad Sci USA. 2021;118:e2020102118.
Yu JS, Cui W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development. 2016;143:3050–60.
Xiang X, Zhao J, Xu G, Li Y, Zhang W. mTOR and the differentiation of mesenchymal stem cells. Acta Biochim Biophys Sin. 2011;43:501–10.
Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020;21:183–203.
Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–34.
Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35.
Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–76.
Nardella C, Carracedo A, Alimonti A, Hobbs RM, Clohessy JG, Chen Z, et al. Differential requirement of mTOR in postmitotic tissues and tumorigenesis. Sci Signal. 2009;2:ra2.
Xu K, Liu P, Wei W. mTOR signaling in tumorigenesis. Biochim Biophys Acta - Rev. 2014;1846:638–54.
Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61.
Murugan AK, editor mTOR: Role in cancer, metastasis and drug resistance. Semin Cancer Biol. 2019: Elsevier.
Dancey J. mTOR signaling and drug development in cancer. Nat Rev Clin Oncol. 2010;7:209–19.
Hasumi Y, Baba M, Ajima R, Hasumi H, Valera VA, Klein ME, et al. Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc Natl Acad Sci USA. 2009;106:18722–7.
Karar J, Maity A. PI3K/AKT/mTOR pathway in angiogenesis. Front Mol Neurosci. 2011;4:51.
Lee D-F, Kuo H-P, Chen C-T, Hsu J-M, Chou C-K, Wei Y, et al. IKKβ suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130:440–55.
Xie X, Hu H, Tong X, Li L, Liu X, Chen M, et al. The mTOR–S6K pathway links growth signalling to DNA damage response by targeting RNF168. Nat Cell Biol. 2018;20:320–31.
Zou Z, Tao T, Li H, Zhu X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020;10:1–11.
Fang J, Xia C, Cao Z, Zheng JZ, Reed E, Jiang B-H. Apigenin inhibits VEGF and HIF‐1 expression via PI3K/AKT/p70S6K1 and HDM2/p53 pathways. FASEB J. 2005;19:342–53.
Liu L-Z, Hu X-W, Xia C, He J, Zhou Q, Shi X, et al. Reactive oxygen species regulate epidermal growth factor-induced vascular endothelial growth factor and hypoxia-inducible factor-1α expression through activation of AKT and P70S6K1 in human ovarian cancer cells. Free Radic Biol Med. 2006;41:1521–33.
Meng Q, Xia C, Fang J, Rojanasakul Y, Jiang B-H. Role of PI3K and AKT specific isoforms in ovarian cancer cell migration, invasion and proliferation through the p70S6K1 pathway. Cell Signal. 2006;18:2262–71.
Zhou Q, Liu L-Z, Fu B, Hu X, Shi X, Fang J, et al. Reactive oxygen species regulate insulin-induced VEGF and HIF-1α expression through the activation of p70S6K1 in human prostate cancer cells. Carcinogenesis. 2007;28:28–37.
Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y. Targeting mTOR for cancer therapy. J Hematol Oncol. 2019;12:1–19.
Nathan N, Keppler-Noreuil KM, Biesecker LG, Moss J, Darling TN. Mosaic disorders of the PI3K/PTEN/AKT/TSC/mTORC1 signaling pathway. Dermatol Clin. 2017;35:51–60.
Armengol G, Rojo F, Castellví J, Iglesias C, Cuatrecasas M, Pons B, et al. 4E-binding protein 1: a key molecular “funnel factor” in human cancer with clinical implications. Cancer Res. 2007;67:7551–5.
Janus A, Robak T, Smolewski P. The mammalian target of the rapamycin (mTOR) kinase pathway: its role in tumourigenesis and targeted antitumour therapy. Cell Mol Biol Lett. 2005;10:479–98.
Bose S, Crane A, Hibshoosh H, Mansukhani M, Sandweis L, Parsons R. Reduced expression of PTEN correlates with breast cancer progression. Hum Pathol. 2002;33:405–9.
Depowski PL, Rosenthal SI, Ross JS. Loss of expression of the PTEN gene protein product is associated with poor outcome in breast cancer. Mod Pathol. 2001;14:672–6.
Braglia L, Zavatti M, Vinceti M, Martelli AM, Marmiroli S. Deregulated PTEN/PI3K/AKT/mTOR signaling in prostate cancer: Still a potential druggable target? Biochim Biophys Acta Mol Cell Res. 2020;1867:118731.
Lim HJ, Crowe P, Yang J-L. Current clinical regulation of PI3K/PTEN/Akt/mTOR signalling in treatment of human cancer. J Cancer Res Clin Oncol. 2015;141:671–89.
Avivar-Valderas A, McEwen R, Taheri-Ghahfarokhi A, Carnevalli LS, Hardaker EL, Maresca M, et al. Functional significance of co-occurring mutations in PIK3CA and MAP3K1 in breast cancer. Oncotarget. 2018;9:21444–58.
Di Malta C, Siciliano D, Calcagni A, Monfregola J, Punzi S, Pastore N, et al. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science. 2017;356:1188–92.
Kraya AA, Maxwell KN, Eiva MA, Wubbenhorst B, Pluta J, Feldman M, et al. PTEN loss and BRCA1 promoter Hypermethylation negatively predict for immunogenicity in BRCA-Deficient ovarian Cancer. JCO Precis Oncol. 2022;6:e2100159.
Lee G, Malietzis G, Askari A, Bernardo D, Al-Hassi H, Clark S. Is right-sided colon cancer different to left-sided colorectal cancer?–a systematic review. Eur J Surg Oncol. 2015;41:300–8.
Whitehead RH, Macrae FA, St. John DJB, Ma J. A colon cancer cell line (LIM1215) derived from a patient with inherited nonpolyposis colorectal cancer. J Natl Cancer Inst. 1985;74:759–65.
Chao M-W, Wang L-T, Lai C-Y, Yang X-M, Cheng Y-W, Lee K-H, et al. eIF4E binding protein 1 expression is associated with clinical survival outcomes in colorectal cancer. Oncotarget. 2015;6:24092–104.
Knight J, Alexandrou C, Skalka GL, Vlahov N, Pennel K, Officer L, et al. MNK Inhibition Sensitizes KRAS-Mutant Colorectal Cancer to mTORC1 Inhibition by Reducing eIF4E Phosphorylation and c-MYC ExpressionTargeting P-eIF4E and mTORC1 in KRAS-Mutant Colorectal Cancer. Cancer Discov. 2021;11:1228–47.
Xu T, Zong Y, Peng L, Kong S, Zhou M, Zou J, et al. Overexpression of eIF4E in colorectal cancer patients is associated with liver metastasis. OncoTargets Ther. 2016;9:815–22.
Zouki DN, Giannopoulou I, Alexandrou PT, Karatrasoglou EA, Pilichos G, Stamopoulos K, et al. MTOR/4EBP1 signaling and MMR status in colorectal cancer: new correlations and arising perspectives. Pathol Res Pr. 2021;228:153655.
Mamane Y, Petroulakis E, Martineau Y, Sato T-A, Larsson O, Rajasekhar VK, et al. Epigenetic activation of a subset of mRNAs by eIF4E explains its effects on cell proliferation. PloS one. 2007;2:e242.
Sun X, Kang H, Yao Y, Chen H, Sun L, An W, et al. Dehydrocostus lactone suppressed the proliferation, migration, and invasion of colorectal carcinoma through the downregulation of eIF4E expression. Anti-Cancer Drugs. 2015;26:641–8.
Miricescu D, Totan A, Stanescu-Spinu I-I, Badoiu SC, Stefani C, Greabu M. PI3K/AKT/mTOR signaling pathway in breast cancer: from molecular landscape to clinical aspects. Int J Mol Sci. 2020;22:173.
Dexter DL, Spremulli EN, Fligiel Z, Barbosa JA, Vogel R, VanVoorhees A, et al. Heterogeneity of cancer cells from a single human colon carcinoma. Am J Med. 1981;71:949–56.
Aldrich T, Hackley B. The impact of obesity on gynecologic cancer screening: an integrative literature review. J Midwifery Women’s Health. 2010;55:344–56.
Andersen BL, Hacker NF. Treatment for gynecologic cancer: A review of the effects on female sexuality. Health Psychol. 1983;2:203–11.
Oza AM, Pignata S, Poveda A, McCormack M, Clamp A, Schwartz B, et al. Randomized phase II trial of ridaforolimus in advanced endometrial carcinoma. J Clin Oncol. 2015;33:3576–82.
Ray-Coquard I, Favier L, Weber B, Roemer-Becuwe C, Bougnoux P, Fabbro M, et al. Everolimus as second-or third-line treatment of advanced endometrial cancer: ENDORAD, a phase II trial of GINECO. Br J Cancer. 2013;108:1771–7.
Slomovitz BM, Lu KH, Johnston T, Coleman RL, Munsell M, Broaddus RR, et al. A phase 2 study of the oral mammalian target of rapamycin inhibitor, everolimus, in patients with recurrent endometrial carcinoma. Cancer. 2010;116:5415–9.
Oh K-J, Kalinina A, Park N-H, Bagchi S. Deregulation of eIF4E: 4E-BP1 in differentiated human papillomavirus-containing cells leads to high levels of expression of the E7 oncoprotein. J Virol. 2006;80:7079–88.
Santiskulvong C, Konecny GE, Fekete M, Chen KY, Karam A, Mulholland D, et al. Dual Targeting of Phosphoinositide 3-Kinase and Mammalian Target of Rapamycin Using NVP-BEZ235 as a Novel Therapeutic Approach in Human Ovarian CarcinomaDual PI3K/mTOR Targeting in Ovarian Carcinoma. Clin Cancer Res. 2011;17:2373–84.
Baffy G, Brunt EM, Caldwell SH. Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. J Hepatol. 2012;56:1384–91.
Vogl TJ, Farshid P, Naguib NN, Zangos S. Thermal ablation therapies in patients with breast cancer liver metastases: a review. Eur Radio. 2013;23:797–804.
Bhat M, Sonenberg N, Gores GJ. The mTOR pathway in hepatic malignancies. Hepatology. 2013;58:810–8.
Chung J-Y, Hong S-M, Choi BY, Cho H, Yu E, Hewitt SM. The expression of phospho-AKT, phospho-mTOR, and PTEN in extrahepatic cholangiocarcinoma. Clin Cancer Res. 2009;15:660–7.
Cui X, Liu X, Han Q, Zhu J, Li J, Ren Z, et al. DPEP1 is a direct target of miR-193a-5p and promotes hepatoblastoma progression by PI3K/Akt/mTOR pathway. Cell Death Dis. 2019;10:701.
Liu P, Calvisi DF, Kiss A, Cigliano A, Schaff Z, Che L, et al. Central role of mTORC1 downstream of YAP/TAZ in hepatoblastoma development. Oncotarget. 2017;8:73433–47.
Chang Y, Nagasue N, Abe S, Kohno H, Kumar D, Nakamura T. alpha Fetoprotein producing early gastric cancer with liver metastasis: report of three cases. Gut. 1991;32:542–5.
Lu Y, Zhu M, Li W, Lin B, Dong X, Chen Y, et al. Alpha fetoprotein plays a critical role in promoting metastasis of hepatocellular carcinoma cells. J Cell Mol Med. 2016;20:549–58.
Lee J-C, Hung H-C, Wang Y-C, Cheng C-H, Wu T-H, Lee C-F, et al. Risk score model for microvascular invasion in hepatocellular carcinoma: the role of tumor burden and alpha-fetoprotein. Cancers. 2021;13:4403.
McHugh PP, Gilbert J, Vera S, Koch A, Ranjan D, Gedaly R. Alpha-fetoprotein and tumour size are associated with microvascular invasion in explanted livers of patients undergoing transplantation with hepatocellular carcinoma. Hpb. 2010;12:56–61.
Wang X-W, Xie H. Alpha-fetoprotein enhances the proliferation of human hepatoma cells in vitro. Life Sci. 1998;64:17–23.
Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67.
Westermarck J, Kähäri VM. Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J. 1999;13:781–92.
Chen Y, Zheng L, Liu J, Zhou Z, Cao X, Lv X, et al. Shikonin inhibits prostate cancer cells metastasis by reducing matrix metalloproteinase-2/-9 expression via AKT/mTOR and ROS/ERK1/2 pathways. Int Immunopharmacol. 2014;21:447–55.
Lemaître V, Dabo AJ, D’Armiento J. Cigarette smoke components induce matrix metalloproteinase-1 in aortic endothelial cells through inhibition of mTOR signaling. Toxicol Sci. 2011;123:542–9.
Guri Y, Colombi M, Dazert E, Hindupur SK, Roszik J, Moes S, et al. mTORC2 promotes tumorigenesis via lipid synthesis. Cancer cell. 2017;32:807–23.e12.
Javary J, Allain-Courtois N, Saucisse N, Costet P, Heraud C, Benhamed F, et al. Liver Reptin/RUVBL2 controls glucose and lipid metabolism with opposite actions on mTORC1 and mTORC2 signalling. Gut. 2018;67:2192–203.
Ricoult SJ, Yecies JL, Ben-Sahra I, Manning BD. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene. 2016;35:1250–60.
Iksen, Pothongsrisit S, Pongrakhananon V. Targeting the PI3K/AKT/mTOR signaling pathway in lung cancer: an update regarding potential drugs and natural products. Molecules. 2021;26:4100.
Schettino C, Bareschino MA, Sacco PC, Maione P, Rossi A, Casaluce F, et al. New molecular targets in the treatment of NSCLC. Curr Pharm Des. 2013;19:5333–43.
Tan AC. Targeting the PI3K/Akt/mTOR pathway in non‐small cell lung cancer (NSCLC). Thorac Cancer. 2020;11:511–8.
Azzariti A, Iacobazzi RM, Di Fonte R, Porcelli L, Gristina R, Favia P, et al. Plasma-activated medium triggers cell death and the presentation of immune activating danger signals in melanoma and pancreatic cancer cells. Sci Rep. 2019;9:4099.
Ilic M, Ilic I. Epidemiology of pancreatic cancer. World J Gastroenterol. 2016;22:9694–705.
Li J, Huang W, Han Q, Xiong J, Song Z. LDLRAD2 promotes pancreatic cancer progression through Akt/mTOR signaling pathway. Med Oncol. 2021;38:1–11.
Ye J, Huang A, Wang H, Zhang A, Huang X, Lan Q, et al. PRDM3 attenuates pancreatitis and pancreatic tumorigenesis by regulating inflammatory response. Cell Death Dis. 2020;11:187.
Zhu X-r, Peng S-q, Wang L, Chen X-y, Feng C-x, Liu Y-y, et al. Identification of phosphoenolpyruvate carboxykinase 1 as a potential therapeutic target for pancreatic cancer. Cell Death Dis. 2021;12:918.
Jiang B-H, Liu L-Z. Role of mTOR in anticancer drug resistance: perspectives for improved drug treatment. Drug Resist Updat. 2008;11:63–76.
Yardley DA, Noguchi S, Pritchard KI, Burris HA, Baselga J, Gnant M, et al. Everolimus plus exemestane in postmenopausal patients with HR+ breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther. 2013;30:870–84.
Debes JD, Tindall DJ. Mechanisms of androgen-refractory prostate cancer. N. Engl J Med. 2004;351:1488–90.
Sun Y, Ai JZ, Jin X, Liu LR, Lin TH, Xu H, et al. IL‐8 protects prostate cancer cells from GSK‐3β‐induced oxidative stress by activating the mTOR signaling pathway. Prostate. 2019;79:1180–90.
Diepgen TL, Mahler V. The epidemiology of skin cancer. Br J Dermatol. 2002;146:1–6.
Franceschi S, Levi F, Randimbison L, La Vecchia C. Site distribution of different types of skin cancer: new aetiological clues. Int J Cancer. 1996;67:24–8.
Dai DL, Martinka M, Li G. Prognostic significance of activated Akt expression in melanoma: a clinicopathologic study of 292 cases. J Clin Oncol. 2005;23:1473–82.
Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr, et al. Braf V600E cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009;41:544–52.
Szabo A, Fekete T, Koncz G, Kumar BV, Pazmandi K, Foldvari Z, et al. RIG-I inhibits the MAPK-dependent proliferation of BRAF mutant melanoma cells via MKP-1. Cell Signal. 2016;28:335–47.
Wellbrock C, Hurlstone A. BRAF as therapeutic target in melanoma. Biochem Pharm. 2010;80:561–7.
Zipser MC, Eichhoff OM, Widmer DS, Schlegel NC, Schoenewolf NL, Stuart D, et al. A proliferative melanoma cell phenotype is responsive to RAF/MEK inhibition independent of BRAF mutation status. Pigment Cell Melanoma Res. 2011;24:326–33.
Halaban R, Zhang W, Bacchiocchi A, Cheng E, Parisi F, Ariyan S, et al. PLX4032, a selective BRAFV600E kinase inhibitor, activates the ERK pathway and enhances cell migration and proliferation of BRAFWT melanoma cells. Pigment Cell Melanoma Res. 2010;23:190–200.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54.
So P-L, Wang GY, Wang K, Chuang M, Chiueh VC, Kenny PA, et al. PI3K-AKT signaling is a downstream effector of retinoid prevention of murine basal cell carcinogenesis. Cancer Prev Res. 2014;7:407–17.
Piérard-Franchimont C, Hermanns-Lê T, Paquet P, Herfs M, Delvenne P, Piérard GE. Hedgehog-and mTOR-targeted therapies for advanced basal cell carcinomas. Future Oncol. 2015;11:2997–3002.
Karayannopoulou G, Euvrard S, Kanitakis J. Differential expression of p-mTOR in cutaneous basal and squamous cell carcinomas likely explains their different response to mTOR inhibitors in organ-transplant recipients. Anticancer Res. 2013;33:3711–4.
Wu N, Du Z, Zhu Y, Song Y, Pang L, Chen Z. The expression and prognostic impact of the PI3K/AKT/mTOR signaling pathway in advanced esophageal squamous cell carcinoma. Technol Cancer Res Treat. 2018;17:1533033818758772.
Chen SJ, Nakahara T, Takahara M, Kido M, Dugu L, Uchi H, et al. Activation of the mammalian target of rapamycin signalling pathway in epidermal tumours and its correlation with cyclin‐dependent kinase 2. Br J Dermatol. 2009;160:442–5.
Lin Z, McDermott A, Shao L, Kannan A, Morgan M, Stack BC Jr, et al. Chronic mTOR activation promotes cell survival in Merkel cell carcinoma. Cancer Lett. 2014;344:272–81.
Kannan A, Lin Z, Shao Q, Zhao S, Fang B, Moreno MA, et al. Dual mTOR inhibitor MLN0128 suppresses Merkel cell carcinoma (MCC) xenograft tumor growth. Oncotarget. 2016;7:6576–92.
Villani A, Fabbrocini G, Costa C, Carmela Annunziata M, Scalvenzi M. Merkel cell carcinoma: therapeutic update and emerging therapies. Dermatol Ther. 2019;9:209–22.
Morrison AS. Advances in the etiology of urothelial cancer. Urol Clin North Am. 1984;11:557–66.
Puzio-Kuter AM, Castillo-Martin M, Kinkade CW, Wang X, Shen TH, Matos T, et al. Inactivation of p53 and Pten promotes invasive bladder cancer. Genes Dev. 2009;23:675–80.
Tao T, Su Q, Xu S, Deng J, Zhou S, Zhuang Y, et al. Down‐regulation of PKM2 decreases FASN expression in bladder cancer cells through AKT/mTOR/SREBP‐1c axis. J Cell Physiol. 2019;234:3088–104.
Wu Y, Zhang Y, Qin X, Geng H, Zuo D, Zhao Q. PI3K/AKT/mTOR pathway-related long non-coding RNAs: roles and mechanisms in hepatocellular carcinoma. Pharm Res. 2020;160:105195.
Foth M, Ahmad I, van Rhijn BW, van der Kwast T, Bergman AM, King L, et al. Fibroblast growth factor receptor 3 activation plays a causative role in urothelial cancer pathogenesis in cooperation with Pten loss in mice. J Pathol. 2014;233:148–58.
Tsuruta H, Kishimoto H, Sasaki T, Horie Y, Natsui M, Shibata Y, et al. Hyperplasia and carcinomas in Pten-deficient mice and reduced PTEN protein in human bladder cancer patients. Cancer Res. 2006;66:8389–96.
Tsui KH, Chiang KC, Lin YH, Chang KS, Feng TH, Juang HH. BTG2 is a tumor suppressor gene upregulated by p53 and PTEN in human bladder carcinoma cells. Cancer Med. 2018;7:184–95.
Ching CB, Hansel DE. Expanding therapeutic targets in bladder cancer: the PI3K/Akt/mTOR pathway. Lab Invest. 2010;90:1406–14.
Kurman Y, Kiliccioglu I, Dikmen AU, Esendagli G, Bilen CY, Sozen S, et al. Cucurbitacin B and cisplatin induce the cell death pathways in MB49 mouse bladder cancer model. Exp Biol Med. 2020;245:805–14.
Svatek RS, Ji N, De Leon E, Mukherjee NZ, Kabra A, Hurez V, et al. Rapamycin Prevents Surgery-Induced Immune Dysfunction in Patients with Bladder CancerRapamycin Prevents Surgery-Induced Immune Dysfunction. Cancer Immunol Res. 2019;7:466–75.
Xu M, Gu M, Zhou J, Da J, Wang Z. Interaction of YAP1 and mTOR promotes bladder cancer progression. Int J Oncol. 2020;56:232–42.
Hansel DE, Platt E, Orloff M, Harwalker J, Sethu S, Hicks JL, et al. Mammalian target of rapamycin (mTOR) regulates cellular proliferation and tumor growth in urothelial carcinoma. Am J Pathol. 2010;176:3062–72.
Gray JM, Pierce JrGB. Relationship between growth rate and differentiation of melanoma in vivo. J Natl Cancer Inst. J Natl Cancer Inst. 1964;32:1201–11.
Mitelman F. The chromosomes of fifty primary Rous rat sarcomas. Hereditas. 1971;69:155–86.
Dexter DL, Kowalski HM, Blazar BA, Fligiel Z, Vogel R, Heppner GH. Heterogeneity of tumor cells from a single mouse mammary tumor. Cancer Res. 1978;38:3174–81.
Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275–91.
Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–8.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–8.
Ailles LE, Weissman IL. Cancer stem cells in solid tumors. Curr Opin Biotechnol. 2007;18:460–6.
Masters JR, Köberle B. Curing metastatic cancer: lessons from testicular germ-cell tumours. Nat Rev Cancer. 2003;3:517–25.
Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11.
Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer stem cells—perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–44.
Maheswaran S, Sequist LV, Nagrath S, Ulkus L, Brannigan B, Collura CV, et al. Detection of mutations in EGFR in circulating lung-cancer cells. N. Engl J Med. 2008;359:366–77.
Barollo S, Pennelli G, Vianello F, Watutantrige Fernando S, Negro I, Merante Boschin I, et al. BRAF in primary and recurrent papillary thyroid cancers: the relationship with 131I and 2-[18F] fluoro-2-deoxy-D-glucose uptake ability. Eur J Endocrino. 2010;163:659–63.
John T, Kohler D, Pintilie M, Yanagawa N, Pham N-A, Li M, et al. The ability to form primary tumor xenografts is predictive of increased risk of disease recurrence in early-stage non–small cell lung cancer. Clin Cancer Res. 2011;17:134–41.
Neff G, Montalbano M, Tzakis A. editors. Ten years of sirolimus therapy in orthotopic liver transplant recipients. Transpl Proc. 2003;35:209S–216S.
Sofroniadou S, Goldsmith D. Mammalian target of rapamycin (mTOR) inhibitors: potential uses and a review of haematological adverse effects. Drug Saf. 2011;34:97–115.
Hart PC, Mao M, De Abreu AL, Ansenberger-Fricano K, Ekoue DN, Ganini D, et al. MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat Commun. 2015;6:6053.
Lai Y, Yu X, Lin X, He S. Inhibition of mTOR sensitizes breast cancer stem cells to radiation-induced repression of self-renewal through the regulation of MnSOD and Akt. Int J Mol Med. 2016;37:369–77.
Rattan R, Ali Fehmi R, Munkarah A. Metformin: an emerging new therapeutic option for targeting cancer stem cells and metastasis. J Oncol. 2012;2012:928127.
Jung J-W, Park S-B, Lee S-J, Seo M-S, Trosko JE, Kang K-S. Metformin represses self-renewal of the human breast carcinoma stem cells via inhibition of estrogen receptor-mediated OCT4 expression. PLoS One. 2011;6:e28068.
Song CW, Lee H, Dings RP, Williams B, Powers J, Santos TD, et al. Metformin kills and radiosensitizes cancer cells and preferentially kills cancer stem cells. Sci Rep. 2012;2:1–9.
Soo JS, Ng C-H, Tan SH, Malik RA, Teh Y-C, Tan B-S, et al. Metformin synergizes 5-fluorouracil, epirubicin, and cyclophosphamide (FEC) combination therapy through impairing intracellular ATP production and DNA repair in breast cancer stem cells. Apoptosis. 2015;20:1373–87.
Baselga J, Campone M, Piccart M, Burris HA, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor–positive advanced breast cancer. N. Engl J Med. 2012;366:520–9.
Rozenblit M, Mun S, Soulos P, Adelson K, Pusztai L, Mougalian S. Patterns of treatment with everolimus exemestane in hormone receptor-positive HER2-negative metastatic breast cancer in the era of targeted therapy. Breast Cancer Res. 2021;23:1–10.
Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–62.
Kolev VN, Wright QG, Vidal CM, Ring JE, Shapiro IM, Ricono J, et al. PI3K/mTOR dual inhibitor VS-5584 preferentially targets cancer stem cells. Cancer Res. 2015;75:446–55.
Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:1–12.
Daniele S, Costa B, Zappelli E, Da Pozzo E, Sestito S, Nesi G, et al. Combined inhibition of AKT/mTOR and MDM2 enhances Glioblastoma Multiforme cell apoptosis and differentiation of cancer stem cells. Sci Rep. 2015;5:1–14.
Sorriento D, Del Giudice C, Bertamino A, Ciccarelli M, Gomez-Monterrey I, Campiglia P, et al. New small molecules, ISA27 and SM13, inhibit tumour growth inducing mitochondrial effects of p53. Br J Cancer. 2015;112:77–85.
Friedman MD, Jeevan DS, Tobias M, Murali R, Jhanwar-Uniyal M. Targeting cancer stem cells in glioblastoma multiforme using mTOR inhibitors and the differentiating agent all-trans retinoic acid. Oncol Rep. 2013;30:1645–50.
Maris JM. Recent advances in neuroblastoma. N. Engl J Med. 2010;362:2202–11.
Bahmad HF, Mouhieddine TH, Chalhoub RM, Assi S, Araji T, Chamaa F, et al. The Akt/mTOR pathway in cancer stem/progenitor cells is a potential therapeutic target for glioblastoma and neuroblastoma. Oncotarget. 2018;9:33549–61.
Mendiburu-Eliçabe M, Gil-Ranedo J, Izquierdo M. Efficacy of rapamycin against glioblastoma cancer stem cells. Clin Transl Oncol. 2014;16:495–502.
Francipane MG, Lagasse E. Selective targeting of human colon cancer stem-like cells by the mTOR inhibitor Torin-1. Oncotarget. 2013;4:1948–62.
Cai Z, Ke J, He X, Yuan R, Chen Y, Wu X, et al. Significance of mTOR signaling and its inhibitor against cancer stem-like cells in colorectal cancer. Ann Surg Oncol. 2014;21:179–88.
Kim M-J, Koo J-E, Han G-Y, Kim B, Lee Y-S, Ahn C, et al. Dual-blocking of PI3K and mTOR improves chemotherapeutic effects on SW620 human colorectal cancer stem cells by inducing differentiation. J Korean Med Sci. 2016;31:360–70.
Chen J, Shao R, Li F, Monteiro M, Liu JP, Xu ZP, et al. PI 3K/Akt/mTOR pathway dual inhibitor BEZ 235 suppresses the stemness of colon cancer stem cells. Clin Exp Pharm Physiol. 2015;42:1317–26.
Fang DD, Zhang CC, Gu Y, Jani JP, Cao J, Tsaparikos K, et al. Antitumor efficacy of the dual PI3K/mTOR inhibitor PF-04691502 in a human xenograft tumor model derived from colorectal cancer stem cells harboring a PIK3CA mutation. PLoS One. 2013;8:e67258.
Yamashita T, Wang XW. Cancer stem cells in the development of liver cancer. J Clin Invest. 2013;123:1911–8.
Galle PR, Forner A, Llovet JM, Mazzaferro V, Piscaglia F, Raoul J-L, et al. EASL clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69:182–236.
Kahraman DC, Kahraman T, Cetin-Atalay R. Targeting PI3K/Akt/mTOR pathway identifies differential expression and functional role of IL8 in liver cancer stem cell enrichment. Mol Cancer Ther. 2019;18:2146–57.
Liang S, Deng X, Lei L, Zheng Y, Ai J, Chen L, et al. The comparative study of the therapeutic effects and mechanism of baicalin, baicalein, and their combination on ulcerative colitis rat. Front Pharm. 2019;10:1466.
Wu R, Murali R, Kabe Y, French SW, Chiang YM, Liu S, et al. Baicalein targets GTPase‐mediated autophagy to eliminate liver tumor–initiating stem cell–like cells resistant to mTORC1 inhibition. Hepatology. 2018;68:1726–40.
Perona R, López-Ayllón BD, de Castro Carpeño J, Belda-Iniesta C. A role for cancer stem cells in drug resistance and metastasis in non-small-cell lung cancer. Clin Transl Oncol. 2011;13:289–93.
Wang J, Li Z-h, White J, Zhang L-B. Lung cancer stem cells and implications for future therapeutics. Cell Biochem Biophys. 2014;69:389–98.
Chen W-L, Huang A-F, Huang S-M, Ho C-L, Chang Y-L, Chan JY-H. CD164 promotes lung tumor-initiating cells with stem cell activity and determines tumor growth and drug resistance via Akt/mTOR signaling. Oncotarget. 2016;8:54115–35.
Lin J, Xu K, Wei J, Heimberger AB, Roth JA, Ji L. MicroRNA-124 suppresses tumor cell proliferation and invasion by targeting CD164 signaling pathway in non-small cell lung cancer. J Gene Ther. 2016;2:6.
Cheng G, Li L, Li Q, Lian S, Chu H, Ding Y, et al. β-elemene suppresses tumor metabolism and stem cell-like properties of non-small cell lung cancer cells by regulating PI3K/AKT/mTOR signaling. Am J Cancer Res. 2022;12:1535–55.
Medicine WUSo. A Phase I Study of SUNITINIB and Rapamycin in Advanced Non-Small Cell Lung Cancer (NSCLC) (SU/Rapamycin). ClinicalTrialsgov. 2016. https://clinicaltrials.gov/study/NCT00555256 .
Puma Biotechnology I. Neratinib With and Without Temsirolimus for Patients With HER2 Activating Mutations in Non-Small Cell Lung Cancer. ClinicalTrialsgov. 2018 https://clinicaltrials.gov/study/NCT01827267.
(NCI) NCI. Sapanisertib in Treating Patients With Stage IV or Recurrent Lung Cancer. ClinicalTrialsgov. 2022. https://clinicaltrials.gov/study/NCT02417701.
Sharma N, Nanta R, Sharma J, Gunewardena S, Singh KP, Shankar S, et al. PI3K/AKT/mTOR and sonic hedgehog pathways cooperate together to inhibit human pancreatic cancer stem cell characteristics and tumor growth. Oncotarget. 2015;6:32039–60.
Miyazaki Y, Matsubara S, Ding Q, Tsukasa K, Yoshimitsu M, Kosai K, Takao S. Efficient elimination of pancreatic cancer stem cells by hedgehog/GLI inhibitor GANT61 in combination with mTOR inhibition. Mol Cancer. 2016;15:1–10.
Schroeder GM, An Y, Cai Z-W, Chen X-T, Clark C, Cornelius LA, et al. Discovery of N-(4-(2-amino-3-chloropyridin-4-yloxy)-3-fluorophenyl)-4-ethoxy-1-(4-fluorophenyl)-2-oxo-1, 2-dihydropyridine-3-carboxamide (BMS-777607), a selective and orally efficacious inhibitor of the Met kinase superfamily. J Med Chem. 2009;52:1251–4.
Zeng J-Y, Sharma S, Zhou Y-Q, Yao H-P, Hu X, Zhang R, et al. Synergistic Activities of MET/RON Inhibitor BMS-777607 and mTOR Inhibitor AZD8055 to Polyploid Cells Derived from Pancreatic Cancer and Cancer Stem CellsSynergism of BMS-777607 and AZD8055 on Polyploid PDAC Cells. Mol Cancer Ther. 2014;13:37–48.
Deng J, Bai X, Feng X, Ni J, Beretov J, Graham P, et al. Inhibition of PI3K/Akt/mTOR signaling pathway alleviates ovarian cancer chemoresistance through reversing epithelial-mesenchymal transition and decreasing cancer stem cell marker expression. BMC cancer. 2019;19:1–12.
Fourneaux B, Bourdon A, Dadone B, Lucchesi C, Daigle SR, Richard E, et al. Identifying and targeting cancer stem cells in leiomyosarcoma: prognostic impact and role to overcome secondary resistance to PI3K/mTOR inhibition. J Hematol Oncol. 2019;12:1–12.
Du J, Liu S, He J, Liu X, Qu Y, Yan W, et al. MicroRNA-451 regulates stemness of side population cells via PI3K/Akt/mTOR signaling pathway in multiple myeloma. Oncotarget. 2015;6:14993–5007.
Andrade NP, Warner KA, Zhang Z, Pearson AT, Mantesso A, Guimaraēs DM, et al. Survival of salivary gland cancer stem cells requires mTOR signaling. Cell Death Dis. 2021;12:108.
Yang C, Zhang Y, Zhang Y, Zhang Z, Peng J, Li Z, et al. Downregulation of cancer stem cell properties via mTOR signaling pathway inhibition by rapamycin in nasopharyngeal carcinoma. Int J Oncol. 2015;47:909–17.
Fan CW, Chen T, Shang YN, Gu YZ, Zhang SL, Lu R, et al. Cancer-initiating cells derived from human rectal adenocarcinoma tissues carry mesenchymal phenotypes and resist drug therapies. Cell Death Dis. 2013;4:e828.
Yan C, Luo L, Goto S, Urata Y, Guo C-Y, Doi H, et al. Enhanced autophagy in colorectal cancer stem cells does not contribute to radio-resistance. Oncotarget. 2016;7:45112–21.
Chen X, Lingala S, Khoobyari S, Nolta J, Zern MA, Wu J. Epithelial mesenchymal transition and hedgehog signaling activation are associated with chemoresistance and invasion of hepatoma subpopulations. J Hepatol. 2011;55:838–45.
Chou M-Y, Hu F-W, Yu C-H, Yu C-C. Sox2 expression involvement in the oncogenicity and radiochemoresistance of oral cancer stem cells. Oral Oncol. 2015;51:31–9.
Dhawan A, Madani Tonekaboni SA, Taube JH, Hu S, Sphyris N, Mani SA, et al. Mathematical modelling of phenotypic plasticity and conversion to a stem-cell state under hypoxia. Sci Rep. 2016;6:1–10.
Yeh D-W, Huang L-R, Chen Y-W, Huang C-YF, Chuang T-H. Interplay between inflammation and stemness in cancer cells: the role of toll-like receptor signaling. J Immunol Res. 2016;2016:4368101.
Mai Y, Su J, Yang C, Xia C, Fu L. The strategies to cure cancer patients by eradicating cancer stem-like cells. Mol Cancer. 2023;22:171.
Lee JW, Lee HY. Targeting cancer stem cell markers or pathways: a potential therapeutic strategy for oral cancer treatment. Int J Stem Cells. 2021;4:386.
Takaishi S, Okumura T, Tu S, Wang SS, Shibata W, Vigneshwaran R, et al. Identifcation of gastric cancer stem cells using the cell surface marker CD44. Stem Cells. 2009;27:1006–20.
Bourguignon LY, Spevak CC, Wong G, Xia W, Gilad E. Hyaluronan-CD44 interaction with protein kinase C(epsilon) promotes oncogenic signaling by the stem cell marker Nanog and the Production of microRNA-21, leading to down-regulation of the tumor suppressor protein PDCD4, anti-apoptosis, and chemotherapy resistance in breast tumor cells. J Biol Chem. 2009;284:26533–46.
Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol. 2003;4:33–45.
Barzegar Behrooz A, Syahir A, Ahmad S. CD133: beyond a cancer stem cell biomarker. J Drug Target. 2019;27:257–69.
O’Flaherty JD, Barr M, Fennell D, Richard D, Reynolds J, O’Leary J, et al. The cancer stem-cell hypothesis: its emerging role in lung cancer biology and its relevance for future therapy. J Thorac Oncol. 2012;7:1880–90.
Mak AB, Nixon AM, Kittanakom S, Stewart JM, Chen GI, Curak J, et al. Regulation of CD133 by HDAC6 promotes beta-catenin signaling to suppress cancer cell differentiation. Cell Rep. 2012;2:951–63.
Munz M, Baeuerle PA, Gires O. The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res. 2009;69:5627–9.
Sun YF, Xu Y, Yang XR, Guo W, Zhang X, Qiu SJ, et al. Circulating stem cell-like epithelial cell adhesion molecule-positive tumor cells indicate poor prognosis of hepatocellular carcinoma after curative resection. Hepatology. 2013;57:1458–68.
Gires O, Pan M, Schinke H, Canis M, Baeuerle PA. Expression and function of epithelial cell adhesion molecule EpCAM: where are we after 40 years? Cancer Metastasis Rev. 2020;39:969–87.
Xu X, Chai S, Wang P, Zhang C, Yang Y, Yang Y, et al. Aldehyde dehydrogenases and cancer stem cells. Cancer Lett. 2015;369:50–7.
Croker AK, Goodale D, Chu J, Postenka C, Hedley BD, Hess DA, et al. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J Cell Mol Med. 2009;13:2236–52.
Zhang H, Fu L. The role of ALDH2 in tumorigenesis and tumor progression: Targeting ALDH2 as a potential cancer treatment. Acta Pharmaceutica Sin B. 2021;11:1400–11.
Ying M, Wang S, Sang Y, Sun P, Lal B, Goodwin CR, et al. Regulation of glioblastoma stem cells by retinoic acid: role for Notch pathway inhibition. Oncogene. 2011;30:3454–67.
Harati M, Rodemann H, Toulany M. Nanog signaling mediates radioresistance in ALDH-positive breast cancer cells. Int J Mol Sci. 2019;20:1151.
Piva M, Domenici G, Iriondo O, Rabano M, Simoes BM, Comaills V, et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol Med. 2014;6:66–79.
Zhang Q, Han Z, Zhu Y, Chen J, Li W. The role and specifc mechanism of OCT4 in cancer stem cells: a review. Int J Stem Cells. 2020;13:312–25.
Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5:8.
Ghielmini M, et al. The effect of Rituximab on patients with follicular and mantle-cell lymphoma. Swiss Group for Clinical Cancer Research (SAKK). Ann Oncol. 2000;11:123–6.
Turner JH, Martindale AA, Boucek J, Claringbold PG, Leahy MF. 131Ianti CD20 radioimmunotherapy of relapsed or refractory non-Hodgkins lymphoma: a phase II clinical trial of a nonmyeloablative dose regimen of chimeric rituximab radiolabeled in a hospital. Cancer Biother Radiopharm. 2003;8:513–24.
Nabhan C, Patton D, Gordon LI, Riley MB, Kuzel T, Tallman MS, et al. A pilot trial of rituximab and alemtuzumab combination therapy in patients with relapsed and/or refractory chronic lymphocytic leukemia (CLL). Leuk Lymphoma. 2004;45:2269–73.
Colnot DR, Roos JC, de Bree R, Wilhelm AJ, Kummer JA, Hanft G, et al. Safety, biodistribution, pharmacokinetics, and immunogenicity of 99mTc-labeled humanized monoclonal antibody BIWA 4 (bivatuzumab) in patients with squamous cell carcinoma of the head and neck. Cancer Immunol Immunother. 2003;52:576–82.
Riechelmann H, Sauter A, Golze W, Hanft G, Schroen C, Hoermann K, et al. Phase I trial with the CD44v6-targeting immunoconjugate bivatuzumab mertansine in head and neck squamous cell carcinoma. Oral Oncol. 2008;44:823–9.
He SZ, Busfield S, Ritchie DS, Hertzberg MS, Durrant S, Lewis ID, et al. A phase 1 study of the safety, pharmacokinetics and anti-leukemic activity of the anti-CD123 monoclonal antibody CSL360 in relapsed, refractory or high-risk acute myeloid leukemia. Leuk Lymphoma. 2015;56:1406–15.
Frankel AE, Woo JH, Ahn C, Pemmaraju N, Medeiros BC, Carraway HE, et al. Activity of SL-401, a targeted therapy directed to interleukin-3 receptor, in blastic plasmacytoid dendritic cell neoplasm patients. Blood. 2014;124:385–92.
Pemmaraju N, et al. Results from phase 2 trial ongoing expansion stage of SL401 in patients with blastic plasmacytoid dendritic cell neoplasm (BPDCN). Blood. 2016;128:342.
Oberneder R, Weckermann D, Ebner B, Quadt C, Kirchinger P, Raum T, et al. A phase I study with adecatumumab, a human antibody directed against epithelial cell adhesion molecule, in hormone refractory prostate cancer patients. Eur J Cancer. 2006;42:2530–8.
Sebastian M, Passlick B, Friccius-Quecke H, Jäger M, Lindhofer H, Kanniess F, et al. Treatment of non-small cell lung cancer patients with the trifunctional monoclonal antibody catumaxomab (anti-EpCAM × anti-CD3): a phase I study. Cancer Immunol Immunother. 2007;56:1637–44.
Niedzwiecki D, Bertagnolli MM, Warren RS, Compton CC, Kemeny NE, Benson AB, et al. Documenting the natural history of patients with resected stage II adenocarcinoma of the colon after random assignment to adjuvant treatment with edrecolomab or observation: results from CALGB 9581. J Clin Oncol. 2011;29:3146–52.
Schmidt M, Scheulen ME, Dittrich C, Obrist P, Marschner N, Dirix L, et al. An open-label, randomized phase II study of adecatumumab, a fully human anti-EpCAM antibody, as monotherapy in patients with metastatic breast cancer. Ann Oncol. 2010;21:275–82.
Venkatesh V, Nataraj R, Thangaraj GS, Karthikeyan M, Gnanasekaran A, Kaginelli SB. et al. Targeting Notch signalling pathway of cancer stem cells. Stem Cell Investig. 2018;5:5.
Pannuti A, Foreman K, Rizzo P, Osipo C, Golde T, Osborne B, et al. Targeting Notch to target cancer stem cells. Clin Cancer Res. 2018;16:3141–52.
Merchant AA, Matsui W. Targeting Hedgehog—a cancer stem cell pathway. Clin Cancer Res. 2010;16:3130–40.
Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015;12:445–64.
Roberts A, Mishra L. Role of TGF-β in stem cells and cancer. Oncogene. 2005;24:5667.
Sakaki-Yumoto M, Katsuno Y, Derynck R. TGF-β family signaling in stem cells. Biochim Biophys Acta-Gen Subj. 2013;1830:2280–96.
Dolatabadi S, Jonasson E, Lindén M, Fereydouni B, Bäcksten K, Nilsson M, et al. JAK–STAT signaling controls cancer stem cell properties including chemotherapy resistance in myxoid liposarcoma. Int J Cancer. 2019;145:435–49.
Park SY, Lee CJ, Choi JH, Kim JH, Kim JW, Kim JY, et al. The JAK2/STAT3/CCND2 Axis promotes colorectal Cancer stem cell persistence and radioresistance. J Exp Clin Cancer Res. 2019;38:1–18.
Yamamoto M, Taguchi Y, Ito-Kureha T, Semba K, Yamaguchi N, Inoue JI. NF-κB non-cell-autonomously regulates cancer stem cell populations in the basal-like breast cancer subtype. Nat Commun. 2013;4:2299.
Quail DF, Taylor MJ, Postovit LM. Microenvironmental regulation of cancer stem cell phenotypes. Curr Stem Cell Res Ther. 2012;7:197–216.
Meurette O, Mehlen P. Notch signaling in the tumor microenvironment. Cancer Cell. 2018;34:536–48.
Fouladi M, Stewart CF, Olson J, Wagner LM, Onar-Thomas A, Kocak M, et al. Phase I trial of MK-0752 in children with refractory CNS malignancies: a pediatric brain tumor consortium study. J Clin Oncol. 2011;29:3529–34.
Hoffman LM, Fouladi M, Olson J, Daryani VM, Stewart CF, Wetmore C, et al. Phase I trial of weekly MK-0752 in children with refractory central nervous system malignancies: a pediatric brain tumor consortium study. Childs Nerv Syst. 2015;31:1283–9.
Mukherjee S, Frolova N, Sadlonova A, Novak Z, Steg A, Page GP, et al. Hedgehog signaling and response to cyclopamine differ in epithelial and stromal cells in benign breast and breast cancer. Cancer Biol Ther. 2006;5:674–83.
Yuan Z, Goetz JA, Singh S, Ogden SK, Petty WJ, Black CC, et al. Frequent requirement of hedgehog signaling in non-small cell lung carcinoma. Oncogene. 2007;26:1046–55.
Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851–6.
Sekulic A, Migden MR, Oro AE, Dirix L, Lewis KD, Hainsworth JD, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl J Med. 2012;366:2171–9.
Dummer R, Guminski A, Gutzmer R, Dirix L, Lewis KD, Combemale P, et al. The 12-month analysis from basal cell carcinoma outcomes with LDE225 treatment (BOLT): a phase II, randomized, double-blind study of sonidegib in patients with advanced basal cell carcinoma. J Am Acad Dermatol. 2016;75:113–25.
Norsworthy KJ, By K, Subramaniam S, Zhuang L, Del Valle PL, Przepiorka D, et al. FDA approval summary: glasdegib for newly diagnosed acute myeloid leukemia. Clin Cancer Res. 2019;25:6021–5.
Ma S, Lee T, Zheng B, Chan K, Guan X. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008;27:1749–58.
Lee K-m, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE, et al. MYC and MCL1 cooperatively promote chemotherapy-resistant breast cancer stem cells via regulation of mitochondrial oxidative phosphorylation. Cell Metab. 2017;26:633–47.e7.
Dingwall S, Lee JB, Guezguez B, Fiebig A, McNicol J, Boreham D, et al. Neoplastic human embryonic stem cells as a model of radiation resistance of human cancer stem cells. Oncotarget. 2015;6:22258–69.
Todaro M, Alea MP, Di Stefano AB, Cammareri P, Vermeulen L, Iovino F, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell. 2007;1:389–402.
Wang L, Huang X, Zheng X, Wang X, Li S, Zhang L, et al. Enrichment of prostate cancer stem-like cells from human prostate cancer cell lines by culture in serum-free medium and chemoradiotherapy. Int J Biol Sci. 2013;9:472–9.
Najafi M, Mortezaee K, Majidpoor J. Cancer stem cell (CSC) resistance drivers. Life Sci. 2019;234:116781.
Eckert F, Zwirner K, Boeke S, Thorwarth D, Zips D, Huber SM. Rationale for combining radiotherapy and immune checkpoint inhibition for patients with hypoxic tumors. Front immunol. 2019;10:407.
Marhold M, Tomasich E, El-Gazzar A, Heller G, Spittler A, Horvat R, et al. HIF1α Regulates mTOR Signaling and Viability of Prostate Cancer Stem CellsHIF1α in Prostate Cancer Stem Cells. Mol Cancer Res. 2015;13:556–64.
Khan C, Pathe N, Fazal S, Lister J, Rossetti JM. Azacitidine in the management of patients with myelodysplastic syndromes. Ther Adv Hematol. 2012;3:355–73.
Brabec V, Kasparkova J. Modifications of DNA by platinum complexes: relation to resistance of tumors to platinum antitumor drugs. Drug Resist Updat. 2005;8:131–46.
Ameli H, Alizadeh N. Targeted delivery of capecitabine to colon cancer cells using nano polymeric micelles based on beta cyclodextrin. RSC adv. 2022;12:4681–91.
Midgley R, Kerr DJ. Capecitabine: have we got the dose right? Nat Clin Pr Oncol. 2009;6:17–24.
Baselga J. The EGFR as a target for anticancer therapy–focus on cetuximab. Eur J Cancer. 2001;37:S16–22.
Schalken J, Fitzpatrick JM. Enzalutamide: targeting the androgen signaling pathway in metastatic castration-resistant prostate cancer. BJU Int. 2016;117:215–25.
Fujino S, Miyoshi N, Ito A, Yasui M, Ohue M, Ogino T, et al. Crenolanib regulates ERK and AKT/mTOR signaling pathways in RAS/BRAF-mutated colorectal cancer cells and organoids. Mol Cancer Res. 2021;19:812–22.
Sanfilippo R, Jones RL, Blay JY, Le Cesne A, Provenzano S, Antoniou G, et al. Role of chemotherapy, VEGFR inhibitors, and mTOR inhibitors in advanced perivascular epithelioid cell tumors (PEComas). Clin Cancer Res. 2019;25:5295–300.
Bocci F, Gearhart-Serna L, Boareto M, Ribeiro M, Ben-Jacob E, Devi GR, et al. Toward understanding cancer stem cell heterogeneity in the tumor microenvironment. Proc Natl Acad Sci USA. 2019;116:148–57.
Kise K, Kinugasa-Katayama Y, Takakura N. Tumor microenvironment for cancer stem cells. Adv Drug Deliv Rev. 2016;99:197–205.
Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest. 2011;121:3804–9.
Buenrostro D, Kwakwa KA, Putnam NE, Merkel AR, Johnson JR, Cassat JE, et al. Early TGF-β inhibition in mice reduces the incidence of breast cancer induced bone disease in a myeloid dependent manner. Bone. 2018;113:77–88.
Reina-Campos M, Shelton PM, Diaz-Meco MT, Moscat J. Metabolic reprogramming of the tumor microenvironment by p62 and its partners. Biochim Biophys Acta - Rev. 2018;1870:88–95.
Hao N-B, Lü M-H, Fan Y-H, Cao Y-L, Zhang Z-R, Yang S-M. Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol. 2012;2012:948098.
Dijkgraaf EM, Heusinkveld M, Tummers B, Vogelpoel LT, Goedemans R, Jha V, et al. Chemotherapy Alters Monocyte Differentiation to Favor Generation of Cancer-Supporting M2 Macrophages in the Tumor MicroenvironmentEffect of Chemotherapy on Tumor Microenvironment. Cancer Res. 2013;73:2480–92.
Boutilier AJ, Elsawa SF. Macrophage polarization states in the tumor microenvironment. Int J Mol Sci. 2021;22:6995.
Ahmad G, Amiji MM. Cancer stem cell-targeted therapeutics and delivery strategies. Expert Opin Drug Deliv. 2017;14:997–1008.
Arner EN, Rathmell JC. Metabolic programming and immune suppression in the tumor microenvironment. Cancer Cell. 2023;41:421–33.
Ikeda-Imafuku M, Gao Y, Shaha S, Wang LL, Park KS, Nakajima M, et al. Extracellular matrix degrading enzyme with stroma-targeting peptides enhance the penetration of liposomes into tumors. J Control Release. 2022;352:1093–103.
Stetler‐Stevenson WG, Liotta LA, Kleiner JrDE. Extracellular matrix 6: role of matrix metalloproteinases in tumor invasion and metastasis. FASEB J. 1993;7:1434–41.
Macciò A, Gramignano G, Cherchi MC, Tanca L, Melis L, Madeddu C. Role of M1-polarized tumor-associated macrophages in the prognosis of advanced ovarian cancer patients. Sci Rep. 2020;10:1–8.
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55.
Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol. 2009;9:324–37.
Hackstein H, Taner T, Zahorchak AF, Morelli AE, Logar AJ, Gessner A, et al. Rapamycin inhibits IL-4-induced dendritic cell maturation in vitro and dendritic cell mobilization and function in vivo. Blood. 2003;101:4457–63.
Woltman AM, de Fijter JW, Kamerling SW, van Der Kooij SW, Paul LC, Daha MR, et al. Rapamycin induces apoptosis in monocyte- and CD34-derived dendritic cells but not in monocytes and macrophages. Blood. 2001;98:174–80.
Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–87.
Zheng Y, Rudensky AY. Foxp3 in control of the regulatory T cell lineage. Nat Immunol. 2007;8:457–62.
Roncarolo MG, Battaglia M. Regulatory T-cell immunotherapy for tolerance to self antigens and alloantigens in humans. Nat Rev Immunol. 2007;7:585–98.
Luo H, Duguid W, Chen H, Maheu M, Wu J. The effect of rapamycin on T cell development in mice. Eur J Immunol. 1994;24:692–701.
Coenen JJ, Koenen HJ, van Rijssen E, Kasran A, Boon L, Hilbrands LB, et al. Rapamycin, not cyclosporine, permits thymic generation and peripheral preservation of CD4+CD25+ FoxP3+T cells. Bone Marrow Transpl. 2007;39:537–45.
Haxhinasto S, Mathis D, Benoist C. The AKT–mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med. 2008;205:565–74.
Shrestha S, Banstola A, Jeong JH, Seo JH, Yook S. Targeting Cancer Stem Cells: Therapeutic and diagnostic strategies by the virtue of nanoparticles. J Control Release. 2022;348:518–36.
Ibraheem D, Elaissari A, Fessi H. Gene therapy and DNA delivery systems. Int J Pharm. 2014;459:70–83.
Choi J-W, Lee J-S, Kim SW, Yun C-O. Evolution of oncolytic adenovirus for cancer treatment. Adv Drug Deliv Rev. 2012;64:720–9.
Lv P, Liu X, Chen X, Liu C, Zhang Y, Chu C, et al. Genetically engineered cell membrane nanovesicles for oncolytic adenovirus delivery: a versatile platform for cancer virotherapy. Nano Lett. 2019;19:2993–3001.
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33.
Aqeilan R, Calin G, Croce C. miR-15a and miR-16-1 in cancer: discovery, function and future perspectives. Cell Death Differ. 2010;17:215–20.
Esquela-Kerscher A, Slack FJ. Oncomirs—microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–69.
Davis ME, Chen Z, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–82.
Byrne JD, Betancourt T, Brannon-Peppas L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv Drug Deliv Rev. 2008;60:1615–26.
Martins JP, Santos HA Microfluidics as a Tool for the Synthesis of Advanced Drug Delivery Systems. Nano-and Microfabrication Techniques in Drug Delivery: Recent Developments and Future Prospects. Springer. 2023;321–64.
Brannon-Peppas L, Blanchette JO. Nanoparticle and targeted systems for cancer therapy. Adv Drug Deliv Rev. 2004;56:1649–59.
Wang AZ, Langer R, Farokhzad OC. Nanoparticle delivery of cancer drugs. Annu Rev Med. 2012;63:185–98.
Ingato D, Edson JA, Zakharian M, Kwon YJ. Cancer cell-derived, drug-loaded nanovesicles induced by sulfhydryl-blocking for effective and safe cancer therapy. ACS Nano. 2018;12:9568–77.
Hong J, Jung M, Kim C, Kang M, Go S, Sohn H, et al. Senescent cancer cell-derived nanovesicle as a personalized therapeutic cancer vaccine. Exp Mol Med. 2023;55:541–54.
Meng Z, Zhang Y, Zhou X, Ji J, Liu Z. Nanovaccines with cell-derived components for cancer immunotherapy. Adv Drug Deliv Rev. 2022;182:114107.
Hong J, Kang M, Jung M, Lee YY, Cho Y, Kim C, et al. T‐cell‐derived nanovesicles for cancer immunotherapy. Adv Mater. 2021;33:2101110.
Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature. 2015;520:368–72.
Coley HM. Mechanisms and strategies to overcome chemotherapy resistance in metastatic breast cancer. Cancer Treat Rev. 2008;34:378–90.
Hickman J. Apoptosis and chemotherapy resistance. Eur J Cancer. 1996;32:921–6.
Zhang J, Jin H, Xu Y, Shan J. editors. Rapamycin modulate Treg/Th17 balance via regulating metabolic pathways: a study in mice. Transpl Proc. 2019;51:2136–40.
Kahn J, Hayman TJ, Jamal M, Rath BH, Kramp T, Camphausen K, et al. The mTORC1/mTORC2 inhibitor AZD2014 enhances the radiosensitivity of glioblastoma stem-like cells. Neuro-Oncol. 2014;16:29–37.
Sunayama J, Matsuda K, Sato A, Tachibana K, Suzuki K, Narita Y, et al. Crosstalk between the PI3K/mTOR and MEK/ERK pathways involved in the maintenance of self-renewal and tumorigenicity of glioblastoma stem-like cells. Stem cells. 2010;28:1930–9.
Wang WJ, Long L-M, Long LM, Yang N, Zhang QQ, Ji WJ, Zhao JH, et al. NVP-BEZ235, a novel dual PI3K/mTOR inhibitor, enhances the radiosensitivity of human glioma stem cells in vitro. Acta Pharmacol Sin 2013;34:681–90.
Sparks C, Guertin D. Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy. Oncogene. 2010;29:3733–44.
Strimpakos AS, Karapanagiotou EM, Saif MW, Syrigos KN. The role of mTOR in the management of solid tumors: an overview. Cancer Treat Rev. 2009;35:148–59.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. RS-2023-00207746, 2021R1C1C1014606). This work was supported by Korea Institute of Planning and Evaluation for Technology in Food, Agriculture and Forestry (IPET) through High Value-added Food Technology Development Program, funded by Ministry of Agriculture, Food and Rural Affairs (MAFRA) (RS-2024-00402798). This work was partly supported by Institute of Information & Communications Technology Planning & Evaluation (IITP) grant funded by the Korea government (MSIT) (No.2020-0-01373, Artificial Intelligence Graduate School Program (Hanyang University)) and the research fund of Hanyang University (HY-2024).
Author information
Authors and Affiliations
Contributions
BS and WL collected the related papers. BS prepared the figures and tables. BS and HHP were the major contributors to the writing of the manuscript. BS and HK drafted the paper, BS and WL revised the manuscript critically for important intellectual content, BS and HK drew the figures. HS and HHP provided the final approval for the version to be published and agreed to be accountable for all aspects of the work in ensuing that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Gennaro Ciliberto
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Son, B., Lee, W., Kim, H. et al. Targeted therapy of cancer stem cells: inhibition of mTOR in pre-clinical and clinical research. Cell Death Dis 15, 696 (2024). https://doi.org/10.1038/s41419-024-07077-8
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41419-024-07077-8
This article is cited by
-
Harnessing nanomedicine to target NF-κB signalling in cancer: at the intersection of inflammatory signalling, metabolic reprogramming, and therapeutic innovation
Cell Communication and Signaling (2026)
-
Cisplatin-mediated activation of NF-κB promotes lung cancer stem cell formation via DNA repair pathways
Journal of Translational Medicine (2025)
-
Biology of stem cell paradox: a double-edged sword-implications for cancer therapy
Cancer Cell International (2025)
-
Posttranslational modifications of YAP/TAZ: molecular mechanisms and therapeutic opportunities
Cellular & Molecular Biology Letters (2025)
-
The role of the mTOR pathway in breast cancer stem cells (BCSCs): mechanisms and therapeutic potentials
Stem Cell Research & Therapy (2025)
