
Abstract
Mitophagy, a form of selective autophagy that removes damaged or dysfunctional mitochondria, plays a crucial role in maintaining mitochondrial and cellular homeostasis. Recent findings suggest that defective mitophagy is closely associated with various diseases, including breast cancer. Moreover, a better understanding of the multifaceted roles of mitophagy in breast cancer progression is crucial for the treatment of this disease. Here, we will summarize the molecular mechanisms of mitophagy process. In addition, we highlight the expression patterns and roles of mitophagy-related signaling molecules in breast cancer progression and the potential implications of mitophagy for the development of breast cancer, aiming to provide better therapeutic strategies for breast cancer treatment.
Similar content being viewed by others
Facts
-
Mitophagy removes damaged or dysfunctional mitochondria and plays a crucial role in maintaining mitochondrial homeostasis.
-
Mitophagy-related signaling molecules are abnormally expressed in breast cancer and are involved in cancer biology.
-
Dysregulation of mitophagy has been implicated in various diseases, including breast cancer progression.
-
The modulation of mitophagy has novel therapeutic potential for breast cancer treatment.
Open questions
-
Does mitophagy increase or decrease the risk of developing breast cancer?
-
In the context of different stages, different molecular types, and different tumor microenvironments of breast cancer, how does mitophagy govern the fate of breast cancer cells (promote or inhibit tumors)?
-
How can adverse effects be weighed and avoided when mitophagy is modulated as a therapeutic approach in breast cancer treatment?
Introduction
Breast cancer is the leading cause of cancer morbidity and mortality in women around the world [1]. Although modern treatments are multimodal and there have been recent advancements in therapeutics, drug resistance is common and remains a major clinical challenge in breast cancer treatment [2, 3]. Therefore, identifying effective therapeutic targets is still an important issue that needs to be solved in clinical practice. Recent studies have indicated that dysfunction of regulated cell death (RCD) is a hallmark feature of cancer and contributes to the mechanisms of tumor progression and drug resistance [4, 5]. Moreover, several new forms of RCD, such as autophagy, ferroptosis, pyroptosis, and cuproptosis, have been extensively studied and may represent promising avenues for enhancing treatment efficacy and overcoming drug resistance in breast cancer [6,7,8,9].
Mitochondrial autophagy, also known as mitophagy, is a form of selective autophagy that serves to maintain mitochondrial and cellular homeostasis. The term “mitophagy” was first proposed in 2005 [10], and subsequently, the classic process of mitophagy was characterized by significant discoveries, particularly in terms of understanding its molecular mechanisms and implications for health and disease [11]. Mitophagy is responsible for the selective degradation and removal of damaged or dysfunctional mitochondria through lysosomes [12]. This process is tightly regulated by a complex network of signaling pathways and molecules. However, uncontrolled mitophagy disrupts the basal mitochondrial turnover and leads to excessive degradation of mitochondria [13].
Recently, accumulating evidence has shown that mitophagy dysregulation has been implicated in various diseases [14, 15], including breast cancer. However, the process and roles of mitophagy in breast cancer development and progression appear to be complex, as mitophagy acts as both a tumor suppressor and a tumor promoter depending on the context, microenvironment, or stage of the cancer. Moreover, the potential of modulating mitophagy as a therapeutic strategy in breast cancer treatment remains to be further addressed. This review aims to present a current overview of the expression patterns and roles of mitophagy-related signaling molecules in breast cancer progression and the implications of mitophagy for the development and treatment of breast cancer.
Mechanisms of mitophagy in brief
Ubiquitin-dependent pathways
Currently, PINK1/Parkin-driven mitophagy is the most characterized pathway. In normal mitochondria, PINK1 is continuously imported into the inner membrane of the mitochondria and then cleaved by the mitochondrial proteases MPP and PARL [16, 17]. However, in the context of mitochondrial damage or depolarization, the import of PINK1 into the inner membrane is blocked, resulting in PINK1 accumulation on the outer mitochondrial membrane (OMM) [18]. This accumulation of PINK1 on the OMM leads to its stabilization and activation and subsequent phosphorylation of several substrates, including ubiquitin (Ub) and E3 ubiquitin ligases [19, 20]. Parkin, a cytosolic E3 ubiquitin ligase, can be phosphorylated at Ser65 by PINK1, which recruits more Parkin to mitochondria and activates its E3 ligase activity to generate more ubiquitin chains [21]. Subsequently, autophagy receptor proteins, including p62, TAX1BP1, NDP52, NBR1, and OPTN, contain both ubiquitin-binding domains (UBDs) that recognize phosphorylated ubiquitin chains, and LC3-interacting regions (LIRs) that facilitate interaction with LC3 family proteins, thereby initiating the autophagosome biogenesis machinery [13]. In addition to Parkin, several other ubiquitin E3 ligases, such as SMURF1, Gp78, MUL1, SIAH1, and ARIH1, also participate in the ubiquitination of mitochondrial proteins and induce mitophagy [14, 21].
Ubiquitin-independent pathways
In addition to ubiquitin-dependent mitophagy, mitochondrial surface proteins can also serve as receptors for mitophagy and facilitate ubiquitin-independent mitophagy. These proteins contain LIRs within their structure, enabling direct binding to LC3 and/or GABARAP (GABA-receptor-associated protein) without ubiquitination, thus initiating mitophagy. BNIP3 (BCL2 interacting protein 3) is an OMM protein, and phosphorylation of Ser17 and Ser24 within its LIR motif is essential for binding to LC3 [22, 23]. BNIP3L (NIP3-like protein X, or NIX) is highly homologous to BNIP3, and phosphorylation at Ser34 and Ser35 within its LIR motif significantly increases its binding affinity to LC3 [11]. Similar to BNIP3 and BNIP3L, the phosphorylation and dephosphorylation of FUNDC1 (FUN14 domain-containing protein 1) are critical for regulating its interaction with LC3 [11]. It was reported that phosphorylation of FUNDC1 at Ser17 by ULK1 enhances its interaction with LC3, whereas dephosphorylation of FUNDC1 at Ser13 by PGAM5 facilitates its binding to LC3, thereby promoting mitophagy [24]. Attractively, an increasing number of autophagy receptor proteins, including BCL2L13, FKBP8, AMBRA1, MCL-1, SAMM50, and others, have been identified and documented in the previous literature [11, 25].
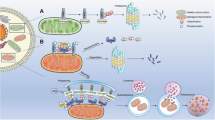
In addition to protein receptors, emerging evidence indicates that certain lipids can also interact with LC3 and mediate lipid-dependent mitophagy. For example, cardiolipin (CL), primarily localized to the inner mitochondrial membrane (IMM) in healthy mitochondria, can translocate to the OMM in response to mitochondrial stress or damage. This translocation of CL facilitates direct binding to LC3 and promotes CL-mediated mitophagy [26, 27]. Additionally, ceramide, a bioactive sphingolipid, has been shown to directly bind LC3 at the OMM. This ceramide-LC3 interaction at the OMM facilitates the recruitment of autophagosomes to mitochondria and subsequently initiates mitophagy [28]. The main pathways involved in mitophagy are summarized and presented in Fig. 1.

The PINK1/Parkin-driven mitophagy is the most extensively analyzed pathway. When PINK1 is stabilized and activated on the OMM, it phosphorylates ubiquitin at serine 65 and Parkin within its ubiquitin-like domain. Subsequently, autophagy receptor proteins recognize phosphorylated Ub chains and initiate autophagosome biogenesis machinery through binding with LC3. Moreover, mitochondrial surface proteins, such as BNIP3, BNIP3L, FUNDC1, and other autophagy receptor proteins, can interact directly with LC3, thus facilitating non-ubiquitin-dependent mitophagy. Additionally, certain lipids, including cardiolipin and ceramide, can also interact with LC3 and mediate lipid-dependent mitophagy. Created with BioRender.com.
Major mitophagy proteins in breast cancer
PINK1 in breast cancer
PTEN-induced kinase-1 (PINK1), a serine/threonine kinase, has been extensively studied in autosomal recessive familial Parkinson’s disease (PD) [29]. However, emerging evidence indicates that PINK1 has roles in other human diseases, such as cancers [30]. In breast cancer, Yaghoobi et al. compared the transcript levels of PINK1 in 54 breast cancer samples as well as their paired nearby tissues and reported that PINK1 mRNA expression was significantly downregulated in tumors compared with neighboring normal tissues [31]. Inconsistently, Li et al. conducted immunohistochemical (IHC) analysis on 150 cases of breast cancer tissues and 18 cases of benign specimens [32] and found a higher level of PINK1 protein expression in breast cancer tissues than in benign specimens [32]. This difference is likely because of the different nature of the tissues or the posttranscriptional regulation of PINK1. More specifically, Berthier et al. first described the subcellular distribution of the PINK1 protein in normal and tumor tissues [33]. Although PINK1 is positively expressed in both normal breast tissue and breast cancer tissue, PINK1 exhibits diffuse cytoplasmic staining along with strong membrane staining in breast cancer, whereas, in normal breast tissue, it shows a granular cytoplasmic pattern and minimal membrane staining [33]. This distinctive staining pattern indicated that the localization of PINK1 might be a new biomarker in breast cancer.
Even induced by the multifunctional tumor suppressor PTEN [34], the biological function of PINK1 in tumors remains controversial. In breast cancer, both the mitochondrial and nonmitochondrial localization of PINK1 have been shown to suppress MCF-7 cell (a luminal A cell line) growth in vitro [33]. In addition, PINK1 also confers resistance to apoptosis triggered by mitochondria-dependent apoptotic death stimuli (H2O2 treatment) [33]. This suggests that PINK1 may play multifaceted roles in tumor biology beyond its role in mitochondrial functions. Consistently, Miyahara et al. reported that PINK1 inhibition induced the expression of BRCA1, thus promoting MCF-7 cell growth [35]. Conversely, another study revealed that the absence of PINK1 inhibits MDA-MB-231 cell growth [32]. This observation supports the tumor-promoting properties of PINK1 in triple-negative breast cell (TNBC) lines. Likewise, in the HER-2 positive breast cancer subtype, inhibition of PINK1 expression has been found to enhance paclitaxel-induced apoptosis in BT-474 breast cancer cells [36]. These findings indicate that PINK1 may exert both tumor-promoting and tumor-restrictive effects on different subtypes of breast cancer cells, but the underlying mechanisms remain unclear. Moreover, cytosolic PINK1 is involved in the PI3K/Akt pathway via the activation of Akt phosphorylation [37, 38]. Since PI3K/Akt signaling is abnormally activated in breast cancer and its activation is associated with cancer progression [39, 40], targeting cytosolic PINK1 might serve as a promising therapeutic approach against breast cancer in the future. Overall, the above studies describe the multifaceted nonmitochondrial roles of PINK1 in breast cancer, and the relationship between cytosolic PINK1 and breast cancer biology is still unclear and requires further study.
Parkin in breast cancer
Parkin, an E3 ubiquitin ligase, is crucial for diverse cellular processes and is implicated in a variety of cancers. In breast cancer, the mRNA and protein levels of Parkin are frequently downregulated [35, 41,42,43]. Moreover, breast cancer patients with high Parkin expression tend to exhibit a lower histological grade, a lower proportion of triple-negative subtypes, decreased lymph node metastasis, and improved patient prognosis [42]. These results imply that Parkin acts as a tumor suppressor in breast cancer progression.
As reported, accumulating evidence indicates that Parkin exerts antitumor effects through various mechanisms in breast cancer. Early research revealed that Parkin has the potential to increase paclitaxel-microtubule interactions, thus enhancing the sensitivity of breast cancer cells to paclitaxel treatment [44]. Interestingly, Tay et al. revealed that Parkin enhances the mRNA and protein expression levels of cyclin-dependent kinase 6 (CDK6) and inhibits the proliferation and metastasis of MCF-7 cells both in vitro and in vivo [41]. However, the underlying mechanism by which Parkin regulates the transcript level of CDK6 remains obscure. Moreover, Liu et al. identified the classical role of Parkin as an E3 ubiquitin ligase and demonstrated its interaction with HIF-1α, facilitating HIF-1α degradation through ubiquitination [45]. This process effectively inhibits breast cancer metastasis [45]. Moreover, a similar relationship has been observed between Parkin and other proteins [35, 43, 46]. Miyahara et al. reported that Parkin directly interacts with BRCA1, leading to the ubiquitination of BRCA1 [35]. Given the significance of BRCA1 mutations in the development and progression of breast cancer [47, 48], these findings have potential prognostic and therapeutic implications for clinical breast cancer management. Phosphoglycerate dehydrogenase (PHGDH), the first rate-limiting enzyme of serine synthesis, is frequently upregulated in breast cancer and promotes tumor progression [49, 50]. However, Parkin was identified as a negative regulator of PHGDH by interacting with it and promoting its degradation through ubiquitination in breast cancer cells [43]. This finding emphasizes the role of Parkin in regulating cellular metabolism and underscores its potential significance in breast cancer pathogenesis.
BNIP3, BNIP3L, and FUNDC1 in breast cancer
BNIP3
BNIP3, also known as BCL2/Adenovirus E1B 19 KDa Protein-Interacting Protein 3, is a member of the Bcl-2 proapoptotic family and is upregulated under hypoxic conditions [51]. An early study conducted IHC analysis of BNIP3 in 81 cases of breast ductal carcinoma in situ (DCIS) and 251 cases of invasive cancer [52]. The results had shown that the localization of BNIP3 was primarily detected in the cytoplasm rather than in the nucleus, and the expression level of cytoplasmic BNIP3 in DCIS and invasive cancer tissues was significantly greater than that in normal breast tissues [52]. Consistent with these findings, recent studies have reported that BNIP3 is upregulated in breast cancer specimens compared with normal specimens [53,54,55]. However, controversial results have been reported regarding the relationship between BNIP3 expression and its potential clinical significance in breast cancer patients. The nuclear expression of BNIP3 in invasive carcinomas was significantly correlated with decreased tumor size, low tumor grade, and estrogen receptor positivity [52]. However, no correlation was found between clinicopathologic variables and cytoplasmic BNIP3 expression in invasive carcinoma [52]. Moreover, breast cancer patients who are positive for BNIP3 expression exhibit lower rates of axillary lymph node metastasis [56], while high expression of BNIP3 in node-negative breast cancer patients is associated with a worse prognosis [57].
Accordingly, the biological function of BNIP3 in breast cancer is controversial. In the absence of external estrogen administration, overexpression of BNIP3 in ER-positive cells (MCF-7 cells) enhanced their tumorigenic potential in a mouse xenograft model, and there was a positive correlation between tumor size and BNIP3 expression intensity [58]. Consistent with these findings, in vitro experiments revealed that knockdown of BNIP3 expression in MCF-7 cells inhibited the malignant phenotypes of breast cancer cells under hypoxic conditions [55]. These results suggest that BNIP3 acts as an oncogene in MCF-7 cells, but the potential mechanism has not been fully identified. However, there is controversy concerning the role of BNIP3 in breast cancer progression. In an MMTV‐PyMT mouse mammary tumor model, loss of BNIP3 promoted tumor growth and accelerated lung metastasis in vivo [59]. Interestingly, compared with wild-type tumors, BNIP3-null tumors in this mouse model exhibited a loss of ER-α expression, and the loss of BNIP3 combined with high HIF‐1α levels was associated with a poor prognosis in TNBC patients [59]. Moreover, another study revealed that BNIP3 inhibits breast tumor growth and progression by activating apoptotic pathways [60]. Overall, BNIP3 exhibits complex and context-dependent functions in breast cancer biology, and the specific functions of BNIP3 in breast cancer progression need further investigation.
BNIP3L
Like BNIP3, BNIP3L (also called NIX) is a member of the Bcl-2 family of proteins and contains a BH3 domain [51, 61]. A previous study assessed the RNA level of BNIP3L and revealed that the expression of BNIP3L in breast cancer cells was similar to that in normal breast epithelial cells [62]. However, in breast tissue samples, researchers have shown that the level of BNIP3L is greater in primary breast cancers than in normal tissues [63]. Interestingly, another study involving IHC analysis of human breast tissues revealed that BNIP3L is abundantly expressed in the stroma of breast cancers but is largely absent from tumor cells [64]. Moreover, the ITGB4 protein derived from breast cancer exosomes has been shown to upregulate BNIP3L expression in cancer-associated fibroblasts (CAFs), thereby inducing BNIP3L-dependent mitophagy in CAFs and providing energy metabolites for breast cancer cells [65]. These findings indicate that BNIP3L may play a significant role in modulating tumor-stromal interactions.
In terms of the biological function of BNIP3L, Pedanou et al. reported that BNIP3 and BNIP3L, which are transcriptionally regulated by KDM3A, can promote anoikis in breast cancer cells [66]. In addition, another study revealed that BNIP3L is also transcriptionally activated by FOXO3A and sensitizes breast cancer cells to chemotherapy-induced apoptosis [67]. These results suggest the potential significance of BNIP3L in preventing cancer metastasis and enhancing chemotherapy efficacy in breast cancer patients.
FUNDC1
FUN14 domain-containing 1 (FUNDC1) is a newly identified mitochondrial outer membrane protein and its dysregulation has been implicated in various human diseases, including cancers [68]. Yang et al. reported that inhibition of FUNDC1 by miR-101 suppressed the proliferation and metastasis of TNBC in vitro and in vivo [69], suggesting that FUNDC1 acts as a tumor-promoting factor in TNBC. Similarly, another study revealed that the expression level of FUNDC1 in breast cancer tissues was significantly greater than that in normal breast tissues, and higher expression of FUNDC1 was related to worse disease progression in breast cancer patients [70]. Furthermore, they also reported that elevated FUNDC1 promotes Ca2+ flux into the cytoplasm, leading to the dephosphorylation of NFATC1. Consequently, this activation of NFATC1 stimulates BMI1 expression at the transcriptional level, thereby promoting the progression of breast cancer [70]. This result highlights the importance of FUNDC1-mediated signaling pathways in breast cancer progression beyond its role in mitophagy.
Mitophagy in breast cancer progression
Mitophagy in tumor growth and metastasis
Accumulating evidence suggests that the accumulation of intracellular reactive oxygen species (ROS) is associated with tumor promotion and progression [71]. However, mitophagy can remove dysfunctional mitochondria and reduce the production of ROS in tumor cells, thus exerting a tumor suppressor effect. For example, Deng et al. proposed that ULK1 depletion in breast cancer cells results in mitophagy deficiency, leading to excessive ROS production. This aberrant ROS accumulation subsequently activates the NLRP3 inflammasome and promotes breast cancer bone metastasis [72]. Consistently, another study provided further evidence supporting the role of BRCA1 as a tumor suppressor by regulating mitophagy [73]. Mechanistically, mitophagy is impaired in BRCA1-deficient cells through the blockade of mitochondrial fission, resulting in the accumulation of damaged mitochondria and increased levels of ROS [73]. This accumulation further triggers NLPR3 inflammasome activation and promotes breast cancer metastasis [73]. Consistent with these findings, Chourasia et al. demonstrated that mitophagy defects resulting from BNIP3 loss can also lead to the accumulation of dysfunctional mitochondria and increased levels of ROS, which in turn enhances the growth and metastasis of TNBC cells [59]. Therefore, reducing ROS production by restoring proper mitophagy may become a therapeutic strategy for treating breast cancer. Interestingly, growing evidence indicates that certain compounds and drugs exert antitumor effects on breast cancer through the activation of mitophagy [74,75,76,77]. These findings further highlight the significance of mitophagy modulation in breast cancer suppression.
However, mitophagy plays dual roles in cancer development in response to various stress conditions [15, 78]. In certain circumstances such as metabolic stress or nutrient deprivation, mitophagy can also promote tumor survival to adapt to unfavorable conditions [79, 80], thereby acting as a tumor-promoting mechanism in cancer development. There is evidence that the upregulation of MRPL52 promotes protective mitophagy through the PINK1/Parkin pathway, which helps breast cancer cells survive under hypoxic conditions [81]. Indeed, cancer cells may enter a state of dormancy to protect themselves against inhospitable microenvironments [82]. Interestingly, Vera-Ramirez et al. revealed the activation of mitophagy in dormant BC cells [83]. These studies suggest that mitophagy can serve as a protective mechanism for breast cancer cells against adverse microenvironmental conditions. Moreover, another study discovered that Mucin1 (MUC1), localized to the mitochondrial outer membrane, can promote PINK1-dependent mitophagy by interacting with the mitochondrial protein ATAD3A [84]. Moreover, the authors showed that inhibition of either MUC1 or mitophagy alone impedes tumor cell proliferation [84]. Remarkably, when these two approaches are combined, they exhibit even more effective inhibitory activity against breast tumor growth both in vivo and in vitro [84]. Therefore, targeting mitophagy has therapeutic potential in breast cancer for certain conditions.
Mitophagy in metabolic reprogramming
In metabolically hostile environments, tumor cells can undergo metabolic reprogramming to ensure their survival despite limited oxygen and nutrient availability [85]. Given the central roles of mitochondria in cellular energy and redox homeostasis, a number of studies have investigated how mitophagy modulates metabolic reprogramming during breast cancer progression [86, 87].
Indeed, cancer cells exhibit a unique metabolic phenomenon known as the Warburg effect, where they preferentially utilize glycolysis for energy production despite the availability of oxygen [88]. Dysfunctional mitochondria and impaired mitophagy may compromise oxidative phosphorylation (OXPHOS) activity, leading to an increased reliance on glycolysis for energy production during cancer development. An associated study revealed that the activation of insulin-like growth factor 1 (IGF-1)/phosphoinositide 3-kinase (PI3K) signaling promotes BNIP3-mediated mitophagy in breast cancer [89]. This mechanism enhances the degradation of dysfunctional mitochondria by upregulating mitophagy, consequently preserving mitochondrial homeostasis to support OXPHOS activity [89]. Moreover, it has been shown that breast cancer cells with acquired resistance to an IGF-1 receptor tyrosine kinase inhibitor exhibit decreased mitophagy and increased dependence on glycolysis rather than OXPHOS for ATP production [89]. In agreement with this, another study revealed that deficiency in BNIP3‐dependent mitophagy leads to the accumulation of dysfunctional mitochondria, which exhibit increased ROS production [59]. These elevated levels of ROS subsequently promote HIF-1α activity, which further induces aerobic glycolysis and reduces OXPHOS activity in breast cancer cells [59]. Interestingly, the loss of BNIP3 is associated with malignant progression in TNBC, and treating BNIP3-null cells with 2‐deoxyglucose (2DG) to inhibit glycolysis has been demonstrated to effectively suppress breast cancer growth in vitro [59]. These findings suggest that targeting glycolysis could represent a highly promising anticancer therapeutic approach in breast cancer, especially when the OXPHOS pathway is already impaired by dysfunctional mitochondria.
Notably, excessive mitophagy can also lead to detrimental effects. Compelling evidence indicates that HIF-1α can promote the transcriptional activity of EBF1 [90]. Conversely, EBF1 interacts with HIF-1α and attenuates its activity by interfering with the activity of p300, which forms a negative feedback loop and maintains mitochondrial homeostasis in TNBC [90]. Intriguingly, the absence of EBF1 causes persistent activation of HIF-1α, resulting in pronounced mitophagy and a metabolic shift from glucose metabolism toward glycolysis in TNBC [90]. Consistent with these findings, Zou et al. revealed that Mdivi-1 could suppress mitophagy in MDA-MB-231 cells [91]. This inhibition leads to an increase in cellular OXPHOS capacity and a reduction in glycolytic activity, ultimately resulting in the suppression of growth in TNBC cells [91]. Moreover, previous studies have shown that Warburg-type metabolism is the most common metabolic phenotype in TNBC [92], suggesting that targeting abnormal metabolic pathways via mitophagy may represent a novel approach for inhibiting TNBC cell progression.
Mitophagy in the tumor microenvironment (TME)
The TME involves a complex network of components, including primary tumor cells, various stromal and immune cells, the extracellular matrix, and cytokines [93]. Considerable progress has already been made in understanding the role of mitophagy in dynamically regulating the TME and its impact on tumor progression and treatment strategies.
Recent investigations have revealed a novel metabolic phenomenon called the reverse Warburg effect, in which CAFs undergo aerobic glycolysis to provide metabolic support for cancer cells [94, 95]. In the breast cancer stromal microenvironment, evidence suggests that cancer cells induce oxidative stress in adjacent CAFs [96, 97]. This oxidative stress then initiates mitophagy in CAFs, leading to the loss of functional mitochondria and a shift toward aerobic glycolysis [96, 97]. Consistent with these findings, a study revealed that ethanol exposure significantly downregulated Cav-1 expression while inducing mitophagy in CAFs [98]. This dysregulation in stromal fibroblasts further promotes the generation of ketone bodies, which serve as energy-rich substrates for breast cancer cells [98]. Notably, several additional studies reported that loss of Cav-1 in breast stromal fibroblasts is a functional biomarker of mitophagy in stromal cells under conditions of oxidative stress [97] and is associated with tumor-stromal metabolic coupling in the TME [99, 100]. Importantly, the absence of Cav-1 in stromal fibroblasts could be used to predict poor clinical outcomes in breast cancer patients [100,101,102]. In addition, Sung et al. elucidated an alternative mechanism by which breast cancer cells transport the ITGB4 protein via the exosome pathway. This process further induces BNIP3L-dependent mitophagy and glycolysis in CAFs [65]. Taken together, these studies highlighted the role of mitophagy in CAFs, which provides high-energy metabolites to tumors through aerobic glycolysis and promotes breast cancer progression [65, 96, 97], and that targeting mitophagy in CAFs could represent a promising therapeutic strategy for inhibiting breast cancer progression by disrupting metabolic interactions between CAFs and cancer cells.
Moreover, mitophagy in immune cells such as macrophages may affect their functions. For example, Zheng et al. demonstrated that urolithin A (UA) treatment activates PINK1/Parkin-driven mitophagy in tumor-associated macrophages, resulting in a decrease in stimulator of interferon genes (STING)-mediated inflammation and subsequently a reduction in the release of inflammatory factors such as IL-6 in the TME [103]. Mechanistically, UA inhibits TFEB ubiquitination-mediated degradation and enhances TFEB nuclear translocation in tumor-associated macrophages, thereby promoting the transcriptional activation of mitophagy-related genes regulated by TFEB [103]. This finding highlights the potential therapeutic significance of targeting mitophagy in immune cells as a strategy to modulate inflammation and reshape the TME, thus exerting antitumor effects on breast cancer.
Specifically, defective mitophagy within cells leads to the accumulation of ROS, which then activates inflammasome and contributes to TME remodeling [104]. Notably, several publications have described the significant roles of ROS in the breast tumor microenvironment [105, 106]; however, whether these ROS are directly derived from dysregulated mitophagy remains an open question. In the context of mitophagy, the evidence indicates that the absence of BRCA1 is associated with impaired mitophagy, resulting in the accumulation of damaged mitochondria and increased ROS levels [73]. Subsequently, this excessive ROS accumulation triggers the activation of the NLRP3 inflammasome and the secretion of mature cytokines such as interleukin-1β (IL-1β) within the TME, which facilitates breast cancer recurrence and metastasis [73]. In addition, Sun et al. applied a CCCP-loaded nanoplatform to induce excessive mitophagy in 4T1 cells [107]. Interestingly, during mitophagy, the authors observed the release of several endogenous DAMPs (such as ATP and HMGB1) and the expression of calreticulin (CRT) on the surface of 4T1 cells, which led to increased tumor-infiltrating lymphocytes (TILs) and elicits robust immune activation within the tumor microenvironment (TME) [107].
Overall, mitophagy plays a multifaceted role across different cellular components within the TME. This includes mediating metabolic reprogramming in CAFs, modulating immune cell function, regulating oxidative stress, and facilitating the release of DAMPs from tumor cells. Notably, exploring the therapeutic implications of targeting mitophagy within the context of TME reprogramming may open up innovative avenues for breast cancer treatment.
Mitophagy in stem-like cells
Breast cancer stem cells (BCSCs) are a small subpopulation of cancer cells within breast tumors that exhibit stem cell-like properties, such as tumor onset, self-renewal, multidirectional differentiation, and treatment resistance [108]. Although previous research has explored the potential significance of mitophagy in several stem cell developmental processes [15, 109,110,111], the role and mechanism of mitophagy in maintaining stemness and regulating differentiation in BSCSs remain to be fully elucidated. Limited data indicate that miR-137 restored cell mitochondrial dysfunction by inhibiting FUNDC1-mediated mitophagy in BCSCs [112]. This intervention significantly attenuated the hypoxia-induced abnormal mitochondrial homeostasis, leading to inhibition of apoptosis and ultimately exerting a protective effect on the survival of BCSCs [112]. In accordance with this, another study demonstrated that overexpression of miR-1 increased the protein levels of LC3-II and PINK1 in BCSCs, thereby inducing mitophagy [113]. This mechanism is associated with the negative regulation of tumorigenesis in cancer stem cells [113]. However, since cancer stem cells are often characterized by their heightened reliance on mitochondrial function for energy production and self-renewal [114], disrupting the integrity of mitochondria through mitophagy induction may be a promising approach to impairing the survival and proliferation of BCSCs.
Mitophagy in apoptosis, ferroptosis, and pyroptosis
RCD, also known as programmed cell death, is the autonomous and orderly death of cells controlled by specific genes [4]. In the context of cancer, several forms of RCD, including apoptosis, ferroptosis, pyroptosis, and cuproptosis have been identified and shown to play crucial roles in cancer development [115]. Interestingly, recent studies indicate a potential interaction between mitophagy and other forms of RCD, which might contribute to stress responses and disease progression in breast cancer. Nonetheless, for simplicity, we will only focus on interactions between three forms of RCD (apoptosis, ferroptosis, and pyroptosis) and mitophagy (Fig. 2).

Mitochondria are central regulators of both mitophagy and other forms of RCD. A By degrading damaged mitochondria, mitophagy prevents the release of ROS and keeps the cell from undergoing intrinsic apoptosis. Moreover, RL2-induced mitophagy blocks TRAIL-mediated extrinsic apoptosis by suppressing core proapoptotic regulators in breast cancer cells. B Treatment with salinomycin blocks the late stages of mitophagy, which leads to an accumulation of damaged mitochondria and makes breast cancer cells more prone to salinomycin-induced ferroptosis. In addition, certain ferroptosis-inducing agents, such as BAY, can induce ferroptosis through the Nrf2-SLC7A11-HO-1 pathway. Meanwhile, upregulation of HO-1 by BAY treatment can also lead to mitochondrial damage and activate mitophagy in breast cancer cells. C Accumulation of mitochondrial ROS (mtROS) can lead to oxidative stress, thereby activating the caspase 3-GSDME pyroptosis pathway. Therefore, mitophagy acts as a protective mechanism that prevents pyroptosis by clearing damaged mitochondria. Created with BioRender.com.
A specific link between mitophagy activity and apoptosis has been suggested by several studies [116,117,118,119]. As mentioned above, suppressing mitophagy increases the level of ROS within the cell, and increased ROS can serve as an indicator of intrinsic apoptotic processes [120, 121]. For example, Liu et al. revealed that FBP1 restrained mitophagy by suppressing the HIF-1α/BNIP3 pathway in breast cancer cells [122]. Accordingly, this inhibition of mitophagy results in cytotoxic ROS accumulation and eventually causes cancer cell apoptosis [122]. Consistently, another study on hypoxic breast cancer cells showed that Sirt3 can activate mitophagy by increasing the interaction of VDAC1 with Parkin. However, Sirt3 inhibition promotes the ROS-dependent proteasomal degradation of the Mcl-1 and survivin proteins, thereby triggering apoptosis in breast cancer cells [123]. Moreover, certain drugs (e.g., silibinin, polyphyllin I, and warangalone) have shown efficacy in inducing apoptosis in breast cancer cells to exert antitumor effects, but these effects are prevented by mitophagy [124,125,126]. Taken together, these data indicate that cancer cells might exploit mitophagy as a mechanism to evade apoptosis [119], and targeting mitophagy therefore represents a promising anticancer strategy against apoptosis-resistant breast cancer cells.
In addition to being related to the intrinsic (mitochondrial) apoptosis pathway, mitophagy also interacts with the extrinsic (cell death receptor) apoptosis pathway. The extrinsic pathway provides a rapid response to external signals, specifically tumor necrosis factor-alpha (TNF-α), Fas ligand (FasL), and TNF-related apoptosis-inducing ligand (TRAIL) [127]. Accordingly, RL2 (recombinant lactaptin 2) serves as a pro-mitophagy factor in breast cancer cells by interacting with TOM70 in mitochondria [128, 129]. However, the effects of RL2/TRAIL cotreatment exhibit opposite outcomes: under short-term stimulation, RL2-induced mitophagy can alleviate TRAIL-mediated extrinsic apoptosis, whereas under long-term costimulation, RL2 enhances TRAIL-induced cell death [128]. In addition, in combination with doxorubicin, RL2 can also promote doxorubicin-induced caspase-3/7 activity in breast cancer cells, thereby increasing the level of apoptosis and amplifying the antitumor effects of doxorubicin [129]. Therefore, through a well-considered combination of drugs affecting mitophagy and apoptosis, these two different types of programmed cell death can cooperate with each other and lead to a stronger antitumor response.
Ferroptosis, which is characterized by intracellular iron accumulation and lipid peroxidation, has been extensively investigated in cancer research in recent years [130]. Morphologically, cells undergoing ferroptosis display distinctive mitochondrial characteristics, including mitochondrial shrinkage, increased membrane density, and decreased or absent mitochondrial cristae [131]. Interestingly, several lines of evidence have shown crosstalk between these two forms of RCD [132,133,134]. For example, Yu et al. reported that the inhibition of O-GlcNAcylation results in extensive mitophagy, which helps release stored iron and renders cell lines more sensitive to ferroptosis [133]. In contrast, Lin et al. revealed that mitophagy could also alleviate cisplatin-induced ferroptosis through the ROS/HO-1/GPX4 axis [135]. Therefore, depending on the specific context, mitophagy can act as both an enhancer and a suppressor of ferroptosis. In breast cancer, researchers revealed that BAY 11–7085 (BAY) induces ferroptosis and suppresses cell viability through the Nrf2-SLC7A11-HO-1 pathway [136]. Additionally, HO-1 also mediates cell mitochondrial damage, leading to subsequent mitophagy [136]. This finding suggested that certain ferroptosis-inducing agents can impair mitochondrial function and activate mitophagy in breast cancer cells. In addition, Cosialls et al. demonstrated that mTOR inhibition could prevent salinomycin-induced ferroptosis in breast cancer stem cells [137]. Mechanistically, they observed that under salinomycin treatment, the late stages of mitophagy were inhibited, resulting in the accumulation of damaged mitochondria [137]. However, treatment with Torin, an mTOR inhibitor, was able to reactivate the late stages of mitophagy [137]. This reactivation of mitophagy helped overcome the accumulation of damaged mitochondria and protected breast cancer stem cells from salinomycin-induced ferroptosis [137]. In this context, mitophagy acts as a protective mechanism to remove damaged mitochondria, thus suppressing ferroptosis in breast cancer cells.
Pyroptosis, an inflammatory form of RCD mediated by GSDMs, is associated with tumor-associated inflammation [138]. Recently, several publications have documented the interplay between mitophagy and pyroptosis [139, 140]. In breast cancer, research has demonstrated that the mitochondrial protein UCP1 inhibits the metastasis and proliferation of TNBC cells in vitro and in vivo [141], and this effect has been attributed to the involvement of GSDME-mediated pyroptosis and PINK1/parkin-induced mitophagy [141]. This finding indicates a synergistic relationship between these two types of RCD in suppressing breast cancer. However, controversy exists regarding the protective cell survival pathways induced by mitophagy, which could hamper pyroptosis. To address this challenge, Deng et al. constructed a biomimetic nanoparticle loaded with pyroptosis-inducing nanoparticles (CG/RH-NPs) and mitophagy-inhibiting nanoparticles (CQ-NPs) [142]. In this system, CG/RH-NPs cause mitochondrial damage in 4T1 breast cancer cells, leading to the generation of substantial mitoROS, thereby activating the caspase 3-GSDME pyroptosis pathway [142]. However, the above process in turn activates compensatory mitophagy and attenuates the antitumor efficacy of CG/RH-NPs by eliminating damaged mitochondria and mitoROS [142]. Notably, the authors also highlighted the contribution of CQ-NPs with biomimetic nanoparticles, as they effectively block compensatory mitophagy, thereby amplifying the antitumor effect of pyroptosis [142]. Therefore, the above data indicate that addressing the complex interaction between mitophagy and pyroptosis pathways produced a novel combination therapy for breast cancer treatment.
Overall, understanding the intricate crosstalk between mitophagy and other forms of RCD provides valuable insights into how breast cancer cells maintain homeostasis, progress, and acquire drug resistance. In breast cancer treatment, promising results may be obtained when combining different types of RCD with mitophagy modulation.
Mitophagy in breast cancer therapy
Chemotherapy offers significant benefits in reducing the risk of breast cancer recurrence and mortality. Notably, doxorubicin is a cornerstone of the standard chemotherapy regimen for the clinical management of breast cancer [143]. Recently, several clinical trials have investigated the role of chloroquine (CQ) and its derivative hydroxychloroquine (HCQ) in enhancing the efficacy of chemotherapy in various cancer types [144], which implies that disrupting the protective autophagic process has the potential to overcome chemoresistance. In terms of mitophagy, emerging evidence also shows that targeting mitophagy during chemotherapy represents a novel anticancer strategy for treating breast cancer. For example, Chang et al. reported that mitochondrial division inhibitor-1 blocks mitophagy initiation and prevents TNBC cells from clearing dysfunctional mitochondria, which enhances breast cancer cell sensitivity to doxorubicin [145]. Similarly, an additional study revealed that liensinine could inhibit mitophagy and lead to excessive accumulation of mitophagosomes, which sensitizes doxorubicin-induced cell death in breast cancer [146]. Consistently, Naso et al. demonstrated that PINK1/Parkin-dependent mitophagy could be activated in response to doxorubicin treatment in breast cancer and that mitophagy inhibition through miR-218-5p expression enhances the sensitivity of breast cancer cells to doxorubicin [147]. Although studies have shown that inhibiting mitophagy can enhance the sensitivity of breast cancer cells to doxorubicin, further investigation is needed to determine whether similar effects can be observed with other chemotherapeutic agents, such as paclitaxel.
Notably, immune checkpoint inhibitors (ICIs) are promising immunotherapies for some cancer types (such as non-small cell lung cancer or melanoma) and brings hope to breast cancer treatment, especially for TNBC. Previous studies have shown that programmed death-ligand 1 (PD-L1) is often upregulated in breast cancer cells as a mechanism of immune evasion, and PD-L1 blockers have emerged as promising therapeutic agents for the treatment of TNBC [148, 149]. Interestingly, Xie et al. revealed that mitophagy-mediated PD-L1 distribution in TNBC samples is correlated with the therapeutic response to ICIs combined with paclitaxel [150]. Specifically, their study revealed that paclitaxel treatment upregulates the level of ATAD3A, which suppresses PINK1 function in TNBC cells [150]. This suppression of PINK1 prevents the recruitment of PD-L1 to mitochondria by binding with PD-L1, thereby inhibiting PD-L1 degradation via mitophagy [150]. However, reduced PD-L1 mitochondrial localization leads to failure of mitophagy-dependent PD-L1 degradation, resulting in excessive accumulation of PD-L1 on the cell surface [150]. This increase in surface PD-L1 can reduce the efficacy of immunotherapy by promoting immune evasion in TNBC. Therefore, upregulating PD-L1 distribution in mitochondria through the PINK1-mediated mitophagy pathway might enhance the efficacy of anti-PD-L1 therapy for breast cancer.
In clinical practice, it has been shown that breast cancer patients with BRCA mutation could benefit from olaparib treatment. Accordingly, Arun et al. reported that mitophagy contributes to olaparib-induced cell death in BRCA1- and BRCA2-mutant breast cancer cell lines [151]. This finding suggests a potential additional mechanism by which olaparib exerts its anticancer effects beyond its role in inhibiting DNA repair. Moreover, the induction of mitophagy might elicit additive or synergistic effects of olaparib in BRCA-mutated breast cancer cells.
Radiotherapy is another important treatment for breast cancer. Zheng et al. constructed a novel fusion protein called TAT-ODD-p53, which consists of wild-type p53 combined with the TAT domain and the minimum oxygen-dependent degradation domain (ODD) of HIF-1α [152]. This innovative fusion protein was further confirmed to sensitize hypoxic breast cells to irradiation by inhibiting Parkin-mediated mitophagy [152]. Mechanistically, p53 can physically bind to Parkin and interfere with its translocation to mitochondria, thereby blocking mitophagy [152]. This finding suggested that mitophagy plays a protective role in cellular homeostasis under radiation. Nevertheless, another study proposed that PINK1-induced mitophagy, triggered by mitochondrial ROS (mitoROS), can initiate the sensitization of breast cancer cells to radiation [153]. Therefore, mitophagy also plays a dual role in radiation therapy for breast cancer treatment, acting as both a protective mechanism for cancer cell survival and a pro-death pathway that increases cancer cell sensitivity to radiation. However, the balance between the protective and damaging effects of mitophagy during the response to radiation presents a dilemma for further therapeutic intervention.
Meanwhile, recent studies have shown that several small-molecule chemicals and medications can regulate mitophagy in breast cancer cells, and provides a promising avenue for cancer therapy. Here, we summarize these agents in Table 1. However, when considering mitophagy as a new therapeutic avenue against breast cancer, it is crucial to fully understand its dual effects on tumors. Generally, basic or moderate mitophagy degrades the damaged mitochondria, which supports cellular energy production and promotes mitochondrial homeostasis. This protective mechanism provides a survival advantage to tumor cells under various stress conditions. In contrast, excessive or persistent mitophagy causes mitochondrial depletion and impairs energy metabolism, which leads to the death of tumor cells [154]. For example, Qiu et al. demonstrated that the absence of EBF1 triggers pronounced mitophagy in TNBC cells, which results in robust metabolic reprogramming, mitochondrial dysfunction, and eventual cell death [90]. Besides, several studies have employed excessive mitophagy as a therapeutic strategy against breast cancer. It has been shown that flubendazole upregulates DRP1 expression and results in excessive mitophagy through PINK1/parkin signaling in breast cancer cells [74]. This excessive mitophagy leads to mitochondrial damage and dysfunction, subsequently inhibiting the proliferation and migration of breast cancer cells [74]. Consistently, acid ground nano-realgar processed product could also suppress breast cancer cell proliferation by activating the p53/BNIP3/NIX mitophagy pathway [76]. Moreover, Shen et al. reported that Guangsangon E inhibited TNBC cell growth and induced non-apoptotic cell death in vitro and in vivo, with its effects strongly associated with the activation of mitophagy [155]. Therefore, excessive mitophagy disrupts mitochondrial homeostasis and impairs energy production, ultimately contributing to the death of tumor cells.
Conclusions and future perspectives
The physiological process and molecular mechanism of mitophagy have been extensively investigated in recent years, particularly in the context of cancer and neurodegenerative diseases [11]. In this review, we first summarize the expression patterns and functions of mitophagy-related molecules in breast cancer. Notably, a comprehensive understanding of these molecules will assist in identifying the occurrence of mitophagy in breast cancer cells and may facilitate the discovery of new therapeutic targets in breast cancer therapy. Besides, we present evidence of mitophagy on breast cancer progression from multiple perspectives, including tumor growth and metastasis, metabolic reprogramming, the tumor microenvironment, and stem-like cell characteristics. These findings will offer valuable insights into the multifaceted roles of mitophagy in breast cancer progression. Moreover, the interactions between mitophagy and other forms of RCD offer promising evidence for developing combined therapeutic strategies for breast cancer treatment.
However, compared with the issues that have been resolved, there is still a gap in the research on translating mitophagy-based mechanistic research into effective breast cancer therapy. Does mitophagy increase or decrease the risk of developing breast cancer? After tumor initiation, how does mitophagy govern the fate of breast cancer cells (promoting or inhibiting tumors) at different stages, different molecular types, and different tumor microenvironments of breast cancer? How can we modulate mitophagy to enhance the efficacy of chemotherapy, radiotherapy, or immunotherapy in breast cancer treatment? As alterations in mitophagy are also related to the development of other diseases, such as neurological disorders, how can we weigh and avoid adverse effects when modulating mitophagy as a therapeutic approach in breast cancer treatment? In clinical practice, some anticancer drugs exert antitumor effects by inducing apoptosis, ferroptosis or pyroptosis; however, breast cancer cells can exploit protective mitophagy mechanisms to survive and develop drug resistance. Therefore, the precise interaction between mitophagy and RCD requires further exploration.
In summary, modulating mitophagy can profoundly impact the progression of breast cancer through tumorigenesis, metastasis, metabolic reprogramming, the tumor microenvironment, stem-like cell characteristics, and RCD (Fig. 3). Moreover, modulating the process of mitophagy might offer novel therapeutic avenues for future breast cancer treatments. Notably, in the context of different stages, different molecular types, and different tumor microenvironments of breast cancer, the effects induced by mitophagy may be contradictory, and a comprehensive understanding of the full spectrum of mitophagy effects is required to develop efficient strategies for breast cancer treatment.

Modulating mitophagy can profoundly impact the progression of breast cancer through tumorigenesis, metastasis, metabolic reprogramming, the tumor microenvironment, stem-like cell characteristics, and regulated cell death. Created with BioRender.com.
References
Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229–63.
Agostinetto E, Gligorov J, Piccart M. Systemic therapy for early-stage breast cancer: learning from the past to build the future. Nat Rev Clin Oncol. 2022;19:763–74.
Ye F, Dewanjee S, Li Y, Jha NK, Chen ZS, Kumar A, et al. Advancements in clinical aspects of targeted therapy and immunotherapy in breast cancer. Mol Cancer. 2023;22:105.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541.
Bedoui S, Herold MJ, Strasser A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat Rev Mol Cell Biol. 2020;21:678–95.
Tyutyunyk-Massey L, Gewirtz DA. Roles of autophagy in breast cancer treatment: target, bystander or benefactor. Semin Cancer Biol. 2020;66:155–62.
Liu Y, Hu Y, Jiang Y, Bu J, Gu X. Targeting ferroptosis, the achilles’ heel of breast cancer: a review. Front Pharm. 2022;13:1036140.
Chen C, Ye Q, Wang L, Zhou J, Xiang A, Lin X, et al. Targeting pyroptosis in breast cancer: biological functions and therapeutic potentials on It. Cell Death Discov. 2023;9:75.
Yang X, Deng L, Diao X, Yang S, Zou L, Yang Q, et al. Targeting cuproptosis by zinc pyrithione in triple-negative breast cancer. iScience. 2023;26:108218.
Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8:3–5.
Wang S, Long H, Hou L, Feng B, Ma Z, Wu Y, et al. The mitophagy pathway and its implications in human diseases. Signal Transduct Target Ther. 2023;8:304.
Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021;40:e104705.
Uoselis L, Nguyen TN, Lazarou M. Mitochondrial degradation: mitophagy and beyond. Mol Cell. 2023;83:3404–20.
Lu Y, Li Z, Zhang S, Zhang T, Liu Y, Zhang L. Cellular mitophagy: mechanism, roles in diseases and small molecule pharmacological regulation. Theranostics. 2023;13:736–66.
Panigrahi DP, Praharaj PP, Bhol CS, Mahapatra KK, Patra S, Behera BP, et al. The emerging, multifaceted role of mitophagy in cancer and cancer therapeutics. Semin Cancer Biol. 2020;66:45–58.
Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet. 2011;20:867–79.
Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, et al. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012;13:378–85.
Jin SM, Youle RJ. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy. 2013;9:1750–7.
Shiba-Fukushima K, Inoshita T, Hattori N, Imai Y. PINK1-mediated phosphorylation of Parkin boosts Parkin activity in Drosophila. PLoS Genet. 2014;10:e1004391.
Zhuang N, Li L, Chen S, Wang T. PINK1-dependent phosphorylation of PINK1 and Parkin is essential for mitochondrial quality control. Cell Death Dis. 2016;7:e2501.
Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. 2018;20:1013–22.
Zhu Y, Massen S, Terenzio M, Lang V, Chen-Lindner S, Eils R, et al. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem. 2013;288:1099–113.
Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287:19094–104.
Teresak P, Lapao A, Subic N, Boya P, Elazar Z, Simonsen A. Regulation of PRKN-independent mitophagy. Autophagy. 2022;18:24–39.
Choubey V, Zeb A, Kaasik A. Molecular mechanisms and regulation of mammalian mitophagy. Cells. 2021;11:38.
Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15:1197–205.
Kagan VE, Jiang J, Huang Z, Tyurina YY, Desbourdes C, Cottet-Rousselle C, et al. NDPK-D (NM23-H4)-mediated externalization of cardiolipin enables elimination of depolarized mitochondria by mitophagy. Cell Death Differ. 2016;23:1140–51.
Sentelle RD, Senkal CE, Jiang W, Ponnusamy S, Gencer S, Selvam SP, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol. 2012;8:831–8.
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–60.
O’Flanagan CH, O’Neill C. PINK1 signalling in cancer biology. Biochim Biophys Acta. 2014;1846:590–8.
Yaghoobi H, Azizi H, Oskooei VK, Taheri M, Ghafouri-Fard S. PTEN-induced putative kinase 1 (PINK1) down-regulation in breast cancer samples in association with mitotic rate. Meta Gene. 2020;25:100748.
Li J, Xu X, Huang H, Li L, Chen J, Ding Y, et al. Pink1 promotes cell proliferation and affects glycolysis in breast cancer. Exp Biol Med. 2022;247:985–95.
Berthier A, Navarro S, Jimenez-Sainz J, Rogla I, Ripoll F, Cervera J, et al. PINK1 displays tissue-specific subcellular location and regulates apoptosis and cell growth in breast cancer cells. Hum Pathol. 2011;42:75–87.
Unoki M, Nakamura Y. Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene. 2001;20:4457–65.
Miyahara K, Takano N, Yamada Y, Kazama H, Tokuhisa M, Hino H, et al. BRCA1 degradation in response to mitochondrial damage in breast cancer cells. Sci Rep. 2021;11:8735.
MacKeigan JP, Murphy LO, Blenis J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat Cell Biol. 2005;7:591–600.
Murata H, Sakaguchi M, Jin Y, Sakaguchi Y, Futami J, Yamada H, et al. A new cytosolic pathway from a Parkinson disease-associated kinase, BRPK/PINK1: activation of AKT via mTORC2. J Biol Chem. 2011;286:7182–9.
Furlong RM, Lindsay A, Anderson KE, Hawkins PT, Sullivan AM, O’Neill C. The Parkinson’s disease gene PINK1 activates Akt via PINK1 kinase-dependent regulation of the phospholipid PI(3,4,5)P(3). J Cell Sci. 2019;132:jcs233221
Ellis H, Ma CX. PI3K inhibitors in breast cancer therapy. Curr Oncol Rep. 2019;21:110.
Miricescu D, Totan A, Stanescu S II, Badoiu SC, Stefani C, Greabu M. PI3K/AKT/mTOR signaling pathway in breast cancer: from molecular landscape to clinical aspects. Int J Mol Sci. 2020;22:173.
Tay SP, Yeo CW, Chai C, Chua PJ, Tan HM, Ang AX, et al. Parkin enhances the expression of cyclin-dependent kinase 6 and negatively regulates the proliferation of breast cancer cells. J Biol Chem. 2010;285:29231–8.
Wahabi K, Perwez A, Kamarudheen S, Bhat ZI, Mehta A, Rizvi MMA. Parkin gene mutations are not common, but its epigenetic inactivation is a frequent event and predicts poor survival in advanced breast cancer patients. BMC Cancer. 2019;19:820.
Liu J, Zhang C, Wu H, Sun XX, Li Y, Huang S, et al. Parkin ubiquitinates phosphoglycerate dehydrogenase to suppress serine synthesis and tumor progression. J Clin Invest. 2020;130:3253–69.
Wang H, Liu B, Zhang C, Peng G, Liu M, Li D, et al. Parkin regulates paclitaxel sensitivity in breast cancer via a microtubule-dependent mechanism. J Pathol. 2009;218:76–85.
Liu J, Zhang C, Zhao Y, Yue X, Wu H, Huang S, et al. Parkin targets HIF-1alpha for ubiquitination and degradation to inhibit breast tumor progression. Nat Commun. 2017;8:1823.
Singh N, Tapader R, Chatterjee S, Pal A, Pal A. Subtilisin from Bacillus amyloliquefaciens induces apoptosis in breast cancer cells through ubiquitin-proteasome-mediated tubulin degradation. Int J Biol Macromol. 2022;220:852–65.
Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2011;12:68–78.
Loibl S, Poortmans P, Morrow M, Denkert C, Curigliano G. Breast cancer. Lancet. 2021;397:1750–69.
Reid MA, Allen AE, Liu S, Liberti MV, Liu P, Liu X, et al. Serine synthesis through PHGDH coordinates nucleotide levels by maintaining central carbon metabolism. Nat Commun. 2018;9:5442.
Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–50.
Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939–46.
Tan EY, Campo L, Han C, Turley H, Pezzella F, Gatter KC, et al. BNIP3 as a progression marker in primary human breast cancer; opposing functions in in situ versus invasive cancer. Clin Cancer Res. 2007;13:467–74.
Naushad SM, Prayaga A, Digumarti RR, Gottumukkala SR, Kutala VK. Bcl-2/adenovirus E1B 19 kDa-interacting protein 3 (BNIP3) expression is epigenetically regulated by one-carbon metabolism in invasive duct cell carcinoma of breast. Mol Cell Biochem. 2012;361:189–95.
Mustafa MF, Saliluddin SM, Fakurazi S, Tizen Laim NMS, Md Pauzi SH, Nik Yahya NH, et al. Expression of autophagy and mitophagy markers in breast cancer tissues. Front Oncol. 2021;11:612009.
Zhang G, Xu Z, Yu M, Gao H. Bcl-2 interacting protein 3 (BNIP3) promotes tumor growth in breast cancer under hypoxic conditions through an autophagy-dependent pathway. Bioengineered. 2022;13:6280–92.
Koop EA, van Laar T, van Wichen DF, de Weger RA, Wall E, van Diest PJ. Expression of BNIP3 in invasive breast cancer: correlations with the hypoxic response and clinicopathological features. BMC Cancer. 2009;9:175.
Petry IB, Fieber E, Schmidt M, Gehrmann M, Gebhard S, Hermes M, et al. ERBB2 induces an antiapoptotic expression pattern of Bcl-2 family members in node-negative breast cancer. Clin Cancer Res. 2010;16:451–60.
Vijayalingam S, Pillai SG, Rashmi R, Subramanian T, Sagartz JE, Chinnadurai G. Overexpression of BH3-only protein BNIP3 leads to enhanced tumor growth. Genes Cancer. 2010;1:964–71.
Chourasia AH, Tracy K, Frankenberger C, Boland ML, Sharifi MN, Drake LE, et al. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. 2015;16:1145–63.
Niu Y, Lin Z, Wan A, Chen H, Liang H, Sun L, et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol Cancer. 2019;18:46.
Mellor HR, Harris AL. The role of the hypoxia-inducible BH3-only proteins BNIP3 and BNIP3L in cancer. Cancer Metastasis Rev. 2007;26:553–66.
Lai J, Flanagan J, Phillips WA, Chenevix-Trench G, Arnold J. Analysis of the candidate 8p21 tumour suppressor, BNIP3L, in breast and ovarian cancer. Br J Cancer. 2003;88:270–6.
Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001;61:6669–73.
Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle. 2011;10:4047–64.
Sung JS, Kang CW, Kang S, Jang Y, Chae YC, Kim BG, et al. ITGB4-mediated metabolic reprogramming of cancer-associated fibroblasts. Oncogene. 2020;39:664–76.
Pedanou VE, Gobeil S, Tabaries S, Simone TM, Zhu LJ, Siegel PM, et al. The histone H3K9 demethylase KDM3A promotes anoikis by transcriptionally activating pro-apoptotic genes BNIP3 and BNIP3L. Elife. 2016;5:e16844.
Real PJ, Benito A, Cuevas J, Berciano MT, de Juan A, Coffer P, et al. Blockade of epidermal growth factor receptors chemosensitizes breast cancer cells through up-regulation of Bnip3L. Cancer Res. 2005;65:8151–7.
Zhang W. The mitophagy receptor FUN14 domain-containing 1 (FUNDC1): a promising biomarker and potential therapeutic target of human diseases. Genes Dis. 2021;8:640–54.
Yang A, Peng F, Zhu L, Li X, Ou S, Huang Z, et al. Melatonin inhibits triple-negative breast cancer progression through the Lnc049808-FUNDC1 pathway. Cell Death Dis. 2021;12:712.
Wu L, Zhang D, Zhou L, Pei Y, Zhuang Y, Cui W, et al. FUN14 domain-containing 1 promotes breast cancer proliferation and migration by activating calcium-NFATC1-BMI1 axis. EBioMedicine. 2019;41:384–94.
Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G, et al. ROS in cancer therapy: the bright side of the moon. Exp Mol Med. 2020;52:192–203.
Deng R, Zhang HL, Huang JH, Cai RZ, Wang Y, Chen YH, et al. MAPK1/3 kinase-dependent ULK1 degradation attenuates mitophagy and promotes breast cancer bone metastasis. Autophagy. 2021;17:3011–29.
Chen Q, Lei JH, Bao J, Wang H, Hao W, Li L, et al. BRCA1 deficiency impairs mitophagy and promotes inflammasome activation and mammary tumor metastasis. Adv Sci. 2020;7:1903616.
Zhen Y, Yuan Z, Zhang J, Chen Y, Fu Y, Liu Y, et al. Flubendazole induces mitochondrial dysfunction and DRP1-mediated mitophagy by targeting EVA1A in breast cancer. Cell Death Dis. 2022;13:375.
Cao J, Ma X, Yan X, Zhang G, Hong S, Ma R, et al. Kaempferol induces mitochondrial dysfunction and mitophagy by activating the LKB1/AMPK/MFF pathway in breast precancerous lesions. Phytother Res. 2023;37:3602–16.
Fang J, Zou X, Gong L, Xi J, Liu Y, Yang X, et al. Acid ground nano-realgar processed product inhibits breast cancer by inducing mitophagy via the p53/BNIP3/NIX pathway. J Cell Mol Med. 2023;27:3478–90.
Sun F, Jiang X, Wang X, Bao Y, Feng G, Liu H, et al. Vincristine ablation of Sirt2 induces cell apoptosis and mitophagy via Hsp70 acetylation in MDA-MB-231 cells. Biochem Pharm. 2019;162:142–53.
Qian H, Chao X, Ding WX. A PINK1-mediated mitophagy pathway decides the fate of tumors-to be benign or malignant? Autophagy. 2018;14:563–6.
Chourasia AH, Boland ML, Macleod KF. Mitophagy and cancer. Cancer Metab. 2015;3:4.
Poole LP, Macleod KF. Mitophagy in tumorigenesis and metastasis. Cell Mol Life Sci. 2021;78:3817–51.
Li X, Wang M, Li S, Chen Y, Wang M, Wu Z, et al. HIF-1-induced mitochondrial ribosome protein L52: a mechanism for breast cancer cellular adaptation and metastatic initiation in response to hypoxia. Theranostics. 2021;11:7337–59.
Park SY, Nam JS. The force awakens: metastatic dormant cancer cells. Exp Mol Med. 2020;52:569–81.
Vera-Ramirez L, Vodnala SK, Nini R, Hunter KW, Green JE. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat Commun. 2018;9:1944.
Li Q, Chu Y, Li S, Yu L, Deng H, Liao C, et al. The oncoprotein MUC1 facilitates breast cancer progression by promoting Pink1-dependent mitophagy via ATAD3A destabilization. Cell Death Dis. 2022;13:899.
Pavlova NN, Zhu J, Thompson CB. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022;34:355–77.
Liu L, Liao X, Wu H, Li Y, Zhu Y, Chen Q. Mitophagy and its contribution to metabolic and aging-associated disorders. Antioxid Redox Signal. 2020;32:906–27.
Ferro F, Servais S, Besson P, Roger S, Dumas JF, Brisson L. Autophagy and mitophagy in cancer metabolic remodelling. Semin Cell Dev Biol. 2020;98:129–38.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33.
Lyons A, Coleman M, Riis S, Favre C, O’Flanagan CH, Zhdanov AV, et al. Insulin-like growth factor 1 signaling is essential for mitochondrial biogenesis and mitophagy in cancer cells. J Biol Chem. 2017;292:16983–98.
Qiu Z, Guo W, Dong B, Wang Y, Deng P, Wang C, et al. EBF1 promotes triple-negative breast cancer progression by surveillance of the HIF1alpha pathway. Proc Natl Acad Sci USA. 2022;119:e2119518119.
Zou P, Liu L, Zheng LD, Payne KK, Manjili MH, Idowu MO, et al. Coordinated upregulation of mitochondrial biogenesis and autophagy in breast cancer cells: the role of dynamin related protein-1 and implication for breast cancer treatment. Oxid Med Cell Longev. 2016;2016:4085727.
Kim S, Kim DH, Jung WH, Koo JS. Metabolic phenotypes in triple-negative breast cancer. Tumour Biol. 2013;34:1699–712.
Bejarano L, Jordao MJC, Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Discov. 2021;11:933–59.
Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984–4001.
Liang L, Li W, Li X, Jin X, Liao Q, Li Y, et al. Reverse Warburg effect’ of cancer‑associated fibroblasts (Review). Int J Oncol. 2022;60:67.
Salem AF, Howell A, Sartini M, Sotgia F, Lisanti MP. Downregulation of stromal BRCA1 drives breast cancer tumor growth via upregulation of HIF-1alpha, autophagy and ketone body production. Cell Cycle. 2012;11:4167–73.
Capparelli C, Guido C, Whitaker-Menezes D, Bonuccelli G, Balliet R, Pestell TG, et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle. 2012;11:2285–302.
Sanchez-Alvarez R, Martinez-Outschoorn UE, Lin Z, Lamb R, Hulit J, Howell A, et al. Ethanol exposure induces the cancer-associated fibroblast phenotype and lethal tumor metabolism: implications for breast cancer prevention. Cell Cycle. 2013;12:289–301.
Sotgia F, Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Lisanti MP. Understanding the Warburg effect and the prognostic value of stromal caveolin-1 as a marker of a lethal tumor microenvironment. Breast Cancer Res. 2011;13:213.
Witkiewicz AK, Kline J, Queenan M, Brody JR, Tsirigos A, Bilal E, et al. Molecular profiling of a lethal tumor microenvironment, as defined by stromal caveolin-1 status in breast cancers. Cell Cycle. 2011;10:1794–809.
Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFkappaB activation in the tumor stromal microenvironment. Cell Cycle. 2010;9:3515–33.
Capparelli C, Whitaker-Menezes D, Guido C, Balliet R, Pestell TG, Howell A, et al. CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle. 2012;11:2272–84.
Zheng B, Wang Y, Zhou B, Qian F, Liu D, Ye D, et al. Urolithin a inhibits breast cancer progression via activating TFEB-mediated mitophagy in tumor macrophages. J Adv Res. 2024;S2090-1232:00153.
Kuo CL, Ponneri Babuharisankar A, Lin YC, Lien HW, Lo YK, Chou HY, et al. Mitochondrial oxidative stress in the tumor microenvironment and cancer immunoescape: foe or friend? J Biomed Sci. 2022;29:74.
Malla R, Surepalli N, Farran B, Malhotra SV, Nagaraju GP. Reactive oxygen species (ROS): critical roles in breast tumor microenvironment. Crit Rev Oncol Hematol. 2021;160:103285.
Oshi M, Gandhi S, Yan L, Tokumaru Y, Wu R, Yamada A, et al. Abundance of reactive oxygen species (ROS) is associated with tumor aggressiveness, immune response, and worse survival in breast cancer. Breast Cancer Res Treat. 2022;194:231–41.
Sun Q, Yang J, Shen W, Lu H, Hou X, Liu Y, et al. Engineering mitochondrial uncoupler synergistic photodynamic nanoplatform to harness immunostimulatory pro-death autophagy/mitophagy. Biomaterials. 2022;289:121796.
Butti R, Gunasekaran VP, Kumar TVS, Banerjee P, Kundu GC. Breast cancer stem cells: biology and therapeutic implications. Int J Biochem Cell Biol. 2019;107:38–52.
Lin Q, Chen J, Gu L, Dan X, Zhang C, Yang Y. New insights into mitophagy and stem cells. Stem Cell Res Ther. 2021;12:452.
Praharaj PP, Patro BS, Bhutia SK. Dysregulation of mitophagy and mitochondrial homeostasis in cancer stem cells: novel mechanism for anti-cancer stem cell-targeted cancer therapy. Br J Pharm. 2022;179:5015–35.
Li D, Peng X, He G, Liu J, Li X, Lin W, et al. Crosstalk between autophagy and CSCs: molecular mechanisms and translational implications. Cell Death Dis. 2023;14:409.
Hu Q, Yuan Y, Wu Y, Huang Y, Zhao Z, Xiao C. MicroRNA‑137 exerts protective effects on hypoxia‑induced cell injury by inhibiting autophagy/mitophagy and maintaining mitochondrial function in breast cancer stem‑like cells. Oncol Rep. 2020;44:1627–37.
Zhang S, Liu C, Zhang X. Mitochondrial damage mediated by miR-1 overexpression in cancer stem cells. Mol Ther Nucleic Acids. 2019;18:938–53.
Zheng XX, Chen JJ, Sun YB, Chen TQ, Wang J, Yu SC. Mitochondria in cancer stem cells: Achilles heel or hard armor. Trends Cell Biol. 2023;33:708–27.
Tong X, Tang R, Xiao M, Xu J, Wang W, Zhang B, et al. Targeting cell death pathways for cancer therapy: recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research. J Hematol Oncol. 2022;15:174.
Wanderoy S, Hees JT, Klesse R, Edlich F, Harbauer AB. Kill one or kill the many: interplay between mitophagy and apoptosis. Biol Chem. 2020;402:73–88.
Ham SJ, Lee D, Yoo H, Jun K, Shin H, Chung J. Decision between mitophagy and apoptosis by Parkin via VDAC1 ubiquitination. Proc Natl Acad Sci USA. 2020;117:4281–91.
Praharaj PP, Naik PP, Panigrahi DP, Bhol CS, Mahapatra KK, Patra S, et al. Intricate role of mitochondrial lipid in mitophagy and mitochondrial apoptosis: its implication in cancer therapeutics. Cell Mol Life Sci. 2019;76:1641–52.
Tang Y, Wang L, Qin J, Lu Y, Shen HM, Chen HB. Targeting mitophagy to promote apoptosis is a potential therapeutic strategy for cancer. Autophagy. 2023;19:1031–3.
Jeong CH, Joo SH. Downregulation of reactive oxygen species in apoptosis. J Cancer Prev. 2016;21:13–20.
Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta. 2016;1863:2977–92.
Liu Y, Jiang Y, Wang N, Jin Q, Ji F, Zhong C, et al. Invalidation of mitophagy by FBP1-mediated repression promotes apoptosis in breast cancer. Tumour Biol. 2017;39:1010428317708779.
Qiao A, Wang K, Yuan Y, Guan Y, Ren X, Li L, et al. Sirt3-mediated mitophagy protects tumor cells against apoptosis under hypoxia. Oncotarget. 2016;7:43390–43400.
Si L, Fu J, Liu W, Hayashi T, Mizuno K, Hattori S, et al. Silibinin-induced mitochondria fission leads to mitophagy, which attenuates silibinin-induced apoptosis in MCF-7 and MDA-MB-231 cells. Arch Biochem Biophys. 2020;685:108284.
Li GB, Fu RQ, Shen HM, Zhou J, Hu XY, Liu YX, et al. Polyphyllin I induces mitophagic and apoptotic cell death in human breast cancer cells by increasing mitochondrial PINK1 levels. Oncotarget. 2017;8:10359–74.
Mao L, Liu H, Zhang R, Deng Y, Hao Y, Liao W, et al. PINK1/Parkin-mediated mitophagy inhibits warangalone-induced mitochondrial apoptosis in breast cancer cells. Aging (Albany NY). 2021;13:12955–72.
Peng F, Liao M, Qin R, Zhu S, Peng C, Fu L, et al. Regulated cell death (RCD) in cancer: key pathways and targeted therapies. Signal Transduct Target Ther. 2022;7:286.
Wohlfromm F, Richter M, Otrin L, Seyrek K, Vidakovic-Koch T, Kuligina E, et al. Interplay Between Mitophagy and Apoptosis Defines a Cell Fate Upon Co-treatment of Breast Cancer Cells With a Recombinant Fragment of Human kappa-Casein and Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand. Front Cell Dev Biol. 2020;8:617762.
Wohlfromm F, Seyrek K, Ivanisenko N, Troitskaya O, Kulms D, Richter V, et al. RL2 enhances the elimination of breast cancer cells by doxorubicin. Cells. 2023;12:2779.
Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022;22:381–96.
Chen X, Kang R, Kroemer G, Tang D. Organelle-specific regulation of ferroptosis. Cell Death Differ. 2021;28:2843–56.
Rademaker G, Boumahd Y, Peiffer R, Anania S, Wissocq T, Liegeois M, et al. Myoferlin targeting triggers mitophagy and primes ferroptosis in pancreatic cancer cells. Redox Biol. 2022;53:102324.
Yu F, Zhang Q, Liu H, Liu J, Yang S, Luo X, et al. Dynamic O-GlcNAcylation coordinates ferritinophagy and mitophagy to activate ferroptosis. Cell Discov. 2022;8:40.
Basit F, van Oppen LM, Schockel L, Bossenbroek HM, van Emst-de Vries SE, Hermeling JC, et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017;8:e2716.
Lin Q, Li S, Jin H, Cai H, Zhu X, Yang Y, et al. Mitophagy alleviates cisplatin-induced renal tubular epithelial cell ferroptosis through ROS/HO-1/GPX4 axis. Int J Biol Sci. 2023;19:1192–210.
Chang LC, Chiang SK, Chen SE, Yu YL, Chou RH, Chang WC. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018;416:124–37.
Cosialls E, Pacreau E, Duruel C, Ceccacci S, Elhage R, Desterke C, et al. mTOR inhibition suppresses salinomycin-induced ferroptosis in breast cancer stem cells by ironing out mitochondrial dysfunctions. Cell Death Dis. 2023;14:744.
Broz P, Pelegrin P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol. 2020;20:143–57.
Yu J, Nagasu H, Murakami T, Hoang H, Broderick L, Hoffman HM, et al. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci USA. 2014;111:15514–9.
Yu X, Hao M, Liu Y, Ma X, Lin W, Xu Q, et al. Liraglutide ameliorates non-alcoholic steatohepatitis by inhibiting NLRP3 inflammasome and pyroptosis activation via mitophagy. Eur J Pharm. 2019;864:172715.
Xia J, Chu C, Li W, Chen H, Xie W, Cheng R, et al. Mitochondrial protein UCP1 inhibits the malignant behaviors of triple-negative breast cancer through activation of mitophagy and pyroptosis. Int J Biol Sci. 2022;18:2949–61.
Deng Y, Jia F, Jiang P, Chen L, Xing L, Shen X, et al. Biomimetic nanoparticle synchronizing pyroptosis induction and mitophagy inhibition for anti-tumor therapy. Biomaterials. 2023;301:122293.
Waks AG, Winer EP. Breast cancer treatment: a review. JAMA. 2019;321:288–300.
Xu R, Ji Z, Xu C, Zhu J. The clinical value of using chloroquine or hydroxychloroquine as autophagy inhibitors in the treatment of cancers: a systematic review and meta-analysis. Medicine. 2018;97:e12912.
Chang JC, Chang HS, Yeh CY, Chang HJ, Cheng WL, Lin TT, et al. Regulation of mitochondrial fusion and mitophagy by intra-tumoral delivery of membrane-fused mitochondria or Midiv-1 enhances sensitivity to doxorubicin in triple-negative breast cancer. Biomed Pharmacother. 2022;153:113484.
Zhou J, Li G, Zheng Y, Shen HM, Hu X, Ming QL, et al. A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy. 2015;11:1259–79.
Naso FD, Bruqi K, Manzini V, Chiurchiu V, D’Onofrio M, Arisi I, et al. miR-218-5p and doxorubicin combination enhances anticancer activity in breast cancer cells through Parkin-dependent mitophagy inhibition. Cell Death Discov. 2024;10:149.
Mittendorf EA, Zhang H, Barrios CH, Saji S, Jung KH, Hegg R, et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): a randomised, double-blind, phase 3 trial. Lancet. 2020;396:1090–1100.
Schmid P, Rugo HS, Adams S, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21:44–59.
Xie XQ, Yang Y, Wang Q, Liu HF, Fang XY, Li CL, et al. Targeting ATAD3A-PINK1-mitophagy axis overcomes chemoimmunotherapy resistance by redirecting PD-L1 to mitochondria. Cell Res. 2023;33:215–28.
Arun B, Akar U, Gutierrez-Barrera AM, Hortobagyi GN, Ozpolat B. The PARP inhibitor AZD2281 (Olaparib) induces autophagy/mitophagy in BRCA1 and BRCA2 mutant breast cancer cells. Int J Oncol. 2015;47:262–8.
Zheng R, Yao Q, Xie G, Du S, Ren C, Wang Y, et al. TAT-ODD-p53 enhances the radiosensitivity of hypoxic breast cancer cells by inhibiting Parkin-mediated mitophagy. Oncotarget. 2015;6:17417–29.
Yu L, Yang X, Li X, Qin L, Xu W, Cui H, et al. Pink1/PARK2/mROS-dependent mitophagy initiates the sensitization of cancer cells to radiation. Oxid Med Cell Longev. 2021;2021:5595652.
Vara-Perez M, Felipe-Abrio B, Agostinis P. Mitophagy in cancer: a tale of adaptation. Cells. 2019;8:493.
Shen Y, Han Z, Wang L, Liang Y, Zhang X, Li W, et al. Guangsangon E triggers mitochondria dysfunction and mitophagy in triple-negative breast cancer and leads to non-apoptotic cell death. Mol Carcinog. 2022;61:1128–42.
Liu HJ, Jiang XX, Guo YZ, Sun FH, Kou XH, Bao Y, et al. The flavonoid TL-2-8 induces cell death and immature mitophagy in breast cancer cells via abrogating the function of the AHA1/Hsp90 complex. Acta Pharm Sin. 2017;38:1381–93.
Han H, Chou CC, Li R, Liu J, Zhang L, Zhu W, et al. Chalcomoracin is a potent anticancer agent acting through triggering Oxidative stress via a mitophagy- and paraptosis-dependent mechanism. Sci Rep. 2018;8:9566.
Yao N, Wang C, Hu N, Li Y, Liu M, Lei Y, et al. Inhibition of PINK1/Parkin-dependent mitophagy sensitizes multidrug-resistant cancer cells to B5G1, a new betulinic acid analog. Cell Death Dis. 2019;10:232.
Hou J, Yun Y, Xue J, Jeon B, Kim S. Doxorubicin-induced normal breast epithelial cellular aging and its related breast cancer growth through mitochondrial autophagy and oxidative stress mitigated by ginsenoside Rh2. Phytother Res. 2020;34:1659–69.
Babak MV, Chong KR, Rapta P, Zannikou M, Tang HM, Reichert L, et al. Interfering with metabolic profile of triple-negative breast cancers using rationally designed metformin prodrugs. Angew Chem Int Ed Engl. 2021;60:13405–13.
Tran VT, Pham DV, Choi DY, Park PH. Mitophagy induction and aryl hydrocarbon receptor-mediated redox signaling contribute to the suppression of breast cancer cell growth by taloxifene via regulation of inflammasomes activation. Antioxid Redox Signal. 2022;37:1030–50.
Xue YW, Itoh H, Dan S, Inoue M. Gramicidin A accumulates in mitochondria, reduces ATP levels, induces mitophagy, and inhibits cancer cell growth. Chem Sci. 2022;13:7482–91.
Shen LW, Jiang XX, Li ZQ, Li J, Wang M, Jia GF, et al. Cepharanthine sensitizes human triple negative breast cancer cells to chemotherapeutic agent epirubicin via inducing cofilin oxidation-mediated mitochondrial fission and apoptosis. Acta Pharm Sin. 2022;43:177–93.
Gu L, Zhang J, Liu D, Chen J, Liu S, Peng Q, et al. Development of artesunate intelligent prodrug liposomes based on mitochondrial targeting strategy. J Nanobiotechnol. 2022;20:376.
Choi YJ, Choi YK, Ko SG, Cheon C, Kim TY. Investigation of molecular mechanisms involved in sensitivity to the anti-cancer activity of costunolide in breast cancer cells. Int J Mol Sci. 2023;24:4009.
Zheng H, Hu C, Quan Y, Ye X, Shi X, Guo Z, et al. A copper complex that combats triple negative breast cancer by restraining angiogenesis. Dalton Trans. 2023;52:7626–34.
Hua F, Xiao YY, Qu XH, Li SS, Zhang K, Zhou C, et al. Baicalein sensitizes triple negative breast cancer MDA-MB-231 cells to doxorubicin via autophagy-mediated down-regulation of CDK1. Mol Cell Biochem. 2023;478:1519–31.
Zhang GD, Wang MM, Su Y, Fang H, Xue XL, Liu HK, et al. Mitochondria-targeted ruthenium complexes can be generated in vitro and in living cells to target triple-negative breast cancer cells by autophagy inhibition. J Inorg Biochem. 2024;256:112574.
Zhang Y, Yang F, Wu J, Huang J, Li P, Huang G. Idebenone exerts anti-triple negative breast cancer effects via dual signaling pathways of GADD45 and AMPK. Nutr Cancer. 2024;76:379–92.
Acknowledgements
Figures were created using BioRender.com. Retrieved from https://app.biorender.com.
Funding
This study was supported by the National Natural Science Foundation of China (Grant No: 82303598), Zhejiang Provincial Natural Science Foundation of China (Grant No: LQ24H160033; No.ZCLQ24H1601).
Author information
Authors and Affiliations
Contributions
Manuscript writing (original draft): CC. Conceptualization and supervision: CC and QY. Manuscript review and editing: AX and XL. Manuscript revise: JG and JL. Figures editing: SH and TR. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
All authors agreed to publication.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, C., Xiang, A., Lin, X. et al. Mitophagy: insights into its signaling molecules, biological functions, and therapeutic potential in breast cancer. Cell Death Discov. 10, 457 (2024). https://doi.org/10.1038/s41420-024-02226-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-024-02226-6
This article is cited by
-
Mitophagy’s impacts on cancer and neurodegenerative diseases: implications for future therapies
Journal of Hematology & Oncology (2025)
-
Repurposing chlorpromazine for the treatment of triple-negative breast cancer growth and metastasis based on modulation of mitochondria-mediated apoptosis and autophagy/mitophagy
British Journal of Cancer (2025)
-
Discovery of a new mitophagy-related gene signature for predicting the outlook and immunotherapy in triple-negative breast cancer
Scientific Reports (2025)
-
Establishment and characterization of a new Chinese extrahepatic cholangiocarcinoma cell line, EBC-X1
Human Cell (2025)
-
Lipid droplet formation induced by icaritin derivative IC2 promotes a combination strategy for cancer therapy
Chinese Medicine (2024)
