
Abstract
Studies of forces driving interlineage variability in the evolutionary rates (both sequence and architecture) of mitochondrial genomes often produce contradictory results. Flatworms (Platyhelminthes) exhibit the fastest-evolving mitogenomic sequences among all bilaterian phyla. To test the effects of multiple factors previously associated with different aspects of mitogenomic evolution, we used mitogenomes of 223 flatworm species, phylogenetic multilevel regression models, and causal inference. Thermic host environment (endothermic vs. ectothermic) had nonsignificant impacts on both sequence evolution and mitogenomic size. Mitogenomic gene order rearrangements (GORR) were mostly positively correlated with mitogenomic size (R2 ≈ 20–30%). Longevity was not (negatively) correlated with sequence evolution in flatworms. The predominantly free-living “turbellaria” exhibited much shorter branches and faster-evolving mitogenomic architecture than parasitic Neodermata. As a result, “parasitism” had a strong explanatory power on the branch length variability (>90%), and there was a negative correlation between GORR and branch length. However, the stem branch of Neodermata comprised 63.6% of the total average branch length. This evolutionary period was also marked by a high rate of gene order rearrangements in the ancestral Neodermata. We discuss how this period of rapid evolution deep in the evolutionary history may have decoupled sequence evolution rates from longevity and GORR, and overestimated the explanatory power of “parasitism”. This study shows that impacts of variables often vary across lineages, and stresses the importance accounting for the episodic nature of evolutionary patterns in studies of mitogenomic evolution.
Similar content being viewed by others


Introduction
Evolutionary rates of mitochondrial genomes (mitogenomes) exhibit remarkable interlineage variability, both in terms of sequence and architecture evolution. Multiple factors have been associated with mitogenomic evolutionary rates, including body size, longevity, the effective population size (Ne), parasitism, metabolic rate, etc. (Martin and Palumbi 1993; Nabholz et al. 2016; Jakovlić et al. 2023). However, results are often inconsistent across different lineages, so there is growing consensus that drivers of mitogenomic evolution in animals are multifactorial, lineage-specific, and in general difficult to predict (Bazin et al. 2006; Thomas et al. 2006; Lanfear et al. 2007; Nabholz et al. 2008a; Nabholz et al. 2008b; Galtier et al. 2009; Allio et al. 2017; Saclier et al. 2018; Jakovlić et al. 2021). Mitochondrial genomes of Platyhelminthes (flatworms) exhibit the highest average amino acid substitution rate among the bilaterian phyla (Bernt et al. 2013; Jakovlić et al. 2023), but the factors underlying this phenomenon remain unknown. They also exhibit a number of other characteristics that make them uniquely suitable to test impacts of various variables putatively associated with mitogenomic evolution, so herein we used this phylum as a model to test six hypotheses related to mitogenomic evolution patterns in animals (Table 1).
Several studies found support for the hypothesis that thermic habitat affects mitochondrial sequence evolution (Lajbner et al. 2018) and that evolutionary rates differ between endotherms and ectotherms (Rand 1994). Two hypotheses have been put forward to explain this association. The “functional constraints” hypothesis proposes that variations in the thermic environment may restrict physiologically acceptable amino acid substitutions, which implies that thermally stable endotherms should have a higher rate of sequence evolution than thermally variable ectotherms due to relaxed purifying selection pressures (Rand 1994). The ”metabolic rate” hypothesis postulates that endothermic animals have higher metabolic rates than ectotherms due to higher energy expenditure, which generates more reactive oxygen species, and results in an increased DNA mutation rate in endotherms (Martin and Palumbi 1993). Thermic habitat may also affect the mitogenomic size: higher replication rates in endothermic animals may result in smaller mitogenomes and reduced size variability via the mechanism described as “race for replication”, wherein shorter mitogenomes will be replicated at higher rates due to mechanistic constraints (Rand 1993). These hypotheses were previously extended to parasites: a host’s thermic habitat might also affect mitogenomic evolution in their parasites (Lagisz et al. 2013). Flatworms are among the rare parasitic lineages that parasitize both endothermic and ectothermic hosts, which makes them a suitable model to further test these hypotheses. However, all flatworms are ectothermic themselves, so they do not have to spend energy to generate heat to maintain the body temperature regardless of the thermic type of the host. Therefore, there should be no direct relationship between their energy consumption and host type with respect to endothermy/ectothermy. This implies that under the functional constraints hypothesis, parasites of endotherms should have faster-evolving mitogenomes than parasites of ectotherms due to the stable thermic environment, but there is no direct reason why their evolutionary rates should be affected by the thermic environment of the host under the metabolic rate hypothesis. There is also no direct reason why their mitogenome sizes should be affected by the thermic environment of the host, unless parasites of endotherms invest much more in increased growth rates and/or fecundity, but the evidence for this hypothesis is weak, inconsistent, and limited to a few helminth lineages (Jackson and Tinsley 1998; Poulin and Latham 2003; Bakke et al. 2007; Benesh et al. 2021). To test these hypotheses (Hypothesis 1: Functional constraints and Hypothesis 2: Race for replication; Table 1), we divided parasitic flatworms (Neodermata) according to the thermic environment of their intermediate and definitive hosts into ectotherms and endotherms.
Some types of architecture rearrangement events (e.g. tandem-duplication-random-loss) cause sequence duplications and result in an increased abundance and size of noncoding intergenic regions in mitogenomes (Boore 2000). In addition, we hypothesise that mitogenome replication and maintenance machinery errors should result in both increased gene reshuffling and sequence duplication errors. Accordingly, our working hypothesis is that mitogenome size should be positively correlated with the gene order rearrangement rate (GORR) (Hypothesis 3: Mitogenome size and GORR; Table 1). Surprisingly, besides the positive correlation between the mitogenome size and gene order rearrangement rate established in nematodes (Lagisz et al. 2013), the correlation between mitogenomic size and other relevant parameters does not appear to have been tested statistically in other animal studies. Flatworms exhibit highly variable mitogenome sizes (13 to 27 Kbp) and architecture rearrangement rates, with (mostly) relatively slow-evolving Neodermata (Zhang et al. 2019) and fast-evolving basal radiation of mostly non-parasitic flatworms (“turbellaria”) (Solà et al. 2015; Rosa et al. 2017). This makes flatworms a good model to test these hypotheses.
As gene order rearrangements and size expansions should be selectively constrained (Lynch et al. 2006; Shtolz and Mishmar 2023), mitogenomic size and rearrangement rate should be positively correlated with sequence evolution. Indeed, there is evidence for this in a range of animal lineages (Shao et al. 2003; Hassanin 2006; Xu et al. 2006; Bernt et al. 2013; Zou et al. 2022b; Struck et al. 2023). However, the existence of several exceptions indicates that the correlation might be lineage-specific (Chong and Mueller 2013; Bernt et al. 2013; Tan et al. 2019), so the hypothesis requires further testing (Hypothesis 4: Sequence evolution and size/GORR; Table 1).
Longevity is another factor putatively associated with mitogenomic evolution, according to the proposal that long-lived animals may have adapted to an increased lifespan by evolving macromolecular components more resistant to oxidative damage, thus reducing their evolutionary rates (Nabholz et al. 2008a; Welch et al. 2008; Galtier et al. 2009). In addition, longevity is positively correlated to generation time in many animals (Jeschke and Kokko 2009), which is also a variable putatively negatively associated with mitogenomic evolutionary rates (Li et al. 1987; Martin and Palumbi 1993; Thomas et al. 2010). Previous studies also found contradictory results with regard to the impact of these two variables (longevity and generation time) on the mitogenomic evolutionary rates (Nabholz et al. 2008a; Nabholz et al. 2008b; Min and Hickey 2008; Nabholz et al. 2009; Thomas et al. 2010; Hua et al. 2015; Allio et al. 2017; Saclier et al. 2018; Struck et al. 2023). Flatworms exhibit a wide variation in longevity: whereas monogeneans on average have very short life spans of several weeks (Bakke et al. 2007), trematodes have maximum lifespans of up to 25 years (Muller and Wakelin 2002), and some non-parasitic planarians are practically somatically immortal (Valenzano et al. 2017). Furthermore, it has been proposed that parasite longevity should be inversely correlated to the thermic environment of the host: at higher temperatures, parasites invest in higher metabolic and reproductive rates, but a shorter life span appears to be an unavoidable trade-off (Bakke et al. 2007). We tested both hypotheses in flatworms (Hypothesis 5: Longevity-dependent selection; Table 1).
Parasitic bilaterian animal lineages exhibit highly elevated mitogenomic evolutionary rates in comparison to non-parasitic lineages, putatively due to the reduced locomotory capacity and metabolic dependence on the host (Jakovlić et al. 2023). Along with Nematoda, Platyhelminthes comprise the largest and medically most important radiation of parasitic animals (Neodermata). The phylum also comprises a substantial (paraphyletic) radiation of predominantly non-parasitic lineages, which makes it one of the few animal phyla that comprise both parasitic and nonparasitic lineages. We tested the impact of parasitism on branch length in flatworms (Hypothesis 6: Parasitism; Table 1).
In addition to the over 20 flatworm mitogenomes so far sequenced by our research team (Supplementary file S1: Table S1), we retrieved all other available flatworm mitogenomes from public databases, adding up to over 200 flatworm species. We divided them into multiple subdatasets according to the lifestyle (parasitic and non-parasitic) and host’s thermic type (endothermic and ectothermic), and relied on multilevel regression models and causal inference to test the six hypotheses outlined above (Table 1).
Materials and Methods
Datasets
We retrieved all flatworm mitogenomes available in the NCBI’s GenBank database (last accessed 25/2/2022), and used PhyloSuite v.1.2.3 (Zhang et al. 2020; Xiang et al. 2023) to extract data from GenBank files. Mitogenomes were available for 223 species, 30 of which belonged to non-parasitic lineages (Supplementary file S2: Dataset S1), comprising the paraphyletic basal flatworm radiation previously classified as “turbellaria” (Rohde 1994). Analyses were conducted on three datasets: 1) full (comprising all Platyhelminthes); 2) non-parasitic (“turbellaria”), and 3) parasitic (Neodermata). We classified parasites according to the definitive host’s thermic type into endotherms (124 species) and ectotherms (55 species). We additionally tested the impact of the intermediate host by further subdividing them according to definitive and intermediate host types into ectotherm-ectotherm (ecto-ecto), endotherm-endotherm (endo-endo), and ectotherm-endotherm (ecto-endo) groups respectively. As the major parasitic radiation Neodermata is monophyletic, we tested hypothesis 6 (Parasitism) with this limitation in mind. All other tested categories comprised multiple lineages of paraphyletic origins. To minimise the impact of sequencing artefacts on the analysis, for all analyses involving the mitogenome size parameter, we removed putatively incomplete mitogenomes (marked as “linear” in GenBank files; 182 species left in the dataset; more details in Supplementary file S1: Text S1). Longevity data were retrieved from a range of sources, listed in Supplementary file S2: Dataset S1. Given the absence of precise longevity data for many flatworm lineages, we first categorised it into six categories, and then coded it as a mean of a range in days: 15 (0–30 days), 105 (30–180 days), 270 (6 months - 1 year), 547 (1–2 years), 1278 (2–5 years), and 2500 (>5 years).
Phylogenetic analyses
PhyloSuite was used to conduct phylogenetic analyses with the help of its plug-in programs using the methodology described before (Jakovlić et al. 2021). We used datasets comprising concatenated amino acid (AAs) sequences of all 12 (atp8 is missing from Neodermata) mitochondrial PCGs. Sequences were aligned using the normal mode of MAFFT (Katoh and Standley 2013) available in PhyloSuite. The amino acid alignments of PCGs were trimmed using trimAI (Capella-Gutiérrez et al. 2009), optimal evolutionary model and mixture models (CAT model, c10…c60) inferred using ModelFinder (Kalyaanamoorthy et al. 2017), and phylogeny inferred using the mtInv+C50 + F + R10 model (C50 can account for compositional heterogeneity) in IQ-TREE, with 50,000 Ultrafast bootstraps (Minh et al. 2020). The most likely sister group to Platyhelminthes, Gastrotricha (Giribet et al. 2000; Struck et al. 2014; Laumer et al. 2019), was used as the outgroup (Nemertea was also tested). We tested and confirmed that branch lengths are highly correlated regardless of the dataset, algorithm, or outgroup used (more details in Supplementary file S1: Text S2). The sequence evolution rate was defined as the root-to-tip sum of branch lengths (Lanfear et al. 2007; Allio et al. 2017), which were extracted using the TreeSuite function of PhyloSuite. The phylogeny was visualised using iTOL (Letunic and Bork 2024) and files generated by PhyloSuite.
Statistical, gene order rearrangement rate, selection pressure, and multivariate regression analyses
To infer the gene order rearrangement rate, we first inferred the putative ancestral gene order for Platyhelminthes using the SPP algorithm of MLGO (Hu et al. 2014) and phylogeny with Gastrotricha as the outgroup, and then used it to calculate distances from it for all species in the dataset using the breakpoint distance score in CREx (Bernt et al. 2007). To test the reliability of this inference, we also used Nemertea as the outgroup. As the ancestral gene orders differed, we confirmed that both ancestral gene orders, as well as the ancestral gene arrangement of Lophotrochozoa (Podsiadlowski et al. 2009), all produce the same conclusions (Supplementary file S1: Text S3). We used the AnalyzeCodonData.bf function in HyPhy (Kosakovsky Pond et al. 2020) to infer branch-specific ω values (dN/dS; details in Supplementary file S1: Text S3) on a dataset comprising concatenated 12 PCGs of all mitogenomes, with the best-fit GY codon model selected by ModelFinder (Kalyaanamoorthy et al. 2017). Selective pressures in target lineages were further tested using the RELAX function in HyPhy, where K > 1 values indicate intensification and K < 1 relaxation of selection (Wertheim et al. 2015). GC skews were proposed as a proxy for estimating the strength of purifying selection pressure in mitogenomes (Jakovlić et al. 2021), so we calculated them using PhyloSuite and the formula (G-C)/(G + C) (Perna and Kocher 1995).
Standard statistical analyses were conducted in R. After conducting standard ANOVA to compare means among groups, for comparisons between specific pairs of groups we used a version of the t-test that controls for multiple comparisons - Tukey’s HSD. As our data violated the assumption of independence of observations, we also applied the ANOVA phylogenetic generalised least squares (PGLS) in the nlme package (Pinheiro and Bates 2006; Pinheiro et al. 2024) to conduct pairwise and multigroup comparisons. For correlation analyses, we used Pearson’s (in cases when we compared two measurement variables), Spearman’s (ranked + measurement), and Polyserial (category + measurement). Multilevel regression analyses were conducted using two different models that can remove the effect of the evolutionary relationships of species when fitting a regression between variables: brms (Bürkner 2018) and lmekin (Therneau 2018). For these, we used a matrix of phylogenetic distances extracted from the phylogram using TreeSuite. To conduct the causal inference analyses, we used dagitty (Textor et al. 2016), and then calculated causal effects using brms and lmekin following the methodology outlined in (Arif et al. 2022) (details in Supplementary file S1: Text S3).
Results
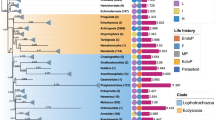
Topologies, mitogenomic architecture and studied variables can be viewed in Fig. 1 and, with details of all studied parameters (exact p-values, etc.), alignments, trees, gene order distance matrices, and alternative topologies provided in Supplementary files S2 – S5. We relied on branch lengths (brl) as indicators of overall evolutionary rates, so we confirmed that they were highly correlated between phylograms regardless of the dataset, methodology, or the outgroup used (all >0.97) (Supplementary file S1: Text S3 and Figure S1; Supplementary file S6). The two multilevel regression algorithms, brms and lmekin, also produced highly correlated results (correlation = 0.981).

Centre-to-edge, the image shows IQ-TREE_NUC phylogram where stars at branches indicate 100% IQ-TREE ultrafast bootstrap support, and with the two basal stems that were removed for some analyses highlighted; species names with GenBank accession numbers and taxonomic identity as coloured ranges; the division of lineages into non-parasitic and parasitic, with the latter further subdivided into endotherms and ectotherms according to the thermic type of definitive (the inner circle) and intermediate (the outer circle) hosts (Life history); longevity; branch length; gene order rearrangement rate (GORR); and the mitogenome size, where 0 was set at 12,000 bp (base pairs). The tree was rooted with one Gastrotricha species.
Hypothesis 1: Functional constraints (thermic habitat and sequence evolution)
Branch lengths were nonsignificantly (p-value = 0.892) higher in parasites of ectotherms compared to parasites of endotherms (Fig. 2A; details of statistical analyses in Supplementary file S2: Dataset S2). When the dataset was divided according to the intermediate host, parasites with both (intermediate and definitive) endothermic hosts exhibited significantly (p = 0) longer branches (Supplementary file S1: Fig. S4). The ω values were nonsignificantly higher (p = 0.057; Fig. 2B) and purifying selection pressure significantly relaxed in endotherms compared to ectotherms (p < 0.0001, K = 0.8). However, multivariate regression analyses indicated that thermic host type was not a significant predictor of the branch length variability (R2 ≤ 1%; Table 2; details of multivariate analyses in Supplementary file S2: Dataset S3).

Neodermata (parasites) were grouped in two ways: by taxonomic lineages and by the thermic type of definitive host (Life history); the corresponding labels are shown beneath the panels, and the parameter compared is shown on the y-axis. In the ‘longevity’ dataset, all flatworms were grouped into six categories according to their maximum life span. Brl is branch length, where “stem” indicates that the ancestral stem branch was removed (see Fig. 1). Omega is the ratio of non-synonymous to synonymous mutations (ω = dN/dS). GORR is the gene order rearrangement rate. The PGLS ANOVA parameter shows the result of ANOVA analysis controlled for phylogenetic relationships among the species using nlme, where *p < 0.05 and ***p < 0.001. Statistically significant (p < 0.05) differences in Tukey HSD comparisons between pairs of groups are indicated by different letters above the box plots.
Hypothesis 2: Race for replication (thermic habitat and mitogenome size)
Mitogenomes were nonsignificantly smaller in parasites of endotherms than in ectotherms (by ≈300 bp; p = 0.62), and the largest in non-parasitic species (Fig. 2C). The intermediate host type did not affect the size (<50 bp difference, p = 0.82; Supplementary file S1: Figs. S4). The ectotherm size range was marginally narrower than the endotherm range (13.4–17.5 Kbp vs. 13.4–17.7 Kbp respectively), with non-parasitic lineages exhibiting by far the widest range: 14.2–27.1 Kbp (outliers were included for all range analyses). The host’s thermic environment explained ≈7% of the mitogenome size variability (a significant predictor; Table 2). When covariates were accounted for in the causal analysis, the causal effect was −0.04 (Fig. 3).

The tested exposure variables are shown on the y-axis. For the first analysis, we used the full Platyhelminthes dataset (data_full_scm) and for the other two, we used the parasitic Neodermata dataset (data_parasitic_scm). Parameter estimates show standardised posterior means and 95% CI (credible intervals). GC is GC skew, omega is the dN/dS ratio, and GORR is the gene order rearrangement rate. “Parasitism” indicates that the Platyhelminthes dataset was divided into non-parasitic turbellaria (0) and parasitic Neodermata (1), and “thermal” indicates that Neodermata were divided into lineages with endothermic (1) and ectothermic (0) hosts.
Hypothesis 3: Mitogenome size and gene order rearrangement rate
Mitogenomes were significantly larger and more rearranged in turbellaria than in Neodermata (both p = 0; Fig. 2C, D). We also confirmed that alternative ancestral gene order patterns produce congruent results (Supplementary file S1: Figure S4). As a result, across the Platyhelminthes, the mitogenome size was significantly positively correlated with the gene order rearrangement rate (0.58), and mitogenome size explained ≈22–24% of the variability in the gene order rearrangement rate, and vice versa (Fig. 4, Table 2). When covariates were accounted for, the causal effect of GORR on mitogenome size was 0.26 (Fig. 3). A similar correlation (0.46) and predictive power (R2) of 20–25% was observed in Neodermata, but the two variables were not correlated in Turbellaria (0.08 and <5% respectively) (Supplementary file S1: Figures S5 and S6).

Above-right of the diagonal are correlation values, also shown as a correlation heatmap (0 = grey; 1 = red, −1 = blue). Below-left of the diagonal are data plots, where dotted/solid trendlines represent nonsignificant/significant (P > 0.05/P < 0.05) correlations respectively. The units on the axes correspond to the variables being compared (genome size = base pairs, longevity = days). G_brl is branch length and G_GORR is gene order rearrangement rate (Gastrotricha was used as the outgroup in both cases).
Hypothesis 4: Sequence evolution and mitogenome size/gene order rearrangement rate
Branch lengths were negatively correlated with gene order rearrangement rate and mitogenome size in Platyhelminthes (−0.74 and −0.29; Fig. 4), but positively in Neodermata (0.50 and 0.27) and turbellaria (0.36 and 0.24) subdatasets, respectively (Supplementary file S1: Figures S5 and S6). This discrepancy was caused by higher gene order rearrangement rates and much shorter branches (both p = 0) in non-parasitic lineages than in parasitic lineages (Fig. 2A, D). Despite this discrepancy, gene order rearrangement rate was a significant predictor of branch length in all three datasets: 57–62% in Platyhelminthes, 30–32% in Neodermata, and ≈14% in turbellaria datasets (Table 2). In all three datasets, mitogenome size explained a smaller but similar proportion (≈7–11%) of branch length variability, with inconsistent significance (Table 2; Supplementary file S2: Dataset S3).
Hypothesis 5: Longevity-dependent selection
Longevity was the lowest in Monopisthocotylea, higher in endotherms than in ectotherms, and had a very wide range in turbellaria (Fig. 2E, F). By far the shortest average branch lengths were exhibited by the relatively short-living category 2, which also comprised most of the turbellaria, whereas all other categories had similar values (Fig. 2G). As a result, longevity was not correlated with branch length in Platyhelminthes and Neodermata (both R2 < 1%), but the two variables were weakly positively correlated in turbellaria (0.16, R2 = 11–12%; Fig. 4, Table 2, Supplementary file S1: Figures S5 and S6, Supplementary file S2: Dataset S3). When covariates were accounted for in the causal analysis, the causal effect was 0.04 (Fig. 3).
Hypothesis 6: Parasitism
Neodermata had more than twice as long (p < 0.001) branches as turbellaria (Fig. 2H). As a result, “parasitism” explained ≈ 90–96% of the branch length variability (Table 2), with a correspondingly large causal effect (0.95; Fig. 3). However, the phylogram indicated that Neodermata had a disproportionately long basal stem, comprising 63.6% of the total average branch length. When we removed it along with the basal stem of the main clade of turbellaria (as the latter clade is paraphyletic, the stem branch was not removed for Catenulida; Fig. 1), turbellaria still had significantly shorter branches, but the difference was much smaller (2.15 vs. 2.27, p = 0.026). The difference was largely driven by the long branch of Polyopisthocotylea (p = 0), as other lineages of Neodermata did not have significantly longer branches than turbellaria (Fig. 2I). We further attempted to look at a number of different parameters that might help us distinguish different selection pressures (purifying vs. directional) from mutation pressure. GC skews were the lowest in turbellaria, and they exhibited a significant correlation with the branch length (0.46). Their explanatory power on branch length was significant but varied strongly across the three datasets: 22–25% in Platyhelminthes, ≈5% in Neodermata; and 36–41% in turbellaria (Fig. 2J; Fig. 4; Table 2; Supplementary file S1: Figs. S4–S6; Supplementary file S6; Supplementary file S2: Dataset S3). The purifying selection was significantly intensified in turbellaria, and relaxed in Neodermata (both p < 0.0001; Kparasitic = 0.90, Knon-parasitic = 1.06). However, ω values did not vary significantly between the two (Fig. 2B), and they were a nonsignificant predictor of branch length in Platyhelminthes and Neodermata (≤4%), but significant in turbellaria (45–50%). dN exhibited a marginally lower predictive power than dS on branch length in Platyhelminthes (≈6% vs. ≈8% respectively), but much higher in Neodermata (≈6% vs. ≈1%) and turbellaria (≈31–35% vs. <4%) (Table 2; Supplementary file S2: Dataset S3).
Discussion
The impact of the host’s thermic environment on sequence evolution and mitogenome size of parasites is small and inconsistent
Similar to the previous observation in nematodes (Lagisz et al. 2013), we found some indications in support of the first working hypothesis (“functional constraints”), such as higher GC skews and relaxed purifying selection pressure in parasites of endothermic hosts, but the effects on branch lengths were minimal and statistical significance of differences between parameters inconsistent. Notably, the effect was stronger in parasites with endothermic intermediate and definitive hosts. As most parasites of endothermic hosts have life history stages during which they are not confined to the thermally stable environment of the host, this may limit the scope of the relaxation of purifying selection pressures.
In agreement with our predictions, indications in support of the “race for replication” hypothesis were weak and statistically largely nonsignificant, and the size range contradicted the predictions of this hypothesis. In addition, mitogenomes were the largest and size most variable in non-parasitic species, which we would expect to have higher metabolic demands compared to parasites due to their higher locomotory capacity and metabolic requirements (Jakovlić et al. 2023). Notably, large mitogenomic noncoding regions often possess repetitive sequences interfering with sequencing and amplification, so this analysis was highly susceptible to sequencing artifacts. Finally, parasites of endotherm hosts had a longer average life span than parasites of ectotherm hosts, which contradicts the hypothesis of a reduced life span at higher temperatures (Bakke et al. 2007). These findings contradict the results of (Lagisz et al. 2013), and indicate that putative differences in metabolic rates between flatworm parasites of endothermic and ectothermic hosts are either non-existent or insufficiently pronounced to have a significant impact on branch length and mitogenome size. Additional discussion in Supplementary file S1: Text S4.
Mitogenomic architecture rearrangements are positively correlated with mitogenomic size in flatworms
We found relatively consistent support for this hypothesis in Platyhelminthes and Neodermata, but not in turbellaria. Notably, the latter analysis was weakened by a comparatively small number of samples. In Nematoda, exceptionally large mitogenomes are commonly associated with a hypervariable structure (Hyman et al. 2011; Zou et al. 2022b), and a study found that compact mitogenomes were more structurally stable in nematodes (Lagisz et al. 2013). However, this is not a rule, as a recent study found that a lineage of enoplean nematodes, Longidoridae, exhibits exceptionally small mitogenomes with rapidly evolving gene order rearrangements (Zou et al. 2022a). A potential explanation for this inconsistency is that some mitogenomic rearrangement mechanisms may not produce sequence duplications (Lunt and Hyman 1997), and that ancient rearrangements may have a negligible impact on the mitogenome size: if mitogenomes are evolving under pressure for a smaller size, purifying selection may eventually remove all non-functional remnants of ancient duplications. This would imply the existence of a correlation at small evolutionary scales and the absence at large scales across bilaterian animals, but patterns discussed above contradict this, with correlation apparently breaking down in isolated lineages at smaller phylogenetic scales. This hypothesis requires further testing.
The long stem branch of Neodermata affects the relationships between sequence evolution, GORR, and longevity
We found strong support for the hypothesis that parasitic lineages evolve faster in flatworms. The difference was striking, as Neodermata exhibited more than twice longer branches than “turbellaria”, which was reflected in a huge predictive power of the categorisation into parasitic and non-parasitic lineages. However, this difference was mostly caused by the exceptionally long stem branch of Neodermata, which prevents us from excluding factors other than nominal parasitism as the major drivers of elevated evolutionary rates of Neodermata. As parasitic flatworm lineages included in our study were monophyletic, we removed the basal stem, re-conducted the analyses, and found that Polyopisthocotylea was the only neodermatan lineage exhibiting significantly longer branches than turbellaria. Notably, this lineage also exhibits a disproportionately elongated stem branch in mitogenomic phylogenies (Zhang et al. 2024). Under the hypothesis that the common ancestor of Neodermata appeared ≈513 million years ago (Perkins 2010), this would imply that the ancestral lineage of contemporary Neodermata underwent a period of highly elevated evolutionary rates in the period following the “Cambrian explosion”. Phylogenies inferred using nuclear genomic data also produced a relatively long stem branch (albeit not as extreme), especially when the CAT-GTR model was applied (Brabec et al. 2023). This discrepancy in the ratio of stem branch to the total branch length between mtDNA and nDNA data could be an artefact caused by the absence of Bothrioplanidae, the most likely sister group to Neodermata (Laumer and Giribet 2014; Brabec et al. 2023), from our dataset. This was likely to result in the overestimation of the stem branch length inferred using mitogenomic data, so an improved turbellarian sampling might produce more congruent results between the two genomic compartments. Despite the discrepancy, both genomic compartments indicate that the transition to obligatory parasitism in the ancestral Neodermata lineage was putatively marked by a long period of high extinction rates, selective sweeps, and population bottlenecks, but this hypothesis needs to be further confirmed using nuclear genomic data.
This scenario might also explain the apparent negative correlation between sequence evolution and gene order rearrangements across the Platyhelminthes. The absence of similarity between the ancestral gene order for Neodermata (Zhang et al. 2019) and gene arrangements of both turbellaria (Solà et al. 2015) and the ancestral gene order of Lophotrochozoa (Podsiadlowski et al. 2009) indicates that the ancestral neodermatans simultaneously underwent a period of elevated architecture and sequence evolution rates. Following this birthing period marked by high evolutionary rates, Neodermata subsequently underwent a remarkably successful radiation into a huge lineage of obligate parasites of practically all vertebrates and many invertebrates (Poulin and Morand 2000; Hahn et al. 2014). If this was reflected in strongly increasing population sizes, it might explain the reestablishment of stringent purifying selection pressures, thus slowing down the sequence evolution and largely restabilising the mitogenomic architecture. This is in agreement with the observation in spirurine nematodes, where sequence and architecture evolution also exhibit episodic but synchronised evolutionary patterns (Zou et al. 2022b). This episodic evolution generated a pattern wherein Neodermata exhibit the fastest sequence evolution rates among all bilaterian lineages (Jakovlić et al. 2023), while their mitogenomic architecture remained largely unchanged over hundreds of millions of years (Zhang et al. 2019). As the lineages of free-living turbellaria continued undergoing occasional architectural rearrangements (Solà et al. 2015; Rosa et al. 2017), this resulted in comparatively highly rearranged and large mitogenomes. Due to their short branches, this caused an inversed relationship between gene order rearrangement rates and branch length across the Platyhelminthes. Indeed, a positive correlation between these two parameters emerged within Neodermata and turbellaria when we analysed them separately, indicating that purifying selection pressures probably do play a major role in the evolutionary dynamics of these two variables, but stochastic and episodic nature of rearrangement events can disrupt their correlation in some cases.
Similarly, the long stem branch may also explain the absence of the explanatory power of longevity at the Platyhelminthes level. As most mutations in the dataset map to the ancestral neodermatans of unknown longevity, this may have strongly reduced the predictive power of this variable. In addition, generation time might be decoupled from longevity in flatworms, thus further confounding the correlation between longevity and sequence evolution (see Supplementary file S1: Text S5 for additional discussion).
In addition, if the ancestral Neodermata lineage underwent a long evolutionary period of extreme bottlenecks and/or selective sweeps during the transition from nonparasitic to parasitic life history strategies, followed by a strong subsequent increase in the effective population size (Ne) this may have decoupled this variable from the overall branch length. This indicates that our analyses may have overestimated the impact of parasitism, and underestimated the impact of Ne (or some other variable not tested herein) on mitogenomic sequence evolution. We attempted to test the impact of Ne, but due to several sources of noise, the results were deemed insufficiently reliable to be included in the main study (see Supplementary file S1: Text S6).
Conclusions
There are multiple limitations of our study, including the limited scope of a single phylum, relatively poor coverage of turbellarian lineages, difficulties in the inference of ancestral mitogenomic architecture for Platyhelminthes, and the absence of several variables previously associated with mitogenomic evolution, such as Ne, locomotory capacity, generation time, the number of germ-line divisions per generation, etc. (Thomas et al. 2006; Saclier et al. 2018; Jakovlić et al. 2023). With these limitations in mind, our results offer a promising explanation for the reasons underlying the recognized difficulties in the prediction of patterns of mitogenomic evolution (Bazin et al. 2006; Thomas et al. 2006; Nabholz et al. 2008b; Allio et al. 2017; Jakovlić et al. 2021). The fact that a large proportion of substitutions in the mitogenomes of Neodermata was accumulated early in the evolutionary history of Neodermata strongly confounded our attempts to parse the impacts of different variables on mitogenomic evolution. In addition, we show that the impacts of many variables often strongly vary across different lineages, which may also help explain inconsistent results across different studies. We outlined a scenario that explains how episodic evolutionary patterns deep in the evolutionary history of Neodermata may have produced contradictory patterns across different flatworm lineages, as well as reduced the predictive power of some variables and overestimated the impact of parasitism on the evolutionary rate. In addition, the volatility of correlations and predictive powers of variables across different datasets make us suspect that pronounced differences in average branch lengths and other parameters between lineages may have resulted in numerous statistical flukes, comprising both type 1 and 2 errors. This stresses the importance of thorough testing of the robustness of statistical significance findings in such studies. Finally, the results also highlight the importance of accounting for the episodic nature of evolutionary patterns (Gillespie 1984) and relying on comprehensive approaches in studies of mitogenomic evolution.
Data archiving
All relevant data are included within the manuscript and its supplementary files. GenBank Accession Numbers for all mitogenomes used in the analyses are available in Supplementary file S2: Dataset S1 (Column “ID”).
References
Allio R, Donega S, Galtier N, Nabholz B (2017) Large variation in the ratio of mitochondrial to nuclear mutation rate across animals: Implications for genetic diversity and the use of mitochondrial DNA as a molecular marker. Mol Biol Evol 34:2762–2772
Arif S, Graham NAJ, Wilson S, MacNeil MA (2022) Causal drivers of climate-mediated coral reef regime shifts. Ecosphere 13:e3956
Bakke TA, Cable J, Harris PD (2007) The biology of Gyrodactylid Monogeneans: The “Russian-Doll Killers”. In: Baker J. R., Muller R., Rollinson D. (eds) Advances in Parasitology, Academic Press Vol 64, pp 161–460.
Bazin E, Glémin S, Galtier N (2006) Population size does not influence mitochondrial genetic diversity in animals. Science 312:570–572
Benesh DP, Parker G, Chubb JC (2021) Life-cycle complexity in helminths: What are the benefits? Evolution 75:1936–1952
Bernt M, Bleidorn C, Braband A, Dambach J, Donath A, Fritzsch G et al. (2013) A comprehensive analysis of bilaterian mitochondrial genomes and phylogeny. Mol Phylogenet Evol 69:352–364
Bernt M, Merkle D, Ramsch K, Fritzsch G, Perseke M, Bernhard D et al. (2007) CREx: Inferring genomic rearrangements based on common intervals. Bioinformatics 23:2957–2958
Boore JL (2000) The Duplication/Random Loss Model for Gene Rearrangement Exemplified by Mitochondrial Genomes of Deuterostome Animals. In: Sankoff D., Nadeau J. H. (eds) Comparative Genomics: Empirical and Analytical Approaches to Gene Order Dynamics, Map Alignment and the Evolution of Gene Families, Springer Netherlands: Dordrecht, pp 133–147
Brabec J, Salomaki ED, Kolísko M, Scholz T, Kuchta R (2023) The evolution of endoparasitism and complex life cycles in parasitic Platyhelminths. Curr Biol 33:4269–4275.e3
Bürkner P-C (2018) Advanced Bayesian multilevel modeling with the R Package brms. R J 10:395–411
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T (2009) trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973
Chong RA, Mueller RL (2013) Evolution along the mutation gradient in the dynamic mitochondrial genome of salamanders. Genome Biol Evol 5:1652–1660
Galtier N, Jobson RW, Nabholz B, Glémin S, Blier PU (2009) Mitochondrial whims: metabolic rate, longevity and the rate of molecular evolution. Biol Lett 5:413–416
Gillespie JH (1984) The molecular clock may be an episodic clock. Proc Natl Acad Sci 81:8009–8013
Giribet G, Distel DL, Polz M, Sterrer W, Wheeler WC (2000) Triploblastic relationships with emphasis on the acoelomates and the position of Gnathostomulida, Cycliophora, Plathelminthes, and Chaetognatha: A Combined Approach of 18S rDNA Sequences and Morphology. Syst Biol 49:539–562
Hahn C, Fromm B, Bachmann L (2014) Comparative genomics of Flatworms (Platyhelminthes) reveals shared genomic features of ecto- and endoparastic Neodermata. Genome Biol Evol 6:1105–1117
Hassanin A (2006) Phylogeny of Arthropoda inferred from mitochondrial sequences: Strategies for limiting the misleading effects of multiple changes in pattern and rates of substitution. Mol Phylogenet Evol 38:100–116
Hu F, Lin Y, Tang J (2014) MLGO: phylogeny reconstruction and ancestral inference from gene-order data. BMC Bioinforma 15:354
Hua X, Cowman P, Warren D, Bromham L (2015) Longevity is linked to mitochondrial mutation rates in Rockfish: A test using Poisson Regression. Mol Biol Evol 32:2633–2645
Hyman BC, Lewis SC, Tang S, Wu Z (2011) Rampant gene rearrangement and haplotype hypervariation among nematode mitochondrial genomes. Genetica 139:611–615
Jackson JA, Tinsley RC (1998) Effects of temperature on oviposition rate in Protopolystoma xenopodis (Monogenea: Polystomatidae). Int J Parasitol 28:309–315
Jakovlić I, Zou H, Chen J-H, Lei H-P, Wang G-T, Liu J et al. (2021) Slow crabs - fast genomes: locomotory capacity predicts skew magnitude in crustacean mitogenomes. Mol Ecol 30:5488–5502
Jakovlić I, Zou H, Ye T, Zhang H, Liu X, Xiang C-Y et al. (2023) Mitogenomic evolutionary rates in Bilateria are influenced by parasitic lifestyle and locomotory capacity. Nat Commun 14:6307
Jeschke JM, Kokko H (2009) The roles of body size and phylogeny in fast and slow life histories. Evol Ecol 23:867–878
Kalyaanamoorthy S, Minh BQ, Wong TKF, Von Haeseler A, Jermiin LS (2017) ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat Methods 14:587–589
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol Biol Evol 30:772–780
Kosakovsky Pond SL, Poon AFY, Velazquez R, Weaver S, Hepler NL, Murrell B et al. (2020) HyPhy 2.5—A customizable platform for evolutionary hypothesis testing using Phylogenies. Mol Biol Evol 37:295–299
Lagisz M, Poulin R, Nakagawa S (2013) You are where you live: parasitic nematode mitochondrial genome size is associated with the thermal environment generated by hosts. J Evol Biol 26:683–690
Lajbner Z, Pnini R, Camus MF, Miller J, Dowling DK (2018) Experimental evidence that thermal selection shapes mitochondrial genome evolution. Sci Rep. 8:9500
Lanfear R, Thomas JA, Welch JJ, Brey T, Bromham L (2007) Metabolic rate does not calibrate the molecular clock. Proc Natl Acad Sci 104:15388–15393
Laumer CE, Fernández R, Lemer S, Combosch D, Kocot KM, Riesgo A et al. (2019) Revisiting metazoan phylogeny with genomic sampling of all phyla. Proc R Soc B Biol Sci 286:20190831
Laumer CE, Giribet G (2014) Inclusive taxon sampling suggests a single, stepwise origin of ectolecithality in Platyhelminthes. Biol J Linn Soc 111:570–588
Letunic I, Bork P (2024) Interactive Tree of Life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res 52:W78–W82
Li W-H, Tanimura M, Sharp PM (1987) An evaluation of the molecular clock hypothesis using mammalian DNA sequences. J Mol Evol 25:330–342
Lunt DH, Hyman BC (1997) Animal mitochondrial DNA recombination. Nature 387:247–247
Lynch M, Koskella B, Schaack S (2006) Mutation pressure and the evolution of organelle genomic architecture. Science 311:1727–1730
Martin AP, Palumbi SR (1993) Body size, metabolic rate, generation time, and the molecular clock. Proc Natl Acad Sci 90:4087–4091
Min XJ, Hickey DA (2008) An evolutionary footprint of age-related natural selection in mitochondrial DNA. J Mol Evol 67:412
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A et al. (2020) IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol 37:1530–1534
Muller R, Wakelin D (2002) Worms and human disease, 2nd edn. CABi Publishing: Wallingford, UK.
Nabholz B, Glémin S, Galtier N (2008a) Strong variations of mitochondrial mutation rate across mammals—the longevity hypothesis. Mol Biol Evol 25:120–130
Nabholz B, Glémin S, Galtier N (2009) The erratic mitochondrial clock: variations of mutation rate, not population size, affect mtDNA diversity across birds and mammals. BMC Evol Biol 9:54
Nabholz B, Lanfear R, Fuchs J (2016) Body mass-corrected molecular rate for bird mitochondrial DNA. Mol Ecol 25:4438–4449
Nabholz B, Mauffrey J-F, Bazin E, Galtier N, Glemin S (2008b) Determination of mitochondrial genetic diversity in mammals. Genetics 178:351–361
Perkins E (2010) Family ties: molecular phylogenetics, evolution and radiation of flatworm parasites (Monogenea: capsalidae). PhD Thesis, University of Adelaide, School of Earth and Environmental Sciences
Perna NT, Kocher TD (1995) Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol 41:353–358
Pinheiro J, Bates D (2006) Mixed-effects models in S and S-PLUS, 1st edn. Springer Science & Business Media: New York, NY.
Pinheiro J, Bates D, R Core Team (2024) nlme: Linear and Nonlinear Mixed Effects Models (https://CRAN.R-project.org/package=nlme)
Podsiadlowski L, Braband A, Struck TH, von Döhren J, Bartolomaeus T (2009) Phylogeny and mitochondrial gene order variation in Lophotrochozoa in the light of new mitogenomic data from Nemertea. BMC Genomics 10:364
Poulin R, Latham ADM (2003) Effects of initial (larval) size and host body temperature on growth in trematodes. Can J Zool 81:574–581
Poulin R, Morand S (2000) The diversity of parasites. Q Rev Biol 75:277–293
Rand DM (1993) Endotherms, ectotherms, and mitochondrial genome-size variation. J Mol Evol 37:281–295
Rand DM (1994) Thermal habit, metabolic rate and the evolution of mitochondrial DNA. Trends Ecol Evol 9:125–131
Rohde K (1994) The origins of parasitism in the Platyhelminthes. Int J Parasitol 24:1099–1115
Rosa MT, Oliveira DS, Loreto ELS (2017) Characterization of the first mitochondrial genome of a catenulid flatworm: Stenostomum leucops (Platyhelminthes). J Zool Syst Evol Res 55:98–105
Saclier N, François CM, Konecny-Dupré L, Lartillot N, Guéguen L, Duret L et al. (2018) Life history traits impact the nuclear rate of substitution but not the mitochondrial rate in isopods. Mol Biol Evol 35:2900–2912
Shao R, Dowton M, Murrell A, Barker SC (2003) Rates of gene rearrangement and nucleotide substitution are correlated in the mitochondrial genomes of insects. Mol Biol Evol 20:1612–1619
Shtolz N, Mishmar D (2023) The metazoan landscape of mitochondrial DNA gene order and content is shaped by selection and affects mitochondrial transcription. Commun Biol 6:1–15
Solà E, Álvarez-Presas M, Frías-López C, Littlewood DTJ, Rozas J, Riutort M (2015) Evolutionary analysis of mitogenomes from parasitic and free-living flatworms. PLOS ONE 10:e0120081
Struck TH, Golombek A, Hoesel C, Dimitrov D, Elgetany AH (2023) Mitochondrial genome evolution in Annelida—A systematic study on conservative and variable gene orders and the factors influencing its evolution. Syst Biol 72:925–945
Struck TH, Wey-Fabrizius AR, Golombek A, Hering L, Weigert A, Bleidorn C et al. (2014) Platyzoan paraphyly based on phylogenomic data supports a noncoelomate ancestry of Spiralia. Mol Biol Evol 31:1833–1849
Tan MH, Gan HM, Lee YP, Bracken-Grissom H, Chan T-Y, Miller AD et al. (2019) Comparative mitogenomics of the Decapoda reveals evolutionary heterogeneity in architecture and composition. Sci Rep. 9:1–16
Textor J, van der Zander B, Gilthorpe MS, Liśkiewicz M, Ellison GT (2016) Robust causal inference using directed acyclic graphs: the R package ‘dagitty’. Int J Epidemiol 45:1887–1894
Therneau T (2018) The lmekin function (https://cran.r-project.org/web/packages/coxme/vignettes/lmekin.pdf) (Accessed 17.7.2024.)
Thomas JA, Welch JJ, Lanfear R, Bromham L (2010) A generation time effect on the rate of molecular evolution in invertebrates. Mol Biol Evol 27:1173–1180
Thomas JA, Welch JJ, Woolfit M, Bromham L (2006) There is no universal molecular clock for invertebrates, but rate variation does not scale with body size. Proc Natl Acad Sci 103:7366–7371
Valenzano DR, Aboobaker A, Seluanov A, Gorbunova V (2017) Non-canonical aging model systems and why we need them. EMBO J 36:959–963
Welch JJ, Bininda-Emonds OR, Bromham L (2008) Correlates of substitution rate variation in mammalian protein-coding sequences. BMC Evol Biol 8:53
Wertheim JO, Murrell B, Smith MD, Kosakovsky Pond SL, Scheffler K (2015) RELAX: Detecting relaxed selection in a phylogenetic framework. Mol Biol Evol 32:820–832
Xiang C, Gao F, Jakovlić I, Lei H, Hu Y, Zhang H et al. (2023) Using PhyloSuite for molecular phylogeny and tree‐based analyses. iMeta 2:e87
Xu W, Jameson D, Tang B, Higgs PG (2006) The relationship between the rate of molecular evolution and the rate of genome rearrangement in animal mitochondrial genomes. J Mol Evol 63:375–392
Zhang D, Gao F, Jakovlić I, Zou H, Zhang J, Li WX et al. (2020) PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour 20:348–355
Zhang D, Jakovlić I, Zou H, Liu F, Xiang C-Y, Gusang Q et al. (2024) Strong mitonuclear discordance in the phylogeny of Neodermata and evolutionary rates of Polyopisthocotylea. Int J Parasitol 54:213–223
Zhang D, Li WX, Zou H, Wu SG, Li M, Jakovlić I et al. (2019) Homoplasy or plesiomorphy? Reconstruction of the evolutionary history of mitochondrial gene order rearrangements in the subphylum Neodermata. Int J Parasitol 49:819–829
Zou H, Chen F-L, Li W-X, Li M, Lei H-P, Zhang D et al. (2022a) Inverted base composition skews and discontinuous mitochondrial genome architecture evolution in the Enoplea (Nematoda). BMC Genomics 23:376
Zou H, Lei H-P, Chen R, Chen F-L, Li W-X, Li M et al. (2022b) Evolutionary rates of mitochondrial sequences and gene orders in Spirurina (Nematoda) are episodic but synchronised. Water Biol Secur 1:100033
Acknowledgements
This work was supported by the National Natural Science Foundation of China (32360927, 32102840); the Key Project of Natural Science Foundation of Tibet (XZ202301ZR0028G); the Open Fund Project of Key Laboratory of Breeding Biotechnology and Sustainable Aquaculture (2023FB07); and the Start-up Funds of Introduced Talent in Lanzhou University (561120206). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We would like to thank Dr. Xiang Liu for helpful discussions and technical assistance.
Author information
Authors and Affiliations
Contributions
IJ: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft. TY, FZ, YS, YM: formal analysis, investigation, methodology, writing – review & editing. HZ: formal analysis, resources, validation, visualization, writing – review & editing. GTW: formal analysis, resources, validation, visualization, supervision, writing – review & editing. WXL: validation, funding acquisition, supervision, writing – review & editing. ZD: data curation, formal analysis, investigation, methodology, resources, software, validation, visualization, writing – review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Research ethics statement
No approval of research ethics committees was required to accomplish the goals of this study because it was conducted on previously sequenced mitogenomes acquired from GenBank and included only unregulated invertebrate species.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Xiangjiang Zhan.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jakovlić, I., Ye, T., Zou, H. et al. Drivers of interlineage variability in mitogenomic evolutionary rates in Platyhelminthes. Heredity 133, 276–286 (2024). https://doi.org/10.1038/s41437-024-00712-2
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41437-024-00712-2
