
Abstract
In recent years, the increase in blood pressure at high altitudes has become an interesting topic among high-altitude researchers. In our animal studies using Wistar rats, we observed the existence of two rat populations that exhibit differential physiological responses during hypoxic exposure. These rats were classified as hypoxia-induced hypertensive rats and nonhypertensive rats. A decrease in nitric oxide levels was reported in different hypertension models associated with increased concentrations of asymmetric dimethylarginine (ADMA) and homocysteine, and we recently described an increase in arginase type II expression under hypoxia. ADMA and homocysteine decrease nitric oxide (NO) bioavailability; however, whether ADMA and homocysteine have a regulatory effect on arginase activity and therefore regulate another NO synthesis pathway is unknown. Therefore, the aim of this study was to measure basal ADMA and homocysteine levels in hypoxia-induced hypertensive rats and evaluate their effect on arginase II activity. Our results indicate that hypoxia-induced hypertensive rats presented lower nitric oxide concentrations than nonhypertensive rats, associated with higher concentrations of homocysteine and ADMA. Hypoxia-induced hypertensive rats also presented lower dimethylarginine dimethylaminohydrolase-2 and cystathionine β-synthase levels, which could explain the high ADMA and homocysteine levels. In addition, we observed that both homocysteine and ADMA had a significant effect on arginase II activation in the hypertensive rats. Therefore, we suggest that ADMA and homocysteine have dual regulatory effects on NO synthesis. The former has an inhibitory effect on eNOS, and the latter has a secondary activating effect on arginase II. We propose that arginase II is activated by AMDA and homocysteine in hypoxia-induced hypertensive rats.
Similar content being viewed by others

Introduction
Systemic arterial hypertension at high altitudes has become an interesting topic among high-altitude researchers. There have been conflicting reports on this issue, with some researchers reporting a slight increase in blood pressure soon after arrival at a high altitude [1, 2] and others reporting no such change [3, 4] or a decrease in blood pressure followed by an increase in blood pressure [5, 6]. In our animal studies using Wistar rats, we observed the existence of two groups of rats that exhibit differential physiological responses during hypoxic exposure [7]. These groups were classified as tolerant and intolerant to hypobaric hypoxia. Intolerant rats typically developed arterial hypertension with average pressure values of 183 ± 3.7 mmHg and hematocrit levels of 74.0 ± 4.0, leading to death when hypoxic exposure persisted. Therefore, rats that are intolerant to our chronic intermittent hypobaric hypoxia model constitute a hypertensive group. In contrast, tolerant rats did not develop arterial hypertension, and their physiological parameters were similar to those of control rats (normoxia), therefore, these rats constitute a nonhypertensive group.
Hypertensive responses can arise as a result of changes in endothelial function [8]. In this context, one of the common causes of deregulated endothelial vasodilator function is the alteration of nitric oxide (NO) synthesis pathways [9, 10]. Our previous results showed that hypoxia-induced hypertensive rats exhibited lower NO production, indicating that the regulation of NO synthesis is crucial to the development of hypertensive hypoxia [7]. In fact, various studies in hypertension models have described that downregulation of the NO synthetic pathway is accompanied by an increase in asymmetric dimethylarginine (ADMA) and homocysteine plasma levels [11,12,13,14,15].
ADMA and homocysteine are inhibitors of nitric oxide synthase (NOS), and ADMA has also been identified as a regulator of NO synthesis in vivo [16]. Increased levels of both of these metabolites are cardiovascular risk factors [14,15,16].
An increase in ADMA was described in rats and humans exposed to hypoxia [17] and was recently shown to be a predictor for the development of hypoxia-induced pulmonary hypertension [18]. On the other hand, we recently demonstrated that low baseline levels of ADMA and homocysteine in llamas (Lama glama), an animal genetically adapted to life at a high altitude, is an adaptive mechanism against the development of hypoxia-induced pulmonary hypertension [19].
Almost 80% of ADMA is degraded by a group of hydrolases called dimethylarginine dimethylaminohydrolases (DDAHs) [20]. There are two types of DDAHs known for their ability to hydrolyze ADMA: DDAH-1 and dimethylarginine dimethylaminohydrolase-2 (DDAH-2). Recently, we reported that DDAH-2 participates in ADMA regulation under hypoxic conditions [19]. In addition, cystathionine β-synthase (CBS) participates in the regulation of homocysteine degradation. Currently, both DDAH-2 and CBS are considered molecular risk targets for the development of cardiovascular diseases due to the existence of polymorphisms in their genes that dysregulate their metabolism [21, 22]. However, whether NO production can also be regulated by l-arginine plasma levels as a result of the competition between NOS and arginase has been less well studied [23,24,25].
Arginase plays an important role in the pathogenesis of several hypertension models. For instance, an increase in arginase activity was associated with several pulmonary and systemic hypertension models [26,27,28]. Similarly, we reported an increase in arginase II expression that led to the development of hypertension associated with decreased plasma NO in intolerant rats [7]. However, no scientific evidence regarding the effect of ADMA and homocysteine on the arginase pathway and the participation of the arginase pathway in the regulation of hypertensive processes is available. Therefore, the main objective of this study was to determine the baseline levels of ADMA and homocysteine in hypertensive and nonhypertensive rats induced by hypobaric hypoxia and to study the effect of ADMA and homocysteine on arginase type II activity. The study of new regulatory NO synthesis mechanisms will help to identify novel therapeutic targets for the pharmacological treatment of hypertension.
Materials and methods
Animal study groups
A total of 40 young and healthy Wistar rats (age: 3.0 ± 0.4 months; weight: 250 ± 25 g) were randomly divided into two groups: the hypobaric hypoxia (2 days of hypoxia, 2 days of normoxia, n = 30) and normoxia (n = 10) groups. Hypobaric hypoxia was simulated in a hypobaric chamber at 428 torr, which is equivalent to an altitude of 4600 m, maintained with an average internal flow of room air of 13.14 L/min (0.438 L/min of air per rat) at 22 ± 2 °C on a 12 h light–dark cycle. Rats were intermittently kept in the hypobaric chamber, spending 2 days inside the chamber (hypoxic conditions) followed by 2 days outside the chamber, for a period of 1 month (30 days). Rats in the control group were placed in the same room under the same conditions to which the hypoxic group was exposed (with the exception of hypoxia). Four animals were housed per cage, and the animals were provided food (10 g/day of pellets per rat) and water using bottles dispensers. The variables weight (W, g) and systolic blood pressure (SBP, mmHg) were measured at sea level under normoxic conditions immediately after the rats had been removed from the hypobaric chamber, starting with basal measurements and every 4 days thereafter for 1 month. Weight was measured using an Acculab V-1200 electronic balance 3 h after rats descended from the chamber to allow for hydration. Blood pressure and heart rate were measured using an inflatable tail-cuff and a pressure sensor (RTBP1003-220, Kent Scientific, and Torrington, CT, USA) as previously described [29]. The signal was sent through a preamplifier to a workbench data acquisition system, RTBP-001DS worksheet (Kent Scientific), and the average value of six consecutive measurements was calculated. Measurements were performed by placing the animals inside a movement-limiting Plexiglas chamber for 10–20 min.
Classification of hypoxia-induced hypertensive and nonhypertensive rats
Hypertension in rats in Wistar rats with adverse outcomes due to hypoxia was determined according to the predictive factors previously described in our laboratory [7]. Briefly, hypoxia-induced hypertensive rats were identified as those that developed arterial hypertension. The criteria used to determine whether rats were hypertensive were blood pressure increased by ≥20% compared to baseline, body weight loss ≥30% compared to baseline, and increased hematocrit ≥20% compared to baseline. Nonhypertensive rats were identified as those that did not develop arterial hypertension and exhibited an increase in body weight and increase in Hct of <20% compared to baseline. Rats were sacrificed when they met these criteria (average days of life: 30 days, n = 10). The rats were handled in accordance with guidelines for the humanitarian treatment of laboratory animals, and all experiments were conducted according to the NIH Guide for the Care and Use of Laboratory Animals (NIH publication No. 8023, revised 1978)
The animals were sacrificed using anesthesia, and the Ethics Committee at the Universidad Católica del Norte, Coquimbo-Chile, approved the design of the protocol.
Enzyme preparation
The hearts were excised, frozen using liquid nitrogen and stored at −80 °C until further use. Heart tissue was homogenized in a solution of 10 mM Tris-HCl (pH 7.5), 0.1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM DTT, and 5 mM EDTA and centrifuged at 5000 × g for 10 min at 4 °C. Subsequently, the enzyme solution (supernatant) was separated by chromatography on a DEAE-cellulose column equilibrated with 5 mM Tris-HCl (pH 7.5). The active eluted fractions obtained by washing the column corresponded to arginase I, and the fractions obtained with a linear gradient of Tris/KCl at 0.10–0.15 M corresponded to arginase II [30]. The presence of arginase I and II in the wash fraction and eluted fraction, respectively, was confirmed by Western blotting. Finally, the active fractions were pooled and stored at −20 °C.
Western blotting
All Western blotting was performed according to standard methods. Briefly, total aortic artery tissues were frozen in liquid nitrogen and then homogenized in homogenization buffer containing 50 mM Tris-HCl, 150 mM NaCl, 100 mM NaF, 1% Triton X-100, 1 mM dithiothreitol (DTT), 0.1 mM PMSF, 1 mM leupeptin, 0.02 M HEPES, 10−3 M EDTA, 10−3 M EGTA, and 20% glycerol, followed by centrifugation to 10000 ×g for 10 min. The supernatant was removed, and protein quantification was carried out by Bradford assay. Equal amounts of protein (25 μg) were resolved by 7.5–12% SDS-PAGE, and the proteins were transferred to PVDF membranes. Nonspecific binding to the membranes was blocked using 5% nonfat dry milk in TBS-T buffer (10 mM Tris-HCl, 150 mM NaCl, 0.05% Tween 20 (pH 7.4) for 1 h. Membranes were incubated with the primary polyclonal antibodies rabbit anti-eNOS, anti-iNOS, (1:500; Santa Cruz Biotechnology, Dallas, Texas, USA), anti-DDAH-2, anti-CBS (1:500; Abcam, Biosonda, Chile), anti-arginase type II (1:5000; Invitrogen, Thermo Scientific, Chile), anti-β actin, and anti-GDPH (1:5000; Sigma Chile Ltd., Chile); washed in TBS with 0.1% Tween; and incubated (1 h, 22 °C) in TBS/0.1% Tween containing horseradish peroxidase-conjugated goat antirabbit or anti-mouse secondary antibodies. Proteins were detected by enhanced chemiluminescence and quantified by densitometry using ImageJ (NIH). Expression levels of eNOS were normalized to GAPDH expression.
Measurement of arginase activity
Specific arginase activity in heart tissues was determined based on urea production using a colorimetric method with α-isonitrosopropiophenone [29]. Briefly, 50 μL of the fractions obtained from DEAE-cellulose chromatography were incubated in 30 mM Tris-HCl (pH 7.5) containing 10 mM l-arginine at 37 °C for 10 min. The reaction was stopped by the addition of 1 ml of an acidic mixture containing 9% (v/v) H3PO4 and 23% (v/v) H2PO4. To evaluate the effect of homocysteine and ADMA on arginase activity, a specific arginase activity assay was performed in the presence of ADMA and homocysteine at physiological concentrations (0.5, 1.0, 3.0, 6.0, and 10.0 μM) in a solution of 50 mM Tris-HCl (pH 7.5).
Protein assays
Total protein was determined by using the Bio-Rad protein assay (Bio-Rad CA, USA) with bovine serum albumin as a standard [31].
Collection of blood and plasma
A 2 ml blood sample was obtained by cardiac puncture under anesthesia (0.3 mg of ketamine). The plasma was then separated from the blood by centrifugation (4500 × g, 12 min, 4 °C) and stored at −80 °C until further analyses to measure ADMA, homocysteine and NO. After this procedure, the rat was comfortably placed in a separate cage under veterinary care. Once awake and able to mobilize, drink water, and eat food, the rat was moved to the chamber.
At the end of the protocol, the rats were euthanized with an overdose of ketamine (7 mg/k), and the hearts and aortic arteries were surgically removed, dissected, immediately frozen in liquid nitrogen and stored at −80 °C.
Detection of NO metabolites
Total plasma nitrate and nitrite (NOx) concentrations were determined using a kit based on the Griess reaction (Cayman Chemical). The kit enabled the detection of NOx (sum of nitrate and nitrite) by providing an index of the NO content. The results are reported as μmol/L NOx.
Measurement of ADMA and homocysteine plasma concentrations
Plasma levels of ADMA (Enzo Life Sciences, Farmingdale, New York, USA) and homocysteine (Alpco Diagnostics, Salem, New Hampshire, USA) were measured by enzyme-linked immunosorbent assay according to the manufacturer’s instructions.
Measurement of endothelial nitric oxide synthase activity
NOS activity was measured by assessing the conversion of L-[3H]-arginine to L-[3H]-citrulline [32]. Briefly, one volume of tissue extract from the aortic artery containing 300–400 mg of total protein in 20 mmol/L CHAPS was added to 200 μL of reaction mixture (50 mmol/L Tris buffer (pH 7.4) containing 10 μmol/L DTT, 10 μmol/L THB4, 10 μg/mL calmodulin, 1 mmol/L NADPH, 4 μmol/L flavin adenine dinucleotide, 4 mol/L flavin mononucleotide, 2 μmol/L l-arginine, and 10−3 mCi/mL l-[3H]-arginine. The assay was carried out at 37 °C and terminated with 2 mL of ice-cold stop buffer (20 mmol/L CH3COONa (pH 5.5) containing 2 mmol/L EDTA, 0.2 mmol/L EGTA, and 1 μmol/L l-citrulline). As l-[3H]-citrulline is neutral at pH 5.5, it was separated from the incubation mixture with cation exchange resin (Dowex AG 50W-X8; Bio-Rad) to which l-[3H]-arginine adhered. After elution with 2 mL of water, l-[3H]-citrulline was quantitated by 16 mL liquid scintillation counting (SL 3000; Intertechnique, Plaisir, France). Background radioactivity (as a percentage of total radioactivity) was measured to assess the quality of the l-[3H]-arginine stock (background radioactivity below 3%). Arginase activity was determined by measuring the formation of l-[3H]-citrulline from the homogenized aortic artery of nonhypertensive and hypoxia-induced hypertensive rats.
Extraction and quantification of oxide endothelial nitric synthase (eNOS), dimethyl-aminohydrolase (DDAH-2), cystathionine β-Synthase (CBS), and arginase Type II (ARG II) mRNA Levels by qRT-PCR
For qRT-PCR analysis, total RNA was prepared from the aortic artery tissue by homogenization of the sample in the presence of TRIzol reagent (Invitrogen Corp., Carlsbad, CA, USA). The RNA concentration and purity were assessed by ultraviolet (UV) spectrophotometry (1.8 < A260/A280 < 2.0). RNA integrity was evaluated using electrophoresis. Reverse transcription was carried out using 4 μg of total RNA and the First Strand Complementary DNA (cDNA) Synthesis Kit (Thermo Scientific, Boston, MA, USA).
To make standard curves, 1 μL of first-strand cDNA was amplified with an AffinityScript cDNA Synthesis Kit, and quantified polymerase chain reaction (PCR) products were used to plot standard curves. Quantitative real-time PCR was carried out using an HT-7500 thermocycler (Applied Biosystems) and Brilliant III Ultra-Fast SYBR Green QPCR Master Mix. The PCR cycling conditions consisted of 95 °C for 3 min, followed by 40 cycles of 95 °C for 30 s and 60 °C for 30 s. GAPDH was used as an internal control. DDAH-2, CBS, eNOS, and ARG II mRNA levels were normalized to GAPDH (Glyceraldehyde 3-phosphate dehydrogenase) mRNA levels to compensate for variations in initial amounts of RNA. Normalization was carried out by dividing the logarithmic of DDAH-2, CBS, and eNOS expression levels by the logarithmic of GAPDH expression levels.
Data analyses and statistics
All statistical analyses were performed using SPSS 12.0.1 software (SPSS Inc., Chicago, Illinois, USA). The mean values and standard errors were calculated for each parameter. The normality of quantitative variables was established using the Kolmogorov–Smirnov test. The statistical validity of differences across all testing conditions was established using one-factor analysis of variance (ANOVA), and repeated measures ANOVA was performed to evaluate differences within the group. Pearson’s correlation was also performed. Linear regression analysis was carried out to establish the association between arginase activity (dependent variable) and the other variables. Significance was established by a p value < 0.05.
Results
Characteristics of hypoxia-induced hypertensive and nonhypertensive rats
After 1 month of hypoxia exposure, ~30% of the rats presented an increase in SBP (140 ± 2.4 vs. 199 ± 1.3 without and with hypoxia exposure, respectively) associated with an increase in hematocrit (42 ± 1.5 vs. 73 ± 2.5%) without and with hypoxia exposure, respectively). These rats were classified as hypoxia-induced hypertension rats. Meanwhile, the remaining rats exposed to hypoxia (~70%) did not show an increase in blood pressure and were therefore classified as nonhypertensive rats (Fig. 1). These characteristics were some of the criteria used to determine tolerance to hypoxia in our model. In addition, control rats (rats under normoxia, data not shown) did not present variations in SBP during the experiment (1 month). The average SBP of nonhypertensive rats after hypoxia exposure was not significantly different than that of control rats after hypoxia exposure (Fig. 1).

Effect of exposure to hypobaric hypoxia on SBP in rats. The mean shows the SBP in rats (n = 30) before exposure to hypoxia and after an exposure time of 30 days. SBP values were significantly higher in the group of hypoxia-induced hypertension rats (n = 10, *p < 0.001). Differences in SBP between hypertensive and nonhypertensive rats after exposure to hypoxia (n = 10, **p < 0.001)
Hypoxia-induced hypertensive rats exhibited a lower NO plasma concentration
As shown in Fig. 2A, hypoxia-induced hypertensive rats exhibited significantly lower NO levels after 1 month of exposure than nonhypertensive rats. In fact, the NO plasma levels in the hypoxia-induced hypertensive rats and nonhypertensive rats were 7.7 ± 0.4 μmol/L and 12.5 ± 0.59 μmol/L, respectively (n = 10, *p < 0.001), nonhypertensive and control rats (normoxia) presented the same NO plasma levels (12 ± 0.59 and 13.5 ± 0.6 μmol/L, respectively; n = 10, *p > 0.05).

Effect of exposure to hypobaric hypoxia on plasma levels of NO, homocysteine, and ADMA in rats. A Plasma NO levels before and after exposure for 30 days. Total NO plasma levels in the hypoxia-induced hypertensive rats were significantly lower than those after exposure to hypoxia (n = 10, *p < 0.001) and significantly lower than those in nonhypertensive rats (n = 10 nonhypertensive rats vs. hypoxia-induced hypertension rats, **p < 0.001). B Plasma ADMA levels before and after exposure for 30 days. Total ADMA plasma levels in hypoxia-induced hypertensive rats were significantly higher than those after exposure to hypoxia (n = 10 *p < 0.001) and significantly higher than those in nonhypertensive rats (n = 10 nonhypertensive rats vs. hypoxia-induced hypertensive rats, **p < 0.001). C Plasma homocysteine levels before and after exposure for 30 days. The total homocysteine plasma levels in the hypoxia-induced hypertensive rats were significantly higher than those in nonhypertensive rats (n = 10 nonhypertensive rats vs. hypoxia-induced hypertensive rats, *p < 0.001)
Hypoxia-induced hypertensive rats exhibited higher plasma ADMA and homocysteine levels
To evaluate whether the decreased NO levels in hypertensive rats were also due to increased levels of NO synthesis inhibitors, plasma levels of homocysteine and ADMA were measured. As seen in Fig. 2B, ADMA plasma concentrations were increased after 1 month of exposure to hypoxia. In the hypertensive rats, a significant increase in ADMA concentration compared to the baseline concentration prior to hypoxia exposure was observed (0.80 ± 0.05 vs. 2.08 ± 0.08, respectively; n = 10, *p < 0.001). In addition, nonhypertensive rats showed a discrete increase in ADMA concentration compared to the baseline concentration prior to exposure (0.80 ± 0.05 vs. 1.53 ± 0.16; n = 10 **p < 0.01). Hypoxia-induced hypertensive rats exhibited a significant increase in homocysteine plasma levels after 1 month of exposure to hypoxia (15.83 ± 0.59 vs. 29.36 ± 1.27 µmol/L before and after hypoxia, respectively; n = 10, *p < 0.001), and homocysteine plasma levels after 1 month of exposure to hypoxia were higher in hypertensive rats than in nonhypertensive rats (29.36 ± 1.27 µmol/L vs. 16.02 ± 0.98 µmol/L, respectively; n = 10, *p < 0.001) (Fig. 2C). The average homocysteine levels in nonhypertensive rats after hypoxia exposure did not significantly differ from those in control rats (under normoxia). In fact, homocysteine plasma levels were 15.83 ± 0.59 µmol/L and 16.02 ± 0.98 µmol/L in the control and nonhypertensive groups, respectively (n = 10, *p > 0.05).
Hypoxia-induced hypertensive rats exhibited decreased eNOS activity
To explain the low levels of NO found in hypoxia-induced hypertensive rats, eNOS activity and mRNA levels were measured. Exposure to hypoxia decreased eNOS activity in hypoxia-induced hypertensive rats (n = 10, *p < 0.001), which showed significantly lower levels of eNOS activity than those found in nonhypertensive rats after 1 month of exposure to hypoxia (Fig. 3A) (n = 10, **p < 0.001). The eNOS activity levels in nonhypertensive rats were not significantly lower after exposure to hypoxia. In contrast, when evaluating eNOS mRNA levels, we observed that exposure to hypoxia induced a slight increase in eNOS expression in nonhypertensive rats (Fig. 3B); however, we did not detect the expression of other NOS isoforms, such as iNOS (data not shown). In addition, the results regarding eNOS mRNA levels correlate with those regarding eNOS protein levels (Western blotting analysis).

Endothelial NOS synthase activity and expression. A Measurement of eNOS activity was performed in a homogenate of the aortic arteries of rats exposed to hypoxia by measuring L-[3H]-citrulline. NOS synthase activity in hypoxia-induced hypertensive rats was significantly lower after exposure to hypoxia (n = 10 *p < 0.001) and significantly higher than that in nonhypertensive rats (n = 10, **p < 0.001 nonhypertensive rats vs. hypoxia-induced hypertension rats). B Effect of hypoxia exposure on eNOS mRNA expression in hypoxia-induced hypertensive and nonhypertensive rats; levels before exposure to hypoxia (white bar) and after 30 days of exposure (black bar) (n = 6 per group, *p < 0.001 and **p < 0.01) in homogenized aortic arteries are shown. The boxes correspond to the Western blot analysis from aortic artery homogenates
Hypoxia-induced hypertensive rats exhibited decreased DDAH-2 and CBS levels and increased arginase type II levels
To explain the high ADMA and homocysteine levels found in the hypoxia-induced hypertensive rats, DDAH-2 and CBS expression levels were evaluated. Hypoxia induced a decrease in DDAH-2 protein and mRNA levels (Fig. 4), and this decrease was more substantial in hypoxia-induced hypertensive rats. To evaluate the potential L-arginine levels available for NO synthesis, arginase type II levels were measured. Hypoxia-induced hypertensive rats exhibited a significant increase in arginase type II expression (Fig. 4C).

Effect of hypobaric hypoxia exposure on mRNA expression and the protein levels of DDAH-2, CBS, and arginase type II. A Quantitative PCR of DDAH-2 in hypoxia-induced hypertensive and nonhypertensive rats before exposure to hypoxia (white bar) and 30 days after exposure (black bar) (n = 6 per group, *p < 0.001 and **p < 0.001). B Quantitative PCR of CBS mRNA expression in hypoxia-induced hypertensive and nonhypertensive rats before exposure to hypoxia (white bar) and after 30 days of hypoxia exposure (black bar) (n = 6 per group, *p < 0.001). (C) Quantitative PCR of arginase type II mRNA expression in hypoxia-induced hypertensive and nonhypertensive rats before exposure to hypoxia (white bar) and after 30 days of exposure to hypoxia (black bar) (n = 6 per group, *p < 0.001). Measurements were made in aortic artery tissue homogenates. The mRNA levels were normalized to GAPDH mRNA levels. The boxes correspond to Western blots analysis of aortic artery homogenates
Arginase activity was increased by homocysteine and ADMA in hypoxia-induced hypertensive rats
Arginase I and II activity was poorly inhibited by ADMA in both control rats and rats exposed to hypoxia (Fig. 5A). Contrary to our expectations, arginase II activity was significantly increased by ADMA (p < 0.01) in hypertensive rats (Fig. 5B).

Effect of ADMA concentrations on arginase type I and II activity in homogenized heart tissue from controls (normoxia) and rats exposed to hypoxia. Control (open triangles), nonhypertensive (open circles), and hypertensive (black circles) rats. A Effect of ADMA concentrations on arginase type I activity. An asterisk indicates a significant difference (p < 0.01) for the control group vs. the hypertensive and nonhypertensive groups. Double asterisk indicates a significant difference (p < 0.01) for the hypertensive group vs. the nonhypertensive group. B Effect of ADMA concentrations on arginase type II activity. An asterisk indicates a significant difference (p < 0.01) for the hypertensive group vs. the control and nonhypertensive groups. Arginase activity is expressed as the percent specific activity in μmol/urea/mg/min. Data are expressed as the mean ± SE (n = 10 per group)
Homocysteine was a potent activator of arginase I in the control rats and rats exposed to hypoxia (Fig. 6A). However, homocysteine had no effect on arginase II activity in the control group (Fig. 6B, triangles, normoxia). Therefore, exposure to hypoxia generated a particular form of arginase II that was sensitive to homocysteine-mediated activation (Fig. 6B).

Effect of homocysteine concentrations on arginase type I and II activity in homogenized heart tissue from control rats (normoxia) and rats exposed to hypobaric hypoxia. Control (open triangles), nonhypertensive (open circles), and hypertensive (black circles) rats. A Effect of homocysteine concentrations on arginase type I activity. An asterisk indicates a significant difference (p < 0.01) for the control group vs. the hypertensive and nonhypertensive groups; Double asterisk indicates a significant difference (p < 0.01) for the nonhypertensive group vs. the hypertensive group. B Effect of homocysteine concentrations on arginase type II activity. An asterisk indicates a significant difference (p < 0.01) for the hypertensive group vs. The nonhypertensive group. Arginase activity is expressed as a percent specific activity in μmol/urea/mg/min. Data are expressed as the mean ± SE (n = 10 per group)
Correlations between homocysteine levels, ADMA levels, arginase activity, and SBP
To determine the relationships between homocysteine and ADMA with relevant molecular and physiological parameters induced by hypobaric hypoxia in rats, we evaluated the correlation between homocysteine levels, ADMA levels, arginase activity, and SBP variables.
ADMA plasma levels exhibited a positive correlation with increases in arginase activity (r = 0.594, r2 = 0.352, p < 0.01) and SBP (r = 0.927, r2 = 0.850, p < 0.0001) (Fig. 7A, B). Likewise, we found that increased homocysteine plasma levels exhibited a positive correlation with arginase activity (r = 0.872, r2 = 0.761, p < 0.001) and SBP (r = 0.950, r2 = 0.904, p < 0.001; Fig. 8A, B).

Correlations between plasma ADMA concentration with arginase II activity and systolic blood pressure (SBP). A Correlation between ADMA and arginase II activity. B Correlation between ADMA and SBP. Control (open triangles), nonhypertensive (open circles), and hypertensive (black circles) rats. Pearson’s r values, r2, and pi values correspond to the hypertensive rat group. No significant difference was found between the nonhypertensive rat group and the control group (n = 10 per group)

Correlations between homocysteine concentration with arginase type II activity and systolic blood pressure (SBP). A Correlation between homocysteine and arginase II activity. B Correlation between homocysteine and SBP. Control (open triangles), nonhypertensive (open circles), and hypertensive (black circles) rats. (Pearson’s r values, r2, and pi values correspond to the hypertensive rat group. No significant difference was found between the nonhypertensive rat group and the control group (n = 10 per group)
Discussion
Hypoxia-induced hypertensive rats exhibited lower NO concentrations
To assess whether hypoxia-induced hypertension is caused by endothelial dysfunction, we measured NO plasma concentrations. Hypertensive rats exhibited lower NO plasma concentrations (Fig. 2A), which suggests decreased NO production due to the inhibition of NOS as a consequence of increased ADMA and homocysteine levels (Fig. 2B, C). This would explain why although eNOS expression was increased (Fig. 3B), NO concentrations were decreased. Specifically, we suggest that hypertensive rats presented an increase in eNOS expression and a decrease in NO synthesis due to inhibition of the enzyme eNOS as a consequence of the increase in ADMA concentrations. The inhibitory mechanisms of these metabolites on eNOS activity are currently unclear. To resolve this, we are currently carrying out kinetic characterization of the inhibitory effect of ADMA and homocysteine on eNOS in hypertensive and nonhypertensive rats.
On the other hand, a decrease in NO production could be explained by a reduction in the l-arginine concentration available for NO synthesis due to increased arginase II expression (Fig. 4C). We recently showed that Lama glama (L, glama) an animal genetically resistant to the development of hypoxia-induced pulmonary hypertension [33], present 10 times less arginase type II than other species, allowing increased NO synthesis. Therefore, low expression of type II arginase in these animals could constitute an adaptive mechanism to the development of hypertension [19]. Our results suggest that increased type II arginase levels are involved in the development of hypoxia-induced hypertension. Although l-arginine levels were not measured in this study, we suggest that the bioavailability of l-arginine for NO synthesis would be lower [25].
Hypoxia-induced hypertensive rats have higher ADMA concentrations and increased arginase type II activity
ADMA is a competitive inhibitor of endogenous eNOS [25] and can inhibit NO synthesis. In this context, ADMA was proposed as a risk marker for cardiovascular disease [34,35,36,37] and other pathologies, such as atherosclerosis, diabetes mellitus, rheumatoid arthritis, and preeclampsia [38,39,40,41]. In this study, we observed a significant increase in ADMA levels in hypertensive rats. According to our results, one possible explanation for the high ADMA levels is a decrease in DDAH-2 expression [20]. According to the literature, dimethylarginine dimethylaminohydrolase-2 (DDAH-2) has been proposed as a “target molecule” due to the presence of a polymorphism that induces the imbalance of ADMA metabolism and therefore contributes to the development of cardiovascular diseases [21].
Contrary to expectations, ADMA proved to be a poor inhibitor of arginase I and II (Fig. 5A, B). However, ADMA was a potent activator of arginase II in hypertensive rats (Fig. 5B). One putative mechanism by which ADMA production was mediated by the activation of arginase II in hypertensive rats may be the existence of polymorphisms in the arginase II gene, which may generate a specific regulatory site for ADMA. In this context, ADMA-induced arginase II activation may result in the decreased bioavailability of l-arginine for NO synthesis, explaining the increased arterial blood pressure observed in hypertensive rats. Nevertheless, the discovery of specific polymorphisms in the arginase II gene opens new avenues into the study of new mechanisms of hypoxia tolerance and predisposition to the development of hypertension triggered by environmental factors.
On the other hand, elevated ADMA plasma concentrations correlated with increases in arginase II activity and systolic blood pressure in hypertensive rats (Figs. 7A and 8B, respectively). We have described for the first time differential responses among rats exposed to hypoxia. Specifically, our results indicate that hypertensive rats have high ADMA concentrations and that arginase type II activity is susceptible to the effect of this metabolite, probably due to the existence of a regulatory site for ADMA.
Hypoxia-induced hypertensive rats exhibited higher homocysteine concentrations and increased arginase activity
A wide range of biological and pathological conditions associated with high levels of plasma homocysteine, such as diabetes, hypertriglyceridemia, endothelial dysfunction, apoptosis, oxidative stress, and renal and hepatic impairment, has been described [14, 42,43,44]. Increased homocysteine was recently described as a risk factor for neurodegenerative and neuropsychiatric diseases (e.g., vascular dementia) [45,46,47]. Nevertheless, most research on homocysteine conducted over the last two decades has focused on its role as a risk factor for cardiovascular [48,49,50,51] and other associated diseases, such as thrombosis, aortic aneurysm, and atherosclerosis [50, 52, 53]. In this study, we describe for the first time an increase in plasma homocysteine levels in hypoxia-induced hypertension (Fig. 2C).
The differences in plasma homocysteine between nonhypertensive and hypertensive rats may be related to differences in the regulation of key enzymes involved in homocysteine metabolism. The presence of genetic polymorphisms in the CBS enzyme gene has been described [52, 54, 55], and these polymorphisms may be associated with increases in plasma homocysteine levels and the risk of cardiovascular diseases. In 2014 [52], Sarov et al. observed an association between mutations in the CBS enzyme and the prevalence of hyperhomocysteinemia and homocystinuria in patients with acute deep vein thrombosis (DVT). Furthermore, patients with DVT completely recovered after treatment with heparin, a methionine-restricted diet, and vitamin B12 supplements.
In the present study, the differences in plasma homocysteine between hypertensive and nonhypertensive rats may be explained by reduced CBS expression during exposure to hypoxic conditions, suggesting that hypoxia may decrease CBS expression in hypertensive rats, probably due to the presence of polymorphisms associated with this gene. Based on these results, we suggest that the increased homocysteine concentrations observed in hypertensive rats may be due to the existence of polymorphisms in the CBS gene, producing changes in the activity and expression of these enzymes with a subsequent increase in plasma homocysteine levels.
In addition, in this study, we describe for the first time the role of homocysteine as an important trigger of arginase isoforms (Fig. 6A, B). However, as mentioned above, homocysteine was not observed to affect arginase II under normoxic conditions (Fig. 6B). These results correlate with those reported by López et al. in 2009, who showed that arginase II activity and arginase I activity displayed different kinetics. Based on these observations, we suggest the existence of a regulatory site for homocysteine in arginase II that leads to a significant increase in its activity.
Previously, we reported that hypoxia-induced hypertensive rats exhibit higher arginase II expression [7], which leads to lower arginine bioavailability for NO synthesis, and that a lower NO concentration leads to the development of arterial hypertension. Based on these observations, we suggest that the insensitivity of homocysteine to arginase II could represent a protective mechanism against endothelial dysfunction under normoxic conditions that avoids alterations to the vasoconstrictor systems dependent on NO. Nevertheless, exposure to hypoxia may generate an arginase II conformation that is sensitive to homocysteine-mediated activation (Fig. 6B). This suggests that under hypoxic conditions, arginase II may adopt a structural conformation with an allosteric site for homocysteine. We are currently undergoing studies in our laboratory to confirm his hypothesis.
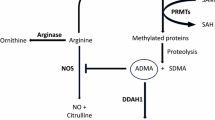
The results from this study reveal that the development of hypertension induced by hypoxia in hypertensive rats may be the result of increased ADMA and homocysteine levels and activation of the arginase II pathway, which in turn could inhibit NO synthesis and reduce l-arginine bioavailability (Fig. 9).

Proposed mechanism for the development of hypoxia-induced hypertension in hypertensive rats
Finally, based on our findings, we propose a model for the development of hypertension induced by hypobaric hypoxia. Hypoxia-induced hypertension is caused by arginase II activation mediated by an increase in homocysteine and ADMA concentrations, suggesting decreased l-arginase bioavailability for the synthesis of NO (Fig. 9). ADMA and homocysteine have dual inhibitory effects on NO synthesis. The first involves the inhibition of eNOS, and the second, described here for the first time, involves the effect of ADMA and homocysteine in activating type II arginase. This is likely due to the existence of regulatory sites for these metabolites, such as those generated by polymorphisms in the arginase type II gene. However, this hypothesis needs to be evaluated.
Finally, our study proposes regulation of the arginase II pathway by homocysteine and ADMA as a new mechanism for NO synthesis regulation and that this regulation may play an important role as a molecular target for clinical applications.
References
Parati G, Bilo G, Faini A. Changes in 24 h ambulatory blood pressure and effects of angiotensin II receptor blockade during acute and prolonged high-altitude exposure: a randomized clinical trial. Eur Heart J. 2014;35:3113–22.
Moraga FA, López I, Morales A, Soza D, Noack J. The effect of oxygen enrichment on cardiorespiratory and neuropsychological responses in workers with chronic intermittent exposure to high altitude (ALMA, 5,050 m). Front Physiol. 2018;23:187. 9
Norboo T, Stobdan T, Tsering N, Angchuk N, Tsering P, Ahmed I, et al. Prevalence of hypertension at high altitude: cross-sectional survey in Ladakh, Northern India. BMJ Open. 2015;5:e007026.
Negi PC, Bhardwaj R, Kandoria A, Asotra S, Ganju N, Marwaha R, et al. Epidemiological study of hypertension in natives of Spiti Valley in Himalayas and impact of hypobaric hypoxemia; a cross-sectional study. J Assoc Physicians India. 2012;60:21–5.
Levine BD, Zuckerman JH, de Filippi CR. Effect of high-altitude exposure in the elderly: the tenth mountain division study. Circulation. 1997;96:1224–32.
Baggish AL, Wolfel EE, Levine BD. Cardiovascular system. In: Swenson ER, Bartsch P, editors. High altitude: human adaptation to hypoxia. New York: Springer; 2014. p. 103–40.
López V, Siques P, Brito J, Vallejos C, Naveas N. Upregulation of arginase expression and activity in hypertensive rats exposed to chronic intermittent hypobaric hypoxia. High Alt Med Biol. 2009;10:373–81.
Landmesser U, Drexler H. Endothelial function and hypertension. Curr Opin Cardiol. 2007;22:316–20.
Ignarro LJ, Cirino G, Casini A, Napoli C. Nitric oxide as a signaling molecule in the vascular system: an overview. J Cardiovasc Pharm. 1999;34:879–86.
Klinger JR, Abman SH, Gladwin MT. Nitric oxide deficiency and endothelial dysfunction in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2013;188:639–46.
Arrigoni FI, Vallance P, Haworth SG, Leiper JM. Metabolism of asymmetric dimethylarginines is regulated in the lung developmentally and with pulmonary hypertension induced by hypobaric hypoxia. Circulation. 2003;107:1195–201.
Pullamsetti S, Kiss L, Ghofrani HA, Voswinckel R, Haredza P. Increased levels and reduced catabolism of asymmetric and symmetric dimethylarginines in pulmonary hypertension. FASEB J. 2005;19:1175–7.
Sasaki A, Doi S, Mizutani S, Azuma H. Roles of accumulated endogenous nitric oxide synthase inhibitors, enhanced arginase activity, and attenuated nitric oxide synthase activity in endothelial cells for pulmonary hypertension in rats. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1480–7.
Wierzbicki AS. Homocysteine and cardiovascular disease: a review of the evidence. Diab Vasc Dis Res. 2007;4:143–50.
Sun L, Sun S, Li Y, Pan W, Xie Y. Potential biomarkers predicting risk of pulmonary hypertension in congenital heart disease: the role of homocysteine and hydrogen sulfide. Chin Med J. 2014;127:893–9.
Böger RH, Maas R, Schulze F, Schwedhelm E. Asymmetric dimethylarginine (ADMA) as a prospective marker of cardiovascular disease and mortality-an update on patient populations with a wide range of cardiovascular risk. Pharm Res. 2009;60:481–7.
Lüneburg N, Siques P, Brito J, De La Cruz JJ, León-Velarde F, Hannemann J, et al. Long-term intermittent exposure to high altitude elevates asymmetric dimethylarginine in first exposed young adults. High Alt Med Biol. 2017;18:226–33.
Siques P, Brito J, Schwedhelm E, Pena E, León-Velarde F, De La Cruz JJ, et al. Asymmetric dimethylarginine at sea level is a predictive marker of hypoxic pulmonary arterial hypertension at high altitude. Front Physiol. 2019;27:651.
López V, Moraga FA, Llanos AJ, Ebensperger G, Taborda MI, Uribe E. Plasmatic concentrations of ADMA and Homocystein in Llama (Lama glama) and Regulation of Arginase Type II: an animal resistent to the development of pulmonary hypertension induced by hypoxia. Front Physiol. 2018;29:606.
Tran CT, Leiper JM, Vallance P. The DDAH/ADMA/NOS pathway. Atheroscler Suppl. 2003;4:33–40.
Xuan C, Xu LQ, Tian QW, Li H, Wang Q, He GW et al. Dimethylarginine dimethylaminohydrolase 2 (DDAH 2) gene polymorphism, asymmetric dimethylarginine (ADMA) concentrations, and risk of coronary artery disease: a case-control study. Sci Rep. 2016;6:33934.
Ding R, Lin S, Chen D. The association of cystathionine β synthase (CBS) T833C polymorphism and the risk of stroke: a meta-analysis. J Neurol Sci 2012;312:26–30.
Pernow J, Jung C. Arginase as a potential target in the treatment of cardiovascular disease: reversal of arginine steal? Cardiovasc Res. 2013;98:334–43.
Huynh NN, Chin-Dusting J. Amino acids, arginase and nitric oxide in vascular health. Clin Exp Pharm Physiol. 2006;33:1–8.
Boger RH. Asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, explains the “L-arginine paradox” and acts as a novel cardiovascular risk factor. J Nutr. 2004;134:2842S–7S.
Johnson FK, Johnson RA, Peyton KJ, Durante W. Arginase inhibition restores arteriolar endothelial function in Dahl rats with salt-induced hypertension. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1057–62.
Holowatz LA, Kenney WL. Up-regulation of arginase activity contributes to attenuated reflex cutaneous vasodilatation in hypertensive humans. J Physiol. 2007;581:863–72.
Sharma S, Kumar S, Sud N, Wiseman DA, Tian J. Alterations in lung arginine metabolism in lambs with pulmonary hypertension associated with increased pulmonary blood flow. Vasc Pharm. 2009;51:359–64.
Archibald RM. Colorimetric measurement of uric acid. Clin Chem. 1957;3:102–5.
Venkatakrishnan G, Shankar V, Reddy SR. Microheterogeneity of molecular forms of arginase in mammalian tissues. Indian J Biochem Biophys. 2003;40:400–8.
Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54.
Bredt DS, Schmidt HW. The citrulline assay, in methods in nitric oxide research. John Wiley & Sons: New York; 1996. p. 249–55.
Herrera EA, Reyes RV, Giussani DA, Riquelme RA, Sanhueza EM, Ebensperger G, et al. Carbon monoxide: a novel pulmonary artery vasodilator in neonatal llamas of the Andean altiplano. Cardiovasc Res. 2008;77:197–201.
Gorenflo M, Zheng C, Werle E, Fiehn W, Ulmer HE. Plasma levels of asymmetrical dimethyl-L-arginine in patients with congenital heart disease and pulmonary hypertension. J Cardiovasc Pharm. 2001;37:489–93.
Schulze F, Lenzen H, Hanefeld C, Bartling A, Osterziel KJ. Asymmetric dimethylarginine is an independent risk factor for coronary heart disease: results from the multicenter Coronary Artery Risk Determination investigating the Influence of ADMA Concentration (CARDIAC) study. Am Heart J. 2006;152:493.e1–8.
Jawalekar SL, Karnik A, Bhutey A. Risk of cardiovascular diseases in diabetes mellitus and serum concentration of asymmetrical dimethylarginine. Biochem Res Int. 2013;18:9430.
Iribarren C, Husson G, Sydow K, Wang BY, Sidney S. Asymmetric dimethyl-arginine and coronary artery calcification in young adults entering middle age: the CARDIA Study. Eur J Cardiovasc Prev Rehabil. 2007;14:222–9.
Landim MB, Casella Filho A, Chagas AC. Asymmetric dimethylarginine (ADMA) and endothelial dysfunction: implications for atherogenesis. Clinics. 2009;64:471–8.
Zaciragic A, Huskic J, Mulabegovic N, Avdagic N, Valjevac A. An assessment of correlation between serum asymmetric dimethylarginine and glycated haemoglobin in patients with type 2 diabetes mellitus. Bosn J Basic Med Sci. 2014;14:21–4.
Spasovski D, Latifi A, Osmani B, Krstevska-Balkanov S, Kafedizska I. Determination of the diagnostic values of asymmetric dimethylarginine as an indicator for evaluation of the endothelial dysfunction in patients with rheumatoid arthritis. Arthritis. 2013;2013:818037.
Alpoim PN, Godoi LC, Freitas LG, Gomes KB, Dusse LM. Assessment of L-arginine asymmetric 1 dimethyl (ADMA) in early-onset and late-onset (severe) preeclampsia. Nitric Oxide. 2013;33:81–2.
Guthikonda S, Haynes WG. Homocysteine: role and implications in atherosclerosis. Curr Atheroscler Rep. 2006;8:100–6.
Suhara T, Fukuo K, Yasuda O, Tsubakimoto M, Takemura Y. Homocysteine enhances endothelial apoptosis via upregulation of Fas-mediated pathways. Hypertension. 2004;43:1208–13.
Baszczuk A, Kopczynski Z, Thielemann A. Endothelial dysfunction in patients with primary hypertension and hyperhomocysteinemia. Postepy Hig Med Dosw. 2014;68:91–100.
Davis G, Baboolal N, Nayak S, McRae A. Sialic acid, homocysteine and CRP: potential markers for dementia. Neurosci Lett. 2009;465:282–4.
Cervellati C, Romani A, Seripa D, Cremonini E, Bosi C. Oxidative balance, homocysteine, and uric acid levels in older patients with Late Onset Alzheimer’s Disease or Vascular Dementia. J Neurol Sci. 2014;337:156–61.
Cervellati C, Romani A, Seripa D, Cremonini E, Bosi C, Magon S, et al. Systemic oxidative stress and conversion to dementia of elderly patients with mild cognitive impairment. Biomed Res Int. 2014;2014:309507.
Marcus J, Sarnak MJ, Menon V. Homocysteine lowering and cardiovascular disease risk: lost in translation. Can J Cardiol. 2007;23:707–10.
Sengwayo D, Moraba M, Motaung S. Association of homocysteinaemia with hyperglycaemia, dyslipidaemia, hypertension and obesity. Cardiovasc J Afr. 2013;24:265–9.
Narayanan N, Tyagi N, Shah A, Pagni S, Tyagi SC. Hyperhomocysteinemia during aortic ane urysm, a plausible role of epigenetics. Int J Physiol Pathophysiol Pharm. 2013;5:32–42.
Van Meurs JB, Pare G, Schwartz SM, Hazra A, Tanaka T. Common genetic loci influencing plasma homocysteine concentrations and their effect on risk of coronary artery disease. Am J Clin Nutr. 2013;98:668–76.
Sarov M, Not A, de Baulny HO, Masnou P, Vahedi K. A case of homocystinuria due to CBS gene mutations revealed by cerebral venous thrombosis. J Neurol Sci. 2014;336:257–9.
Zhou S, Zhang Z, Xu G. Notable epigenetic role of hyperhomocysteinemia in atherogenesis. Lipids Health Dis. 2014;21:134–8.
Alessio AC, Siqueira LH, Bydlowski SP, Hoehr NF, Annichino-Bizzacchi JM. Polymorphisms in the CBS gene and homocysteine, folate and vitamin B12 levels: association with polymorphisms in the MTHFR and MTRR genes in Brazilian children. Am J Med Genet. 2008;15:2598–602.
Yakub M, Moti N, Parveen S, Chaudhry B, Azam I. Polymorphisms in MTHFR, MS and CBS genes and homocysteine levels in a Pakistani population. PLoS ONE. 2012;7:33222–5.
Acknowledgements
We acknowledge CONICYT for a scientific research grant from FONDECYT (11075096).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
López, V., Uribe, E. & Moraga, F.A. Activation of arginase II by asymmetric dimethylarginine and homocysteine in hypertensive rats induced by hypoxia: a new model of nitric oxide synthesis regulation in hypertensive processes?. Hypertens Res 44, 263–275 (2021). https://doi.org/10.1038/s41440-020-00574-1
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41440-020-00574-1
Keywords
This article is cited by
-
Arginase: shedding light on the mechanisms and opportunities in cardiovascular diseases
Cell Death Discovery (2022)
