
Abstract
The development of electrocatalysts capable of efficient reduction of nitrate (NO3−) to ammonia (NH3) is drawing increasing interest for the sake of low carbon emission and environmental protection. Herein, we present a CuCo bimetallic catalyst able to imitate the bifunctional nature of copper-type nitrite reductase, which could easily remove NO2− via the collaboration of two active centers. Indeed, Co acts as an electron/proton donating center, while Cu facilitates NOx− adsorption/association. The bio-inspired CuCo nanosheet electrocatalyst delivers a 100 ± 1% Faradaic efficiency at an ampere-level current density of 1035 mA cm−2 at −0.2 V vs. Reversible Hydrogen Electrode. The NH3 production rate reaches a high activity of 4.8 mmol cm−2 h−1 (960 mmol gcat−1 h−1). A mechanistic study, using electrochemical in situ Fourier transform infrared spectroscopy and shell-isolated nanoparticle enhanced Raman spectroscopy, reveals a strong synergy between Cu and Co, with Co sites promoting the hydrogenation of NO3− to NH3 via adsorbed *H species. The well-modulated coverage of adsorbed *H and *NO3 led simultaneously to high NH3 selectivity and yield.
Similar content being viewed by others


Introduction
Nitrate anions (NO3−), widely present in industrial and agricultural wastewater, pose a real potential threat to human health and ecological balance, especially their incomplete conversion into nitrites (NO2−) which are thought to be cancerogenic by inducing liver damage and methaemoglobinaemia1. The conventional biological treatments for NO3− removal into nitrogen (N2) gas, involving nitrification and denitrification processes, are energy intensive (~11.7 to 12.5 kWh kg N−1)2. Actually, the reduction of NO3− into NH3 has become of great interest from an industrial point of view since NH3 is a highly important industrial chemical for the synthesis of pharmaceuticals, fertilizers, dyes, plastic, etc.3, and is also considered for hydrogen storage/release as a carbon-free hydrogen carrier4. To date, the industrial synthesis of NH3 relies heavily on the non-sustainable and eco-unfriendly Haber–Bosch route, which requires harsh conditions i.e., high temperature (400–600 °C) and high pressure (200–350 atm), and heavily relies on fossil energy3. The total amount of CO2 produced during the Haber–Bosch process accounts for roughly 1.2% of the global annual CO2 emissions, more than any other industrial chemicals synthesis5.
Electrocatalytic reduction of NO3− into NH3, powered by green energy, has drawn increasing attention and is considered as a sustainable complementary process to the Haber–Bosch process2,6,7, since this technology can simultaneously meet the 21st session of the Conference of the Parties (COP) two-degree scenario (2DS) target for NH3 and protect the environment from the eutrophic water pollution. The rational design of novel electrocatalysts with both high activity and selectivity is crucial for reducing NO3− (NO3−RR) and achieving large-scale applications, and satisfying high industrial demands. Since Nature has developed sophisticated and efficient machinery such as enzymes8,9, it is interesting to design catalysts by studying the mechanism of enzymatic reduction. Indeed, biocatalytic reduction of NO3− to NH3 widely exists inside many microorganisms, where NO3− ions are firstly reduced into NO2− by NO3− reductases that accept electrons from quinone10. The generated NO2− is further converted to NH3 by nitrite reductases (NIRs) using quinone as electron donors. Among various NIRs, the Cu-type NIRs (Cu-NIRs) found in Rhizobium is one of the most important enzymes for N2 fixation. Cu-NIRs are trimeric proteins composed of 3 identical subunits, and each monomer has two types of Cu atomic active centers, acting as electron-donating centers (T1Cu) and catalytic centers (T2Cu), respectively10. The reported mechanism proposes that *NO2− (where * denotes an adsorbed specie) is bound to the T2Cu in a bidentate form via two oxygen atoms. Due to the electrons transfer from T1Cu to T2Cu, the T2Cu oxidation state decreases from (II) to (I), facilitating *NO2− association with T2Cu in a bridging nitro binding form. Meanwhile, the aspartate acid besides the T2Cu provides proton to one oxygen and extends the N-O bond length, leading to the breaking of the N-O bond11. According to the mechanism of NO2− reduction on T2Cu and T1Cu, it can be speculated that moderate affinity with NO3−, protons availability and electrons provision are the key factors for the high efficiency in NO3−RR to NH3. Jimmy C. Yu et al.12 proved that hydrogen adsorption (*H), with moderate adsorption energy on catalysts, can behave as an important reactive species for the hydrogenation of NOx intermediates to NH3 whilst suppressing the hydrogen evolution reaction (HER) competition. Matthew J. Liu et al.13 also found that the NO3−RR activity and product selectivity highly depended on the *H coverage on the titanium surface at different potentials with the increase of NO3−RR overpotential.
Very recently, Schuhmann and his colleagues14 reported a tandem mechanism for PO43−-modified CuCo binary metal sulfides. They proposed that NO2− intermediates were preferentially formed on Cu-based phases followed by splitting over nearby Co-based phases; however, the role of *H during NO3−RR was unclear and the effect of the electronic structure of alloyed catalysts on the adsorption of reaction intermediates lacked further discussions in the proposed mechanism14. In this study, inspired by the bifunctional nature of the Cu-NIRs, we prepared CuCo alloy nanosheets via a one-step electrodeposition route. The CuCo alloy could well mimic the behavior of the two catalytic centers in Cu-NIRs. The Co species could efficiently provide the electrons and generate the hydrogen protons to the nearby Cu species with high adsorption of NO3− and its derivatives. The as-prepared CuCo nanosheet exhibited superior catalytic performances, as (1) the lowest η of 290 mV for ammonia production (at 0.4 V vs. reversible hydrogen electrode (RHE), all electrode potentials mentioned below were provided with respect to RHE unless specially stated); (2) an ampere-level current density of 1035 mA cm−2 with a 100% Faradaic efficiency for NH3 generation at an overpotential of 890 mV, and a corresponding \({{{{{\rm{Yield}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\) of 4.8 mmol cm−2 h−1; (3) a wide potential window (300 mV, from −0.1 to −0.4 V) for NH3 generation with >90% Faradaic efficiency (FE), which is one of the most advanced catalysts for NO3−RR so far. Electrochemical in situ Fourier transform infrared spectroscopy (FTIR) and in situ shell-isolated nanoparticle enhanced Raman spectroscopy (SHINERS) associated with density functional theory (DFT) calculations were conducted to clarify the pathways and mechanisms of NO3−RR, with the aim to contribute to subsequent catalyst optimization and scaling up.
Results
Preparation and characterization of CuCo bimetallic electrocatalysts
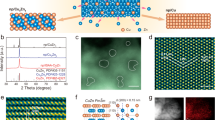
CuCo bimetallic materials were synthesized by co-electrodeposition of Cu and Co on Ni foams’ surfaces (Fig. 1a and Method section). Ni foam is widely used as a supporting substrate for nanostructured electrocatalysts due to its smooth surface and good conductivity, benefiting the electrodeposition by an efficient electron transfer15,16,17. Meanwhile, Ni was proved as a relatively inert material for NO3−RR18,19, without affecting the CuCo catalysts’ performance. The Cu/Co molar ratio was determined using an inductively coupled plasma-optical emission spectrometer (ICP-OES) (Supplementary Table S1). The catalyst with a Cu/Co molar ratio of ca. 50/50 was named Cu50Co50 and was considered as a reference in this study. The alloying of Cu and Co in Cu50Co50 was confirmed by X-ray diffraction (XRD) and high-resolution transmission electron microscopy (HRTEM). Indeed, in the diffractogram (Fig. 1b and Supplementary Fig. S1) shifts in the Cu (111) and Cu (200) diffraction peaks toward higher degrees were observed after the addition of Co to Cu, which were attributed to the shrinkage of the lattice spacing caused by the partial alloying of Co atom with a smaller diameter compared to Cu atom20. Furthermore, the lattice spacing of the Cu (111) plane contracted to 0.208 nm after alloying Cu with Co (Fig. 1c), when it was 0.212 nm for the Cu (111) plane for the pure Cu catalyst (Supplementary Fig. S2a)15. Scanning electron microscopy (SEM) was applied to examine the morphologies of Cu, Co and CuxCoy catalysts and showed micro-pines structure for all the catalysts (Supplementary Fig. S3a–j). At the nanoscale level, small bump structures on the surface of the micro-pines were observed on pure Cu (Supplementary Fig. S3a, b). After the incorporation of Co, a nanosheet structure emerged on the micro-pine’s surface of the Cu50Co50 catalyst (Fig. 1d and Supplementary Fig. S3e, f), very similar to the structure of pure Co (Supplementary Fig. S3i, j). The thickness of the Cu50Co50 nanosheet was evaluated by atomic force microscopy (AFM) and was around 10 nm (Fig. 1e). Besides, the EDS mapping analysis disclosed an even distribution of Cu and Co in Cu50Co50 (Fig. 1f).

a Schematic diagram of CuCo alloy electrodeposition on nickel foam’s surface. b XRD spectra of Cu50Co50, Cu, and Co. HRTEM image (c), SEM image (d), linear topography profiles from AFM images (e), and EDS mapping analysis (f) of Cu50Co50. XPS peaks spectra of Cu 2p (g) and Co 2p (h) of Cu50Co50.
The electronic properties of the Cu50Co50 nanosheet were explored by X-ray photoelectron spectroscopy (XPS). Cu2+ 2p peaks were observed in the high-resolution Cu 2p spectra (Fig. 1g), and was due to the partial oxidation of the alloy’s surface exposed to air, and the same observation was made for Co (Fig. 1h). The decrease of the Cu 2p binding energy, compared with pure Cu (Supplementary Fig. S4a) and the notable increase of the Co 2p binding energy, compared with pure Co (Supplementary Fig. S4b), revealed a redistribution of the electrons between Cu and Co after their alloying21, leading to the movement of the d band towards the Fermi level22 comparing with the pure Cu catalyst. X-ray absorption spectroscopy (XAS) analysis was also performed to check the redistribution of electrons between Cu and Co. Extended X-ray absorption fine structure (XANES) spectra depicted a negative shift of the absorption edge position for the Cu K-edge after interacting with Co (indicated by the red arrow in the inset) (Supplementary Fig. S5), illustrating the electron density transfer from Co to Cu23. In addition, according to the Bader charge analysis (Supplementary Fig. S6), compared with monometallic Cu and Co, a charge redistribution on CuCo(111) was observed, where the Cu center displayed a higher electrons density compared to the Co center (Supplementary Fig. S7)24. The correlation between electrons redistribution within Cu and Co and the adsorption energy of *H, *NO3 and the *NOx intermediates25 are discussed further in the mechanistic study section.
Electrochemical activity and kinetics of NO3 −RR
NO3−RR was first investigated by linear sweep voltammetry (LSV) at a low scan rate of 1 mV s−1 on all the prepared electrocatalysts. The reduction of NOx species contributed mainly to the current density (solid curve), indicating a high catalytic activity toward NO3−RR for all the catalysts (Fig. 2a). The substrate of Ni foam was inactive for NO3−RR compared with the electrodeposited catalysts (Supplementary Figs. S8, S9). The overpotential η at 10 mA cm−2 (denoted \({\eta }_{{@{{{{{\rm{10mA}}}}}}{{{{{\rm{cm}}}}}}}^{-2}}\), η = Eo – E, where E is the potential with a current density of 10 mA cm-2, Eo = 0.69 V), where NO3−RR is under kinetic control, was used as a criterion to compare the catalysts’ activity. Cu50Co50 and Cu exhibited a lower energy barrier for NO3−RR, with \({\eta }_{{@{{{{{\rm{10mA}}}}}}{{{{{\rm{cm}}}}}}}^{-2}}\) of 498 and 503 mV, respectively, compared to Co, that displayed a \({\eta }_{{@{{{{{\rm{10mA}}}}}}{{{{{\rm{cm}}}}}}}^{-2}}\) of 690 mV. In fact, two reduction current peaks (peak S1 and S2) were observed in the curves of Cu and Cu50Co50. At the initial stage of the reaction, Cu and Cu50Co50 seemed to display a similar behavior toward NO3−RR, suggesting the important role of Cu at this stage. According to previous studies26,27, the peak S1 near 0.08 V was assigned to *NO3− (adsorbed NO3−) reduction into *NO2− (1) following a 2-electrons transfer process, while the peak S2 was allocated to the *NO2− reduction into *NH3 (2) following a 6-electrons transfer process. In the peak S2 region, Cu50Co50 displayed a positive potential shift of 67 mV, compared to Cu, suggesting a significant synergy between Co and Cu and a drop of the barrier energy of *NO2− reduction to *NH3, as the applied potential increases.

a j-E curve (80% iR corrected) over Cu50Co50, pure Cu, and pure Co modified Ni foams (catalysts loading was 5 mg cm−2) in 1 M KOH solution containing 100 mM KNO3 (solid lines) or in the absence of KNO3 (dotted line) at a scan rate of 1 mV s−1 (the red dash line presenting the j of 10 mA cm−2, the shading S1 and S2 presenting the peak around 0.2 to 0.05 V and 0.05 to −0.15 V, respectively). b j-E curve (80% iR corrected) at 400 rpm and electron transfer numbers at different potentials calculated by the K–L equation for Cu50Co50 on RDE in 100 mM KNO3 + 1 M KOH electrolyte at a scan rate of 10 mV s−1 (catalysts loading was 0.25 mg cm−2). Tafel slopes in the potential range of peak S1 (c) S2 (d). e j-E curves over Cu50Co50 modified Ni foam in 1 M KOH solution containing 100 mM KNO3 at different scan rates without agitation (solid line) and at a scan rate of 5 mV s−1 with agitation (catalysts loading was 5 mg cm−2). f Time-dependent current density curves over Cu50Co50, Cu, Co modified Ni foam at −0.2 V with a magnetic stirring speed of 1000 rpm (catalysts loading was 5 mg cm−2).
In the aim to investigate the reaction routes for all the catalysts, the number of electrons transferred (n) during the NO3− reduction reaction was estimated from the slope of the Koutecký–Levich (K–L) plots (Supplementary Fig. S10). For the Cu catalyst (Supplementary Fig. S11a), n was 2 at −0.1 V and a quasi-first-order reaction relationship between j and the NO3− concentration was obtained (Supplementary Fig. S12), validating the rate-determinate step (RDS) as the reduction route described by Eq. (1). The n value increased to 6 at −0.25 V, validating the reduction route described in Eq. (2). In comparison, a direct 8-electrons transfer process was observed on Cu50Co50 in the potential region between the peaks S1 and S2 (Fig. 2b), revealing a strong alloying effect promoting simultaneously both routes. Though an 8-electrons transfer process also occurred on the pure Co catalyst at a far less negative potential of ca. −0.45 V (Supplementary Fig. S11b), i.e., at a higher barrier energy.
In the potential region of peak S1, the Tafel slopes derived from the j-E curves (Fig. 2c), for Cu50Co50 was 205.75 mV decade−1, which was slightly lower than Cu (232.43 mV decades−1), indicating that the addition of Co could promote the electrons transfer over the catalyst/electrolyte interface during *NO3− reduction to *NO2−. This result was supported by EIS data (Supplementary Fig. S13), demonstrating a smaller charge-transfer resistance on Cu50Co50 compared to Cu (3.51 vs. 3.81 Ω). The Co catalyst had minor electrocatalytic activity for NO3− reduction at this relatively positive working potential, so its Tafel slop in this potential region was not determined. In the potential region of peak S2, the Tafel slope (Fig. 2d) of Cu50Co50 (148.95 mV decades−1) was significantly lower than Cu (229.75 mV decades−1); this was explained by an electron redistribution between Cu and Co over Cu50Co50 and was emphasized in the characterization section. The Co catalyst displayed the lowest Tafel slope value of 94.24 mV decades−1, but at a more negative potential, i.e., 95 mV shift, than Cu50Co50. This latter result suggested that after crossing a high energy barrier, Co displayed better kinetics’ performances toward the NO3−RR for NH3 production compared to the Cu-based catalysts. Alloying Co to Cu implemented a faster electron transfer rate and improved kinetics’ performances toward the NO3−RR. In addition to being affected by the properties of the catalysts, the NO3−RR was also diffusion-controlled (Fig. 2e). To further evaluate the NO3−RR activity of the catalysts under steady-state conditions, potentiostatic electrolytic reduction of NO3− in a homemade H-type electrolytic cell (Supplementary Fig. S14) was carried out by electrodepositing the catalysts on Ni foams’ surfaces. A magnetic stirring rate of 1000 rpm was used to minimize the effect of diffusion and fresh electrolyte solution was constantly supplemented to maintain a constant NO3− ion concentration. In these conditions, the current densities obtained on the Cu50Co50 catalyst reached an Ampere level at −0.2 V vs. RHE (Fig. 2f).
NH3 and NO2− were quantitatively detected by the Nessler Reagent method and ion chromatography, respectively (Supplementary Fig. S15)28,29. The gas products were analyzed during the reaction by gas chromatography and online electrochemical mass spectrometry (OEMS)30. There was no N2 production detected and the amount of the stripped NH3 in this work was negligible. (Supplementary Fig. S16) In order to figure out the effect of Co content on the NO3−RR activity, CuCo bimetallic catalysts with different Cu/Co ratios were prepared, i.e., Cu65Co35 and Cu15Co85. As shown in Fig. 3a, when a potential of 0 V was applied, the Faraday efficiency of NH3 (\({{{{{\rm{FE}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\)) and NO2− (\({{{{{\rm{FE}}}}}}_{{{{{{\rm{NO}}}}}}_{2}^{-}}\)) on pure Cu were 32% and 60%, respectively. The corresponding molar ratio of NO2− to NH3 was seven times higher (Fig. 3c), suggesting an accumulation of NO2− due to the low kinetic of *NO2− hydrogenation to *NH3 on the Cu surface. With the addition of Co to Cu, both the \({{{{{\rm{FE}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\) and the geometrically normalized current density of NH3 production (\({j}_{{{{{{\rm{NH}}}}}}_{3}}\)) increased significantly (Fig. 3a, b). A volcano shape was observed and the highest \({j}_{{{NH}}_{3}}\) was obtained on Cu50Co50 (i.e., 347 mA cm−2 \({j}_{{{{{{\rm{NH}}}}}}_{3}}\) and 88% \({{{{{\rm{FE}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\)), and was ten times higher than monometallic Cu (i.e., 34 mA cm−2 and 32% \({{{{{\rm{FE}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\)), and nearly 17 times higher than monometallic Co catalyst (i.e., 21 mA cm−2 and 84% \({{{{{\rm{FE}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\)). Furthermore, the molar ratio of NO2− to NH3 exhibited a dramatic drop from 7 to 0.6 as the Co content was raised from 0 to 100% (Fig. 3c). This observation suggested that the incorporation of Co enhanced further the *NO2− hydrogenation into NH3. Moreover, when the ratio of Co was increased by over 50%, the \({{{{{\rm{FE}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\) was remained constant, but the \({{{{{\rm{j}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\) declined dramatically, indicating that a moderate Cu/Co ratio was important to maintain high catalytic performance for NH3 production. The electrochemically active surface area (ECSA) was also measured (Supplementary Figs. S17f, S18b), and all the catalysts had comparable ECSA. The maximum ECSA normalized current density for NH3 production (\({j}_{{{{{{\rm{NH}}}}}}_{3}({{{{\rm{ECSA}}}}})}\)) was obtained on Cu50Co50 (Supplementary Fig. S19), indicating that this catalyst had the highest intrinsic activity for NH3 production.

\({{{\mbox{FE}}}}_{{{{\mbox{NH}}}}_{3}}\)and \({{{\mbox{FE}}}}_{{{{\mbox{NO}}}}_{2}^{-}}\) of NO3−RR (a), bias-current density and products yield for NH3 (b), and the ratio of NO2− to NH3 generated (c) for different Cu/Co ratio at 0 V in 100 mM KNO3 + 1 M KOH electrolyte (catalysts loading was 5 mg cm−2). \({{{\mbox{FE}}}}_{{{{\mbox{NH}}}}_{3}}\) (d) and NH3 product yield (e) at different electrode potentials on Cu50Co50, pure Cu and pure Co catalysts modified Ni foam (catalysts loading was 5 mg cm−2). Comparison of the electrocatalytic NO3-RR performances of Cu50Co50 modified Ni foam with other extensively reported electrocatalysts (f). \({{{\mbox{FE}}}}_{{{{\mbox{NH}}}}_{3}}\) and \({{{\mbox{Yield}}}}_{{{\mbox{mass}}}-{{{\mbox{NH}}}}_{3}}\) on Cu50Co50/Ni foam under the applied potential of −0.2 V during 10 periods of 1 h electrocatalytic NO3-RR (g) (catalysts loading was 5 mg cm−2). The time-dependent concentration of NO3−, NO2− and NH3 and corresponding FE over Cu50Co50 modified Ni foam at −0.1 V (h) (catalysts loading was 5 mg cm−2). Error bars represent the standard deviations calculated from three independent measurements.
To clarify the possible 14N pollution, the electrolysis in the electrolyte free of NO3− ions was performed and little NH3 was produced (Supplementary Fig. S20). \({j}_{{{{{{\rm{NH}}}}}}_{3}}\) in the solution with NO3− ions was over 150-fold higher than that in the electrolyte free of NO3−. The content of 14NH4+ was also analyzed by 1H NMR test29,31 and was ca. 4.62 mmol cm−2 h−1. The obtained results were close to the one from the spectrophotometric analysis (4.75 mmol cm−2 h−1) (Supplementary Fig. S21d), and confirmed the reliability of the quantification methods used in this work. The typical two peaks of 15NH4+ after the electrolysis of 15NO3− also suggested that the NH3 product indeed came from the electrocatalytic reduction of NO3− (Supplementary Fig. S21e).
It is known that the applied potential influences the products’ selectivity13, so we investigated the effect of the applied potential toward the NO3−RR (Fig. 3d, e and Supplementary Fig. S22). The \({{{{{\rm{FE}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\) was about 51% at a η of 290 mV (at 0.4 V) on Cu50Co50. The η was comparatively much lower than that of the state-of-the-art catalysts reported in the literature (Fig. 3f and Supplementary Table S2). When the η reached 590 mV (at 0.1 V), the \({{{{{\rm{FE}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\) got about 65% on Cu50Co50 (Fig. 3d and Supplementary Fig. S23). In comparison, the \({{{{{\rm{FE}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\) was around 21% on Cu at 0.1 V (Fig. 3d), with NO2− as the main product. As the electrode potential shifted negatively, the intermediate NO2− was rapidly reduced (Supplementary Fig. S23). At −0.2 V, the \({{{{{\rm{FE}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\) on Cu50Co50 achieved 100 ± 1%, and \({j}_{{{{{{\rm{NH}}}}}}_{3}}\) reached 1035 mA cm−2 (Supplementary Fig. S24b), corresponding to an NH3 production rate of 4.8 mmol cm−2 h−1 that is about two and eight times higher than the ones obtained on monometallic Co and Cu (Fig. 3e and Supplementary Fig. S25), respectively. Based on the charge consumed during the CuCo electrodeposition on the Ni foam substrate, the mass activity of NH3 yield (\({{{{{\rm{Yield}}}}}}_{{{{{\rm{mass}}}}}-{{{{{\rm{NH}}}}}}_{3}}\)) on Cu50Co50 was estimated roughly to 960 mmol gcat−1 h−1 at 0.2 V. The obtained \({{{{{\rm{Yield}}}}}}_{{{{{\rm{mass}}}}}-{{{{{\rm{NH}}}}}}_{3}}\) was slightly underestimated since hydrogen evolution was observed during CuCo electrodeposition. It should be noted that the production of NH3 from electrocatalytic NO3−RR is currently at a laboratory scale. More follow-up pilot tests and scale-up work are required to meet the industrial demands. Based on the preliminary calculations, we consider that our work inspired by the bifunctional nature of nitrite reductase, provides a new expectation and shows a great prospect, and after further development could compete with the well-established Haber–Bosch process12,32 which currently shows a \({{{{{\rm{Yield}}}}}}_{{{{{\rm{mass}}}}}-{{{{{\rm{NH}}}}}}_{3}}\) of ca. 200 mmol gcat−1 h−1 at industrial scale. The potential window for \({{{{{\rm{FE}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\) above 90% was wide and ranged from −0.1 to −0.4 V (Fig. 3d). Ten cycles of electrolysis at constant potential were performed to check the stability of Cu50Co50 (Fig. 3g) and the results displayed a stable \({{{{{\rm{FE}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\) exceeding 90% over the cycles. According to the SEM analysis, the decay of the yield for NH3 would be due to the nanosheet agglomeration after the consecutive recycling tests (Supplementary Fig. S26a, b). Furthermore, XRD (Supplementary Fig. S26c) and XPS (Supplementary Fig. S26d, e) analysis was also performed on the samples after the consecutive recycling tests, and the results demonstrated negligible changes in the chemical compositions and oxidation states, which confirmed the excellent stability of the catalyst.
The concentration of NO3− in real wastewater can vary from 0.88 mmol L−1 to 1.95 mol L−1 33. Therefore, NO3−RR on Cu50Co50 was performed at a wide range of NO3− concentrations (1–100 mmol L−1). The \({{{{{\rm{FE}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\) was maintained above 95% in the whole range of NO3− concentrations (Supplementary Fig. S27) at −0.1 V. The NO3−RR in neutral conditions was carried out on Cu50Co50 catalysts at −0.2 V in an electrolyte solution of 0.1 M KNO3 + 0.5 M K2SO4. The current density of NO3−RR on Cu50Co50 in a neutral condition was higher than the one on monometallic Cu and Co, and the \({{{{{\rm{FE}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\) over Cu50Co50 was more than 90% (Supplementary Fig. S28). These experiments demonstrated the excellent property of Cu50Co50 toward NO3− recovery in various environmental wastewater systems. In batch conditions with an initial nitrate’s concentration of 100 mmol L−1 (ca. 6200 ppm) at a reduction potential of −0.1 V and after 10 h, the nitrate’s removal efficiency reached 99.5%, with a \({{{{{\rm{FE}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\) of 96% (Fig. 3h). The remaining NO3− in the solution was 31 ppm which was much lower than the limitations fixed by the World Health Organization for drinking water, (i.e., 50 ppm)34. Several processes can be then considered for further extracting NH3, such as air stripping, ion exchange, struvite precipitation, etc.35.
Electrochemical in situ FTIR, SHINERS and DFT calculations were conducted to elucidate the reaction mechanism as well as the origin of the different activities observed between the catalysts.
Electrochemical in situ FTIR analysis of NO3 −RR
The electrochemical thin-layer in situ FTIR can track intermediates in solution within the thin-layer (thickness around 10 μm) between the electrode and IR window and species adsorbed on the electrode surface36. A reference spectrum (RRef) at reference potential (ER, 0.4 V) was firstly acquired, and then the potential was stepped to studied potentials (ES) and to collect working spectra (RS). The resulting spectra were represented as relative changes in the reflectance: ΔR/R = (RS-Rref)/RRef. As a result, the downward band in the resulting spectra indicated the formation of NO3− intermediates at ES, while the upward band referred to the consumption of NO3−. The FTIR peaks observed on Cu50Co50, Cu, and Co are compiled in Supplementary Table S3. As illustrated in Fig. 4, in the potential range from 0.4 to −0.2 V, the absorption bands were assigned to intermediates present in the electrolyte, since the wavenumbers of all the absorption bands were independent of the working potential37. In Fig. 4a, five obvious absorption bands appeared in the infrared spectra of Cu50Co50 viz. (1) At the working potential of 0.2 V, close to the onset potential of the LSV curve, the upward absorption bands at 1392 and 1354 cm−1 were ascribed respectively to N-O symmetric and asymmetric stretching vibration of NO3−38, indicating consumption of NO3− species in the thin layer; (2) at the same time, the downward band at 1236 cm−1 appeared and was attributed to N-O antisymmetric stretching vibration of NO2−39, indicating NO2− formation from NO3− reduction; (3) with potential negatively moving to 0.1 V, another intermediate observed around 1110 cm−1 was ascribed to -N-O- stretching vibration of hydroxylamine (NH2OH)39,40, which was a key intermediate for NH3 formation; (4) The upward band around 1638 cm−1 was attributed to water electrolysis responsible of hydrogen generation involved in the hydrodeoxidation of NO3− in the solution of thin-layer41.

Electrochemical thin-layer in situ FTIR spectra of NO3−RR on Cu50Co50 (a), Cu (b), and Co (c) in 100 mM KNO3 + 1 M KOH. d \(\frac{{{{{{\rm{I}}}}}}_{{{{{{\rm{NO}}}}}}_{2}^{-}}}{{{{{{\rm{I}}}}}}_{{{{{{\rm{NH}}}}}}_{2}{{{{\rm{OH}}}}}}+{{{{{\rm{I}}}}}}_{{{{{{\rm{NO}}}}}}_{2}^{-}}}\) ratio at different electrode potentials. ATR-FTIR spectra on Cu50Co50 (e), Cu (f), and Co (g) in 1 M KOH.
The FTIR spectra collected on Cu (Fig. 4b) were very similar to the Cu50Co50 catalyst’s ones, indicating that the NO3−RR behaviors were similar for both catalysts. However, the NO3− consumption on Cu at a potential of 0.1 V was 100 mV more negative than the one obtained on Cu50Co50, indicating a better kinetic with the latter one. In addition, with the negative shift of the working potential, the band intensity of NO2− relative to the sum intensity of NO2− and NH2OH production (i.e.,\(\frac{{{{{{\rm{I}}}}}}_{{{{{{\rm{NO}}}}}}_{2}^{-}}}{{{{{{\rm{I}}}}}}_{{{{{{\rm{NH}}}}}}_{2}{{{{\rm{OH}}}}}}+{{{{{\rm{I}}}}}}_{{{{{{\rm{NO}}}}}}_{2}^{-}}}\)) on Cu50Co50 dropped down sharply, while the \(\frac{{{{{{\rm{I}}}}}}_{{{{{{\rm{NO}}}}}}_{2}^{-}}}{{{{{{\rm{I}}}}}}_{{{{{{\rm{NH}}}}}}_{2}{{{{\rm{OH}}}}}}+{{{{{\rm{I}}}}}}_{{{{{{\rm{NO}}}}}}_{2}^{-}}}\) ratio was almost independent of the potential on Cu (Fig. 4d). The two phenomena, i.e., the appearance of NH2OH after the formation of NO2− and the increase of NH2OH at the expense of the consumption of NO2−, suggested that *NH2OH was obtained by the deep hydrogenation of *NO2 42. The \(\frac{{{{{{\rm{I}}}}}}_{{{{{{\rm{NO}}}}}}_{2}^{-}}}{{{{{{\rm{I}}}}}}_{{{{{{\rm{NH}}}}}}_{2}{{{{\rm{OH}}}}}}+{{{{{\rm{I}}}}}}_{{{{{{\rm{NO}}}}}}_{2}^{-}}}\) ratio on Cu50Co50 was much lower than the one on Cu catalyst, demonstrating that alloying Cu with Co could deeply enhance the hydrogenation of *NO2 to the final product NH3 on Cu/Co alloy catalysts. Thereby we speculated that Co sites were responsible for the variation of the \(\frac{{{{{{\rm{I}}}}}}_{{{{{{\rm{NO}}}}}}_{2}^{-}}}{{{{{{\rm{I}}}}}}_{{{{{{\rm{NH}}}}}}_{2}{{{{\rm{OH}}}}}}+{{{{{\rm{I}}}}}}_{{{{{{\rm{NO}}}}}}_{2}^{-}}}\) ratio, owing to their excellent protons’ adsorption (*H) capacity, as it will be demonstrated in the next section, promoting the hydrodeoxidation of *NO2 to *NH2OH according to Eqs. (3) and (4):43
On pure Co (Fig. 4c), no spectra bands of NO3−RR were detected until the working potential negatively shifted to 0 V. The absence of NO2− on the Co catalyst suggested a low NO2− accumulation in the thin layer solution, corroborating very well first the results obtained from the K–L equation where the Co catalyst was more inclined to perform continuous hydrogenation of NO3− to NH3 via an 8-electrons transfer and second the lower Tafel slope in the Peak S2 region data.
In comparison with thin-layer in situ FTIR, attenuated total reflection in situ FTIR analysis (ATR-FTIR) is more sensitive to the signal of adsorbed species on catalysts’ surfaces44. Two weak vibration bands of adsorbed NO in two different adsorption modes (“bridge” and “on top”) were detected at 1557 and 1639 cm−1 on the Cu50Co50 catalyst (Supplementary Fig. S29a)38,45. Interestingly, a downward band center at 2109 cm−1 at 0.5 V was observed in NO3–free electrolyte on Cu50Co50 (Fig. 4e), and the band center was negatively shifted to 2085 cm−1 at −0.4 V, yielding a Stark turn rate of 26.7 cm−1 V−1. These IR features could be attributed to adsorbed *H on the Cu50Co50 surface, and indicated also the enhancement of *H adsorbed on the Cu50Co50 surface at a higher η, which is consistent with the reported studies46,47. The fact that *H was observed on Cu50Co50 and Co (around 2110 cm−1) (Fig. 4e, g and Supplementary Fig. S30c) while being absent on Cu (Fig. 4f and Supplementary Fig. S30b), indicated that *H on Cu50Co50 was mainly attributed to water dissociation on Co sites. When the potential negatively shifted, the intensity of Co-H gradually raised, demonstrating that more *H were generated which in turn enhanced the hydrodeoxidation reaction and gave a lower \(\frac{{{{{{\rm{I}}}}}}_{{{{{{\rm{NO}}}}}}_{2}^{-}}}{{{{{{\rm{I}}}}}}_{{{{{{\rm{NH}}}}}}_{2}{{{{\rm{OH}}}}}}+{{{{{\rm{I}}}}}}_{{{{{{\rm{NO}}}}}}_{2}^{-}}}\) ratio value. The wavenumber attributed to *H on Co was slightly higher than the one on Cu50Co50, indicating a stronger affinity of *H for pure Co catalyst. In the presence of NO3−, Co-H was still present in the spectra of pure Co (Supplementary Fig. S30c), while it was vanished in the spectra of Cu50Co50 (Supplementary Fig. S30a). This could be explained by the fact that *H could adsorb quickly on Co sites inhibiting the adsorption of NOx, then the adsorbed hydrogen can react with the equivalent amount of *NOx on nearby Cu sites giving a high reaction kinetics. In the case of monometallic Co, the active sites are majorly occupied by *H species preventing the adsorption of NOx onto the catalyst’s surface, leading to weak activity. The presence of active hydrogen (H*) in the reaction process was also verified by electron paramagnetic resonance (EPR) analysis on Cu50Co50 catalyst (Supplementary Fig. S31) using 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a spin trap48. The intensity of the EPR signal of the DMPO-H adduct on the Cu50Co50 catalyst decreased when nitrate was added into the electrolyte, indicating the consumption of active hydrogen during NO3−RR. These results were consistent with the ATR-FTIR analysis. Based on the coupling of thin-layer in situ FTIR and ATR-FTIR analysis, therefore, we proposed the following pathway for the NO3−RR on Cu50Co50: NO3− → *NO3 → *NO2 → *NH2OH → *NH3, where *H on Co sites can promote the deep hydrodeoxidation of NO2− to NH2OH.
SHINERS analysis of NO3 −RR
The reaction intermediates provided by electrochemical in situ FTIR spectra were still insufficient to figure out the overall roadmap of NO3−RR to NH3, in this aim, SHINERS spectra of the catalysts were collected to probe the catalysts’ surface during the reaction. Supplementary Table S4 compiled the Raman scattering peaks observed during NO3− reduction on Cu50Co50, Cu, and Co, which were not detected on Au@SiO2 (Supplementary Fig. S32). The wavenumber below 750 cm−1 corresponds mainly to the chemical properties of the catalyst’s surface43. SHINERS spectra of Cu50Co50, Cu, and Co, in this section, were summarized and shown in Fig. 5. At 0.6 V, a characteristic band at 625 cm-1 associated with Cu2O49 was observed on Cu and Cu50Co50 (Fig. 5a, b), indicating partial oxidation of the catalysts surface due to air exposition before NO3−RR; these results were consistent with the XPS data. As the working potential decreased to 0.3 V, the band intensity of Cu2O gradually shrank, and a peak at 714 cm−1 emerged in replacement and was associated with the bending mode of free Cu-OHad50,51, indicating a gradual reduction of Cu before NO3−RR started to occur at 0.2 V. Besides, the band at 431 cm−1 can be assigned to Cu-Ox due to the adsorption of oxynitride on Cu surface51, and the band’s intensity increased as the potential moved negatively. For Co (Fig. 5c), a band at 568 cm−1 was also observed, associated with the formation of Co-Ox caused by the same oxynitride species adsorbed on the catalyst’s surface52.

SHINERS spectra between 230–750 cm−1 on Cu50Co50 (a), Cu (b), and Co (c). SHINERS spectra between 750–1700 cm−1 on Cu50Co50 (d) in 100 mM KNO3 + 10 mM KOH during cathodic polarization from 0.7 to −0.1 V.
In addition, the signals of NO3− and its reduced intermediates adsorbed on the catalysts’ surface were observed in the spectra between 750 and 1700 cm−1. As the potential decreased from 0.7 to −0.1 V, several peaks appeared in sequences on Cu50Co50 (Fig. 5d) viz. (1) At open circuit potential (OCP) and 0.7 V, the only obvious peak viewed at around 1049 cm−1 was attributed to the symmetric NO3− stretching from solution NO3− species53. (2) When the potential was decreased to 0.4 V, the NO3− species in the solution started to adsorb on the surface of Cu50Co50 since four peaks appeared in three different forms as NO stretching vibration from the unidentate nitrate near 998 cm−1, symmetric and antisymmetric stretching vibration of the NO2 group from the chelated nitro configuration around 1125 and 1254 cm−154, and the N=O stretching vibration of the bridged nitro group closed to 1439 cm−1, respectively55. (3) As the working potential shifted negatively further to 0.2 V, the symmetric NO3− stretch of *NO3− appeared around 1028 cm−1 56,57 and symmetric bending vibrations of the HNH near 1315 and 1374 cm−1 were apparent;55,57 N=O stretch of HNO56 was visible around 1540 cm−1. (4) When the potential negatively shifted further to 0.1 V, the HNO peak near 1540 cm−1 disappeared quickly; meanwhile, the antisymmetric bending vibration of the HNH of NH3 at 1591 cm−1 came out58, which indicated an efficient formation of NH3 from HNO hydrodeoxidation.
The SHINERS spectra on Cu and Co catalysts (Supplementary Fig. S33a,b) were similar to the one of Cu50Co50, but a new peak close to 800 cm−1 related to the bending vibration of NO2− group was only observed on Cu (Supplementary Fig. S33a)26. This could be due to the accumulation of NO2− species near by the Cu surface due to its poor ability for deep NO2− hydrodeoxidation properties. The intensity of \({\nu }_{{{{{{\rm{s}}}}}}({{{{{\rm{N}}}}}}{O}_{3}^{-})}\) from NO3− species in solution on Co at low overpotential was similar to the one on Cu50Co50 and Cu, however, the band’s intensities of all the intermediates formed on Co were very weak (Supplementary Fig. S33b), meaning that a low amount of *NO3− species and its derivates were adsorbed on the Co surface. It can be rationally speculated that the affinity of *H species to the Co surface was such strong that the active sites on the Co catalyst were majorly occupied by *H species leading to low coverage of *NO3− species, bringing a \({{{{{\rm{FE}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\) close to 85% (Fig. 3b) at the expense of a low current density of NO3−RR of 25.4 mA cm−2 (Supplementary Fig. S24a) at 0 V. Similarly, *H species affinity for Cu surface was so weak that most active sites were occupied by *NO3− species, leading to a relatively high current density up to 104.8 mA cm−2 (Supplementary Fig. S24a) and yet an unsatisfactory \({{{{{\rm{FE}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\) of 32% (Fig. 3b). Therefore only if a balanced coverage of *H and *NO3 species is achieved, a satisfactory \({{{{{\rm{FE}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\) and j can be simultaneously obtained, which is the case with Cu50Co50 where \({{{{{\rm{FE}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\) and j were 88% 394.6 mV respectively.
With combined electrochemical in situ FTIR and SHINERS spectroscopic analysis, the NO3−RR pathway on Cu, Co and CuCo was proposed as a series of deoxygenation reactions according to the following path, NO3− → *NO3 → *NO2 → *NO, accompanied by a series of hydrogenation reactions: *NO → *NOH → *NH2OH → * NH3 → NH3.
DFT calculations
Based on all the aforementioned observations and to shed light on the NO3−RR mechanism on Cu, Co, and CuCo at the atomic level, the density functional theory (DFT) calculations were conducted and the Gibbs free energies (ΔG) of *NO3 species and their derivates on Cu(111), Co(111) and CuCo(111) surface were presented in Fig. 6a.

Reaction-free energies for different intermediates of NO3-RR (a) and HER (b) at −0.2 V vs. RHE on CuCo(111), pure Cu(111), and pure Co(111) surface, respectively.
In terms of thermodynamics, the rate-determining step (RDS) on Cu(111), Co(111) and CuCo(111) was the initial *NO3 adsorption step and lower energy was required on CuCo(111) (0.17 eV), compared to Cu(111) (0.61 eV) and Co(111) (0.34 eV), indicating stronger adsorption of NO3− species on CuCo(111)15,59. Interestingly, the ΔG of the initial *NO3 species on Co(111) was lower than the one obtained on Cu(111), which should lead to better NO3−RR performances. However, this result contradicted the fact that \({\eta }_{{@10{{{{{\rm{mA}}}}}}{{{{{\rm{cm}}}}}}}^{-2}}\) of NO3−RR on Cu was 503 mV more positive than on Co (\({\eta }_{{@10{{{{{\rm{mA}}}}}}{{{{{\rm{cm}}}}}}}^{-2}}\) of 690 mV). This inconformity could be explained by a strong competition of *H species on the catalyst sites’ surface compared to nitrogenous species, as proved by in situ FTIR and SHINERS analysis. Indeed, Co was capable of promoting hydrogenation during the NO3−RR by transferring in situ *H species13, because of its excellent adsorption for *H species60. However, the excessive adsorption of *H can reduce the coverage of *NO3 species and their intermediates, thereby weakening the electrochemical activity toward NO3−RR. Compared with the ΔG of *NO3 and *H on Co(111) (Fig. 6b and Supplementary Table S11), the adsorption for *H was stronger, but the adsorption for NO3− was poor. With the incorporation of the Cu atom, the electronic structure changed, enhancing the adsorption of NO3− species on CuCo(111) and furthermore, the partial replacement of Co sites with Cu sites also reduced the coverage of *H species.
Based on the comprehension of the results discussed above, the redistribution of electrons in Cu50Co50 facilitated the electrons transfer rate and uplifted the catalytic kinetic of the NO3−RR (Supplementary Figs. S6, S7). The fair coverage of *H and *NO3 species were important to obtain simultaneously a high \({{{{{\rm{FE}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\) and \({j}_{{{{{{\rm{NH}}}}}}_{3}}\). Co-adjustment of Cu and Co sites on Cu50Co50 can balance the adsorption energy between *H and *NO3 species, not only by lowering the energy barrier of NO3− reduction but also by improving the hydrogenation capability with the enhanced *H adsorption compared with pure Cu, and at the same time avoiding the excessive *H occupation of the active sites compared to pure Co.
To conclude, we presented a novel Cu-based catalyst able to mimic the Cu core center of the Cu-NIR for the electrocatalytic NO3− reduction. The addition of Co to Cu formed a micro-pine structure with nanosheets and enhanced dramatically the proton availability over the surface of the catalyst from water electrolysis, resulting in an almost full Faraday efficiency of NH3 production at an ampere-level current density of 1035 mA cm−2 at −0.2 V. This process exhibited a high activity and could be proposed as a sustainable and eco-friendly complementary route of NH3 production. In batch conditions, the catalyst was able to achieve a \({{{{{\rm{FE}}}}}}_{{{{{{\rm{NH}}}}}}_{3}}\) of 96% and a nitrate’s removal of 99.5%, from an initial concentration of 6,200 ppm, reaching a final concentration of 31 ppm after 10 h of reaction, lower than the limitations fixed by the World Health Organization for drinking water. A mechanism was established by combining in situ FTIR and SHINERS spectroscopic investigations and DFT calculations. The synergy between Cu and Co can reduce the high energy barrier of the rate-determining step of the initial *NO3 adsorption step on Cu, due to a regulation of the electronic structure. The ΔG of the hydrogenation of the intermediate species, *NOx, could also be reduced due to the facile adsorption of *H species on Co(111) compared to Cu(111). We proposed that a rational control of the *H adsorption over the surface of the bimetallic catalyst is the key to managing further adsorption of intermediates from NO3−RR to achieve excellent performances. This discovery can provide an additional dimension to research into surface adsorption-modulated bimetallic catalysts for highly reactive hydrodeoxidation reactions and can provide a novel strategy for the development of multi-component heterogeneous catalysts for efficient NH3 production and wastewater treatment.
Methods
Preparation of CuxCoy, Cu, and Co catalysts
The catalysts were prepared via an electrodeposition process under the current density of 50 mA cm−2 for 300 s in a two-electrodes system where a platinum plate was used as the counter electrode and the Ni foam as the working electrode. Ni foams were pretreated with acetone and ethanol, and then repeatedly rinsed with ultrapure water and dried under a heating lamp. The deposition electrolyte (50 mL) comprised of 0.015 M trisodium citrate pentahydrate solutions and 50 mM CuSO4 + CoSO461,62. The CuxCoy catalysts with Cu:Co ratios of 65:35, 50:50, and 15:85, denoted as Cu65Co35, Cu50Co50, and Cu15Co85, were obtained in the deposition solutions with the ratio of CuSO4 to CoSO4 as 60:40, 45:55, and 10:90, respectively. The Cu and Co catalysts were prepared. The preparation for pure Cu and Co catalysts followed the same steps as the CuxCoy synthesis, except that only CuSO4 or CoSO4 solution was present in the electroplating solution. The catalysts were finally rinsed with ultrapure water and dried under the protection of Ar.
Material characterization
Morphology and elemental composition were characterized using a scanning electron microscope (SEM, ZEISS Sigma) with an energy dispersive X-ray spectrometer at an operating voltage of 15 kV. The lattice arrangement was observed using a high-resolution transmission electron microscope (HRTEM, FEI-Tecnai G2 F20) at an accelerated voltage of 200 kV. The chemical composition was analyzed by an inductively coupled plasma emission spectrometer (ICP-OES, Thermo Fisher iCap 7000). X-ray diffractometer (XRD, SmartLab-SE) with Cu-Kα X-ray source was used for crystal material structure analysis. Al Ka X-ray excited Thermo Fisher Scientific Nexsa X-ray Photoelectron spectrometer (XPS, Nexsa) was used for chemical state analysis. All XPS spectra were corrected with a C 1 s spectral line of 284.8 eV. X-ray absorption fine structure (XAFS) spectra at Cu K-edge and Co K-edge were obtained on the 1W1B beamline of Beijing Synchrotron Radiation Facility (BSRF) operated at 2.5 GeV and 250 mA. Standard data processing, including energy calibration and spectral normalization of the raw spectra was performed using Athena software.
Electrochemical test
The electrochemical measurements were performed using a three-electrodes system connected to the CHI 760E workstation (Chenhua, Shanghai) in a homemade H-type cell (separated by Nafion 117 membrane; with magnetic stirring of 1000 rpm). The Nafion 117 was preprocessed according to the reported procedures. The CuxCoy/Ni foams (0.5 cm2 × 0.5 cm2) were used as working electrodes, and platinum plate and Hg/HgO electrode (filled with 1 M KOH solution) were used as counter and reference electrodes, respectively. Before the testing, all the catalysts were electro-reduced at −0.2 V vs. RHE for 600 s in 1 M KOH solution to eliminate surface oxidation. 1 M KOH aqueous solution containing different KNO3− concentrations (5, 10, 50, and 100 mM) were used as an electrolyte (30 ml). The electrolyte was bubbled with Ar to remove O2 and N2 for 10 min before the experiment. The electrochemical linear voltammetry (LSV) curves were obtained in a single cell. The current density was normalized to the geometric electrode area (0.25 cm2) unless otherwise specified. The cyclic voltammetry curves in electrochemical double-layer capacitance (Cdl) determination were measured in a potential window where no Faradaic process occurred in an electrolyte of 1 M KOH at different scanning rates of 20, 40, 60, 80, and 100 mV s−1. All the potentials were converted to the RHE reference scale by E (V vs. RHE) = E (V vs. Hg/HgO) + 0.0591 × pH + 0.098. Note that NH3 volatilization in the 1 M KOH electrolytes (pH 13.6) is negligible during the 1-h electrolysis.
Kinetic evaluation
The electrochemical kinetic analysis of NO3−RR was performed based on the Koutecký–Levich (K–L) equation, as shown in Eq. (5):
Where im is the test current; iK is the kinetic current of NO3− reduction; n is the number of electrons transferred in the reaction; F is the Faraday constant, 96485 C mol−1; D represents the effective diffusion coefficient of 0.1 mol L−1 NO3− at 25 °C, 1.4 × 10−5 cm2 s−1; υ represents the kinematic viscosity of water at 25 °C, 1 × 10−6 m2 s−1; C is NO3− concentration, mmol L−1;ω is the electrode speed, rpm.
The working electrode was prepared as follows: (1) 5 mg of catalyst powder dropped down from CuxCoy/Ni foam and was dispersed in the solution of 600 μL isopropanol + 380 μL ultrapure water + 20 μL 5% Nafion solution, and then sonicated for at least 1 h to get a homogeneous ink; (2) 10 μL ink was drop-casted onto the rotating disk electrode (Ø, 0.196 cm2) with the loading of 0.255 mgcatalyst cm−2 for the further LSV analysis at different speeds (100, 225, 400, and 625 rpm) with a scan rate of 10 mV s−1. A platinum electrode and an Hg/HgO electrode were used as counter electrode and reference electrode, respectively. An aqueous solution of 1 mol L−1 KOH containing 100 mmol L−1 KNO3 was used as the electrolyte. Ar was used to purge the dissolved O2 and N2 from the electrolyte.
Product detection and efficiency calculation
The NH3 concentration was quantified by the Nessler Reagent method28. The electrolytes sampled after electrolysis for 1 h were first neutralized with 0.5 M H2SO4 and then mixed with 6.25 mL ultrapure water and 0.25 mL of Nessler reagent for the chromogenic reaction. The absorbance of the mixed solution was measured at a wavelength of 420 nm after keeping it at room temperature for 30 min. The quantitation of NH3 was performed by the standard curves, which was built using standard NH4Cl solutions in 1 M KOH. The 1H NMR (500 MHz) determination31 was also carried out to quantify the 14NH4+ and 15NH4+ after electrolysis at −0.2 V vs. RHE for 1 h. The electrolytes were mixed with 0.4 M H2SO4 at a ratio of 500:125 to ensure adequate protonation of NH3. Then, 125 μl of the diluted electrolytes or standard solution were mixed with 125 μl of 10 mM maleic acid in DMSO-D6, 50 μl of 4 M H2SO4, and 300 μl of H2O.
The NO3− and NO2− in the solution were quantitatively determined by ion chromatography (IC). The possible gas products of NO3−RR, such as H2, N2, NO, NO2, N2O, and NH3(g) were analysed using a gas chromatography (GC) and online electrochemical mass spectrometry (OEMS)30.
The Faradaic efficiency was calculated according to the following equation:
where c represents the concentration of the product, mol cm-3; Vcatholyte is the volume of catholyte, mL; Q is the total amount of charge consumed, C.
The yield rate of NH3 was calculated according to the following equations:
where S is the area of the geometrical cathode, cm-2; m is the mass of the catalyst on the cathode; t is the time of the electrolysis.
Electrochemical in situ FTIR reflection analysis
Electrochemical in situ FTIR reflection spectroscopy44. Electrochemical thin-layer in situ FTIR spectroscopy measurements were performed on a Nicolet Nexus 8700 FTIR spectrometer equipped with a liquid N2-cooled system and MCT-A detector. The glassy carbon electrode loading with catalysts was used as the working electrode, which was pressed vertically on the CaF2 window plate to form a thin liquid layer with a thickness of about 10 μm. A platinum foil and an Hg/HgO electrode (filled with 1 M KOH solution) were used as the counter electrode and reference electrode, respectively. The incoming infrared beam was approximately aligned with the normal electrode surface. Unless otherwise noted, the sample spectra were averaged from 200 interference spectra with a resolution of 8 cm−1. Reference spectrum (RRef) were collected at 0.4 V, and sample spectra (RS) were collected in the potential region from 0.4 V to −0.2 V and stepped by 100 mV. The spectra were reported as ΔR/R = (RS − RRef)/ RRef.
Attenuated Total Reflection in situ FTIR reflection spectroscopy36. The gold-plated Si prism with catalysts were assembled into a homemade spectral-electrochemical cell, which contained a carbon sheet as a counter electrode and Hg/HgO electrode as a reference electrode. It was then fixed in a homemade optics system built in the chamber of a Nicolet Nexus 8700 FTIR spectrometer for electrochemical ATR-FTIR measurements at an incidence angle of ca. 65°. The ATR-FTIR spectra were reported in the same way of thin-layer in situ FTIR, except that RRef was taken at 0.5 V and RS were collected in the potential region from 0.5 to −0.4 V.
SHINERS analysis
SHINERS spectra were recorded in a custom-made in situ Raman spectroelectrolysis cell using an XplorA confocal microprobe Raman spectrometer (HORIBA Jobin Yvon)63. The excitation wavelength of the laser was 637.8 nm and came from a He-Ne laser with a power of about 6 mW. The electrochemically polished gold electrode (diameter 3 mm) was modified by 10 μL catalyst ink with 10 μL homemade shell-isolated gold nanoparticle, which was provided by Prof. Jian-Feng Li at Xiamen University, China, and applied as the working electrode. Hg/HgO electrode was used as the reference electrode, and platinum wires was used as the counter electrode. A long-focus objective (8 mm) of A × 50 magnification was used. A Si wafer (520.6 cm−1) was used to calibrate the Raman frequency before the experiment. The SHINERS spectra were obtained using the cumulative results of four tests for 30 s each.
EPR Experiments
5,5-dimethyl-1-pyrroline N-oxide (DMPO) was used to capture the instable hydrogen radical to form the DMPO-H adduct to generate EPR spectra64. In the experiments, 5 ml electrolyte was mixed with 10 μL DMPO and was deoxygenated by bubbling Ar. The potentiostatic electrolysis was carried out for 5 min in the H-type cell under the protection of Ar. EPR measurement was performed by Bruker EMX-10/12 spectrometer operating at a frequency near 9.5 GHz, sweep width of 200 G and power of 20 mW.
DFT calculations
All DFT calculations in this work were carried out with the Vienna Ab initio Simulation Package (VASP)65. And the projector augmented-wave (PAW) pseudopotential was selected to deal with the core-valence interaction66. The generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE) was used to account for the exchange and correlation of electronics and the cut-off energy of plane-wave was 600 eV67. The energy convergence criterion was within 10-5 eV and the Hellmann–Feynman force was smaller than 0.01 eV Å−1 on each atom. The converged unit cell models of Cu (3.64 × 3.64 × 3.64 Å3), Co (3.52 × 3.52 × 3.52 Å3), and CuCo (3.78 × 3.49 × 3.49 Å3) were used in DFT calculations, respectively. The dimension of a 2 × 2 supercell of Cu (111) (8.91 × 10.28 Å2), a 2 × 2 supercell of Co (111) (8.62 × 9.95 Å2) and a 2 × 2 supercell of CuCo (111) (9.03 × 9.87 Å2) were used, respectively. These supercells were constructed and contained three layers and a sufficient vacuum layer of 15 Å thicknesses. For the structural optimization, the bottom two layers were fixed and the top layer was fully relaxed68. For unit cell geometry optimization, an 8 × 8 × 8 k-point analysis was used. A grid of 3 × 3 × 1 k-point mesh was used for these supercell calculations15. The calculations of all molecules and intermediate species on Co(111) and CuCo(111) were performed with spin polarization69. Dipole corrections in the z direction were included in all computations to minimize inaccuracies in the total energy because of simulated slab interactions. The spin polarization was not taken into account in the calculations of intermediate species on Cu(111) due to the spin polarization did not affect the Cu (111) calculations.
Data availability
The raw data of the figures in the main manuscript are available in figshare with the identifier(s) https://doi.org/10.6084/m9.figshare.21671075. All other data needed to evaluate the conclusions in the paper are present in the paper and the Supplementary Information or can be obtained from the corresponding authors on reasonable request.
Code availability
The code used in this work can be obtained from the corresponding authors on reasonable request.
References
Yao, F. et al. Highly selective electrochemical nitrate reduction using copper phosphide self-supported copper foam electrode: performance, mechanism, and application. Water Res 193, 116881 (2021).
Gao, J. et al. Electrocatalytic upcycling of nitrate wastewater into an ammonia fertilizer via an electrified membrane. Environ. Sci. Technol. 56, 11602–11613 (2022).
Ye, S. et al. Elucidating the activity, mechanism and application of selective electrosynthesis of ammonia from nitrate on cobalt phosphide. Energy Environ. Sci. 15, 760–770 (2022).
Nishina, K. New ammonia demand: ammonia fuel as a decarbonization tool and a new source of reactive nitrogen. Environ. Res. Lett. 17, 021003 (2022).
Liu, X., Elgowainy, A. & Wang, M. Life cycle energy use and greenhouse gas emissions of ammonia production from renewable resources and industrial by-products. Green. Chem. 22, 5751–5761 (2020).
Liu, H. et al. Efficient electrochemical nitrate reduction to ammonia with copper-supported rhodium cluster and single-atom catalysts. Angew. Chem. Int. Ed. 61, e202202556 (2022).
Wu, Z. Y. et al. Electrochemical ammonia synthesis via nitrate reduction on Fe single atom catalyst. Nat. Commun. 12, 2870 (2021).
Min, Y. et al. Integrating single-cobalt-site and electric field of boron nitride in dechlorination electrocatalysts by bioinspired design. Nat. Commun. 12, 303 (2021).
Li, Y. et al. Enzyme mimetic active intermediates for nitrate reduction in neutral aqueous media. Angew. Chem. Int. Ed. 59, 9744–9750 (2020).
Kuypers, M. M. M., Marchant, H. K. & Kartal, B. The microbial nitrogen-cycling network. Nat. Rev. Microbiol. 16, 263–276 (2018).
Sundararajan, M., Hillier, I. H. & Burton, N. A. Mechanism of nitrite reduction at T2Cu centers: electronic structure calculations of catalysis by copper nitrite reductase and by synthetic model compounds. J. Chem. Phys. B 111, 5511–5517 (2007).
Li, J. et al. Efficient ammonia electrosynthesis from nitrate on strained ruthenium nanoclusters. J. Am. Chem. Soc. 142, 7036–7046 (2020).
Liu, M. J. et al. Catalytic performance and near-surface X-ray characterization of titanium hydride electrodes for the electrochemical nitrate reduction reaction. J. Am. Chem. Soc. 144, 5739–5744 (2022).
He, W. et al. Splicing the active phases of copper/cobalt-based catalysts achieves high-rate tandem electroreduction of nitrate to ammonia. Nat. Commun. 13, 1129 (2022).
Wang, Y. et al. Enhanced nitrate-to-ammonia activity on copper-nickel alloys via tuning of intermediate adsorption. J. Am. Chem. Soc. 142, 5702–5708 (2020).
Yuan, C. et al. Growth of ultrathin mesoporous Co3O4 nanosheet arrays on Ni foam for high-performance electrochemical capacitors. Energy Environ. Sci. 5, 7883–7887 (2012).
Zhang, G. Q., Wu, H. B., Hoster, H. E., Chan-Park, M. B. & Lou, X. W. Single-crystalline NiCo2O4 nanoneedle arrays grown on conductive substrates as binder-free electrodes for high-performance supercapacitors. Energy Environ. Sci. 5, 9453–9456 (2012).
Zhang, Y. et al. Exhaustive conversion of inorganic nitrogen to nitrogen gas based on a photoelectro-chlorine cycle reaction and a highly selective nitrogen gas generation cathode. Environ. Sci. Technol. 52, 1413–1420 (2018).
Zhang, Y. et al. Electrochemical reduction of nitrate via Cu/Ni composite cathode paired with Ir-Ru/Ti anode: High efficiency and N2 selectivity. Electrochim. Acta 291, 151–160 (2018).
Baricco, M., Rizzi, P. & Enzo, S. X-ray diffraction study of nanocrystalline phases formation in metallic systems. Mater. Sci. Forum 278-281, 559–564 (1998).
Hsieh, H. H. et al. Electronic structure of Ni-Cu alloys: the d-electron charge distribution. Phys. Rev. B 57, 15204–15210 (1998).
Wang, C. et al. Regulation of d-band electrons to enhance the activity of Co-based non-noble bimetal catalysts for hydrolysis of ammonia borane. ACS Sustain. Chem. Eng. 8, 8256–8266 (2020).
Smith, M. L., Campos, A. & Spivey, J. J. Reduction processes in Cu/SiO2, Co/SiO2, and CuCo/SiO2 catalysts. Catal. Today 182, 60–66 (2012).
Ma, F. et al. Graphene-like two-dimensional ionic boron with double dirac cones at ambient condition. Nano Lett. 16, 3022–3028 (2016).
Bhattacharjee, S., Waghmare, U. V. & Lee, S. C. An improved d-band model of the catalytic activity of magnetic transition metal surfaces. Sci. Rep. 6, 35916 (2016).
Bae, S.-E., Stewart, K. L. & Gewirth, A. A. Nitrate adsorption and reduction on Cu(100) in acidic solution. J. Am. Chem. Soc. 129, 10171–10180 (2007).
Molodkina, E. B. et al. Electroreduction of nitrate ions on Pt(111) electrodes modified by copper adatoms. Electrochim. Acta 56, 154–165 (2010).
Zhao, Y. et al. Ammonia detection methods in photocatalytic and electrocatalytic experiments: how to improve the reliability of NH3 production rates? Adv. Sci. 6, 1802109 (2019).
Andersen, S. Z. et al. A rigorous electrochemical ammonia synthesis protocol with quantitative isotope measurements. Nature 570, 504–508 (2019).
Shen, S. et al. Tuning electrochemical properties of Li-rich layered oxide cathodes by adjusting Co/Ni ratios and mechanism investigation using in situ X-ray diffraction and online continuous flow differential electrochemical mass spectrometry. ACS Appl. Mater. Interfaces 10, 12666–12677 (2018).
Hodgetts, R. Y. et al. Refining universal procedures for ammonium quantification via rapid 1H NMR analysis for dinitrogen reduction studies. ACS Energy Lett. 5, 736–741 (2020).
Humphreys, J., Lan, R. & Tao, S. Development and recent progress on ammonia synthesis catalysts for Haber–Bosch process. Adv. Energy Sustain. Res. 2, 2000043 (2020).
van Langevelde, P. H., Katsounaros, I. & Koper, M. T. M. Electrocatalytic nitrate reduction for sustainable ammonia production. Joule 5, 290–294 (2021).
World Health Organization. Water, S. A. H. T. Guidelines for Drinking-Water Quality (World Health Organization, 2022).
Xiang, S. et al. New progress of ammonia recovery during ammonia nitrogen removal from various wastewaters. World J. Microbiol. Biotechnol. 36, 144 (2020).
Zhou, Z.-Y., Wang, Q., Lin, J.-L., Tian, N. & Sun, S.-G. In situ FTIR spectroscopic studies of electrooxidation of ethanol on Pd electrode in alkaline media. Electrochim. Acta 55, 7995–7999 (2010).
Ye, J.-Y. et al. Ammonia electrooxidation on dendritic Pt nanostructures in alkaline solutions investigated by in-situ FTIR spectroscopy and online electrochemical mass spectroscopy. J. Electroanal. Chem. 819, 495–501 (2018).
Figueiredo, M. C., Souza-Garcia, J., Climent, V. & Feliu, J. M. Nitrate reduction on Pt(111) surfaces modified by Bi adatoms. Electrochem. Commun. 11, 1760–1763 (2009).
Pérez-Gallent, E., Figueiredo, M. C., Katsounaros, I. & Koper, M. T. M. Electrocatalytic reduction of nitrate on copper single crystals in acidic and alkaline solutions. Electrochim. Acta 227, 77–84 (2017).
Rosca, V., Beltramo, G. L. & Koper, M. T. M. Hydroxylamine electrochemistry at polycrystalline platinum in acidic media: a voltammetric, DEMS and FTIR study. J. Electroanal. Chem. 566, 53–62 (2004).
Ataka, K., Yotsuyanagi, Takao & Osawa, M. Potential-dependent reorientation of water molecules at an electrode/electrolyte interface studied by surface-enhanced infrared absorption spectroscopy. J. Chem. Phys. 100, 10664–10672 (1996).
de Vooys, A. C. A., Beltramo, G. L., van Riet, B., van Veen, J. A. R. & Koper, M. T. M. Mechanisms of electrochemical reduction and oxidation of nitric oxide. Electrochim. Acta 49, 1307–1314 (2004).
Le, Ru,E. C. & Etchegoin, P. G. Single-molecule surface-enhanced Raman spectroscopy. Annu. Rev. Phys. Chem. 63, 65–87 (2012).
Zhou, Z.-Y., Tian, N., Chen, Y.-J., Chen, S.-P. & Sun, S.-G. In situ rapid-scan time-resolved microscope FTIR spectroelectrochemistry: study of the dynamic processes of methanol oxidation on a nanostructured Pt electrode. J. Electroanal. Chem. 573, 111–119 (2004).
Agrawal, V. K. & Trenary, M. An infrared study of NO adsorption at defect sites on Pt(111). Surf. Sci. 259, 116–128 (1991).
Yao, Y., Zhu, S., Wang, H., Li, H. & Shao, M. A spectroscopic study on the nitrogen electrochemical reduction reaction on gold and platinum surfaces. J. Am. Chem. Soc. 140, 1496–1501 (2018).
Zhu, S. et al. The role of ruthenium in improving the kinetics of hydrogen oxidation and evolution reactions of platinum. Nat. Catal. 4, 711–718 (2021).
Liu, R. et al. Defect sites in ultrathin Pd nanowires facilitate the highly efficient electrochemical hydrodechlorination of pollutants by H*ads. Environ. Sci. Technol. 52, 9992–10002 (2018).
Deng, Y., Handoko, A. D., Du, Y., Xi, S. & Yeo, B. S. In situ Raman spectroscopy of copper and copper oxide surfaces during electrochemical oxygen evolution reaction: identification of CuIII oxides as catalytically active species. ACS Catal. 6, 2473–2481 (2016).
Bodappa, N. et al. Early stages of electrochemical oxidation of Cu(111) and polycrystalline Cu surfaces revealed by in situ Raman spectroscopy. J. Am. Chem. Soc. 141, 12192–12196 (2019).
Zhao, Y. et al. Speciation of Cu surfaces during the electrochemical CO reduction reaction. J. Am. Chem. Soc. 142, 9735–9743 (2020).
Orlovskaya, N. et al. Detection of temperature- and stress-induced modifications of LaCoO3 by micro-Raman spectroscopy. Phys. Rev. B 72, 014122 (2005).
Zhou, Z., Huang, G. G., Kato, T. & Ozaki, Y. Experimental parameters for the SERS of nitrate ion for label-free semi-quantitative detection of proteins and mechanism for proteins to form SERS hot sites: a SERS study. J. Raman. Spectrosc. 42, 1713–1721 (2011).
GOODGAME, D. M. L. & HITCHMAN, M. A. Studies of nitro and nitrito complexes. II. Complexes containing chelating NO2 groups. Inorg. Chem. 4, 721–725 (1964).
Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds (John Wiley & Sons, 2008).
Ling, Y., Mills, C., Weber, R., Yang, L. & Zhang, Y. NMR, IR/Raman, and structural properties in HNO and RNO (R = alkyl and aryl) metalloporphyrins with implication for the HNO-myoglobin complex. J. Am. Chem. Soc. 132, 1583–1591 (2010).
Butcher, D. P. & Gewirth, A. A. Nitrate reduction pathways on Cu single crystal surfaces: Effect of oxide and Cl−. Nano Energy 29, 457–465 (2016).
Wen, N. & Brooker, M. H. Ammonium carbonate, ammonium bicarbonate, and ammonium carbamate equilibria: a Raman study. J. Chem. Phys. 99, 359–368 (1995).
Hu, T., Wang, C., Wang, M., Li, C. M. & Guo, C. Theoretical insights into superior nitrate reduction to ammonia performance of copper catalysts. ACS Catal. 11, 14417–14427 (2021).
Sheng, W., Myint, M., Chen, J. G. & Yan, Y. Correlating the hydrogen evolution reaction activity in alkaline electrolytes with the hydrogen binding energy on monometallic surfaces. Energy Environ. Sci. 6, 1509–1512 (2013).
Silva, F. L. G. et al. Response surface analysis to evaluate the influence of deposition parameters on the electrodeposition of Cu–Co alloys in citrate medium. J. Appl. Electrochem. 38, 1763–1769 (2008).
Wu, W., Yang, F., Luo, M., Tian, Z. & Zhou, S. Electrodeposition of copper in a citrate bath and its application to a micro-electro-mechanical system. Acta Phys. Chim. Sin. 26, 2625–2632 (2010).
Su, M., Dong, J.-C. & Li, J.-F. In-situ Raman spectroscopic study of electrochemical reactions at single crystal surfaces. Electrochemistry 26, 54–60 (2020).
Wang, Y. et al. Structurally disordered RuO2 nanosheets with rich oxygen vacancies for enhanced nitrate electroreduction to ammonia. Angew. Chem. Int. Ed. 61, e202202604 (2022).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 78, 1396–1396 (1997).
Chen, G.-F. et al. Electrochemical reduction of nitrate to ammonia via direct eight-electron transfer using a copper–molecular solid catalyst. Nat. Energy 5, 605–613 (2020).
Prieto, G. et al. Design and synthesis of copper-cobalt catalysts for the selective conversion of synthesis gas to ethanol and higher alcohols. Angew. Chem. Int. Ed. 53, 6397–6401 (2014).
Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (NSFC) (Nos. 22002131, Y.-Y.L.) and China Postdoctoral Science Foundation (Grant No 2020M671963, Y.-Y.L.). X.-Y.H. thanks R.O. from Seoul National University for the fruitful discussions. We are thankful to the Beijing Synchrotron Radiation Facility (1W1B, BSRF) for their help with the characterizations.
Author information
Authors and Affiliations
Contributions
Y.-Y.L., J.-Y.F., & S.-G.S. contributed to the conception of the study. J.-Y.F. performed the experiments. S.-N.H. provided assistance with the HRTEM analysis. K.-M.Z. helped with the SHINERS analysis. G.L. helped with the electrochemical in situ FTIR analysis. Q.-Z.Z. performed the DFT calculations analysis. J.-Y.F. & Y.-Y.L. performed the data analyses and wrote the manuscript. X.-Y.H., O.A., & S.-G.S. contributed significantly to the analysis and manuscript preparation. The project was supervised by Y.-Y.L. and S.-G.S. All authors helped perform the analysis with constructive discussions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Chuan-Xin He, Jeonghoon Lim and the other anonymous reviewer(s) for their contribution to the peer review of this work. Peer review reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fang, JY., Zheng, QZ., Lou, YY. et al. Ampere-level current density ammonia electrochemical synthesis using CuCo nanosheets simulating nitrite reductase bifunctional nature. Nat Commun 13, 7899 (2022). https://doi.org/10.1038/s41467-022-35533-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-022-35533-6
This article is cited by
-
Urea electrosynthesis via an integrated Pd1-Cu interface strategy
Science China Materials (2026)
-
Boron-Insertion-Induced Lattice Engineering of Rh Nanocrystals Toward Enhanced Electrocatalytic Conversion of Nitric Oxide to Ammonia
Nano-Micro Letters (2026)
-
Cobalt-Based Electrocatalysts for Sustainable Nitrate Conversion: Structural Design and Mechanistic Advancements
Nano-Micro Letters (2026)
-
Efficient Neutral Nitrate-to-Ammonia Electrosynthesis Using Synergistic Ru-Based Nanoalloys on Nitrogen-Doped Carbon
Nano-Micro Letters (2026)
-
Multivariate covalent organic frameworks with tailored electrostatic potential promote nitrate electroreduction to ammonia in acid
Nature Communications (2025)
