
Abstract
Medicinal compounds from plants include bicyclo[3.3.1]nonane derivatives, the majority of which are polycyclic polyprenylated acylphloroglucinols (PPAPs). Prototype molecules are hyperforin, the antidepressant constituent of St. John’s wort, and garcinol, a potential anticancer compound. Their complex structures have inspired innovative chemical syntheses, however, their biosynthesis in plants is still enigmatic. PPAPs are divided into two subclasses, named type A and B. Here we identify both types in Hypericum sampsonii plants and isolate two enzymes that regiodivergently convert a common precursor to pivotal type A and B products. Molecular modelling and substrate docking studies reveal inverted substrate binding modes in the two active site cavities. We identify amino acids that stabilize these alternative binding scenarios and use reciprocal mutagenesis to interconvert the enzymatic activities. Our studies elucidate the unique biochemistry that yields type A and B bicyclo[3.3.1]nonane cores in plants, thereby providing key building blocks for biotechnological efforts to sustainably produce these complex compounds for preclinical development.
Similar content being viewed by others
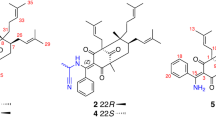

Introduction
The medicinal value of plants of the Hypericaceae and Clusiaceae families is linked to a variety of bioactive meroterpenes, which arise from the conjunction of terpene and polyketide biosyntheses. The meroterpenes include polycyclic polyprenylated acylphloroglucinols (PPAPs), a diverse molecular family, which is chemically characterised by the complex bicyclo[3.3.1]nonane scaffold. Since the first report of a PPAP (hyperforin) in 19711,2, this class has rapidly expanded as contemporary research continues to unravel new members with distinct structural elements3. The acyl group of PPAPs is either aliphatic or aromatic. PPAPs bearing the benzoyl group, i.e., phlorbenzophenone derivatives, account for the largest proportion of elucidated structures4,5. They are abundant in the traditional Chinese medicinal plant Hypericum sampsonii6. Clusianone and nemorosone, alongside their C-7 epimers, are among the most intensively studied PPAPs with a phlorbenzophenone core (Fig. 1a). Their established in vitro medicinal properties feature anti-viral7,8, anti-microbial9,10, anti-inflammatory11, and cytotoxic activities11,12. Similar bioactivities are exerted by their caged derivatives, hyperibone K and plukenetione A, respectively8,13,14. Aiming at harnessing their potential as valuable pharmacophores, pioneering studies demonstrated the total syntheses of these compounds in the previous decade15,16,17,18. Yet, innovative efforts to improve the access to the coveted bicyclo[3.3.1]nonane scaffold continue to impact the recent literature of organic chemistry19,20,21.

a Examples of PPAPs originating from phlorbenzophenone. b Hypothetic scheme depicting the biogenic conversion of phlorbenzophenone to bridged polycyclic derivatives4. Red dashed arrows indicate ring closure reactions. The newly added prenyl group and the polycyclic ring systems are highlighted in blue and orange, respectively. All homoadamantane-type PPAPs so far isolated have type A structures.
Despite the promising pharmacological outlook, biosynthesis of the complex PPAPs is largely obscure. Important milestones at the gene level were the assembly of the basic phloroglucinol core22 and the detection of the first plant-derived aromatic prenyltransferase (PT)23. However, the nature of the enzymatic machinery that catalyses the formation of the bicyclo[3.3.1]nonane skeleton remained unanswered. The following hypothesis for the biosynthetic assembly of the bridged polycyclic compounds was proposed4,24,25 (Fig. 1b). After polyprenylation of the phlorbenzophenone scaffold catalysed by aromatic PTs, one of the geminal prenyl side chains is activated by head-to-middle prenylation, resulting in the formation of a reactive carbocation intermediate. Subsequent ring closure between this carbocation and either the C-1 or the C-5 position via electrophilic addition yields the bridged bicyclic type A and B scaffolds, respectively. Whether these reactions are catalysed by PTs with additional cyclase functionality (‘prenylative cyclization’) or by PTs in concert with independent cyclases remained ambiguous. The position of the introduced C-7 prenyl group is either endo or exo with respect to the C-9 carbonyl. However, only the endo configuration is thought to be involved in the formation of metabolites with rare adamantane and homoadamantane scaffolds, which appear to be achieved by intramolecular cyclization between C-3 and C-27 or C-28, respectively24. An array of such caged tricyclic PPAPs was identified in H. sampsonii6,24.
Here we detect and characterise two bifunctional PTs from H. sampsonii, which catalyse regiodivergent prenylative cyclizations of the same gem-diprenylated phorbenzophenone substrate to yield the type A and B products 7-epi-nemorosone and 7-epi-clusianone, respectively. Substitution of a prenyl with a geranyl group results in the formation of nemosampsone, the undescribed but anticipated geranylated type A precursor of many bi- and tricyclic PPAPs in H. sampsonii. We use a combination of computational and mutational studies to investigate the underlying reaction mechanisms. Inverted substrate binding modes control the regiospecific reactions in the two active site cavities. Identification of a set of amino acid residues that stabilise the alternative binding modes allows for reciprocal mutagenesis and results in the conversion of the type A-forming enzyme into a type B cyclase by ninefold mutation. Furthermore, we use the sequence information obtained to identify a homologous enzyme in Hypericum perforatum, which forms hyperforin and secohyperforin from substrates bearing an aliphatic rather than aromatic acyl group.
Results and discussion
H. sampsonii plants contain type A and B PPAP prototypes
Reversed-phase high-performance liquid chromatography (RP-HPLC) of dichloromethane extracts from fresh leaf material of in vitro propagated and pot-cultivated H. sampsonii plants revealed a tractable signal landscape consisting of four major constituents and a few minor metabolites (Fig. 2a). Isolation and NMR spectroscopic elucidation disclosed the structures of the well-studied type A and B PPAP congeners 7-epi-nemorosone 3 and 7-epi-clusianone 5 (Supplementary Figs. 1–2). Interestingly, the analysis of another major constituent led to the discovery of a yet undescribed PPAP. The 1D and 2D NMR spectra of this compound closely resembled those of 3, except for the signals of an additional prenyl group at C-26. Because of this similarity, the compound is hereafter referred to as nemosampsone 4. The complete elucidation data of 4 is available in the Supplementary Information (Supplementary Table 1 and Supplementary Figs. 3–10). The fourth major constituent exhibited absorption in the visible spectrum, which was characteristic of hypericin. This naphthodianthrone is typical of Hypericum species but was not further analysed here. Crucially, examination of the minor extract components identified the putative direct precursors of the aforementioned PPAPs, namely grandone 1 and kolanone 2 (Supplementary Figs. 11–12). These intermediates were utilised as acceptor substrates in subsequent PT assays.

a HPLC-DAD analysis of a dichloromethane extract from H. sampsonii leaves. b Enzyme assays containing HsCPTa, DMAPP, and either grandone 1 (blue chromatogram) or kolanone 2 (orange chromatogram) as acceptor substrates yielded the products 7-epi-nemorosone 3 and nemosampsone 4, respectively. c Enzyme assays containing HsCPTb, DMAPP, and grandone 1 (blue chromatogram) yielded 7-epi-clusianone 5. Kolanone 2 (orange) was not transformed. d No conversions were observed in control assays containing microsomes from empty vector-transformed yeast cells. The enzyme assays are shown at their respective optimal absorbance wavelengths between 272–360 nm, whereas the leaf extract chromatogram was recorded at 292 nm. *, hypericin (naphthodianthrone).
Interestingly, all PPAPs elucidated here, including the newly described compound 4, were type A and B prototypes without further chemical functionalisation. In contrast, many PPAPs reported for H. sampsonii comprise secondary ring closures, which were hypothesised to require the formation of activated epoxide intermediates24,26. The resulting pyran-fused or adamantyl-like consolidations are structural elements of many elucidated hypersampsone26 and sampsonione type6,24 molecules (Supplementary Fig. 13). In vitro propagated and pot-cultivated H. sampsonii plants used in this study contained high levels of 3 and 4, which are direct precursors of the adamantane PPAPs plukenetione A and otogirinin A, respectively. This accumulation is presumably due to the downregulation of the yet unidentified late pathway steps leading to caged PPAPs. Compounds 3 and 4 are supposed to have a high turnover in the plant’s metabolism, given the high number of isolated compounds postulated as their putative downstream products. In contrast, 5 has been commonly identified in H. sampsonii extracts6, which might be related to the comparatively low diversity of reported type B derivatives.
Previous rigorous NMR studies pointed out that close examination of 1H and 13C shifts provides valuable information about the relative configuration of the C-7 prenyl group of PPAPs27. According to the 13C shifts of C-7 and the geminal methyl groups attached to C-8, along with the 1H shifts at the diastereotopic C-6 position, all PPAPs investigated here (3–5) have the C-7 endo configuration (Supplementary Table 2). These findings further support the theory that C-7 endo stereochemistry is important for the assembly of caged scaffolds, as spatial proximity to C-3 may be required for intramolecular cyclization.
Conspicuous hybrid aspartate-rich motif suggests target sequences
A transcriptomic database was constructed de novo using H. sampsonii RNA. Based on the working hypothesis that bifunctional PTs, which catalyse both prenylation and cyclization, may be the target catalysts, HsPT8px served as a probe to identify homologous amino acid sequences. HsPT8px and HsPTpat are the only aromatic PTs characterised from H. sampsonii28(NCBI accession numbers AZK16224.1 and AZK16225.1, respectively). They catalyse the successive gem-diprenylation of tetrahydroxyxanthone, which is a direct descendant of the phlorbenzophenone framework (Supplementary Fig. 14). Phylogenetic analysis of the 13 transcriptomically selected homologues suggested a xanthone-specific clade that accommodated HsPT8px but did not help uncover putative bifunctional PT candidates (Supplementary Fig. 15). However, closer examination of the two conserved aspartate-rich loop regions responsible for the coordination of metal ion cofactors was able to guide the selection of two PT candidate sequences (Fig. 3). These conserved motifs are present in all plant PTs and known to comprise distinct canonical sequences in aromatic PTs of the homogentisate (HG) and p-hydroxybenzoate (PHB) families29. Interestingly, all homologues assembled in this study shared the expected HG-specific motifs, with the exception of candidates 2948c0g1 and 2948c0g2. These two sequences had unique hybrid motifs, which included a PHB-like sequence in loop 2 and retained the HG consensus in loop 6, making them stand out from previously characterised PTs. Their shared amino acid sequence identity was 64.7% at 96.2% coverage (69.7% identity at 100% coverage outside the first 100 residues including the plastidial signal peptide), while the nearest putative PT homologue shared 51.4% and 51.0% sequence identity with 2948c0g1 and 2948c0g2, respectively.

a Representative PT topology with highlighted metal ion binding motifs (aspartate-rich regions). b All transcriptomically selected candidates had the expected canonical sequences of homogentisate (HG) PTs, except for two (red), which contained an unusual combination of a p-hydroxybenzoate (PHB)-like sequence as the first motif and a HG-like sequence as the second. The PHB enzymes listed are examples of previously studied family members.
HsCPTa and HsCPTb catalyse prenylative cyclizations to yield type A and B PPAPs
The two cDNA sequences 2948c0g1 and 2948c0g2 were cloned into the episomal vector pESC-URA, which enables gene expression in yeast (Saccharomyces cerevisiae) under the control of the inducible GAL1 promoter. Microsomal fractions containing the recombinant proteins were incubated in PT assays. Both enzymes catalysed prenylative cyclization, using 1 as acceptor substrate and dimethylallyl pyrophosphate (DMAPP) as donor substrate (Fig. 2b, c). Interestingly, the regiospecificities of the cyclization reactions differed. The type A product 3 was formed by the translation product of transcript 2948c0g1 and the enzyme was named HsCPTa (Fig. 2b). In contrast, the type B product 5 was formed by the translation product of transcript 2948c0g2 and the enzyme was named HsCPTb (Fig. 2c). HsCPTa was also able to accept substrate 2, resulting in the formation of 4 as another type A product. Both enzymes failed to accept geranyl pyrophosphate (GPP) as the donor substrate. Control assays containing microsomes from empty vector-transformed yeast cells lacked enzyme activity (Fig. 2d). Furthermore, assays with phlorbenzophenone confirmed that HsCPTa and HsCPTb, unlike other aromatic PTs, failed to catalyse the transfer of a prenyl group to the aromatic core, underlining their specific role in the catalysis of alkene-intercepted prenylation. All enzymatic products were identified by comparing their retention times, UV spectra, and tandem-MS fragmentation patterns with those of the structure-elucidated constituents of H. sampsonii leaves (Supplementary Fig. 16).
These findings highlight that plants are able to regulate the biosynthesis of type A and B molecules by employing highly specialised enzymes. Their kinetic characterisation revealed similar parameters for the common acceptor substrate, grandone 1, which were comparable to the data reported for the patulone forming enzymes, HsPT8px and HsPTpat, used above as probes. HsCPTa preferentially accepted grandone 1 with a KM matching that of DMAPP (62.01 ± 10.06 µM and 65.44 ± 6.63 µM, respectively). The affinity to the secondary acceptor substrate, kolanone 2, was lower (114.24 ± 20.03 µM; Supplementary Fig. 17). HsCPTb exclusively accepted grandone 1 with an affinity comparable to that of HsCPTa (40.36 ± 5.06 µM). It had a relatively low affinity towards DMAPP (297.79 ± 29.49 µM), which is still in the range reported for aromatic PTs30. While optimising the assay parameters, the preferred pH values were found to differ. While HsCPTa functioned best at pH 7, HsCPTb reached the highest activity at pH 10 (Supplementary Fig. 17). This prompted us to investigate whether the enzymes might be present in different subcellular compartments in planta. However, localisation of translational reporter fusions in transiently transformed Nicotiana benthamiana leaves provided evidence that both enzymes are associated with the chloroplast envelope (Supplementary Fig. 18). Although the outer or inner membrane of the chloroplast envelope may be differentially targeted by the enzymes, the observed discrepancy in the pH optima is likely to be an artifact resulting from the in vitro reconstruction of the enzymatic reactions. In fact, there is evidence that some aromatic PTs are involved in heteromeric enzyme complexes31, potentially adding to other effects of their specific membranous microenvironment.
To confirm the detected activities of HsCPTa and HsCPTb in the plant environment, we incubated disk sections of transiently transformed N. benthamiana leaves in the presence of acceptor substrate 1. Consistent with the results of the in vitro assays, the type A product 3 was exclusively detected in leaves expressing HsCPTa (Fig. 4a), whereas the type B product 5 was solely formed in samples expressing HsCPTb (Fig. 4b). No enzymatic activity was observed in leaves infiltrated with a GFP-encoding control construct (Fig. 4c). Moreover, no significant differences in overall product yields were detected between leaves expressing HsCPTa or HsCPTb genes, as confirmed by three biological repeats.

The HsCPTa and HsCPTb genes were transiently expressed in N. benthamiana leaves, and transgenic disk sections were incubated with the acceptor substrate grandone 1. a, b Extracted ion chromatograms (m/z [M–H]– = 501.3010 ± 0.005) confirmed the formation of 7-epi-nemorosone 3 and 7-epi-clusianone 5 in leaves expressing HsCPTa and HsCPTb, respectively. c Neither product was observed in leaves infiltrated with the GFP-encoding control construct. d Authentic reference compounds.
Inverted substrate binding modes result in type A and B PPAPs
In molecular docking simulations on refined in silico structures generated by AlphaFold2, the binding cavities predicted for HsCPTa and HsCPTb were too small to accommodate the extended geranyl chain of acceptor substrate 2 to generate docked conformations that could effectively explain the experimental results. To overcome this problem, we referenced the crystal structures of homologous UbiA-type PTs (PDB ID: 4tq3–6, 6m31, 6m34, 7BPU, 5OON). While differences in the core structure of the crystals (transmembrane helices αII–VII) are minimal, the relative positions of αI (N-terminal) and αVIII–IX (C-terminal) adapt to changes in substrate size. Thus, to allow the docking of 2 with minimal adjustments, we modelled the N- and C-terminal helices after 4tq3, which contains a hydrophobic tunnel for the putative binding of polyprenyl chains (Supplementary Fig. 19)32. We constructed the binding of Mg2+ ions and DMAPP based on the highly conserved aspartate-rich motifs of 4tq3 (Supplementary Fig. 20). According to the known initiation of prenyl transfer reactions, the dimethylallyl cation was dissociated from the pyrophosphate moiety, setting the C-O distance to 2.8 Å, which is common for such reaction transition states33,34. In addition, electron densities of the activated intermediate obtained from quantum mechanical calculations at the B3LYP-D3/(6-31 G + (d)) level in the gas phase indicated that a nucleophilic attack of C-1 and C-5 on the prenyl cation requires the deprotonation of a hydroxy group (C-4 or C-6). Consequently, we constrained putative hydrogen bonds between the hydroxy groups and potentially assisting deprotonation residues in the subsequent induced-fit docking procedure.
The binding modes of the acceptor substrate 1, which fit best the chemical logic of the reactions, specified binding energies of −105.76 kcal mol−1 and −85.48 kcal mol−1 in HsCPTa and HsCPTb, respectively. The binding poses of 1 rotate around the C-3 to C-6 axis of the phloroglucinol ring in the two enzymes, presenting opposing prenyl residues of the C-3 gem-diprenyl group towards the prenyl donor (Fig. 5 a, b). The trans-like conformation of the benzoyl moiety at C-1 and the prenyl residue at C-5 is characteristic of both binding modes. It ensures the regiospecificity of the cyclization reaction by restricting the access to the carbon with the upward-facing substituent. The OH group at C-6 of the acceptor substrate forms a hydrogen bond with the N153 residue of HsCPTa and the H142 residue of HsCPTb to facilitate the deprotonation. Furthermore, the specific binding mode in HsCPTa appears to be stabilised by π-π interactions between the benzoyl group of the substrate and the F111 side chain of the protein. In HsCPTb, the benzoyl group may turn towards the internal hydrophobic region of the binding cavity due to the polarity difference of the C100 residue.

Simulation included the aspartate-mediated coordination of Mg2+ ions (spheres), the interacting pyrophosphate moiety (orange), and the dissociated isoprenyl residue (turquoise) (for details of the conserved donor substrate binding, see Supplementary Fig. 20). a, b Inverted binding poses of acceptor substrate 1 in the active site cavities of HsCPTa (green) and HsCPTb (red). The two C-3 gem-prenyl residues involved in the reactions are distinguished by light and deep blue colours. Steric hindrance is introduced by the trans-like conformation of the benzoyl (C-1) and prenyl groups (C-5) with respect to the phloroglucinol ring plane, leading to the regiospecific ring closure reactions. c Docking of acceptor substrate 2 into the HsCPTa pocket allows the same reaction principle, which is not affected by the elongated geranyl group (deep blue). d In the inverted binding mode of HsCPTb, the upward-facing geranyl chain interferes with the binding of the donor substrate, preventing the reaction from proceeding. White surface, binding cavity; dashed red arrows, reaction sequence; dashed yellow lines, distance measure in Å.
Docking of substrate 2 into HsCPTa suggests a similar binding mode and reaction principle (Fig. 5c). The elongated C-3 geranyl chain does not affect the reaction mechanism, as it is buried in the hydrophobic part of the binding tunnel and thus faces away from the prenyl donor. However, in the inverted binding mode of HsCPTb (Fig. 5d), the bulky geranyl chain interferes with the binding of the donor substrate and no reaction proceeds. This simulation is in good agreement with the experimental results, given that 2 was only converted by HsCPTa.
The outlined binding modes are supported by the previously established relationships between the absolute configurations and the specific optical rotations of type A and B PPAPs19,35,36,37. Depending on which of the two C-3 gem-prenyl residues of the acceptor substrate is activated by prenyl transfer, the subsequent cyclization yields different enantiomers by bridging opposite sides of the phloroglucinol ring (Supplementary Fig. 21). Crucially, type A and B ring closures on opposite faces of the ring yield products with equal optical activities. As the relative configuration of (+)-3 can only be inferred from the type A framework of a synthetic (–)-nemorosone precursor19, studies of the Porco group, in combination with their earlier work on the synthesis of (–)-clusianone36, were critical to support the described relationships. The specific rotations measured for the H. sampsonii PPAPs 3, 4 and 5 were [α]D = +82.4, +110.3 and +46.7, respectively. Thus, the binding orientations of the acceptor substrate in HsCPTa and HsCPTb must be inverted to form the specific product rotamers.
Specific amino acid residues control the regiospecificities of the prenylative cyclizations
Mutational studies were designed to identify key amino acid residues that confer regiospecificity to the reactions. In a first holistic approach, we gradually transformed HsCPTb into HsCPTa by C-terminal exchange of incrementally larger fragments (Fig. 6a and Supplementary Fig. 22). Interestingly, up to 192 residues could be replaced without affecting catalytic efficiency and regiospecificity (chimera stages 1–3). Further substitutions rendered the chimeric HsCPTb inactive (stages 4–5). The substitution of 291 C-terminal residues gave rise to a chimera with restored functionality and switched regiospecificity (stage 6). Notably, the stage 3 inversed chimera confirmed that 123 N-terminal residues of both HsCPTb and HsCPTa are sufficient for the selective formation of their respective regiomers. These experiments highlighted a stretch of 96 amino acids as the primary region of interest. This was further substantiated by modelling and docking simulations with reciprocal HsCPTa and HsCPTb chimeras comprising the exchanged 96 residues region, which predicted inverted substrate binding modes similar to the docked conformations in their wild-type counterparts (Supplementary Fig. 23).

a Progressive C-terminal exchange of HsCPTb residues with corresponding fragments of HsCPTa identified a 96 amino acids region essential for regiospecificity (box). b Sequence alignment of this region of interest, its location in the predicted HsCPTa fold (opaque model), and its interface to the central cavity (grey surface). c Progressive conversion of the HsCPTa regiospecificity into the HsCPTb regiospecificity using expanding sets of amino acid substitutions. Total amounts of type A and B products obtained in single standardised reactions are indicated. Data are means ± SD of three biological replicates.
Subsequent site-directed mutagenesis focused on the divergent residues in this region, particularly those directly associated with the modelled binding pocket (Fig. 6b). An overview of all tested mutants is provided in Supplementary Fig. 24. The most insightful combinations of amino acid substitutions, which underpin the complete transformation of HsCPTa as a type A cyclase into a type B cyclase (Fig. 6c), are as follows. A single point mutation, F111C, located at the beginning of the first transmembrane helical segment (αI) completely abolished the regiospecificity of HsCPTa. While the incorporation of three additional mutations Y107F, T112S and Q115H (αI) slightly shifted the conversion in favour of the type B product, introduction of N153H into the second transmembrane helix (αII) solidified this change in selectivity. The overall diminished activity of the mutant was partially restored by including the additional point mutation M149V (αII). To ultimately produce a mutant activity equivalent to HsCPTb, the incorporation of three further mutations, I156M, E161D and V167I was found to be essential. The latter two were located at the first aspartate-rich motif of the enzyme.
Of the nine mutations identified to control the catalytic transformation of HsCPTa, seven (excluding M149V and V167I) were predicted to interact directly with the substrate binding pocket. While each of those seven substitutions cooperatively contributed to the stabilisation of the type B specific binding pose, the most dramatic effect was observed for mutations in the F111/C100 position. As hypothesised based on the obtained docking results, the F111 residue appears to be critical for ensuring type A regiospecificity, most likely by interacting directly with the C-1 benzoyl moiety of the acceptor substrate. In the F111C point mutant, this missing interaction seems to enable both binding orientations at the cost of diminished activity. The same observation could not be made in the completely inactivated HsCPTb C100F mutant. It appears that the bulky phenyl ring restricts the volume of the binding pocket, as the relatively small size of C100 would normally allow the C-5 prenyl chain of the substrate to protrude beyond this residue in the wild-type enzyme (Fig. 5b, d). Although not in direct proximity to the binding pocket, the two remaining mutations, M149V and V167I, are likely to influence the helix spacing between αI and αII or helix loops HL23 and HL67, respectively. Rather than affecting the regiospecificity, these substitutions appear to influence the overall performance of the enzyme.
HsCPTa and its H. perforatum homologue HpCPTa1 catalyse the formation of hyperforin analogues
PPAPs with aliphatic acyl groups, such as hyperforin and its derivatives, are renowned constituents of Hypericum perforatum (St. John’s wort, Supplementary Fig. 25), but uncommon in H. sampsonii. Metabolic profiling of H. sampsonii root, stem, and leaf extracts revealed the presence of varying levels of phlorbenzophenone derivatives in all tissues (Supplementary Fig. 26a, b). Interestingly, the root extract additionally contained two compounds, 6 and 7, with mass and UV signatures consistent with the well-studied phlorisobutyrophenone-derived PPAPs secohyperforin and hyperforin, respectively. However, comparison with available reference compounds revealed different retention times. Isolation and structure elucidation of compound 6 yielded NMR data very similar to those reported for the type A PPAP secohyperforin38 (Supplementary Table 3 and Supplementary Fig. 27). Crucially, the chemical shifts that differed from the reference values were associated with the critical positions C-6, C-7, and C-8. They were consistent with the values expected for endo-PPAPs (Supplementary Table 2). Thus, the structure of 6 was established as 7-epi-secohyperforin. In contrast, compound 7 that shared the same molecular mass as hyperforin was not unambiguously elucidated in this study due to its instability and limited amount. However, its MS/MS data resembled the pattern produced by nemosampsone 4 and clearly differed from that of hyperforin (Supplementary Fig. 28). Therefore, its structure is postulated to be the aliphatic nemosampsone analogue 7. Given the absence of further transcriptomic CPT candidates, we speculated that HsCPTa may be responsible for the formation of PPAPs with an aliphatic acyl group in the roots of H. sampsonii. This hypothesis was supported by RT-qPCR results, revealing a higher relative expression of HsCPTa than HsCPTb in the roots (Supplementary Fig. 26c).
To identify HsCPTa homologues from H. perforatum potentially involved in the final step of the biosynthesis of hyperforins (Supplementary Fig. 25), the HsCPTa sequence was used as a query to search the Medicinal Plant Genomics Resource database (MPGR, mpgr.uga.edu). The first BLAST hit (hpa_locus_470_iso_1_len_1384_ver_2), named HpCPTa1, shared 64.5% amino acid sequence identity with HsCPTa. It also contained the same variation in the metal ion binding motifs. Interestingly, the transcripts of HpCPTa1 were upregulated in flower tissues, as evidenced by the FPKM values (Supplementary Table 4), aligning with the expected expression profile of hyperforin biosynthetic genes. Notably, hyperforin accumulation in flowers and fruits of H. perforatum is well-established39.
In vitro assays with recombinant yeast microsomes confirmed that both HsCPTa and HpCPTa1 accepted the prenyl donor DMAPP to convert the triprenylated phlorisobutyrophenone substrate, colupulone, into distinct products (Fig. 7). Two secohyperforin epimers were formed, which eluted less than 30 seconds apart and produced identical MS/MS fragments. In agreement with the isolated metabolites, the H. sampsonii enzyme catalysed the formation of 7-epi-secohyperforin (endo configuration), whereas the H. perforatum enzyme yielded secohyperforin (exo configuration). Both enzymes also accepted the geranyl-bearing precursor and catalysed reactions with different cyclization modes. In analogy to the conversion of acceptor substrate 2, HsCPTa directed the initial prenyl transfer to the C-3 prenyl group of the precursor, whereas HpCPTa1 prenylated the C-3 geranyl group to form hyperforin. No activity was detected in HsCPTb-containing assays and control assays with microsomes from empty-vector transformed cells.

HPLC-DAD analysis of enzyme assays containing recombinant yeast microsomes. HsCPTa and HpCPTa1 catalysed the conversion of triprenylated phlorisobutyrophenone to secohyperforin endo and exo epimers, respectively. HsCPTa transformed diprenylated-monogeranylated phlorisobutyrophenone to the aliphatic nemosampsone analogue 7, whereas HpCPTa1 catalysed a different mode of cyclization that resulted in the formation of hyperforin. No conversions were observed in control assays containing microsomes from empty vector-transformed yeast cells. S, substrate peak.
These results demonstrate that H. sampsonii roots are another source of phlorisobutyrophenone-derived PPAPs, which are structurally different from the metabolites found in H. perforatum. To the best of our knowledge, no example of a type B PPAP with an aliphatic acyl group has been reported in the literature. This finding is consistent with the inability of HsCPTb to act on the corresponding substrate. The kinetic characterisation of colupulone in HsCPTa-catalysed reactions revealed parameters similar to those of substrate 2 (KM, 105.55 ± 9.86 µM; Supplementary Fig. 17), reconfirming grandone 1 as the preferred acceptor substrate.
In conclusion, we present the discovery of two enzymatic blueprints, which confirm the biosynthetic scenario of prenylative cyclization in H. sampsonii, and characterise their sophisticated mechanisms, which enable H. sampsonii to regulate the regiodivergent formation of diverse type A and B PPAP compounds. The sequence information obtained, especially for the aspartate-rich regions, will help elucidate the biosynthetic machinery that yields other valuable PPAPs in the Hypericaceae and Clusiaceae families, as exemplified here by detection of the (seco)hyperforin-forming enzyme in H. perforatum. These discoveries are anticipated to contribute to deciphering further biosynthetic steps via phylogenetic and co-expression analyses. Gaining access to these biosynthetic gateway reactions is paramount to the development of synthetic biology approaches aiming at the production of medicinal PPAPs. The differences in the metabolomes of in vitro propagated and field-grown H. sampsonii plants need confirmation by more extensive analysis but may provide a rare opportunity to study the biosynthesis of caged PPAPs with adamantane and homoadamantane scaffolds via comparative transcriptomics.
Methods
Plant material
In vitro-propagated Hypericum sampsonii Hance (Hypericaceae) plants were established at the Institute of Botany, the Chinese Academy of Sciences, Beijing, China40. Seeds were surface sterilised and germinated on MS medium (Duchefa Biochemie, Haarlem, The Netherlands) supplemented with 2% sucrose and 0.7% agar. Seedlings were cultured on the same medium containing 0.5% activated charcoal to keep roots in the dark and absorb harmful root exudates. For propagation, shoots were cut into segments with at least one axillary bud forming plantlets on the same medium within two months. Following one-week-acclimatisation, rooted plantlets were transferred to pots containing commercial soil for garden plants and cultivated in a whole-plant culture room (Institute of Pharmaceutical Biology, Braunschweig) at 22 °C, 70% relative humidity, and 16/8 (light/dark) photoperiod.
Extraction and isolation of prenylated benzophenone derivatives
Leaves of four-month-old plants (10 g dry weight) were lyophilised and homogenised (M20 Universal mill; IKA, Staufen, Germany). Eight aliquots of the resulting powder were transferred to 50 mL conical reaction tubes and extracted three times with 50 mL dichloromethane. The combined extract was centrifuged, the supernatant filtrated through filter paper, and the solvent evaporated under N2 gas. The residue (1 g) was fractionated using an Isolera One Flash Purification Chromatography system (Biotage, Uppsala, Sweden), equipped with a Chromabond Flash BT cartridge (SiOH, 40–63 µm, 40 g; Macherey-Nagel, Düren, Germany). Separation was stretched over 23 column volumes (CV), using n-hexane (A) and ethyl acetate (B) as mobile phase (gradient: 2 CV 0% B, 6 CV 0–10% B, 15 CV 10–60% B). A total of 30 fractions a 45 mL were collected, automatically controlled by detection of UV signals in the λ-all mode. After solvent evaporation under N2 gas, the fractions were passed through a Chromabond HR-X SPE column (6 mL, 55–60 µm; Macherey-Nagel) to remove chlorophyll. Lipophilic target compounds were isolated by semi-preparative HPLC on a normal-phase EC NUCLEODUR column (100-5 silica, 4.6×250 mm, 5 µm; Macherey-Nagel) with a flow rate of 3 mL min−1. The mobile phase consisting of n-hexane (A) and ethyl acetate (B) was used for isocratic elution at 3% B for 16 min. Separation of fractions 16 and 17 yielded pure nemosampsone 4 (7 mg), while fraction 18 yielded 7-epi-nemorosone 3 (6 mg), compound 4 (4 mg), and 7-epi-clusianone 5 (3 mg). More polar target compounds were purified on a reversed-phase EC NUCLEODUR column (π², 10 × 250 mm, 5 µm; Macherey-Nagel) at a flow rate of 3 mL min−1 using 0.1% formic acid in water (A) and acetonitrile (B) as mobile phase (80–92% B from 0–15 min). Separation of fractions 23–28 resulted in the isolation of grandone 1 (5 mg) and kolanone 2 (2 mg). 7-epi-secohyperforin 6 (0.28 mg) was isolated on the above normal-phase HPLC system from fresh H. sampsonii roots used for RNA isolation.
NMR spectroscopy
Measurements were carried out on either a Bruker Advance III HD 500 MHz NMR spectrometer (Bruker Biospin, Karlsruhe, Germany) or a Bruker Advance III HD 700 MHz spectrometer, both equipped with cryoplatforms and TCI cryoprobes. The 1D and 2D spectral data were obtained from 1H NMR, 13C NMR, DEPTQ, 1H-1H COSY, 1H-1H ROESY, 1H-13C HSQC, and 1H-13C HMBC experiments. Spectrometer control and data processing were accomplished using the Bruker TopSpin software v3.6.1. Standard Bruker pulse programs were used for NMR data acquisition.
Polarimetry experiments
Specific optical rotations were measured on a Jasco P-2000 polarimeter (Jasco, Gross-Umstadt, Germany), equipped with a cylindrical quartz cell (3.5 mm diameter, 100 mm optical path length). Measurements were carried out at 20 °C using sodium light at 589 nm, setting the source aperture to 3 mm. Sample concentrations (w/v %) of 7-epi-nemorosone 3, nemosampsone 4, and 7-epi-clusianone 5 were 0.22, 0.20, and 0.06, respectively.
RNA isolation and sequencing
Shoots and roots of two-month-old H. sampsonii plants were homogenised in liquid nitrogen using mortar and pestle. Total RNA was extracted from the fine powders (~100 mg) using the InviTrap Spin Plant RNA mini kit (Invitek Molecular, Berlin, Germany). After removal of residual genomic DNA by digestion with DNAse I (Thermo Fisher, Waltham, USA), RNA was purified using the Monarch RNA Cleanup kit (New England Biolabs, Ipswich, USA). Quality and quantity of total RNA were determined on a Bioanalyzer 2100, paired with the RNA 6000 Nano LabChip kit (both Agilent, Santa Clara, USA). Samples with RIN > 7.0 were accepted for further work-up. Library preparation, RNA-sequencing, and transcriptome assembly were performed at LC Sciences (Houston, USA). Poly(A) mRNA was isolated from total RNA (∼1 μg) using oligo(dT)-coupled magnetic beads (Thermo Fisher) and fragmented into small pieces using divalent cations at elevated temperature. RNA fragments were reverse-transcribed to prepare the final strand-specific cDNA library according to the dUTP method41. The average insert size for the paired-end libraries was 300 ± 50 bp. Paired-end 2 × 150 bp sequencing was performed on an Illumina Hiseq 4000 platform (Illumina, San Diego, USA), following the vendor recommended protocol. 54 to 57 million RNA-Seq reads were collected per sample.
De novo transcriptome assembly, annotation, and evaluation
Trimmomatic42 software was used for adaptor trimming and removal of reads containing low quality or undetermined bases. Quality control was performed by FastQC43. The clean data of ensured quality was used for downstream analysis. De novo assembly of the transcriptome was performed using the Trinity44 method. The longest transcript in each Trinity cluster was defined as the unigene. All assembled unigenes were functionally annotated by alignment against non-redundant (NR) NCBI protein database, Gene ontology (GO), SwissProt, Kyoto Encyclopedia of Genes and Genomes (KEGG), and EggNOG, using DIAMOND45 software with default parameters (e ≤ 1–5). A translated nucleotide BLAST search (tblastn) identified homologous sequences, employing the amino acid sequence of HsPT8px as query (NCBI accession number AZK16224.1). Amino acid sequence alignment utilised the MUSCLE46 algorithm. Phylogenetic analysis relied on MEGA1147 software using the maximum-likelihood method (JTT + F G5)48, as recommended by the in-built evaluation tool. The quality of the phylogenetic tree was inferred for each node by computation of 500 bootstrap replicates.
Molecular biology essentials
Our molecular cloning toolbox comprised an assortment of commercial products and services. Synthesis of modified and unmodified primers and sequencing of DNA samples were performed by Eurofins Genomics (Ebersberg, Germany) and Microsynth Seqlab (Göttingen, Germany), respectively. PCR reactions were conducted using either PCRBIO HiFi polymerase (PCR Biosystems, London, UK) or PfuTurbo Cx HotStart DNA polymerase (Agilent), if encountering uracil stalling. Clean-up of DNA fragments from agarose gels or PCR reactions was done using the innuPREP DOUBLEpure kit (Analytik Jena, Jena, Germany). Restriction enzymes for cloning and control digestions and T4 DNA ligase for ligation of blunt or sticky ends were purchased from Thermo Fisher. Plasmid DNA was isolated using the QIAprepTM Spin Miniprep kit (Qiagen, Hilden, Germany). Unless indicated otherwise, all reactions using commercial products were performed according to the manufacturers’ guidelines.
Gene expression analysis by RT-qPCR
Total H. sampsonii root, stem, and leaf RNA (1 µg) served for cDNA synthesis using the iScript Reverse Transcription Supermix for RT-qPCR (Bio-Rad, Hercules, USA). RT-qPCR reactions were set up using the iTaq Universal SYBR Green Supermix (Bio-Rad) according to the manufacturer’s guidelines and run on a CFX Connect Real-Time PCR Detection System (Bio-Rad). The 2-step PCR method comprised an initial denaturation step at 95 °C for 2 min, followed by 40 cycles at 95 °C for 5 s and 60 °C for 30 s, recording plate data after each annealing/extension step. Melting curves ensuring the amplification specificity were recorded after each run from 65 to 95 °C in 0.5 °C increments over 5 s. Efficiencies of primers designed for the quantification of HsCPTa, HsCPTb and the housekeeping genes ACT2 and TUB-B (Supplementary Table 5) were tested against a serial dilution made with pooled cDNA of all tissues. Data analysis was performed using the Bio-Rad CFX Manager v3.1 software (Bio-Rad).
Functional expression in Saccharomyces cerevisiae
The binary pESC-URA vector allowed inducible gene expression in S. cerevisiae and efficient cloning and plasmid propagation in Escherichia coli. Construct assembly was achieved by a standard restriction-ligation technique, utilising MCS2 for the insertion of target genes under control of the yeast GAL1 promoter. Inserts were amplified using overhang primers introducing the desired restriction sites (Supplementary Table 5). Both backbone and insert were digested with Bam HI and Kpn I prior to assembly. Reaction products were directly used for transformation of E. coli DH5α by the common CaCl2 heat-shock method and transformants were recovered on LB agar plates (0.5% yeast extract, 1% peptone, 1% NaCl) supplemented with 100 µg mL−1 ampicillin. After ensuring correct assembly by sequencing, expression constructs were introduced into S. cerevisiae INVsc1 (Thermo Fisher), using the Frozen-EZ Yeast Transformation II kit (Zymo Research Europe, Freiburg, Germany). S. cerevisiae clones were selected on SGI plates (0.67% yeast nitrogen base, supplemented with amino acids, 2% glucose) lacking uracil as an auxotrophic marker. For protein production, single yeast colonies were suspended in 5 mL uracil-deficient SGI medium (100 mL flasks). After 24 h incubation at 30 °C and 200 rpm, around 2 mL were used to start 150 mL main cultures (1 L flasks, OD600 = 0.01) in YPEG medium (1% yeast extract, 1% peptone, 3% ethanol, 0.5% glucose). After incubation for 28–30 h, heterologous gene expression was induced by adding 2% galactose and cultures were incubated for another 16 h to accumulate heterologous enzymes. Cells were harvested by centrifugation at 2500 x g for 5 min and rinsed twice with 10 and 5 mL TEK buffer (50 mM Tris-HCl pH 7.4, 1 mM EDTA, 100 mM KCl), prior to mechanical disruption by vigorous vortexing (30 min, 4 °C) with glass beads (3 g, 0.45 mm) in 3 mL TES buffer (50 mM Tris-HCl, pH 7.4, 1 mM EDTA, 600 mM sorbitol). To preserve enzyme stability, all following steps were conducted at 4 °C. After centrifugation at 2500 x g for 5 min, the protein-containing supernatant was collected and the remaining cell debris extracted two more times with 3 ml TES buffer. Membrane fractions were pelleted by ultra-centrifugation of the combined extract at 100,000 x g for 1 h and the pellet was resuspended in 2 mL buffer A (50 mM Tris-HCl pH 7.4, 300 mM NaCl, 10% glycerol).
Enzyme assays and characterisation
Standard reactions for activity screening (125 µL) contained 100 µg microsomal protein, 5 mM MgCl2, 400 µM DMAPP, 200 µM acceptor substrate, buffered by 100 mM Tris-HCl pH 8. DMAPP was synthesised as previously described49. Assays were incubated at 37 °C for 1 h at constant agitation, stopped with 17.5 µL 2 N HCl and extracted with 500 µL ethyl acetate. The solvent was evaporated in vacuo, the residue taken up in 100 µL methanol, and 50 µL analysed by HPLC. For enzyme characterisation, assay parameters were individually investigated: temperature (25–50 °C), pH (6–11), protein concentration (10–500 µg per assay), and reaction time (2–200 min). Under optimum conditions for each enzyme, the KM values were determined at DMAPP saturation, while eight concentrations of 1 and 2 ranged from 0.1 to 3 times KM. Maximum velocities were deduced from Michaelis Menten kinetics in analogy to Vmax. Curve-fitting was managed using OriginPro2022 (OriginLab, Northampton, USA) with the provided Enzyme Kinetics plugin. Means and standard deviations were calculated from three technical replicates, for which exact amounts of microsomal protein from the same batch were provided. The product yield of all HsCPTa and HsCPTb mutants created was determined by single standardised reactions using 200 µg microsomal protein to convert 15 pmol donor and acceptor substrates in Tris-HCl pH 8.5 buffer for 45 min at 40 °C. To account for differences in the relative protein amounts obtained from yeast cultivation and induction procedure, reactions were repeated three times using membrane fractions from three individual cultivations.
Transient gene expression in Nicotiana benthamiana
Assembly of pCambia 2300 u 35 S expression constructs was achieved by adapting the USERTM strategy50. One-pot reactions were set up using uracil-containing PCR products and vectors in a 10:1 ratio. Restriction enzyme Pac I, nicking enzyme Nt.Bbv CI (NEB), and USER enzyme (NEB) worked simultaneously in rCutSmart buffer (NEB). One-pot excision and hybridisation were facilitated at alternating temperatures (20 min at 37 °C, 15 cycles of 5 min at 37 °C and 2 min at 25 °C, 20 min at 25 °C). Sequence-confirmed constructs were transferred into Agrobacterium tumefaciens EHA105 cells, using the established freeze-thaw technique. After selection of positive transformants, pre-cultures were inoculated with a single colony in 10 mL LB medium (50 µg mL−1 kanamycin, 50 µg mL−1 rifampicin) and incubated overnight at 28 °C and 220 rpm. On the next day, 5 mL were used to start 50 mL main-cultures (100 mL in case of P19 helper-strain), which were incubated for an additional 24 h. Agrobacteria were pelleted by centrifugation at 2000 x g for 10 min, briefly washed with water and diluted to an OD600 of 0.5 with infection medium (10 mM MES, 10 mM MgSO4, 100 µM acetosyringone pH 5.5). Virulence was induced for 2 h at 25 °C and constant agitation. All tested strains were mixed 1:1 with a helper-strain expressing the commonly used P19 inhibitor of gene silencing51. 4–5-week-old N. benthamiana plants were infected by syringe-infiltration into the abaxial side of the two youngest fully expanded leaves. Plants were left to dry in the dark and subsequently transferred to the above-mentioned whole-plant culture room.
Nicotiana benthamiana leaf disk experiments
To confirm the activities of HsCPTa and HsCPTb in planta, 1 cm diameter disk sections of transiently transformed N. benthamiana leaves were collected 4 days after Agrobacterium infiltration. Based on the previously described protocol52, the leaf sections were transferred to a 48-well plate containing 250 µL Tris-HCl (100 mM, pH 7) and 20 µM acceptor substrate. The plate was sealed with parafilm to prevent buffer evaporation and incubated at plant growth conditions (22 °C, 70% relative humidity, 16/8 light/dark cycles). After 48 h, leaf discs were placed in 2 mL screw cap tubes, flash-frozen in liquid nitrogen and homogenised using a MM400 Tissuelyser (Retsch, Haan, Germany) and 0.7 mm garnet beads. Metabolites were extracted with 500 µL ethyl acetate and cell debris was removed by repeated centrifugation at 19,000 x g for 10 min. After solvent evaporation under N2 gas, residues were taken up in 100 µL MeOH for LC-MS analysis.
Analytical procedures
HPLC analysis was performed on a VWR Hitachi LaChrom Elite machine, equipped with a L-2130 pump, L-2200 autosampler, and L-2455 DAD detector. Samples were run on a ZORBAX Eclipse Plus column (C18, 4.6 × 100 mm, 3.5 µm; Agilent) at a flow rate of 1 mL min−1, using 0.1% formic acid in water (A) and acetonitrile (B) as mobile phase (gradient: 50–70% B from 0–5 min, 70–92% B from 5–10 min, 92–96% B from 10–28 min). For HRESI-MS characterisation, the HPLC system was coupled to a Bruker Compact QToF mass spectrometer (Bruker Daltonics, Bremen, Germany), operated via the Compass HyStar control platform (Bruker Daltonics). Measurements were performed in negative ionisation mode at 30,000 m Δm−1 resolving power, recording ions in the full scan range of m/z 50–1300. The m/z values obtained were recalibrated using the expected cluster ion m/z values of a sodium formate-isopropanol solution, which was injected at the beginning of each run. For LC-MS/MS analysis, the multiple reaction monitoring (MRM) mode of the instrument was used, setting the collision energy to 30, 35, and 40 eV.
Enzyme engineering
Stepwise substitution of the HsCPTb C-terminus with corresponding amino acids of HsCPTa was performed using the one-pot fusion PCR method53. HsCPTa fragments designated for substitution were amplified and purified from agarose gel. In subsequent fusion PCR, they functioned as megaprimers for the initial amplification of chimerical sequences, using gene-specific pESC-URA forward primers and HsCPTb as the template. After 5 cycles, pESC-URA reverse primers were added to increase the fidelity of the reaction and introduce the required restriction sites. To eliminate any residual template DNA, PCR products were digested with Dpn I (2–3 h). The resulting hybrid genes were cloned into pESC-URA, as described above. Site-directed mutagenesis served to introduce point mutations to HsCPTa and HsCPTb constructs by full replication of the plasmid, using tailored pairs of partially overlapping complementary primers (Supplementary Table 5), as previously described54. After removal of parental DNA by Dpn I digestion and transfer to E. coli, the nicked plasmids were repaired by virtue of the bacterial DNA maintenance machinery.
Homology modelling and substrate docking
The initial structures of HsCPTa and HsCPTb were modelled using AlphaFold2 (DeepMind, London, England) and further refined with Prime (Schrödinger, New York, USA), utilising implicit membrane and OPLS4 force fields. The resulting structures were validated using the SAVES6 server (saves.mbi.ucla.edu). Protein-ligand interactions were investigated using Schrödinger’s suite of molecular modelling tools (Schrödinger Release 2018-2) by employing the induced-fit docking (IFD) approach, which accounts for ligand and protein flexibility during docking simulations. To accommodate the substrate in the smaller binding pocket during the docking process, both the substrate and the ligand van der Waals radii scaling factor were set to 0.3. Taking into account the size of the ligand, the docking box size was set to 15 Å with the docking centre defined by the centroid of the C-alpha atoms of four residues that envelop the binding site. After docking, clustering of the results was performed and a representative conformation from each cluster was selected as the starting structure for further docking refinement, using the extra precision (XP) mode. Upon completion of the docking, we assessed the binding free energies of each binding conformation using the generalised Born surface area (MM/GBSA) method55 and selected the most favourable binding conformation. The LigPrep module (Schrödinger) was used to generate ligand 3D structures with optimised geometries and appropriate ionisation states at the preferred pH values of 7 and 10 for computations involving HsCPTa and HsCPTb, respectively. The representative membrane topology of H. sampsonii PTs was visualised based on transcript 8613c0g2 using MembraneFold56.
Subcellular localisation
Two versions of the original binary plant-expression vector pCambia 2300 u 35 S were utilised, designated for translational fusion to either N- or C-terminus of YFP reporter. Four days after N. benthamiana leaf infiltration, localisation of YFP-fused enzymes was determined by confocal laser scanning microscopy. Simple preparations of abaxial epidermal cells were mounted in water and analysed on the confocal laser scanning microscope cLSM 880 (Zeiss, Oberkochen, Germany), equipped with a Plan-Apochromat 20x/0.8 air objective (Zeiss). Excitation with a 488 nm argon laser allowed comparison of chlorophyll autofluorescence to the emissions of YFP-fused localisation markers. Results observed were confirmed in three biological replicates.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The nucleotide sequences of HsCPTa, HsCPTb, and HpCPTa1 are deposited in the National Center for Biotechnology (NCBI) GenBank under the accession numbers OR464113, OR464114, and PP538028. respectively. The raw reads obtained from the RNA sequencing of H. sampsonii root and shoot samples are deposited in the NCBI Sequence Read Archive (SRA) under the BioProject accession PRJNA1010338. Source data are provided with this paper.
References
Li, X.-X. et al. Hyperforin: a natural lead compound with multiple pharmacological activities. Phytochemistry 206, 113526 (2023).
Gurevich, A. I., Dobrynin, V. N., Kolosov, M. N., Popravko, S. A. & Riabova, I. D. Antibiotik giperforin iz Hypericum perforatum. Antibiotiki 16, 510–513 (1971).
Huang, L. et al. Hypersampones A-C, Three nor-polycyclic polyprenylated acylphloroglucinols with lipid-lowering activity from Hypericum sampsonii. Org. Lett. 24, 5967–5971 (2022).
Yang, X.-W., Grossman, R. B. & Xu, G. Research progress of polycyclic polyprenylated acylphloroglucinols. Chem. Rev. 118, 3508–3558 (2018).
Grossman, R. B. Living Database of PPAPs. uky.edu/~rbgros1/PPAPs/allPPAPs.html (2024).
Xiao, Z. Y., Mu, Q., Shiu, W. K. P., Zeng, Y. H. & Gibbons, S. Polyisoprenylated benzoylphloroglucinol derivatives from Hypericum sampsonii. J. Nat. Prod. 70, 1779–1782 (2007).
Piccinelli, A. L. et al. Structural revision of clusianone and 7-epi-clusianone and anti-HIV activity of polyisoprenylated benzophenones. Tetrahedron 61, 8206–8211 (2005).
Díaz-Carballo, D. et al. Antiretroviral activity of two polyisoprenylated acylphloroglucinols, 7-epi-nemorosone and plukenetione A, isolated from Caribbean propolis. Int. J. Clin. Pharmacol. Ther. 48, 670–677 (2010).
Monzote, L. et al. Antimicrobial evaluation of the polyisoprenylated benzophenones nemorosone and guttiferone A. Phytother. Res. 25, 458–462 (2011).
De Barros, M. et al. Antibacterial activity of 7-epiclusianone and its novel copper metal complex on Streptococcus spp. isolated from bovine mastitis and their cytotoxicity in MAC-T cells. Molecules 22, 823 (2017).
Taylor, W. F. et al. 7-epi-clusianone, a multi-targeting natural product with potential chemotherapeutic, immune-modulating, and anti-angiogenic properties. Molecules 24, 4415 (2019).
Simpkins, N. S., Holtrup, F., Rodeschini, V., Taylor, J. D. & Wolf, R. Comparison of the cytotoxic effects of enantiopure PPAPs, including nemorosone and clusianone. Bioorg. Med. Chem. 22, 6144–6147 (2012).
Díaz-Carballo, D., Malak, S., Bardenheuer, W., Freistuehler, M. & Peter Reusch, H. The contribution of plukenetione A to the anti-tumoral activity of Cuban propolis. Bioorg. Med. Chem. 16, 9635–9643 (2008).
Huang, C. Y., Chang, T. C., Wu, Y. J., Chen, Y. & Chen, J. J. Benzophenone and benzoylphloroglucinol derivatives from Hypericum sampsonii with anti-inflammatory mechanism of otogirinin A. Molecules 25, 4463 (2020).
Biber, N., Möws, K. & Plietker, B. The total synthesis of hyperpapuanone, hyperibone L, epi-clusianone and oblongifolin A. Nat. Chem. 3, 938–942 (2011).
Zhang, Q., Mitasev, B., Qi, J. & Porco, J. A. Total synthesis of plukenetione A. J. Am. Chem. Soc. 132, 14212–14215 (2010).
Zhang, Q. & Porco, J. A. Total synthesis of (±)-7-epi-nemorosone. Org. Lett. 14, 1796–1799 (2012).
Qi, J., Beeler, A. B., Zhang, Q. & Porco, J. A. Catalytic enantioselective alkylative dearomatization-annulation: total synthesis and absolute configuration assignment of hyperibone K. J. Am. Chem. Soc. 132, 13642–13644 (2010).
Wen, S., Boyce, J. H., Kandappa, S. K., Sivaguru, J. & Porco, J. A. Regiodivergent photocyclization of dearomatized acylphloroglucinols: asymmetric syntheses of (-)-nemorosone and (-)-6-epi-garcimultiflorone A. J. Am. Chem. Soc. 141, 11315–11321 (2019).
Shen, X., Ting, C. P., Xu, G. & Maimone, T. J. Programmable meroterpene synthesis. Nat. Commun. 11, 508 (2020).
Sanchez, A. & Maimone, T. J. Taming shapeshifting anions: total synthesis of ocellatusone C. J. Am. Chem. Soc. 144, 7594–7599 (2022).
Liu, B., Falkenstein-Paul, H., Schmidt, W. & Beerhues, L. Benzophenone synthase and chalcone synthase from Hypericum androsaemum cell cultures: cDNA cloning, functional expression, and site-directed mutagenesis of two polyketide synthases. Plant J. 34, 847–855 (2003).
Sasaki, K., Mito, K., Ohara, K., Yamamoto, H. & Yazaki, K. Cloning and characterization of naringenin 8-prenyltransferase, a flavonoid-specific prenyltransferase of Sophora flavescens. Plant Physiol. 146, 1075–1084 (2008).
Hu, L. H. & Sim, K. Y. Sampsoniones A–M, a unique family of caged polyprenylated benzoylphloroglucinol derivatives, from Hypericum sampsonii. Tetrahedron 56, 1379–1386 (2000).
Boubakir, Z., Beuerle, T., Liu, B. & Beerhues, L. The first prenylation step in hyperforin biosynthesis. Phytochemistry 66, 51–57 (2005).
Tian, W. J. et al. Hypersampsones S–W, new polycyclic polyprenylated acylphloroglucinols from Hypericum sampsonii. RSC Adv. 6, 50887–50894 (2016).
Grossman, R. B. & Jacobs, H. On the structures of plukenetiones B, D, and E and their relationships to other polycyclic polyprenylated acylphloroglucinols. Tetrahedron Lett. 41, 5165–5169 (2000).
Nagia, M. et al. Sequential regiospecific gem-diprenylation of tetrahydroxyxanthone by prenyltransferases from Hypericum sp. New Phytol. 222, 318–334 (2019).
Bruijn, W. J. C., de, Levisson, M., Beekwilder, J., van Berkel, W. J. H. & Vincken, J. P. Plant aromatic prenyltransferases: tools for microbial cell factories. Trends Biotechnol. 38, 917–934 (2020).
Munakata, R. et al. Molecular evolution of parsnip (Pastinaca sativa) membrane-bound prenyltransferase for linear and/or angular furanocoumarin biosynthesis. New Phytol. 211, 332–344 (2016).
Li, H. et al. A heteromeric membrane-bound prenyltransferase complex from hop catalyzes three sequential aromatic prenylations in the bitter acid pathway. Plant Physiol. 167, 650–659 (2015).
Huang, H. et al. Structure of a membrane-embedded prenyltransferase homologous to UBIAD1. PLoS Biol. 12, e1001911 (2014).
Yao, J., Chen, F. & Guo, H. QM/MM free energy simulations of the reaction catalysed by (4S)-limonene synthase involving linalyl diphosphate (LPP) substrate. Mol. Simul. 44, 1158–1167 (2018).
Major, D. T. & Weitman, M. Electrostatically guided dynamics-the root of fidelity in a promiscuous terpene synthase? J. Am. Chem. Soc. 134, 19454–19462 (2012).
Christian, O. E. et al. Redetermination and absolute configuration of (+)-7-epiclusianone. Acta Crystallogr. E68, o3222–o3223 (2012).
Boyce, J. H. & Porco, J. A. Asymmetric, stereodivergent synthesis of (-)-clusianone utilizing a biomimetic cationic cyclization. Angew. Chem. Int. Ed. 53, 7832–7837 (2014).
Horeischi, F., Guttroff, C. & Plietker, B. The enantioselective total synthesis of (+)-clusianone. Chem. Commun. 51, 2259–2261 (2015).
Charchoglyan, A. et al. Differential accumulation of hyperforin and secohyperforin in Hypericum perforatum tissue cultures. Phytochemistry 68, 2670–2677 (2007).
Tekel’ová, D., Repcák, M., Zemková, E. & Tóth, J. Quantitative changes of dianthrones, hyperforin and flavonoids content in the flower ontogenesis of Hypericum perforatum. Planta Med. 66, 778–780 (2000).
Huang, L. et al. Differential expression of benzophenone synthase and chalcone synthase in Hypericum sampsonii. Nat. Prod. Commun. 7, 1615–1618 (2012).
Parkhomchuk, D. et al. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 37, e123 (2009).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Andrews S. FastQC: A quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797 (2004).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Jones, D. T., Taylor, W. R. & Thornton, J. M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282 (1992).
Woodside, A. B., Huang, Z. & Poulter, C. D. Trisammonium geranyl diphosphate: diphosphoric acid, mono (3,7-dimethyl-2,6-octadienyl) ester (E)-, trisammonium salt. Org. Synth. 66, 211 (2003).
Bitinaite, J. et al. USER friendly DNA engineering and cloning method by uracil excision. Nucleic Acids Res. 35, 1992–2002 (2007).
Lakatos, L., Szittya, G., Silhavy, D. & Burgyán, J. Molecular mechanism of RNA silencing suppression mediated by p19 protein of tombusviruses. EMBO J. 23, 876–884 (2004).
Kamileen, M. O. et al. Recycling upstream redox enzymes expands the regioselectivity of cycloaddition in pseudo-aspidosperma alkaloid biosynthesis. J. Am. Chem. Soc. 144, 19673–19679 (2022).
Büttner, M. & Barleben, L. One-pot fusion polymerase chain reaction for combinatorial synthesis of DNA from several cassettes. Anal. Biochem. 421, 797–798 (2012).
Liu, H. & Naismith, J. H. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 8, 91 (2008).
Genheden, S. & Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 10, 449–461 (2015).
Gutierrez, S., Tyczynski, W. G., Boomsma, W., Teufel, F. & Winther, O. MembraneFold: visualising transmembrane protein structure and topology. bioRxiv https://doi.org/10.1101/2022.12.06.518085 (2022).
Acknowledgements
The authors thank Prof. Paraskev T. Nedialkov of the Medical University of Sofia, Bulgaria for graciously providing the 7-epi-clusianone reference compound, Dr. Veit Grabe of the Max Planck Institute for Chemical Ecology, Jena, Germany for his kind assistance in operating the confocal laser scanning microscope, and Dr. Till Beuerle, TU Braunschweig, for kindly handling the tandem mass spectrometry infusions. This research was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, LI 3400/4-1), the National Key Research and Development Program of China (2018YFA0900300), and Tianjin Synthetic Biotechnology Innovation Capacity Improvement Projects (TSBICIP-PTJS-002). H.M.B.S. acknowledges support from the German Egyptian Research Long-Term Scholarship (GERLS) programme funded by the Egyptian Ministry of Higher Education and the Deutscher Akademischer Austauschdienst (DAAD). Open access funding enabled and organised by Project DEAL.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
L.E., L.B., I.E-A., and B.L. devised the experimental design. L.E. conducted cloning, expression, and characterisation of biosynthetic genes in S. cerevisiae and N. benthamiana, generated reciprocal mutants, and performed activity assays. H.L. extracted and purified metabolites from H. sampsonii and performed NMR experiments. H.L. and C.P. interpreted NMR data. P.L. performed molecular modelling and docking simulations and analysed the results. H.M.B.S. and I.E-A. carried out synthesis and purification of donor substrates. H.M.B.S. and T.M. identified and assayed HpCPTa1. H.M. provided advice and assisted with bioinformatic analyses. L.E., L.B., and B.L. wrote the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jungui Dai and Yang Qu for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ernst, L., Lyu, H., Liu, P. et al. Regiodivergent biosynthesis of bridged bicyclononanes. Nat Commun 15, 4525 (2024). https://doi.org/10.1038/s41467-024-48879-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-48879-w
