
Abstract
Electrocatalytic nitrate reduction to ammonia holds great promise for developing green technologies for electrochemical ammonia energy conversion and storage. Considering that real nitrate resources often exhibit low concentrations, it is challenging to achieve high activity in low-concentration nitrate solutions due to the competing reaction of the hydrogen evolution reaction, let alone considering the catalyst lifetime. Herein, we present a high nitrate reduction performance electrocatalyst based on a Co nanosheet structure with a gradient dispersion of Ru, which yields a high NH3 Faraday efficiency of over 93% at an industrially relevant NH3 current density of 1.0 A/cm2 in 2000 ppm NO3- electrolyte, while maintaining good stability for 720 h under −300 mA/cm2. The electrocatalyst maintains high activity even in 62 ppm NO3- electrolyte. Electrochemical studies, density functional theory, electrochemical in situ Raman, and Fourier-transformed infrared spectroscopy confirm that the gradient concentration design of the catalyst reduces the reaction energy barrier to improve its activity and suppresses the catalyst evolution caused by the expansion of the Co lattice to enhance its stability. The gradient-driven design in this work provides a direction for improving the performance of electrocatalytic nitrate reduction to ammonia.
Similar content being viewed by others


Introduction
Nitrate anions (NO3−), widely known for their detrimental effects on the environment and human health, pose a significant threat as an industrial and agricultural pollutant1,2. The reduction of NO3− into a green and valuable product, i.e., ammonia (NH3), is a sustainable method because NH3 is a widely used industrial chemical with immense global significance, playing a crucial role in supporting human growth and development3,4. To date, the main route for NH3 synthesis is the Haber-Bosch process, which operates at high temperatures (between 400 and 500 °C) and high pressures (between 130 and 170 bar). This process accounts for ~1.4% of global energy consumption and contributes to ~1% of global energy-related CO2 emissions4,5. The electrochemical synthesis of NH3 from NO3− by using renewable electricity to supply energy has attracted increasing attention as a promising alternative to the traditional Haber-Bosch process.
High NH3 yield rates of electrocatalysts for electrocatalytic nitrate reduction to ammonia (NRA) are achieved under a relatively high NO3− concentration (usually greater than 6192 ppm)6,7,8,9,10,11,12,13,14,15. Given that NO3− resources in reality often exhibit lower concentrations, often ~1000–2000 ppm in typical industrial wastewater3,16,17,18,19, developing efficient electrocatalysts for nitrate reduction at low NO3− concentration is crucial yet challenging. This challenge arises due to the inevitable competition from hydrogen evolution reaction (HER), which reduces the yield rate20,21,22. Due to the susceptibility of cobalt (Co) sites to adsorb NO3−, Co-based materials are regarded as superior catalysts for NO3− electroreduction to NH3 under low nitrate concentrations8,23,24, as they enable a spontaneous reduction process at a positive potential, thus avoiding competition from the HER. To further improve catalytic activity, doping precious metals in Co-based materials accelerate the protonation process during the electroreduction of nitrite (NO2−) to NH3 and promote the generation of high-valence Co species9,25. However, due to the difficulty in reducing the reconstructed high-valence Co species to metallic Co, which accelerate the spontaneous reduction reaction with NO3− during NRA, such high activity was quickly lost26. The stability directly affects the service life and reaction efficiency of catalysts, which also determines the production cost and efficiency of commercial catalysts. Thus, under low nitrate concentration conditions, improving the stability of the catalyst while maintaining high activity is challenging but essential for real applications.
Here, we report a highly active and stable NRA electrocatalyst with a gradient doping structure to solve the above challenges. Inspired by the concentration gradient materials that previously explored for their application in coatings and interface layers for lithium batteries to simultaneously achieve superior electrochemical performance and excellent cycling stability27,28,29, we synthesized a concentration gradient electrocatalyst of Ru atoms in Co nanosheets. This was accomplished through the Ru cation exchange method with a gradually decreasing Ru concentration from the surface to the interior. The catalyst exhibits a high NH3 Faradaic efficiency (FE) of over 93% within a wide potential range of +0.088 to −0.136 V vs. the reversible hydrogen electrode (RHE) while delivering an industrially relevant NH3 current density of 1.0 A/cm2 in a low NO3− concentration of typical industrial wastewater (2000 ppm). The high performance can be maintained for 720 h at −300 mA/cm2, surpassing the previously reported performance of NRA catalysts6,11,12,30,31,32(Supplementary Table 1). The electrocatalyst maintains high activity even in 62 ppm NO3− electrolyte with an excellent NH3 current density of 71 mA/cm2 at −0.095 V vs. RHE, which is superior to the reported performance of catalysts in low concentrations of nitrate13,33,34(Supplementary Table 2). In situ electrochemical Raman and Fourier-transformed infrared spectroscopy (FTIR), electron paramagnetic resonance (EPR) characterization, and density functional theory (DFT) calculations confirmed that the high activity results from the decreased reaction energy with the gradient concentration of Ru in Co lattice. Inductively coupled plasma–optical emission spectroscopy and X-ray diffractometry (XRD) characterization indicated that the improvement in stability is due to the inhibition of in situ reconstruction of the Co lattice by gradient doping.
Results
Preparation and characterization of G-RuCo electrocatalysts
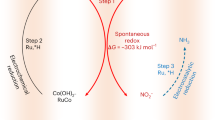
Recent research has found that Ru, Rh, Pd, Ir, and Pt possess moderate adsorption energies for hydrogen atoms35. Ru is considered the optimal doping element due to its low cost. Therefore, we take the view that Co-based NRA catalysts with gradient-doped Ru atoms (G-RuCo catalysts) are promising candidates for high NRA activity and stability. We tried to prepare the G-RuCo catalyst by gradually decreasing Ru concentrations from the surface to the interior using the following steps: In the first step, Ru-Co(OH)2/Co was prepared by cation exchange reaction through immersing electrodeposited Co(OH)2/Co loaded on nickel foam (Ni foam) in RuCl3 solution; Subsequently, by annealing in air, Ru-Co(OH)2/Co was oxidized to Ru-Co3O4/Co; Finally, Ru-Co3O4/Co was electrochemically reduced to G-RuCo (Fig. 1; see “Methods” section). Here, Ni foam was employed as a self-supporting substrate for the nanostructured electrocatalysts due to its low cost, large geometric surface area, and high electrical conductivity36(Supplementary Fig. 1). Notably, the pure Ni foam has no contribution to the NRA process (Supplementary Fig. 2), consistent with other reported results37,38. The catalysts’ crystal structural transformation and morphological changes during the preparation process were tracked by XRD and scanning electron microscopy (SEM) (Supplementary Figs. 3 and 4). Control samples of the non-gradient Ru-doped Co-based catalyst (NG-RuCo catalyst), pure Co and Ru were synthesized and characterized for comparison (Supplementary Figs. 5 and 6, see “Methods” section).

The golden, red, and green spheres refer to nitrogen, oxygen, and hydrogen atoms, respectively.
The crystalline structure of our G-RuCo catalyst was investigated by XRD, and a slight left shift of the Co (002) diffraction peak was observed upon introducing Ru into Co lattice (Supplementary Fig. 7). This shift can be attributed to the expansion of the lattice spacing resulting from the substitution of larger diameter Ru atoms for Co atoms39. Transmission electron microscopy shows that the lattice spacing of the Co (100) plane increased from 0.21 nm in the pure Co catalyst to 0.22 nm after atomic exchange with Ru (Supplementary Figs. 8 and 9). To further explore the crystal structure of G-RuCo, we conducted grazing incident X-ray diffraction (GIXRD) measurements to estimate the residual strain at different depths of the catalysts40,41. By tuning the incidence angle α from 0.1° to 0.5°, the diffraction peaks gradually shift to a higher degree for G-RuCo, while it maintains the same degree for NG-RuCo (Fig. 2a). The crystal plane distance of G-RuCo is larger at the top surface compared to the interior, a feature not present in NG-RuCo. This suggests that more Ru atoms with a larger diameter are doped into the Co lattice at the top surface than in the interior for G-RuCo. In contrast, a constant concentration of Ru atoms is doped into the Co lattice for NG-RuCo. Ar+ sputtering-assisted X-ray photoelectron spectroscopy (XPS) in depth was applied to clearly show the difference of element distribution between the G-RuCo and NG-RuCo (Fig. 2b–g). With increasing Ar+ etching depth from 0 nm to 24 nm, the content of Ru in G-RuCo decreases as the content of Co increased, while the content of Ru in NG-RuCo remains unchanged (Fig. 2b, c, e, f). The calculated atomic ratio of Ru/Co further confirms the gradient distribution of Ru from the surface to the interior in G-RuCo, compared with the constant atomic ratio of Ru/Co in NG-RuCo (Fig. 2d, g). In addition, the elemental composition of Ru and Co in the G-RuCo electrocatalyst was further revealed by energy-dispersive X-ray spectrometry (Supplementary Fig. 10). Figure 2h, i shows the time of flight secondary ion mass spectrometry results for G-RuCo and NG-RuCo sputtered with ~25 nm of Ar+, which also confirms the concentration gradient distribution of Ru in G-RuCo. The reconstructed three-dimensional distribution of the selected species is shown in the inset figures, which agree well with the depth profiling results and clearly show the structural difference of G-RuCo and NG-RuCo.

a GIXRD spectra of G-RuCo and NG-RuCo. b–d XPS depth etching of Ru 3d (c) and Co 2p (d) and variation in the atomic ratio of Ru/Co (e) as a function of Ar+ etching depth from 0 nm to 24 nm for the G-RuCo catalyst. e–g XPS depth etching for the NG-RuCo catalyst. h, i Time-of-flight secondary ion mass spectrometry depth profiles of Ru and Co ion fragments for the G-RuCo and NG-RuCo catalysts. The inset pictures show the reconstructed 3D spatial fragment distributions of the selected ions.
The electronic structures of NG-RuCo and G-RuCo were further investigated by XPS (Supplementary Fig. 11). The peaks corresponding to Ru0 3d5/2 and Ru0 3d3/2 in NG-RuCo and G-RuCo is basically consistent with those in pure Ru, indicating that the Ru elements in these two catalysts exhibit metallic properties42. According to the high-resolution Co 2p spectrum, there are no significant shifts in the peaks corresponding to metallic cobalt (Co0) in NG-RuCo and G-RuCo compared to pure Co, indicating that cobalt cations in these catalysts have been reduced to metallic cobalt23. This metal feature can be further demonstrated using extended X-ray absorption fine structure (EXAFS). The EXAFS spectrum of the Co L3-edge shows that the Co-O bonds disappear in the catalyst after the electrochemical reduction reaction, and a Co-Co or Co-Ru bonds appear, further confirming that the catalyst has been reduced (Supplementary Fig. 12). Moreover, in the wavelet transform of Co L3-edge EXAFS spectrum, a wavelet aggregation peak appears at R ≈ 2.5 Å, k ≈ 12.8 Å−1, which is absent in Co foil and can be attributed to Co-Ru scattering.
Electrochemical activity and stability of NRA
The electrochemical NRA performance of the G-RuCo and NG-RuCo catalysts was investigated using a standard three-electrode H-type cell under ambient temperature and pressure conditions (details including the experimental set-up are provided in the Methods). In this study, ultraviolet‒visible (UV‒vis) spectrophotometry and calibration curves were used to quantify NH3, NO2− and NO3− (Supplementary Figs. 13–15). The quantification accuracy of NH3 is also confirmed by 1H nuclear magnetic resonance (NMR) of 15NH4+ and 14NH4+ detection14 (Supplementary Fig. 16). Furthermore, a continuous electrolyte flow system was employed to maintain a typical industrial wastewater level NO3− concentration of 2000 ppm, thereby reducing the impact on the measured catalytic performance. G-RuCo exhibits a maximum NH3-FE of 99.7% at −0.042 V vs. RHE and maintains a high FE plateau across a wide potential range from +0.088 to −0.136 V vs. RHE, comparable to the performance of NG-RuCo but notably surpassing that of pure Co or Ru (Fig. 3a, Supplementary Figs. 17–20). Moreover, the activity of the G-RuCo catalyst shows superior performance in the electrochemical NRA process compared to the NG-RuCo catalyst (Fig. 3b, c and Supplementary Fig. 21). The G-RuCo catalyst requires a positive potential of only 0.055 V vs. RHE to achieve a high current density of −410 mA/cm2. Importantly, no notably competitive HER occurs at such a positive potential, ensuring a high NH3-FE of 94.5%. Notably, the I-V curves and corresponding NH3-FE values indicate that the catalytic performance of G-RuCo (−1135 mA/cm2 at −0.136 V vs. RHE, 93.1%) is superior to that of NG-RuCo (−884 mA/cm2 at −0.136 V vs. RHE, 90.3%). The G-RuCo catalyst exhibits a high NH3 partial current of −1057 mA/cm2 with an ammonia generation rate of 5.564 mmol/h/cm2, i.e. 94,000 μg/h/cm2, not only surpassing the NG-RuCo catalyst with 71961 μg/h/cm2 but also outperforming previously reported catalysts6,11,12,30,31,32,43,44,45,46,47,48,49,50,51 (Supplementary Fig. 22 and Supplementary Table 1). We also tested the catalytic performance of G-RuCo catalyst at different nitrate concentrations to demonstrate its widespread application. Different concentrations of NO3− electrolytes, such as 6192 ppm, 1000 ppm, 500 ppm, 100 ppm, and 62 ppm, were selected to cover possible nitrate concentration ranges in heavy industry wastewater, textile wastewater, and contaminated groundwater3 (Supplementary Fig. 23 and Supplementary Tables 2 and 3). Surprisingly, G-RuCo preformed well even under a low concentration of NO3− (62 ppm) and delivered the NH3 current density of 71 mA/cm2 with the NH3-FE of ~88.0%. This performance also far exceeds NG-RuCo and currently reported NRA catalysts13,33,34 (Supplementary Table 2). Electrochemical double-layer capacitance and the corresponding redox peak testing were further conducted to probe the catalyst’s inherent activity to normalize the NH3 current density based on the electrochemical surface area (ECSA) (Supplementary Figs. 24–27 and Supplementary Table 4). G-RuCo exhibits a higher NH3 current density after the ECSA normalization, indicating the excellent intrinsic activity of G-RuCo for NH3 generation. The superior NRA activity of the G-RuCo catalyst compared to the NG-RuCo catalyst confirms the impact of the gradient element distribution on catalyst’s performance.

a, b NH3-FEs and corresponding current density-voltage curves of G-RuCo, NG-RuCo, Co and Ru electrocatalysts at different potentials with 50% iR correction (the value of solution resistance is 1.8 Ω). Measurements were taken at least three times and the average FE valves are presented with the standard deviation as error bars. c Corresponding NH3 production rate and partial current density of G-RuCo and NG-RuCo. d Redependence of nitrate concentration on the reaction time on G-RuCo at −0.1 V vs. RHE. e Long-term electrocatalytic stability test of the NRA over G-RuCo at −300 mA/cm2 using a continuous-flow system in an H-cell. Black arrows indicate the renewal of fresh electrolytes.
To assess the NO3− removal capability of G-RuCo in low-concentration NO3− solution, we performed batch conversion tests with an initial 2000 ppm NO3− and measured the remaining products (Fig. 3d). The selectivity of NO3− conversion to NH3 reaches 97.8%, while the overall NH3-FE remains above 98.9% (Supplementary Fig. 28). One hour after electrolysis, the residual nitrate concentration of 9.77 μg/mL and nitrite concentration of 0.20 μg/mL prove that the NO3− and NO2− concentrations are significantly lower than the World Health Organization guidelines for drinking water25. These results demonstrate the promising application potential of the G-RuCo catalyst for the complete removal and/or transformation of NO3−. To highlight the NRA performance of the G-RuCo catalyst, we further evaluated its NRA stability under high current densities. The G-RuCo catalyst maintains stable electrolysis for over 720 h at −300 mA/cm2 with NH3-FE over 81.9% (Fig. 3e). After prolonged operation, the measured decay rate of the G-RuCo catalyst shows only 0.451 mV/h, which is much better than that of most recently reported NRA catalysts, indicating its potential as a commercial catalyst9,23,26(Supplementary Fig. 29). In contrast, NG-RuCo exhibits considerably poorer stability with a higher decay rate of 3.034 mV/h, demonstrating the significant improvement in stability achievable through a gradient distribution of elements (Supplementary Table 1). In addition, at an industrially relevant current density of −1000 mA/cm2, the G-RuCo catalyst also shows 381 h of durability and a stability decay rate of only 1.090 mV/h (Supplementary Fig. 30). The comparison of SEM and ECSA between NG-RuCo and G-RuCo before and after stability testing further proves the improved stability of G-RuCo (Supplementary Figs. 31 and 32). Through inductively coupled plasma–optical emission spectroscopy, the loss of active Co and Ru in NG-RuCo is more significant than that in G-RuCo (Supplementary Fig. 33). This may be due to the inhibition of in situ reconstruction of the Co lattice by Ru gradient doping. To further observe the crystal reconstruction after the reaction, XRD characterization was performed, and the results show that Co(OH)2 is reconstructed in situ on the surface of NG-RuCo after the reaction. In contrast, the crystal structure of G-RuCo remains stable (Supplementary Fig. 34).
Mechanistic studies
The NRA reaction process of the G-RuCo and NG-RuCo catalysts in the electrolyte were monitored through electrochemical studies, Raman spectroscopy, and a mixed isotope labeling experiment (Fig. 4a–e and Supplementary Figs. 35–39). After soaking G-RuCo and NG-RuCo in the electrolyte solution (2000 ppm KNO3), nitrate is partially converted to nitrite (Supplementary Fig. 35). This indicates that it is the spontaneous oxidation-reduction between Co0 and NO3− on the catalyst that produces NO2−. Therefore, it can be inferred that both G-RuCo and NG-RuCo catalysts can spontaneously absorb NO3−, and Co will undergo oxidation by NO3− to form Co(OH)2, rather than the oxidation of Ru to Ru(OH)2 (Eq. (1)). This reaction is further proved by the observed oxidation-reduction peak of Co/Co(OH)2 from the I–V curves of RuCo catalysts tested in 1.0 M KOH solution after being immersed in an electrolyte solution with 1.0 M KOH and 2000 ppm KNO3 without applied voltage (Supplementary Fig. 36). In this case, the Co sites in the catalyst can spontaneously convert NO3− to NO2− at a more positive potential, which is consistent with the results in Supplementary Figs. 18 and 20. The oxidization of Co by NO3− to Co(OH)2 of RuCo catalysts (Eq. (1)) can also be tracked by Raman spectroscopy (Supplementary Fig. 37). The Raman spectrum of G-RuCo after soaking in the electrolyte at the open circuit voltage displays three peaks at 479, 522, and 688 cm−1. The characteristic peaks at 479 and 522 cm−1 may be attributed to the Co-O bond stretching vibrations of Co(OH)29,52. The peak at 688 cm−1 might be associated with the Co-O bond vibration modes of Co(OH)2 or Co3O453. In contrast, Ru does not exhibit any characteristic Raman peaks, proving its stability in the electrolyte.

a–d Electrochemical in situ Raman spectra of NRA (1.0 M KOH with 2000 ppm NO3− electrolyte) on G-RuCo (a), NG-RuCo (b), Co (c) and Ru (d) (the value of solution resistance is 1.8 Ω). e Schematic of the three-step relay mechanism for the NRA and the difference of phase-transition energy between G-RuCo and NG-RuCo. f, g In situ FTIR of G-RuCo and NG-RuCo from 0.5 to −0.5 V vs. RHE. Shaded regions represent the area where the characteristic peaks appear. Dotted lines and numbers represent the peak position and valve.
The conversion of nitrite to ammonia driven by Co sites mainly driven by Co sites can be clearly seen by the in situ Raman spectra (Fig. 4a–d). Furthermore, with the decreased voltage, the Co-O characteristic peak (688 cm−1) disappears, indicating that Co(OH)2 species, derived from redox reactions, can be electrochemically reduced in situ to metallic Co0 (Eq. (2)). The disappeared voltage of G-RuCo was 0 V vs. RHE, which is more positive than that of NG-RuCo (−0.1 V vs. RHE) and Co (−0.2 V vs. RHE) (Fig. 4a–c). It indicates that the presence of a gradually Ru-doped structure promotes the electrochemical reduction process to metallic Co0, which promotes the recycling of Co (Eq. (2)). For Ru catalysts, no significant oxidation peak can be observed, proving that Ru does not undergo a redox process (Fig. 4d). In this case, the products of Eq. (1) are further transferred to the final product of NH3, while the catalysts are reduced to the original valance. To further prove the effect of gradually doped structure, we further calculated the energy barrier for the reconstructed Co(OH)2 phase in the catalyst to be converted back into Co phase (Fig. 4e). The phase-transition energy of the G-RuCo catalyst (0.48 eV) is much lower than the NG-RuCo catalyst (0.96 eV), indicating that the electrochemical reduction from Co(OH)2 to Co0 in G-RuCo is much easier than in NG-RuCo. On the contrary, the transition from Co0 to Co(OH)2 in NG-RuCo is more rapid, which hinders the occurrence of Eq. (2) and breaks the recycling mechanism of Co. This process can be confirmed by experimental results, and it can be observed that the spontaneous oxidation process on NG-RuCo was faster (Supplementary Figs. 35 and 36).
As the applied voltage changes from positive to negative, NO2− is completely transformed into NH3 (Eq. (3)). The NO2− electroreduction reaction instead of NO3− to produce NH3 was further investigated using a mixed isotope labeling experiment (Supplementary Fig. 38). The results further confirm that NO2− was more easily reduced to NH3 than NO3−. Notably, in Eqs. 2 and 3, the formed Co(OH)2 and NO2− are converted to Co and NH3, respectively, through electrochemical reduction and electrocatalytic reduction with the participation of active H26,54. Therefore, the generation of active H is crucial for the cyclic conversion mechanism. In general, in the NRA process, the dynamic evolution mechanism of the catalyst is as follows (Fig. 4e): NO3− first spontaneously undergoes redox at Co sites; Co is oxidized to Co(OH)2; and NO3− is reduced to NO2− (Eq. (1)). As the reaction progresses, when a more negative potential is applied, Co(OH)2 is electrochemically reduced to Co, and the active H provided at Ru sites promotes this process (Eq. (2)), achieving a valence state cycle of Co. At the same time, the NO2− adsorbed on Co sites is further reduced to NH3, while the active H provided on Ru sites accelerates the protonation process (Eq. (3)).
The moderate adsorption capacity of metallic Ru for hydrogen atoms benefits the production of active H. The electrochemical quasi-in-situ EPR results further demonstrates this inference. Nine peaks are displayed in the EPR results (Supplementary Fig. 39), and the ratio of peak intensities is 1:1:2:1:2:1:2:1:1, which are characteristic peaks of the hydrogen-free radical signal6,55. The characteristic peaks of the hydrogen-free radical signal for Ru are the strongest, and compared to Co, the characteristic peak of the hydrogen-free radical signal for G-RuCo is significantly enhanced. The results indicate that the introduction of Ru positively influences the formation of active hydrogen. In this case, Co in G-RuCo is beneficial for converting NO3− to NO2−, while Ru in G-RuCo is more effective in promoting the conversion of NO2− to NH3, which can be further confirmed through testing the electrochemical behaviors of Ru and Co in KNO2 solution (Supplementary Fig. 40).
NRA reaction pathway analysis
To further understand the mechanism of our catalysts during the NRA reaction process, we performed in situ FTIR to track intermediates in solution. As shown in Fig. 4f, g, in the potential range from 0.5 V to −0.5 V, five obvious absorption bands can be observed due to the present intermediates in the electrolyte during the reaction process15,23. The upward absorption bands at 3215 cm−1 correspond to *NH2, while those at 1625 cm−1 correspond to *NO and H2O. Additionally, the upward absorption bands at 1219 cm−1 correspond to *NO2, and those at 1115 cm−1 correspond to the stretching vibration of –N-O- in *NH2OH. The FTIR spectra of G-RuCo and NG-RuCo are similar, which reveals that the NRA processes are similar. However, the intensity of *NH2OH in G-RuCo is higher than in NG-RuCo, indicating that both G-RuCo and NG-RuCo undergo a reaction pathway from *NO to *NH2OH, and the reaction process of G-RuCo is faster. Therefore, we propose the following pathway for the NRA reaction process on G-RuCo and NG-RuCo: NO3−→ *NO3 → *NO2 → *NO → *NOH → *NHOH → *NH2OH → *NH2 → *NH3 → NH3, and the introduction of Ru positively influences the formation of *H, promoting the hydrogenation process of *NO to *NH2OH.
To verify the influence mechanism of the gradient element distribution and reaction active site on the high NRA activity of G-RuCo, the minimum energy path of G-RuCo and NG-RuCo was calculated by DFT according to the results of FTIR. Before calculating the energy path, the optimal adsorption site is determined by calculating the adsorption energies of *NO3 on different sites of the catalyst (Supplementary Fig. 41). The results imply that pure Co or Ru may not be the optimal adsorption site, and the *NO3 adsorption energy is the lowest on the Co-Ru bridge site, indicating that the joint promotion of Co and Ru in the actual reaction process promotes the NRA reaction on the catalyst. We then built different models on the crystal plane Co (100) observed in the results of TEM characterization. Based on XPS spectral results, a three-layer gradient model of Ru4Co121st/Ru2Co142nd/Ru1Co153rd for G-RuCo and a non-gradient model of Ru4Co121st for NG-RuCo were set up (the atomic coordinate information of the models can be found in the Supplementary Data 1) (Supplementary Fig. 42 and Table 5). The adsorption configuration of the reaction intermediate was optimized before the thermodynamic calculations (see details at the bottom of each energy level diagram in Fig. 5a, Supplementary Tables 6 and 7). The overall reaction pathway of NO3− conversion into NH3 occurred under 0 V vs. RHE at pH 14. The energy level diagram shows that the adsorption and reduction processes of *NO3 for all models may be exergonic in free energy, which indicates that the conversion of NO3− to the *NO intermediate is spontaneous. It suggests that the formation of NO2− products is perhaps thermodynamically unfavorable, which is consistent with the experimentally observed negligible NO2−-FE (see “Methods” section, Supplementary Table 8). It was further found that the subsequent protonation process of the seven models was slightly hindered, which may be due to the difficulty in capturing *H species derived from the dissociation of water molecule under alkaline conditions. Given this, we calculated the corresponding free energy, indicating that the potential determining step (PDS) of the three-layer gradient model for G-RuCo is probably the desorption of NH3, with an energy barrier of 0.622 eV (Fig. 5a). In contrast, the PDS of non-gradient model for NG-RuCo is also the desorption of NH3, with an energy barrier of 0.652 eV. Obviously, the faster NRA reaction of G-RuCo may prove its high NRA activity. In the pure Co models, the PDS of the Co is still the protonation process of *NO → *NOH. The PDS of G-RuCo migrates to the desorption of NH3 gas, which may be attributed to the gradient distribution of Ru in the G-RuCo, reducing the energy potential barrier of the initial PDS by providing abundant *H intermediates.

a Free energy diagrams of the NRA process. b PDOS diagrams of the different (100) facets-*NH3 adsorption models. c Reaction-free energies of the HER at 0 V vs. RHE.
To further investigate the relationship between the local electron distribution and the NH3 desorption ability of G-RuCo, we calculated the projected density of state (PDOS) and d-band centers of the different (100) facets–*NH3 adsorption models for the G-RuCo and NG-RuCo (Fig. 5b). The results show that compared to the *NH3-NG-RuCo (100) adsorption model, the d-band center of the *NH3-G-RuCo (100) adsorption model is further away from the Fermi level, suggesting a weaker interaction between the *NH3 intermediate and G-RuCo, which just requires a lower energy barrier to desorb and generate NH3. These results are also consistent with the observation that the d-orbitals of the metal atoms with the p-orbitals of nitrogen atoms in NG-RuCo have a greater overlap area (overlap area of d-p orbitals is 1.834) than that of in G-RuCo (overlap area of d-p orbitals is 1.831) (Supplementary Fig. 43). Meanwhile, the d-band centers of the Ru active centers for adsorbing NH3 on the two models were also calculated. It may be found that the d-band center of the Ru active center after adsorbing NH3 on G-RuCo is also further away from the Fermi level than that of NG-RuCo, which also confirms the above results (Supplementary Table 9). Furthermore, G-RuCo minimizes competitive HERs than NG-RuCo, given that the adsorption for *H became stronger with increasing doping gradient, but the adsorption for *NO3 remains unchanged (Fig. 5c and Supplementary Table 7). These calculation results may demonstrate that a gradient element distribution optimizes the adsorption of the reaction intermediate, changes the PDS, reduces the reaction energy barrier, and thus promotes the reaction pathway of electrochemical reduction of NO3− to NH3.
To achieve the practical application of electrochemical NRA of Co-based materials for large-scale industrial NH3 production under ambient conditions, we assembled an NRA-OER alkaline electrolyser device using G-RuCo as the cathode and Co2P as the anode (Fig. 6a, see the Methods section for details). The anodic electrolyte contains 1.0 M KOH, while the cathodic electrolyte comprised 1.0 M KOH and 2000 ppm NO3−. The membrane electrode assembly (MEA) device can provide industrial current densities up to 1.5 A/cm2 with a cell voltage of merely 2.5 V, achieving high-efficiency industrial NH3 production performance (Fig. 6b). More importantly, the device can operate continuously for at least 100 h at 1.2 A/cm2 while maintaining a stable voltage (Fig. 6c). To further validate the system’s potential for practical production, we collected the product aqueous ammonia after electroreduction of nitrate using an acid trap, and calculated the collection rate of aqueous ammonia (Supplementary Fig. 44). The results demonstrate that in the actual conversion process, the collection rate of aqueous ammonia can reach close to 90%.

a Schematic illustration of MEA configuration. b Steady-state chronopotentiometry curves of the MEA (using G-RuCo as the cathode and Co2P as the anode). c Stability of the G-RuCo electrode in the MEA water electrolysis device at 1.2 A/cm2.
Discussion
In summary, we propose a high-performance NRA catalyst with a gradient distribution of Ru atoms on Co nanosheets, which provides industrially relevant NH3 generation current while maintains a high FE (over 93.1% within a wide potential range of +0.088 to −0.136 V vs. RHE) and high stability (720 h at −300 mA/cm2) in a 2000 ppm NO3− electrolyte. The electrocatalyst maintains its high activity even in a 62 ppm NO3− electrolyte with an excellent NH3 current density of 71 mA/cm2 at −0.095 V vs. RHE. Electrochemical testing and in situ electrochemical Raman, in situ FTIR and EPR characterization confirmed that the introduction of Ru is beneficial for the formation of active H, thereby promoting the electrocatalytic reduction of NO2− to NH3 and the electrocatalytic reduction of Co(OH)2 to Co. DFT calculations further verified that gradient doping of Ru changed the PDS for the NRA reaction and optimized the reaction energy barrier. In addition, the MEA with the G-RuCo electrocatalyst as the cathode provides industrial-grade current density, indicating that it is a promising commercial catalyst. This work will boost the development of highly active and stable NRA catalysts, promoting large-scale industrial electroreduction of NO3− for NH3 production.
Methods
Materials and reagents
The nickel foam (NF) (0.58 g/cm2) was purchased from Kunshan Dessco Electronics Co. Ltd. (Kunshan, China). The reagents hydrochloric acid (HCl, 38.0%), ethanol (C2H5OH, 99.7%), cobalt chloride (CoCl2·6H2O, 99.5%), ammonium chloride (NH4Cl, 99.0%), Ruthenium(III) chloride (RuCl3·xH2O, 37% Ru basis), potassium nitrate (KNO3, 90%) and potassium hydroxide (KOH, 90%) were purchased from Shanghai Macklin Biochemical Co., Ltd. All reagents were of analytical purity and used without further purification.
Preparation of G-RuCo
The preparation of G-RuCo on Ni foam was based on a combined method of electrodeposition, cation exchange, and electrochemical reduction. A 0.5 × 0.5 cm2 piece of Ni foam was washed sequentially in ethanol, 0.1 M HCl, and deionized water using an ultrasonic bath to remove surface oxides. Co(OH)2/Co nanosheets were first prepared via an electrodeposition process under a -3 A/cm2 current density for 120 s in a three-electrode system consisting of a graphite rod as the counter electrode, Ag/AgCl electrode (saturated KCl solution) as the reference electrode, and Ni foam as the working electrode. The electrodeposition solution was an aqueous mixture of 0.12 M cobalt chloride, 1.5 M ammonium chloride, and 100 mL deionized water. The obtained Co(OH)2/Co nanosheets were washed with deionized water, and then cation exchange was conducted by soaking in 30 mM RuCl3 solution under ambient conditions for 40 h. The resultant catalyst was first dried at 70 °C for 1 h and then annealed in an oven at 240 °C for 3 h to convert it into Ru-Co3O4/Co. Finally, an in situ electrochemical prereduction step was performed using the chronopotentiometry method at −800 mA/cm2 for 1 h to obtain the final G-RuCo catalyst.
Preparation of Co
The as-prepared Co(OH)2/Co nanosheets on Ni foam described above were directly annealed in an oven at 240 °C for 3 h to convert them into Co3O4/Co nanosheets without cation exchange. The Co catalyst was finally obtained after electrochemical reduction using the chronopotentiometry method at −800 mA/cm2 for 1 h.
Preparation of Ru
In the electrolytic cell, an electrolyte of RuCl3 (60 mL, 2 mg/mL), a working electrode of 0.5 × 0.5 cm2 Ni foam, and a counter electrode of a Pt plate were used. Electrodeposition was performed for 15 min at −30 mA/cm2. Subsequently, calcination was carried out at 700 °C for 2 h in a tubular furnace with 10% H2/Ar mixed gas and a heating rate of 10 °C/min to further improve the crystallinity of the Ru product.
Preparation of NG-RuCo
In the electrolytic cell, an electrolyte of RuCl3 (60 mL, 2 mg/mL) and CoCl2·6H2O (23 mg/mL), a working electrode of as-prepared Co on Ni foam, and a counter electrode of a graphite rod were used. Electrodeposition was performed for 15 min at −50 mA/cm2. Finally, an in situ electrochemical prereduction step was performed using the chronopotentiometry method at −800 mA/cm2 for 1 h to obtain the final NG-RuCo on Ni foam catalyst.
Characterizations
The morphology and elemental composition of catalysts were analyzed using a scanning electron microscope (SEM, ZEISS Sigma) equipped with an energy-dispersive X-ray spectrometer at a working voltage of 15 kV. The lattice arrangement of the catalyst was characterized by transmission electron microscopy (TEM, JEM-2100F, Japan) at an operating voltage of 200 kV; the tested catalyst comes from powder samples scraped off the nickel substrate. The crystal structure of the catalyst was analyzed using an X-ray diffractometer (XRD, Rigaku, Japan) with a Cu-Kα X-ray source (λ = 1.5418 Å). TOF-SIMS (PHI Nano TOF II Time-of-Flight SIMS) also was applied to investigated gradient elements distributions of G-RuCo. The sputter etching was performed using an Ar+ beam (3 kV 100 nA) to obtain a depth profile. The surface valence state and chemical composition of the catalyst were studied via X-ray photoelectron spectroscopy (XPS; Thermo ESCALAB 250) using monochromatic Al-Kα radiation (1486.6 eV). All XPS spectra were calibrated by shifting the detected carbon C 1 s peak to 284.8 eV. XAFS experiments were performed at the 1W1B beamline of the Shanghai Synchrotron Radiation Facility. The XAFS spectra were analyzed with the Athena software package. The k-weighting was set to 2 for the Fourier transforms.
Electrochemical measurements
All electrochemical tests were performed under environmental conditions using a three-electrode system, and the results were recorded by an electrochemical workstation (CS310, Wuhan Kesite) in a customized H-type cell with an anion exchange membrane (separated by a Nafion 117 membrane; magnetic stirring at 1500 rpm). A figure of the experimental set-up was provided in the Supplementary Fig. 45. Unless otherwise specified, G-RuCo on Ni foam (0.5 × 0.5 cm2, the loading of the catalyst is about 8 mg/cm2) catalyst was typically employed as the working electrode, with platinum wire and a Hg/HgO electrode (filled with 1.0 M KOH solution) serving as the counter electrode and reference electrode, respectively. And the hydrogen reversible reaction was used to calibrate the reference electrode. In addition, a solution of 2000 ppm NO3− in 1.0 M KOH was used as the electrolyte, with the initial electrolyte volume set at 30 mL for the H-cell measurements. The electrolyte solution was bubbled with Ar gas for 10 min before the experiment to remove O2 and N2. Before testing, all catalysts were first electrochemically reduced at −0.2 V vs. RHE for 600 s in a 1.0 M KOH solution to eliminate surface oxidation. Electrochemical NRA measurements were performed using linear sweep voltammetry polarization curves via the potential dynamic method at a scanning rate of 1 mV/s in 1.0 M KOH electrolyte (the pH value was 13.7). All potentials were calibrated to the RHE by the equation:
All measured potentials were 50% iR-compensated by the solution resistance, unless otherwise specified. Electrochemical impedance spectroscopy (EIS) was performed in the frequency range of 0.1 Hz–200 kHz with the amplitude of 10 mV at the overpotentials of −60 mV vs. RHE under the NRA operating condition. Rs is related to the solution resistance. Rct denotes the charge transfer resistance. Long-term stability was examined through chronopotentiometry tests at −300 and −1000 mA/cm2 in a flow-system H-cell with a 30 mL/min electrolyte flow rate.
ECSA analysis
For the electrochemical active surface area (ECSA), we used the double-layer capacitance method in an electrolyte of 1.0 M KOH in the non-Faradaic potential range with different scanning rates of 10, 20, 30, 40, 50 and 60 mV/s. The ECSA of the working electrodes was calculated according to the following equations:
where Ic represents the charging current at different scan rates, ν is the scan rate, Cdl is the double-layer capacitance, and Cs is the specific capacitance for a flat metallic surface, which is generally in the range of 20–60 μF/cm2 (we assume a value of 40 μF/cm2 here)37,38.
FE, yield rate and current density determination
The FE of NRA for NH3 and NO2− was calculated according to:
where n is the electron-transfer number (8 for 1 mol NH3, 2 for 1 mol NO2−), V is the volume of the catholyte of the cathode chamber (30 mL), c represents the concentration of the outlet products (M), F is the Faraday constant (96,485 C/mol), and Q represents the applied overall coulomb quantity (C).
The yield rate and current density of NH3 were calculated according to the following equations:
where S is the area of the geometrical cathode and t is the electrolysis time.
Ammonia calculation
The concentration of NH3 was spectrophotometrically determined using the indophenol blue method56,57. First, 2 mL of the diluted electrolyte solution was mixed with 2 mL of chromogenic agent (a mixture of 1.0 M KOH solution, 0.36 M salicylic acid, and 0.18 M sodium citrate). Then, 100 μL of 0.05 M NaClO solution (containing 4.00–4.99% effective chlorine) was added, followed by 0.2 mL of 0.034 M (1 wt.%) sodium nitrite ferrocyanide solution (stored at 4 °C) to initiate the color reaction. After allowing the mixture to stand at room temperature for 1 h, the absorbance spectrum was measured using a UV-vis spectrophotometer, and the formation of indophenol blue was determined at a wavelength of 655 nm. A standard concentration–absorbance calibration curve was prepared in advance using a range of NH4Cl (≥99.5%) solutions; the concentration of the NH3 product was then calculated based on the measured absorbance and standard curve.
Nitrite detection
The nitrite concentration was detected using UV‒vis spectrophotometry10. Initially, the collected electrolyte was diluted to the detection range. Next, 1 mL of 1.0 M HCl was added to 5 ml of diluted electrolyte, followed by adding 0.1 mL of a NO2−-specific color developing agent (a mixed solution of 0.2 g N-(1-naphthyl) ethylenediamine hydrochloride, 4.0 g sulfanilamide and 10 mL phosphoric acid (85 wt.% in H2O) in 50 ml deionized water). The absorbance intensity at a 540 nm wavelength was tested using UV‒vis spectrophotometry after allowing the resultant solution to react for 20 min at room temperature. A concentration–absorbance calibration curve was obtained by linear fitting of a series of standard potassium nitrite solutions, and the nitrite concentration was calculated based on the measured absorbance and standard curve.
Nitrate detection
The nitrate concentration was detected using UV‒vis spectrophotometry58. First, 4 ml of diluted electrolyte was mixed with 1 mL of 1.0 M HCl and 0.1 mL of sulfamic acid (0.8 wt.%) to form a mixed solution. Following a 20-minute reaction at room temperature, the absorption intensities at 220 nm and 275 nm wavelengths were recorded using UV‒vis spectrophotometry. The final absorbance (A) was calculated with the following equation: A = A220nm−A275nm. A concentration–absorbance calibration curve was established by linear fitting of a series of standard potassium nitrate solutions, and the nitrate concentration was calculated based on the measured absorbance and standard calibration curve.
Determination of ammonia by 1H NMR
To detect the FE of 14NH4+ after 1 h of electrolysis at −0.1 V (vs. RHE) in 2000 ppm K14NO3, a calibration curve of 1H NMR (400 MHz) measurements was constructed using a series of 14NH4Cl standard solutions with specified concentrations (0, 10, 20, 30, and 40 mM). Subsequently, 0.5 mL of electrolyte, mixed with 15 mM maleic acid, 50 μl of 4 M H2SO4, and 50 μL DMSO-d6, was sealed into an NMR tube for 1H NMR. Next, 2000 ppm K15NO3 was used to qualitatively determine the source of NH3. Electrolysis is performed for 1 h at −0.1 V (vs. RHE), and 15NH4+ in the electrolyte is detected with 1H NMR59.
H2 detection
For gaseous products (HER, OER), the FE has been monitored by measuring the volume of gas collected from the working electrode in an inverted burette or graduated cylinder60. The H2-FE measurement in the work was based on the water drainage method at different potentials for 1 h for different catalysts. Firstly, connect the 50 ml inverted burette to the electrolyte (2000 ppm NO3− and 1.0 M KOH) containing electrolysis chamber on one side of the working electrode through a gas guide tube. Then, apply an external potential and conduct an electrolysis reaction at a constant potential. The generated hydrogen gas enters the top of the inverted burette through the gas guide tube. Finally, after 1 h of reaction, record the volume of the hydrogen product generated.
In situ Raman spectroscopy
In situ Raman measurements were carried out using a Raman microscopy system and an electrochemical workstation. Raman spectroscopy was conducted with a Lab-RAM HR Raman microscopy system (Horiba Jobin Yvon, HR550) equipped with a 532 nm laser as the excitation source, a water immersion objective (Olympus LUMFL, 50×), a monochromator (1800 grooves/mm grating), and a Synapse charge-coupled device (CCD) detector. The electrolytic cell was made of polytetrafluoroethylene, and the working electrode was immersed into the electrolyte through the cell wall, with its plane remaining perpendicular to the incident laser. Platinum wire and Ag/AgCl electrodes were used as the counter and reference electrodes, respectively. Electrochemical intermittent in situ Raman spectroscopy was performed with a Renishaw InVia Qontor Raman system at 0.1 V intervals over a potential range of +0.5 to −0.2 V vs. RHE. After obtaining the first Raman spectrum, we added 0.1 mL of 2000 ppm KNO3 solution to the electrolyte. Each spectrum is an average of five continuously acquired spectra, with a collection time of 50 s for each collection. The cycle test was repeated four times.
EPR experiments
5,5-Dimethyl-1-pyrroline N-oxide (DMPO) was used to capture unstable hydrogen radicals by forming DMPO-H adducts, and the resulting EPR spectra were analyzed to detect the hydrogen radical signals produced by the catalyst during the reaction process44. In an H-type cell, the catalyst served as the working electrode, and constant electrolysis was performed at −0.1 V vs. RHE in a solution of 1.0 M KOH and 2000 ppm KNO3 for 5 min. After the reaction, 5 mL of the electrolyte solution was collected, and 10 μL of DMPO capturing agent was added, followed by Ar2 degassing. EPR measurements were carried out using a Bruker EMX-10/12 spectrometer under a frequency of ~9.5 GHz, a sweep width of 200 G, and a power of 20 mW.
In situ FTIR spectroscopy
Electrochemical in situ FTIR spectroscopy measurements were collected using Nicolet Nexus 8700 FTIR spectrometer equipped with a liquid N2-cooled system and MCT-A detector23,61. The Hg/HgO electrode and platinum foil electrode (Area~2 cm2) were used as the reference and counter electrode, respectively. The working electrode was prepared by depositing our developed catalysts as an active material over the glassy carbon electrode. The uniform thin layer (~10 μm) on the working electrode was obtained by vertically pressing it on the CaF2 window. The working electrode surface was set perpendicular to incoming infrared beam for obtaining the maximum response signals during electrochemical NO3− reduction. The catalyst’s in situ IR spectra (Rs) were obtained in the potential range of 0.5 V to −0.5 V at a scan rate of 100 mV/s. All the spectrums were reported after using the relation: ΔR/R = (RS−RRef)/ RRef, where the spectrum obtained at 0.5 V were considered as reference RRef. 62.
GIXRD
In the GIXRD configuration, the incident X-ray beam with a wavelength of 0.6877 Å and an energy of 18 keV is kept at a small angle α concerning the sample surface, and the distance of samples to the detector was 315 mm. The incident X-ray beam with a wavelength of 0.8266 Å and an energy of 15 keV is kept at 0.1°, 0.2°, 0.3°, 0.4°, 0.5° concerning sample surface, and the distance of samples to the detector was 1946 mm63.
Computational methods
All calculations were carried out using density functional theory with dispersion correction D3 (DFT-D3), and the projected augmented wave (PAW) scheme was implemented in the Vienna ab initio simulation software package (VASP)64,65,66. For the structural relaxation and energy calculations, the generalized gradient approximation with Perdew-Burke-Ernzerhof (PBE) parameterization was used. The cut-off energy of the plane wave function was 500 eV. For the converged unit cell models of Co (2.49 × 2.05 × 4.02 Å3), the Brillouin zone was sampled with a 15 × 15 × 6 Γ-point centered Monk horst–Pack mesh, the energy convergence criterion was within 10−5 eV, and the force tolerance was smaller than 0.01 eV Å−1 on each atom. The (100) surface of Co was used as the catalytic substrate, and the Ru-Co gradient model was constructed based on Co (100). That is because it was observed in the experiment that the Co (100) crystal plane was exposed and was the lowest energy crystal plane67. The constructed (100) surface models contained six layers; the bottom two layers were fixed, and the top four layers were fully relaxed for geometry optimization. We applied a vacuum layer of at least 20 Å in the Z-direction of the slab models to prevent interactions between the slabs in the vertical direction. The energy convergence criteria were set to 10−4 eV, and the force tolerance of each atom was smaller than 0.02 eV/Å. The Brillouin zone was sampled by a k-point mesh of 3 × 3 × 168. The calculations involving all molecules and intermediate species on the Co (100) and Ru-Co (100) substrates were conducted with spin polarization.
The Gibbs free energy change for the adsorbed *NO2 on an electrode surface to nitrite in aqueous solution (forming NO2−(l)) was calculated in three steps using the thermodynamic cycle shown in equations (Supplementary Table 8)69,70. The formation energy of NO2− on G-RuCo and NG-RuCo is calculated with the according to the following formula:
\({G}_{{gas}}\left({H}_{2}\right)\) and \({G}_{{gas}}\left({{HNO}}_{2}\right)\) are the corresponding Gibbs free energies of H2 and HNO2 molecules in the gas phase at 300 K and 1 atm. The entropic (ΔS) and enthalpic (ΔH) contributions to the free energy of the gaseous species were obtained from the NIST database. Nørskov’s computational hydrogen electrode model is used in the calculations71.
Potential industrial application
For the scaled-up NH3 production process, we used chronopotentiometry to demonstrate the potential industrial application. A homemade NRA-OER industrial electrolytic cell in an MEA flow reactor was assembled. Here, the Co2P catalyst on Ni foam was selected as the anode electrode due to the potential of Co2P material in hydrogen evolution and oxygen evolution72,73. Self-supported G-RuCo on Ni foam catalysts (1 cm × 1 cm) and self-supported Co2P on Ni foam catalysts (1 cm × 1 cm) were directly used as cathodes and anodes, respectively, with anion exchange membranes (Nafion AMI-7001S) as separators. The catholyte was 1.0 M KOH and 2000 ppm KNO3 mixed electrolyte, while the anolyte was 1.0 M KOH with an electrolyte flow rate of 50 mL/min. Polarization curves and chronopotentiometry were used to evaluate the prospects of industrial nitrate electroreduction for ammonia production.
Data availability
The data that support the conclusions of this study are available from the corresponding authors upon request. Source data are provided with this paper.
References
Zhai, S. et al. Control of particulate nitrate air pollution in China. Nat. Geosci. 14, 389–395 (2021).
Abascal, E., Gómez-Coma, L., Ortiz, I. & Ortiz, A. Global diagnosis of nitrate pollution in groundwater and review of removal technologies. Sci. Total Environ. 810, 152233 (2022).
van Langevelde, P. H., Katsounaros, I. & Koper, M. T. M. Electrocatalytic nitrate reduction for sustainable ammonia production. Joule 5, 290–294 (2021).
van der Ham, C. J. M., Koper, M. T. M. & Hetterscheid, D. G. H. Challenges in reduction of dinitrogen by proton and electron transfer. Chem. Soc. Rev. 43, 5183–5191 (2014).
Smith, C., Hill, A. K. & Torrente-Murciano, L. Current and future role of Haber–Bosch ammonia in a carbon-free energy landscape. Energy Environ. Sci. 13, 331–344 (2020).
Li, J. et al. Efficient ammonia electrosynthesis from nitrate on strained ruthenium nanoclusters. J. Am. Chem. Soc. 142, 7036–7046 (2020).
Liu, M. J. et al. Catalytic performance and near-surface X-ray characterization of titanium hydride electrodes for the electrochemical nitrate reduction reaction. J. Am. Chem. Soc. 144, 5739–5744 (2022).
He, W. et al. Splicing the active phases of copper/cobalt-based catalysts achieves high-rate tandem electroreduction of nitrate to ammonia. Nat. Commun. 13, 1129 (2022).
Han, S. et al. Ultralow overpotential nitrate reduction to ammonia via a three-step relay mechanism. Nat. Catal. 6, 402–414 (2023).
Wang, Y., Zhou, W., Jia, R., Yu, Y. & Zhang, B. Unveiling the activity origin of a copper-based electrocatalyst for selective nitrate reduction to ammonia. Angew. Chem. Int. Ed. 59, 5350–5354 (2020).
Chen, G.-F. et al. Electrochemical reduction of nitrate to ammonia via direct eight-electron transfer using a copper–molecular solid catalyst. Nat. Energy 5, 605–613 (2020).
Wang, Y. et al. Enhanced nitrate-to-ammonia activity on copper–nickel alloys via tuning of intermediate adsorption. J. Am. Chem. Soc. 142, 5702–5708 (2020).
Jia, R. et al. Boosting selective nitrate electroreduction to ammonium by constructing oxygen vacancies in TiO2. ACS Catal. 10, 3533–3540 (2020).
Wu, Z.-Y. et al. Electrochemical ammonia synthesis via nitrate reduction on Fe single atom catalyst. Nat. Commun. 12, 2870 (2021).
Huang, Y. et al. Pulsed electroreduction of low-concentration nitrate to ammonia. Nat. Commun. 14, 7368 (2023).
Chauhan, R. & Srivastava, V. C. Electrochemical denitrification of highly contaminated actual nitrate wastewater by Ti/RuO2 anode and iron cathode. Chem. Eng. J. 386, 122065 (2020).
Duca, M. & Koper, M. T. M. Powering denitrification: the perspectives of electrocatalytic nitrate reduction. Energy Environ. Sci. 5, 9726–9742 (2012).
Guo, S. et al. Insights into nitrate reduction over indium-decorated palladium nanoparticle catalysts. ACS Catal. 8, 503–515 (2018).
Fernández-Nava, Y., Marañón, E., Soons, J. & Castrillón, L. Denitrification of wastewater containing high nitrate and calcium concentrations. Bioresour. Technol. 99, 7976–7981 (2008).
Li, P., Jin, Z., Fang, Z. & Yu, G. A single-site iron catalyst with preoccupied active centers that achieves selective ammonia electrosynthesis from nitrate. Energy Environ. Sci. 14, 3522–3531 (2021).
Deng, X., Yang, Y., Wang, L., Fu, X.-Z. & Luo, J.-L. Metallic Co nanoarray catalyzes selective NH3 production from electrochemical nitrate reduction at current densities exceeding 2 A·cm−2. Adv. Sci. 8, 2004523 (2021).
Zhang, Y. et al. Conjugated coordination polymer as a new platform for efficient and selective electroreduction of nitrate into ammonia. Adv. Mater. 35, 2209855 (2023).
Fang, J.-Y. et al. Ampere-level current density ammonia electrochemical synthesis using CuCo nanosheets simulating nitrite reductase bifunctional nature. Nat. Commun. 13, 7899 (2022).
Ye, S. et al. Elucidating the activity, mechanism and application of selective electrosynthesis of ammonia from nitrate on cobalt phosphide. Energy Environ. Sci. 15, 760–770 (2022).
Chen, F.-Y. et al. Efficient conversion of low-concentration nitrate sources into ammonia on a Ru-dispersed Cu nanowire electrocatalyst. Nat. Nanotechnol. 17, 759–767 (2022).
Zhu, W. et al. Weakened d–p orbital hybridization in in situ reconstructed Ru/β-Co(OH)2 heterointerfaces for accelerated ammonia electrosynthesis from nitrates. Energy Environ. Sci. 16, 2483–2493 (2023).
Yu, H. et al. Surface enrichment and diffusion enabling gradient-doping and coating of Ni-rich cathode toward Li-ion batteries. Nat. Commun. 12, 4564 (2021).
Kong, D. et al. Ti-gradient doping to stabilize layered surface structure for high performance high-Ni oxide cathode of Li-ion battery. Adv. Energy Mater. 9, 1901756 (2019).
Wu, J. et al. Gradient design for high-energy and high-power batteries. Adv. Mater. 34, 2202780 (2022).
Wang, J. et al. Electrocatalytic reduction of nitrate to ammonia on low-cost ultrathin CoOx nanosheets. ACS Catal. 11, 15135–15140 (2021).
Zhang, R. et al. Efficient ammonia electrosynthesis and energy conversion through a Zn-nitrate battery by iron doping engineered nickel phosphide catalyst. Adv. Energy Mater. 12, 2103872 (2022).
Zhang, S. et al. Fe/Cu diatomic catalysts for electrochemical nitrate reduction to ammonia. Nat. Commun. 14, 3634 (2023).
Song, Z. et al. Efficient electroreduction of nitrate into ammonia at ultralow concentrations via an enrichment effect. Adv. Mater. 34, 2204306 (2022).
Li, J. et al. Boosted ammonium production by single cobalt atom catalysts with high Faradic efficiencies. Proc. Natl Acad. Sci. USA 119, e2123450119 (2022).
Jaramillo, T. F. et al. Identification of active edge sites for electrochemical H2 evolution from MoS2 nanocatalysts. Science 317, 100–102 (2007).
Yuan, C. et al. Growth of ultrathin mesoporous Co3O4 nanosheet arrays on Ni foam for high-performance electrochemical capacitors. Energy Environ. Sci. 5, 7883–7887 (2012).
Xue, Z.-H. et al. Boronization of nickel foam for sustainable electrochemical reduction of nitrate to ammonia. ACS Energy Lett. 8, 3843–3851 (2023).
Fan, K. et al. Active hydrogen boosts electrochemical nitrate reduction to ammonia. Nat. Commun. 13, 7958 (2022).
He, T. et al. Mastering the surface strain of platinum catalysts for efficient electrocatalysis. Nature 598, 76–81 (2021).
He, S. et al. Residual strain reduction leads to efficiency and operational stability improvements in flexible perovskite solar cells. Mater. Adv. 3, 6316–6323 (2022).
Luo, C. et al. Engineering the buried interface in perovskite solar cells via lattice-matched electron transport layer. Nat. Photon. 17, 856–864 (2023).
Yao, Y. et al. Engineering the electronic structure of single atom Ru sites via compressive strain boosts acidic water oxidation electrocatalysis. Nat. Catal. 2, 304–313 (2019).
Fang, Z. et al. Porous two-dimensional iron-cyano nanosheets for high-rate electrochemical nitrate reduction. ACS Nano 16, 1072–1081 (2022).
Wang, Y. et al. Structurally disordered RuO2 nanosheets with rich oxygen vacancies for enhanced nitrate electroreduction to ammonia. Angew. Chem. Int. Ed. 61, e202202604 (2022).
Guo, Y. et al. Pd doping-weakened intermediate adsorption to promote electrocatalytic nitrate reduction on TiO2 nanoarrays for ammonia production and energy supply with zinc–nitrate batteries. Energy Environ. Sci. 14, 3938–3944 (2021).
Zhang, N. et al. Governing interlayer strain in bismuth nanocrystals for efficient ammonia electrosynthesis from nitrate reduction. ACS Nano 16, 4795–4804 (2022).
Hu, Q. et al. Reaction intermediate-mediated electrocatalyst synthesis favors specified facet and defect exposure for efficient nitrate–ammonia conversion. Energy Environ. Sci. 14, 4989–4997 (2021).
Yang, J. et al. Potential-driven restructuring of Cu single atoms to nanoparticles for boosting the electrochemical reduction of nitrate to ammonia. J. Am. Chem. Soc. 144, 12062–12071 (2022).
Zhang, N. et al. Lewis acid Fe-V pairs promote nitrate electroreduction to ammonia. Adv. Funct. Mater. 33, 2211537 (2023).
Chen, D. et al. Tailored p-orbital delocalization by diatomic Pt-Ce induced interlayer spacing engineering for highly-efficient ammonia electrosynthesis. Adv. Energy Mater. 13, 2203201 (2023).
Ke, Z. et al. Selective NOx– electroreduction to ammonia on isolated Ru sites. ACS Nano 17, 3483–3491 (2023).
Yang, J., Liu, H., Martens, W. N. & Frost, R. L. Synthesis and characterization of cobalt hydroxide, cobalt oxyhydroxide, and cobalt oxide nanodiscs. J. Phys. Chem. C 114, 111–119 (2010).
Yeo, B. S. & Bell, A. T. Enhanced activity of gold-supported cobalt oxide for the electrochemical evolution of oxygen. J. Am. Chem. Soc. 133, 5587–5593 (2011).
He, D. et al. Regulation of the electrocatalytic nitrogen cycle based on sequential proton–electron transfer. Nat. Catal. 5, 798–806 (2022).
Feng, X. et al. Hydrogen radical-induced electrocatalytic N2 reduction at a low potential. J. Am. Chem. Soc. 145, 10259–10267 (2023).
Wang, J. et al. Ambient ammonia synthesis via palladium-catalyzed electrohydrogenation of dinitrogen at low overpotential. Nat. Commun. 9, 1795 (2018).
Zhao, Y. et al. Ammonia detection methods in photocatalytic and electrocatalytic experiments: How to improve the reliability of NH3 production rates? Adv. Sci. 6, 1802109 (2019).
Bastian, R., Weberling, R. & Palilla, F. Ultraviolet spectrophotometric determination of nitrate: application to analysis of alkaline carbonates. Anal. Chem. 29, 1795–1797 (1957).
Hodgetts, R. Y. et al. Refining universal procedures for ammonium quantification via rapid 1H NMR analysis for dinitrogen reduction studies. ACS Energy Lett. 5, 736–741 (2020).
Kempler, P. A. & Nielander, A. C. Reliable reporting of Faradaic efficiencies for electrocatalysis research. Nat. Commun. 14, 1158 (2023).
Zhou, Z.-Y., Tian, N., Chen, Y.-J., Chen, S.-P. & Sun, S.-G. In situ rapid-scan time-resolved microscope FTIR spectroelectrochemistry: study of the dynamic processes of methanol oxidation on a nanostructured Pt electrode. J. Electroanal. Chem. 573, 111–119 (2004).
Zhou, Z.-Y., Wang, Q., Lin, J.-L., Tian, N. & Sun, S.-G. In situ FTIR spectroscopic studies of electrooxidation of ethanol on Pd electrode in alkaline media. Electrochim. Acta 55, 7995–7999 (2010).
Huang, W. J. et al. He2+ irradiation induced microstructure evolution in sub-surface layer of the coarse-grained tungsten accessed by synchrotron GIXRD and GISAXS. Appl. Surf. Sci. 593, 153461 (2022).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Hafner, J. Ab-initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251–14269 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Tian, B. et al. Carboxyl-assisted synthesis of Co nanorods with high energy facet on graphene oxide sheets for efficient photocatalytic hydrogen evolution. Appl. Catal. B: Environ. 203, 789–797 (2017).
Monkhorst, H. J. & Pack, J. D. Special points for brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Liu, J.-X., Richards, D., Singh, N. & Goldsmith, B. R. Activity and selectivity trends in electrocatalytic nitrate reduction on transition metals. ACS Catal. 9, 7052–7064 (2019).
Ma, L. et al. A high-energy aqueous Zn‖NO2 electrochemical cell: a new strategy for NO2 fixation and electric power generation. Energy Environ. Sci. 16, 1125–1134 (2023).
Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886–17892 (2004).
Kandel, M. R. et al. Unique heterointerface engineering of Ni2P−MnP nanosheets coupled Co2P nanoflowers as hierarchical dual-functional electrocatalyst for highly proficient overall water-splitting. Appl. Catal. B: Environ. 331, 122680 (2023).
Li, H. et al. Retracted: colloidal cobalt phosphide nanocrystals as trifunctional electrocatalysts for overall water splitting powered by a zinc–air battery. Adv. Mater. 30, 1705796 (2018).
Acknowledgements
This work acknowledges the funding support from the State Key Laboratory of Urban Water Resources & Environment (Harbin Institute of Technology) (No. 2022TS36 (J.Z.)), National Natural Science Foundation of China (52103300 (S.H.)), Guangdong Basic and Applied Basic Research Foundation (2023A1515010572 (S.H.), 2023A1515011332 (J.Z.)), Shenzhen Science and Technology Program (JCYJ20210324132806017 (S.H.), GXWD20220811163904001 (S.H.), GXWD20231130100010001 (J.Z.)). The XAFS was carried out at the BL14W1 beamline, Shanghai Synchrotron Radiation Facility (SSRF). We thank Qisheng Yan, Wenzhe Niu (Fudan University), Prof. Yuhang Wang (Soochow University), Prof. Fengwang Li (University of Sydney), Sibo Li and Prof. Longbin Qiu (Southern University of Science and Technology) for discussions and equipment assistance.
Author information
Authors and Affiliations
Contributions
S.H. and J.Z. conceived the idea and supervised the project. S.H. and X.C. designed the experiments. X.C. synthesized the materials and carried out electrochemical measurements. J.Z. and Y.C. performed theoretical calculations. X.C. carried out in situ experiments and explored the reaction mechanism. S.H. and X.C. drew the schematic diagram. S.H., J.Z., B.Z., and X.C. wrote the paper, with comments from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, X., Cheng, Y., Zhang, B. et al. Gradient-concentration RuCo electrocatalyst for efficient and stable electroreduction of nitrate into ammonia. Nat Commun 15, 6278 (2024). https://doi.org/10.1038/s41467-024-50670-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-50670-w
This article is cited by
-
Recent advances in mechanistic studies and catalyst development for electrochemical nitrate reduction to ammonia
Communications Chemistry (2025)
-
Revealing and modulating catalyst reconstruction for highly efficient electrosynthesis of ammonia
Nature Communications (2025)
-
Stabilizing Cu0-Cuδ+ sites via ohmic contact interface engineering for ampere-level nitrate electroreduction to ammonia
Nature Communications (2025)
-
Sustainable ammonia production beyond Haber-Bosch: a review of nitrogen reduction pathways from diverse feedstocks
Science China Chemistry (2025)
-
Nanoporous surface high-entropy alloys enable tandem catalysis and ultrafast hydrogen spillover for efficient nitrate electroreduction to ammonia
Science China Chemistry (2025)
