
Abstract
Ni-based catalysts are highly reactive for dry reforming of methane (DRM) but they are prone to rapid deactivation due to sintering and/or coking. In this study, we present a straightforward approach for anchoring dispersed Ni sites with strengthened metal-support interactions, which leads to Ni active sites embedded in dealuminated Beta zeolite with superior stability and rates for DRM. The process involves solid-state grinding of dealuminated Beta zeolites and nickel nitrate, followed by calcination under finely controlled gas flow conditions. By combining in situ X-ray absorption spectroscopy and ab initio simulations, it is elucidated that the efficient removal of byproducts during catalyst synthesis is conducted to strengthen Ni–Si interactions that suppress coking and sintering after 100 h of time-on-stream. Transient isotopic kinetic experiments shed light on the differences in intrinsic turnover frequency of Ni species and explain performance trends. This work constructs a fundamental understanding regarding the implication of facile synthesis protocols on metal-support interaction in zeolite-supported Ni sites, and it lays the needed foundations on how these interactions can be tuned for outstanding DRM performance.
Similar content being viewed by others
Introduction
Dry reforming of methane (DRM) provides a promising route for converting methane and carbon dioxide into a valuable industrial feedstock, syngas, with equimolar H2/CO ratio, which is ideally suited for Fischer–Tropsch synthesis of long-chain hydrocarbons1. Rising concerns about climate change, primarily driven by the accumulation of atmospheric greenhouse gases, have reinvigorated research interest in DRM. The consumption of two prevalent greenhouse gases makes DRM an intrinsically greener approach compared to steam methane reforming (SMR). However, the endothermic nature of DRM (ΔH298K = 247 kJ mol−1) requires a high energy input and operating temperature (>600 °C) to achieve practical conversion and yield. In an optimized scenario, DRM at a relatively lower temperature could reduce energy input, thereby enhancing the energy efficiency and economic viability of the process1,2. Unfortunately, at temperatures between 580 °C and 710 °C, catalysts undergo quick deactivation caused by severe coke formation3,4 and active phase sintering5. Thus, the rational design of efficient and stable catalysts for low-temperature DRM is a challenging but vital task in both applied and academic research.
Ni-based solid catalysts are traditionally employed for DRM due to their high reactivity and low cost6. Unfortunately, the catalysts suffer from rapid deactivation, especially caused by coke deposition at lower temperatures (~650 °C)7,8. It is found that the C-C bond formation rate shows apparent size dependency on metallic Ni surfaces, where larger particles favor coke formation9,10. Therefore, it is paramount to find ways to stabilize dispersed Ni sites by either physical confinement or metal-support interactions. Ni-loaded zeolites have served as effective catalysts in reactions under harsh conditions, including CO2 hydrogenation11, alkene dimerization12,13, and DRM1. The microporous structure not only provides physical confinement to limit sintering but also offers a unique coordination environment for anchoring Ni species within micropores14,15. Up to now, Ni-supported zeolites have been synthesized via both in situ and post-synthesis methods, including hydrothermal synthesis with sol gel16,17, seed-directed crystallization18, inter zeolite transformation19,20, impregnation21,22 and decomposition of volatile organic metal precursors23. The consensus is that the interactions between Ni2+ and the framework enhance catalyst stability during DRM24. Successful anchoring of atomically dispersed Ni2+ has been revealed as a key step to creating stable catalysts. Therefore, maintaining isolated Ni2+ ions during synthesis is desirable. Strategies to stabilize Ni2+ include the use of organic ligands/solvents during hydrothermal synthesis or impregnation16,21, and the use of vapor-phase organic nickel precursors for metal incorporation23. In addition, ion-exchange sites22 and defects formed during hydrothermal synthesis, dealumination25, or deboronation26 are effective in encapsulating isolated species and clusters. However, despite efforts in selecting metal precursors and anchoring sites, the Ni2+ species loaded onto zeolites still exhibit a broad size distribution, from isolated cations and clusters to nanoparticles (NPs), even when using similar supports21,23.
All in all, the underlying chemistry of Ni–Si interaction in zeolites for the creation of stable catalytic sites is poorly understood, and thus, catalyst optimization defaults to inefficient trial-and-error. In this study, we establish a fundamental understanding regarding the impact of catalyst synthesis mechanisms on catalyst properties, and ultimately on catalyst performance for DRM. We employ a solid-state grinding protocol to anchor highly dispersed Ni2+ sites onto dealuminated Beta (dBeta) zeolite supports. Notably, the dispersion and Ni–zeolite interaction can be precisely controlled by adjusting the airflow during calcination, allowing for tunable metal dispersion ranging from NPs to isolated sites within the framework. A combination of infrared (IR) spectroscopy, X-ray absorption spectroscopy (XAS), and microscopy allowed us to characterize the dispersion of the synthesized Ni species and their interaction with the support. Structure-performance correlations demonstrated that the finely tuned synthesis method leads to catalysts with significantly enhanced stability during DRM. Interestingly, transient isotopic kinetic studies showed that metallic Ni sites in NPs have higher turnover frequency (TOF) than those on dispersed sites (clusters), explaining the observed macroscopic conversion of reactants. More importantly, in situ XAS reveals the distinct mechanisms dominating NP and isolated Ni formation under varied flow conditions. The fundamental understanding of the precision synthesis of active sites and their intrinsic activity opens up a gateway to rational catalyst design. The superior catalyst designed in this work challenges or surpasses the stability and reaction rate of reported best-performing catalysts for moderate temperature DRM.
Results and discussion
High air flow during catalyst synthesis leads to enhanced stability during DRM
Nickel nitrate hexahydrate is widely used as a precursor in the synthesis of Ni-zeolite catalysts and the chemistry of nitrate decomposition can provide guidance for precision synthesis of Ni active sites, from NP to clusters or isolated sites. Based on the literature27,28, the incorporation of Ni species into the zeolitic support is postulated to occur through four key steps: (1) solvation of Ni(NO3)2 after losing chemically bonded water below 100 °C27, (2) solution diffusion into pores by capillary effect28, (3) decomposition of nitrate to hydroxide accompanied with water and NOx desorption at elevated temperature27, and (4) condensation of Ni hydroxide with silanol on zeolitic support27. Thus, it is hypothesized that enhancing the removal of decomposition by-products, through high airflow, can strengthen Ni-support interaction.
In our synthesis procedure, dBeta was mixed with nickel nitrate hexahydrate and subjected to grinding in a mortar. The subsequent calcination step was conducted using a stepwise heating procedure under varying flow conditions, as depicted in Figs. 1a and S1 and S2. This mechanochemical method predominantly involves the decomposition of nitrate in the solid phase, circumventing the complexities of speciation and interactions in the solution phase or sol-gel process. The as-obtained catalysts are anticipated to exhibit a metal dispersion trend correlating with the gas flow rate: Ni-dBeta HD (HD for highly dispersed, 6300 mL min−1 g−1) > Ni-dBeta D (D for dispersed, 2000 mL min−1 g−1) > Ni/dBeta D (1200 mL min−1 g−1) > Ni/dBeta NP (NP for NPs, static air).

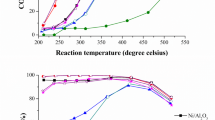
a Gas flow condition during precursor calcination to tune the metal dispersion. The synthesis of Ni-dBeta D and Ni-dBeta HD is performed in a tubular furnace and U-shape reactor, respectively. The configuration of reactors for synthesizing Ni-dBeta D and HD is shown in Fig. S2. b CH4 conversion is measured at 650 °C, 12.3 kPa CO2, 12.3 kPa CH4 balanced by 111.1 kPa Ar, overall WHSV = 1056 L g−1cat h−1. c The normalized methane conversion is evaluated by normalizing conversion by the peak value. d Stability is compared by TOS when normalized methane conversion drops by 8%.
Catalyst particles were sieved to 45–75 µm to avoid internal mass transfer limitations (Fig. S3). The catalysts were pretreated via calcination at 550 °C and reduction at 750 °C to attain the most stable structures (named as Ni/dBeta NP_red, Ni/dBeta D_red, Ni-dBeta D_red, Ni-dBeta HD_red) before conducting DRM at 650 °C (see Figs. S4–S7 and associated discussion). During catalytic tests, methane conversion was intentionally kept below equilibrium (61% CH4 conversion at 650 °C) to accurately assess real deactivation trends. Under enhanced gas flow conditions in a fixed-bed configuration (Figs. 1a and 3), both Ni-dBeta samples demonstrated slightly higher initial methane conversions and H2 to CO ratios compared to Ni/dBeta (Figs. 1b and S8). Normalized values of CH4 conversion (Fig. 1c) and TOS to reach equal deactivation (Fig. 1d) show a clear stability trend, ranking from least to most stable as: Ni/dBeta NP_red < Ni/dBeta D_red < Ni-dBeta D_red < Ni-dBeta HD_red. This ranking relates to the gas flow rate used during synthesis (Fig. 1a). The range of particle sizes of the precursors plays no significant role in the catalytic performance (Fig. S9). Temperature-programmed oxidation (TPO) of the spent samples was conducted to measure the coke deposited after conducting DRM (at reaction conditions shown in Fig. 1), as depicted in Fig. S10 and Table S1. TPO profiles show that all the samples exhibit a minor peak between 200–300 °C and a broader peak around 650–750 °C, typically ascribed to soft and hard coke respectively29. Quantitative analysis of CO2 formation revealed carbon deposits of 1.15 wt% for Ni/dBeta NP_red and 0.68 wt% for Ni/Beta D_red. Both the resilient Ni-dBeta D_red and Ni-dBeta HD_red samples present minor coke formation, accounting for 0.32 and 0.21 wt%, respectively (Table S1). The direct correlation between carbon deposition and catalyst stability indicates that enhanced gas flow conditions during synthesis might hinder sintering and coke formation in moderate-temperature DRM. The best-performing catalyst, Ni-dBeta HD_red, shows comparable stability and reaction rate to the reported best-performing catalysts for moderate temperature DRM. A summary of best-performing zeolite-based catalysts is presented in Table S2. However, the deactivation of the Ni-dBeta HD_red catalyst is more pronounced at higher reaction temperatures (Fig. S11). In the following sections, we thoroughly characterize the local environment of the Ni sites; further, we provide a clear link between the mechanisms of catalyst synthesis and the catalytic properties of the active sites created.
Ni speciation in as-synthesized samples
The preservation of the Beta zeolite structure during synthesis is confirmed by X-ray diffraction (XRD) patterns (Fig. S12) and N2 adsorption–desorption isotherms (Fig. S13). For Ni/dBeta NP, diffraction patterns for NiO were observed. Ni loading is consistent across all samples, ranging from 5.1 wt% to 5.6 wt% (Table S3). High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images (Figs. S14–S16) reveal that Ni particles, visible as bright dots due to the higher atomic weight of Ni compared to Si, are highly dispersed on Ni/dBeta D and Ni-dBeta D, with particle sizes of 1.98 nm and 1.87 nm, respectively. In contrast, Ni/dBeta NP shows noticeably larger Ni particles (20.8 ± 5.2 nm, Figs. S14 and S17a), which agrees with XRD patterns (Fig. S12).
Ex situ XAS spectra were collected on the as-synthesized samples to investigate the local electronic and coordination environment of Ni. The Ni K edge XANES spectra of all samples and NiO (Fig. 2a) display the characteristic pre-edge absorption peak of Ni2+ at 8330.4 eV, corresponding to the dipole-forbidden 1 s → 3d transition30. This fingerprint of Ni2+ and the alignment of the absorption edge for all samples with NiO suggest the predominant Ni2+ sites. Nonetheless, the k space EXAFS spectrum of Ni/dBeta NP is quite different from the other Ni-zeolite samples, as shown in Fig. S18. The k space EXAFS spectrum of Ni/dBeta NP is very similar to that of NiO, suggesting that NiO NPs are the dominant Ni species in the as-synthesized Ni/dBeta NP. Interestingly, the other three samples with higher dispersion show different k-space spectral features compared with NiO and Ni foil, indicating that the highly dispersed Ni2+ species have a unique local bonding environment generated by controlling the gas flow condition (Fig. 1a). Thus, the peaks in the range of 2–3 Å in the R-space spectra of Ni/dBeta D and two Ni-dBeta samples (Fig. 2b) are not primarily due to Ni–Ni contribution as in NiO. Wavelet transform EXAFS analysis performed on Ni/dBeta D (as described in Fig. S19) indicates that the second-shell peak is originated from Ni–Si, different from Ni–Ni in Ni/dBeta NP. Fitting results of the EXAFS (Fig. S20) further confirm the 4 coordinated O at the first shell as well as 4 Si at the second shell for the dispersed Ni2+ center, which is evidence for the incorporation in the zeolite framework. It is worth noting that synthesis in flowing Ar, instead of flowing air, has negligible influence on Ni dispersion; rather, the flow conditions play the main role (Fig. S21).

Ni K edge ex situ XAS spectra of calcined samples, NiO and Ni foil reference. a XANES, b k2-Weighted Fourier-transformed (FT) EXAFS. c DRIFTS spectra of CO adsorption at −150 °C on calcined samples after 5 min CO adsorption. d Normalized DRIFTS spectra regarding the highest peak intensity for 5 min adsorption were plotted to compare the change of fraction for different components.
However, according to particle size distribution derived from STEM images, the observed Ni2+ clusters in Ni/dBeta D show a mean diameter of about 1.98 nm, exceeding the size for both silanol nests and micropores of Beta zeolites. A potential explanation is that there is a considerable amount of silanol-grafted Ni2+ sites below the detection limit of STEM characterization (Fig. S15a), and they make a dominant contribution to the second-shell signal in the EXAFS spectra. The same phenomenon has also been observed on Pt/SiO231. For now, it is safe to declare that more highly dispersed Ni species in Ni/dBeta D are linked to the zeolite support with a Ni–Si interaction, compared to Ni/dBeta NP. These chemically bonded species can either present as isolated sites beyond the detection limit of STEM, or exist at the interface between NiO clusters and the zeolite support. Similar XANES and EXAFS features are shared by two Ni-dBeta samples, but both samples present a discernible shift toward higher absorption energy, compared to Ni/dBeta D (insert in Fig. 2a). Notably, the second-shell scattering peaks in the R space EXAFS spectra of Ni-dBeta D and HD are apparently displaced to an increased bonding distance (see Fig. 2b) compared to Ni/dBeta D, originating from the longer distance of Ni–Si (Ni–Si distance labeled by the red dash line in Fig. 2b) compared to second shell Ni–Ni path. EXAFS fitting also reveals the presence of a Ni–Si path for the second-nearest neighbor (Table S4). Accordingly, Ni-dBeta samples could have more framework-associated Ni2+ sites compared with Ni/dBeta D. Thus, the potential metal-support interaction should be stronger.
To gain further insights into the chemical properties of Ni sites in dBeta, diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) of CO was performed on the as-synthesized catalysts. All samples were calcined at 400 °C before CO adsorption, and the spectra were measured at −150 °C. For comparison, spectra of the dBeta and NiO were also recorded. As shown in Fig. 2c, CO spectra recorded after 5 min adsorption show vibrational modes at 2193 cm−1, 2156 cm−1, and 2135 cm−1 across all Ni-embedded Beta samples. The vibrational mode at 2193 cm-1 can be ascribed to CO adsorbed on grafted Ni2+ sites in silanol nests32. The vibrational mode at 2135 cm−1 is also observed for CO adsorbed on the NiO reference. Peaks at 2156 cm−1 and 2132 cm−1 in dBeta mainly arise from silanol-group-adsorbed and physically adsorbed CO33, respectively. These vibration modes indicate a complex surface speciation including grafted Ni2+ sites, surface Ni2+ in NPs/clusters, and unoccupied silanol groups. Normalized IR spectra (Figs. 2d and S22) undoubtedly show a greater contribution of Ni2+ in NiO for Ni/dBeta NP than for Ni/dBeta D. Further, more silanol groups are available for CO adsorption on Ni/dBeta NP compared to Ni/dBeta D, suggesting that more silanol groups are occupied by anchored Ni2+ species in Ni/dBeta D.
In conjunction with the XAS findings, it is inferred that Ni/dBeta NP has fewer Ni2+ sites associated with the framework, leaving more unoccupied silanol nests on the dBeta support. In contrast, more highly dispersed Ni2+ species are anchored by silanol groups in Ni/dBeta D and two Ni-dBeta samples in the form of grafted isolated sites or framework-associated interfacial species. Therefore, the interactions between the silanol-enriched Beta support and the metal precursor are postulated to enhance the anchoring of the dispersed Ni2+ sites—a phenomenon promoted by increased airflow during calcination. Tuning the calcination procedure allowed us to tune the bonding and electronic environment of Ni in dBeta support, from dispersed Ni (Ni/dBeta D, Ni-dBeta D, and Ni-dBeta HD) to Ni NP (Ni/dBeta NP).
Mechanism of Ni2+ anchoring in dBeta
It has been shown that enhanced airflow during the calcination of the precursor mixture (ground dBeta and nickel nitrate hexahydrate) dramatically increases the dispersion of Ni sites on the dBeta support with a stronger Ni–Si interaction. In addition, catalytic performance shows that catalysts with more dispersed Ni2+ sites are more stable than NPs during DRM. Therefore, in situ characterization will provide valuable insight into the interactions between Ni nitrate and dBeta support with the varied flow. Herein, in situ XAS was performed while ramping the temperature and using two distinct airflow conditions (100 mL min−1 vs static air) to study mechanisms of Ni anchoring during synthesis (Fig. 3). In situ XANES spectra for the synthesis of Ni/dBeta D and Ni/dBeta NP (Fig. S23a–c), respectively) show a decline in the white-line peak intensity, which is attributed to thermal motion and loss of chemically bonded water. In Fig. S23a, the circled isosbestic points along the heating process indicate a smooth transition between the initial and final states under flowing air (synthesis of Ni/dBeta D)34, corresponding to a stepwise decomposition of Ni(NO3)2 into Ni(OH)x intermediates, which subsequently condense with the silanol groups on dBeta27 without aggregation of isolated Ni2+ species (Fig. 4a). In contrast, under static air (Figs. 3b and 4b, c), the phase transition can be separated at 400 °C as indicated by the change of isosbestic points (Fig. S23b, c) and emerging new features at 8361 eV. At temperatures above 400 °C, the sudden shift of XANES spectra involves the increasing of white line peak intensity, accompanied by the rapid growth of second-shell Ni–Ni scattering (Fig. 4c). It has been reported that nickel nitrate hexahydrate becomes anhydrous at 205 °C under flowing air35, thus, during synthesis in flowing air, H2O and NOx desorb and Ni(OH)x is formed, followed by anchoring of Ni. However, in static air, basic nickel intermediates, represented as Ni(NO3)x·Ni(OH)y·H2O, form around 225 °C, due to inefficient removal of reaction intermediates like H2O and NOx27,35. The sluggish desorption of H2O and NOx can be exacerbated by the confined micropore structure, thus, the generation of Ni(OH)x will occur at higher temperatures. A hydrated framework lowers the energetic barrier required for the sintering of Ni, as demonstrated computationally later in Section: “Enhanced metal-support interaction and intrinsic activity of Ni sites”, which promotes the formation of bulk NiO NP.

a Schematic of Ni2+ anchoring during synthesis in flowing air and b schematic of NiO NP formation during synthesis in static air.

a–c EXAFS spectra of in situ XAS characterization for Ni/dBeta precursor calcination under a 100 mL min−1 air and b, c static air. Evolution of Ni species during calcination with d 100 mL min−1 air and e static air according to the fitting of in situ XANES. f Desorption of water during in situ XAS characterization. The red dashed lines are temperature while the solid line represents the percentage of different states of H2O in (d, e), and (f). The linear fitting method has been developed and standardized in the Demeter software suite which is widely used for XAS analysis49.
Further linear combination fitting reveals two distinct reaction pathways. Principal component analysis of XANES spectra indicates that spectral changes can be represented predominantly by two states when air is flown and three states under static air conditions (Fig. 4d, e). For the air flow case, a linear combination of two spectra, one recorded at 100 °C (t = 15 min) and one recorded at 550 °C (t = 226 min) (representing the initial and final states) offers a good fit for the observed XANES spectra. For the case of static air, it was not possible to describe the array of XANES spectra as a linear combination of only two spectra at the initial and final stages. Instead, a linear combination of three spectra, recorded at 100 °C (t = 15 min), 400 °C (t = 165 min), and 550 °C (t = 226 min) (representing an initial, intermediate, and final state) offers a good fit for the observed XANES spectra. The intermediate temperature corresponds to the beginning of the increase in Ni–Ni second-shell scattering in NiOx. In the synthesis of Ni/dBeta NP under static air, initially, the metal nitrates slowly transit to intermediates over a temperature range of 50 °C to 400 °C. Next, these intermediates exhibit rapid decomposition (400–460 °C, 195–207 min in Fig. 4e), accompanied by a marked increase in the contribution of the “final state”. Notably, a swift phase transition between 400 °C and 500 °C is also coupled with a discernible rise in the water signal as measured by the residual gas analyzer (RGA) (Fig. 4f). This suggests that water desorption plays a crucial role in the rapid phase transition. This analysis characterizes the distinct decomposition chemistry of nitrate under both dynamic and static air conditions27,35. As a result, the Ni–Si interaction is significantly impacted by the desorption of synthesis by-products, and in turn, dramatically affects the stability of Ni sites for DRM.
Enhanced metal-support interaction and intrinsic activity of Ni sites
To confirm the stability of Ni2+ in the dehydrated dBeta structure, we carried out density functional theory (DFT) calculations to determine the energy required to remove a Ni atom from the surface (Erm). Erm = ENi + Esurface − ENi_on_surface, where ENi is the energy of Ni bulk, Esurface is the energy of the surface after removing a Ni atom, and ENi_on_surface is the energy of the surface with Ni anchored on it. The more positive Erm, the more stable Ni on the surface. Three cases were studied: removal of Ni from NiO surface (and bulk), removal of Ni integrated into the framework of a hydrated dBeta structure, and removal of Ni integrated into the framework of a dehydrated dBeta structure. As shown in Fig. S24, Erm is significantly more positive for dehydrated Ni-dBeta than for all NiO surface models and the hydrated Ni-dBeta. These findings support the exceptional stability of framework-incorporated Ni2+ sites against reduction or sintering in our experimental observations. In the following section, we show how enhanced catalyst stability during DRM can be linked to strengthened Ni–Zeolite interaction during catalyst synthesis.
To illustrate the evolution of Ni species, XAS were measured on reduced and spent samples. After H2 treatment at 750 °C, the reduction of Ni2+ in all catalysts was confirmed via XAS (Figs. S25–S27, and adjacent discussion). Nearly all the NiO NPs within Ni/dBeta NP are reduced to metallic form. However, the catalyst synthesized using flowing air retains some cationic Ni species. Considering the ex situ measurement could result in the reoxidation of metallic clusters, CO DRIFTS at −100 °C on in situ reduced samples (4% H2/Ar, 500 °C, 2 h) was performed to elucidate the electronic state of Ni species (Fig. 5a). The vibrational mode at 2196 is assigned to CO adsorbed on grafted Ni2+32. Modes between 2169 cm−1 and 2094 cm−1 are assigned to CO–Ni1+32,36. Vibrational modes attributed to CO adsorption on metallic Ni are 2050 cm−1 (Ni0–(CO)x), 2029 cm−1 (a-top CO on Ni0), and 1980 cm−1 (CO adsorbed on Ni–Ni with a bridge configuration)37. The greater relative intensity of the oxidized Ni sites versus metallic Ni sites in the Ni/dBeta D_red sample undoubtedly reveals its enhanced resistance to Ni reduction when compared with the Ni/dBeta NP_red sample.

a CO DRIFTS at −100 °C measured on Ni/dBeta samples and NiO reference pretreated with 4% H2/Ar at 500 °C, after 10 min CO adsorption and 10 min desorption by Ar flushing. b Resident time (τ) measured by SSITKA at 450 °C, reactant flow of 66 mL min−1, 88 mL min−1, and 110 mL min−1 9.1% CO2, 9.1% H2 balanced by He (1% Ar as internal standard).
The average particle size estimated from the coordination number (CN) of Ni–Ni (Table S1) in the reduced samples indicates that the dispersed Ni2+ sites within the three high-dispersion samples are reduced to metallic clusters with similar average size of around 1.1 nm, smaller than those formed on Ni/dBeta NP. Interestingly, the initial conversion of Ni/dBeta NP_red and Ni/dBeta D_red was almost identical (Fig. 1b), despite the difference in a number of exposed Ni species according to the particle size estimated from XAS (Table S1). This suggests that Ni NPs have higher turnover frequencies than Ni clusters. To estimate TOF for the catalysts under operando conditions, steady-state isotopic transient kinetic analysis (SSITKA) was employed. During the SSITKA experiment, DRM was conducted over Ni/dBeta NP_red and Ni/dBeta D_red catalysts for 2 h to reach a pseudo-steady state (Fig. S28). Next, the reactants were switched from CH4 + CO2 to CH4 + 13CO2, and the transient evolution of CO and 13CO was monitored to estimate the surface residence time (τ), which can be related to TOF (τ ~ 1/TOF for pseudo-first-order reactions)38. Initially, the SSITKA experiment was conducted at 650 °C but τ of the surface species was shorter than the time-resolution of our apparatus. Then, we conducted the SSITKA experiment at 450 °C, the temperature at which τ was measurable with our apparatus. At 450 °C, Ni/dBeta D_red exhibited a τ0 of 0.75 s (Fig. 5b), corresponding to a TOF of 1.3 s−1. Ni/dBeta NP_red showed an almost negligible τ0, implying a larger TOF than for Ni/dBeta D_red (beyond the experimental detection limit). Therefore, SSITKA shows a structure sensitivity, where larger Ni metallic NPs (2.5 nm) have higher intrinsic activity than smaller metallic Ni NP/clusters (1.1 nm), agreeing with findings on SiO2-supported Ni catalysts10. Despite more exposed sites on Ni/dBeta D_red, the higher intrinsic reactivity of larger Ni NPs on Ni/dBeta NP_red leads to comparable methane conversion (Fig. 1).
Particle size derived from XAS analysis on the spent Ni/dBeta NP_red (50 h DRM), Ni/dBeta D_red (50 h DRM), Ni-dBeta D_red (53 h DRM) and Ni-dBeta HD_red (60 h DRM) are summarized in Table S1. The major portion of the Ni2+ sites within all samples are reduced to metallic form and some undergo gradual sintering during DRM. As predicted, the NiO NP within the Ni/dBeta NP catalyst exhibits the most aggregation to larger particles, and presents the most coke deposition. Metallic Ni clusters in Ni/dBeta D_red, Ni-dBeta D_red and Ni-dBeta HD_red had similar particle sizes according to the detection limit of XAS39. More importantly, the most stable Ni-dBeta HD catalyst shows no apparent sintering after a 60 h reaction, along with negligible coke deposition according to TPO experiments.
The summary of the CN from XAS fitting reveals a clear trend in the coordination environment evolution across samples subjected to different treatments and reaction conditions. During synthesis, enhanced gas flow conditions appear to favor the formation of highly dispersed Ni species within the zeolite, as evidenced by the significant reduction in the Ni–Ni second shell CN when comparing Ni/dBeta NP with Ni/dBeta D (Fig. 6a). Concurrently, an increase in Ni–Si second shell interactions suggests a rise in framework or framework-associated Ni sites. Analysis of the reduced samples (Fig. 6b) provides further insight into the chemical nature of these highly dispersed entities. Post-reduction in H2 at 750 °C for 2 h, the retention of Ni–O features is most pronounced in Ni-dBeta D_red and Ni-dBeta HD_red, followed by Ni/dBeta D_red (Fig. 6b). In contrast, the Ni/dBeta NP sample exhibits a near-complete reduction of oxidized sites. Following prolonged DRM reaction (Fig. 6c), the Ni-dBeta HD_red sample, which possesses the most robust metal-support interactions, retains the greatest number of Ni–O features and even some Ni–Si coordination in the second shell. The preservation of cationic species and particle size of metallic Ni in both reduced and spent samples suggests that strong Ni–Si interaction prohibits the complete reduction of framework Ni2+ sites and slows down the aggregation of metallic Ni clusters. The unreduced Ni species could be located at the interface between the metallic phase and supports, stabilizing the as-formed metallic clusters similar to the Cu(II)/Cu(I) interface in reduced Cu-MFI40. Thus, Ni-dBeta D shows the higher fraction of cationic species than Ni/dBeta D in the reduced and spent samples (Fig. 6b, c), contributing to the enhanced stability (Fig. 1b). Enhancing the desorption of decomposed byproducts during catalyst synthesis promotes strengthened metal-support interaction between Ni and dBeta, and this interaction is directly correlated with the ability of the catalyst to withstand the harsh conditions during DRM and provide unbeaten stability towards moderate temperature DRM.

a As-synthesized samples, b reduced samples, and c spent samples after 50–60 h DRM at 650 °C.
In conclusion, in this study, we developed a straightforward method to fine-tune the dispersion and metal-support interactions of Ni2+ species within dBeta zeolites. Advanced techniques such as STEM, XAS, and CO DRIFTS confirm that enhanced water removal during the calcination of the catalyst precursors promotes a strong Ni–Si interaction that ultimately leads to catalysts more resistant to sintering and carbon deposition during DRM. The catalyst synthesized under the highest gas space velocity (6300 mL min−1 g−1), Ni-dBeta HD, exhibits at least a fivefold increase in stability to the catalyst synthesized in static air, Ni/dBeta NP. Despite dispersed Ni sites being more resistant to sintering and coke during DRM, we show by means of operando isotopic transient kinetic analysis that this enhanced stability comes at the expense of intrinsic TOF since Ni/dBeta NP has higher TOF. In situ XAS characterization of the synthesis protocol reveals that efficient removal of decomposition byproducts during Ni nitrate decomposition enhances the interaction between Ni(OH)x intermediates and silanol nests on dBeta. This enhanced interaction frustrates Ni–Ni sintering to form NiO NPs, which are less stable for DRM. Our work constructs a fundamental understanding regarding the implication of facile synthesis protocols on metal-support interaction in zeolite-supported Ni sites, and how these interactions can be tuned for outstanding DRM performance. Further optimization of our synthesis protocol promises to push the boundaries of catalyst stability for DRM.
Methods
Materials and catalysts
Ammonium form Beta zeolite was purchased from Zeolyst (NH4-Beta, SAR = 12.5). Nickel nitrate hexahydrate (98%), nitric acid (69–70%), calcium fluoride, and quartz sand were purchased from Sigma Aldrich. All the gas cylinders are purchased from Airgas.
First, the Beta zeolite support was dealuminated. For dealumination, 1 g of commercial H-Beta (obtained by calcining NH4-Beta at 550 °C in 600 mL min−1 air flow for 12 h) was mixed with 25 mL concentrated nitric acid in the Teflon jar (Savillex) and heated to 80 °C in oil bath for 16 h. The solid was recovered by centrifuging and washing, then dried in an oven at 90 °C overnight. The Ni-embedded zeolite samples were prepared using a solid-state grinding method. Nickel nitrate hexahydrate was combined with dBeta zeolite in a mortar, and the mixture was ground for 20 min to achieve a homogeneous fine powder, characterized by a uniform light green color. The nickel loading was targeted to be 6 wt% based on the metal content. Next, the mixture was calcined. For the synthesis of Ni/dBeta D with dispersed Ni sites, the resultant mixture was placed in a ceramic boat, with a typical sample mass of about 1 g. The sample was then subjected to a controlled temperature program, initially being heated to 100 °C at a rate of 1 °C min−1 and maintained at this temperature for 3 h. Subsequently, the temperature was increased to 600 °C at the same ramping rate and held for 6 h. The calcination was conducted under a continuous air flow of 1200 mL min−1 (1200 mL min−1 g−1). In contrast, for the synthesis of Ni/dBeta NP, intended to support nickel NPs, an identical synthesis protocol was followed, except that the calcination occurred in static air. For the synthesis of Ni-dBeta D, to enhance the gas flow through the precursor bed, 0.2 g of the precursor mixture was pelletized, then crushed and sieved to achieve a particle size of 125–180 µm. This material was then packed into an ½ inch quartz tube reactor arranged in a fixed-bed configuration. The calcination procedure for this sample mirrored that of the Ni/dBeta D, except with a reduced airflow of 400 mL min−1 (2000 mL min−1 g−1). Finally, the Ni-dBeta HD sample was synthesized by packing 6.2 mg of pelletized catalyst precursor, sized between 45 µm and75 µm, along with 50 mg of quartz sand. This sample was loaded in a 4 mm U-shape reactor with a fixed-bed configuration and calcined using a commercial AMI-300 instrument from Altamira Instruments, Inc. The calcination protocol for these samples was consistent with that of the other dispersed samples, but the flow was controlled to 40 mL min−1 of 21% O2/He (6300 mL min−1 g−1).
Catalyst characterization
Powder XRD patterns of as-synthesized catalysts were measured by PANalytical X′Pert Pro MPD equipped with an X′Celerator solid-state detector (Cu Kα source, λ = 0.1542 nm, 45 kV, 40 mA). The 2θ scan ranged from 5° to 70°, with an incremental step size of 0.017 °, and the scan rate was set to 0.03° s−1. Metal compositions were ascertained through inductively coupled plasma atomic emission spectroscopy (ICP-AES), conducted by Galbraith Laboratories, Inc. Nitrogen adsorption–desorption isotherms of the samples (powder form, 125–180 μm) were determined at 77 K on a Quantachrome Autosorb iQ. Prior to these measurements, the samples were degassed under a vacuum of 3 Torr at 623 K for a duration of 6 h. HAADF-STEM was performed on Ni/dBeta NP, Ni/dBeta D, and Ni-dBeta D to characterize the morphology of zeolite particles as well as dispersion of Ni species on an aberration-corrected Hitachi HD2700C STEM operated at 200 kV. The zeolite samples were loaded on cooper grids with carbon-coated film.
DRIFTS was conducted on both as-synthesized and in situ reduced samples using a Nicolet Nexus 670 spectrometer with an MCT detector. CO was employed as the probe molecule, sourced from a gas cylinder containing a 2% CO/He mixture. For the as-synthesized samples, 6–10 mg of material was placed into a porous ceramic cup and then positioned in the DRIFTS cell (Pike technologies). The sample was heated to 400 °C at 5 °C min−1 under 30 mL min−1 3.5% O2 balanced with Ar and He and held at 400 °C for 45 min. Following this, the cell was purged with 30 mL min−1 of Ar to evacuate any residual O2 and then cooled to −150 °C. At this temperature, background spectra were collected. The adsorption spectra for CO were then recorded by introducing a gas mixture of 30 mL min−1 0.67% CO balanced with He and Ar for 5 min. Then, the gas was switched to 30 mL min−1 Ar for 20 min for desorption. For the characterization of the reduced catalysts and Ni metal powder, CaF2 was blended with the sample to enhance the signal intensity. Typically, 1 mg of the sample was blended with 5 mg of CaF2 before being loaded into the DRIFTS cell. The mixture was heated to 400 °C at 5 °C min−1 under 30 mL min−1 3.5% O2 balanced with Ar and He and held for 45 min. After cooling down to 50 °C, the cell was flushed with 30 mL min−1 of Ar and then with 30 mL min−1 of 4% H2/Ar for 30 min. Subsequently, the sample was heated to 500 °C and reduced with 30 mL min−1 4% H2/Ar for 45 min. After flushing with 30 mL min−1 Ar for 45 min, the sample was cooled down to −100 °C. The procedure for measuring the CO spectra for the reduced sample was the same as that used for the as-synthesized sample.
The Ni K edge XAS spectra were measured at beamline QAS 7-BM of National Synchrotron Light Source II at Brookhaven National Laboratory (BNL) and at beamline BL 4-1 of the Stanford Synchrotron Radiation Lighthouse (SSRL) at SLAC National Laboratory. In situ XAS measurements were performed at BNL. For the in situ experiments, a disc of the precursor mixture (ground dBeta and nickel nitrate hexahydrate) was loaded in a Nashner–Adler cell and the temperature was increased stepwise to 100 °C, 250 °C, 400 °C, and 550 °C, each held for 30–60 min. In one experiment, air flowed through the cell (100 mL min−1), and in another experiment, air was static in the cell. The exhaust from the cell was analyzed using a RGA. Spectra were measured in transmission and fluorescence modes, focusing on the K edges of Ni. Concurrently with the sample measurements, energy calibration was executed using a third ion chamber, which was equipped with Ni foil standards. The data analysis and reduction process were carried out utilizing the Demeter software suite41.
Catalytic performance
For the reactivity measurement, the catalysts were pelletized, crushed, and sieved to a particle size from 45 µm to 75 µm to avoid internal mass transfer limitations. Five milligram of the sieved catalyst sample was mixed with 50 mg of 90–125 µm quartz sand and loaded into a U-shape quartz reactor of 4 mm inner diameter. The sample was held by two pieces of quartz wool with a thermocouple on top to measure the temperature of the catalyst bed. The reactor was installed in a commercial AMI-300 instrument (Altamira Instruments, Inc.) for catalytic tests. It was confirmed that the quartz wool and quartz sand dilution did not provide background DRM reactivity. Before any catalytic test, the catalysts were calcined in situ using 40 mL min−1 of 5% O2/He at 550 °C for 1 h to remove adsorbed moisture and organics. After flushing with 50 mL min−1 Ar for 30 min at room temperature, the DRM reaction was performed with a gas mixture containing 88 mL min−1 9.1% CH4, 9.1% CO2, and 81.8% Ar at 650 °C (ramping rate 5 °C min−1) and the pressure is around 135.7 kPa. When H2 pretreatment was performed, the sample was first calcined at 550 °C as described before, and then cooled down to 50 °C under 40 mL min−1 Ar before exposing the sample to 40 mL min−1 of 4% H2/Ar. Then, the reactor temperature was ramped at 5 °C min−1 to 750 °C under 40 mL min−1 of 4% H2/Ar and held for 2 h. After cooling to 650 °C in the 40 mL min−1 4% H2/Ar flow, the gas line was flushed with 50 mL min−1 Ar for 30 min and 88 mL min−1 9.1% CH4, 9.1% CO2 and 81.8% Ar were introduced. The identity and quantity of products were analyzed by a gas chromatograph (Buck Scientific. Inc.) equipped with a thermal conductivity detector and an online mass spectrometer (MS, Pfeiffer Vacuum).
The conversion of CH4 and CO2 was calculated using Eq. (1):
where \({X}_{{{\rm{reactant}}}}\) (%) is the conversion for CH4 and CO2, \({C}_{{{\rm{inlet}}}}\) is the concentration of reactant feeding into the reactor, \({C}_{{{\rm{outlet}}}}\) is the concentration of reactant in the product stream, \({F}_{{{\rm{inlet}}}}\) is the flow rate of the feed and \({F}_{{{\rm{outlet}}}}\) is the flow rate of the outlet stream.
Where \({C}_{{H}_{2},{outlet}}\) and \({C}_{{CO},{outlet}}\) are H2 and CO concentrations in the product stream, respectively.
The carbon balance of all experiments is in the range of 95–102%.
TPO of spent samples was performed on the AMI-300 instrument.
After the reaction, the sample was flushed with 50 mL min−1 Ar and cooled down to 50 °C. Then, the gas was switched to 40 mL min−1 5% O2/He for 30 min before ramping to 750 °C at 10 °C min−1. The signal of CO2 was measured by an online MS.
SSITKA experiments
SSITKA measurements were performed on a homemade instrument with two sets of gas lines42: regular lines and labeled lines (with isotopically labeled reactants). The signals of CO2 (44), 13CO2 (45), CH4 (16), H2 (2), CO (28), 13CO (29), Ar (40), and O2 (32) at the outlet of the reactor were monitored by an online MS. The catalyst amount and dilution used were the same as in the catalytic tests described above. SSITKA measurements were performed at 450 °C. The 88 mL min−1 of 9.1% CH4, 9.1% CO2, and 0.91% Ar (internal standard) balanced with He was fed to the reactor, and the reaction was allowed to stabilize for 2 h. Upon stabilization, the feed gas was switched to 88 mL min−1 9.1% CH4, 9.1% 13CO2 balanced with He. During the switch, the pressure in the regular and labeled line was held constant at ~48.3 kPa (gauge pressure), using back-pressure regulators. The signals of all gases before, during, and after the switch were monitored by MS. At least three isotopic switches were performed to assess uncertainty in the measurements. The SSITKA experiment was performed using three total volumetric flow rates to account for the readsorption of species: 66 mL min−1, 88 mL min−1, and 110 mL min−1.
DFT calculation
The periodic DFT calculations were performed using the Vienna ab initio Simulation Package43,44. The Perdew–Burke–Ernzerhof45 functional generalized-gradient approximation (GGA) was used for electron exchange-correlation. The on-site Coulomb interaction was included using the DFT + U method of Dudarev, et al. 46. using a Hubbard parameter U of 5.3 eV for Ni, following a previous study47. The Brillouin zone was sampled using a 3 × 3 × 1 Monkhorst–Pack scheme. A kinetic energy cutoff of 500 eV was used for the plane waves. Based on the model construction approach by Sazama et al. 48, we built a structure for dBeta zeolite with a composition of Si64O128 and cell parameters of a = 12.66 Å, b = 12.66 Å, and c = 26.41 Å of a tetragonal lattice. Then we replaced some Si atoms with Ni, balanced with –OH groups, to create Ni dBeta structures with an extensive refinement and optimization process where around ten different configurations are tested before arriving at the final most stable Ni/dBeta model with a composition of Ni4Si60O128H8.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information file. Should any raw data files be needed in another format they are available from the corresponding author upon request.
References
Velty, A. & Corma, A. Advanced zeolite and ordered mesoporous silica-based catalysts for the conversion of CO2 to chemicals and fuels. Chem. Soc. Rev. 52, 1773–1946 (2023).
Lee, J., Bae, Y., Hong, K. & Hong, J. Comparative evaluation of Ni‐based bimetallic catalysts for dry reforming of methane at low temperature: the effect of alloy itself on performance. Int. J. Energy Res. 46, 11228–11249 (2022).
Gadalla, A. M. & Bower, B. The role of catalyst support on the activity of nickel for reforming methane with CO2. Chem. Eng. Sci. 43, 3049–3062 (1988).
Wang, S., Lu, G. & Millar, G. J. Carbon dioxide reforming of methane to produce synthesis gas over metal-supported catalysts: state of the art. Energy Fuels 10, 896–904 (1996).
Moulijn, J. A., Van Diepen, A. & Kapteijn, F. Catalyst deactivation: Is it predictable?: What to do? Appl. Catal. A Gen. 212, 3–16 (2001).
Al-Fatesh, A. S. et al. Reforming of methane: effects of active metals, supports, and promoters. Catal. Rev. https://doi.org/10.1080/01614940.2023.2211447 (2023).
Bradford, M. & Vannice, M. CO2 reforming of CH4. Catal. Rev. 41, 1–42 (1999).
Pakhare, D. & Spivey, J. A review of dry (CO2) reforming of methane over noble metal catalysts. Chem. Soc. Rev. 43, 7813–7837 (2014).
Kim, J.-H., Suh, D. J., Park, T.-J. & Kim, K.-L. Effect of metal particle size on coking during CO2 reforming of CH4 over Ni–alumina aerogel catalysts. Appl. Catal. A Gen. 197, 191–200 (2000).
Vogt, C., Kranenborg, J., Monai, M. & Weckhuysen, B. M. Structure sensitivity in steam and dry methane reforming over nickel: activity and carbon formation. ACS Catal. 10, 1428–1438 (2019).
Gac, W., Zawadzki, W., Słowik, G., Kuśmierz, M. & Dzwigaj, S. The state of BEA zeolite supported nickel catalysts in CO2 methanation reaction. Appl. Surf. Sci. 564, 150421 (2021).
Ehrmaier, A. et al. Dimerization of linear butenes on zeolite-supported Ni2+. ACS Catal. 9, 315–324 (2018).
Jaegers, N. R. & Iglesia, E. Theoretical assessment of the mechanism and active sites in alkene dimerization on Ni monomers grafted onto aluminosilicates:(Ni–OH)+ Centers and C–C coupling mediated by Lewis acid–base pairs. J. Am. Chem. Soc. 145, 6349–6361 (2023).
Pang, H., Yang, G., Li, L. & Yu, J. Toward the production of renewable diesel over robust Ni nanoclusters highly dispersed on a two-dimensional zeolite. NPG Asia Mater. 15, 24 (2023).
Yasuda, S. et al. Zeolite-supported ultra-small nickel as catalyst for selective oxidation of methane to syngas. Commun. Chem. 3, 129 (2020).
Cheng, Q. et al. Highly efficient and stable methane dry reforming enabled by a single-site cationic Ni catalyst. J. Am. Chem. Soc. 145, 25109–25119 (2023).
Wang, J. et al. Design of a carbon-resistant Ni@ S-2 reforming catalyst: controllable Ni nanoparticles sandwiched in a peasecod-like structure. Appl. Catal. B Environ. 282, 119546 (2021).
Zhu, Q. et al. Enhanced CO2 utilization in dry reforming of methane achieved through nickel-mediated hydrogen spillover in zeolite crystals. Nat. Catal. 5, 1030–1037 (2022).
Liu, Y. et al. Embedding high loading and uniform Ni nanoparticles into silicalite-1 zeolite for dry reforming of methane. Appl. Catal. B Environ. 307, 121202 (2022).
Kweon, S. et al. Defect-stabilized nickel on beta zeolite as a promising catalyst for dry reforming of methane.Catal. Sci. Technol. 12, 3106–3115 (2022).
Meloni, M. et al. Nano-sized metallic nickel clusters stabilized on dealuminated beta-zeolite: a highly active and stable ethylene hydrogenation catalyst. J. Phys. Chem. C. 126, 21213–21222 (2022).
Luengnaruemitchai, A. & Kaengsilalai, A. Activity of different zeolite-supported Ni catalysts for methane reforming with carbon dioxide. Chem. Eng. J. 144, 96–102 (2008).
He, D. et al. Dynamic trap of Ni at elevated temperature for yielding high-efficiency methane dry reforming catalyst. Appl. Catal. B Environ. 346, 123728 (2024).
Xu, S. et al. Developing silicalite-1 encapsulated Ni nanoparticles as sintering-/coking-resistant catalysts for dry reforming of methane. Chem. Eng. J. 446, 137439 (2022).
Tamura, K. et al. Dry reforming of methane with suppressed carbon deposition over Cr-and Ni-loaded dealuminated β zeolites. Energy Fuels https://doi.org/10.1021/acs.energyfuels.3c02837 (2023).
Kweon, S. et al. Defect-induced formation of nickel silicates on two-dimensional MWW-type catalysts promoting catalytic activity for dry reforming of methane. Microporous Mesoporous Mater. 332, 111683 (2022).
Cochran, E. A., Woods, K. N., Johnson, D. W., Page, C. J. & Boettcher, S. W. Unique chemistries of metal-nitrate precursors to form metal-oxide thin films from solution: materials for electronic and energy applications. J. Mater. Chem. A 7, 24124–24149 (2019).
Lee, S.-Y. & Aris, R. The distribution of active ingredients in supported catalysts prepared by impregnation. Catal. Rev. Sci. Eng. 27, 207–340 (1985).
de Sousa, F. F. et al. Nanostructured Ni-containing spinel oxides for the dry reforming of methane: effect of the presence of cobalt and nickel on the deactivation behaviour of catalysts. Int. J. Hydrog. Energy 37, 3201–3212 (2012).
Pickering, I. J., George, G. N., Lewandowski, J. T. & Jacobson, A. J. Nickel K-edge X-ray absorption fine structure of lithium nickel oxides. J. Am. Chem. Soc. 115, 4137–4144 (1993).
Li, Y. et al. Complex structural dynamics of nanocatalysts revealed in Operando conditions by correlated imaging and spectroscopy probes. Nat. Commun. 6, 7583 (2015).
Penkova, A., Dzwigaj, S., Kefirov, R., Hadjiivanov, K. & Che, M. Effect of the preparation method on the state of nickel ions in BEA zeolites. A study by fourier transform infrared spectroscopy of adsorbed CO and NO, temperature-programmed reduction, and X-ray diffraction. J. Phys. Chem. C. 111, 8623–8631 (2007).
Mintova, S. et al. Variation of the Si/Al ratio in nanosized zeolite Beta crystals. Microporous mesoporous Mater. 90, 237–245 (2006).
Moliner, M. et al. Reversible transformation of Pt nanoparticles into single atoms inside high-silica chabazite zeolite. J. Am. Chem. Soc. 138, 15743–15750 (2016).
Elmasry, M., Gaber, A. & Khater, E. Thermal decomposition of Ni(II) and Fe(III) nitrates and their mixture. J. Therm. Anal. Calorim. 52, 489–495 (1998).
Aleksandrov, H. A. et al. Precise identification of the infrared bands of the polycarbonyl complexes on Ni–MOR zeolite by 12C16O–13C18O coadsorption and computational modeling. J. Phys. Chem. C. 116, 22823–22831 (2012).
He, Y., Song, Y. & Laursen, S. The origin of the special surface and catalytic chemistry of Ga-rich Ni3Ga in the direct dehydrogenation of ethane. ACS Catal. 9, 10464–10468 (2019).
Polo-Garzon, F. et al. CH4 activation over perovskite catalysts: true density and reactivity of active sites. ACS Catal. 12, 11845–11853 (2022).
Finzel, J. et al. Limits of detection for EXAFS characterization of heterogeneous single-atom catalysts. ACS Catal. 13, 6462–6473 (2023).
Pang, J. et al. Hierarchical echinus-like Cu-MFI catalysts for ethanol dehydrogenation. ACS Catal. 10, 13624–13629 (2020).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541 (2005).
Polo-Garzon, F., Pakhare, D., Spivey, J. J. & Bruce, D. A. Dry reforming of methane on Rh-doped pyrochlore catalysts: a steady-state isotopic transient kinetic study. ACS Catal. 6, 3826–3833 (2016).
Kresse, G. & Furthmüller, J. Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA + U study. Phys. Rev. B 57, 1505–1509 (1998).
Jiang, S. & Mushrif, S. H. Determining surface-specific Hubbard-U corrections and identifying key adsorbates on nickel and cobalt oxide catalyst surfaces. Phys. Chem. Chem. Phys. 25, 8903–8912 (2023).
Sazama, P. et al. Acid and redox activity of template-free Al-rich H-BEA* and Fe-BEA* zeolites. J. Catal. 318, 22–33 (2014).
Wang, Q., Hanson, J. C. & Frenkel, A. I. Solving the structure of reaction intermediates by time-resolved synchrotron X-ray absorption spectroscopy. J. Chem. Phys. 129, 234502 (2008).
Acknowledgements
This research was sponsored by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division, Catalysis Science program. Some of the work including synthesis and XRD was conducted as part of a user project at the Center for Nanophase Materials Science (CNMS), which is a U.S. DOE, Office of Science User Facility located at Oak Ridge National Laboratory. This research used resources of the Center for Functional Nanomaterials and National Synchrotron Light Source II, which are U.S. DOE, Office of Science User Facilities located at BNL under Contract No. DE-SC0012704. Beamline operations were supported in part by the Synchrotron Catalysis Consortium (U.S. DOE, Office of Basic Energy Sciences, Grant DE-SC0012335). This research used resources of the Stanford Synchrotron Radiation Lightsource (SSRL) of SLAC National Accelerator Laboratory supported by Basic Energy Sciences under contract No.DE-AC02-76SF00515. This research used resources of the National Energy Research Scientific Computing Center, a U.S. DOE, Office of Science User Facility supported by the Office of Science of the U.S. DOE under contract no. DE-AC02-05CH11231. Notice: this manuscript has been authored by UT-Battelle, LLC under contract no. DE-AC05-00OR22725 with the U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
Author information
Authors and Affiliations
Contributions
J. Zhang: conceptualization, investigation, methodology, data curation, formal analysis, and writing—original draft. Y. Li: methodology, formal analysis, and writing—reviewing and editing. H. Song: DFT calculation and writing—reviewing and editing. L. Zhang: investigation and methodology. Y. Wu: investigation. Y. He: investigation. L. Ma: investigation and methodology. J. Hong: investigation. A. Tayal: investigation and methodology. N. Marinkovic: investigation and methodology. D. Jiang: DFT calculation and writing—reviewing and editing. Z. Li: conceptualization and writing—reviewing and editing. Z. Wu: methodology and writing—reviewing and editing. F. Polo-Garzon: conceptualization, investigation, methodology, formal analysis, writing—reviewing and editing, supervision, and project administration. All authors have given approval to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
Oak Ridge National Laboratory has submitted a US patent application serial number 63/550.647, February 7, 2024, file date, titled, “Stable Beta Zeolite Catalyst,” on which J.Z. and F.P. are listed as inventors. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Liang Wang and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, J., Li, Y., Song, H. et al. Tuning metal-support interactions in nickel–zeolite catalysts leads to enhanced stability during dry reforming of methane. Nat Commun 15, 8566 (2024). https://doi.org/10.1038/s41467-024-50729-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50729-8
