
Abstract
Filovirus-host interactions play important roles in all stages of the virus lifecycle. Here, we identify LATS1/2 kinases and YAP, key components of the Hippo pathway, as critical regulators of EBOV transcription and egress. Specifically, we find that when YAP is phosphorylated by LATS1/2, it localizes to the cytoplasm (Hippo “ON”) where it sequesters VP40 to prevent egress. In contrast, when the Hippo pathway is “OFF”, unphosphorylated YAP translocates to the nucleus where it transcriptionally activates host genes and promotes viral egress. Our data reveal that LATS2 indirectly modulates filoviral VP40-mediated egress through phosphorylation of AMOTp130, a positive regulator of viral egress, but more surprisingly that LATS1/2 kinases directly modulate EBOV transcription by phosphorylating VP30, an essential regulator of viral transcription. In sum, our findings highlight the potential to exploit the Hippo pathway/filovirus axis for the development of host-oriented countermeasures targeting EBOV and related filoviruses.
Similar content being viewed by others

Introduction
Ebola (EBOV) and Marburg (MARV) viruses are emerging pathogens of the Filoviridae family that cause sporadic and global outbreaks of acute hemorrhagic fever in humans, resulting in mortality rates as high as 90%. Filoviruses can establish chronic/persistent infections in immunologically privileged sites and their re-emergence can cause long-term sequelae or death1,2. As current preventative and therapeutic treatments are limited, more effective antivirals are urgently needed to curb these deadly infections3,4. The identification and characterization of filovirus-host interactions that impact the virus lifecycle can be exploited as potential targets for the development of host-oriented countermeasures and represent a potential way forward5. The PPxY Late domain motifs conserved in the VP40 matrix proteins of EBOV and MARV drive the egress of infectious virions by interacting with select host WW-domain-containing proteins that either positively (e.g., Nedd4, Itch, and WWP1)6,7,8 or negatively (e.g., BAG3 and WWOX)9,10,11 regulate filovirus egress and spread. For example, we demonstrated recently that the host mTORC1/CASA (chaperone-assisted selective autophagy) axis regulates EBOV egress, as suppression of mTORC1 signaling inhibits viral egress through selective autophagic degradation of VP40 via the BAG3-mediated CASA complex12.
Intriguingly, we identified the Hippo pathway downstream effectors YAP/TAZ, as WW-domain-containing interactors with the PPxY motifs from both EBOV and MARV VP4013,14. The Hippo pathway is a pivotal host pathway that controls cell proliferation, differentiation, and migration and is itself regulated in part by PPxY/WW-domain interactions. Specifically, the core Hippo kinases LATS1 and LATS2 bear PPxY motifs that interact with YAP/TAZ, and the ensuing phosphorylation prevents shuttling of YAP/TAZ from the cytoplasm (Hippo ON) into the nucleus where they function as transcriptional coactivators (Hippo OFF)15,16,17. Additionally, YAP/TAZ activity can be regulated by peripheral Hippo components, such as PPxY-containing AMOTp13018. The potential competitive battle between the filoviral PPxY motif (VP40), and host PPxY motifs (LATS and AMOTp130) for binding to the WW domains of YAP/TAZ is likely to have a significant biological impact on both the virus and host.
In this study, we characterize a previously undescribed intersection between the EBOV lifecycle and key Hippo pathway components LATS1/2 and YAP. Specifically, we find that Hippo signaling-regulated YAP localization and activity shape the cellular environment towards one that either inhibits (Hippo ON) or promotes (Hippo OFF) viral egress. Our findings also suggest that LATS2 indirectly regulates egress by phosphorylating AMOTp130, and more importantly, LATS1/2 kinases directly regulate EBOV transcription by interacting with EBOV NP and phosphorylating EBOV VP30. In sum, our results reveal mechanistic insights into the multifaceted Hippo pathway-mediated regulation of multiple stages of the EBOV lifecycle, which may lead to the identification and exploitation of antiviral targets for developing host-oriented countermeasures against EBOV and related pathogens.
Results
Egress of EBOV is controlled by Hippo pathway effectors YAP/TAZ
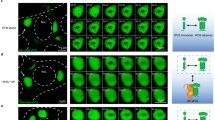
We demonstrated previously that transient expression of cytoplasmically localized YAP inhibited egress of VP40 VLPs13,14. Since the phosphorylated status of YAP regulates its cytoplasmic-nuclear shuttling19,20, we investigated the effects of phosphorylation and intracellular localization of YAP on VP40-mediated egress. As expected, egress of eVP40 VLPs was inhibited by exogenously expressed WT-YAP localized primarily in the cytoplasm (Fig. 1a and S1a, b)13,14. Interestingly, we found that the expression of YAP-S127A, a nonphosphorylated nuclear-localized YAP mutant, did not inhibit the egress of eVP40 VLPs. In contrast, the phosphomimetic mutant YAP-S127D that remains in the cytoplasm21, robustly inhibited eVP40 VLP egress (Fig. 1a), indicating that the phosphorylation and localization status of YAP dictates its ability to regulate eVP40 VLP egress.

a YAP differentially regulates EBOV VP40 VLP egress in a phosphorylation-dependent manner. eVP40 VLP budding assay at 24 h.p.t. in HEK 293 cells expressing eVP40 alone, or with exogenous flag-tagged WT-YAP, or YAP mutants S127A or S127D. b eVP40 VLP budding assay at 24 h.p.t. in WT and Y/T-dKO cells. c Representative confocal images of WT and Y/T-dKO cells showing eVP40 (green) and endogenous YAP (magenta). Scale bar = 10 µm. Three times experiments were repeated independently with similar results. d eVP40 VLP budding assay at 24 h.p.t. in WT or Y/T-dKO cells, or Y/T-dKO cells expressing exogenous WT-YAP, or YAP mutants S127A or S127D. e Working model showing Hippo “ON” vs. Hippo “OFF” conditions and differential regulation of VP40 VLP egress by YAP/TAZ. f Schematic diagram showing the EBOV infection protocol in WT and Y/T-dKO cells. g Representative images and quantification of EBOV-GFP (MOI = 0.05) infected WT and Y/T-dKO cells at 24- and 48- h.p.i. The infection efficiency was determined as a ratio of GFP-positive (infected) cells to total cell nuclei. h Representative images and quantification (data were normalized to the infection efficiency in corresponding HEK 293 cells) of Vero cells infected with the virus isolated from the supernatants of WT and Y/T-dKO cells in panel (g). Scale bar = 200 µm. Data in (a, b) and (d) are presented as mean ± SD (n = 3). Data in (g, h) are presented as mean ± SD (n = 8). Statistical analyses were performed by one-way ANOVA in (a, d), or by two-tailed unpaired t-test in (b, g, h). Source data are provided as a Source Data file.
To further delineate the role of YAP/TAZ in VP40-mediated egress, we assessed VP40 VLP budding in WT and YAP/TAZ double knockout (Y/T-dKO) HEK 293 cells. Surprisingly, egress of eVP40 VLPs was reduced significantly in the Y/T-dKO cells compared to that from WT cells (Fig. 1b). It is worth noting that a single knockout of YAP or TAZ did not lead to a reduction in VLP egress (Fig. S1c), due most likely to their redundant functions as Hippo pathway downstream effectors22. We then visualized the subcellular localization of eVP40 and endogenous YAP in the WT and Y/T-dKO cells. As expected, eVP40 was distributed in abundant, well-organized projections around the cell periphery, indicative of robust VLP budding in WT cells. In contrast, eVP40 projections were reduced significantly in the Y/T-dKO cells, leading to the accumulation of eVP40 on the plasma membrane (Fig. 1c). Live cell imaging confirmed the dynamic and efficient formation of eVP40 VLPs in WT cells and revealed condensed and tangled eVP40 projections in the Y/T-dKO cells (Video S1).
We next determined whether VLP egress could be rescued in the Y/T-dKO cells by trans-complementation of YAP-WT or mutants. We observed that expression of nuclear-localized YAP-S127A, but not cytoplasmic localized YAP-S127D, rescued egress of eVP40 VLPs (Fig. 1d). Interestingly, trans-expression of WT-YAP in Y/T-dKO cells also restored egress of eVP40 VLPs back to WT levels (Fig. 1d), due most likely to a substantial amount of exogenous YAP localizing to the nucleus in Y/T-dKO cells (Fig. S1a, b). These findings suggest that YAP can toggle its effect on VP40-mediated egress, with phosphorylated cytoplasmic YAP inhibiting egress, and nonphosphorylated, nuclear YAP promoting egress. In our working model, we speculate that under Hippo “OFF” conditions, activated nuclear YAP cooperates with TEAD to enhance the transcription of target genes that promote cell proliferation/migration and establish a pro-virus egress environment (Fig. 1e, I). In contrast, under Hippo “ON” conditions, inactive cytoplasmic YAP binds and sequesters VP40 away from the plasma membrane to inhibit egress and establish an anti-virus egress environment (Fig. 1e, II). The double knockout of YAP/TAZ abolishes their activity and ability to shape the cellular environment into one that is conducive to productive EBOV infection (Fig. 1e, III).
Notably, results from live EBOV infection in WT and Y/T-dKO cells strongly support our working model (Fig. 1f). Specifically, EBOV infection in the Y/T-dKO cells was inhibited significantly compared to that observed in WT cells (Fig. 1g). More importantly, the virus yield from the Y/T-dKO cells was reduced by 80-95% compared to that from the WT cells (Fig. 1h) at both 24- and 48-hours post-infection (h.p.i.). This inhibitory effect was consistent over a wide range of MOIs, as validated by the analyses of intracellular and extracellular viral RNA levels (Figs. S1d, e). These findings suggest that expression of endogenous YAP/TAZ is critical for both early (infectivity) and late (egress) stages of the EBOV lifecycle.
YAP/TAZ transcriptional activity is required for a productive EBOV infection
As transcriptional coactivators and effectors of the Hippo pathway, YAP/TAZ can influence the cellular environment by regulating the expression of a diverse set of target genes23. As some of these genes may play a mechanistic role in the reduction of EBOV infectivity and egress (Fig. 1g, h), we next identified differentially expressed genes (DEGs) in WT vs. Y/T-dKO cells using RNA Seq. We identified 518 genes that were significantly downregulated in the Y/T-dKO cells, and only 93 genes were upregulated (Fig. S2a). Notably, we identified downregulated DEGs associated with the composition and biology of the extracellular matrix (ECM) network, the construction of basolateral and apical plasma membranes (PM), and the formation and organization of cell projections (Fig. 2a–d). Changes in ECM composition and cytoskeletal dynamics at the PM would be predicted to affect both PM-associated stages of EBOV entry and egress. As such, we asked whether the abolishment of YAP/TAZ activity would result in alterations of F-actin organization at the cell periphery and subsequent inhibition of virus egress. Indeed, we observed that while WT cells showed well-organized and robust F-actin-based cell projections, particularly across the cell-cell interface, these projections were largely reduced in the Y/T-dKO cells (Fig. 2e and S2b). Concurrently, the vigorous eVP40 VLP budding in WT cells along the cell projections was reduced significantly in the Y/T-dKO cells (Fig. 2e and Videos S2, S3).

a–c Gene Ontology (GO) enrichment analysis of significantly downregulated DEGs in Y/T-dKO cells clustered in three sub-ontologies as Cellular Component, CC (a), Biological Process, BP (b), and Molecular Function, MF (c), the top15 enriched GO terms in each sub-ontologies are shown based on P values and gene counts. P values are based on a two-sided test. d Heatmap showing the top 20 significantly downregulated DEGs in Y/T-dKO cells clustered in the GO term of Cell projection organization, in descending order by fold change. e Representative confocal images of WT and Y/T-dKO cells showing eVP40 (green), endogenous F-actin (Phalloidin, red), and endogenous YAP (magenta). Scale bar = 5 µM. Three times experiments were repeated independently with similar results. f Heatmap showing significantly downregulated DEGs in Y/T-dKO cells clustered in the GO term of Virus entry into the host cell, in descending order by fold change. g Representative confocal images showing internalized EBOV VP40 + GP VLPs (green) in WT and Y/T-dKO cells. Scale bar = 10 µM. Three times experiments were repeated independently with similar results. h Quantification of entry efficiency of VSV pseudotypes encoding the luciferase reporter gene in WT and Y/T-dKO cells. Data in (h) were presented as mean ± SD (n = 6). Statistical analyses were performed by a two-tailed unpaired t-test. Source data are provided as a Source Data file.
We also identified DEGs that could be linked to virus entry (Fig. 2b). Of particular interest was the YAP/TAZ-target gene AXL24 (Fig. 2f), which facilitates entry of EBOV by enhancing macropinocytosis25,26. To determine whether macropinocytosis was impaired in the Y/T-dKO cells, we monitored the EBOV VLP internalization and indeed observed a substantial decrease in VLP uptake in Y/T-dKO cells, compared to that in WT cells (Fig. 2g). To corroborate this observation, we quantified entry of VSV pseudotypes encoding the luciferase reporter and either EBOV GP (eGP), MARV GP (mGP), or VSV-G as a control. We identified a significant decrease in macropinocytosis-dependent entry27 of VSV-eGP or -mGP in Y/T-dKO cells as compared to that in WT cells. However, we did not detect a difference in the entry of VSV-G pseudotypes, which enter cells primarily in a macropinocytosis-independent manner28 (Fig. 2h). Together, these results suggest that a productive EBOV infection may be dependent, in part, on a favorable cellular environment established by nuclear YAP/TAZ and a concomitant, specific pattern of YAP/TAZ-targeted gene expression that favors both viral entry and egress.
Modulation of YAP activity regulates VP40-mediated VLP and live EBOV egress
YAP activity is positively and negatively regulated by EGF and verteporfin, respectively. EGF activates YAP by promoting its shuttling into the nucleus, where it co-activates transcription of target genes29. Conversely, verteporfin targets the nuclear YAP-TEAD complex and inhibits its transcriptional activity30. To determine whether EGF-mediated activation or verteporfin-mediated inhibition of YAP would affect VP40 VLP egress, Huh7 cells expressing eVP40 were treated with vehicle, EGF, or verteporfin followed by quantification of VLP egress. We observed that EGF treatment enhanced, whereas verteporfin treatment inhibited, egress of eVP40 VLPs in a dose-dependent manner (Fig. 3a, b). Consistent with a previous report31, we observed reduced levels of endogenous YAP upon verteporfin treatment (Fig. 3b). Similar results were observed in Huh7 cells expressing mVP40 from MARV and treated with EGF or verteporfin (Fig. 3c, d). Importantly, these results were not specific to Huh7 cells, as we observed identical findings in Hela cells (Fig. S3a, b). Visualization of eVP40 and endogenous YAP localization in Huh7 cells following treatment with EGF revealed a similar pattern of eVP40 distribution and robust VLP budding in both the control and EGF-treated cells, as well as substantial staining of nuclear YAP (Fig. 3e, Control and EGF). In contrast, YAP was detected primarily in cytoplasmic plaques in verteporfin-treated cells, with virtually no YAP being detected in the nuclei. Notably, eVP40 was more dispersed and localized to cytoplasmic puncta (Fig. 3e, Verteporfin), which correlated with a reduction in eVP40 VLP.

a, b eVP40 VLP budding assay at 24 h.p.t. in Huh7 cells that were mock-treated, treated with 5 or 20 ng/ml EGF (a), or treated with 1 or 5 µM Verteporfin (b). c, d mVP40 VLP budding assay at 24 h.p.t. in Huh7 cells that were mock-treated, treated with 5 or 20 ng/ml EGF (c), or treated with 1 or 2.5 µM Verteporfin (d). e Representative confocal images showing eVP40 (green) and endogenous YAP (magenta) in Huh7 cells that were mock-treated, treated with 20 ng/ml EGF, or treated with 5 µM Verteporfin. Scale bar = 5 µm. Three times experiments were repeated independently with similar results. f Heatmap showing the top 20 significantly downregulated DEGs in Y/T-dKO cells clustered in the GO term of Growth factor binding and activity, in descending order by fold change. g Schematic diagram showing the EBOV infection and treatment protocol in MDMs. h, i Quantification of virus infectivity and production in EBOV-GFP infected (MOI = 0.2) human MDMs pre-treated (h), or post-treated (i) with Verteporfin at 24 h.p.i. Virus production from MDMs was titrated on Vero cells and was normalized to the infection efficiency in corresponding MDMs. Data in (a–d) and (h, i) are presented as mean ± SD (n = 3). Statistical analyses were performed by one-way ANOVA in (a–d) or two-way ANOVA in (h, i). Source data are provided as a Source Data file.
Interestingly, our RNA Seq. analysis revealed a transcriptional downregulation of EGF, as well as a variety of growth factors in the Y/T-dKO cells (Figs. 2c, 3f), which agree with the above findings and our working model (Fig. 1e). We postulate that YAP and growth factors are likely part of a positive feedback loop that promotes cell proliferation and a cellular environment beneficial for EBOV infection32. In contrast, verteporfin inhibits YAP-TEAD transcriptional activity and leads to cytoplasmic sequestration of YAP, resulting in a cellular environment that is unfavorable for viral egress. As further evidence to support our working model, human monocyte-derived macrophages (MDMs) were pre-treated (−1 h.p.i.) or post-treated (1 h.p.i.) with verteporfin and infected with live EBOV (Fig. 3g). EBOV infectivity and the release of progeny virions into the supernatants were quantified. We found that while verteporfin treatment did not affect EBOV infectivity (Fig. 3h, i), it consistently reduced EBOV release from either pre- or post-treated MDMs by 75% (Fig. 3h, i) in a dose-dependent manner. These results suggest that YAP activity is critical for a productive EBOV infection in biologically relevant MDMs33,34.
Hippo pathway LATS1/2 kinases differentially regulate VP40 VLP egress
As LATS1/2 are core kinases that directly regulate YAP/TAZ activity, we next examined their role in modulating VLP egress. Interestingly, we found that LATS2, but not LATS1, significantly inhibited egress of both eVP40 and mVP40 VLPs (Fig. 4a, b). Since we demonstrated previously that YAP and AMOTp130 regulate filovirus egress13,14, and both YAP (Ser127)19,20 and AMOTp130 (Ser175)35,36 are phosphorylated by LATS1/2 kinases on their consensus HXRXXS motifs (Fig. 4c), we next asked whether YAP and/or AMOTp130 may be involved in LATS2-mediated inhibition of VLP egress. To begin, our co-IP assay identified that LATS1 and LATS2 interacted equally well with YAP; however, LATS2 interacted more strongly with AMOTp130 than did LATS1 (Fig. 4d). Additionally, we observed typical localization patterns for eVP40 and AMOTp130 in control cells and in cells co-expressing LATS1, with some colocalization of eVP40 and AMOTp130 occurring at the VLP budding sites on the PM (Fig. 4e, Control and LATS1). In contrast, cells co-expressing LATS2 revealed significant alterations in the localization patterns of both eVP40 and AMOTp130, in that AMOTp130 lost its tubular-like pattern and eVP40 projections were reduced (Fig. 4e, LATS2). Moreover, we found that increasing amounts of exogenous AMOTp130 counteracted LATS2-mediated inhibition and rescued egress of both eVP40 (Fig. 4f) and mVP40 (Fig. S4a) VLPs.

a, b eVP40 (a) and mVP40 (b) VLP budding assay at 24 h.p.t. in HEK 293 cells expressing VP40 alone, or with exogenous myc-tagged LATS1 or LATS2. c Schematic diagram showing the LATS kinase-mediated key phosphorylation sites within the consensus motifs HXRXXS of AMOTp130 and YAP. d Co-IP assay with myc-tagged LATS1 or LATS2 and endogenous AMOTp130 and YAP in HEK 293 cells. e Representative confocal images of HEK 293 cells expressing eVP40 (green), AMOTp130 (red), and myc-tagged LATS1 or LATS2 (magenta). Scale bar = 10 µm. f eVP40 VLP budding assay at 24 h.p.t. in HEK 293 cells expressing eVP40 alone, or with constant amounts of exogenous LATS2, and increasing amounts of AMOTp130. g eVP40 VLP budding assay at 24 h.p.t. in HEK 293 cells expressing eVP40 alone, or with exogenous WT LATS2 or LATS2 mutants T1041A or T1041E. h Representative confocal images of shAMOT HEK 293 cells expressing eVP40 (green), and exogenous WT AMOTp130, or AMOTp130 mutants S175A or S175D (red), and endogenous F-actin (Phalloidin, cyan). Scare bar = 10 µm. Three times experiments (d–h) were repeated independently with similar results. i, j eVP40 (i) and mVP40 (j) VLP budding assay and quantification in WT, LATS1-KO, or LATS2-KO cells. Data in (a, b) and (i, j) are presented as mean ± SD (n = 3). Statistical analyses were performed by one-way ANOVA. Source data are provided as a Source Data file.
LATS2 activity is controlled by upstream kinases MST1/2 and MAP4K37, which phosphorylate LATS2 on Thr1041. To determine whether this kinase cascade contributes to LATS2-mediated inhibition of VLP egress, we compared the VLP egress efficiency in HEK 293 cells expressing eVP40 alone or with LATS2-WT, nonphosphorylated mutant T1041A, or phosphomimetic mutant T1041E. We found that egress of both eVP40 (Fig. 4g) and mVP40 (Fig. S4b) VLPs was inhibited by LATS2-WT and T1041E, but not the nonphosphorylated T1041A mutant. In addition, LATS2-WT and mutants interacted equally well with AMOTp130 (Fig. S4c); however, AMOTp130 was phosphorylated by LATS2-WT and T1041E, but not by T1041A mutant (Fig. S4d). To further assess how the phosphorylation status of AMOTp130 impacts VLP egress, shAMOT HEK 293 cells were transfected with eVP40 plus plasmids encoding AMOTp130-WT, S175A, or S175D mutants. We observed similar tubular-like patterns of AMOTp130-WT and S175A mutant, as well as typical eVP40 projections (Fig. 4h, WT and S175A). In contrast, AMOTp130 S175D mutant was distributed more diffusely and formed cytoplasmic puncta38, indicative of its inability to bind to F-actin35,36. Moreover, eVP40 projections were fewer, yet longer, suggesting that eVP40 was unable to bud efficiently (Fig. 4h, S175D).
Lastly, we quantified VLP egress from WT, LATS1-KO, or LATS2-KO HEK 293 cells (Fig. S5a) expressing eVP40 or mVP40, to assess the role of endogenous LATS1 and LATS2 in regulating VLP egress. We observed a significant enhancement of both eVP40 and mVP40 VLP egress in LATS2-KO but not LATS1-KO cells, as compared to WT cells (Fig. 4i, j). Together, these results suggest that Hippo pathway kinase LATS2, but not LATS1, inhibits EBOV and MARV VLP egress indirectly by targeting the positive regulator of filovirus egress, AMOTp130, thereby disrupting its ability to promote VP40-mediated egress.
LATS1/2 kinases affect multiple stages of the EBOV lifecycle
To evaluate the biological impact of the LATS1/2 kinases during EBOV infection, we infected WT, LATS1-KO, LATS2-KO, and LATS1/2-dKO HEK 293 cells with live EBOV and assessed the infectivity and progeny virus release at 24- and 48- h.p.i. (Fig. 5a). Surprisingly, EBOV infectivity at 24 h.p.i. was reduced by 50% and 35% in the LATS1-KO and LATS2-KO cells, respectively, compared to that in WT cells (Fig. 5b), and this reduction was even more pronounced at 48 h.p.i. (Fig. 5c). Strikingly, barely 2% infectivity was observed for EBOV in the LATS1/2-dKO cells at 24 h.p.i., and infectivity was almost completely suppressed at 48 h.p.i., compared to that in WT cells (Fig. 5b, c).

a Schematic diagram of the live EBOV-GFP infection protocol. b, c Representative images and quantification of EBOV-GFP (MOI = 0.05) infected WT, LATS1-KO, LATS2-KO, and LATS1/2-dKO cells at 24- (b) and 48- (c) h.p.i. The infection efficiency was determined by a ratio of GFP-positive cells to total cell nuclei. d, e Representative images and quantification (data was normalized to the infection efficiency in b and c, respectively) of Vero cells infected with the supernatants isolated from WT, LATS1-KO, LATS2-KO, and LATS1/2-dKO cells in panels (b) and (c). Scale bar = 200 µm. f Schematic diagram of the EBOV minigenome assay. g–i EBOV minigenome transcription determined by quantification of eGFP reporter (g) or luciferase reporter (h) in WT, LATS1-KO, LATS2-KO, and LATS1/2-dKO cells. Detection of the indicated viral RNP proteins in HEK 293 cells expressing the EBOV minigenome system (i). Three times experiments were repeated independently with similar results. Data in (b–e, n = 7) and (h, n = 10) are presented as mean ± SD. Statistical analyses were performed by one-way ANOVA. Source data are provided as a Source Data file.
In live EBOV studies, the two LATS kinase paralogs had distinct effects on virus egress at both 24- and 48- h.p.i., with egress inhibited to a greater extent in LATS1-KO and LATS1/2-dKO cells at 24 h.p.i. than in the LATS2-KO cells (Fig. 5d). At 48 h.p.i., viral yields from LATS2-KO cells were almost equivalent to those of WT cells, whereas viral yields from LATS1-KO and LATS1/2-dKO were still significantly inhibited (Fig. 5e). These results correlated well with our VP40 VLP budding data in that expression of LATS2, but not LATS1, restricted VLP egress (Fig. 4). However, the apparent inhibition of release of EBOV in LATS1/2-dKO cells was likely due to the reduced infectivity that we observed (Fig. 5b, c), as we did not observe any inhibition of VLP egress in the LATS1/2-dKO cells (Fig. S5b). This remarkable suppression of EBOV infection in cells lacking LATS1/2 kinases was consistent over a wide range of MOIs, which was validated by the analyses of intracellular and extracellular viral RNA levels (Fig. S5c, d), suggesting that expression of LATS1/2 kinases is likely important for an earlier stage of the EBOV lifecycle.
To identify the early stage of the EBOV lifecycle impacted by the LATS kinases, we first assessed viral entry. However, there was no difference in VLP internalization/uptake among the WT and LATS-KO cells (Fig. S5e). To assess viral transcription, we used the EBOV minigenome assay in which EBOV NP, VP30, VP35, and L plasmids were transfected into WT or LATS-KO cells along with either the eGFP or Luc reporter plasmids (Fig. 5f)39. Interestingly, we observed strong reporter expression in WT, LATS1-KO and LATS2-KO cells; that was reduced significantly in the LATS1/2-dKO cells transfected with either the eGFP reporter plasmid (Fig. 5g) or the Luc reporter plasmid (Fig. 5h). Expression levels of all viral proteins were consistent among all cell types (Fig. 5i). Based on these surprising findings, we speculate that LATS1/2 kinases regulate EBOV transcription in a redundant manner, potentially by interacting with and regulating one or more EBOV RNP proteins.
LATS1/2 kinases target EBOV VP30 for phosphorylation to regulate viral transcription
EBOV VP30 is a dynamically phosphorylated protein that regulates viral transcription, and Ser29 is the critical serine residue for modulating its transcriptional activity40,41,42. Notably, a LATS kinase consensus motif of 24HVRARS29 (HXRXXS) that includes this residue is present in EBOV (Zaire) VP30, and there is a similar motif of 35HPRARS40 in MARV VP30 (Fig. 6a). We therefore assessed the Ser29 phosphorylation of eVP30 in the cells harboring EBOV minigenome system using a specific antibody41. We detected modest, yet equivalent levels of Ser29-phosphorylated eVP30 in WT, LATS1-KO, and LATS2-KO cells; that was reduced significantly in the LATS1/2-dKO cells (Fig. 6b), suggesting that both LATS1 and LATS2 kinases can phosphorylate eVP30 at Ser29. We next isolated total RNA from EBOV-infected WT and LATS1/2-dKO cells at 1-, 5-, and 10- h.p.i. and performed an RT-qPCR to determine the kinetics of eGP genome equivalents (GE). Our analysis revealed a change in eGP GE kinetics between WT and LATS1/2-dKO cells during the window of 5–10 h.p.i. (Fig. 6c), which corresponds with the time of viral transcription43. In addition, we observed that exogenous expression of LATS1 or LATS2 in LATS1/2-dKO cells restored EBOV minigenome activity to WT levels (Fig. 6d and S6a) and restored the modest degree of Ser29 phosphorylation of eVP30 (Fig. 6e). Moreover, trans-complementary expression of LATS1 or LATS2 also rescued EBOV infectivity in the LATS1/2-dKO cells (Fig. 6f and S6b, c). Together, these results suggest a critical role for LATS kinases in phosphorylating eVP30 on Ser29 and regulating EBOV transcription.

a LATS kinase targets the Serine within the consensus HXRXXS which is present in EBOV VP30. b Western blot to detect and quantify eVP30 Ser29 phosphorylation levels in WT, LATS1-KO, LATS2-KO, and LATS1/2-dKO cells. Ser29 phosphorylation intensity was normalized to the total level of eVP30. c RT-qPCR to detect and quantify EBOV GP genome equivalent (GE) kinetics in 1, 5, 10 h.p.i. windows in WT and LATS1/2-dKO cells infected with EBOV-GFP (MOI = 0.01). d, e EBOV minigenome assay (d) and Western blot to detect eVP30 Ser29 phosphorylation (e) in WT and LATS1/2-dKO cells, or LATS1/2-dKO cells expressing exogenous LATS1 or LATS2. f Quantification of EBOV-GFP (MOI = 0.05) infected WT, LATS1/2-dKO, and LATS1/2-dKO cells trans-complemented with exogenous LATS1 or LATS2 at 48 h.p.i. g–h Co-IP assay with eNP and exogenous myc-tagged LATS1 or LATS2 (g), or endogenous LATS (h) in HEK 293 cells. i Co-IP assay showing the association of eNP and eVP30 with endogenous LATS in HEK 293 cells. j, k Representative confocal images of Huh7 cells expressing myc-tagged LATS1/2 (green) and flag-tagged eVP30 (magenta) in the absence (j) or presence (k) of eNP (red). Insert panels in (k) showing the colocalization of LATS1/2, eVP30, and eNP in IBs. Scale bar = 10 µm in (j) and 5 µm in (k). Three times experiments (g–k) were repeated independently with similar results. l Detection of eVP30 Ser29 phosphorylation and the EBOV minigenome assay in HEK 293 cells that were mock-treated or treated with 2.5 or 5 µM LATS kinase inhibitor TDI-011536. m Detection of eVP30 Ser29 phosphorylation and the EBOV minigenome assay in HEK 293 cells that mock-treated, treated with 5 µM TDI-011536, treated with 5 µM SRPK kinase inhibitor SPHINX31, or in combination. n Quantification of virus production from EBOV-GFP (MOI = 0.2) infected MDMs pre-treated or post-treated with TDI-011536 at 24 h.p.i. Data in (b, c, n = 4), (d, l, n = 8), (f, m, n = 9), and (n, n = 3) are presented as mean ± SD. Statistical analyses were performed by one-way ANOVA or two-way ANOVA in (c). Source data are provided as a Source Data file.
Next, we used a co-IP assay to determine whether LATS1/2 kinases interact with eVP30 or other RNP proteins. While we did not detect a direct interaction between LATS and eVP30 (Fig. S6d), we did detect an interaction between eNP and exogenously expressed LATS1 and LATS2, as well as endogenous LATS in HEK 293 cells (Fig. 6g, h). Moreover, we confirmed the eNP and LATS interaction in cells expressing the EBOV trVLP system and detected a low level of eVP30 in the eNP/LATS precipitates (Fig. 6i). In addition, we observed a consistent interaction between eNP and eVP30 in both WT and LATS1/2-dKO cells (Fig. S6e). Since we detected a direct interaction between eNP and LATS, we reasoned that eNP may recruit LATS to cytoplasmic inclusion bodies (IBs) where viral RNA synthesis takes place43. To examine this possibility, we observed the spatial relationship among LATS1/2, eVP30, and eNP. While both eVP30 and LATS1/2 distributed diffusely in the cytoplasm (Fig. 6j and S6f), the typical IBs were observed upon co-expression of eNP, and both eVP30 and LATS1/2 were recruited into the IBs by eNP (Fig. 6k and S6g), implying that eNP may serve as a scaffold to orchestrate the association between eVP30 and LATS1/2 kinases within IBs, thereby enabling LATS-mediated phosphorylation of eVP30 on Ser29.
We next utilized the LATS kinase inhibitor, TDI-011536, to determine whether the EBOV minigenome phenotype recapitulated that observed in the LATS1/2-dKO cells. As expected, Ser127 phosphorylation of YAP was inhibited in TDI-011536-treated HEK 293 cells44, and we observed a similar suppression of Ser29 phosphorylation of eVP30 (Fig. 6l), strongly suggesting that LATS kinase phosphorylates the HXRXXS consensus motifs within both YAP and eVP30. This effect of TDI-011536 was observed in Huh7 cells as well (Fig. S7a). Importantly, we found that EBOV minigenome activity was reduced significantly in both HEK 293 and Huh7 cells treated with TDI-011536 (Fig. 6l and S7a). As the SRPK kinase also phosphorylates Ser29 within the 26RxxS29 motif of eVP3040, we used SRPK inhibitor SPHINX3145 to determine whether inhibition of SRPK kinase would also repress EBOV minigenome activity. Indeed, we found that treatment with SPHINX31 alone, or in combination with TDI-011536, equally reduced eVP30 Ser29 phosphorylation and repressed EBOV minigenome activity (Fig. 6m).
To further validate the regulatory role of LATS kinases in the EBOV lifecycle, human MDMs were pre-treated (-1 h.p.i.) or post-treated (1 h.p.i.) with TDI-011536 and infected with EBOV in the presence of the compound. We observed that the generation of infectious EBOV virions was reduced by 90% (pre-treated) and 80% (post-treated) in TDI-011536 treated cells as compared to mock-treated cells (Fig. 6n), strongly suggesting that LATS activity is required for efficient propagation of EBOV. As viral infectivity (Fig. S7b) and VLP egress (Fig. S7c) were unaffected by TDI-011536, the reduction in virus yield was most likely due to impairment of viral transcription by TDI-011536-mediated inhibition of LATS kinases.
Loss of YAP/TAZ and LATS1/2 kinases impairs EBOV propagation in 3D spheroids
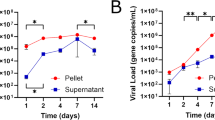
Lastly, we sought to use an advanced infection model to further verify our findings46. Toward this end, we generated 3D spheroids of WT, YAP/TAZ-dKO, and LATS1/2-dKO cells (Fig. 7a and S8a) and infected them with live EBOV. We observed that depletion of YAP/TAZ or LATS1/2 kinases specifically inhibited propagation of EBOV in the 3D spheroids (Fig. 7b), but not that of the VSV control (Fig. S8b–d). Notably, viral RNA loads were reduced by 91% in Y/T-dKO spheroids compared to that in WT. Furthermore, the loss of LATS1/2 kinases potently suppressed propagation of EBOV in 3D spheroids, resulting in a 96% and 92% reduction of viral RNA for 103 and 104 I.U., respectively (Fig. 7c, d).

a Schematic diagram showing the generation of 3D spheroids and the EBOV infection protocol. b Representative images of mock and EBOV-GFP (103 and 104 I.U.) infected 3D spheroids of WT, YAP/TAZ-dKO, and LATS1/2-dKO cells at 48 h.p.i. Scale bar = 300 µm. c, d RT-qPCR to detect and quantify viral genome equivalent (GE) from the spheroids infected with 103 (c) and 104 (d) I.U. of EBOV-GFP. Data were presented as mean ± SD (n = 8). Statistical analyses were performed by one-way ANOVA. Source data are provided as a Source Data file.
Discussion
Here, we describe an in-depth intersection between Hippo signaling and multiple stages of the EBOV lifecycle, allowing the potential exploitation of the Hippo pathway/filovirus axis for the development of therapeutic countermeasures to target this deadly hemorrhagic fever virus. We not only reveal that YAP activity is required for a productive EBOV infection, but also that the LATS1/2 kinases modulate EBOV transcription and egress. As such, our study has yielded a working model of the Hippo pathway/filovirus axis (Fig. 8). Notably, recent studies revealed crosstalk between Hippo signaling and host antiviral defenses47,48, and a dysregulation of Hippo signaling during infection by Zika49 and SARS-CoV-250, suggesting that the Hippo pathway may be involved in the lifecycles of multiple emerging viruses.

a Target genes of YAP/TAZ regulate the composition and function of the ECM/PM. b LATS1/2 kinases interact with NP and phosphorylate VP30 to regulate EBOV transcription. c Involvement of LATS1/2, YAP/TAZ, as well as AMOT in regulating EBOV/MARV egress.
Utilizing WT and functional YAP mutants, as well as pharmacological activation and inhibition of YAP activity, we demonstrate that YAP-mediated regulation is a double-edged sword, both promoting and inhibiting VP40-mediated viral egress. Our findings reveal that while cytoplasmic YAP sequesters VP40 away from PM to inhibit viral egress13,14, transcriptionally active, nuclear-localized YAP promotes viral egress (Fig. 8c). In support, our RNA Seq and comparative analyses identified numerous genes related to the composition and function of ECM/PM were significantly downregulated in the absence of YAP and TAZ (Y/T-dKO cells). Notably, the ECM controls cell stiffness and tension by interacting with cytoskeletal architecture at the PM, and subsequent mechanotransduction signals can modulate YAP/TAZ activity16,23. As both viral entry and egress involve the ECM and PM and are inhibited in the Y/T-dKO cells, it is not surprising that we identified a series of ECM/PM-related genes that were downregulated. We recently reported that the knockdown of ECM-associated and YAP-responsive proteins filamin A/B, differentially regulates the entry of live EBOV and MARV51. It will be of interest to undertake further studies to interrogate the functional link between ECM/PM/Hippo signaling and both the early and late stages of filovirus infection (Fig. 8a, c).
In addition to ECM components, soluble growth factors such as EGF and VEGF trigger YAP nuclear translocation and transcriptional activation of its target gene profile29,52. Notably, EBOV infection stimulates TGF-β and VEGF signaling, leading to the disruption of cellular barriers, induction of EMT, and disease progression53,54. Additionally, comparative transcriptomics indicated that EBOV infection robustly upregulated the expression of a variety of growth factors, including PDGF, TGFB, VEGF, and CTGF32. Therefore, we speculate that EBOV infection induces a positive feedback loop in which expression of growth factors that stimulate YAP/TAZ activity help shape a cellular environment conducive to a productive infection15,52,55. In support, our transcriptomic analysis revealed significant downregulation of CTGF, PDGF, EGF, and multiple TGFB-related genes in the Y/T-dKO cells, suggesting that depletion of YAP/TAZ disrupts the positive feedback loop elicited by YAP/TAZ activating growth factors expression, thereby disabling a host cell response15,52,55 conducive to EBOV infection32,53,54.
We reported previously that AMOTp130 coordinates F-actin dynamics at the PM leading to efficient egress and spread of EBOV13. Here, we found that LATS2, but not LATS1, inhibited VLP egress by phosphorylating AMOTp130 and disabling its ability to bind to F-actin35,36. These non-redundant roles for LATS1 and LATS2 were observed during egress of live EBOV as well, agreeing with previous studies suggesting that the two paralogs have divergent interactomes and, therefore, carry out non-redundant functions56,57. While the impact of LATS2 on viral egress was somewhat predictable based on its interaction with YAP and AMOTp130, we surprisingly found that both LATS1 and LATS2 were critical modulators of EBOV transcription. Our data suggest a model by which LATS1/2 kinases interact with eNP and are recruited to viral IBs. Here, they phosphorylate Ser29 within the LATS kinase 24HXRXXS29 consensus motif in eVP30, an essential activator of vial transcription, thereby modulating EBOV transcription.
Dynamic phosphorylation of eVP30 is crucial for the EBOV lifecycle5,41,42,58,59. Indeed, eVP30 is phosphorylated in the virion60,61, and a basal phosphorylation level of eVP30 was detected in EBOV-infected cells62. While Ser29-phosphorylated eVP30 is required for viral primary transcription40, weakly phosphorylated or dephosphorylated eVP30 activates secondary transcription63, and phosphorylated eVP30 contributes to viral genome replication58,59. Importantly, among the clustered N-terminal serine residues of eVP30, a single Ser29 is sufficient to modulate eVP30 transcriptional activity and to generate a recombinant EBOV42. As eVP30 contains a single 24HXRXXS29 motif, LATS kinase can specifically target the Ser29, leading to a modest level of phosphorylated eVP30. Our results showed that inhibitors targeting either the LATS or SRPK kinases impaired Ser29 phosphorylation of eVP30 and thus repressed EBOV minigenome activity. In addition, since the LATS1/2 kinases interact directly with eNP, and since eNP interacts with and recruits phosphatases PP2A and PP164,65,66, we hypothesize that eNP plays a central role in coordinating the phosphorylation status of eVP30 within viral IBs to regulate RNA synthesis (Fig. 8b).
Collectively, our study reveals how key effector YAP and core kinases LATS1/2 of the Hippo pathway intersect with the EBOV lifecycle. Our data suggest that a productive EBOV infection prefers a Hippo “OFF” environment in which active YAP/TAZ promotes transcription of a wide range of target genes that are “pro-viral” (Fig. 8a, c). Moreover, LATS1/2-mediated Ser29 phosphorylation of eVP30 is required for efficient EBOV transcription (Fig. 8b). This emerging intersection of the Hippo pathway with the lifecycles of EBOV and perhaps other related viruses as well, will open new areas of investigation into the biology and pathogenesis of these emerging viruses, and reveal antiviral targets for the development of host-oriented countermeasures.
Methods
Plasmids, antibodies, and reagents
Plasmids
The plasmids encoding eVP40 and GFP-eVP40 were described previously67. Flag-tagged mVP40-WT was kindly provided by Dr. Stephan Becker (Philipps University of Marburg). The pCMV flag-YAP-WT, flag-YAP-S127A, and flag-YAP-S127D plasmids were kindly provided by Dr. Marius Sudol (Icahn School of Medicine at Mount Sinai). The myc-LATS1 and myc-LATS2 plasmids were kindly provided by Dr. Jixin Dong (University of Nebraska). The flag-LATS2-WT/T1041A/T1041E, HA-AMOTp130-WT/S175A/S175D plasmids were kindly provided by Dr. Dannel McCollum (University of Massachusetts). The EBOV minigenome system that includes the pCAGGS-eNP, pCAGGS-eVP35, pCAGGS-eL, pCAGGS-eVP30, and pCAGGS-3E5E-eGFP or pGAGGS-3E5E-LUC were gifts from Dr. Elke Mühlberger (Addgene plasmid # 103049-103055). The EBOV trVLP system that includes the plasmids encoding EBOV NP, VP35, L, VP30, and the plasmid pCAGGS-T7 encoding T7 promoter were kindly provided by Dr. Christopher F. Basler (Icahn School of Medicine at Mount Sinai).
Antibodies
Anti-eVP40 (0301-010), anti-eVP30 (0301-048), anti-eNP (0301-012), and anti-eVP35 (0301-040) polyclonal antibodies were from IBT Bioservices. The anti-pSer29-eVP30 polyclonal antibody was kindly provided by Dr. Christopher F. Basler (Icahn School of Medicine at Mount Sinai) and Dr. Stephan Becker (Philipps University of Marburg). The anti-YAP (13584-1-AP) and anti-LATS (17049-1-AP, this polyclonal antibody recognized both LATS1 and LATS2 in our study) polyclonal antibodies, and the anti-flag (66008-4-Ig), anti-HA (66006-2-Ig) and anti-β-actin (66009-1-Ig) monoclonal antibodies were from Proteintech. The anti-AMOTp130 monoclonal antibody (sc-166924) was from Santa Cruz. The anti-pSer175-AMOTp130 (18H4L17) monoclonal antibody was from Invitrogen. The anti-myc monoclonal antibody (05-724) was from EMD Millipore. The anti-LATS1 (3477), anti-LATS2 (5888), and the anti-pSer127-YAP (13008) monoclonal antibodies were from Cell Signaling Technology. The secondary antibodies used in this study include mouse IgG HRP linked whole Ab (NA931), rabbit IgG HRP linked whole Ab (NA934) from EMD Millipore; and goat anti-rabbit IgG- Alexa Fluor™ 488/568/647, goat anti-mouse IgG- Alexa Fluor™ 488/555/647 from Invitrogen. The VeriBlot for IP detection reagent (HRP) (ab131366) was from Abcam. The CoraLite® Plus 647-conjugated flag (CL647-80010) and CoraLite® Plus 488-conjugated MYC (CL488-67447) antibodies were from Proteintech.
Reagents
X-tremeGENE™ HP DNA Transfection Reagent (6366236001, Roche). TRIzol RNA Isolation Reagent (Invitrogen, 15596026). RNeasy Plus Micro Kit (Qiagen, 74034). Rhodamine Phalloidin (Invitrogen, R415). Phalloidin-iFluor 405 Reagent (Abcam, ab176752). Mounting Medium with DAPI (Abcam, ab104139). NucBlue™ Live ReadyProbes™ Reagent (Invitrogen, R37605). Human EGF Recombinant Protein (Gibco, PHG0315). Verteporfin (MedChemExpress, HY-B0146), SPHINX31 (MedChemExpress, HY-117661). CellTiter-Glo® Luminescent Cell Viability Assay system (Promega, G7570), Luciferase Assay System (Promega, E4030), ONE-Glo™ Luciferase Assay System (Promega, E6110). TDI-011536 (Selleckchem, E1314).
Cells lines
The WT, YAP/TAZ double knockout, LATS1-KO, LATS2-KO, and LATS1/2-dKO HEK 293A cells were kindly provided by Dr. Kun-Liang Guan (University of California San Diego). The HEK 293, Huh7, Hela, and Vero cells were maintained in DMEM (Corning), supplemented with 10% FBS (Gibco), penicillin (100 U/ml)/streptomycin (100 μg/ml) (Invitrogen), and the cells were grown at 37 °C in a humidified 5% CO2 incubator.
Virus strain
EBOV stock. All experiments with infectious EBOV were performed in the biosafety level 4 (BSL-4) laboratory at the Texas Biomedical Research Institute, San Antonio, TX. The recombinant EBOV variant Mayinga expressing GFP (EBOV-GFP) (NCBI accession number KF_990213) was grown and titrated as described previously68. Briefly, the amplified virus was serially diluted and then incubated with Vero cells for 24 h, then stained with Hoechst 33342 dye (Thermo Fisher Scientific) to identify nuclei and photographed using Nikon Ti-Eclipse microscope running high-content analysis software (Nikon, Tokyo, Japan). The numbers of GFP-expressing (infected) cells and nuclei were determined by CellProfiler software (Broad Institute). VSV-GFP stock. The recombinant VSV expressing GFP (VSV-GFP)69 titer was determined by standard plaque assays of serially diluted samples on BHK-21 cells.
Filoviral VP40 VLP budding assay
HEK 293, Huh7, or Hela cells seeded in six-well plates were transfected with eVP40 or mVP40 alone, or plus with plasmids as indicated. Filovirus VP40 VLP budding assays have been described 70. eVP40 and mVP40 proteins in VLPs and cell extracts were detected by Western blotting and quantified using NIH Image-J software.
Confocal microscopy
HEK 293 or Huh7 cells were seeded into a 35 mm glass bottom MatTek dish on the day before transfection. At 24 h after transfection, cells were washed with cold PBS and fixed with 4% PBS-buffered paraformaldehyde for 20 min at room temperature, then permeabilized with 0.1% Triton X in PBST for 10 min. After blocking, cells were stained with indicated primary antibodies at 4 °C overnight, then stained with appropriated secondary antibodies at room temperature for 1 h, and then were mounted with the Mounting Medium with DAPI (Abcam). The Confocal laser scanning microscopy was performed with a Lecia SP5 confocal laser scanning microscope fitted with a 100x (NA 1.46) objective lens. Images were subsequently deconvolved with Huygens Essential deconvolution software (Scientific Volume Imaging) and generated with the Lecia Application Suite X software (Lecia).
Live cell imaging
HEK 293 cells were transfected with GFP-eVP40 alone or plus with the LifeAct-RFP to visualize the F-actin dynamics. Cell nuclei were stained by NucBlue™ Live ReadyProbes™ Reagent (Invitrogen) at 20 h post-transfection. Cells were then monitored via a Leica SP5 inverted confocal microscope with a 100x (NA 1.46) objective lens using sequential scanning. The videos were obtained with the Lecia Application Suite X software (Lecia).
EBOV infection assays
To test the infectivity and egress of the HEK 293-WT, Y/T-dKO, LATS1-KO, LATS2-KO, and LATS1/2-dKO phenotypes, cells seeded in wells of a 96-well plate were incubated with EBOV-GFP at an MOI of 0.05, in triplicate or quadruplicate, for 1 h, then washed and overlaid with fresh medium. Cells were fixed at 24- or 48-h post-infection, and supernatants were titrated onto Vero cells to quantify the release of infectious virus. All cell monolayers were fixed in a 10% buffered formalin solution in accordance with Institutional SOPs and approved Biohazard and Safety Committee protocols to inactivate the virus. Subsequently, cells were stained with Hoechst 33342 dye and photographed using a Nikon Ti-Eclipse microscope running high-content analysis software (Nikon, Tokyo, Japan). The numbers of GFP-expressing (infected) cells and nuclei were determined by CellProfiler software (Broad Institute). Infection efficiency in all samples was determined as a ratio of GFP-positive (infected) cells and nuclei and reported relative to the values in the WT control.
Bulk RNA sequencing
HEK 293-WT and YAT/TAZ-dKO cells were lysed in Trizol Reagent (Invitrogen) and total RNA was extracted using the RNeasy Plus Micro Kit (Qiagen) following the manufacturer’s instructions. Total RNA was examined for quantity and quality before Bulk RNA Sequencing. Raw reads were mapped to the human reference transcriptome using Kallisto, version 0.46.2. After read-mapping with Kallisto, TxImport was used to read Kallisto outputs into the R environment. Annotation data from Biomart was used to summarize data from transcript level to gene level. All graphics and data wrangling were handled using the tidyverse suite of packages. All packages used are available from the Comprehensive R Archive Network (CRAN), Bioconductor.org, or Github.
EBOV VLP internalization
The GFP-tagged eVP40 and eGP were expressed in HEK 293 cells. At 24 h post-transfection, the supernatant was collected. EBOV VLPs were obtained via ultracentrifugation71. For VLP internalization, cells seeded on a 35 mm MatTek dish were incubated with VLPs at 4 °C for 10 min to synchronize the macropinocytosis, then shifted to 37 °C for 1 h. The cells were then washed with cold PBS and fixed with 4% PBS-buffered paraformaldehyde for 20 min at room temperature, then mounted with DAPI reagent (Abcam). Samples were then observed via a Leica SP5 inverted confocal microscope with a 100x (NA 1.46) objective lens. Images were generated with the Lecia Application Suite X software (Lecia).
VSV pseudotype entry assay
VSV pseudotypes with EBOV GP, MARV GP, or VSV G expressing luciferase were generated71. HEK 293-WT and YAP/TAZ-dKO cells seeded in 96-well plates were transduced with the indicated pseudotype for 16 h at 37 °C. Luciferase activity was assayed using with ONE-Glo™ Luciferase Assay System (Promega) following the manufacturer’s instructions and measured via Biotek Synergy HT Microplate Reader (Biotek).
Pharmacological treatment targeting YAP
Huh7 and Hela cells seeded in six-well plates were transfected with eVP40 or mVP40 plasmids alone, and then mock-treated or treated with EGF and Verteporfin at the indicated concentrations for 24 h. Cell extracts and supernatants were harvested for Western blotting analysis and VLP budding assay. The cytotoxicity of the drug in each treatment condition was determined using the CellTiter-Glo® Luminescent Cell Viability Assay system (Promega) following the manufacturer’s instructions.
Immunoprecipitation assay
HEK 293 cells were transfected with the indicated combination of plasmids. At 24 h post-transfection, cells were harvested and lysed. Cell extracts were incubated with either normal mouse IgG (sc-2025, Santa Cruz) or rabbit IgG (#2729, Cell Signaling Technology), and appropriate antibodies as indicated, followed by gentle rotating overnight at 4 °C. Protein A/G plus agarose beads (sc-2003, Santa Cruz) were then added to the mixtures and incubated for 5–8 h at 4 °C. After incubation, beads were collected via centrifugation and washed five times, eluted with 2 x SDS loading buffer with DTT, and boiled for 10 min. Target proteins were then detected by Western blotting with specific antibodies as indicated.
EBOV minigenome assay
The HEK 293 or Huh7 cells seeded in 12-well plates were transfected with the EBOV minigenome system, including the pCAGGS-NP, pCAGGS-VP35, pCAGGS-VP30, pCAGGS-L, and pCAGGS-pol-II-minigenome plasmids that encoding either the eGFP or Luc reporters39. The minigenome activity was assayed at 48 hours post-transfection, either by examining the eGFP expression using a fluorescent microscope or by measuring luciferase activity. The luciferase activity was quantified using the Luciferase Assay System (Promega, E4030), and the values were calculated as fold induction over the L synth- (-L) values in each identical condition.
Pharmacological inhibition of kinase activity
HEK 293 and Huh7 cells seeded in 12-well plates were transfected with the EBOV minigenome system (± L). At 12 h post-transfection, cells were mock-treated (DMSO) or treated with the LATS or SRPK kinase inhibitors as indicated. After 48 h, the cells were lysed for Western blotting analysis or assayed for luciferase activity as described above. The cytotoxicity of the drug in each treatment condition was determined using the CellTiter-Glo® Luminescent Cell Viability Assay system (Promega) following the manufacturer’s instructions.
EBOV GP RT-qPCR
The WT, YAP/TAZ-dKO, and LATS1/2-dKO cells were infected with EBOV-GFP at MOIs of 0.01, 0.05, 0.5, or 5.0. Cells were lysed in Trizol Reagent (Invitrogen) at the indicated time points post-infection, and total RNA was extracted using the Trizol Reagent protocol following the manufacturer’s instructions. The specific primers for reverse transcribing the eGP gene were forward-GGCCAACGAGACGACTCAA (550 nM) and reverse-GGTGCGTAGCTCGGTTGTG (900 nM). Subsequently, the One-step RT-qPCR was performed with the FAM/MGB-probe: CTCTTCAACTGTTCCTGAGAG (250 nM) using TaqPath 1-Step RT-qPCR Master Mix (Applied Biosystems, # A15299) and QuantStudio RT-PCR systems (Applied Biosystems). Viral GP RNA copy numbers, expressed as genome equivalents per total cell RNA (intracellular viral RNA) or per mL of cell supernatants (extracellular viral RNA), were quantified by extrapolating from the standard curve plot of Ct values. The Ct plot was generated using reactions containing in vitro transcribed tenfold serially diluted viral GP transcript.
EBOV infection in monocyte-derived macrophages
The preparation of MDMs was described previously72. MDMs were maintained in RPMI medium supplemented with 10% autologous serum and grown at 37 °C in a humidified 5% CO2 incubator. Verteporfin or TDI-011536 treatments were added to MDMs seeded in 96-well plates 1 h before, or after infection with EBOV-GFP at an MOI of 0.2 in triplicate. Cells were fixed at 24 h post-infection, and supernatants were titrated onto Vero cells to quantify the release of infectious virus. All monolayers were treated with Hoechst dye (Thermo Fisher) to stain nuclei, photographed by a Nikon automated system, and analyzed by CellProfiler software to quantify infected cells and nuclei (GFP positive). Infection efficiency in each sample was determined as a ratio of infected cells and nuclei and reported relative to mock-treated samples.
3D spheroid generation and virus infection
The WT, YAP/TAZ-dKO, and LATS1/2-dKO were seeded in Nunclon Sphera 96-well U-bottom ultra-low attachment 3D cell culture plate (Thermo Scientific) with an initial density of 10,000 cells/well. The spheroids were well developed and compacted at 72 h post-seeding. The culture medium was carefully removed, and the spheroids were infected with EBOV-GFP or VSV-GFP for 2 h and then washed 3X. Viral infection was detected by GFP expression at 48 h post-infection, and total RNA was isolated from mock-infected and EBOV-GFP-infected spheroids for analysis by qRT-PCR to assess infectivity. Supernatants from mock-infected and VSV-GFP-infected spheroids were harvested, and the released virus was titrated on fresh monolayers of Vero cells.
Statistics and reproducibility
All statistical analyses were performed by the data from at least three independent experiments and are shown as mean ± SD, sample size was indicated. The one-way/two-way ANOVA analyses and two-tailed unpaired t-test were used as indicated in figure legends. All exact P values were provided in the figures. Source data is provided with the paper.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated in this study are available within the paper and its supplementary information. The Raw RNA sequencing data were available on NCBI BioProject ID: PRJNA1116575; SRA accession: SRX24703878, SRX24703879, SRX24703880, SRX24703881, SRX24703882, and SRX24703883. Source data are provided with this paper.
References
Subissi, L. et al. Ebola virus transmission caused by persistently infected survivors of the 2014-2016 outbreak in West Africa. J. Infect. Dis. 218, S287–s291 (2018).
Coffin, K. M. et al. Persistent Marburg virus infection in the testes of nonhuman primate survivors. Cell Host Microbe 24, 405–416.e403 (2018).
Hoenen, T., Groseth, A. & Feldmann, H. Therapeutic strategies to target the Ebola virus life cycle. Nat. Rev. Microbiol. 17, 593–606 (2019).
Jacob, S. T. et al. Ebola virus disease. Nat. Rev. Dis. Primers 6, 13 (2020).
Bodmer, B. S., Hoenen, T. & Wendt, L. Molecular insights into the Ebola virus life cycle. Nat. Microbiol. https://doi.org/10.1038/s41564-024-01703-z (2024).
Harty, R. N., Brown, M. E., Wang, G., Huibregtse, J. & Hayes, F. P. A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: implications for filovirus budding. Proc. Natl Acad. Sci. USA 97, 13871–13876 (2000).
Han, Z. et al. ITCH E3 ubiquitin ligase interacts with Ebola virus VP40 to regulate budding. J. Virol. 90, 9163–9171 (2016).
Han, Z. et al. Ubiquitin ligase WWP1 interacts with Ebola virus VP40 to regulate egress. J. Virol. https://doi.org/10.1128/jvi.00812-17 (2017).
Liang, J. et al. Chaperone-mediated autophagy protein BAG3 negatively regulates Ebola and Marburg VP40-mediated egress. PLoS Pathog. 13, e1006132 (2017).
Liang, J. et al. Angiomotin counteracts the negative regulatory effect of host WWOX on viral PPxY-mediated egress. J. Virol. https://doi.org/10.1128/jvi.00121-21 (2021).
Liang, J., Ruthel, G., Freedman, B. D. & Harty, R. N. WWOX-mediated degradation of AMOTp130 negatively affects egress of filovirus VP40 virus-like particles. J. Virol. 96, e0202621 (2022).
Liang, J., Djurkovic, M. A., Shtanko, O. & Harty, R. N. Chaperone-assisted selective autophagy targets filovirus VP40 as a client and restricts egress of virus particles. Proc. Natl Acad. Sci. USA 120, e2210690120 (2023).
Han, Z. et al. Angiomotin regulates budding and spread of Ebola virus. J. Biol. Chem. 295, 8596–8601 (2020).
Han, Z. et al. Modular mimicry and engagement of the Hippo pathway by Marburg virus VP40: Implications for filovirus biology and budding. PLoS Pathog. 16, e1008231 (2020).
Hansen, C. G., Moroishi, T. & Guan, K. L. YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol. 25, 499–513 (2015).
Meng, Z., Moroishi, T. & Guan, K. L. Mechanisms of Hippo pathway regulation. Genes Dev. 30, 1–17 (2016).
Ma, S., Meng, Z., Chen, R. & Guan, K. L. The Hippo pathway: biology and pathophysiology. Ann. Rev. Biochem. 88, 577–604 (2019).
Yu, F. X. & Guan, K. L. The Hippo pathway: regulators and regulations. Genes Dev. 27, 355–371 (2013).
Zhao, B. et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 21, 2747–2761 (2007).
Zhao, B., Li, L., Tumaneng, K., Wang, C. Y. & Guan, K. L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 24, 72–85 (2010).
Oka, T., Mazack, V. & Sudol, M. Mst2 and Lats kinases regulate apoptotic function of Yes kinase-associated protein (YAP). J. Biol. Chem. 283, 27534–27546 (2008).
Plouffe, S. W. et al. The Hippo pathway effector proteins YAP and TAZ have both distinct and overlapping functions in the cell. J. Biol. Chem. 293, 11230–11240 (2018).
Totaro, A., Panciera, T. & Piccolo, S. YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol. 20, 888–899 (2018).
King, B., Araki, J., Palm, W. & Thompson, C. B. Yap/Taz promote the scavenging of extracellular nutrients through macropinocytosis. Genes Dev. 34, 1345–1358 (2020).
Shimojima, M., Ikeda, Y. & Kawaoka, Y. The mechanism of Axl-mediated Ebola virus infection. J. Infect. Dis. 196, S259–S263 (2007).
Hunt, C. L., Kolokoltsov, A. A., Davey, R. A. & Maury, W. The Tyro3 receptor kinase Axl enhances macropinocytosis of Zaire ebolavirus. J. Virol. 85, 334–347 (2011).
Nanbo, A. et al. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathog. 6, e1001121 (2010).
Cureton, D. K., Massol, R. H., Saffarian, S., Kirchhausen, T. L. & Whelan, S. P. Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog. 5, e1000394 (2009).
Fan, R., Kim, N. G. & Gumbiner, B. M. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc. Natl Acad. Sci. USA 110, 2569–2574 (2013).
Liu-Chittenden, Y. et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 26, 1300–1305 (2012).
Wang, C. et al. Verteporfin inhibits YAP function through up-regulating 14-3-3σ sequestering YAP in the cytoplasm. Am. J. Cancer Res. 6, 27–37 (2016).
Wynne, J. W. et al. Comparative transcriptomics highlights the role of the activator protein 1 transcription factor in the host response to Ebolavirus. J. Virol. https://doi.org/10.1128/jvi.01174-17 (2017).
Zeng, X. et al. Identification and pathological characterization of persistent asymptomatic Ebola virus infection in rhesus monkeys. Nat. Microbiol. 2, 17113 (2017).
Martines, R. B., Ng, D. L., Greer, P. W., Rollin, P. E. & Zaki, S. R. Tissue and cellular tropism, pathology and pathogenesis of Ebola and Marburg viruses. J. Pathol. 235, 153–174 (2015).
Chan, S. W. et al. Actin-binding and cell proliferation activities of angiomotin family members are regulated by Hippo pathway-mediated phosphorylation. J. Biol. Chem. 288, 37296–37307 (2013).
Dai, X. et al. Phosphorylation of angiomotin by Lats1/2 kinases inhibits F-actin binding, cell migration, and angiogenesis. J. Biol. Chem. 288, 34041–34051 (2013).
Meng, Z. et al. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat. Commun. 6, 8357 (2015).
Wang, L. et al. Multiphase coalescence mediates Hippo pathway activation. Cell 185, 4376–4393.e4318 (2022).
Nelson, E. V. et al. An RNA polymerase II-driven Ebola virus minigenome system as an advanced tool for antiviral drug screening. Antiviral Res. 146, 21–27 (2017).
Takamatsu, Y. et al. Serine-arginine protein kinase 1 regulates Ebola virus transcription. mBio https://doi.org/10.1128/mBio.02565-19 (2020).
Lier, C., Becker, S. & Biedenkopf, N. Dynamic phosphorylation of Ebola virus VP30 in NP-induced inclusion bodies. Virology 512, 39–47 (2017).
Biedenkopf, N., Lier, C. & Becker, S. Dynamic phosphorylation of VP30 is essential for Ebola virus life cycle. J. Virol. 90, 4914–4925 (2016).
Hoenen, T. et al. Inclusion bodies are a site of Ebolavirus replication. J. Virol. 86, 11779–11788 (2012).
Kastan, N. R. et al. Development of an improved inhibitor of Lats kinases to promote regeneration of mammalian organs. Proc. Natl. Acad. Sci. USA 119, e2206113119 (2022).
Yaron, T. M. et al. Host protein kinases required for SARS-CoV-2 nucleocapsid phosphorylation and viral replication. Sci. Signal. 15, eabm0808 (2022).
Monteil, V. et al. Identification of CCZ1 as an essential lysosomal trafficking regulator in Marburg and Ebola virus infections. Nat. Commun. 14, 6785 (2023).
Muñoz-Wolf, N. & Lavelle, E. C. Hippo interferes with antiviral defences. Nat. Cell Biol. 19, 267–269 (2017).
Zhang, Q. et al. Hippo signalling governs cytosolic nucleic acid sensing through YAP/TAZ-mediated TBK1 blockade. Nat. Cell Biol. 19, 362–374 (2017).
Garcia, G. Jr. et al. Hippo signaling pathway has a critical role in Zika virus replication and in the pathogenesis of neuroinflammation. Am. J. Pathol. 190, 844–861 (2020).
Garcia, G. Jr. et al. Hippo signaling pathway activation during SARS-CoV-2 infection contributes to host antiviral response. PLoS Biol. 20, e3001851 (2022).
Shepley-McTaggart, A. et al. Contrasting effects of filamin A and B proteins in modulating filovirus entry. PLoS Pathog. 19, e1011595 (2023).
Wang, X. et al. YAP/TAZ orchestrate VEGF signaling during developmental angiogenesis. Dev. Cell 42, 462–478.e467 (2017).
Kindrachuk, J. et al. Ebola virus modulates transforming growth factor β signaling and cellular markers of mesenchyme-like transition in hepatocytes. J. Virol. 88, 9877–9892 (2014).
Gao, J. et al. Ebola virus disrupts the inner blood-retinal barrier by induction of vascular endothelial growth factor in pericytes. PLoS Pathog. 19, e1011077 (2023).
Smoot, R. L. et al. Platelet-derived growth factor regulates YAP transcriptional activity via Src family kinase dependent tyrosine phosphorylation. J. Cell. Biochem. 119, 824–836 (2018).
Furth, N. & Aylon, Y. The LATS1 and LATS2 tumor suppressors: beyond the Hippo pathway. Cell Death Differ. 24, 1488–1501 (2017).
Tang, F. et al. LATS1 but not LATS2 represses autophagy by a kinase-independent scaffold function. Nat. Commun. 10, 5755 (2019).
Martinez, M. J. et al. Role of VP30 phosphorylation in the Ebola virus replication cycle. J. Infect. Dis. 204, S934–S940 (2011).
Biedenkopf, N., Hartlieb, B., Hoenen, T. & Becker, S. Phosphorylation of Ebola virus VP30 influences the composition of the viral nucleocapsid complex: impact on viral transcription and replication. J. Biol. Chem. 288, 11165–11174 (2013).
Brauburger, K., Deflubé, L. R. & Mühlberger, E. in Biology and Pathogenesis of Rhabdo- and Filoviruses (eds. Pattnaik, A. K. & Whitt, M. K.) Chapter #20, (World Scientific, 2014).
Takamatsu, Y. et al. Role of VP30 phosphorylation in Ebola virus nucleocapsid assembly and transport. J. Virol. 96, e0108322 (2022).
Batra, J. et al. Non-canonical proline-tyrosine interactions with multiple host proteins regulate Ebola virus infection. EMBO J. 40, e105658 (2021).
Kolesnikova, L., Nanbo, A., Becker, S. & Kawaoka, Y. in Marburg- and Ebolaviruses: From Ecosystems to Molecules (eds Mühlberger, E., Hensley, L. L. & Towner, J. S.) (Springer International Publishing, 2017).
Ilinykh, P. A. et al. Role of protein phosphatase 1 in dephosphorylation of Ebola virus VP30 protein and its targeting for the inhibition of viral transcription. J. Biol. Chem. 289, 22723–22738 (2014).
Kruse, T. et al. The Ebola virus nucleoprotein recruits the host PP2A-B56 phosphatase to activate transcriptional support activity of VP30. Mol. Cell 69, 136–145.e136 (2018).
Ahmad, A. et al. Ebola virus NP binding to host protein phosphatase-1 regulates capsid formation. Res. Sq. https://doi.org/10.21203/rs.3.rs-2963943/v1 (2023).
Licata, J. M. et al. Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: involvement of host proteins TSG101 and VPS-4. J. Virol. 77, 1812–1819 (2003).
Shtanko, O. et al. Retro-2 and its dihydroquinazolinone derivatives inhibit filovirus infection. Antiviral Res. 149, 154–163 (2018).
Fernandez, M., Porosnicu, M., Markovic, D. & Barber, G. N. Genetically engineered vesicular stomatitis virus in gene therapy: application for treatment of malignant disease. J. Virol. 76, 895–904 (2002).
McCarthy, S. E., Johnson, R. F., Zhang, Y. A., Sunyer, J. O. & Harty, R. N. Role for amino acids 212KLR214 of Ebola virus VP40 in assembly and budding. J. Virol. 81, 11452–11460 (2007).
Miller, M. E., Adhikary, S., Kolokoltsov, A. A. & Davey, R. A. Ebolavirus requires acid sphingomyelinase activity and plasma membrane sphingomyelin for infection. J. Virol. 86, 7473–7483 (2012).
Rogers, K. J. et al. Frontline science: CD40 signaling restricts RNA virus replication in Mϕs, leading to rapid innate immune control of acute virus infection. J. Leukoc. Biol. 109, 309–325 (2021).
Acknowledgements
We thank Leslie B. King for her comments and for editing the manuscript. We thank K-L. Guan, S. Becker, C. F. Basler, M. Sudol, D. McCollum, E. Mühlberger, and J. Dong for kindly providing reagents. The data of confocal microscopy and bulk RNA sequencing were collected at Penn Vet Imaging Core (PVIC) and Cell-Host Microbial Interactions Center (CHMI), respectively. Funding was provided in part by National Institutes of Health grants AI138052, AI139392, and AI153815 to R.N.H., and AI154336 and AI151717 to O.S. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
J.L. conceived the study and wrote the paper under the supervision of O.S. and R.H.; J.L., O.S., and R.H. designed the experiments, J.L., M.A.D., C.G.L., and O.S. performed the experiments; J.L., M.A.D., C.G.L., O.S., and R.H. analyzed the data, O.S. and R.H. supervised the data acquisition. All authors edited and approved the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liang, J., Djurkovic, M.A., Leavitt, C.G. et al. Hippo signaling pathway regulates Ebola virus transcription and egress. Nat Commun 15, 6953 (2024). https://doi.org/10.1038/s41467-024-51356-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-51356-z
