
Abstract
Membrane active peptides are known to porate lipid bilayers, but their exact permeabilization mechanism and the structure of the nanoaggregates they form in membranes have often been difficult to determine experimentally. For many sequences at lower peptide concentrations, transient leakage is observed in experiments, suggesting the existence of transient pores. For two well-know peptides, alamethicin and melittin, we show here that molecular mechanics simulations i) can directly distinguish equilibrium poration and non-equilibrium transient leakage processes, and ii) can be used to observe the detailed pore structures and mechanism of permeabilization in both cases. Our results are in very high agreement with numerous experimental evidence for these two peptides. This suggests that molecular simulations can capture key membrane poration phenomena directly and in the future may develop to be a useful tool that can assist experimental peptide design.
Similar content being viewed by others

Introduction
Membrane-permeabilizing peptides are common in all forms of life, and have been the focus of numerous studies over the last decades with the goal of rational design for pharmaceutical or biotechnological application1,2. However, only for very few peptides, the detailed knowledge of the molecular mechanism of permeabilization, or explicit pore structures have been determined experimentally3,4. What we know of the vast majority of natural and de-novo designed membrane permeabilizing peptides is usually restricted to activity: leakage potency and efficacy in targeting various organisms. Membrane permeabilization is often rapid and involves formation of dynamic and highly heterogeneous nanostructure aggregates of peptides in the membrane, so employing conventional structural biology tools is difficult.
In the last years, fully detailed all atomistic simulations have led to some successes in filling in the missing structural detail, for peptides thought to form stable pores in equilibrium5,6,7,8,9,10,11,12,13,14,15,16. However, simulations have not been successful in determining stable equilibrium pores for most other membrane active peptides. This actually agrees with the experiments: Dye leakage measurements of many antimicrobial peptides (AMPs) are not consistent with stable pores17. What is instead observed, is that shortly after peptides are added, the membrane becomes permeable and most leakage occurs in a burst. The system then quickly transitions from rapid leakage to little or none, and the membranes are no longer highly permeable18,19,20,21. There is no change in the number of peptides bound to the membrane, and no change in peptide secondary structure, but no longer any pronounced leakage. A plateau is reached with the vesicle not completely emptied. Leakage can usually be restarted by addition of more peptide. All these observations are inconsistent with stable transmembrane pores.
A simple model has been suggested to explain this transient leakage: Stochastic dissipation of the transbilayer peptide asymmetry through a spontaneous membrane poration event22,23,24,25,26,27,28. After peptides are added to the solution containing vesicles, the peptides rapidly adsorb to the outer membrane leaflet. In absence of monomeric peptide translocation, an imbalance in mass, charge, and surface pressure builds up across the bilayer, which ultimately is the driving force for permeabilization. The pressure imbalance is thought to be relieved in rapid, catastrophic stochastic events that porate the membrane and enable release of all or some of the vesicle contents. Peptides can then translocate through these membrane defects and become equally distributed between the monolayers of the bilayer. In addition, the membrane becomes briefly permeable to polar molecules and ions. After the dissipation of the transbilayer asymmetry, the membrane defects or pores dissolve and the membrane seals. Permeability is again greatly reduced. Key to this model is that there is no equilibrium leakage. It only occurs when there is a transbilayer asymmetry of peptides, and it occurs only during the dissipation of the transbilayer asymmetry.
In this study, we demonstrate that this non-equilibrium transient leakage can be directly simulated and distinguished from equilibrium pore formation. For this, we chose two of the most widely studied membrane active peptides: The first is alamethicin, produced by the fungus Trichoderma viride, a role model for equilibrium ‘barrel-stave’ pore formation29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45. The second peptide is the honey bee venom membrane-permeabilizing peptide melittin46,47,48,49,50,51,52,53,54,55,56,57. Melittin is widely unselective: it can permeabilize eukaryotic membranes (e.g. erythrocytes), Gram negative and Gram positive bacterial, and viral membranes4. Its membrane permeabilization mechanism depends strongly on concentration. Under many experimental conditions at lower concentration, mellitin is seen to follow the transient leakage model17,21,25,48,58,. Stable pores have also been detected in the micromolar range55,56,57,59,60.
We show here that (i) alamethicin forms equilibrium pores of barrel-stave type, (ii) melittin does not, and (iii) that melittin (at low concentration) can permeabilize membranes by a transient mechanism involving temporary, unstable transmembrane pores of a toroidal shape whose structures greatly differ from those of alamethicin. The simulations illustrate the difference between ‘equilibrium’ pore formation, i.e. pores that form by freely aggregating from individual peptides, and ‘non-equilibrium’ toroidal pores that do not form by aggregation but by a cooperative nucleation event.
At our peptide concentration, we show that these pores are transient, which strongly agree with the numerous experimental observations of transient leakage25. The physiologically relevant activity of melittin, with a minimal inhibitory concentration in the micromolar range, likely involves additional macroscopic effects such as membrane thinning61. Under these conditions, stable pores have been reported, allowing their structure to be determined56,57,59,60. The pores are also toroidal, with a close match in size and structure to our predicted transient toroidal structures.
Many other membrane active peptides are thought to follow similar pathways as alamethicin and melittin23. This suggests that molecular mechanics simulations may develop into a powerful tool that in the future can guide and assist the design process of new membrane active peptides.
Results
Peptide adsorption and insertion
All MD simulations were run in synthetic bilayers composed of pure or simple lipid mixtures, in order to allow for direct, unambiguous comparison to the vast amount of experiments performed in exactly these simple membranes. Real cell membranes are much more complex, and quantitative comparison would be critically dependant on getting the complicated lipidomics right. Bacterial outer membranes would be especially difficult to model correctly. So we stick for now to simple model membranes. Many experiments for alamethicin have used synthetic bilayers29,30,35,37,39,52,62, and the same is true for melittin4,21,48,53,63,64,65,66. To stay as close as possible for comparison, we performed simulations of alamethicin in zwitterionic DOPC, POPC, and anionic DPPC:DPPG 3:1 and DMPC:DMPG 3:1 mixtures. In order to initiate our simulations from completely unbiased starting structures, single peptides of alamethicin and melittin were initially placed into the water phase a distance from the membrane (POPC, DMPC, Fig. 1a, b. For other membranes: Fig. S1). The peptides rapidly adsorb to the membrane interface into a surface alignment first, which we have shown previously via folding/partitioning simulations for melittin67,68. Melittin remains on the surface, while alamethicin, driven by its larger hydrophobicity, inserts transmembrane. No further transitions are seen. These structures were used as the starting conformations for subsequent simulations, by assembling them into a 4 × 4 grid. Aggregation simulations are then initiated (Fig. 1c).

a Single alamethicin adsorption and insertion. Side-view of the single peptide trajectories and peptide center of mass insertion (membrane = gray bar; red dotted line = membrane center; blue dotted lines = interfacial center-of-mass location of the peptides; membrane interface phase = light blue shading, trans-membrane inserted phase = light red shading). b Single melittin adsorption. c Alamethicin aggregation. The initial state is a 4×4 grid of the final snapshot of the single peptide simulation. d Oligomerization state (S = surface, yellow-black: increasing transmembrane oligomer size). e Membrane water number density.
Alamethicin: transmembrane pore bundles
For alamethicin in DOPC, peptides are seen to rapidly aggregate into bundles of 2-6 helices (Fig. 1c). The oligomerization plot (Fig. 1d) reveals that after 2 µs, the system has stabilized to a hexamer, two tetramers and a dimer. Arrows point to the formation of the last tetramer from a trimer and a monomeric peptide. The observed channels are circular and water filled, as can be seen by the steady increase in the water content of the membrane (Fig. 1e). They closely match the experimental ‘barrel-stave’ structures found for alamethicin. Unfortunately, the kinetics of assembly and pore formation is very slow at this temperature (333 K). No further transitions take place after 2 µs. Little spikes in the oligomer plot occur when bundles briefly touch and do not signify pore dissolution or change of bundle size. Experimental kinetic studies show life- and transition times of alamethicin channels on multi-millisecond scales at room temperature33,34,69,70. This cannot yet be reached via simulations. So to get better convergence on the pore equilibrium, and give the system the chance to find higher order bundles, the temperature was further increased in steps of 30 K and the simulations repeated. (Fig. 2a, Fig. S5). Alamethicin is seen to rapidly aggregate into transmembrane bundles of 4-12 peptides. Some of these aggregates are dry, but a significant portion is seen to form water filled barrel-stave pores (Fig. 2d: octamer), resulting in large spikes in both ion and water flux (Fig. 2c). At the higher temperatures, the pores are not always perfectly symmetric, and lifetimes are reduced (Fig. 2b) as compared to the simulation shown in Fig. 1c. However, due to the accelerated kinetics, higher order pores such as the octamer are found in a reasonable simulation time (Fig. 2d). We can also observe that individual peptides join or leave channels as suggested by Boheim in the 1970s34. From the peptide-peptide free energy profile, alamethicin is seen as a rapid equilibrium pore former with a dimer contact minimum of ΔG = −1.2 kcal/mol, irrespective of temperature. The behavior of alamethicin in the other bilayers is almost identical (Figs. S2–S5).

a, g Topview snapshots of the simulation box. b, h Population of oligomers formed throughout the trajectories. e, k Overall oligomer distribution (S = surface, yellow-black: increasing transmembrane oligomer size). c, i Transmembrane flux of water (top) and ions (bottom). d, j Representative structures of the largest pore encountered for alamethicin (water-filled octamer) and the most dominant oligomer populated for melittin (dimer, no pores). f, l Dimerization free energy profile obtained from the peptide-peptide, 2D (in membrane plane) radial distribution function. (Colors indicate different temperatures, P = parallel, AP = anti-parallel).
Melittin: no transmembrane bundles
If all peptides behaved like alamethicin, then the question how membranes are permeablized would be trivial. However, a very different result is seen when trying to repeat these simulations with melittin. The peptide setup, with transmembrane inserted helices, is identical to alamethicin. POPC is used, to be able to compare to key experiments on melittin65. However, peptides never form pores at all (Fig. 2g). In fact, melittin appears self-avoiding, with peptides preferring to stay single or occasionally dimerize, but higher bundles of 3-4 peptides only forming infrequently and rapidly dissolving. No water ever enters the membrane, and no water channel opens up. One reason is obvious: melittin has so many cationic sidechains and a large polar face, so that peptides do not like to touch. This is independent of the inserted orientation, whether all peptides are initially parallel (P), or 50:50 parallel:anti-parallel (AP), (Fig. 2l), and independent of temperature. The free energy profile strongly suggests that melittin is not a barrel-stave pore former, or any type of transmembrane pore aggregator at all.
Melittin: No poration by aggregation
It could be that pores do exist, but they simply do not assemble by spontaneous aggregation of transmembrane inserted helices, as show for alamethicin. There could also be unknown entropic barriers. Unfavorable kinetics could prevent formation to be observed in microsecond timescales. Alternatively, membrane perturbation and pore assembly could initiate from surface aligned states. Like many membrane permeabilizing peptides, melittin, by virtue of its large positive charge and amphipathicity, is expected to initially bind to the membrane in an alignment parallel to the surface67,68. In addition, aggregates could form from a mix of surface aligned and transmembrane inserted states.
To test this, we designed 4 alternative initial configurations for melittin (Fig. 3): (i) symmetrically loaded (meaning both leaflets) all surface aligned. (ii) asymmetrically surface aligned. (iii) mixed surface aligned and transmembrane inserted. (iv) Mixed upper-leaflet surface aligned and transmembrane inserted. In order to increase stability of the simulations, bilayers with asymmetric loading of peptides had 2 lipids removed per peptide to compensate for the excess peptide mass. In addition, a thinner membrane (DMPC) was used to speed up membrane permeabilization. However, no pore formation was observed. As can be seen in Fig. 3a–d from peptide insertion, oligomeric state and transmembrane water flux, the surface aligned peptides never aggregate or insert. And if some peptides were initially already transmembrane inserted, this did not assist further insertion of other peptides or cause poration. For all 4 initial configurations, peptides do not transition from surface to transmembrane or vice versa, and the membrane remains completely dry.

a–d Symmetric or weakly asymmetric mass distributions that do not lead to membrane poration: a Initial mass density distribution and snapshot for (top-to-bottom): symmetrically loaded all surface aligned; asymmetrically loaded, but with lipids removed in the upper leaflet to compensate for excess peptide mass; mixed surface aligned and transmembrane inserted; mixed upper-leaflet surface aligned and transmembrane inserted, with lipids removed in upper leaflet to compensate the excess peptide mass (red = peptide [P], gray = lipids [L], dark gray = total mass density, blue dotted line = mirrored total mass density to visualize assymmetry). b Peptide insertion depth (gray = membrane). c Oligomer population (blue: S = surface, yellow: monomeric transmembrane inserted). d Transmembrane water flux. e Snapshots of spontaneous pore formation by an asymmetric initial mass distribution (orange beads = phosphate of DMPC). f Initial and final mass density distribution, and peptide mass density equilibration as a function of simulation time (red = high; blue = low). g Peptide insertion depth as a function of simulation time. h Pore opening as seen by water flux for three different trajectories.
Transient pore formation
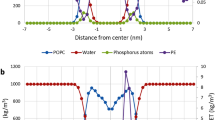
These results (and the one shown in Fig. 2) strongly suggest that melittin does not form pores by an ‘equilibrium’ process that has peptides aggregating. Instead, a different nucleation condition is needed. The 4 configurations shown in Fig. 3, as well as the all-transmembrane configuration shown in Fig. 2 had the total mass (peptides + lipids) either symmetrically distributed across the membrane, or an initial mass imbalance (and thus membrane strain) was relieved by removing lipids in the bilayer leaflet containing the excess peptide. We concluded that the likely mechanism may then be due to a much stronger initial mass asymmetry, and thus the stress exerted on the membrane71,72,73. Melittin has been known to act as a wedge due to its interfacial binding position, causing local membrane thinning and a surface area difference across the bilayer74. This model has been suggested by many as best fit to the numerous experimental evidence22,23,24,25,26,27,28. Indeed, when the bilayer is loaded strongly asymmetrically (by not removing any lipids), there is a sufficient driving force to open a large membrane pore (Fig. 3e, f). The peptides loiter a while on the surface, but the large mass imbalance and membrane strain causes the bilayer to rupture within 500 ns as the mass equilibrates across the membrane (Fig. 3e, h). Some peptides immediately insert transmembrane and form the pore lining (Fig. 3e). But the pore is not of a barrel-stave geometry. There are always interlocuting lipids between peptides, in what has been termed a ‘toroidal’ structure in the literature (Fig. 4a). Typically, 6–12 peptides are involved and the water channel is large, with a fluctuating diameter of 20–40 Å. Melittin peptides can translocate through the pore and are involved in the pore lining both in transmembrane inserted and surface aligned orientation (Fig. 4a). The many cationic sidechains can be accommodated in the water channel or the membrane interface, and there is large water and ion flux, selective for anions due to the large number of cationic sidechains pointing into the channel (Fig. 5d).

a Pictures of the toroidal melittin pore, with lipid molecules in-between peptides lining a large water channel made up of 6–12 peptides. Plot of the pore radius, until pore closure. b Pictures of the barrel-stave pores formed by alamethicin. Plot of the total water number density in the membrane interior, due to presence of several simultaneous pores.

a Snapshots of the final moments of the transmembrane pore of melittin, from one trajectory. Only 4 peptides remain, supporting a tiny water channel that ultimately dissolves (orange beads = lipid phosphate, water molecules = blue, blue sidechains = lysines). b Number of peptides in the pore Npore and the inner pore diameter dpore, throughout the simulation. The red dotted line indicates the pore closure event and gray area the non-pore simulation phase. c Peptide mass density distribution. d Ion and water flux. (green = CL-, purple = Na + ). e Center of mass insertion depth of all peptides. f Tilt angles of all peptides. S1, S2, TM1, TM2 denote the final peptide orientations after pore closure.
Pore dissolution
The formed toroidal pores are unstable. The pore diameter fluctuates over many microseconds, but ultimately, the pore will dissolve stochastically (Figs. 4, 5; Supplementary Movies 1–3). Once water has left the membrane, each melittin peptide remains in one of 4 final positions, two on the surface of each bilayer leaflet (S1, S2), and two transmembrane (TM1, TM2). Pictures of these 4 final positions are shown (Fig. 6a). One simulation was extended for some time after pore closure (Fig. 5) to visualize the 4 distinct minima in a 2D (tilt angle/insertion) free energy surface (ΔG = -kBT ln(p)), as shown in Fig. 6d. The overall population averaged over all simulations is shown in Fig. 6e (error bars are SEM). No further aggregation or poration occurs, which mirrors the observations from the equilibrium simulations shown in Fig. 1, 2. The key reason is that the mass imbalance of the bilayer has been sufficiently relieved, removing the driving force for further poration. Peptide mass is not 100% equilibrated, with the upper leaflet retaining a higher proportion of peptides, but the remaining imbalance is not enough to cause another membrane rupture.

a Snapshots illustrate the mechanism of melittin membrane poration observed in all simulations (peptides colored blue to red from N- to C-terminus). b Water flux at various temperatures. Different colors denote different trajectories (n = 3) at the same temperature. Pore size fluctuations are similar at all temperatures, and all pores ultimately dissolve. c The pore evaporation rate fits to a simple Arrhenius model (error bars = SEM from the n = 3 simulations performed at each temperature, red = estimated lifetime τ at 25 °C). d 2D free energy surface as a function of membrane insertion and tilt angle. The 4 final orientations of the peptides after the pore has closed are denoted S1/S2 = upper/lower leaflet surface aligned. TM1/TM2 = transmembrane up/down. e Final orientation population, averaged over all n = 15 simulations (individual data points shown, error bars = SEM). f Illustration of the definition of the tilt angle τ (gray shading = membrane).
While all simulations lead to poration and, ultimately, pore dissolution, the timescales are strongly dependent on the system temperature. In Fig. 6b, the water flux over the course over all simulations is plotted. Different colors denote different trajectories at the same temperature. Overall, the pore diameter, fluctuation and water flux is similar across all simulations, with no dependence on the temperature at all. But the lifetime strongly is. Indeed, it follows simple exponential kinetics, as shown in the Arrhenius plot in Fig. 6c (r2 = 0.95, error bars = SEM). The kinetic barrier to pore dissolution can be extracted from the fit as ΔHdissolve = 22.4 kcal/mol, indicating a reasonable cohesive force holding the pore together. The increasingly long timescales prevented us observing poration at T < 333 K, but the strong Arrhenius behavior allows for a confident extrapolation of the evaporation timescale to 25 °C, with an estimate of about t = 0.85 ms.
Discussion
Transient vs equilibrium leakage
Our results demonstrate that all-atomistic MD simulations (i) can distinguish between equilibrium poration, and non-equilibrium toroidal transient leakage processes, (ii) can be used to observe the detailed pore structures and mechanism of permeabilization directly, and (iii) provide results that are in very high agreement with the vast experimental evidence.
Alamethicin is seen to form stable equilibrium pores, long predicted for this peptide16,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,62,75,76. We see pores assemble rapidly from transmembrane inserted peptides. The pores are highly dynamic and change size, with the observed flux rising with increasing size of the channel. This matches closely to leakage experiments, which report 100% leakage of probes and permanently permeable vesicles, and Electrochemical impedance spectroscopy (EIS) data where no recovery of bilayer resistance was observed17,77,78.
In contrast, for melittin, we see transient leakage caused by toroidal pores. Understanding melittin leakage is more complicated, because it is known to behave differently depending on its concentration, geometry and thickness of the lipid bilayer, and membrane composition such as charge (i.e. zwitterionic or anionic lipids). Thus, its mechanism is suspected to vary strongly with the experimental conditions: In many studies, predominantly monomeric and interfacially bound states are detected for melittin, rather than pore models55,64,77,79,80,81. This include electron paramagnetic resonance82, oriented circular dichroism and x-ray diffraction77,80, and Fourier-transform infrared spectroscopy79. In other cases at high concentration, melittin can form equilibrium toroidal pores, whose detailed structures have been solved55,56,57,59,60,79. In many other experiments, transient leakage is observed. Evidence of transient permeabilization in erythrocyte membranes treated with melittin has already been reported 40 years ago83. Many further experiments have suggested that membrane permeabilization by melittin can be a transient phenomenon that occurs only immediately after addition of melittin, and there is no equilibrium poration25,48,66,82,84,85. This has been most visible in vesicle leakage assays, which have typically shown an initial burst of leakage followed by a slow phase during which leakage stops, even though the vesicles have not yet released all of their entrapped contents17,48,55,77,85. More recently, measurements from EIS have ruled out membrane-spanning, equilibrium pores but found rapid, transient membrane permeabilization, with a decrease in bilayer resistance after addition of peptide and final equilibrium resistance close to the initial value, independent on the peptide concentration78. Recent bacterial permeabilization experiments also reveal rapid membrane resealing25,86. Prior MD simulations of hypothetical melittin pores were likely significantly too short ( < 1 µs) to observe pore dissolution. However, that melittin transmembrane bundle pores are often not stable has been shown before for hypothetical hexamers of melittin via long-scale MD simulations9,10,15. We have shown this is true for all the pores we have encountered, irrespective of size and number of peptides. The lifetimes of the pores correlate strongly with temperature and indeed follow closely to an Arrhenius model (Fig. 6), with t ≈ 0.85 ms estimated at 25 °C in pure DMPC. This appears a bit shorter, but of the same magnitude as the stopping time in leakage assays: For example, Matsuzaki et al. and Rex et al. report 6–10 ms in the absence of a potential58,85. One reason is that we report timings for a single channel, rather than for an ensemble of vesicles. In addition, the limited number of 16–18 peptides in our simulation box allows only for transient pores of that size. A size dependence on pore lifetime is expected. Thirdly, we have a very thin bilayer, and lifetime is expected to strongly depend on lipid type and bilayer thickness. Melittin clearly behaves differently in each environment.
In addition, lifetime, mechanism and activity of melittin at even higher (i.e. physiologically relevant) concentrations is thought to lead to additional macroscopic effects, such as large-scale membrane thinning61. Under such conditions, very long lifetimes and stable poration has been detected, allowing determination of the structure of the pores as toroidal56,57,59,60. Such macroscopic effects are currently challenging to reproduce in MD due to size limitations in the computational model.
Structure of the pores
Our atomistic simulations reveal the direct ab-initio assembly of 3D pore structures from fragments without any prior assumptions. Alamethicin is seen to follow closely the barrel-stave model (Fig. 4b), where peptides align parallel transmembrane inserted and laterally enclose a water-filled channel, with no participation of lipids29,30,31,33,37,76,87. As predicted long ago, the channels are stabilized by the close peptide-peptide packing of the large hydrophobic face dominated by AIB residues, while the polar Gln7 residues point into the channel pore31,33. The pores change their number of peptides on microsecond scale, mainly by individual peptides separating from one bundle, or single peptides joining an existing bundle, so channels grow or shrink in single peptide steps. Such a pattern of successive conductance levels has been known since the earliest alamethicin studies33,34,45,70,88. An ideal, circular barrel-stave pore of N peptides of radius R has the inner diameter \(d=2R(\sin {\left(\frac{p}{N}\right)}^{-1}-1)\)33. For one of the largest water channels encountered during the simulations (Fig. 2d, Fig. 4b), with N = 8, this matches the measured d ≈ 17 Å, in line with experimental estimates89. However, most pores are not perfectly circular, but assume a variety of deformed shapes and sizes, fluctuating from 4-12 peptides, with 4–8 being the most common (Fig. 2). The agreement with experimental estimates of 3-12 peptides per pore is excellent29,31,33,35,89. Some deformed aggregates are dry, without a central water channel. Then, rearrangement of the peptides can spontaneously open such a channel. Similarly, open channels close on the order of ~100 ns to 500 ns. It has been noted that higher order (N > 8) bundles of alamethicin may be distorted from a circular towards a collapsed ‘torpedo’ shape33,34. In our simulation, we find the high temperatures used to accelerate the kinetics, as well as the constant collision of channels are the main cause of deformations. At the lowest temperature (Fig. 1), we see almost circular channels only, in agreement with prior MD studies of isolated alamethicin channels5,16.
Determining the pore structure of melittin has remained much more challenging despite more than 50 years of research. One reasons is that melittin likely assumes a different form or aggregate in each specific environment, and in general seems to prefer a surface alignment63. Only under a certain conditions, evidence of membrane spanning pores has been found8,56,57,59,60,65,90,91. Several experimental results suggest that melittin can self-assemble into toroidal pores, as also found for related magainins8,56,57,59,60,79,92,93,94,95. With this geometry, both peptides and lipid molecules line the perimeter of a toroidal pore of high curvature (Fig. 4a). In contrast to the barrel-stave type, there are only marginal peptide-peptide contacts, and due to the participation of the lipids, significantly fewer peptides are required to sustain a pore of similar size. The transient pores found in our simulations closely matches this model (Fig. 4a). The pores are large, with the water channel diameter fluctuating around 20–40 Å. Our observed pores do not adhere to ideal, symmetric toroidal geometry, where peptides are distributed equally around the rim and always separated by the same number of lipids. Instead, the pores are quite dynamic, with some of the perimeter made up of peptides, transmembrane inserted or surface attached, and other parts dominated by lipids. Peptides join or leave, and typically 6–12 peptides are involved. Comparing melittin directly to alamethicin as shown in Fig. 4b, it is also obvious why melittin does not form barrel-stave pores. It has a much larger polar face, or polar angle, so tight-bundled hydrophobic peptide-peptide aggregation is hindered. Pores need to have a much larger radius for the same number of peptides to accommodate this large polar face (Fig. 4a). Both pore diameter and number of peptides have been debated and range from early estimates of only 4 peptides90,96, to larger pores observed in vesicle leakage experiments, such as 10–60 Å (6–20 peptides)97, 13–24 Å (10–11 peptides)58, 25–30 Å (10–15 peptides)65. The last one by Ladokhin et al. actually matches our results most closely. In some of these assays, the number of peptides was estimated by assuming barrel-stave geometry, which we can rule out here, so some numbers are likely overestimates. Other experimental estimates are 44 Å from neutron scattering studies57, 35–45 Å (4–8 peptides) from transmission electron microscopy98, and 43 ± 15 Å from atomic force microscopy (AFM) on bilayers supported on gold electrodes99, and a larger value of 87 Å for AFM on supported lipid monolayers100.
Effect of membrane composition
Under certain experimental conditions, melittin pores have been found to be very stable, so their structure could be determined experimentally: They are toroidal, closely matching to what we find in our simulations56,57,59,60. We report results for thin DMPC here only. Thicker membranes, or anionic membranes will almost certainly affect the pore formation mechanism or pore lifetimes. In preliminary simulations in a purely anionic membrane (DOPG), we have not yet detected large toroidal pores yet, because the exact conditions for their formation (bilayer loading imbalance, P/L, box size, temperature, time-scale of poration) are likely different from zwitterionic PC membranes (Fig. S7, S8). Major differences between melittin leakeage in PG (or mixtures) and purely PC membranes have also been found in numerous experimental studies56,63,78,82,84,101,102,103. In contrast, we see the same equilibrium barrel-stave alamethicin pores in thin and thick bilayers, and in zwitterionic and anionic membranes (DOPC, POPC, DMPC: DMPG 3:1, DPPC:DPPG 3:1, Figs. S2–S5). Full convergence of the distribution of oligomers and transmembrane flux is not yet reached. However, the simulations in the ~10 µs range do not suggest a dramatic effect of lipid type on the channel equilibrium. Melittin on the other hand behaves very differently in each environment. It will take considerably more computational effort to reproduce its poration in other membranes and lipid environments.
Ion selectivity
Not only water is flowing through the predicted pores, but also ions. Although we do not apply any voltage, the simulation times are long enough to capture numerous ion transitions through both the stable alamethicin channel and the transient melittin pore. Both are selective, and closely match experimental evidence. Alamethicin both conducts Na+ cations and Cl- anions, with a slight preference for cations, in agreement with the mild cation-selectivity found experimentally76,89. In contrast, we find the melittin transient pore to be strongly anion-selective, a consequence of the large number of cationic side-chains that point into the pore. Anion-selectivity has also been reported experimentally90.
Equilibrium states of melittin after pore closure
All melittin pores in our simulations ultimately dissolve, leaving peptides in an equilibrium state where they can coexist in both transmembrane and surface states: Some peptides will remain trapped in transmembrane conformations, with the cationic sidechains snorkeling and preventing a quick exit of the peptide to the surface, similar what we have reported for the peptide PGLa13. As shown in Figs. 2g, 6, these configurations can be very stable. Such a coexistence of surface and transmembrane states without pores or leakage has been suspected for a long time as the best explanation to oriented circular dichroism measurements of melittin63, and been observed from vibrational spectroscopy with attenuated total reflection-Fourier transform infrared spectroscopy in supported lipid bilayers103.
The results for both alamethicin and melittin show that two very different peptide membrane permeabilization mechanisms can be fully resolved atomistically in silico, with a high level of convergence, reproducibility, and excellent agreement to experimental measurements. Previously, we have shown that there are also other pathways that neither follow the two mechanisms illustrated here, for example for maculatin, which forms a whole ensemble of dynamic nanostructures in the membrane12. Also, a ‘silent’ pathway, in which peptides translocate monomerically without forming pores, can be observed via MD under certain conditions13. We believe atomistic simulations have the potential to become a major tool in assisting the design of new sequences for specific membrane permeabilization in pharmaceutical or biotechnological applications. However, the simulations shown here are just a first step. They demonstrate that the major permeabilization mechanisms can in principle be observed in a reasonable computational time, and without making any initial assumptions. The next step will be to employ more realistic cell membrane models in molecular mechanics. Ultimately, simulations could prove to be an easy to use computational tool, assisting rational peptide design by allowing the pre-screening of sequence libraries to reduce the amount of work needed for experimental assays.
Methods
Molecular dynamics simulations
All-atom MD simulations were performed and analyzed using GROMACS 2020.4 (www.gromacs.org)104, Hippo/Atomix (http://www.biowerkzeug.com)105, and VMD 1.93 (http://www.ks.uiuc.edu/Research/vmd/)106. MD simulations were using the CHARMM36 force field107, in conjunction with the TIP3P water model108. Electrostatic interactions were computed using PME, and a cut-off of 10 Å was used for van der Waals interactions. Bonds involving hydrogen atoms were constrained using LINCS109. The integration time-step was 2 fs and neighbor lists were updated every 5 steps. All simulations were performed in the NPT ensemble, without any restraints or biasing potentials. Water, ions, lipids and the protein were each coupled separately to a heat bath with a time constant τT = 0.5 ps using velocity rescale temperature coupling. The atmospheric pressure of 1 bar was maintained using weak semi-isotropic pressure coupling with compressibility κz = κxy = 4.6 · 10−5 bar−1 and time constant τP = 1 ps.
System configuration and setup
Initial systems were build as peptide/membrane/water boxes, with a salt concentration of 100 mM NaCl using CHARMM-GUI110. Peptides were initially placed in the aqueous phase or transmembrane inserted (TM). Alamethicin is so hydrophobic that S-state peptide immediately insert transmembrane, so simulations only with transmembrane states were considered for this peptide. Bilayers were designed to be symmetric, with equal number of lipids in each leaflet. For some simulations were peptide mass was asymmetrically distributed, several lipid molecules were removed to accommodate the peptides. Individual simulations (Tables S1–S3) were run for 2–41 µs.
Peptide thermostability
Peptides embedded in membranes are resistant to thermal denaturation even at 95 °C12,105,111,112,113,114,115, allowing simulations to be carried out at elevated temperatures, which significantly enhances sampling. We have previously demonstrated that elevating the temperature does not change conformational equilibria or partitioning free energies of helical membrane-active peptides, provided they are stable against thermal denaturation; however, the vast increase in sampling kinetics at high temperatures allows for rapid simulation of bilayer partitioning, translocations between bilayer leaflets, and pore assembly. In this study, it is again confirmed that higher temperatures lead to faster kinetics but identical pore structures, pore dissolution mechanisms, and equilibria.
Choice of bilayer lipids and P/L
Synthetic bilayers were chosen, rather than realistic lipid mixtures mimicking bacterial membranes. The key reason is to stay as close as possible to the vast experimental data sets (e.g. leakage, CD, ssNMR, electrophysiology) available for exactly those synthetic bilayers to enable direct comparison. For actual organisms, often only sterilization data, rather than structural information on poration, is available. Such assays have timescales 7–9 orders of magnitude larger than what MD can do (minutes vs. microseconds), making comparison tenous. Chosen bilayers were pure zwitterionic POPC, DOPC and DMPC, as well as anionic membranes (DOPG) and mixtures such as DMPC:DMPG 3:1 and DPPC:DPPG 3:1, which are often employed in ssNMR experiments. To foster pore formation, a relatively high P/L of 1/12–1/20 was used, resulting in typical simulation boxes of ~120 × 120 × 100 Å3, containing 16–18 peptides and 192–360 lipids (Tables S1–S4). Voltage was not applied, again in order to match experimental conditions (e.g. vesicle leakage assays). In total, ~240 µs of simulations were performed for this study.
Oligomer population and permutational clustering
In order to reveal the most populated pore assemblies during the simulations, a complete list of all oligomers was constructed for each trajectory frame. An oligomer of order n was considered any set of n peptides that are in mutual contact, defined as a heavy-atom (N, C, O) minimum distance of <3.5 Å. This works well for barrel-stave pores where peptides touch. For toroidal pores, where peptides are separated by lipids, a cutoff of 5–6 Å was used. The oligomeric state is often overestimated due to numerous transient surface-bound (S-state) peptides that are only loosely attached to the transmembrane inserted peptides that make up the core of the oligomer. These S-state peptides frequently change position or drift on and off the stable part of the pore. To focus the analysis on true longer-lived transmembrane pores, a cut-off criterion of 75° was introduced for the tilt angle τ of the peptides. Any peptide with τ ≥ 75° was considered in the S-state and removed from the oligomeric analysis. This strategy greatly reduced the noise in the oligomeric clustering algorithm by focusing on the true longer-lived pore structures. Population plots of the occupation percentage of oligomer n multiplied by its number of peptides n were then constructed. These reveal how much peptide mass was concentrated in which oligomeric state at any point in the simulation. Oligomers of the same order n were subjected to conformationally clustering with a backbone RMSD similarity cutoff criterion of 4 Å. As structurally similar oligomers can be made up of different peptides, all n! permutations of peptide arrangements, generated by Heap’s algorithm, were considered in the clustering.
Pore radii
All pores, either barrel-stave or toroidal, were found to fluctuate in size and shape, and often were not perfectly circular, but could also be slightly deformed or ellisoid. This effect is mainly due to the use of elevated temperatures. In order to asses pore sizes by a single representative number, the pore ‘radius’ rpore was defined by counting all water molecules inside the pore and calculating the radius of a cylinder containing an equal amount of water molecules. The radius then corresponds to the circular mean of the various pore geometries.
Transmembrane flux
Water and ion flux through membrane pores were calculated by determining the total instantaneous flux through the whole bilayer patch. Two planes orthogonal to the membrane normal were considered at z = -10 Å and z = +10 Å, with all transition events that cross the planes counted. The flux was then obtained by dividing the transition counts by the area of the membrane patch and the elapsed time for each trajectory frame. The final flux is the average of the individual up and down fluxes recorded. Curves were subsequently smoothed by averaging over 1000 frames.
Free energy surface
An estimate of the free energy surface of the final post-pore state of melittin was calculated from one trajectory that was continued for 3 µs after pore closure. The surface was calculated via ΔG = -kBT ln(p), where p is the population in each bin. ΔG was shifted so that the bin with the highest population has ΔG = 0. No peptide transitions occurred during these final 3 µs.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Due to the large size ( > 1TB), the MD simulations datasets (e.g., pdb and xtc files) are not publicly available but are available from the authors upon request. The source data underlying Figs. 1–6, and Supplementary Figs. are provided as a Source Data file. Source data are provided with this paper.
Code availability
Common MD analysis tools and programs were used to analyse the MD trajectories. Sample scripts are available from the authors upon request.
References
Bayley, H. Designed membrane channels and pores. Curr. Opin. Biotechnol. 10, 94–103 (1999).
Hancock, R. E. W. & Sahl, H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24, 1551–1557 (2006).
Sani, M.-A. & Separovic, F. How membrane-active peptides get into lipid membranes. Acc. Chem. Res. 49, 1130–1138 (2016).
Guha, S., Ghimire, J., Wu, E. & Wimley, W. C. Mechanistic landscape of membrane-permeabilizing peptides. Chem. Rev. https://www.ncbi.nlm.nih.gov/pubmed/30624911 (2019).
Perrin, B. S. J. & Pastor, R. W. Simulations of membrane-disrupting peptides I: Alamethicin pore stability and spontaneous insertion. Biophys. J. 111, 1248–1257 (2016).
Perrin, B. S. J., Fu, R., Cotten, M. L. & Pastor, R. W. Simulations of membrane-disrupting peptides II: AMP piscidin 1 favors surface defects over pores. Biophys. J. 111, 1258–1266 (2016).
Chen, C. H. et al. Simulation-guided rational de novo design of a small pore-forming antimicrobial peptide. J. Am. Chem. Soc. 141, 4839–4848 (2019).
Sengupta, D., Leontiadou, H., Mark, A. E. & Marrink, S.-J. Toroidal pores formed by antimicrobial peptides show significant disorder. Biochim. Biophys. Acta 1778, 2308–2317 (2008).
Leveritt, J. M., Pino-Angeles, A. & Lazaridis, T. The structure of a melittin-stabilized pore. Biophys. J. 108, 2424–2426 (2015).
Sepehri, A., PeBenito, L., Pino-Angeles, A. & Lazaridis, T. What makes a good pore former: A study of synthetic melittin derivatives. Biophys. J. 118, 1901–1913 (2020).
Leontiadou, H., Mark, A. E. & Marrink, S. J. Antimicrobial peptides in action. J. Am. Chem. Soc. 128, 12156–12161 (2006).
Wang, Y., Chen, C. H., Hu, D., Ulmschneider, M. B. & Ulmschneider, J. P. Spontaneous formation of structurally diverse membrane channel architectures from a single antimicrobial peptide. Nat. Commun. 7, 13535 (2016).
Ulmschneider, J. P. Charged antimicrobial peptides can translocate across membranes without forming channel-like pores. Biophys. J. 113, 73–81 (2017).
Hong, J. et al. How melittin inserts into cell membrane: Conformational changes, inter-peptide cooperation, and disturbance on the membrane. Mol. Basel Switz. 2019, 24. https://doi.org/10.3390/molecules24091775.
Pino-Angeles, A. & Lazaridis, T. Effects of peptide charge, orientation, and concentration on melittin transmembrane pores. Biophys. J. 114, 2865–2874 (2018).
Tieleman, D. P., Breed, J., Berendsen, H. J. & Sansom, M. S. Alamethicin channels in a membrane: Molecular dynamics simulations. Faraday Discuss 111, 209–223 (1998).
Krauson, A. J., He, J. & Wimley, W. C. Determining the mechanism of membrane permeabilizing peptides: Identification of potent, equilibrium pore-formers. Biochim. Biophys. Acta BBA - Biomembr. 1818, 1625–1632 (2012).
Hristova, K., Selsted, M. E. & White, S. H. Critical role of lipid composition in membrane permeabilization by rabbit neutrophil defensins *. J. Biol. Chem. 272, 24224–24233 (1997).
Wimley, W. C., Selsted, M. E. & White, S. H. Interactions between human defensins and lipid bilayers: evidence for formation of multimeric pores. Protein Sci. 3, 1362–1373 (1994).
Lau, Q. Y. et al. Elucidating the bactericidal mechanism of action of the linear antimicrobial tetrapeptide BRBR-NH2. Biochim. Biophys. Acta BBA - Biomembr. 1860, 1517–1527 (2018).
Wimley, W. C. & Hristova, K. The mechanism of membrane permeabilization by peptides: Still an enigma. Aust. J. Chem. 73, 96–103 (2019).
Rathinakumar, R. & Wimley, W. C. Biomolecular engineering by combinatorial design and high-throughput screening: small, soluble peptides that permeabilize membranes. J. Am. Chem. Soc. 130, 9849–9858 (2008).
Wimley, W. C. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem. Biol. 5, 905–917 (2010).
Kauffman, W. B., Guha, S. & Wimley, W. C. Synthetic molecular evolution of hybrid cell penetrating peptides. Nat. Commun. 9, 2568 (2018).
Yang, Z., Choi, H. & Weisshaar, J. C. Melittin-induced permeabilization, re-sealing, and re-permeabilization of E. coli membranes. Biophys. J. 114, 368–379 (2018).
Heerklotz, H. Membrane stress and permeabilization induced by asymmetric incorporation of compounds. Biophys. J. 81, 184–195 (2001).
Shai, Y. & Oren, Z. From “Carpet” mechanism to de-novo designed diastereomeric cell-selective antimicrobial peptides. Bact. Anti Bact. Pept. 22, 1629–1641 (2001).
Wheaten, S. A., Ablan, F. D. O., Spaller, B. L., Trieu, J. M. & Almeida, P. F. Translocation of cationic amphipathic peptides across the membranes of pure phospholipid giant vesicles. J. Am. Chem. Soc. 135, 16517–16525 (2013).
He, K., Ludtke, S. J., Worcester, D. L. & Huang, H. W. Neutron scattering in the plane of membranes: Structure of alamethicin pores. Biophys. J. 70, 2659–2666 (1996).
Qian, S., Wang, W., Yang, L. & Huang, H. W. Structure of the alamethicin pore reconstructed by X-ray diffraction analysis. Biophys. J. 94, 3512–3522 (2008).
Fox, R. O. J. & Richards, F. M. A voltage-gated ion channel model inferred from the crystal structure of alamethicin at 1.5-A resolution. Nature 300, 325–330 (1982).
Huang, H. W. & Wu, Y. Lipid-alamethicin interactions influence alamethicin orientation. Biophys. J. 60, 1079–1087 (1991).
Sansom, M. S. Structure and function of channel-forming peptaibols. Q. Rev. Biophys. 26, 365–421 (1993).
Boheim, G. Statistical analysis of alamethicin channels in black lipid membranes. J. Membr. Biol. 19, 277–303 (1974).
He, K., Ludtke, S. J., Huang, H. W. & Worcester, D. L. Antimicrobial peptide pores in membranes detected by neutron in-plane scattering. Biochemistry 34, 15614–15618 (1995).
Bertelsen, K. et al. Residue-specific information about the dynamics of antimicrobial peptides from (1)H-(15)N and (2)H solid-state NMR spectroscopy. J. Am. Chem. Soc. 131, 18335–18342 (2009).
Cafiso, D. S. Alamethicin: A peptide model for voltage gating and protein-membrane interactions. Annu. Rev. Biophys. Biomol. Struct. 23, 141–165 (1994).
Bechinger, B. et al. 15N and 31P solid-state NMR investigations on the orientation of Zervamicin II and alamethicin in phosphatidylcholine membranes. Biochemistry 40, 9428–9437 (2001).
Bak, M. et al. Conformation of alamethicin in oriented phospholipid bilayers determined by (15)N solid-state nuclear magnetic resonance. Biophys. J. 81, 1684–1698 (2001).
Bertelsen, K. et al. Membrane-bound conformation of peptaibols with methyl-deuterated alpha-amino isobutyric acids by 2H magic angle spinning solid-state NMR spectroscopy. J. Am. Chem. Soc. 129, 14717–14723 (2007).
Bertelsen, K., Dorosz, J., Hansen, S. K., Nielsen, N. C. & Vosegaard, T. Mechanisms of peptide-induced pore formation in lipid bilayers investigated by oriented 31P solid-state NMR spectroscopy. PloS One 7, e47745 (2012).
Salnikov, E. S. et al. Structure and alignment of the membrane-associated peptaibols ampullosporin A and alamethicin by oriented 15N and 31P solid-state NMR spectroscopy. Biophys. J. 96, 86–100 (2009).
Barranger-Mathys, M. & Cafiso, D. S. Membrane structure of voltage-gated channel forming peptides by site-directed spin-labeling. Biochemistry 35, 498–505 (1996).
North, C. L., Barranger-Mathys, M. & Cafiso, D. S. Membrane orientation of the N-terminal segment of alamethicin determined by solid-state 15N NMR. Biophys. J. 69, 2392–2397 (1995).
Gordon, L. G. M. & Haydon, D. A. The unit conductance channel of alamethicin. Biochim. Biophys. Acta BBA - Biomembr. 255, 1014–1018 (1972).
Smith, R. et al. Structure and orientation of the pore-forming peptide melittin, in lipid bilayers. J. Mol. Biol. 241, 456–466 (1994).
Smith, R., Separovic, F., Bennett, F. C. & Cornell, B. A. Melittin-induced changes in lipid multilayers. A solid-state NMR study. Biophys. J. 63, 469–474 (1992).
Wimley, W. C. How does melittin permeabilize membranes? Biophys. J. 114, 251–253 (2018).
Habermann, E. Bee and wasp venoms. Science 177, 314–322 (1972).
Dempsey, C. E. The actions of melittin on membranes. Biochim. Biophys. Acta 1031, 143–161 (1990).
Terwilliger, T. C. & Eisenberg, D. The structure of melittin. II. Interpretation Struct. J. Biol. Chem. 257, 6016–6022 (1982).
Bechinger, B. Structure and functions of channel-forming peptides: Magainins, cecropins, melittin and alamethicin. J. Membr. Biol. 156, 197–211 (1997).
Jamasbi, E., Mularski, A. & Separovic, F. Model membrane and cell studies of antimicrobial activity of melittin analogues. Curr. Top. Med. Chem. 16, 40–45 (2016).
Chen, E. H.-L. et al. Visualizing the membrane disruption action of antimicrobial peptides by cryo-electron tomography. Nat. Commun. 14, 5464 (2023).
Lee, M.-T., Hung, W.-C., Chen, F.-Y. & Huang, H. W. Mechanism and kinetics of pore formation in membranes by water-soluble amphipathic peptides. Proc. Natl Acad. Sci. Usa. 105, 5087–5092 (2008).
Lee, M.-T., Sun, T.-L., Hung, W.-C. & Huang, H. W. Process of inducing pores in membranes by melittin. Proc. Natl. Acad. Sci. USA 110, 14243–14248 (2013).
Yang, L., Harroun, T. A., Weiss, T. M., Ding, L. & Huang, H. W. Barrel-stave model or toroidal model? a case study on melittin pores. Biophys. J. 81, 1475–1485 (2001).
Matsuzaki, K., Yoneyama, S. & Miyajima, K. Pore formation and translocation of melittin. Biophys. J. 73, 831–838 (1997).
Sun, Y., Sun, T.-L. & Huang, H. W. Mode of action of antimicrobial peptides on E. Coli spheroplasts. Biophys. J. 111, 132–139 (2016).
Faust, J. E., Yang, P.-Y. & Huang, H. W. Action of antimicrobial peptides on bacterial and lipid membranes: A direct comparison. Biophys. J. 112, 1663–1672 (2017).
Lee, M.-T., Chen, F.-Y. & Huang, H. W. Energetics of pore formation induced by membrane active peptides. Biochemistry 43, 3590–3599 (2004).
He, K., Ludtke, S. J., Heller, W. T. & Huang, H. W. Mechanism of alamethicin insertion into lipid bilayers. Biophys. J. 71, 2669–2679 (1996).
Ladokhin, A. S. & White, S. H. Detergent-like” permeabilization of anionic lipid vesicles by melittin. Biochim. Biophys. Acta 1514, 253–260 (2001).
Ladokhin, A. S. & White, S. H. Folding of amphipathic alpha-helices on membranes: energetics of helix formation by melittin. J. Mol. Biol. 285, 1363–1369 (1999).
Ladokhin, A. S., Selsted, M. E. & White, S. H. Sizing membrane pores in lipid vesicles by leakage of co-encapsulated markers: pore formation by melittin. Biophys. J. 72, 1762–1766 (1997).
Guha, S. et al. Applications and evolution of melittin, the quintessential membrane active peptide. Biochem. Pharmacol. 193, 114769 (2021).
Andersson, M., Ulmschneider, J. P., Ulmschneider, M. B. & White, S. H. Conformational states of melittin at a bilayer interface. Biophys. J. 104, L12–L14 (2013).
Chen, C. H., Wiedman, G., Khan, A. & Ulmschneider, M. B. Absorption and folding of melittin onto lipid bilayer membranes via unbiased atomic detail microsecond molecular dynamics simulation. Biochim. Biophys. Acta 1838, 2243–2249 (2014).
Sakmann, B. & Boheim, G. Alamethicin-induced single channel conductance fluctuations in biological membranes. Nature 282, 336–339 (1979).
Mak, D. O. & Webb, W. W. Two classes of alamethicin transmembrane channels: Molecular models from single-channel properties. Biophys. J. 69, 2323–2336 (1995).
Huang, H. W., Chen, F.-Y. & Lee, M.-T. Molecular mechanism of peptide-induced pores in membranes. Phys. Rev. Lett. 92, 198304 (2004).
Huang, H. W. Free energies of molecular bound states in lipid bilayers: Lethal concentrations of antimicrobial peptides. Biophys. J. 96, 3263–3272 (2009).
Campelo, F., McMahon, H. T. & Kozlov, M. M. The hydrophobic insertion mechanism of membrane curvature generation by proteins. Biophys. J. 95, 2325–2339 (2008).
Terwilliger, T. C., Weissman, L. & Eisenberg, D. The structure of melittin in the form i crystals and its implication for melittin’s lytic and surface activities. Biophys. J. 37, 353–361 (1982).
Pan, J., Tristram-Nagle, S. & Nagle, J. F. Alamethicin aggregation in lipid membranes. J. Membr. Biol. 231, 11–27 (2009).
Hall, J. E., Vodyanoy, I., Balasubramanian, T. M. & Marshall, G. R. Alamethicin. A rich model for channel behavior. Biophys. J. 45, 233–247 (1984).
Krauson, A. J., He, J. & Wimley, W. C. Gain-of-function analogues of the pore-forming peptide melittin selected by orthogonal high-throughput screening. J. Am. Chem. Soc. 134, 12732–12741 (2012).
Wiedman, G., Herman, K., Searson, P., Wimley, W. C. & Hristova, K. The electrical response of bilayers to the bee venom toxin melittin: evidence for transient bilayer permeabilization. Biochim. Biophys. Acta 1828, 1357–1364 (2013).
Frey, S. & Tamm, L. K. Orientation of melittin in phospholipid bilayers. A polarized attenuated total reflection infrared study. Biophys. J. 60, 922–930 (1991).
Hristova, K., Dempsey, C. E. & White, S. H. Structure, location, and lipid perturbations of melittin at the membrane interface. Biophys. J. 80, 801–811 (2001).
Sharma, V. K. et al. Dynamical and phase behavior of a phospholipid membrane altered by an antimicrobial peptide at low concentration. J. Phys. Chem. Lett. 7, 2394–2401 (2016).
Gordon-Grossman, M., Zimmermann, H., Wolf, S. G., Shai, Y. & Goldfarb, D. Investigation of model membrane disruption mechanism by melittin using pulse electron paramagnetic resonance spectroscopy and cryogenic transmission electron microscopy. J. Phys. Chem. B 116, 179–188 (2012).
DeGrado, W. F., Musso, G. F., Lieber, M., Kaiser, E. T. & Kézdy, F. J. Kinetics and mechanism of hemolysis induced by melittin and by a synthetic melittin analogue. Biophys. J. 37, 329–338 (1982).
van den Bogaart, G., Guzmán, J. V., Mika, J. T. & Poolman, B. On the mechanism of pore formation by melittin. J. Biol. Chem. 283, 33854–33857 (2008).
Rex, S. & Schwarz, G. Quantitative studies on the melittin-induced leakage mechanism of lipid vesicles. Biochemistry 37, 2336–2345 (1998).
Gee, M. L. et al. Imaging the action of antimicrobial peptides on living bacterial cells. Sci. Rep. 3, 1557 (2013).
Oren, Z. & Shai, Y. Mode of action of linear amphipathic alpha-helical antimicrobial peptides. Biopolymers 47, 451–463 (1998).
Harriss, L. M., Cronin, B., Thompson, J. R. & Wallace, M. I. Imaging multiple conductance states in an alamethicin pore. J. Am. Chem. Soc. 133, 14507–14509 (2011).
Menestrina, G., Voges, K. P., Jung, G. & Boheim, G. Voltage-dependent channel formation by rods of helical polypeptides. J. Membr. Biol. 93, 111–132 (1986).
Tosteson, M. T. & Tosteson, D. C. The sting. melittin forms channels in lipid bilayers. Biophys. J. 36, 109–116 (1981).
Irudayam, S. J. & Berkowitz, M. L. Binding and reorientation of melittin in a POPC bilayer: Computer simulations. Biochim. Biophys. Acta 1818, 2975–2981 (2012).
Matsuzaki, K., Murase, O., Fujii, N. & Miyajima, K. An antimicrobial peptide, magainin 2, induced rapid flip-flop of phospholipids coupled with pore formation and peptide translocation. Biochemistry 35, 11361–11368 (1996).
Ludtke, S. J. et al. Membrane pores induced by magainin. Biochemistry 35, 13723–13728 (1996).
Kokot, G., Mally, M. & Svetina, S. The dynamics of melittin-induced membrane permeability. Eur. Biophys. J. EBJ 41, 461–474 (2012).
Allende, D., Simon, S. A. & McIntosh, T. J. Melittin-induced bilayer leakage depends on lipid material properties: Evidence for toroidal pores. Biophys. J. 88, 1828–1837 (2005).
Vogel, H. & Jähnig, F. The structure of melittin in membranes. Biophys. J. 50, 573–582 (1986).
Rex, S. Pore formation induced by the peptide melittin in different lipid vesicle membranes. Biophys. Chem. 58, 75–85 (1996).
Park, S.-C. et al. Investigation of toroidal pore and oligomerization by melittin using transmission electron microscopy. Biochem. Biophys. Res. Commun. 343, 222–228 (2006).
Juhaniewicz, J. & Sek, S. Interaction of melittin with negatively charged lipid bilayers supported on gold electrodes. Electrochim. Acta 197, 336–343 (2016).
Giménez, D., Sánchez-Muñoz, O. L. & Salgado, J. Direct observation of nanometer-scale pores of melittin in supported lipid monolayers. Langmuir 31, 3146–3158 (2015).
Benachir, T. & Lafleur, M. Study of vesicle leakage induced by melittin. Biochim. Biophys. Acta BBA - Biomembr. 1235, 452–460 (1995).
Hincha, D. K. & Crowe, J. H. The lytic activity of the bee venom peptide melittin is strongly reduced by the presence of negatively charged phospholipids or chloroplast galactolipids in the membranes of phosphatidylcholine large unilamellar vesicles. Biochim. Biophys. Acta BBA - Biomembr. 1284, 162–170 (1996).
Chen, X., Wang, J., Boughton, A. P., Kristalyn, C. B. & Chen, Z. Multiple orientation of melittin inside a single lipid bilayer determined by combined vibrational spectroscopic studies. J. Am. Chem. Soc. 129, 1420–1427 (2007).
Pronk, S. et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinforma. Oxf. Engl. 29, 845–854 (2013).
Ulmschneider, J. P., Smith, J. C., White, S. H. & Ulmschneider, M. B. In silico partitioning and transmembrane insertion of hydrophobic peptides under equilibrium conditions. J. Am. Chem. Soc. 133, 15487–15495 (2011).
Humphrey, W., Dalke, A. & Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Huang, J. & MacKerell, A. D. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput Chem. 34, 2135–2145 (2013).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating. Liq. Water J. Chem. Phys. 79, 926–935 (1983).
Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. G. E. M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997).
Lee, J. et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput 12, 405–413 (2016).
Ulmschneider, M. B. et al. Spontaneous transmembrane helix insertion thermodynamically mimics translocon-guided insertion. Nat. Commun. 5, ncomms5863 (2014).
Ulmschneider, J. P. & Ulmschneider, M. B. Molecular dynamics simulations are redefining our view of peptides interacting with biological membranes. Acc. Chem. Res. 51, 1106–1116 (2018).
Gumbart, J. C., Ulmschneider, M. B., Hazel, A., White, S. H. & Ulmschneider, J. P. Computed free energies of peptide insertion into bilayers are independent of computational method. J. Membr. Biol. 251, 345–356 (2018).
Ulmschneider, M. B., Doux, J. P. F., Killian, J. A., Smith, J. C. & Ulmschneider, J. P. Mechanism and kinetics of peptide partitioning into membranes from all-atom simulations of thermostable peptides. J. Am. Chem. Soc. 132, 3452–3460 (2010).
Ulmschneider, J. P., Smith, J. C., White, S. H. & Ulmschneider, M. B. The importance of the membrane interface as the reference state for membrane protein stability. Biochimica et. Biophys. Acta (BBA) - Biomembranes 1860, 2539–2548 (2018).
Acknowledgements
J.P.U. was supported by the National Natural Science foundation of China (Grant No. 12174253), the Shanghai Municipal Natural Science Fund (Grant No. 21ZR1428700), and by the Science and Technology Commission of Shanghai Municipality (Grant No. 21JC1403700). J.P.U. was also supported by “the Fundamental Research Fund for the Central Universities” and the Key Laboratory of Scientific and Engineering Computing (Ministry of Education). M.B.U was supported by a BBSRC standard grant (BB/S017844/1), a KCL IP & Licensing Wellcome Trust Genesis Fund grant (IP501/2712), and a BBSRC accelerator grant (BB/Y514159/1). We are grateful to the UK Materials and Molecular Modeling Hub for computational resources, which is partially funded by EPSRC (EP/T022213/1, EP/W032260/1 and EP/P020194/1). We would like to thank Kalina Hristova and William C. Wimley for helpful discussions.
Author information
Authors and Affiliations
Contributions
J.P.U and M.B.U. designed the study. J.P.U. performed and analyzed the molecular dynamics simulations. J.P.U. and M.B.U. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Huey Huang, Alexey Ladokhin, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ulmschneider, J.P., Ulmschneider, M.B. Melittin can permeabilize membranes via large transient pores. Nat Commun 15, 7281 (2024). https://doi.org/10.1038/s41467-024-51691-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51691-1
