
Abstract
We report bio-structural, bio-chemical and bio-physical evidence demonstrating how small molecules can bind to both wild-type and mutant IDH1, but only inhibit the enzymatic activity of the mutant isoform. Enabled through x-ray crystallography, we characterized a series of small molecule inhibitors that bound to mutant IDH1 differently than the marketed inhibitor Ivosidenib, for which we have determined the x-ray crystal structure. Across the industry several mutant IDH1 inhibitor chemotypes bind to this allosteric IDH1 pocket and selectively inhibit the mutant enzyme. Detailed characterization by a variety of biophysical techniques and NMR studies led us to propose how compounds binding in the allosteric IDH1 R132H pocket inhibit the production of 2-Hydroxy glutarate.
Similar content being viewed by others


Introduction
Mutations in the isocitrate dehydrogenase 1 (IDH1) gene have been observed in multiple human tumor types, with the highest prevalence seen in low-grade glioma (LGG) and secondary glioblastoma (GBM)1,2,3,4. For example, a common subtype of LGG, astrocytoma, is up to 70% IDH1 mutant. In addition, 5–7% of AML patients harbor an IDH1 mutation, which, like in LGG and GBM, most commonly occurs on codon 132 where an arginine is replaced by a histidine (R132H). Prognosis in AML is adversely affected by IDH1 mutations with a trend for shorter overall survival, shorter progression-free survival, and a higher cumulative risk for relapse5. Other less common tumor types have also been shown to have high percentages of IDH1 mutations (e.g., Chondrosarcoma) or low percentages in more common tumors (e.g., melanoma)6.
Mutations in IDH1 typically occur at arginine 132 and a histidine (R132H) alteration is by far the most frequently observed amino acid alteration in patients. These mutations lead to a neomorphic gain of function, resulting in the production of (R)−2-Hydroxyglutarate (2HG) which is found in very high intratumoral and intracellular levels7. High 2HG levels, often found at mM levels intracellularly, have been shown to competitively inhibit dioxygenase enzymes which are involved in regulating epigenetic mechanisms associated with histone and DNA methylation, thereby promoting tumorigenesis8. The epigenetic dysregulation imparted by such IDH1 mutations is apparent in the hypermethylation phenotypes observed in tumor types bearing these alterations9,10.
Inhibiting mutant IDH1 has been clinically validated, with ivosidenib (AG-120)11 approved by the FDA for the treatment of relapsed/refractory AML12. Several other small molecule mutant IDH1 inhibitors, including IDH1/2 dual-inhibitor AG-88113, BAY-143603214, GSK32115, IDH30516, FT-210217, and others18,19, have also shown efficacy in various preclinical and clinical studies, potentially offering new treatment options for different tumor types carrying IDH1 mutations.
We previously reported in vivo target validation studies in a BT142 orthotopic patient-derived glioma model, in which we saw a significant survival benefit in mice treated with our proprietary mutant IDH1 inhibitor MRK-A20.
Here we report characterization of screening hits and their more potent analogs to better understand how they selectively inhibit mutant IDH1 but not the wild-type enzyme in biochemical and cellular assays. Data from binding studies, NMR spectroscopy, biochemical studies, and x-ray crystallography allow us to derive an allosteric mode of action for the inhibition of IDH1 R132H. In addition, we compare the binding of the MRK-A family to the binding of AG120, also known as Ivosidenib (Tibsovo®).
Results
Identification of IDH1R132H -selective inhibitors
To identify selective inhibitors of IDH1R132H, we executed a high throughput screen of our company’s full 2 M compound screening library configured to identify inhibitors agnostic of their biochemical mechanism. We elected to screen the full collection of compounds available at MSD due to the anticipated challenge of finding mutant-selective inhibitors for a mutation where an arginine has been replaced by another basic amino acid residue, a histidine. The high affinity of IDH1R132H for NADPH (Kd ~ 50 nM) made it challenging to establish an assay sensitive to NADPH-competitive inhibitors while avoiding substrate depletion. To achieve this, we utilized two variations of an enzyme coupling system that allows continuous regeneration of NADPH via an NADPH-utilizing glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from spinach. The 1,3-Bisphosphoglyceric acid formed in the NADPH regeneration reaction is then coupled to the formation of ATP via phosphoglycerate kinase (PGK). The ATP formed in the PGK reaction was then detected by firefly luciferase (Fig. 1a). An alternative form of this assay also utilized the NADPH regeneration system but directly detected 2HG using a high-throughput mass spectrometry system (Agilent Rapid Fire, Fig. 1a). This screening campaign led to the identification of compound 1, (Fig. 1b) a molecule that inhibited reductive 2HG production by IDH1R132H (IC50 = 48 ± 59 nM, N = 16) but did not affect the activity of wild-type IDH1 in either the reductive (ICT formation) or oxidative (aKG formation) direction (IC50 of both reactions > 20,000 nM). Compound 1 also inhibited 2HG in MOG-G-UVW cells, stably transduced to overexpress IDH1R132H (IC50 = 0.7 µM, N = 1). Of note, compound 1 existed in our collection with high purity, but as an inseparable mixture of isomers stemming from the original synthetic route to this compound. Our studies commenced utilizing compound 1 but soon transitioned to compounds 2-4 (see compound optimization section).

a Assay description, (b) Structure of compound 1.
Compound 1 thermally stabilizes IDH1R132H
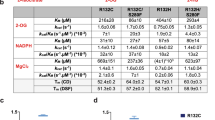
The interaction of compound 1 with IDH1R132H and IDH1wt was investigated using a thermal shift protein unfolding assay21. The inclusion of compound 1 (20 µM) stabilized IDH1R132H by 5.3 degrees C, similar to NADPH but less than NADP + (Table 1). Neither isocitrate nor aKG provided appreciable stabilization to thermal denaturation. However, when compound 1 was included along with NADPH or NADP +, the melting temperature was increased by > 4.4 degrees Celsius beyond that provided by either substrate alone, suggesting compound 1 and NADP + or NADPH can bind IDH1R132H simultaneously. In contrast, compound 1 did not thermally stabilize IDH1wt, however, strong stabilization was provided by isocitrate, NADP +, and NADPH. In stark contrast to IDH1R132H, the thermal stabilization of IDH1wt by substrates was not altered by the presence of 20 µM compound 1.
Compound 1 binds IDH1R132H and IDH1wt
Compound 1 interaction with IDH1R132H and IDH1wt was further studied using a solution-based affinity selection mass spectrometry binding assay termed ALIS22. Titration of compound 1 into a solution containing 2 µM IDH1wt or IDH1R132H, surprisingly, showed saturable binding to both proteins (Fig. 2). Inclusion of isocitrate in the solution showed 86% displacement of compound 1 from IDH1wt but not from IDH1R132H. Similarly, the addition of 10 mM aKG resulted in the displacement of 67% of compound 1 from IDH1wt but not from IDH1R132H. Additional binding studies using Surface Plasmon Resonance (SPR) showed that compound 1 binds to IDH1R132H and IDH1wt with a Kd of 3 µM and 6 µM, respectively (Supplementary Note 1). SPR demonstrated similar binding of compound 1 to apo-IDH1R132H and apo-IDH1wt but the affinities are lower than from the biochemical data and Mass Spec binding data. SAR for compound optimization was guided by functional data from biochemical and cell-based assays.

a Compound 1 titration showing (blue) binding to IDH1R132H that is unaffected by the addition of 10 mM ICT (red). b Compound 1 titration showing (blue) binding to IDH1wt that is decreased by the addition of 10 mM ICT (red). c Percent change of compound 1 MS response when binding to IDH1R132H or IDH1wt in the presence of 10 mM ICT or aKG, when compared to compound 1 MS response for binding to apo-proteins. The above MS binding data confirms compound 1 binds both apo-IDH1wt and apo-IDH1 R132 but shows that compound 1 binding is affected by substrate binding only for IDH1wt and not mutant IDH1 R132.
Optimization of compound 1
We began a medicinal chemistry campaign to optimize compound 1 both in terms of potency as well as overall pharmaceutical properties. Upon examination of the available SAR from our initial screening as well as an analysis of our follow-up exploration with close similars of compound 1 in our collection, we soon appreciated that the dimethyl substitution, which led to the mixture of isomers, was unnecessary for affinity. Envisioning a simplified tricyclic core scaffold, we also sought to eliminate unnecessary hydrogen bond donors in our molecules and were pleased to find that compound 2, which replaced the urea with an amide moiety, retained the selectivity and activity for IDH1R132H as evident from the data in Table 2. With compound 2 in hand as a simplified mutant IDH1 inhibitor, we set out to optimize both potency and overall pharmaceutical properties such as solubility and pharmacokinetic profiles. In addition, we were hoping to furnish an additional tool molecule for NMR studies that contained fluorine atoms to allow for 19F detection upon binding (for detailed results, see the NMR section). Through extensive SAR studies, we arrived at potent compounds such as compound 3, which boasted a significantly improved biochemical and cellular potency. While the introduction of compounds like 3 resulted in a boost in potency, it came at the expense of balanced physicochemical properties and pharmacokinetic profiles. We profiled compound 3 quite extensively and have referred to it as MRK-A in our 2018 publication that reported the in vivo target validation studies we have conducted in an orthotopic patient-derived glioma model20.
To arrive at a more beneficial overall profile, we investigated orthogonal opportunities to gain biochemical potency while achieving a more balanced overall profile. We learned that the introduction of certain substituents in the 8-position of the tricyclic core resulted in potency gains and a reduction of the lipophilicity of the amide substituent. In addition, we could install certain hindered alcohols where their hydrogen-bond-donor ability was sufficiently limited to eliminate undesired properties such as p-glycoprotein recognition. Optimized compound 4 ultimately combined all salient features and provided an advanced tool compound with good overall properties, potency, and a trifluoromethyl group to aid 19F NMR explorations. Figure 3 shows the progression from screening hit 1 to tool compounds 2–4. Subsequent optimization & characterization of this series of compounds as a lead series with salient pharmaceutical properties has been reported elsewhere23. Table 2 summarizes the biochemical and cell activity data for compounds 1–4. Representative inhibition curves are shown in Supplementary Fig. 1. The synthesis of compounds 2–4 is described in Supplementary Note 2.

Compound 1, identified from high-throughput screening, was evolved into more potent analogs 2-3 and an NMR tool compound 4.
NMR binding and quenched functional assays
Direct IDH1 binding of weak high-throughput screening hits was confirmed using saturation transfer difference NMR spectroscopy (STD NMR). High-affinity compounds do not give an STD NMR signal; their binding was assessed through competition studies with low-affinity analogs, cofactors, and substrates. In one study (Fig. 4a, top) compound 1 affected IDH1R132H -NADP + interactions but did not compete directly with substrates or cofactors, consistent with an allosteric binding mechanism affecting the organization of the IDH1R132H active site. While compound 1 and close analogs showed potent IDH1R132H inhibition without affecting IDH1wt activity, ALIS binding studies confirmed that compound 1 binds to both IDH1wt and IDH1R132H, prompting further biochemical, biophysical, and structural studies.

a compound 1 binding to IDH1R132H affects NADP+ interactions using ligand-detected STD NMR. b Changes in NADPH concentration are measured to assess IDH1R132H inhibition by compound 1. c All substrates and products from IDH1R132H reduction and oxidation reactions can be monitored simultaneously using 1H NMR in “one pot” reactions. d Addition of compound 1 to the IDH1R132H oxidation reaction has little effect on ICT-to-aKG conversion (wild-type reaction) but inhibits aKG-to-2HG reduction. e Multicomponent IDH1wt/R132H reaction monitoring shows aKG pooling. f 19F NMR studies of compound 4 complexes with IDH1wt (black), IDH1R132H (red), IDH1wt/132H (blue) enzymes and free in solution (green). g Differences in deuterium incorporation measured by 1H NMR suggest differences in processing aKG by IDH1wt and IDH1R132H enzymes.
1H NMR analysis of products and reactants from quenched IDH1 reactions was used as a label-free, orthogonal secondary assay to confirm functional IDH1R132H inhibition, which might be complicated in coupled functional assays. Reactions monitored by NMR take place in deep well-plates or microliter scale sample tubes, can be initiated by enzyme, cofactor, or substrate addition, and quenched by heating or with the addition of a potent inhibitor. Solutions are transferred to an NMR tube and analyzed using 1H NMR spectroscopy. Changes to products and reactants are quantitatively compared to control samples with no protein and samples with no ligand (DMSO only). Quenched functional NMR assays were used to confirm the activity of several IDH1R132H hits and test for IDH1wt selectivity. Results for compound 1 are shown in Fig. 4b, where 1 µM of IDH1R132H enzyme was incubated with 20 µM inhibitor, 200 µM NADPH cofactor and initiated by aKG (alpha keto glutarate) substrate addition. In this experiment, compound 1 was confirmed to be a potent IDH1R132H inhibitor with > 20-fold selectivity over the IDH1wt reaction.
NMR of multicomponent oxidation-reduction reactions
NMR spectroscopy allows for individual small molecules to be detected and identified, and therefore provides a unique way to directly observe real-time, simultaneous structural changes to small molecule products and reactants in evolving IDH1 reactions (Fig. 4c). IDH1R132H inhibition was tested in functional NMR assays where the time-dependent changes to the NMR signals of all reaction components (NADP +, NADPH, ICT, aKG, 2HG) were simultaneously detected and the concentration of each molecule was measured in “one-pot”. The results from several one-pot IDH1R132H reactions are shown in Fig. 4d. While ICT is the natural substrate for IDH1wt; it is also processed into aKG by IDH1R132H in the samples containing DMSO and compound 1. IDH1R132H is performing the wild-type reaction in the presence of R132H inhibitors. Samples with no inhibitor convert aKG into 2HG; 2HG production is blocked in the sample incubated with compound 1. IDH1R132H inhibitors act by preventing the reduction of aKG and have little effect on ICT-to-aKG conversion. One pot IDH1wt reactions yield similar results, rapidly producing aKG, then slowly producing 2HG. 2HG production from IDH1wt has been previously noted24. Rendina et al. ran IDH1wt reactions using NADPH as a cofactor and aKG as a substrate. The addition of CO2 yielded facile isocitrate production from aKG oxidation; when CO2 was withheld, slow conversion of aKG to 2HG was observed by LCMS. The reaction proceeds slowly, producing 20 nM 2HG/min. We monitored the IDH1wt reaction (aKG → 2HG) by NMR for 60 h, which allowed for enough 2HG to accumulate for NMR detection. Earlier studies measured NADPH depletion in IDH1wt reactions with aKG for 10 min and saw no measurable activity7. From our experiments, the IDH1R132H enzyme, rather than being a gain of function mutation, appears to select for a conformation of the wild-type enzyme that favors 2HG production. NMR and biochemical data suggest that allosteric IDH1R132H inhibitors bind and stabilize a wild-type-like conformation that is unfavorable for the reduction reaction.
Heterodimer NMR studies
Due to the presence of both mutant and wild-type alleles, IDH1wt/R132H heterodimers are likely to be the predominant mutant IDH1 enzyme in the cell. NMR studies were carried out on IDH1wt/R132H to investigate compound binding and heterodimer reaction details. One concern was the potential accelerated usage of aKG produced in the wild-type subunit that would be consumed locally by the IDH1R132H subunit. One-pot reactions using the heterodimer enzyme (Fig. 4e) showed aKG pooling followed by consumption; local use7 was not dominant in the reactions that were studied. Direct ligand binding to IDH1wt/R132H was studied by using the intrinsic 19F NMR signal of compound 4 as a local environment/conformational sensor. Figure 4f shows 19F NMR data collected from compound 4 complexes with IDH1wt, IDH1R132H, and IDH1wt/R132H proteins. The presence of one well-defined 280 Hz peak centered at − 81.1 ppm for compound 4 binding to IDH1R132H contrasts sharply with the two peaks observed for compound 4 binding to IDH1wt; a major 160-Hz wide peak at − 80.8 PPM and a minor 320-Hz wide peak centered at − 81.1 PPM. More details are given in Supplementary Fig. 2 and Supplementary Note 3. These data are consistent with the fluorinated ligand sensing two IDH1wt environments which could represent two conformations and one IDH1R132H environment, consistent with a single conformation. Compound 4 interactions with IDH1wt and IDH1R132H are preserved in binding to IDH1wt/R132H; no additional environmental or conformational complexity is introduced in the heterodimeric enzyme. While 19F NMR is well-suited as a probe of binding site differences, protein structures or protein NMR binding studies are needed to confirm that the ligand 19F NMR signal differences are related to protein conformation differences.
Deuterium incorporation NMR studies
A detailed mechanistic understanding of classical enzymatic kinetic studies of IDH1 enzymes is beyond the scope of our work. Nonetheless, some reaction details were revealed by performing IDH1 reactions in D2O (Fig. 4g) where product deuteration occurs via transfer from solvent and protonation is cofactor-dependent. A series of proton 1H NMR spectra show time-dependent changes to NMR peaks as the substrate is converted to a product. Reactions that involve protonation due to rearrangements and/or transfer from a protonated cofactor can be directly detected; water protons will incorporate deuterium instead of hydrogen and will not be detected by 1H NMR. Typical experiments show decreases in 1H NMR signals due to direct deuterium replacement; changes to the splitting patterns of nearby J-coupled protons can also occur. From deuterium incorporation differences when compared with IDH1wt, we propose that the IDH1R132H mechanism could involve the formation of an enol from aKG, followed by non-stereo selective protonation (Fig. 4g, right). These observations are consistent with transition state disruption that results in substrate/product release followed by non-specific deuteration from the solvent.
X-Ray Crystal structures of compound 1 and AG120
Multiple IDH1R132H inhibitors of different classes have been reported in recent years, most of them bind to an allosteric pocket floored by β-sheet β4, β5, and β12, surrounded by the loop connecting β4-β5, helix α9, and the beginning portion of β13, and covered by helix α10 (Regulatory Element, Segment 2) on the top (Fig. 5a). Here we report the crystal structures of compound 1 in complex with IDH1R132H homodimers at 2.2 Å (PDB: 8T7N), Fig. 5. One molecule of compound 1 binds to one protomer of IDH1R132H homodimer. The overall structure of compound 1:IDH1R132H complex is in an open/inactive state25. Compound 1 is a T-shaped molecule with a tricyclic core (dimethyl-5,11-dihydro-6 H-benzopyrido-1,4-diazepine) connected with a t-butylphenyl moiety at the center of the molecule. The core of compound 1 wedges in a parallel fashion into a groove formed by side chain walls of β strands (β4 and β5), and the interaction is stabilized by two pairs of hydrogen bonds between the nitrogen atoms of the core and the backbone amide NH and carbonyl group of Ile128. The tricyclic core is further sandwiched by long hydrophobic side chains from both sides of the groove, including Met291, Arg109, Ile113, Ile128, and Ile130. The pyridine ring of the tricyclic core inserts into a hydrophobic sub-pocket underneath the loop connecting β4 and β5. The limited size of this sub-pocket and the bidentate hydrogen bond interactions force the tricyclic core to align in a way only with the dimethyl-6H-benzo ring pointing toward Arg109 and His132. Only the 7,8-dimethyl isomer of compound 1 is found in the electron density map. The 9,10-dimethyl isomer, in contrast, would cause steric clashes with the β sheet. The t-butylphenyl sidechain of compound 1 sits perpendicularly to the tricyclic core and reaches out toward helix α9, engaging in mostly hydrophobic interactions with Leu120, Trp124, Met259, Val255, and Trp267. The highly dynamic helix α10 or the Seg2 is not resolved from the electron density map (neither is Seg1), probably due to lack of stable interaction with the compound and the intrinsic dynamic nature of the segment.

a The allosteric inhibitor binding pocket of compound-1:IDH1R132H. b The hydrophobic interactions of compound 1. c The bidentate interaction of compound 1 with IDH1R132H.
Even though compound 1 has a low inhibitory potency but a reasonable binding affinity against IDH1wt, we managed to determine the structure of compound 1 in complex with IDH1wt homodimer at 3.4 Å through co-crystallization at a high concentration of compound 1 (PDB: 8T7D). The binding mode of compound 1 in the IDH1wt structure is in essence, identical to the one in IDH1R132H (Supplementary Fig. 3). Both structures confirm that neither His132 nor Arg132 is directly involved in a binding interaction with compound 1. The X-ray crystal structure of IDH1R132H in complex with compound 4 was also solved (PDB: 9B81) and an overlay with the compound 1 structure confirmed that two molecules bind nearly identically to the same pocket (Supplementary Fig. 4).
To further the understanding of the structural basis of the IDH1R132H inhibitory mechanism by small molecules, we also determined the structure of IDH1R132H in complex with AG-120 (Ivosidenib) at 2.05 Å (PDB: 8T7O). The structure shows that AG-120 also binds at the allosteric pocket that is adjacent to the compound 1 binding site. However, there are two significant differences in the AG-120 binding mode compared with other allosteric inhibitors such as compound 1. One molecule of AG-120 binds to one dimer of IDH1R132H (Fig. 6a). AG-120 sits between the dimer interface, engaging in extensive hydrophobic interactions with Ile130, Trp124, Met259, Val255, and Trp267 of the protomer B. The regulatory element (Seg2) from protomer A forms a short helix (α10), and packs against AG-120 with hydrogen bonding and hydrophobic interaction. Seg2 of protomer A plays a pivotal role in bridging hydrophobic interactions between one AG-120 molecule and sidechains from helix α9 of two IDH1 protomers. AG-120 seems to form a hydrogen bond with the side chain of Ser280 of chain A and is also within hydrogen bond distance of the side chain of Ser277 (chain A) (Fig. 6b). The electron density around the ligand AG-120 is well resolved, except for the nitrile moiety (Fig. 6c). The cyanide pyridine end of AG-120 is intriguingly situated next to the side chain of Cys269 and Asn271 of chain B (Fig. 6b, d), leading to the possibility that AG-120 might covalently modify Cys269 through its nitrile moiety, though we don’t have enough experimental evidence besides x-ray crystallography (Fig. 6d). The other end (the difluorocyclobutyl moiety) of AG-120 reaches out the helix α9 of protomer A (Met259), breaks the dimer symmetry of IDH1 and might prevent the 2nd copy of AG-120 binding to chain A, as observed in the binding mode of compound 1 in IDH1R132H in which two copies of compound 1 bind to the protein. Comparisons of the binding mode of AG-120 with that of compound 1 are shown in Fig. 6e. The two copies of compound 1 exhibit essentially identical binding modes in chain A and chain B separately, while the AG-120 molecule spreads out and contacts both chain A and chain B.

a AG-120 sits between the IDH1R132H dimer interface, engaging in extensive hydrophobic interactions. b AG-120 forms a hydrogen bond with the side chain of Ser280 of chain A and is also within the hydrogen bond distance of the side chain of Ser277 (chain A). c The surface representation of AG-120 binding pocket. d The cyanide pyridine end of AG-120 is situated next to the side chain of Cys269 of chain B, leading to the possibility that AG-120 might covalently modify Cys269 through its nitrile moiety (shown in yellow). e Comparisons of AG-120:IDH1R132H and compound-1:IDH1R132H crystal structures.
Discussion
MRK-A belongs to a class of mutant IDH1 inhibitors, which we derived from high throughput screening (HTS). We and others in the field found that a wide range of chemotypes had the ability to bind to an allosteric pocket in IDH1R132H and inhibit the mutant enzyme selectively over the wild type, which was unanticipated. To build our understanding of how such molecules appeared to selectively inhibit only the mutant but not the wild-type reaction in biochemical and cellular assay settings, we embarked on a biophysical and structural exploration that included binding studies via our Automated Ligand Identification System (ALIS) platform, NMR evaluation, biochemical studies, and x-ray crystallography.
We found early on that compounds from the MRK-A series can bind to both the wild-type as well as the mutant enzyme, but only inhibit the neomorphic enzymatic activity of the mutant. Binding to the wild-type and mutant enzyme was confirmed by ALIS, SPR, and NMR as well as by x-ray crystallography. In addition, in all crystal structures we obtained with this series, compounds are always bound to the wild-type and the mutant enzyme in the exact same binding mode. While several compounds in this series, such as MRK-A, possess functional groups that project towards the mutated residue, no direct interaction was apparent that could explain the selectivity observed. Furthermore, compounds that lacked such substitution patterns or bound at a significant distance from the mutant residue still appeared to be selective inhibitors. Faced with such unanticipated findings, we sought to better understand how these compounds bind to IDH1 and under which conditions. We established, with competition experiments, how substrates can compete off compounds such as compound 1 from the allosteric pocket of the wild type, but not the mutant enzyme. Therefore, while compounds could bind in the absence of substrates, under physiologically relevant conditions, the wild-type enzyme regains catalytic activity by competitively displacing the allosteric ligand, whereas the mutant cannot. In the mutant context, the allosteric ligand can bind in the presence of substrates and further stabilize the protein.
To better understand the unexpected behavior of these mutant-selective inhibitors, we conducted comprehensive NMR and biochemical/biophysical studies with both the wild type, mutant, and importantly also with the dimer consisting of one wild type and one mutant copy, a scenario that more closely resembles a cellular setting where wild type and mutant enzymes co-exist. We confirmed how mutant-selective inhibitors affect the interconversion of the substrate to the product over time, which has allowed us to obtain detailed information not only on how the homodimeric mutant and wildtype enzyme behave but importantly also on how the heterodimeric units of one wildtype and one mutant have the ability to perform the wild-type and mutant reaction, respectively. Our NMR studies explain how the heterodimeric IDH1 dimer first oxidatively processes ICT to form 2-OG which then serves as the substrate for the mutant counterpart to ultimately produce 2HG. We were able to show under such conditions that our mutant-specific inhibitors were able to effectively suppress the mutant reaction, but still allowed for the processing of ICT to 2-OG. This data is consistent with our enzymology studies, which similarly confirmed our inhibitors, such as compound 1 or MRK-A only inhibit the mutant ‘reductive’ reaction of the heterodimeric enzyme, whereas the ‘oxidative’ wild-type reaction was not affected.
Lastly, we studied the binding of our compounds to the allosteric binding site by x-ray crystallography, both in the wild type and the mutant enzyme, and further explored by NMR spectroscopy. 19F NMR characterized environmental differences, suggesting conformational changes to wild-type and mutant IDH1 enzymes. Data from these orthogonal techniques is consistent with the wild-type enzyme sampling multiple conformations while the R132H mutation selects a conformation that is favorable for compound 2 binding. Conformational information comes indirectly from a comparison of compound 4 CF3 signals bound to wt, R132H, and heterodimer proteins. We obtained high-quality x-ray crystallographic data for several structural classes we had identified through our medicinal chemistry campaign, which bound in different pockets of the large allosteric binding site of the IDH1 protein. We compared the binding of our compounds from the MRK-A series to the binding of AG120, also known as Ivosidenib (Tibsovo®) for which we determine the x-ray crystal structure, which was previously unknown. It has long been speculated that AG-120 directly binds to Cys269, however, to date only indirect evidence is available. While in our crystallographic analysis, we observe a shortened distance between AG-120 and Cys269, which is consistent with a bond that has formed between the sulfur of Cys269 and the heterocyclic nitrile in AG-120, we do not have biochemical or cellular data to support the relevance of this observation. Interestingly, while compounds represented by 1–4 bind quite distal to the AG-120 binding site, they similarly confer high mutant selectivity.
Taking our biochemical, -physical, and -structural studies together led us to derive a proposed mode of action for the mutant IDH1 enzyme that is consistent with all our data and the emerging reports from others in the field. While inhibitors of the mutant enzyme can bind both wild-type and mutant enzymes, the highly dynamic nature of the wild-type in the presence of substrates does not allow for the formation of the closed inactive conformation, substrates rather displace the ligand from the wild-type enzyme. Binding to the mutant enzyme in contrast leads a stable closed protein conformation that can no longer bind substrates and is thus not catalytically competent.
Methods
Protein production
Genes encoding IDH1(1-414) and IDH1(1-414 with R132H mutation) were cloned into pET41a vector and expressed in E. coli BL21(DE3) cells as C-terminal His-tagged proteins. Each protein was purified by Ni-NTA column followed by gel-filtration chromatography (Superdex S200 column). Heterodimer protein was produced using 6His-IDH1(1-414) and IDH1(1-414)R132H-FLAG plasmids that were cloned into pET41a vector and expressed in E. coli BL21(DE3) cells. Cells were incubated at 37 °C until reaching an OD600 = 0.6 ~ 0.8, transferred to 16 °C, induced with 0.5 mM IPTG, and incubated overnight. After pelleting, cells were suspended in a 50 mM Tris-HCl buffer, pH 7.5, 500 mM NaCl, 10% glycerol, 5 mM βME, and EDTA-free protease inhibitor cocktail tablets, then lysed using a microfluidizer, passing through twice to ensure complete cell lysis, then centrifuged at 28,000 × g for 45 min at 4 °C. The protein was loaded onto a Ni-NTA column, washed with wash buffer (50 mM Tris-HCl, pH 7.5, 500 mM NaCl, 10% glycerol, 5 mM βME, and 10 mM imidazole), and eluted with elution buffer (50 mM Tris-HCl, pH 8.3, 150 mM NaCl, 10% glycerol, 5 mM βME, and 500 mM imidazole. Fractions were pooled, bound to an anti-FLAG M2 column, washed with 50 mM Tris-HCl, pH 7.5, 500 mM NaCl, 10% glycerol, 5 mM βME, and eluted with FLAG elution buffer (50 mM Tris-HCl, pH 8.3, 150 mM NaCl, 10% glycerol, 5 mM βME and 100 μg/mL FLAG peptide). Fractions were pooled and run on a Superdex 75 26/60 SEC column using the 50 mM HEPES, pH 7.5, 200 mM NaCl, 10% glycerol, 10 mM MgCl2, and 2 mM DTT storage buffer.
Crystallization
IDH1wt (10 mg/ml) was co-crystallized with compound 1 (at 1:10 molar ratio) by hanging-drop vapor diffusion method using a reservoir containing 0.1 M Sodium Cacodylate, pH6.5, 0.2 M NaCl, 2 M Ammonium Sulfate. IDH1R132H (18 mg/ml) was co-crystallized with compound 1 or AG-120 (at 1:10 molar ratio) by hanging-drop vapor diffusion method using a reservoir containing 25% PEG3350, 0.45 M Di-Ammonium Tartrate. Crystals were cryoprotected with 25% (v/v) sterile glycerol before being snap-frozen in liquid nitrogen.
X-ray data collection, processing & structure determination
X-ray diffraction data were collected at beamline 17-ID of the Advanced Photon Source at Industrial Macromolecular Crystallography Association (IMCA), Argonne National Laboratory at a wavelength of 1.0 Å. All data were integrated using either HKL200026 or XDS27 and merged and scaled using either HKL200026, autoPROC28, and Aimless in the CCP429 suite of programs.
The structures for IDH1wt with compound 1, IDH1R132H with compounds 1 and 4, and the IDH1R132H:AG-120 complex were determined by molecular replacement with the program Phaser30 using the previously reported structures of IDH1 (PDB: 3MAP) as the search models respectively. The molecular models were built manually using Coot31 and completed using iterative rounds of refinement and rebuilding. The structures were refined using REFMAC32 as implemented in CCP429 and Buster33(GlobalPhasing Ltd.). The final models all have favorable R/R free values, Molprobity34 scores, and good geometry and stereochemistry. The final structures have been deposited with the RCSB protein data bank. Structure determination statistics are provided in Supplementary Table 1. Figures were prepared using the program PyMOL (Schrödinger LLC, version 1.8).
Thermal shift assay
The thermal denaturation of both IDH1wt and IDH1R132H enzymes were evaluated in their apo-form, with and without compound 1 and in combination with and without substrate: isocitrate, NADP +, NADPH, and alpha-ketoglutarate. In a buffer consisting of 50 mM Tris, pH 7.5, 250 mM sodium chloride, and 5 mM magnesium chloride, a 5 µL mixture containing 4 µM IDH1wt or 2 µM IDH1R132H, DMSO or 20 µM compound 1 and 200 µM 1,8-ANS (1-anilinonaphthalene-8-sulfonic acid) dye was dispensed into a 384-well plate. All substrates were tested at 1 mM. To the top of each well, 3 µL of mineral oil was added. The samples were subjected to a continuous increase of 1 °C/min from 35 °C to 80 °C on the ThermoFluor® 384ELS System (Johnson & Johnson). The fluorescence intensity was plotted vs temperature to calculate the melting temperature for each sample. Data represents the average of 4 replicates ± standard deviation.
NMR spectroscopy
STD and 1D NMR spectra were collected on a Bruker DRX 500 MHz spectrometer equipped with a 5 mm TCI CryoProbe. 19 F NMR spectra were collected on a Bruker Avance III 500 MHz spectrometer using a 5 mm QCI CryoProbe. All datasets were acquired at a sample temperature of 303 K and processed using Bruker TopSpin software, v2.1 (or higher), or MestreNova 14.2.
STD NMR
Ligand binding was detected by saturation transfer difference (STD) NMR spectroscopy. We used a 1D proton STD experiment (std19slsp) with 3-9-19 WATERGATE solvent suppression and shaped pulse saturation (50 ms Gaussian pulses, 3 sec) applied at on- and off-resonance saturation frequencies of − 120 Hz (− 0.234 PPM) and 20,000 Hz, respectively. Key experimental parameters include: 1280 scans, 8 K total data points, 8012 Hz (16 PPM) sweep width, 3 sec relaxation delay, 511 ms acquisition time, and a total experiment time (expt) of 2 hours. The time domain data was multiplied by a Lorentzian window function (EM, 1 Hz) prior to transformation. The effect of compound binding on NADP + (Fig. 4a) was assessed on a sample consisting of 2 µM IDH1R132H, 130 µM NADP +, and 10 mM MgCl2 in 50 mM phosphate pH 7.0 buffer, 50 mM NaCl.
19 F NMR
Direct IDH1 binding of fluorinated compounds (Fig. 4f) was detected by 19 F NMR, using a proton decoupled, 19 F detected experiment (zgfhigqn). Key experimental parameters include: 1280 scans, 32 K total data points, 37.5 kHz (80 PPM) sweep width, 2.8 s relaxation delay, 436.9 ms acquisition time, and a total experiment duration of 7 hrs. 16 min. The time domain data was multiplied by a Lorentzian window function (EM, 30 Hz) prior to transformation. Samples consisted of 10 µM compound added to 9.2 µM IDH1R132H in a 25 mM TRIS pH 7.1 buffer, 100 mM NaCl.
NMR Inhibitor studies
Quenched reaction monitoring to detect enzyme inhibition by screening hits (Fig. 4b) was accomplished using a conventional proton-detected experiment with presaturation solvent suppression (zgpr). Reactions were carried out in a 1.5 ml Eppendorf tube. 20 µM compound was added to a reaction mixture containing 2 µM IDH1R132H, 200 µM NADPH, 200 µM aKG in 50 mM phosphate pH 7.0 H2O/D2O buffer, 50 mM NaCl. The reaction was initiated by the addition of 5 mM MgCl2 and quenched after 100 min by heating. The contents were transferred to an NMR tube for analysis. Percent inhibition was calculated using NADPH integrals, relative to a sample with no enzyme. A weak aKG-competitive inhibitor of IDH1R132H, NOG (N-oxalylglycine), was used as a positive control.
NMR continuous “one pot” reaction monitoring
Unlike quenched experiments, evolving reactions take place in an NMR tube and are monitored directly in a series of 1D proton experiments. Continuous reaction monitoring was accomplished using a conventional proton-detected zgpr experiment. Key experimental parameters include: 40 scans, 32 K total data points, 6666 Hz (13.3298 PPM) sweep width, 2.0 s relaxation delay, 2.45 sec acquisition time, and a total experiment duration of 10 min. The time domain data was multiplied by a Lorentzian window function (EM, 1 Hz), transformed, and a baseline correction was applied using a 5th order polynomial function.
One pot IDH1R132H reactions (Fig. 4c, d) were carried out in the NMR tube, at 303 K. The zgpr experiment was used to monitor the 1H NMR spectrum for all molecules in the reaction mixture for 120 min at 10 min intervals. The reaction mixture includes: 2 µM IDH1R132H, 200 µM NADPH, 200 µM aKG in 50 mM phosphate pH 7.0 H2O/D2O buffer, and 50 mM NaCl. The reaction was initiated by the addition of 5 mM MgCl2 to the NMR sample tube, The tube was then placed in the magnet and shimmed, resulting in a dead time of ~ 6 min. Integrals for products and reactants are plotted versus time, using the starting reactant integrals as references for reactant/product concentrations.
One pot IDH1wt/R132H reactions (Fig. 4e) were carried out in the NMR tube, at 303 K. Reaction progression was monitored for 150 min. at 10-minute intervals by 1H NMR. The reaction mixture includes: 100 nM IDH1wt/R132H, 200 µM NADPH, 3 mM aKG in 50 mM phosphate pH 7.0 H2O/D2O buffer, and 50 mM NaCl. The reaction was initiated by the addition of 5 mM MgCl2 to the NMR sample tube. Sample loading, acquisition and data treatment were the same as was used for the IDH1R132H experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Structural data has been deposited with the Protein Data Bank. Accession codes are 8T7N for IDH1R132H-compound 1, 8T7D for IDH1wt-compound 1, 9B81 for IDH1R132H-compound 4 and 8T7O for IDH1R132H-AG120 complexes. Data collection and refinement statistics are included in Supplementary Table 1. Representative dose-response curves for compounds 1–4 are in Supplementary Fig. 1. SPR binding data and protocols are in Supplementary Note 1. Synthesis of compounds 2–4 are in Supplementary Note 2. NMR experimental details are in Supplementary Note 3 and Supplementary Fig. 2. Protocols for biochemical and cell assays are in Supplementary Note 4. Ligand electron densities are shown in Supplementary Fig. 3. Source data are provided in this paper.
References
Balss, J. et al. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 116, 597–602 (2008).
Cairns, R. A. & Mak, T. W. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov. 3, 730–741 (2013).
Parsons, D. W. et al. An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812 (2008).
Yan, H. et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773 (2009).
Schnittger, S. et al. IDH1 mutations are detected in 6.6% of 1414 AML patients and are associated with intermediate risk karyotype and unfavorable prognosis in adults younger than 60 years and unmutated NPM1 status. Blood 116, 5486–5496 (2010).
Cimino, P. J., Kung, Y., Warrick, J. I., Chang, S. H. & Keene, C. D. Mutational status of IDH1 in uveal melanoma. Exp. Mol. Pathol. 100, 476–481 (2016).
Dang, L. et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744 (2009).
Xu, W. et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30 (2011).
Cooper, L. A. et al. The proneural molecular signature is enriched in oligodendrogliomas and predicts improved survival among diffuse gliomas. PLoS ONE 5, e12548 (2010).
Noushmehr, H. et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17, 510–522 (2010).
Popovici-Muller, J. et al. Discovery of AG-120 (Ivosidenib): A first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med. Chem. Lett. 9, 300–305 (2018).
Dhillon, S. Ivosidenib: first global approval. Drugs 78, 1509–1516 (2018).
Konteatis, Z. et al. Vorasidenib (AG-881): A first-in-class, brain-penetrant dual inhibitor of mutant IDH1 and 2 for treatment of glioma. ACS Med. Chem. Lett. 11, 101–107 (2020).
Chaturvedi, A. et al. Pan-mutant-IDH1 inhibitor BAY1436032 is highly effective against human IDH1 mutant acute myeloid leukemia in vivo. Leukemia 31, 2020–2028 (2017).
Okoye-Okafor, U. C. et al. New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat. Chem. Biol. 11, 878–886 (2015).
Cho, Y. S. et al. Discovery and evaluation of clinical candidate IDH305, a brain penetrant mutant IDH1 inhibitor. ACS Med. Chem. Lett. 8, 1116–1121 (2017).
Caravella, J. A. et al. Structure-based design and identification of FT-2102 (Olutasidenib), a potent mutant-selective IDH1 inhibitor. J. Med. Chem. 63, 1612–1623 (2020).
Lin, J. et al. Discovery and optimization of quinolinone derivatives as potent, selective, and orally bioavailable mutant isocitrate dehydrogenase 1 (mIDH1) inhibitors. J. Med. Chem. 62, 6575–6596 (2019).
Rohde, J. M. et al. Discovery and optimization of 2H-1lambda(2)-Pyridin-2-one inhibitors of mutant isocitrate dehydrogenase 1 for the treatment of cancer. J. Med. Chem. 64, 4913–4946 (2021).
Kopinja, J. et al. A brain penetrant mutant IDH1 inhibitor provides in vivo survival benefit. Sci. Rep. 7, 13853 (2017).
Zhang, R. & Monsma, F. Fluorescence-based thermal shift assays. Curr. Opin. Drug Discov. Devel. 13, 389–402 (2010).
Annis, D. A., Nickbarg, E., Yang, X., Ziebell, M. R. & Whitehurst, C. E. Affinity selection-mass spectrometry screening techniques for small molecule drug discovery. Curr. Opin. Chem. Biol. 11, 518–526 (2007).
Huang, C. et al. Diminishing GSH-adduct formation of tricyclic diazepine-based mutant IDH1 inhibitors. ACS Med. Chem. Lett. 13, 734–741 (2022).
Rendina, A. R. et al. Mutant IDH1 enhances the production of 2-hydroxyglutarate due to its kinetic mechanism. Biochemistry 52, 4563–4577 (2013).
Yang, B., Zhong, C., Peng, Y., Lai, Z. & Ding, J. Molecular mechanisms of “off-on switch” of activities of human IDH1 by tumor-associated mutation R132H. Cell Res. 20, 1188–1200 (2010).
Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997).
Kabsch, W. Xds. Acta Crystallogr D Biol. Crystallogr. 66, 125–132 (2010).
Vonrhein, C. et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr D Biol. Crystallogr. 67, 293–302 (2011).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol. Crystallogr. 67, 235–242 (2011).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol. Crystallogr. 60, 2126–2132 (2004).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol. Crystallogr. 67, 355–367 (2011).
Bricogne, G. et al. BUSTER version 2.11.6. (Cambridge, United Kingdom: Global Phasing Ltd., 2016).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol. Crystallogr. 66, 12–21 (2010).
Acknowledgements
The authors wish to acknowledge the assistance with experiments (Brian Lacey, Peter Spacciapoli, Anthony Donofrio, Xiaohua Huang, Gopal Parthasarathy, Pravien Abeywickrema) and discussions of the results (Astrid Kral, John Lampe, Elliott Nickbarg, Daniel Klein, Christine Andrews) described in the publication.
Author information
Authors and Affiliations
Contributions
J.L., M.M., R.M., S.S., M.L., and C.F. were involved in experimental design. J.L., M.M., R.M., M.L., and C.F. contributed to data collection and/or interpretation. J.L., M.M., and C.F. contributed to writing the paper.
Corresponding author
Ethics declarations
Competing interests
J.L., M.M., R.M., S.S., M.L., and C.F. are current or former employees of Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA and may own stock or stock options in Merck & Co., Inc., Rahway, NJ, USA.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
McCoy, M.A., Lu, J., Richard Miller, F. et al. Biostructural, biochemical and biophysical studies of mutant IDH1. Nat Commun 15, 7877 (2024). https://doi.org/10.1038/s41467-024-51692-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-51692-0
