
Abstract
The remarkable capacity of bacteria to adapt in response to selective pressures drives antimicrobial resistance. Pseudomonas aeruginosa illustrates this point, establishing chronic infections during which it evolves to survive antimicrobials and evade host defenses. Many adaptive changes occur on the P. aeruginosa cell surface but methods to identify these are limited. Here we combine phage display with high-throughput DNA sequencing to create a high throughput, multiplexed technology for surveying bacterial cell surfaces, Phage-seq. By applying phage display panning to hundreds of bacterial genotypes and analyzing the dynamics of the phage display selection process, we capture important biological information about cell surfaces. This approach also yields camelid single-domain antibodies that recognize key P. aeruginosa virulence factors on live cells. These antibodies have numerous potential applications in diagnostics and therapeutics. We propose that Phage-seq establishes a powerful paradigm for studying the bacterial cell surface by identifying and profiling many surface features in parallel.
Similar content being viewed by others

Introduction
Clinical care relies on effective antimicrobial therapy. This is threatened by the remarkable capacity of bacteria to develop multi-drug resistance (MDR). Among MDR organisms, Pseudomonas aeruginosa presents particular challenges. Patients with structural lung damage (e.g. cystic fibrosis, bronchiectasis, chronic obstructive pulmonary disease) frequently develop chronic P. aeruginosa infections that are difficult to eradicate. Many selective pressures act on naïve environmentally-acquired bacteria during years of chronic infection: host metabolites, the immune system, antibiotics. In response, P. aeruginosa acquires many mutations1,2, evolving towards common phenotypes that increase in-host fitness3,4. Often, this involves the modification or change in expression of a cell-surface structure that mediates within-host virulence, surface attachment, or biofilm formation1,4,5,6,7,8,9.
In this study, we explore phage display as a tool for interrogating bacterial surface antigens10,11. Our phage library displays VHH recognition domains (“Nanobodies”) derived from camelid heavy chain-only immunoglobulins; VHHs have comparable antigen specificity to full-length immunoglobulins but are smaller, more stable, and more soluble12. Inspired by recent work13,14,15, we combine phage display with high-throughput sequencing (HTS) for greater insight into the dynamics of selection, an approach not—to the best of our knowledge—previously reported in bacteria.
Phage-seq is a high-throughput, highly-multiplexed technology for unbiased profiling and quantification of bacterial surface antigens. Phage-seq produces both a dataset—describing the bacterial surface-ome of a given strain or population—as well as VHH reagents useful for further study. Importantly, Phage-seq does not require antigens to be known in advance and decouples profiling from antigen identification. This technique is well-suited for studying bacteria when mutations are frequent and readily observed, but their phenotypic consequences are unpredictable. Our construction of a phage-displayed VHH library against P. aeruginosa antigens enables the identification of VHHs against multiple P. aeruginosa antigens via both conventional biopanning and Phage-seq. Datasets generated by Phage-seq capture biologically-important information about the bacterial cell surface. We anticipate the methods, dataset, and reagents generated herein will be useful for other applications that require profiling of the P. aeruginosa cell surface, including studies of virulence, antibiotic resistance, and longitudinal bacterial adaptation in chronic infection. These reagents can also guide the de novo choice of therapies such as lytic phage or anti-pseudomonal biologics. Finally, we anticipate that Phage-seq can be used to study micro-organisms beyond P. aeruginosa.
Results
Phage display panning identifies VHHs specific to purified P. aeruginosa proteins
A VHH phage display library was constructed from an alpaca (Vicugna pacos) immunized with a mixture of soluble and membrane proteins from several P. aeruginosa laboratory strains cultured under various conditions. We estimated the library’s diversity at ~80,000 clones by HTS (Supplementary Fig. 1).
This phage display library was panned against purified P. aeruginosa flagella and Type IV Pili (T4P) (Fig. 1a) and yielded numerous VHH clones recognizing these antigens—either as purified proteins (Fig. 1b) or on intact P. aeruginosa cells fixed to microplates with methanol (Fig. 1c). These VHHs exhibited similar specificity when expressed as soluble recombinant VHHs (“rVHHs,” Supplementary Fig. 4a, b). However, when tested for binding to intact, live bacterial cells—both by live-cell dot blot (Supplementary Fig. 5) and flow cytometry (FCM; Supplementary Fig. 6)—these rVHHs showed weak or absent staining. Cell fixation restored rVHH staining as analyzed by FCM (Fig. 1e). This suggested that panning against surface-immobilized antigens had selected VHHs specific for non-native conformations of these antigens.

a Schematic of experiment. Flagellin and pilin were purified from P. aeruginosa using standard methods. In each case, a mock purification was carried out on isogenic mutants lacking the antigen of interest; this “antigen–” sample was handled and diluted similarly to the “antigen+” sample. Wells of a 96-well plate were coated with purified antigen and phage display panning was performed for 4 rounds. Phage were first applied to the antigen– (counter-selection) wells, then transferred to the antigen+ (selection) wells. b Clonal ELISAs. After the final round of selection, eluted phage from the antigen+ wells was plated and 15 clones were picked and amplified; phage-displayed VHHs were tested for binding to the antigen of interest (orange circles), or the mock, antigen− prep (blue circles) by ELISA. “Ab YU573” and “Ab YU586” are positive controls using polyclonal rabbit antiserum raised against the indicated antigen. “No 1°” is a negative control where buffer replaces the primary antibody. n = 3 independent replicates per sample. c Cell-based ELISA using phage-displayed VHHs: P. aeruginosa cells were fixed to a plate with methanol and dried, then phage-displayed VHHs were tested for binding by ELISA as in panel (b). n = 3 independent replicates per sample. d Bacterial dot blot using rVHHs: VHHs were cloned into a mammalian expression vector, expressed, and purified. P. aeruginosa cells were serially diluted and spotted onto dry nitrocellulose membranes. Total protein was estimated by Ponceau S staining (right column). Blots were then blocked, probed with rVHH or polyclonal antisera, appropriate secondary antibody, and developed with enhanced chemiluminescence (left column). e Flow cytometry of P. aeruginosa cells fixed with methanol, then stained with rVHHs or polyclonal antiserum and PE-conjugated secondary antibody. Orange traces show antigen-positive cells and blue traces show antigen-negative cells. Source data for panels (b, c) are provided as a Source Data file. Raw data for panel (e) is available at https://doi.org/10.5281/zenodo.12826667. BSA bovine serum albumin.
Phage-seq reveals different dynamics of phage display selection against bacterial cells
Based on these results, we performed phage display panning against live P. aeruginosa cells in suspension, with the goal of selecting VHHs that recognized surface antigens in their native conformations (Fig. 2a). Selection cells expressed flagella or T4P, while isogenic mutants deleted for the antigen of interest (i.e. flagellin or pilin) served as counter-selection cells.

a Schematic of experiment. For each antigen, a pair of P. aeruginosa strains were chosen to serve as “selection” (e.g. wild type) and “counter-selection” (e.g. isogenic mutant lacking the antigen of interest) strains. Phage display panning was performed by applying the phage library to the counter-selection cells, collecting un-bound phage (supernatant), applying those phage to the selection cells, washing, and eluting bound phage with acid and base. Eluted phage were amplified in E. coli and the process repeated for 4 rounds. After each round, titers were measured and VHH populations were determined by Illumina sequencing. b, c Rank-abundance (Whittaker) plots of VHH pools at each round of selection. b Selections against purified proteins (as displayed in Supplementary Fig. 1). c Selections against intact bacterial cells. Traces in the top-right corner represent samples with higher diversity, while less diverse samples appear as traces in the bottom-left. Progression of traces down and to the left over the course of rounds of selection suggests populations of VHHs are becoming less diverse. d, e Bar plots showing relative abundance of VHH CDR3 clonotypes, for each round of selection. Each colored bar represents one of the 40 most abundant CDR3 clonotypes in that selection. Each CDR3 clonotype was identified by MD5 hash of its sequence, and the first six hexadecimal digits of this identifier (e.g. e06677) are used to identify and color that clonotype in figure marks. Colors and labels are comparable, with the same color identifying the same CDR3 clonotype in different panels and figures. Each plot shows a different selection. d Selections against purified proteins. e Selections against intact bacterial cells with corresponding antigens. For panels (b–e), source data are provided as a Source Data file.
Samples generated by both cell-based and solid-phase panning were analyzed by HTS of the complementarity-determining regions (CDRs) 1–3. We identified between ~2500 to >40,000 VHH clones per sample, with more clones encountered at greater sequencing depth. Given the dominant contribution of CDR3 to antibody binding specificity16,17, we pooled reads with the same CDR3 sequence (“CDR3 clonotype”) in our analysis. The number of CDR3 clonotypes per sample was ~100–2500 (Fig. 2b).
Panning should enrich a library for VHH clones that bind the antigen of interest while depleting all other clones. Simultaneously, VHH fusions expressed and amplified efficiently in the E. coli host will be advantaged over others. Both types of selective pressure would result in a smaller number of VHH clones at relatively higher abundance. Whittaker plots (rank abundance curves) showed that richness (number of clones) and evenness (relative rarity of each clone) of CDR3 clonotypes per sample both declined over rounds of panning (Fig. 2b, c). For solid-phase selections, a large difference was observed after the second round of selection (Fig. 2b); for cell-based selections, this change was more gradual (Fig. 2c). Richness and evenness were higher at the end of the cell-based versus the solid-phase campaign. Barplots revealed that, in solid-phase selections, a small number of clones dominated the library after 1–2 rounds of selection and persisted during subsequent rounds (Fig. 2d). By contrast, in cell-based selections the number of clones gradually decreased over four rounds of selection (Fig. 2e). A few CDR3 clonotypes were highly abundant in the final rounds of both solid phase and cell-based selections targeting the same antigen (e.g. clones e06677, 97861c, 0cf8fb, and 97861c). Usually, however, the most abundant clones differed between cell-based vs. solid-phase selections against the same antigen.
Overall, these data suggested that cell-based panning, like solid-phase panning, narrowed the population of VHHs, albeit more slowly. This could reflect the greater diversity of antigens available for binding and/or the less stringent conditions used for selection in the cell-based campaign compared to the solid-phase campaign.
High-throughput Phage-seq biopanning
We next panned on 180 pairs of P. aeruginosa genotypes, testing the performance of Phage-seq on a diversity of antigens and in a 96-well format (Fig. 3a). Most selections were carried out using isogenic wild-type/mutant pairs differing for a single antigen or structure; growth conditions were selected to promote expression of the antigen(s) of interest (Fig. 3b; Supplementary Fig. 7; Supplementary Data 3). In this experiment, the relationship between the selection pair and antigen was many-to-many, rather than one-to-one. Multiple antigens were predicted to differ within some pairs of selection and counter-selection cells (e.g. all proteins comprising a Type 3 secretion apparatus). In other instances, antigens differed across more than one selection (e.g. mutants in structural and regulatory elements both leading to the absence of the same antigen). Expectations of antigen presence vs. absence are summarized for each pair of selection and counter-selection cells (Fig. 3b, Supplementary Data 3). Selections were considered positive for an antigen expressed by selection cells but not by counter-selection cells, and negative if the antigen was either missing from both cell types or more abundant on the counter-selection cells. Selections were labeled “unknown” if no a priori prediction could be made about the antigen status of a pair.

a Whole cell panning (Fig. 2) was adapted to a 96-well format and performed on ~180 distinct pairs of bacterial genotypes, summarized in panel c. Eleven selections from this experiment were continued in triplicate for three further rounds. b Antigen matrix for a subset of conditions. Each genotype pair enriched for one or more antigens. If the antigen was present on selection cells but absent from counter-selection cells, that selection condition was considered positive for the antigen (orange circles). If the antigen was absent from the selection cells, the selection condition was negative for that antigen (blue circles). The selections labeled in bold and bulleted were continued for three rounds of extended panning. Full set of conditions described in Supplementary Data 3, and full antigen matrix is shown in Supplementary Fig. 7. c Ordination showing selections at each round of high-throughout and extended panning. Abundances were normalized to library size using the scran package. PCA (via TSVD, first 100 components) was performed and the first three components are plotted. Each point represents the input phage for a single selection at a single round. Round 1 input corresponds to the raw library (Supplementary Fig. 1). d–g VHH population structure across rounds of panning for selections targeting the flagellar hook-basal body. d, f Relative abundance of VHH CDR3 clonotypes, as in Fig. 2d–e. e, g Rank-abundance (Whittaker) plots of VHH pools, as in Fig. 2b–c. d, e first four rounds of panning, for three replicate selections targeting the flagellar hook-basal body. Similar plots for all selections in the experiment are shown in Supplementary Figs. 10-16. f, g three rounds of extended panning against the same antigen. Each plot in the bottom row represents the extended panning for the same selection shown in the top row. Similar plots for all selections in the experiment are shown in Supplementary Figs. 19–22. h Distribution of VHH enrichment values, for each selection condition. Enrichment for a given VHH was defined as (relative abundance at final round) ÷ (relative abundance at initial round). Individual selections are displayed along the X axis and grouped according to the predominant antigen expected to differ between selection and counter-selection cells. Each point represents a distinct CDR3 which was significantly enriched in that selection. For panels (c–h), source data are provided as a Source Data file. CPA common polysaccharide antigen, EPS Exopolysaccharide, LPS lipopolysaccharide, OSA O-specific antigen, QS quorum sensing.
High-throughput sequencing of phage over four rounds of selection showed progressive changes from the input library to the final round of selection as visualized by principal components analysis (PCA; Fig. 3c, Supplementary Fig. 8). In contrast to the previous small-scale selections (Fig. 2), library composition did not change substantially throughout this selection campaign (Supplementary Figs. 9–17); in most selections, the 50–100 most abundant clones comprised <25% of the reads in the final round. Many selections showed no clear changes in the final abundance of the top 40 clones or in the richness and/or evenness of the samples (e.g. Fig. 3d–e).
Panning in 96 well format necessitated protocol modifications, e.g. reduced cell numbers and wash steps, and altered methods for cell/phage resuspension and separation. To test whether these affected selections, we extended our campaign for a subset of genotype pairs (Fig. 3b, bolded/bulleted). Over three additional rounds of selection we (1) increased wash stringency; (2) increased cell number; and (3) carried out 3 sequential counter-selection steps per round before selection on antigen-positive cells. In an additional set of selections, we tested whether a higher cell-to-phage ratio would lead to stronger convergence of phage populations by diluting input phage 100-fold before applying to counter-selection cells in rounds 6 and 7. Titers of phage eluted from selection cells increased after round 7 of these extended selection campaigns as compared to counter-selection eluate titers, with the greatest difference seen for those selections where the input phage was diluted 100-fold (Supplementary Fig. 18e).
HTS of these extended selections revealed marked changes in library diversity (Fig. 3f–g; Supplementary Figs. 19–22), suggesting that more stringent washing and counter-selection had successfully tightened the selection bottleneck. Most selections nonetheless remained relatively diverse, rather than converging on a small number of CDR3 clonotypes.
Two closely-related CDR3 clonotypes, ad6f8f and 2c7c51, became highly abundant in numerous samples, suggesting these clones were non-specific binders and/or very effectively replicated in the E. coli host. Samples where ad6f8f and 2c7c51 comprised > 80% of the reads were excluded from further analysis.
Selections with a 1/100x bottleneck showed more pronounced changes in library composition (Supplementary Figs. 20, 22), suggesting that higher cell-to-phage ratios favored convergence. In aggregate, these data indicated that high-throughput cell-based selections converged on VHH clones with affinity for our antigens of interest.
Phage-seq identifies VHHs specific to the P. aeruginosa cell surface
The goal of our cell-based campaign was to select VHHs specific for native-conformation antigens on live cells; we predicted that these VHHs could then be identified from our HTS data, expressed and experimentally tested. We searched for VHHs that were enriched (i.e. increased in abundance over rounds of selection), both because selections had not converged on a few high-abundance VHHs and because previous studies of peptide phage display showed no correlation between final round clone abundance and binding strength to the target antigen18.
We developed several metrics of VHH enrichment in HTS data to guide our choice of VHHs to resynthesize (Supplementary Figs. 23, 24). Ultimately, a composite of several metrics was used to shortlist CDR3 clonotypes enriched in multiple antigen-positive samples (Supplementary Methods), as illustrated for the MDR efflux porin OprM (Fig. 4a–d); a similar analysis was conducted for several antigens (Supplementary Figs. 25–29). Each candidate CDR3 was then manually inspected across all samples, to assess distribution of enrichment and final abundances across antigen-positive, antigen-negative, and antigen-unknown samples (Fig. 4e–f). We favored CDR3s for which enrichment and abundance were highest in the antigen-positive selections and disregarded those with very high enrichment in many samples (e.g. ad6f8f and 2c7c51). For each chosen CDR, a gene fragment encoding the consensus full-length VHH amino acid sequence was designed, synthesized and expressed as a fusion to human IgG1-Fc.

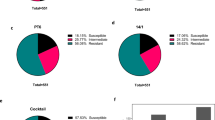
a–d Method of selecting VHHs for resynthesis. Plots show all VHHs identified in selections containing the antigen OprM; each colored point is a distinct CDR3 clonotype. Points within yellow regions represent VHHs likely to recognize the antigen of interest. Opaque points are VHHs chosen for resynthesis. a Binary enrichment probability. For a VHH significantly enriched in N antigen+ selections, indicates the probability of observing this VHH to be significantly enriched in N antigen– selections; see methods. b Percentile sum for antigen+ and antigen– selections; see methods. c Geometric mean of the enrichment for antigen+ and antigen– selections; see methods. d Geometric mean of the starting abundance and ending abundance within antigen+ selections. e, f Behavior of each VHH candidate was examined across all selections; this inset shows CDR3 #7a08ae, which was tested in Fig. 5b below. e Trace of CDR3 abundance at each round of selection; each trace represents a different selection. Traces are colored according to whether the selection was OprM+ (orange), OprM– (blue) or OprM status was unknown (gray). f Ending abundance vs. enrichment plot; each point is a distinct selection, colored using the same scheme as the left panel. g We selected 83 VHHs targeting 5 distinct antigens (the flagellar filament “FliC”, the flagellar hook-basal body “FlgEHKL”, and the efflux pump-associated outer membrane porins OprM, OprN, and OprJ) for resynthesis. Of these, 55 were successfully expressed as fusions to human IgG1-Fc and tested for staining of P. aeruginosa by flow cytometry. Of these, 28 were validated by flow cytometry to have the predicted specificity. For panels (a–f), source data are provided as a Source Data file.
Ultimately we resynthesized VHHs targeting five antigens: the flagellar filament (FliC); the flagellar hook-basal body (HBB, “FlgEHKL”); and three efflux-associated outer-membrane porins (OprM, OprN, and OprJ). Of 83 recombinant VHHs (rVHHs) cloned (12–20 rVHHs per antigen), 55 were successfully expressed (Fig. 4g). Expressed rVHHs were assayed by FCM using unfixed antigen-positive and antigen-negative cells as targets (Fig. 5a–d, Supplementary Fig. 30).

Flow cytometry of P. aeruginosa stained with selected rVHHs, anti-Human IgG1-PE secondary antibody (0.5 µg per well), and the nuclear stain SYTO9. Events were gated by FSC/SSC (Supplementary Fig. 30) and SYTO9+ events were interpreted as bacterial cells. Each rVHH was used to stain antigen+ cells (orange) and antigen– cells (blue). The rVHH+ gate was drawn for fluorescence above the 99th percentile of the antigen– cells; the percent of rVHH+ antigen+ cells is shown in the corner of each plot. The rightmost panel(s) shows the same scheme for cells stained with free IgG1-Fc. a rVHHs targeting the flagellar hook-basal body apparatus. PAK ΔflhA does not produce any part of the flagellar apparatus. PAK ΔfliC ΔfleN produces multiple flagellar hook-basal bodies but no flagellar filament. rVHH per well: D2, 0.073 µg; E1, 0.226 µg; E2, 0.271 µg; F1, 0.201 µg. Additional rVHHs shown in Supplementary Fig. 31. b–d rVHHs targeting efflux-associated outer membrane porins OprM (H), OprJ (I), or multiple efflux porins (J). PAO1 “Δefflux” lacks all Resistance Nodular Division (RND)-type multidrug efflux systems. The antigen-positive strains (“+mexAB +oprM”, etc.) are Δefflux but express the indicated efflux pump in trans from plasmid mpMQ72. All cells were induced overnight with 1% arabinose. b. rVHHs targeting OprM. rVHH per well: A6, 9.7 µg; H5, 9.2 µg; C6, 3.4 µg; IgG1-Fc, 8.8 µg. Additional rVHHs shown in Supplementary Fig. 33. c rVHHs targeting OprJ. rVHH per well: D9, 24.0 µg; E9, 16.7 µg; F9, 5.2 µg; IgG1-Fc, 8.8 µg. Additional rVHHs shown in Supplementary Fig. 34. d rVHHs targeting multiple efflux-associated porins. rVHH per well: 10 µg; IgG1-Fc, 8.8 µg. Additional rVHHs shown in Supplementary Fig. 36. Raw data for panel (e) is available at https://doi.org/10.5281/zenodo.12826667.
Our method identified 8 rVHHs (of 11 expressed) recognizing the flagellar hook-basal bod (HBB) (Fig. 5a; Supplementary Fig. 31). Most stained PAK ΔfliC ΔfleN cells (which express several hook-basal bodies but no flagellar filaments) with an intensity ~1000-fold higher than aflagellate PAK ΔflhA cells. Clone 13c8b9 (E1) stained PAK ΔfleN cells (which express several entire flagella) more strongly than PAK ΔfliC ΔfleN cells; clone 99f9fc (E2) exhibited the opposite staining pattern (Supplementary Fig. 32a). These two rVHHs may bind different HBB epitopes or be sensitive to HBB conformations specific to the presence of the flagellar filament.
Twenty rVHHs recognized the various outer membrane porins (OprM, OprJ, and OprN) associated with Resistance Nodular Devision (RND)-type MDR efflux systems (Fig. 5b–d; Supplementary Figs. 33–35)7. Three rVHHs were enriched in multiple selections against different efflux porins and found to recognize both OprM and OprJ (Fig. 5b; Supplementary Fig. 36). Several rVHHs were tested against P. aeruginosa clinical isolates and stained those with MDR phenotypes (Supplementary Fig. 3819).
Altogether, this data demonstrated that phage-displayed panning coupled with analysis of Phage-seq HTS data could yield numerous VHHs that selectively bound P. aeruginosa antigens in their native conformations on live cells, including clinical isolates.
Mapping the P. aeruginosa cell surface with Phage-seq
Since our extended panning successfully yielded selective VHHs, we re-examined our large high-throughput panning dataset to test whether Phage-seq had captured information about the structure of bacterial cell surfaces during earlier rounds of selection.
Principal components analysis (PCA) of final round abundances showed that panned samples were well-separated from the input library along the first axis, then separated according to growth conditions along the second and third axis (Fig. 6a). Among selections carried out on surface-grown cells, those targeting exopolysaccharides, the related transporter CdrAB, and small colony variants20 formed a distinct manifold, whereas others (e.g. targeting T4P and the type VI secretion system) were contiguous with selections from exponential-phase liquid cultures (Fig. 6a). A rough clustering was apparent, with efflux pump selections appearing close together, while selections for flagella and pili also clustered together (Fig. 6b; Supplementary Fig. 39). Notably, selections on clinical isolates did not separate from other selections against P. aeruginosa laboratory strains. Canonical correspondence analysis (CCA) on the enrichment matrix revealed that the presence or absence of biological features (e.g. flagella, OprM, type III secretion system) could explain variance in VHH enrichments across samples (Fig. 6c).

a, b First 3 principal components (truncated single value decomposition [TSVD]) of VHH relative abundance matrix at final round of selection. Each point represents a distinct selection or biological replicate, or a sample of the non-panned input library. a Selections (or library) colored according to growth condition. b Selections (or library) colored according to the category of antigen targeted by the selection. c Canonical correspondence analysis (CCA) of selections and a subset of the antigen matrix. CCA projects samples (selections) and features (antigens) into the same lower-dimensional space. Antigens are depicted as vectors from the origin, and selections are shown as points. Each plot depicts the same 3D space with a different antigen vector and cells positive for that antigen highlighted in separate panels. Left, flagellar hook-basal body; center, OprM; right, type III secretion system (T3SS). d–g For each antigen shown in Fig. 3c with > 10 example selections, a classifier was trained to predict whether that antigen was present or absent in the relevant selection. Repeated stratified 5-fold cross-validation was performed and area under the receiver-operator characteristic curve (AUROC) was calculated for each antigen. As a control, classifiers were fit against the same selections with the labels randomly permuted. Trained classifiers were used to predict whether each antigen was present or absent for all samples in the selection. \(p\)-values, Wilcoxon rank-sum test (one-sided; H1: AUROC > 0.5). Source data are provided as a Source Data file. EPS Exopolysaccharide, LPS lipopolysaccharide, QS quorum sensing.
To further evaluate our dataset’s ability to characterize an isolate, we trained a series of classifiers to distinguish antigen-positive versus antigen-negative selections (Fig. 6d–g). Our goal was to test whether Phage-seq data could recapitulate aspects of well-characterized isolates in our selection campaign, not to predict antigens of unknown isolates. A separate ensemble of classifiers was trained for each antigen where >5 antigen-positive and antigen-negative selections had been carried out. Data were repeatedly divided into 5 folds, with four of five folds used for training and the fifth used to evaluate classifier performance. The performance of all classifiers was measured to estimate their average, best- and worst-case performance. As a control, we conducted a similar procedure on the same set of selections after randomly permuting the antigen-positive/antigen-negative labels. This shuffled-labels control showed that the models were not excessively flexible (i.e. they did not overfit to distinguish arbitrary subsets of the data). Our classifiers performed acceptably, with mean area under the receiver-operator characteristic curves (AUROC) ranging from 0.7–0.86. By contrast, AUROCs for the shuffled controls were ≤0.52, indicating performance similar to or worse than chance.
Overall, this data suggests that VHH populations measured by Phage-seq reflect biological differences between selection conditions.
Discussion
In this study, phage display selection and high-throughput sequencing were combined to create a tool for probing the surface of living bacterial cells. Our approach identified VHHs that recognize multiple virulence-associated P. aeruginosa antigens, including T4P, flagellin, the flagellar hook-basal body, and efflux-associated outer membrane porins. The high-dimensional VHH abundance datasets generated by this approach also captured biologically relevant information about the bacterial cell surface.
Our study built on two lines of prior work: combining phage display with HTS and using bacterial cells as bait for panning. Phage display studies have used HTS to estimate library diversity13,21, identify novel antigens15, perform affinity maturation22, or quantify many antigens in parallel14; however, these techniques have not been applied to bacteria. Prior panning against bacterial cells sought to identify antibodies recognizing any antigen on a particular strain or species23,24,25. By contrast, our study employed selection/counter-selection between bacterial mutants, allowing us to link genotype to phenotype (antigen expression). This strategy aligned well with bacterial genetics, where knockout and overexpression mutants in many genes are readily available, and the antigen products of these genes could be investigated with Phage-seq. We demonstrated the promise of this method by discovering VHHs against several membrane-associated protein complexes which stain live, intact cells. Our approach is not limited to genes directly encoding surface proteins; it could also be used discover epitopes that differ between pairs of pleiotropic regulators or between clinical isolates before and after exposure to antibiotics or a host immune system.
Phage display is usually performed on purified, surface-immobilized antigens. Using this approach, we identified VHHs specific to immobilized antigen but blind to the same antigen on live cells. Surface-immobilized protein antigens are likely denatured, and this is known to affect antibodies selected by phage display26, particularly VHHs which are highly conformation-sensitive27. Our solid phase-selected VHHs recognized methanol-fixed, antigen-positive cells, suggesting that methanol somewhat mimicked the effects of surface-binding on these antigens. Others have demonstrated that immunization with non-denatured antigens is critical to generate antibodies against epitopes distributed thoughout an antigen surface28. We used relatively crude bacterial extracts as immunogens, prepared without denaturation and adjuvanted only with alum (rather than complete Freund’s); this may have favored presentation of native-conformation antigens to the alpaca, ultimately resulting in VHHs capable of recognizing intact antigens on the bacterial cell surface.
Our study identified key variables affecting the performance of phage display panning on intact bacterial cells, which converged gradually on larger sets of candidate VHHs than those obtained from solid-phase selections. This was likely due, in part, to the relative complexity of antigens presented by a cell surface compared to a purified protein. More critically, the ratio of bait antigen to phage was lower in cell-based selections. Most of a cell’s surface presented irrelevant biomass—in contrast to selections against purified protein, where nearly 100% of well binding capacity was occupied by target antigen. This ratio of antigen to phage was even lower in high-throughput experiments (Fig. 3) than in initial cell-based pannings (Fig. 2) and likely contributed to the slow convergence of selections in the high-throughput format. Experimental manipulation of this ratio in the extended high-throughput pannings, either by doubling the number of cells per well or by diluting the input phage 1/100x, resulted in more dramatic changes in VHH population composition than in the prior four rounds of selection.
HTS datasets generated by Phage-seq posed unique problems. VHHs do not map to an existing ontology, unlike 16S amplicons to taxa or RNA-seq reads to genes. Yet statistical challenges relevant to microbiome or RNA-seq data also existed for Phage-seq data: sparsity, zero inflation29, high dimensionality30, compositionality31, and sensitivity of diversity measures to sequencing depth32. VHH sequences could be mapped to multiple feature spaces—e.g. each CDR3 clonotype is reflected in multiple VHH amino acid sequences, each in turn encoded by multiple nucleic acid sequences. Conversion between these feature spaces was necessary for our work: e.g., resynthesizing a VHH required identifying all VHH sequences encoding a given CDR3 clonotype and calculating a consensus. Interactive querying for CDR3 or VHH sequences similar to a given clone was also useful.
Ultimately, we built a custom pipeline and Python package to correct sequencing errors33, map reads to VHH amino acid sequences, identify CDR and framework regions, and calculate VHH abundance and enrichment. Considering the statistical pathologies above (Supplementary Methods), we settled on a naïve approach of calculating relative abundances (dividing counts by library size per sample), dividing these relative abundances to calculate enrichments, and ranking VHHs according to their enrichments in several samples. Though more sophisticated statistical methods will surely improve future work, our simple methods were vindicated by successful identification of VHHs with specific binding.
The majority of rVHHs that we identified and successfully expressed demonstrated specificity by FCM. Many of the VHHs, particularly those against RND-type MDR efflux system components, hold promise as tools for diagnostics, therapeutics, and research. Several rVHHs also stained MDR clinical isolates by FCM. Monoclonal antibodies against OprM, OprJ, and OprN could be used to rapidly recognize MDR P. aeruginosa isolates. These antibodies could also be used to target antimicrobials to MDR organisms, or even as direct cytotoxic agents.
In its current form, Phage-seq is an indirect tool for bacterial surface -omics. However, our modeling suggests that even early high-throughput pannings captured biologically meaningful information about the cell surface, which could be leveraged to generate useful reagents and insights about the cell surface. Many thousands of VHHs against numerous bacterial genotypes were enriched in our high-throughput panning experiments; we thus expect this library and technique will be a rich source of additional reagents in further work. This would enable an approach more akin to PhaNGS14, CITE-seq34, or REAP-seq35, in which antibodies labeled with nucleic acid markers enable highly multiplexed extracellular antigen profiling for unknown P. aeruginosa isolates. Second, as we build a larger library of Phage-seq profiles for distinct selection pairs, more sophisticated modeling approaches may be able to assign putative binding partners to a wider range of VHHs in the population. This could lead to new hypotheses about the phenotypic consequences of mutation during host adaptation and elucidate the selective pressures at work during infection.
Methods
Ethical statement
Alpaca immunizations were supervised by Dr. Charles Shoemaker at Tufts Cummings Veterinary School Campus. All animal experiments were conducted in accordance with protocols approved by both Yale and Tufts universities’ Institutional Animal Care and Use Committees (Yale IACUC protocol no. 10584 and Tufts IACUC protocol no. G2011-08).
Statistics and reproducibility
No statistical method was used to predetermine sample size. In general, ELISA experiments used a minimum of three biological replicates and flow cytometry results shown are representative of at least two independent experiments. Recombinant VHH transfection and purification were repeated at least once. The extended high-throughput phage display panning experiment was conducted using three biological replicates per selection condition; other panning experiments used only one replicate and were not repeated. As discussed in the main text, we excluded samples where two closely-related CDR3 clonotypes, ad6f8f and 2c7c51, comprised >80% of the reads; the round 8 input sample for each of the following selections was excluded from the indicated figure and was not used to choose rVHHs for resynthesis: 1.A2, 1.C2, 1.B6, 1.C5 (Supplementary Fig. 25, OprM); 1.A4,1.C4 (Supplementary Fig. 27, OprN); 1.A3,1.B3 (Supplementary Fig. 26, OprJ). The underlying reads are included in the SRA submission and in the processed data packages deposited in Zenodo. The experiments were not randomized. The Investigators were not blinded to allocation during experiments and outcome assessment.
Bacterial strains and growth conditions
Bacterial strains discussed in the main text are shown in Table 1. Strains used in each phage display selection are shown in Supplementary Data 1–4. The following abbreviations are used in the Supplementary Data tables:
-
Strain MB5890 “∆efflux” has genotype ∆(mexAB-oprM) ∆(mexCD-oprJ) ∆mexJKL ∆(mexHI-opmD) ∆opmH36
-
Strain PA0397 “∆efflux” has genotype ∆(mexAB-oprM) nfxB ∆(mexCD-oprJ) ∆mexJKL ∆mexXY ∆opmH362 ∆(mexEF-oprN)37
-
Strains marked “::(ZTP)” carry the ZTP riboswitch reporter described by38 and have the genotype ∆exsD attB::PexoT::ZTPlacZ
-
If not otherwise noted in Table 1 or in the tables below, the strains mentioned in the tables below are first described in this study.
Luria Broth (LB) agar and LB broth were used for growth of P. aeruginosa unless noted otherwise. Bacteria were streaked from glycerol stocks onto LB agar plates, then liquid cultures inoculated from well-isolated single colonies and grown overnight. Bacterial growth was at 37 °C, with liquid cultures shaking at 220 rpm.
Unless noted, washes of P. aeruginosa cells were performed in PBS + MC (PBS pH 7.4 plus 0.9 mM calcium, 0.9 M magnesium). Pelleted cells were resuspended by “racking” (gently dragging a tube across a plastic tube rack) or by shaking at 1200 rpm for 30 s (for cells in V-bottom 96-well plates).
Alpaca immunization
Four P. aeruginosa laboratory strains (PA14 [serotype O10], PAO1 [O5], PAK [O6] and PA103 [O11]) were grown to exponential phase in liquid LB and overnight on LB agar. Cells were collected by centrifugation or scraping plates and resuspended in 20 mM HEPES, 150 mM NaCl, 10 mM KCl, 1 mM phenylmethylsulfonyl fluoride (PMSF). Cells were lysed by french press ( >14,000 psi) and the lysate clarified by centrifugation (5000 × g for 10 min). Membrane proteins were separated from soluble proteins by centrifugation (20,000 × g for 30 min).
Membrane proteins were enriched by centrifugation through sucrose density gradients; membrane proteins were collected from the interface of a 60%/25% sucrose step gradient. Additionally, PA14 and PAO1 were grown as static biofilms in Petri dishes in both LB and M9 minimal media at 30 °C for 36 h, harvested by scraping, and lysed as above. The soluble proteins, enriched membrane proteins, and biofilm extracts were pooled to generate a mixed antigen preparation and stored in single-use aliquots at −80 °C.
A single adult male alpaca (Vicugna pacos) was immunized with the P. aeruginosa antigen (2 mg per injection) adjuvanted with alum. Four subcutaneous immunizations were administered at 3–4 week intervals. Serum was collected prior to each immunization and four days after the final immunization. Peripheral blood lymphocytes were harvested from the final bleed, and RNA was prepared from one aliquot of fresh peripheral blood lymphocytes (PBLs) using TRI Reagent LS (Molecular Research Center, Inc.) according to the manufacturer’s protocol, and as previously described39. RNA was column-purified using a RNeasy Mini Kit following the manufacturer’s protocol (Qiagen). The yield (87 µg/mL) was calculated in a spectrophotometer at 260 and 280 nm, and RNA was stored at −80 °C.
ELISA wells were coated with the P. aeruginosa antigen preparation used for immunization (10 µg mL−1), then incubated with 2–5 fold dilutions of alpaca serum (beginning at 1:100), followed by secondary incubation with anti-llama IgG-HRP conjugate. Protein preparations from the different P. aeruginosa strains were prepared as described above, then separated by SDS-PAGE (1 µg per lane), blotted to PVDF, and incubated with alpaca pre-immunization and post-immunization serum (diluted 1:5000).
Construction of a VHH phage display library
Plasmids and primers used in this study are listed in Table 2 and Table 3, respectively. Three cDNA synthesis reactions were performed in parallel using Superscript III First-Strand Kit (Invitrogen), each with different primers: random hexamers, oligo dT, and alpaca gene-specific primers (Al.CH2, AlCH2.2) described previously39. cDNA was amplified by PCR, using AlVHH-F1 as the forward primer and either AlVHH-shR1 or AlVHH-lhR1 as the reverse primer; these primers recognize the alpaca VHH cDNA at conserved sites within the FR1 domain and the short or long hinge domains respectively. Replicate PCRs were pooled and amplicons purified by silica column cleanup (Qiagen), then restriction-digested with NotI/AscI and the digest gel-purified.
The phagemid vector pD was derived from pCANTAB (GE Healthcare) via pJSC. The cloning site of this vector is in-frame with the C-terminus of phage coat protein P3; downstream from the cloning site is an E-epitope tag, separated by an amber stop codon. The vector was prepared, digested as above, and gel purified. Vector and insert were ligated using T4 ligase (NEB), cleaned up (Geneclean Turbo column, MPBio), and transformed into electrocompetent TG1 (Agilent). An aliquot of the transformation reaction was serially-diluted to estimate a titer of 4 × 106 transformants; the remaining transfomants were plated on a large area of selective media, scraped, combined, aliquotted, and stored at −80 °C.
A total of 96 clones from the transformed library were randomly selected; plasmid preparation and Sanger sequencing were performed by a vendor (Beckman-Coulter). Chromatograms were manually trimmed. A multiple sequence alignment was constructed using MUSCLE and the consensus sequence was used as a reference sequence in downstream analysis.
To prepare infectious phage from the library, 1 mL of frozen TG1 cells containing library phagemid transformants were diluted into 25 mL of 2YT media with carbenicillin. This culture was grown for two hours, M13KO7 added to 1 × 1010 pfu mL−1 and grown for 1 h further. This culture was added to 1 L of 2YT plus carbenicillin and kanamycin and grown overnight. Cells were removed by centrifugation; ~5 × 109 cells were collected and phagemids isolated for sequencing; the supernatant was PEG/NaCl-precipitated as described below. Aliquots of this library were considered “passage 1.” One aliquot from passage 1 was amplified as described below to ~1 L to form passage 2. All subsequent experiments were performed using aliquots of passage 2.
Isolation of flagella and pili
Flagella were isolated following40. 10 mL of exponential-phase cells were harvested by centrifugation, 3000 × g for 10 min at 4 °C and resuspended in 100 mL of flagella buffer (50 mM sodium phosphate [pH 7], 10 mM magnesium chloride). Flagella were sheared in a Waring blender for 30 s, then the pulse repeated. Loss of swimming was confirmed by microscopy. Cells were removed by centrifugation at 20,000 × g for 30 min at 4 °C. Flagella were collected from this supernatant by ultracentrifugation at ~105,000 × g for 1 h. Pellets were resuspended in a total volume of 3 mL Tris-NaCl (150 mM NaCl, 50 mM Tris pH 7.6).
Pili were prepared from solid media and removed by vortexing41; cells were grown overnight on 2–3 150 cm2 plates, then collected using a cell scraper and resuspended in 10 mL total volume of TPM buffer (10 mM Tris HCl [pH 7.5], 1 mM KPO4 [pH 7], 8 mM MgSO4). Cells were vortexed for 3 min, then cells removed by centrifugation at 20,000 × g for 5 min at 4 °C. Supernatants were transferred to microfuge tubes, magnesium chloride added to a final concentration of 100 mM, and incubated on ice overnight. Pili were harvested by centrifugation >20,000 × g for 15 min at 4 °C. The supernatant was resuspended in a total volume of 5 mL Tris-NaCl. Large insoluble aggregates were removed by centrifugation 13,000 × g for 5 min at 4 °C.
Protein preparations were kept on ice and quantified by SDS-PAGE and/or bicinchoninic acid (BCA) assay the same or next day, then diluted to single-use aliquots, snap frozen, and stored at −20 °C until ready for use.
Phage display panning
Terminology
“Selection” refers to enrichment of a distinct population of phage-displayed VHHs via multiple rounds of panning against a particular pair of antigen conditions (e.g. counter-selection and selection bacterial cells, antigen-negative and antigen-positive protein-coated wells, etc.). An observation of that population at one point in time is a “sample.” The high-titer phage population, prior to round N of panning, is the “round N input phage”; phage eluted from the wells/cells after that round are the “round N output phage.” The round N output phage are expanded in E. coli to produce the round (N + 1) input phage, and so on. Unless otherwise noted, sequencing data is obtained for high-titer “input” phage populations, as sequencing library preparation is most consistent from these templates. Therefore in all figures showing sequencing data, round N phage refers to the round N input phage, i.e. the result of performing (N-1) rounds of panning.
Common phage display methods
OmniMAX 2 T1R E. coli were used as the host strain for all phage display experiments. OmniMAX E. coli were grown in 2YT media supplemented with tetracycline 10 µg mL−1 to maintain the F episome. Kanamycin 25 µg mL−1 was added when necessary to maintain the M13KO7 helper phage. Carbenicillin 100 µg mL−1 was added when necessary to maintain the phagemid. E. coli was grown overnight in 2YT + tetracycline, then subcultured 1/50–1/100x in the same and grown to exponential phase. Before use, E. coli were routinely tested for maintenance of the F episome and for pre-infection with phagemid or helper phage by confirming an exponential phase culture could grow in 2YT plus tetracycline and could not grow in 2YT plus carbenicillin or kanamycin.
For each selection, one aliquot of library at 1 × 1013 pfu mL−1 was used; library was PEG-precipitated after thawing from −80 °C. Sufficient quantity of library for a given experiment was thawed and pooled; four volumes of ice-cold PBT (PBS plus 0.9 mM calcium, 0.9 M magnesium, 0.5% w/v bovine serum albumin [BSA], 0.05% v/v Tween-20, filter sterilized) and one volume ice cold sterile PEG-NaCl solution (20% w/v PEG-8000, 2.5 M NaCl) were added to one volume library. Phage were PEG-precipitated on ice for 20 min, harvested by centrifugation at 20,000 × g for 20 min; the phage pellet was resuspended in PBT and stored on ice. Immediately before use, phage solutions were spun as before for 5 min to remove insoluble material.
Preparation of bait, washes, and elution are described below. After elution, phage were amplified by infecting E. coli. One volume of neutralized phage eluate was added to nine volumes (solid-phase and small-scale cell-based panning) or two volumes (high-throughput panning) of exponential-phase E. coli (OD 0.3–0.6). Infected cultures were grown for 45 min; helper phage M13KO7 (NEB) was added to 1010 pfu mL−1 final concentration and cultures were grown for 1 h. Finally, one volume culture was added to five volumes (solid-phase and small-scale) or one volume (high-throughput) 2YT, supplemented with carbenicillin and kanamycin to the proper final concentration. Cultures were grown for at least 16 h.
After amplification, cells were removed by centrifugation at 3000 × g for 10 min. The supernatant was transferred to a new tube and one volume ice cold sterile PEG-NaCl solution was added to five volumes culture. Phage were PEG-precipitated on ice for 20 min (solid-phase and small-scale) or 1 h (high-throughput), then harvested at 20,000 × g for 20 min (solid-phase and small-scale) or 5500 × g for 1 h (high-throughput). Phage pellets were resuspended and spun before use as described above.
Phage titers were measured by preparing serial 10-fold dilutions of phage particles in PT buffer (PBS plus calcium, magnesium, and 0.05% v/v Tween-20, filter sterilized), then transferring one volume of phage to 10 volumes of exponential phase E. coli. Infected cells were incubated shaking for 15–30 min, then 10 µL of each dilution was spotted onto LB plus carbenicillin and LB plus kanamycin plates. Colonies were enumerated using Ilastik 1.4.0 in object density mode42. The greatest dilution with more than 10 visible colonies was used to calculate the titer.
To assay individual clones, the phagemid-infected E. coli culture was plated on LB agar plus carbenicillin. Individual colonies were picked to 1 mL of 2YT plus carbenicillin plus M13KO7 at 1 × 1010 pfu mL−1 and grown shaking overnight.
Solid-phase panning
Solid phase panning protocol was adapted from10. Briefly, for each antigen, four wells of a high-binding ELISA plate (Nunc MaxiSorp, Invitrogen) were coated with 0.5 µg of purified protein (or mock purification) diluted in sodium carbonate buffer (50 mM, pH 9.6) overnight at 4 °C. Wells were decanted and blocked for 90 min in blocking buffer (PBS plus 0.5% w/v BSA). 100 µL of input library at 1013 pfu ml−1 was added to each counter-selection well and incubated for 1 h at room temperature nutating. Phage were transferred from counter-selection to selection wells and incubated for 2 h. Phage were decanted and wells washed 10 times with PT buffer. Phage were eluted by adding 100 µL per well HCl (100 mM), incubating 5 min, then neutralizing with 1/8 volume (50 µL) of Tris 1 M, pH 11. Eluted phage were rescued and amplified as described above.
Cell-based panning
P. aeruginosa cells were grown overnight, then subcultured 1/50x into 5 mL of LB and grown to mid-exponential phase (OD 0.4–0.6); subculture, washing, and blocking of selection cells was staggered one hour later than counter-selection cells. Cells were harvested by centrifugation at 3000 × g for 5 min, then washed twice by resuspending in 1 mL PBS + MC and centrifuging at 2700 × g in a microcentrifuge. 1 × 107 cells were transferred to a 2 mL microcentrifuge tube, pelleted, resuspended in 2 mL blocking buffer, and incubated rocking for 1 h at RT. Counter-selection cells were collected by centrifugation, resuspended in 1 mL of re-precipitated, cleared library at 1 × 1013 pfu ml−1, and incubated for 1 h rocking at room temperature. Counter-selection cells were pelleted and selection cells resuspended in the supernatant; selection cells were incubated with the phage library for 2 h. Selection and counter-selection cells were pelleted, then washed four times in 1 mL PT buffer; cells were pelleted and supernatant decanted thoroughly. Phage were eluted by addition of 800 µl of 0.1 N HCl to cells and incubation for 5 min; cells were removed by centrifugation at 18,000 × g at 4 °C for 5 min; the supernatant was neutralized in 100 µL 1 M Tris-HCl, pH 11. Residual phage attached to cells were eluted by addition of 640 µL per well 0.1 M triethylamine (TEA) and incubation for 5 min. Cells were removed as before and supernatant neutralized in 260 µL per well of Tris pH 6.8. The neutralized supernatants from the acid elution and the base elution steps were combined to form the final eluate. 450 µL, approximately 1/4 of this volume, was expanded as above to generate the input library for subsequent rounds.
High-throughput cell-based panning
P. aeruginosa cells were arranged and stored in glycerol in 96-well format; 96-well microplates containing 150 µl per well of sterile LB agar were prepared. Two days prior to the experiment, 10 µL per well of sterile LB broth was added to each well and a multichannel pipette was used to inoculate this semi-solid master plate from the arrayed glycerol stocks. For bacteria panned after growth on solid media, a flame-sterilized loop was used to densely streak from this solid master plate 1/8th of a 100 mm petri dish containing LB agar; these plates were incubated overnight. For bacteria panned in liquid, a multichannel pipette was used to inoculate duplicate liquid cultures of 1.2 mL per well from the solid media microplate. These cultures were incubated overnight. For bacteria panned in exponential phase, the stationary phase cultures were diluted 1/50 in appropriate media and incubated for 3 h. Solid-media cultures were scraped into 1.2 mL per well of LB; solid-media and stationary-phase cultures were transferred to a single microplate along with exponential-phase cultures. Duplicate plates were combined and optical density was measured but not standardized. Subculture, wash, and blocking of the selection cells was staggered by 2 h after the counter-selection cells. Cells were harvested by centrifugation at 3000 × g for 10 min, washed twice with 1 mL per well PBT, and resuspended by shaking 750 rpm × 5 min in 100 µl per well of PBT. 250 µL of re-precipitated, cleared library at 1 × 1013 pfu ml−1 was added to counter-selection cells and incubated at RT shaking 750 rpm for 1 h. Counter-selection cells were removed by centrifugation 3000 × g for 15 min at RT; the supernatant was transferred to the selection cells and resuspended by gently pipetting up and down. Selection cells were incubated with phage for 1 h shaking, then 1 h nutating. Selection cells were pelleted, supernatant decanted, then selection and counter-selection cells were washed three times as above. Phage were eluted from selection and counter-selection cells by both acid and base as above, with the following modifications: add 200 µL per well 0.1 N HCl; shake 750 rpm for 10 min; add 25 µL 1 M Tris-HCl, pH 11 to neutralize; spin 3000 × g for 10 min; the supernatant was passed through a 0.22 µm PVDF filter (Millipore MSGVS2210). We found this filter was particularly effective at removing residual bacteria while allowing phage to pass through without clogging. The pellet was eluted again with base: add 200 µl per well of 0.1 M TEA; incubate shaking 750 rpm for 10 min; add 80 µL per well Tris pH 6.8 to neutralize; spin 3000 × g for 10 min; pass supernatant through a 0.22 µm filter. Filtered acid- and base-eluted eluates were combined, titered, and amplified.
Phage were expanded as described above; 200 µl per well of eluate phage was added to 400 µl per well of exponential-phase E. coli in 96-well format, incubated, M13KO7 was added to a final concentration of 1 × 1010 pfu ml−1, incubated, and 2YT plus kanamycin and carbenicillin were added to a final volume of 1.2 mL per well. This culture was grown overnight. Cells were removed by centrifugation 6000 × g for 10 min and the supernatant transferred to a clean microplate on ice. PEG-precipitation was performed in this microplate; the PEG-precipitated phage were resuspended by shaking at 750 rpm for 10 min, then stored on ice overnight until the next round of panning was to be conducted.
Extended cell-based panning
Prior to subsequent rounds of panning, the round 5 input phage from the high-throughput panning were expanded as follows: 200 µL of phage at ~1 × 1013 pfu ml−1 was added to a 4.5 mL culture of OmniMAX E. coli cells in 2YT plus tetracycline grown to OD 0.3–0.6; cultures were incubated shaking at 37 °C for 30 min, then M13KO7 added to 1 × 1010 pfu ml−1 and incubated for 45 min. The entire volume of culture was added to a final volume of 30 mL 2YT plus carbencillin and kanamycin and incubated overnight. Phage particles were harvested by PEG-precipitation as described earlier for the small-scale panning experiments.
Subsequent rounds of phage display panning were performed as above, except that three separate sets of counter-selection cells were prepared identically. Phage were incubated with the first set of counter-selection cells for 30 min, then cells removed and the supernatant transferred to the second set of counter-selection cells. This process was repeated once more for a total of three counter-selection phases. After applying phage to selection cells, the number of washes was increased from three (rounds 1–4) to five (round 5), seven (round 6), and twelve (round 7).
Library preparation for high-throughput sequencing
For initial high-throughput sequencing of the input library, phagemid DNA was purified, digested using NotI/AscI, and gel purified. The sequencing library was prepared by end-repair and blunt-end ligation of sequencing adapters using a commercial kit (Illumina Nextera). Paired-end 300 bp sequencing was performed on an Illumina MiSeq.
Subsequent HTS libraries were prepared by polymerase chain reaction (PCR). 2 µL of amplified phage particles at ~1 × 1013 pfu ml−1, or 10 µL of phage eluate were used as template for an initial PCR (Phusion, NEB) with primers CDR123-seq-F/CDR123-seq-R which added Illumina R1 and R2 primer binding sites. Silica column cleanup was performed and an additional PCR added combinatorial dual indexes. Yield and size of all products was verified by gel electrophoresis and individual reactions repeated as necessary. Amplicons were pooled using Just-a-Plate 96 PCR Normalization (Charm Biotech). The pooled library was column- and then gel purified. Library purity and concentration was verified by automated electrophoresis (e.g. Fragment Analyzer, Agilent) and qPCR. Paired-end 150 bp sequencing was performed in an Illumina NovaSeq S4 to a target depth of 100,000 reads per round per selection.
High-throughput sequencing data analysis
Primer sequences were removed with Cutadapt43, and reads lacking a primer sequence were discarded. Reads were deduplicated and denoised using DADA233, with each sample processed independently. Distinct read sequences were aligned to the reference sequence described above using Bowtie 244. For paired-end 150 nt sequencing experiments, the reads were not expected to overlap in all cases, as the full-length sequenced amplicon was greater than 300 nt; to ensure that a given read pair captured the full length of the variable portion of the VHH gene, the forward read was required to contain the entirety of CDR1 and CDR2, plus at least 3 nt, while the reverse read was required to contain the entirety of CDR3 plus at least 3 nt. Reads which did not meet this criteria were discarded. Overlapping read pairs were stitched together, with any disagreements being resolved in favor of the forward read. For read pairs which passed the filter but did not overlap, the gap between the two reads was filled with corresponding bases from the reference sequence. The merged read pairs were translated to amino acid sequences (amber stop codons UAG were translated as Gln). Amino acid sequences were grouped into zero edit-distance clusters (i.e. two or more reads with 100% identity and overlap beyond a threshold length were grouped together) using the linclust routine45 from the MMseqs2 package46. Amino acid sequences were then aligned to a reference sequence in order to identify the boundaries of CDR1–3. Reads were filtered according to minimum lengths for CDR1 (≥4 aa), CDR2 (≥6 aa), CDR3 (≥3 aa), and FR4 (≥2 aa) and the full-length amino acid sequences (≥69 aa). Two feature tables (contingency tables of sample vs. feature) were constructed in the biom format47, one where the columns (features) were distinct amino acid sequences (e.g. all nucleic acid sequences with the same translation were summed) and one where the columns were distinct CDR3 sequences. Feature tables from multiple sequencing runs, if applicable, were summed. Relative abundance was calculated by dividing counts by the sum for each sample (row). Enrichment was calculated for each feature for each selection by dividing the ending abundance by the starting abundance. The starting abundance was defined as either: the relative abundance of that feature in the input library or, if undefined, the relative abundance of that feature in the first round of selection where the feature was observed. The ending abundance was the relative abundance of that feature in the last round of selection.
Diversity analysis
The number of distinct full-length VHH amino acid sequences (encompassing the region beginning with CDR1 and ending after CDR3) in the input library was estimated using the breakaway method from the breakaway R package48.
VHH selection metrics
Let \({X}_{i,\, j,r}\) be the relative abundance of VHH \(j\) in selection \(i\) at round \(r\). Define \({E}_{i,j}={X}_{i,\, j,R}/{X}_{i,\, j,1}\) as the enrichment of VHH \(j\) in selection \(i\) during a selection campaign of \(R\) rounds.
We defined the enrichment probability, \(P\big({E}_{i,\, j} \, > \, e{{{\rm{|}}}}{X}_{i,\, j,0}=x\big)\), as the probability of observing an enrichment \(e\), given a starting abundance of \(x\), due only to experimental noise unrelated to the selective pressures of phage display panning. To estimate this quantity, we repeatedly re-sequenced two passages of the input library. This sample captures both variation due to library preparation and due to changes in phage proportion during expansion in E. coli, precipitation of the phage, etc. We obtained \(K=12\) samples of the library and calculated a table of enrichments and initial abundances: \({\hat{E}}_{i,j}={X}_{k1,j}/{X}_{k2,\, j}{{{\rm{;}}}}\, {\hat{X}}_{i,j,0}={X}_{k1,\, j},\, \forall k1,\,k2\in \left(1...K\right)\times \left(1...K\right),\, \, j\in \left(1...J\right),\, i\in \big(0...{K}^{2}\big)\). For all further calculations, we took the logarithm of both abundance and enrichment. We visually confirmed this joint distribution was unimodal. We then performed a Gaussian kernel density estimate on this table to estimate a joint probability mass function \(f\left(e,\, x\right)=P\big({\hat{E}}_{i,\, j}=e\cap {\hat{X}}_{i,\, j,0}=x \big)\). We evaluated this function on a 2D grid of abundances and enrichments, then summed over the first axis to calculate the marginal probability of abundance (e.g. \(P\big({\hat{X}}_{i,j,0}=x\big)={\sum }_{e}\, f\left(e,\, x\right)\)). We divided the discretized joint PMF by the marginal distribution of abundance to create a conditional PMF. We took the cumulative sum of these quantities over the first axis to calculate a conditional cumulative distribution function. Then we fitted a 2D spline to this surface in order to estimate the enrichment probabilities for unknown values.
To calculate the binary enrichment probability, \(P\left({N}_{k}^{+} < \, {N}_{k}^{-}\right)\), we used simulations. For a given antigen, assume there are \(k\) antigen-positive selections, and a particular VHH is significantly enriched in \({N}_{k}^{+}\) of those \(k\) selections. Among \(k\) random antigen-negative selections, let \({N}_{k}^{-}\) be the number of antigen-negative selections where the VHH is significantly enriched. \({N}_{k}^{-}\) is a random variable with a support of \(\left(0...k\right)\). We estimated this probability by repeatedly choosing \(k\) antigen-negative selections, counting \({\hat{N}}_{k}^{-}\) for those selections, then counting the fraction of these simulations where \({N}_{k}^{+} \, < \, {\hat{N}}_{k}^{-}\). We only performed a finite number of simulations, so if \({N}_{k}^{+} \, < \, {\hat{N}}_{k}^{-}\) in all simulations, we say \(P\left({N}_{k}^{+} \, < \, {N}_{k}^{-}\right)\, < \,1/q\), where \(q\) was the number of simulations.
To calculate the normalized rank: Let \({E}_{i,j}\) be the enrichment of enrichment of VHH \(j\) in selection \(i\); let \({E}_{i,\left(0\right)} \, < \, {E}_{i,\left(j\right)} \, < \, {E}_{i,\left(J\right)}\) be the ranks of VHHs in selection \(i\) where \(J\) VHHs were observed. The normalized rank of VHH \(j\) in selection \(i\) is the rank divided by the number of VHHs in a sample, i.e. \({\mbox{NR}}\left(i,\, j\right)={E}_{i,\left(j\right)}/J\). The normalized rank sum for VHH \(j\) in a group of selections \(S=\{{s}_{0},\, {s}_{1},\ldots,\, {s}_{N}\}\), is the sum of the normalized ranks, \({\mbox{NR}}\left(S,\, j\right)={\sum }_{i\in s}{{{\rm{NR}}}}\left(i,j\right)\).
Ordination
Relative abundances at the final round of selection were normalized for sequencing depth with the scran package49,50. truncated single value decomposition (TSVD) was performed using the scikit-learn package51 with the top 100 components. For CCA, ordination was performed on the log-transformed enrichment matrix using the scikit-bio package52.
Machine learning and antigen predictions
Classifiers were gradient-boosted trees trained using XGBoost via the scikit-learn package51. Models were evaluated with repeated stratified \(k\)-fold cross-validation, with \(k=5\), and \(n=15\) repeats, and scored by mean AUROC over all test folds. The effect of learning rate \(\eta\) was evaluated for several antigens and optimal values (the fastest learning rate that still achieved maximum mean AUROC) found to be ~0.01–0.05. For each antigen, models were trained using three datasets: enrichment matrix only, enrichment plus final round abundance, or enrichment plus abundance for all rounds; the dataset with highest performance was chosen per-antigen. Additional model hyperparameters (e.g. min_child_weight, subsample, colsample_bytree, gamma, reg_alpha) were chosen by Bayesian optimization using the BayesSearchCV class from the scikit-optimize package53. Using this final set of hyperparameters, models were trained and evaluated using the cross-validation procedure described above; receiver-operator characteristic curves (ROC) for each training fold, plus a mean over all folds, are shown in Fig. 6d–g. \(p\)-value for mean accuracy determined by one-sided Student’s \(t\)-test. \(p\)-value for mean AUROC >0.5 was determined by Mann-Whitney U test.
Expression of recombinant VHHs in bacteria
Phagemid dsDNA was purified from overnight cultures of phagemid-infected E. coli, digested with NotI/AscI, and the gel-purified insert was ligated with gel-purified, NotI/AscI-digested pJEG3, then transformed into DH5α. Clones were verified by Sanger sequencing, then transformed into BL21(DE3) for expression. BL21(DE3) cells were grown overnight in 2YT plus carbenicillin plus 2% w/v glucose, subcultured 1:20 in 100 mL of the same, then media removed and cells transferred to 200 mL 2YT plus carbenicillin and 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Cells were grown 19 h shaking at 27 °C. Cells were harvested at 17,650 × g in JA-10 rotor (10,000 rpm) and resuspended in a lysis buffer (50 mM sodium phosphate [pH 8.0], 300 mM NaCl, 10 mM imidazole, 1x cOmplete™, EDTA-free Protease Inhibitor Cocktail [Sigma], 1 mM PMSF). Cells were spheroplasted for 30 min at RT by addition of lysozyme to ~1 mg ml−1 and DNaseI to 5 µg ml−1, then lysed by three freeze-thaw cycles. Lysates were cleared by centrifugation 20,000 × g at 4 °C for 20 min. Immobilized metal affinity chromatography (IMAC) was performed; Ni-NTA resin (2.5–5 mg protein/1 mL slurry) were diluted 1/5x in sample and lysis buffer. Slurry was packed on a gravity filtration column and washed with eight bed volumes of wash buffer (50 mM sodium phosphate [pH 8.0], 300 mM NaCl, 20 mM imidazole) and 1/2 bed volume of 100 mM imidazole. Protein was eluted with 2 bed volumes of 250 mM imidazole, and buffer exchanged to PBS + MC using PD-10 desalting columns (Cytiva). Samples were flash-frozen and stored at −20 °C. Yield was measured by BCA assay and purity monitored by SDS-PAGE.
Expression and purification of recombinant VHHs in human cells
Nucleic acid sequences were synthesized as gene fragments (Twist Biosciences), reconstituted, and amplified by PCR. Unpurified amplicons were cloned using NEBuilder HiFi DNA Assembly system into the pCER243 mammalian expression vector, which had previously been linearized with XhoI and gel purified. pCER243 is modified from pD2610-v12 (ATUM Bio), adding an upstream H7 leader sequence and downstream an in-frame (GGGGS)3 linker and human IgG1 Fc with the N297A mutation54. Assembly products were transformed into DH5-alpha competent E. coli (NEB), plated on solid selective media, scraped, and grown overnight in liquid; plasmids were purified. For each VHH clone, presence of the insert was verified by NotI/XbaI digestion. rVHH-Fcs were expressed by transient transfection of the pCER243-rVHH plasmid using the Expi293F Transfection Kit (Gibco), following the manufacturer’s instructions. Briefly, Expi293F cells were grown in Expi293 media in shaking at 125 rpm in non-baffled, vent-cap polystyrene Erlenmeyer flasks at 37 °C, 8% CO2 to a density of 3–5 × 106 cells ml−1. Cells were seeded to 2 × 106 cells ml−1 and grown shaking overnight. In a 2 mL, V-bottom 96-well plate, 0.5 µg plasmid DNA was diluted in Opti-MEM media (Gibco) to 25 µL final volume. A master mix of 1.35 µl per well Expifectamine and 23.65 µl per well Opti-MEM media was prepared, then 25 µL per well of this mixture was added to each well and incubated at room temperature for 20 min to form DNA-Expifectamine complexes. Expi293F cells were diluted to 2 × 106 cells ml−1 and 425 µl cells were added dropwise to this mixture. Cells were grown shaking at 1200 rpm in conditions described above. After at least 20 h of growth, 2.5 µL of transfection enhancer 1 and 25 µL of transfection enhancer 2 were added to each well, and cells were returned to grow for 4 days further (5 days total).
rVHHs were purified from clarified supernatants using protein A magnetic beads. Cultures were clarified by centrifugation at 600 × g for 10 min at room temperature. 12.5 µL protein A magnetic beads (Lytic Solutions) were added to supernatants and supernatants shaken at 750 rpm for 2 h at 4 °C. Beads were harvested, washed three times in PBS, then rVHHs eluted in 100 µL glycine (100 mM, pH 3.0–3.2). Eluates were neutralized with 10 µL tris (1 M, pH 8) and 100 µL PBS. Concentration was measured using the Pierce BCA assay kit (Thermo Scientific). rVHHs with yields > 100 ng/µL in final eluate were tested for activity.
Standard ELISAs
MaxiSorp plates were coated with antigen and blocked as described above for solid-phase phage display. Each condition was performed in triplicate. Individual phage clones were grown as above; cells removed by centrifugation 3000 × g for 10 min, then supernatants diluted 1/3x in PBT. Rabbit antisera was used as a positive control; YU573 is rabbit polyclonal antisera raised against PAK flagella55; YU586 is polyclonal antisera raised against PAK pilin56. Antisera was diluted 1/10,000 in PBT. 100 µL per well of diluted phage or antisera was applied to coated wells and incubated 1 h, nutating. Liquid was decanted and wells washed 4 times with 100 µL per well PT buffer. Secondary antibodies (Goat anti-rabbit::HRP [Bio-Rad Cat #170-6515, RRID:AB_11125142], Mouse anti-M13::HRP [Sino Biological Cat #11973-MM05T, RRID:AB_2857926]) were diluted 1/3000x in PBT; 100 µl per well was applied and incubated 45 min at RT. Wells were washed 5 times, then plates blotted dry on paper towel. 3,3’,5,5’-Tetramethylbenzidine (TMB), 100 µL per well was added and incubated at RT until the control wells containing no primary antibody began to turn blue. 100 µL per well 1 N H2SO4 was used to quench the reaction, and absorbance read at 450 nm.
Cell-based ELISAs
Plates were coated with cells and fixed with methanol following the method of57, with modifications. Overnight cultures of P. aeruginosa were subcultured 1/50x and grown to mid-exponential phase, harvested by centrifugation 3000 × g × 10 min, resuspended and washed twice in PBS + MC. Cells were diluted to OD 0.3 and 100 µL per well (~3 × 107) were added to a microplate. Cells were incubated at 37 °C for 2 h. 100 µL per well of ice cold methanol was added and cells were incubated rocking at room temperature for 10 min. Methanol was removed by gentle aspiration from the wall of the well, and plates were dried 10 min uncovered in the fume hood. PEG-precipitated phage clones at ~1 × 1013 pfu ml−1 were diluted 1/100 in PBT; antisera were diluted 1/20,000x in PBT; 100 µL per well of primary antibody was added and incubated 2 hours with fixed cells. Wells were washed five times with PT, then stained with secondary antibody and developed as above.
Cell-based ELISAs with bacterial rVHHs were performed as above, except purified rVHHs at 2 mg ml−1 were diluted 1/100 and used as primary antibodies. For brVHHs, rabbit anti-E-tag (Novus Biologicals Cat# NB600-527, RRID:AB_10001463), 1 mg ml−1 was used at 1/500x dilution as the secondary antibody; cells were incubated 30 minutes and washed three times. Goat anti-rabbit::HRP (Bio-Rad Cat #170-6515, RRID:AB_11125142) was used as the tertiary antibody for detection.
Live cell-based ELISAs
For cell-based ELISAs performed on live cells, overnight cultures were subcultured 1/50x and grown to mid-exponential phase, then harvested by centrifugation 3000 × g for 5 min. Cells were washed once in PBS, then ~1 × 109 cells were resuspended in PBS + 0.5% BSA + 0.05% Tween-20, added to a microcentrifuge tube, and incubated rocking for 1 h. Cells were pelleted, then resuspended in 1 mL of PBS + 0.5% BSA + primary antibody (either antiserum at 1/100x dilution or brVHH @ ~40 µg ml−1 final concentration) + SYTO9 at 1/250x and incubated rocking for 1 h protected from light. Cells were pelleted and washed once; brVHH samples were resuspended in secondary antibody rabbit anti-E-tag (Novus Biologicals Cat# NB600-527, RRID:AB_10001463), 1 mg ml−1 at 1/500x dilution in 1 mL PBS + 0.5% BSA + 0.05% Tween-20; antisera samples were resuspended in buffer; samples were incubated 1 h, then pelleted and washed once. Cells were resuspended in Goat anti-rabbit::HRP (Bio-Rad Cat #170-6515, RRID:AB_11125142) tertiary antibody at 1/1000x and incubated 1 h, then pelleted and washed once. Cells were resuspended in 100 µL of PBS, then diluted 1/2x, 1/4x, or 1/8x; each dilution of cells was added to the plate. OD600, SYTO9 fluorescence, and A450 were measured. TMB and H2SO4 were added as with standard ELISA. A450 signal saturated detection in some wells, so samples were diluted 1/2x in PBS and read.
Bacterial dot blots
P. aeruginosa cells in stationary phase were harvested, washed once in PBS, and diluted to 2 × 1010 cells ml−1 (nominal OD = 20). Serial 5-fold dilutions were performed in PBS. 3 µL of cells were spotted at each dilution on a 2.5 × 2.5 cm nitrocellulose membrane. Cells were allowed to dry 30 min, then stained 5 min in 0.1% w/v Ponceau S stain in 1% glacial acetic acid and destained 2 min in water. Blots were blocked and fully destained in 1x TBST (Tris HCl pH 8, 100 mM; NaCl, 1.5 M; Tween-20, 0.05% v/v) plus 5% non-fat milk. Blots were washed once with TBST and probed with primary antibody. 3 µg of purified rVHH was diluted in 100 µL of TBST, spotted into a clean 6-well plate, and the membrane placed face down in this puddle. Plates were sealed with tape and incubated overnight at 4 °C. Blots were washed three times for 5 min with 2 mL TBST. Secondary antibodies (Goat anti-Human IgG::HRP, Secondary Antibody, Invitrogen Cat# SA5-10283, RRID:AB_2868331 @ 1 mg ml−1) were diluted 1/5000x in 500 µL TBST and applied to blots; blots were incubated 30 min at RT and washed three times. Enhanced chemiluminescence (ECL) substrate was prepared (100 mM Tris pH 8.5, 1.25 mM luminol, 225 µM coumaric acid, 0.00001% H2O2); 1 mL ECL substrate was added to each blot, then blots were imaged for 3 min using a Chemidoc MP (Bio-Rad).
Live cell dot blots
Stationary phase cells were washed once in PBS and diluted to 1.5 × 108 cells ml−1, then 7.5 × 106 cells (50 µL) per well were added to a 2 mL V-bottom 96-well plate. SYTO9 was diluted to a final concentration of 1/250x and added along with 300 ng per well of rVHH to cells for in a total volume of 100 µl per well. Pre-cleared polyclonal antisera YU573 and YU586 were used at a final concentration of 1/100x. Cells were incubated, protected from light, for 40 min, then washed twice by addition of 500 µL per well PBS, centrifugation 4000 × g for 10 min @ 10 °C, and decanting. Cells were resuspended in 50 µL PBS by pipetting, then 5 µL per well was spotted onto a nitrocellulose membrane and allowed to dry for 30 min protected from light. The membrane was blocked in 5% nonfat milk + TBST at 4 °C overnight, then rinsed and stained with secondary as above. Before addition of the enhanced chemiluminescenc (ECL) substrate, blot was exposed for fluorescence in the AlexaFluor 488 channel (Ex = 460 − 490 nm, Em = 518–546 nm, 0.05 s). Blot was developed and imaged for ECL (10 min exposure).
Bacterial flow cytometry
All solutions used for FCM were passed through a 0.22 µm filter on the day of the experiment.
Bacterial cells in stationary phase (16 h of growth) were harvested in round-bottom culture flasks by centrifugation 3500 × g for 10 min at room temperature. Supernatants were decanted, the pellet disrupted by dragging the tube across a rack (“racking”), cells resuspended in 1 mL FACS buffer (PBS supplemented with 0.05% v/v Tween-20 and 0.5% w/v BSA, filter sterilized), and suspension transferred to 1.7 mL microcentrifuge tubes. Cells were centrifuged at 3500 × g, room temperature for 5 min, decanted, pellet disrupted by racking, then resuspended in 1 mL FACS buffer by flicking and inverting for 1–5 min. We found gentle handling (disrupting pellet by racking, resuspension by flicking and inverting rather than vortexing, low speed centrifugation) was critical to achieving reproducible staining of P. aeruginosa. Cells were resuspended to an OD of 0.2 and 50 µL of cells (1 × 107 cells) per condition added to a 2 mL 96-well V-bottom plate (Corning). The primary antibody (or VHH) was diluted in FACS buffer to 2x final concentration, then 50 µL primary antibody added to cells. Cells were incubated with primary antibody at room temperature on a microplate shaker (750 rpm) for 30 min. Soluble IgG1-Fc served as a negative control. To wash, 500 µL per well of FACS buffer was added; bacteria were collected by centrifugation at 4000 × g, 10 °C for 10 min and buffer was decanted. The wash was repeated once. A mixture of secondary antibody (PE Goat polyclonal anti-Human IgG Fc, eBioscience, [Thermo Fisher Scientific Cat# 12-4998-82, RRID:AB_465926], 1 µg per well; or PE Donkey anti-rabbit IgG [BioLegend Cat# 406421, RRID:AB_2563484], 0.125 µg per well) and SYTO9 nuclear stain at 1/500x dilution was prepared in FACS buffer; 50 µL per well of this mixture was added to equal volume of cells and mixed by gently pipetting up and down 10 times with a multichannel pipette. Cells were incubated with the secondary antibody as above, for 15 min. The cells were washed once as above.
Cells were fixed in 1% paraformaldehyde (PFA) to prevent efflux of the nuclear stain SYTO9. Briefly, paraformaldehyde (Electron Microscopy Solutions) 16% w/v, was diluted to 4% in filter-sterilized PBS and stored at −20 °C with minimal headroom (to prevent oxidation). Single-use aliquots of 4% w/v PFA were thawed and diluted to 1% final concentration in sterile PBS. Cells were incubated with 1% PFA at room temperature as above for 20 min. Excess aldehydes were quenched by addition of 0.75 M Tris pH 8 and cells incubated for 10 min further. Cells were washed by addition of 500 µL FACS buffer, centrifuged, and decanted as above. Cells were resuspended in 250 µL FACS buffer (5x dilution compared to their original density), then 200 µL cells were passed through a 40 µm filter mesh. Cells were analyzed on a CytoFLEX LX flow cytometer (Beckman-Coulter) with the following settings: flow rate, 15 µL/min; event rate-setting, high; threshold, side-scatter height (SSC-H) > 10,000; voltages, FSC = 165, SSC = 400, B525-FITC = 150, Y585-PE = 500–1000. Data was collected for at least 20 s or until >50,000 events were recorded in the gate identifying bacteria.
Data were gated to identify single bacterial cells (see Supplementary Fig. 30). To distinguish intact bacteria from similarly-sized debris, events with SYTO9 fluorescence greater than that of unstained cells were marked as bacteria. Analysis was performed using FlowJo.
For each rVHH, we drew an “rVHH +” gate starting at the 99th percentile of fluorescence for the antigen-negative cells stained with that rVHH; we then calculated the fraction of antigen-positive cells that were rVHH-positive, as well as the mean fluorescence intensity for both antigen-negative and antigen-positive cells. We considered an rVHH successful if the %rVHH+ for antigen-positive cells was at least three times higher than the %rVHH+ for antigen-negative cells. This threshold was established by performing the same gating procedure on the isotype control and observing that the %rVHH+ cells for the antigen-positive population was never greater than three times that for the antigen-negative cells.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw sequencing data has been deposited to the National Center for Biotechnology Information (NCBI) Short Read Archive (SRA) under BioProject accession number PRJNA1073972. Processed sequencing data has been deposited at Zenodo: https://doi.org/10.5281/zenodo.11246657. Raw and processed flow cytometry data have been deposited at Zenodo: https://doi.org/10.5281/zenodo.12826667. Source data are provided with this paper.
Code availability
Code to process sequencing data and generate figures appearing in the manuscript is available at https://github.com/caseygrun/phage-seq58. Code for the nbseq library is available at https://github.com/caseygrun/nbseq59.
References
Marvig, R. L., Sommer, L. M., Molin, S. & Johansen, H. K. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat. Genet. 47, 57–64 (2015).
Klockgether, J., Cramer, N., Fischer, S., Wiehlmann, L. & Tümmler, B. Long-term microevolution of Pseudomonas aeruginosa differs between mildly and severely affected cystic fibrosis lungs. Am. J. Respir. Cell Mol. Biol. 59, 246–256 (2018).
Bartell, J. A. et al. Evolutionary highways to persistent bacterial infection. Nat. Commun. 10, 269 (2019).
Winstanley, C., O’Brien, S. & Brockhurst, M. A. Pseudomonas aeruginosa evolutionary adaptation and diversification in cystic fibrosis chronic lung infections. Trends Microbiol. 24, 1–11 (2016).
Mahenthiralingam, E., Campbell, M. E. & Speert, D. P. Nonmotility and phagocytic resistance of Pseudomonas aeruginosa isolates from chronically colonized patients with cystic fibrosis. Infect. Immun. 62, 596–605 (1994).
Luzar, M. A., Thomassen, M. J. & Montie, T. C. Flagella and motility alterations in Pseudomonas aeruginosa strains from patients with cystic fibrosis: Relationship to patient clinical condition. Infect. Immun. 50, 577–582 (1985).
Li, X.-Z., Plésiat, P. & Nikaido, H. The challenge of efflux-mediated antibiotic resistance in gram-negative bacteria. Clin. Microbiol. Rev. 28, 337–418 (2015).
Maldonado, R. F., Sá-Correia, I. & Valvano, M. A. Lipopolysaccharide modification in gram-negative bacteria during chronic infection. FEMS Microbiol. Rev. 40, 480–493 (2016).
Jain, M. et al. Type III secretion phenotypes of Pseudomonas aeruginosa strains change during infection of individuals with cystic fibrosis. J. Clin. Microbiol. 42, 5229–5237 (2004).
Tonikian, R., Zhang, Y., Boone, C. & Sidhu, S. S. Identifying specificity profiles for peptide recognition modules from phage-displayed peptide libraries. Nat. Protoc. 2, 1368–1386 (2007).
Stark, Y., Venet, S. & Schmid, A. Whole cell panning with phage display. Methods Mol. Biol. 1575, 67–91 (2017).
Muyldermans, S. Nanobodies: natural single-domain antibodies. Annu. Rev. Biochem. 82, 775–797 (2013).
Rouet, R., Jackson, K. J. L., Langley, D. B. & Christ, D. Next-generation sequencing of antibody display repertoires. Front. Immunol. 9, 1315 (2018).
Pollock, S. B. et al. Highly multiplexed and quantitative cell-surface protein profiling using genetically barcoded antibodies. Proc. Natl. Acad. Sci. USA 115, 2836–2841 (2018).
Nixon, A. M. L. et al. A rapid in vitro methodology for simultaneous target discovery and antibody generation against functional cell subpopulations. Sci. Rep. 9, 842 (2019).
Xu, J. L. & Davis, M. M. Diversity in the CDR3 region of VH is sufficient for most antibody specificities. Immunity 13, 37–45 (2000).
Mitchell, L. S. & Colwell, L. J. Comparative analysis of nanobody sequence and structure data. Proteins 86, 697–706 (2018).
Derda, R. et al. Diversity of phage-displayed libraries of peptides during panning and amplification. Molecules 16, 1776–1803 (2011).
Ledizet, M. et al. The ability of virulence factor expression by Pseudomonas aeruginosa to predict clinical disease in hospitalized patients. PLoS One 7, e49578 (2012).
Malone, J. G. Role of small colony variants in persistence of Pseudomonas aeruginosa infections in cystic fibrosis lungs. Infect. Drug Resist. 8, 237–247 (2015).
Ravn, U. et al. Deep sequencing of phage display libraries to support antibody discovery. Methods 60, 99–110 (2013).
Hu, D. et al. Effective optimization of antibody affinity by phage display integrated with high-throughput dna synthesis and sequencing technologies. PLoS One 10, e0129125 (2015).
DiGiandomenico, A. et al. Identification of broadly protective human antibodies to Pseudomonas aeruginosa exopolysaccharide Psl by phenotypic screening. J. Exp. Med. 209, 1273–1287 (2012).
Close, D. W. et al. Using phage display selected antibodies to dissect microbiomes for complete de novo genome sequencing of low abundance microbes. BMC Microbiol. 13, 270 (2013).
Wang, Q. et al. Target-agnostic identification of functional monoclonal antibodies against Klebsiella pneumoniae multimeric MrkA fimbrial subunit. J. Infect. Dis. 213, 1800–1808 (2016).
Lam, K. et al. Probing the structure and function of the protease domain of botulinum neurotoxins using single-domain antibodies. PLoS Pathog. 18, e1010169 (2022).
Pardon, E. et al. A general protocol for the generation of Nanobodies for structural biology. Nat. Protoc. 9, 674–693 (2014).
Paus, D. & Winter, G. Mapping epitopes and antigenicity by site-directed masking. Proc. Natl. Acad. Sci. 103, 9172–9177 (2006).
Silverman, J. D., Roche, K., Mukherjee, S. & David, L. A. Naught all zeros in sequence count data are the same. Comput. Struct. Biotechnol. J. 18, 2789–2798 (2020).
Armstrong, G. et al. Applications and comparison of dimensionality reduction methods for microbiome data. Front. Bioinforma. 2, 821861 (2022).
Gloor, G. B., Macklaim, J. M., Pawlowsky-Glahn, V. & Egozcue, J. J. Microbiome datasets are compositional: and this is not optional. Front. Microbiol. 8, 57 (2017).
Willis, A. D. Rarefaction, alpha diversity, and statistics. Front. Microbiol. 10, (2019).
Callahan, B. J. et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Stoeckius, M. et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017).
Peterson, V. M. et al. Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol. 35, 936–939 (2017).
Singh, S. B. et al. Kibdelomycin is a bactericidal broad-spectrum aerobic antibacterial agent. Antimicrob. Agents Chemother. 59, 3474–3481 (2015).
Chuanchuen, R., Murata, T., Gotoh, N. & Schweizer, H. P. Substrate-dependent utilization of OprM or OpmH by the Pseudomonas aeruginosa MexJK efflux pump. Antimicrob. Agents Chemother. 49, 2133–2136 (2005).
Urdaneta-Páez, V. et al. Identification of efflux substrates using a riboswitch-based reporter in Pseudomonas aeruginosa. mSphere 8, e0006923 (2023).
Maass, D. R., Sepulveda, J., Pernthaner, A. & Shoemaker, C. B. Alpaca (Lama pacos) as a convenient source of recombinant camelid heavy chain antibodies (VHHs). J. Immunol. Methods 324, 13–25 (2007).
Montie, T. C., Craven, R. C. & Holder, I. A. Flagellar preparations from Pseudomonas aeruginosa: isolation and characterization. Infect. Immun. 35, 281–288 (1982).
Jain, R., Sliusarenko, O. & Kazmierczak, B. I. Interaction of the cyclic-di-GMP binding protein FimX and the Type 4 pilus assembly ATPase promotes pilus assembly. PLoS Pathog. 13, e1006594 (2017).
Berg, S. et al. Ilastik: interactive machine learning for (bio)image analysis. Nat. Methods 16, 1226–1232 (2019).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Steinegger, M., & Söding, J. Clustering huge protein sequence sets in linear time. Nat. Commun. 9, 2542 (2018).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
McDonald, D. et al. The biological observation matrix (BIOM) format or: how I learned to stop worrying and love the ome-ome. GigaScience 1, 2047-217X-1-7 (2012).
Willis, A. & Bunge, J. Estimating diversity via frequency ratios. Biometrics 71, 1042–1049 (2015).
Lun, A., Bach, K., Kim, J. K. & Scialdone, A. scran: methods for single-cell RNA-seq data analysis. https://doi.org/10.18129/B9.bioc.scran (2023).
Lun, A. T. L., McCarthy, D. J. & Marioni, J. C. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with bioconductor. F1000 Res. 5, 2122 (2016).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
The scikit-bio development team. scikit-bio: a bioinformatics library for data scientists, students, and developers. (2022).
Head, T., Kumar, M., Nahrstaedt, H., Louppe, G. & Shcherbatyi, I. Scikit-optimize/scikit-optimize. https://doi.org/10.5281/zenodo.5565057 (2021).
Rosen, C. Discovery of host-microbiota interactions. (Yale Graduate School of Arts; Sciences Dissertations, 2021).
Schniederberend, M., Abdurachim, K., Murray, T. S. & Kazmierczak, B. I. The GTPase activity of FlhF is dispensable for flagellar localization, but not motility, in Pseudomonas aeruginosa. J. Bacteriol. 195, 1051–1060 (2013).
Jain, R., Behrens, A.-J., Kaever, V. & Kazmierczak, B. I. Type IV pilus assembly in Pseudomonas aeruginosa over a broad range of cyclic di-GMP concentrations. J. Bacteriol. 194, 4285–4294 (2012).
Comolli, J. C. et al. Pseudomonas aeruginosa gene products PilT and PilU are required for cytotoxicity in vitro and virulence in a mouse model of acute pneumonia. Infect. Immun. 67, 3625–3630 (1999).
Grun, C. caseygrun/phage-seq: v1.0.0. https://doi.org/10.5281/zenodo.12863464 (2024).
Grun, C. caseygrun/nbseq: v1.1.0. https://doi.org/10.5281/zenodo.12814410 (2024).
Chuanchuen, R., Narasaki, C. T. & Schweizer, H. P. The MexJK efflux pump of Pseudomonas aeruginosa requires OprM for antibiotic efflux but not for efflux of triclosan. J. Bacteriol. 184, 5036–5044 (2002).
Schniederberend, M. et al. Modulation of flagellar rotation in surface-attached bacteria: a pathway for rapid surface-sensing after flagellar attachment. PLoS Pathog. 15, e1008149 (2019).
de Kerchove, A. J. & Elimelech, M. Impact of alginate conditioning film on deposition kinetics of motile and nonmotile Pseudomonas aeruginosa strains. Appl. Environ. Microbiol. 73, 5227–5234 (2007).
Liu, P. V. The roles of various fractions of Pseudomonas aeruginosa in its pathogenesis: III. Identity of the lethal toxins produced in vitro and in vivo. J. Infect. Dis. 116, 481–489 (1966).
Schmidt, F. I. et al. A single domain antibody fragment that recognizes the adaptor ASC defines the role of ASC domains in inflammasome assembly. J. Exp. Med. 213, 771–790 (2016).
Jiang, W. et al. Generation of a phage-display library of single-domain camelid VHH antibodies directed against Chlamydomonas reinhardtii antigens, and characterization of VHHs binding cell-surface antigens. Plant J. 76, 709–717 (2013).
Acknowledgements
We wish to thank George O’Toole, Stephen Lory, Matthew Parsek, John Mattick, Fred Ausubel, Verónica Urdaneta-Paez, Deanna Hausman, and Miguel López-Rivera for kindly sharing bacterial strains. We thank Hidde Ploegh for assistance with phage library construction and gift of vectors. We appreciate Aaron Ring’s assistance with recombinant VHH expression and gift of vectors. We are grateful to the Yale Center for Genome Analysis (supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number 1S10OD030363-01A1), particularly Chris Castaldi for technical support. We thank the Yale Center for Research Computing for guidance and use of the research computing infrastructure.
Author information
Authors and Affiliations
Contributions
C.N.G. and B.I.K. conceived the study. C.N.G., B.N., C.B.S., and B.I.K. designed the experiments. C.N.G., R.J., and M.S. conducted the experiments. C.N.G. performed data analysis. C.N.G. and B.I.K. wrote the manuscript. All authors edited and approved of the manuscript.
Corresponding author
Ethics declarations
Competing interests
A provisional patent application encompassing aspects of this work has been filed by Yale University, listing C.N.G., M.S., R.J. and B.I.K. as inventors. The other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Grun, C.N., Jain, R., Schniederberend, M. et al. Bacterial cell surface characterization by phage display coupled to high-throughput sequencing. Nat Commun 15, 7502 (2024). https://doi.org/10.1038/s41467-024-51912-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-51912-7
This article is cited by
-
Bacillus subtilis surface display technology: applications in bioprocessing and sustainable manufacturing
Biotechnology for Biofuels and Bioproducts (2025)
