
Abstract
2,5-Furandicarboxylic acid (FDCA), a critical polymer platform molecule that can potentially replace terephthalic acid, coupled hydrogen coproduction holds great prospects via electrolysis. However, the electrosynthesis of FDCA faces challenges in product separation from complex electrolytes and unclear electrochemical and nonelectrochemical reactions during the 5-hydroxymethylfurfural (HMF) oxidation. Herein, an electrochemical/chemical integrated system of alkaline HMF-H2O co-electrolysis is proposed, achieving distillation-free synthesis of high-purity FDCA by acidic separation/purification and hydrogen coproduction. This system achieves ampere-level current densities of 812 and 1290 mA cm−2 at potentials of 1.50 and 1.60 V, with nearly 100% FDCA yield and HMF conversion in only 6 min at 1.50 V. The electrooxidation of HMF involves a coupling of electrochemical and nonelectrochemical reactions, wherein the aldehyde group is dehydrogenated and oxidized, followed by dehydrated and oxidized of the hydroxyl group, ultimately forming FDCA. Concurrently, nonelectrochemical reactions of intermolecular electron transfer occur in HMF and aldehyde group-containing intermediates.
Similar content being viewed by others
Introduction
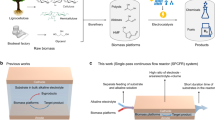
2,5-Furandicarboxylic acid (FDCA) is a valuable platform chemical for the polymer industry, possessing a market size of 1.8 million tons per year, and can potentially replace terephthalic acid for the synthesis of biomass-based polyethylene furanoate (PEF)1,2,3. The PEF, a 100% renewable polyester with outstanding properties such as thermal resistance and gas barrier, is considered one of the promising alternatives to petroleum-based polyethylene terephthalate (PET, with production of more than 80 million tons per year)4,5,6. Currently, in traditional industrial methods, the FDCA is mainly produced by oxidation of 5-hydroxymethylfurfural (HMF) at temperatures of 80–150 °C and high-pressure oxygen (0.3–4.0 MPa) using noble metals (e.g., Pt, Au, and Pd) as catalysts (Fig. 1a)7. Despite the maturity and stability of this approach, the process inevitably relies on fossil energy for FDCA production and the removal of impurities like by-products, oxidants, organic solvents via separation and distillation, resulting in significant energy waste and environmental pollution8. Additionally, capital investment increases significantly due to precious metal catalysts and complex chemical processes. Therefore, there is an urgent need to develop a more environmental and ecological process for FDCA production.

a The traditional process and b the electrocatalytic process.
The electrocatalytic refinery is a novel and attractive process that can efficiently convert renewable substances (biomass, water, carbon dioxide, etc.) into value-added fuels and chemicals at ambient conditions and promote efficient hydrogen production (Fig. 1b)9,10,11. For instance, the theoretical potential of the HMF oxidation reaction (HMFOR, 0.30 V vs. RHE) is significantly lower than that of the oxygen evolution reaction (OER, 1.23 V vs. RHE), being a promising replacement anodic reaction12,13,14,15. The recognized electrooxidation of HMF has two reaction pathways and a variety of intermediates are produced. Therein, the intermediate 2,5-diformylfuran (DFF) without α-H rapidly occur spontaneous intermolecular cannizzaro reaction (nonelectrochemical reaction) under alkaline conditions to generate carboxylic acid and alcohol, respectively, increasing the difficulty in analyzing the HMF reaction process and subsequent separation of the downstream target products. Detailedly, in DFF pathway, the intermediate DFF contains highly reactive aldehyde groups on both sides, making it highly susceptible to mutually interfering electrochemical and nonelectrochemical reactions under applied potential, which poses a challenge for the detection of intermediate DFF and determination of the electrooxidation pathway. In 5-hydroxymethyl-2-furancarboxylic acid (HMFCA) pathway, the HMFCA lacks aldehyde groups and is not expected to be nonelectrochemically reactive during electrochemical reaction. To date, few in-depth studies have investigated the effect of the cannizzaro reaction involving intermediates during the reaction process on the reaction pathway and the conversion process, which could limit the ampere-level electrolysis of HMF and H2O16,17,18. Therefore, a thorough and systematic analysis of HMF and its intermediates by multiple approaches is necessary. More importantly, the intertwined yet unidentified electrochemical and nonelectrochemical reactions during HMF oxidation make it difficult to identify the rate/limiting steps in the reaction pathways, presenting the challenges to the rational design of electrocatalysts and comprehending the molecular-level conversion mechanism.
The ultimate target of the electro-refinery is to increase yields at high current densities and to successfully separate and purify the products, which in turn meet the needs of the downstream industry. Multiple attempts have been made by researchers recently to develop more stable and efficient catalysts toward FDCA, achieving satisfactory FDCA yields19,20,21,22. Nevertheless, electrooxidation at industrial-level current densities (>500 mA cm−2) to achieve the efficient FDCA production under near-industrial conditions is still limited in exploration. Additionally, for this system, one of the key obstacles is the subsequent separation and purification of FDCA. The mixing with complex electrolytes, the easy generation of polymers during the production process and the high boiling point (over 400 °C) increase the difficulty of crystallization and distillation of the product FDCA, which has always been one of the major obstacles to the industrialization of a catalytic refinery system. As known, the solubility of FDCA in water is 0.1 g/100 mL H2O, and it is nearly insoluble under acidic solutions, which presents the possibility and potential for crystallization and separation of FDCA from the complex electrolytes without the energy-intensive distillation processes. Hence, achieving sustainable production of value-added chemicals through combining electro-refinery with traditional chemical engineering unit operation methods, facilitating tandem reactions within an integrated system, holds an immense significance.
Here, we for the first time decouple and present the two intertwined electrochemical and nonelectrochemical reactions through a spatially separated reaction process. The aldehyde group of HMF is firstly dehydrogenated and oxidized, followed by dehydrated and oxidized of the hydroxyl group, and finally electrooxidized into FDCA. During this period, nonelectrochemical reactions (intermolecular cannizzaro reaction) of intermolecular electron transfer occur in HMF and aldehyde group-containing intermediates. Such a conversion process of the aldehyde side chain involves key HMFCA intermediate, which is also identified and demonstrated by in situ ATR-FTIR and density functional theory (DFT) calculations over low-crystallinity A-Co-Ni2P catalyst. An electrochemical/chemical integrated system of alkaline HMF-H2O co-electrolysis is present, where the FDCA can be produced by acidic crystallization separation/purification, finally achieving distillation-free synthesis of FDCA and hydrogen coproduction (Fig. 1b). This system operates an ampere-level current density of 812 mA cm−2 at a potential of 1.50 V and achieves nearly 100% yield of FDCA and 100% conversion of HMF in just 6 min. With Co, the catalyst captures electrons on HMF and improves the adsorption of OH*, promoting the reaction kinetics in the HMFOR process. Finally, we constructed a simple system for both crystallization and purification, resulting in the crystallization of pale yellow FDCA under acidic conditions, followed by its decolorization using N, N-Dimethylacetamide to obtain white high-purity solid FDCA without the energy-intensive distillation processes. This work identifies the molecular-level conversion mechanisms of HMFOR and offers avenues for the separation, purification, and scaling up of value-added chemicals.
Results
Preparation and characterization of HMFOR materials
Electrocatalysts with low crystallinity have more abundant defects and unsaturated coordinating atoms, enabling sufficient exposure of active sites, and are proven to be effective catalysts in various electrochemical reactions23,24,25. Focusing on the earth-abundant Co and Ni, we synthesized low-crystallinity Co-doped nickel phosphide (A-Co-Ni2P) and nickel phosphide (Ni2P) supported on nickel foam by tandem hydrothermal and phosphating processes. The typical nanoneedle structure are presented in A-Co-Ni2P (Scanning electron microscopy images, Supplementary Fig. 1). Subsequently, the high-resolution transmission electron microscopy (HR-TEM) and corresponding inverse fast Fourier transformation (IFFT) patterns of A-Co-Ni2P were measured. A small number of lattice fringes are exhibited in the A-Co-Ni2P with the spacing of 0.192 and 0.221 nm, coinciding with the (210) and (111) planes of Ni2P26, respectively (Supplementary Fig. 2a, b). High-angle annular dark-field scanning-TEM and the mapping analysis show the uniform distribution of Ni, Co, P, and O elements in the electrocatalyst (Supplementary Fig. 2c). For comparison, HR-TEM and corresponding IFFT of Ni2P without Co were also tested. Numerous lattice fringes are displayed in Ni2P catalyst, with only a minor presence of amorphous structures at the edges (Supplementary Fig. 3). These findings indicate that the crystallinity of A-Co-Ni2P is significantly lower than that of Ni2P, which also confirms that Co doping into the A-Co-Ni2P lattice reduces the crystallinity27. Subsequently, X-ray diffraction (XRD) suggests that the peaks of Ni2P and A-Co-Ni2P at 40.9°, 47.3° and 54.9° are attributed to Ni2P (Supplementary Fig. 4). The relative crystallinity of the catalysts with and without Co was calculated to be 82.2 ± 0.7% and 87.6 ± 1.1%, respectively. The doping of Co causes a decrease in the peak intensity of A-Co-Ni2P at 40.9° and 54.9°, indicating an increase in the degree of amorphization.
X-ray photoelectron spectroscopy (XPS) was utilized to evaluate the effect of Co doping on the electronic state of A-Co-Ni2P. For the Ni 2p XPS spectra, the peaks at 853.1 eV and 870.1 eV are ascribed to the Ni–P bonds26, respectively, as depicted in Supplementary Fig. 5. The peaks at 855.2 eV and 872.8 eV are identified as Ni2+, whereas the peaks at 856.9 eV and 874.6 eV are assigned to Ni3+ in the nickel oxides28. Significantly, the ratios of Ni3+/Ni2+ in Ni2P and A-Co-Ni2P are 1.54 and 2.78, respectively, suggesting an elevated proportion of high-valence Ni in A-Co-Ni2P. In the Co 2p XPS spectrum of A-Co-Ni2P, the peak at 779.2 and 781.5 eV is attributed to 2p3/2 of Co (Supplementary Fig. 6)29. Two peaks located near 129.9 eV are identified as metal phosphides from the P 2p XPS spectra (Supplementary Fig. 7). Compared with Ni2P, the P–O peak of A-Co-Ni2P is negatively shifted by about 0.3 eV, indicating the occurrence of intramolecular electron transfer. Overall, the XPS results clearly demonstrate that the Co species within A-Co-Ni2P tunes and optimizes the electronic structure of Ni species.
Reaction mechanism studies towards HMFOR
The OER and HMFOR effects of Ni2P and A-Co-Ni2P were first compared by linear sweep voltammetry (LSV). For OER without HMF substrate in the electrolyte, the potential of oxidation peak (Ni2+/Ni3+) for A-Co-Ni2P that is near 1.35 V vs. RHE shifts toward lower potential compared to that over Ni2P, as depicted in Supplementary Fig. 8. The current density of A-Co-Ni2P is about 2.9 times than that of Ni2P at 1.60 V vs. RHE. For HMFOR, achieving 100 mA cm−2 over the A-Co-Ni2P (1.33 V vs. RHE) only needs a lower potential than Ni2P (1.37 V vs. RHE). The A-Co-Ni2P exhibits the maximum current density of more than 800 mA cm−2 at a potential of 1.50 V compared to Ni2P, demonstrating the good electrocatalytic performance of HMFOR. Further, a comprehensive investigation of the electrochemical and nonelectrochemical reactions involving HMF and its intermediates, along with the corresponding reaction pathways, was conducted over A-Co-Ni2P catalyst via tracer experiment, where high performance liquid chromatography (HPLC), ATR-FTIR, and in situ ATR-FTIR techniques were employed.
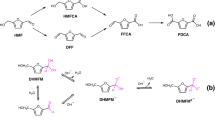
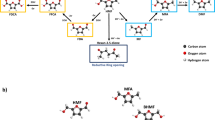
There are two possible pathways for the electrooxidation of HMF21,30, as shown in Fig. 2g. In pathway I, the hydroxyl group on HMF is dehydrated and oxidized to form an aldehyde group, thereby obtaining DFF. In pathway II, the aldehyde group on HMF is dehydrogenated and further oxidized to HMFCA. Subsequently, the DFF and HMFCA intermediates are converted into the same intermediate 2-formyl-5-furancarboxylic acid (FFCA), culminating in the synthesis of FDCA31. The amperometric measurement was performed at 1.50 V vs. RHE using the A-Co-Ni2P as the anodic electrocatalyst. The signals of the intermediates and products during HMFOR were monitored by HPLC and calibrated by calibration curves (Supplementary Fig. 9). As shown in Fig. 2a, the signal of HMF at 37.8 min decreases rapidly with increasing electrolysis time, while the signal of the product FDCA (21.9 min) gradually increases and levels off at about 4 min. Also, the intermediates HMFCA (retention time 25.8 min) and FFCA (retention time 30.3 min) are able to be detected and eventually oxidized. The signal of DFF at 47.2 min is negligible. Simultaneously, the color of the electrolyte changes from initially yellow to brown and finally to light yellow within the electrolysis time of 0–6 min (the inset images in Fig. 2a). Thus, the electrooxidation of HMF can proceed through reaction pathway II shown in Fig. 2g. Also, the DFF intermediate containing highly reactive aldehyde groups is spontaneously and rapidly transformed into other substances through the intermolecular cannizzaro reaction in alkaline electrolytes without external potential. This situation may involve rapid conversion of the generated DFF into HMF and FFCA by HMFOR16,17,18. To effectively reveal the possibility mechanism, a thorough and systematic analysis of the two intertwined processes between the electrochemical and nonelectrochemical reactions of intermediates is required.

a HPLC chromatograms for the electrochemical reaction of HMF obtained at a constant potential of 1.50 V vs. RHE over A-Co-Ni2P catalyst in 1 M KOH containing 0.1 M HMF. b HPLC chromatograms for the nonelectrochemical reaction of DFF obtained without applying potential over A-Co-Ni2P catalyst. c ATR-FTIR spectra of 0.1 M DFF and 0.1 M HMFCA without applying potential as a function of time and reference spectra of HMF, DFF, HMFCA, FFCA and FDCA. d Comparison of DFF and HMFCA in nonelectrochemical reactions under alkaline conditions. e In situ electrochemical ATR-FTIR spectra of the electrolyte (1 M KOH + 0.1 M HMF) over A-Co-Ni2P catalyst recorded within 120 s at 1.50 V vs. RHE. f Corresponding Pseudo-color mapping image. g Two possible electrooxidation pathways for the synthesis of FDCA from HMF. a. u. arbitrary units.
The spatially separated reaction process is employed to decouple the two intertwined electrochemical and nonelectrochemical reactions. The signals in the two intermediates were monitored using HPLC and ATR-FTIR by only adding DFF or HMFCA to the electrolyte, respectively. Firstly, without applied potential, the nonelectrochemical reaction of DFF in 1 M KOH electrolyte using A-Co-Ni2P catalyst was first evaluated and the component of electrolyte was detected by HPLC, as shown in Fig. 2b. After adding DFF to the electrolyte, HMF and FFCA can be detected at initial state, with their signals increasing with time and stabilizing at about 15 min. This indicates that the DFF characterized by aldehyde groups is highly unstable in alkaline electrolytes and disproportionates rapidly via the cannizzaro reaction, yielding a carboxylic acid molecule and an alcohol molecule. With the increase of time, the signals of HMFCA and FDCA all increase to different degrees, while the signal of DFF gradually decreases. The inset of Fig. 2b displays that the color of the electrolyte changes from yellow to dark red within 30 min. To easily compare and analyze the standard spectra of five pure compounds, ATR-FTIR spectroscopy was employed (Supplementary Fig. 10). The components of the electrolyte were further probed using ATR-FTIR to verify the evolution of intermediates over time, as presented in Fig. 2c. Without applied potential, the peak at 1397 cm−1 is enhanced with time when DFF was added to the electrolyte over A-Co-Ni2P catalyst, corresponding to the C–H bonds of the aldehyde in HMF and FFCA, further verifying that a nonelectrochemical reaction occurs for DFF. However, the changes of HMFCA are different from the DFF. Without applied potential, after adding HMFCA to the electrolyte, the signal remains constant in 1 M KOH solution within 30 min, with no detection of any other substances in HPLC (Supplementary Fig. 11). Also, the color of the solution unchanged (the inset images in Supplementary Fig. 11). Furthermore, no change for the peak of HMFCA is monitored by ATR-FTIR, indicating that the hydroxyl group in HMFCA does not participate in intermolecular redox reactions in 1 M KOH (Fig. 2c). Therefore, a clear route under the nonelectrochemical reaction is summarized in Fig. 2d. A nucleophilic addition reaction occurs between DFF without α-H and OH− to form an anionic intermediate. Under alkaline conditions, the hydrogen atom on the anionic intermediate attacks the oxygen on another DFF molecule, causing the oxygen protonation and thus being rapidly disproportionated into HMF and FFCA. In contrast, HMFCA remains unchanged under alkaline conditions. Finally, under the constant potential, the changes of the two intermediates were evaluated and analyzed. As illustrated in Supplementary Fig. 12, HMFCA is rapidly electrooxidized to FDCA via the intermediate of FFCA at 1.50 V vs. RHE within 6 min as derived from HPLC data. DFF can be finally electrooxidized to FDCA via the HMF and FFCA pathways (Supplementary Fig. 13). Notably, the generated HMF is obtained by a nonelectrochemical reaction of DFF, as discussed in Fig. 2b. The electrochemical and nonelectrochemical reaction pathways of DFF and HMFCA intermediates are also clearly summarized in Fig. 2g.
To further decouple the oxidation pathway of the whole process from HMF to FDCA, the signals of intermediates during the electrooxidation of HMF over A-Co-Ni2P were probed by in situ ATR-FTIR spectroscopy, as illustrated in Fig. 2e. These spectra illustrate the evolution of electrooxidation within the time range of 0 to 120 s. The peaks at 1268 and 1390 cm−1 are assigned to the C–O stretching and point to the formation of HMFCA. The intensity of the peak at 1268 cm−1 increases with time, indicating the accumulation of HMFCA. The peak at wavenumber 1187 cm−1 is assigned to the C–H vibration and correlates well with the reference spectra of FDCA and HMF (Supplementary Fig. 10). This particular wavenumber presents an upward pointing trend and increases with increasing time, which indicates that it belongs to the characteristic peak of FDCA. The peaks at 1515, and 1672 cm−1 are ascribed to the C=C ring and C=O bond of the aldehyde, respectively, and both peaks are closely related to the reference spectra of FFCA. The intensity of these peaks increases with the electrooxidation time, indicating the gradual accumulation of FFCA. Also, the intensity of the peak at the wavenumber of 1649 cm−1 (the C=O bond) decreases with increasing time and shows a downward pointing trend, indicating the gradual consumption of HMF. No related peaks for DFF are detected during the in situ monitoring of HMFOR, which also indicated that the process of electrooxidation is dominated by pathway II. As depicted in Fig. 2f, the changes are readily apparent in the pseudo-color mapping images. Thus, the in situ ATR-FTIR results and the associated analyses of the electrochemical and nonelectrochemical reactions of the intermediates demonstrate that the electrooxidation of HMF follows the aldehyde side chain (pathway II, Fig. 2g).
Process optimization of HMFOR
During HMFOR, the calculated HMF conversion increases with time and level off at 4 min, ultimately reaching about 99.4% at 6 min, as presented in Fig. 3a. The A-Co-Ni2P catalyst also can achieve a 99.2% yield of FDCA and carbon balance in 100 mM HMF solution (Supplementary Fig. 14). This demonstrates that the degradation of HMF in this system is negligible6. Also, the yield is relatively high compared to existing literature, as shown in Supplementary Table 1. For this system, the mixing with complex electrolytes and the high boiling point (>400 °C) increase the difficulty of crystallization and distillation of the product FDCA, which is one of the barriers limiting the industrial-scale application. Previous reports primarily considered the electrosynthesis of the FDCA, with little study of subsequent separation and purification. It is known that the FDCA has a low solubility in pure water (0.1 g/100 mL H2O) and is almost insoluble in acidic solutions, which provides the possibility for crystallization and separation of FDCA from electrolytes. As a result, we present, for the first time, an electrochemical/chemical integrated system combining alkaline HMF-H2O co-electrolysis with acidic crystallization separation and purification. As shown in Fig. 3b, the pH of the electrolyte is adjusted to ~3.0 using HCl. After cooling at 0 °C for some time, the FDCA precipitates at the bottom of the electrolyte. After filtration and collection, the FDCA appears as a pale yellow solid, as shown in Supplementary Fig. 15. Subsequently, the FDCA is further decolorized with N, N dimethylacetamide (DMAC, with a mass ratio of FDCA: DMAC = 1:15) and kept at 0 °C for 6 h. Ultimately, the white FDCA is collected by filtration and confirmed by XRD, ATR-FTIR, 1H and 13C NMR, and high resolution mass spectra (Fig. 3c–e and Supplementary Figs. 16–18). Therein, the characteristic peaks at wavenumbers 1187 and 1571 cm−1 of the self-prepared FDCA in the ATR-FTIR results are attributed to the C–H and C=C vibrations on the furan ring, respectively, which coincided with those of the pure FDCA. The characteristic peak of the self-prepared FDCA at 7.2 ppm is attributed to –CH on the furan ring in the 1H NMR spectra, which matches well with the pure FDCA. This is also confirmed by 13C NMR and mass spectra. Additionally, the purity of the self-prepared FDCA is determined to be 99.2% using NMR and HPLC. Except for the ability of the integrated system to operate at industrial-level current densities (>800 mA cm−2), it can also avoid distillation processes to minimize energy waste. Overall, energy-efficient production of value-added FDCA, separation and purification, and simultaneous hydrogen generation are successfully performed via this integrated system, as shown in Fig. 3f. And, the FDCA has the potential to replace terephthalic acid in many polyester industries. Integrating electro-refinery with traditional chemical unit operation methods can offer a potential route toward the future clean and sustainable production of value-added chemicals, even high-purity electronic chemicals.

a The changes for HMF conversion and the yield of product at 1.50 V vs. RHE. The error bars correspond to the standard deviation of three independent measurements. b Photo pictures for the separation and purification process of FDCA from electrolyte. The red dashed box and red arrow represent the precipitated solid FDCA. c XRD patterns, (d) ATR-FTIR and (e) 1H NMR spectra of self-prepared and pure FDCA. f Illustration of the proposed integrated system for HMFOR process and simultaneous hydrogen production. DMAC: N, N dimethylacetamide, HCl: hydrochloric acid. a. u. arbitrary units.
The universality of other alcohol, aldehyde and ketone substrates over A-Co-Ni2P catalyst
The universality of various substrates, including natural molecules, biomass-derived compounds, and industrial feedstocks containing alcohol, aldehyde, and ketone groups such as benzyl alcohol, furfuryl alcohol, cyclohexanol, benzaldehyde, furfural, and cyclohexanone, was evaluated. As illustrated in Supplementary Fig. 19, industrial-level current densities (292–746 mA cm−2) can be achieved over A-Co-Ni2P catalyst at 1.5 V vs. RHE for all organic substrates essentially. High yields (75–98%) of carboxylic acid-containing products (Table 1) are also realized. Then, the products obtained by crystallization and separation processes were compared with the pure compounds using the 1H NMR, 13C NMR and mass spectra, as shown in Fig. 4a–c and Supplementary Figs. 20 and 21. After analysis, it was found that the obtained self-prepared carboxylic acid products all match with the characteristic peaks of the pure compounds, which confirms the successful synthesis of the corresponding products. These results indicate that the electrocatalyst is great promise for producing various carboxylic acids, featuring a wide universality.

1H NMR spectra of (a) pure benzoic acid and self-prepared benzoic acid obtained from benzyl alcohol and benzaldehyde, (b) pure furoic acid and self-prepared furoic acid obtained from furfuryl alcohol and furfural, and (c) pure adipic acid and self-prepared adipic acid obtained from cyclohexanol and cyclohexanone. a. u. arbitrary units.
Electrochemical activity and application studies
The OER and HMFOR electrochemical activities at different potentials were then compared by LSV over A-Co-Ni2P catalyst. As illustrated in Fig. 5a and Supplementary Fig. 22, for HMFOR, the current densities over A-Co-Ni2P electrocatalyst can easily reach 362, 583, 812, and 1290 mA cm−2 at 1.40, 1.45, 1.50 and 1.60 V vs. RHE, respectively. And, the OER does not occur until after 1.50 V vs. RHE. As depicted in Fig. 5b and Supplementary Table 2, the A-Co-Ni2P catalyst exhibits the superior electrocatalytic activity for HMFOR at relatively low potentials compared with most of the reported noble metal/non-noble metal-based electrocatalysts. The stability of the A-Co-Ni2P catalyst for electrocatalytic HMF oxidation was assessed at 1.50 V vs. RHE (Supplementary Fig. 23). After 8 cycles at a constant potential, the current density only slightly changes. Moreover, both the conversion of HMF and the yield of FDCA are close to 100% (Fig. 5c), demonstrating the high stability of A-Co-Ni2P catalyst for HMF oxidation. The high yield and conversion are attributed to the fact that this system can efficiently oxidize the hydroxyl and aldehyde groups in HMF, without causing over-oxidation of the carboxyl groups in the FDCA product by controlled experiments at a constant potential (Supplementary Fig. 24). Electrochemical impedance spectroscopy (EIS) tests were conducted to analyze the ability of charge transfer for HMFOR over the as-prepared catalysts. The equivalent circuit is derived from the Nyquist plots, as illustrated in the inset Supplementary Fig. 25. The charge transfer resistance (Rct) of A-Co-Ni2P is ~2.5 Ω, significantly lower than that of Ni2P (∼5.4 Ω). The A-Co-Ni2P catalyst, characterized by its lower crystallinity, exhibits the superior charge transfer properties. The electrochemical surface area (ECSA) of the catalyst was further tested (Supplementary Fig. 26) using cyclic voltammetry (CV). The current density of A-Co-Ni2P is significantly higher than that of Ni2P, and the Cdl of A-Co-Ni2P and Ni2P are 22.9 and 12.7 mF cm−2, respectively (Supplementary Fig. 27), which indicates that Co doping promotes a significant increase in the ECSA of A-Co-Ni2P catalyst. These results imply that the A-Co-Ni2P has more active sites and better charge transfer compared to Ni2P, significantly improving the electrocatalytic performance32.

a The LSV curves of A-Co-Ni2P catalyst without iR compensation (internal resistance: 0.7 ± 0.2 Ω). b Comparison of the electrocatalytic organic oxidation performance for the developed catalyst in this work with recently reported electrocatalysts. c The conversion of HMF and the yield of FDCA. The error bars correspond to the standard deviation of three independent measurements. d The values of WE and \({W}_{{{\mbox{H}}}_{2}}\) during the process of HMFOR and the OER over A-Co-Ni2P catalyst. e Digital photo of the MEA electrolyzer. f The visualized MEA setup for HMFOR and HER (AEM: anion exchange membrane). g The LSV curves of A-Co-Ni2P catalyst in the MEA (internal resistance: 0.3 ± 0.1 Ω). h Stability measurements for the MEA electrolyzer at 1.8 V over A-Co-Ni2P in 1 M KOH containing 0.1 M HMF.
Next, an evaluation was conducted on the consumed electricity (WE) to reach 100 mA cm−2 and the required energy to produce 1 m3 of hydrogen (\({W}_{{{\mbox{H}}}_{2}}\)) during the process of HMFOR and the OER over the A-Co-Ni2P catalyst. As presented in Fig. 5d, the electricity consumption of HMFOR at 100 mA cm−2 is ~0.132 Wh, being lower than that of OER as calculated from Equation S4 in the supporting information. Also, the values of \({W}_{{{\mbox{H}}}_{2}}\) at the same current density are 3.435 and 2.888 kWh m−3 H2 when the electrolyte without and with 0.1 M HMF, respectively, according to Equation S5. This represents a saving of ~ 16% (0.547 kWh) in electricity for HMFOR compared to the case without HMF, underscoring the superiority of electro-refinery coupled hydrogen production. Subsequently, a zero-gap membrane electrode assembly (MEA) electrolyzer is designed for the electrooxidation of HMF coupled-hydrogen production to evaluate the activity of the catalyst in close to practical situations with A-Co-Ni2P as the anode and Pt-coated Ti as the cathode (Fig. 5e, f). Without HMF, the MEA electrolyzer requires an input cell voltage of up to 1.94 V to achieve 200 mA, as shown in Fig. 5g. Encouragingly, the introduction of HMF accelerates the reaction kinetics by shifting the polarization curves toward lower voltages by 400 mV at a current density of 200 mA. More importantly, industrial-level current densities of 460 and 700 mA are achieved at cell voltages of 1.8 and 2.0 V, respectively. The operating stability of HMFOR was evaluated in the assembled MEA at a voltage of 1.8 V over the A-Co-Ni2P electrode (Fig. 5h). As a result, this MEA can maintain stable electrolysis for 60,000 s with current density decay due to the reduced HMF concentration which can be recovered by refreshing the electrolyte (every 7200 s). The catalyst after the reaction was analyzed and characterized in detail by SEM, XRD and XPS. As illustrated in the SEM images, the A-Co-Ni2P electrocatalyst retains the overall nanoneedle morphology (Supplementary Fig. 28), with surface roughness caused by electrochemical reconstruction33. The A-Co-Ni2P catalyst after the reaction still shows a Ni2P crystalline phase (Supplementary Fig. 29). Additionally, the Ni 2p XPS spectra of the A-Co-Ni2P after the reaction displays a Ni3+/Ni2+ ratio of 3.19 (Supplementary Fig. 30), being higher than that of A-Co-Ni2P before the reaction (2.78), confirming the partial oxidation of the catalyst34. This phenomenon can also be observed on Ni2P catalysts (Supplementary Figs. 31 and 32). Accordingly, the amount of detected H2 experimentally matches well with the theoretical calculation results (Supplementary Fig. 33), indicating that the Faradaic efficiency is close to 100%. Overall, this integrated system not only significantly reduces the cell voltage but also facilitates the simultaneous production of FDCA and H2, which can open the door to potential future practical industrial production.
Revealing the HMF oxidation mechanism
To reveal the reaction mechanism of electrocatalytic oxidation, the reaction kinetics of the A-Co-Ni2P during the electrocatalytic process was explored by in situ EIS. As shown in Fig. 6a, without HMF, the peak decrease in the frequency region of 100–102 at 1.35 V vs. RHE represents the oxidation of the electrocatalyst (Ni2+ to Ni3+). Another peak begins to appear in the low-frequency region (10−1–100 Hz) and the peak shifts toward a lower phase degree with increasing potential, indicating the onset of OER at 1.50 V vs. RHE1. In contrast, with HMF (Fig. 6b), the peak appears at 1.30 V vs. RHE in the low-frequency region, corresponding to the occurrence of HMFOR. An additional peak related to OER is observed at 1.55 V vs. RHE. This also suggests that the reaction kinetics of the HMFOR is more favorable than that of OER at the electrode interface. Further, to reveal the catalyst phase or surface construction over the A-Co-Ni2P electrocatalysts during the OER and HMFOR, in situ Raman spectra were acquired using a custom-made electrolytic cell (Supplementary Fig. 34). As depicted in Fig. 6c, during the OER, two typical bands are identified at 475 and 554 cm−1, corresponding to the Ni-O vibration in Co-NiOOH at 1.35 V vs. RHE35. In contrast, as illustrated in Fig. 6d, for HMFOR, the signal of Ni-O is not monitored at 1.35 V vs. RHE. This suggests the presence of a dynamic conversion process between the electrocatalyst and organic substrate36. The signal of Ni-O is not observed until 1.45 V vs. RHE, and its intensity is noticeably weaker compared with that of the OER process. This further implies that the formed NiOOH is quickly consumed due to the occurrence of HMF oxidation. During this process, the NiOOH phase formed by the catalyst can efficiently capture the HMF for electron transfer and is identified as the active species of the catalyst37.

Bode phase plots of the in situ EIS over A-Co-Ni2P without (a) and with (b) HMF. In situ Raman spectra of A-Co-Ni2P without (c) and with (d) HMF. a. u. arbitrary units. e Charge density difference and bader charge analysis of HMF adsorbed on the NiOOH and Co-NiOOH (the yellow and cyan colors represent charge accumulation and depletion, respectively, the iso-surface value is 0.0008 e Å−3). f Free energies diagram of HMFOR step via pathway II on NiOOH and Co-NiOOH, and corresponding adsorption configurations intermediates for HMFOR on Co-NiOOH. The blue and red shading represent the rate-determining step for different catalysts. The numerical symbols represent the reaction steps.
To further elucidate the reaction pathways and rate/limiting steps of HMF oxidation, DFT calculations were performed. The optimized structural models of NiOOH and Co-NiOOH were established (see Supplementary Data 1 and Supplementary Fig. 35) according to the in situ Raman results. Also, the adsorption configurations of HMF on these models were simulated and optimized, as shown in Supplementary Figs. 36 and 37. After optimization, the HMF is more easily bound to two electrocatalysts through the O atoms of the aldehyde and hydroxyl groups. For the NiOOH structure, the bridge-on adsorption pattern of HMF is the most stable adsorption configuration with an adsorption energy of −1.60 eV, where both the aldehyde and hydroxyl groups of HMF are bound to Ni sites. After introducing Co, the furan ring of HMF is adsorbed on the Co-NiOOH surface through the bridge-on adsorption pattern with an adsorption energy increase to −1.62 eV, where the aldehyde group of HMF attaches to the Ni site, and the hydroxyl group binds to the Co site. This implies the relatively strong adsorption of Co-NiOOH toward HMF. As indicated by the charge density difference and bader charge analysis in Fig. 6e, compared to NiOOH (0.22 e), more electrons are transferred through the O atoms on HMF to Co-NiOOH (0.35 e). This means that HMF is more likely to lose electrons and be oxidized over Co-NiOOH38,39. The possible reaction pathways for HMFOR on the Co-NiOOH surface are presented in Supplementary Fig. 38. The energy barriers for each elementary step in the two pathways on Co-NiOOH were calculated. In the DFF pathway (pathway I), compared to other elementary steps, the step from DFF* to DFF* + OH* exhibits the maximum energy barrier of 0.54 eV. And, this step is also identified as the rate-determining step (RDS). In contrast, in the HMFCA pathway (pathway II), the energy barrier for the RDS (FFCA* → FFCA* + OH*) drops to 0.47 eV. This result suggests that the electrooxidation of HMF follows the HMFCA pathway, aligning with our experiment results. For comparison, the energy barriers on NiOOH were further calculated in the HMFCA pathway. Resultantly, the RDS changes to the step of HMFCA* to HMFCA* + OH*, and the energy barrier increases to 1.15 eV for NiOOH, as presented in Fig. 6f and Supplementary Fig. 39. The results show that the introduced Co species can improve the adsorption of OH* and promote the kinetics of the RDS in the HMFOR process compared to NiOOH.
Discussion
In summary, both intertwined electrochemical and nonelectrochemical reactions of HMF electrooxidation are decoupled through a spatially separated reaction process. The aldehyde group of HMF is first dehydrogenated and oxidized, followed by dehydrated and oxidized of the hydroxyl group, and finally electrooxidized into FDCA. During this period, nonelectrochemical reactions of intermolecular electron transfer occur in HMF and aldehyde group-containing intermediates. Such a conversion process includes key intermediates is also identified by in situ ATR-FTIR and DFT calculation results. An electrochemical/chemical integrated process of alkaline HMF-H2O co-electrolysis and acidic separation and purification is presented, achieving the distillation-free synthesis of high-purity FDCA and hydrogen coproduction. The ampere-level current densities of 812 and 1290 mA cm−2 can be achieved at potentials of 1.50 and 1.60 V, and nearly 100% yield of FDCA and 100% conversion of HMF are achieved for just 6 minutes at 1.50 V in three-electrode system. The established MEA electrolyzer also achieves an industrial-level current density of 700 mA at the cell voltage of 2.0 V and operates stably for 60,000 s. About 16% of electricity can be saved for producing per m3 of H2 through HMFOR compared to the case without HMF. The enhanced performance stems from the fact that the introduced Co species improve the capture of electrons on HMF by the catalyst and the adsorption of OH*, accelerating the kinetics of the RDS in the HMFOR process. Finally, the pale yellow FDCA is crystallized and separated in an acidic environment, followed by decolorization using N, N-Dimethylacetamide to obtain white high-purity solid FDCA. This identifies and visualizes the reaction pathways of HMF oxidation involving electrochemical and nonelectrochemical reactions, and offers a guidance for product separation and purification of value-added chemical production.
Methods
Synthesis of catalysts
Ni2P: 1 mmol of nickel nitrate and 3 mmol of urea were added to 40 mL of water. The NF and the above solution were put into the reactor and hydrothermally heated at 120 °C for 10 h to obtain the precursor. The precursor and 1.0 g of NaH2PO2⋅H2O were placed in a porcelain boat, where the precursor located downstream in the tube furnace. Subsequently, the samples were heated in Ar atmosphere at 300 °C for 1 h to obtain Ni2P40. A-Co-Ni2P: 0.9 mmol of nickel nitrate, 0.1 mmol of cobalt nitrate and 3 mmol of urea were added to 40 mL of water. The rest of the synthesis method was also similar to the procedure for preparing the Ni2P electrocatalyst.
Electrochemical measurements
The electrocatalytic activity was determined by linear scanning voltammetry (LSV) without iR compensation using a standard three-electrode configuration, with the prepared catalyst, Ag/AgCl electrode, and Pt sheet electrode as the working, reference, and counter electrodes, respectively (internal resistance: 0.7 ± 0.2 Ω). The area of the electrode is 1 cm2 with a loading of 2.3 ± 0.2 mg cm−2. The resistance of the electrochemical cell was measured utilizing the resistance measurement function on the CHI 1140C workstation. Both cyclic voltammograms (CV) and LSV data were collected using the CHI 1140C workstation. Before the formal measurements, all the fresh catalysts underwent activation by CV scanning until the electrocatalysts were stable. A-Co-Ni2P electrode with a size of 2.0 × 2.0 cm2 was used as the anodic catalyst and Pt-coated Ti as the cathodic catalyst for the membrane electrode assembly electrolyzer (internal resistance: 0.3 ± 0.1 Ω). The size of the membrane (FAB-PK-130) is 16 cm2 and the thickness is 130 μm, without additional treatment. The calibration of the Ag/AgCl reference immersed in the solution relative to the reversible hydrogen electrode was performed, with two Pt sheets serving as the working and counter electrodes. The difference value between the H+/H2 equilibrium potential and the Ag/AgCl reference electrode was determined by identifying the central point of the two potential values corresponding to zero current in the CV curve. All potentials in this study were referenced to the RHE using the equation:
In situ measurements
In situ ATR-FTIR tests were conducted with an INVENIO-R spectrometer containing a spectro-electrochemical cell. Meanwhile, the in situ ATR-FTIR measurement was triggered with a resolution of 16 cm−1 in the range of 800–3700 cm−1, and corresponding spectra were recorded by the OPUS software. In situ EIS tests were performed using a Energylab instrument over the frequency range from 100 kHz to 0.1 Hz at various potentials in 1 M KOH with or without HMF. In situ Raman spectra were probed on Raman spectroscopy (LABRAM HR EVO). All measurements were performed in an in-situ electrolysis cell containing 1 M KOH with standard three-electrode equipment, and a physical diagram of the equipment is shown in Supplementary Fig. 34.
Data availability
Additional data are available from the corresponding authors on reasonable request. The source data underlying Figs. 2–6 and Supplementary Figs. 4–40 are provided as a Source Data file. Source data are provided with this paper.
References
Ye, F. et al. The role of oxygen-vacancy in bifunctional indium oxyhydroxide catalysts for electrochemical coupling of biomass valorization with CO2 conversion. Nat. Commun. 14, 2040 (2023).
Lei, D. et al. Facet effect of single-crystalline Pd nanocrystals for aerobic oxidation of 5-hydroxymethyl-2-furfural. ACS Catal. 7, 421–432 (2017).
Wang, C. et al. A novel electrode for value‐generating anode reactions in water electrolyzers at industrial current densities. Angew. Chem. Int. Ed. 135, e202215804 (2023).
Van Putten, R.-J. et al. Hydroxymethylfurfural, a versatile platform chemical made from renewable resources. Chem. Rev. 113, 1499–1597 (2013).
Barwe, S. et al. Electrocatalytic oxidation of 5‐(hydroxymethyl) furfural using high‐surface‐area nickel boride. Angew. Chem. Int. Ed. 57, 11460–11464 (2018).
Chen, D. et al. Highly efficient biomass upgrading by a Ni‐Cu electrocatalyst featuring passivation of water oxidation activity. Angew. Chem. Int. Ed. 62, e202309478 (2023).
Wan, X. et al. Base-free aerobic oxidation of 5-hydroxymethyl-furfural to 2, 5-furandicarboxylic acid in water catalyzed by functionalized carbon nanotube-supported Au–Pd alloy nanoparticles. ACS Catal. 4, 2175–2185 (2014).
Jiang, L. et al. PEF plastic synthesized from industrial carbon dioxide and biowaste. Nat. Sustain. 3, 761–767 (2020).
Sheng, H. et al. Linear paired electrochemical valorization of glycerol enabled by the electro-Fenton process using a stable NiSe2 cathode. Nat. Catal. 5, 716–725 (2022).
Chong, L. et al. La-and Mn-doped cobalt spinel oxygen evolution catalyst for proton exchange membrane electrolysis. Science 380, 609–616 (2023).
Cheng, Q., Huang, M., Ye, Q., Deng, B. & Dong, F. Indium-based electrocatalysts for CO2 reduction to C1 products. Chin. Chem. Lett. 35, 109112 (2023).
Wang, T. et al. Combined anodic and cathodic hydrogen production from aldehyde oxidation and hydrogen evolution reaction. Nat. Catal. 5, 66–73 (2022).
Yang, Y. & Mu, T. Electrochemical oxidation of biomass derived 5-hydroxymethylfurfural (HMF): pathway, mechanism, catalysts and coupling reactions. Green Chem. 23, 4228–4254 (2021).
Zeng, L. et al. Cooperative Rh-O5/Ni (Fe) site for efficient biomass upgrading coupled with H2 production. J. Am. Chem. Soc. 145, 17577–17587 (2023).
Lin, L. et al. Low-temperature hydrogen production from water and methanol using Pt/α-MoC catalysts. Nature 544, 544 (2017).
You, B., Jiang, N., Liu, X. & Sun, Y. Simultaneous H2 generation and biomass upgrading in water by an efficient noble‐metal‐free bifunctional electrocatalyst. Angew. Chem. Int. Ed. 55, 9913–9917 (2016).
You, B., Liu, X., Jiang, N. & Sun, Y. A general strategy for decoupled hydrogen production from water splitting by integrating oxidative biomass valorization. J. Am. Chem. Soc. 138, 13639–13646 (2016).
Heidary, N. & Kornienko, N. Operando vibrational spectroscopy for electrochemical biomass valorization. Chem. Commun. 56, 8726–8734 (2020).
Yang, G. et al. Unraveling the mechanism for paired electrocatalysis of organics with water as a feedstock. Nat. Commun. 13, 3125 (2022).
Gao, Y. et al. Electrocatalytic refinery of biomass-based 5-hydroxymethylfurfural to fine chemicals. ACS Catal. 13, 11204–11231 (2023).
Lu, Y. et al. Integrated catalytic sites for highly efficient electrochemical oxidation of the aldehyde and hydroxyl groups in 5-hydroxymethylfurfural. ACS Catal. 12, 4242–4251 (2022).
Yang, Z. et al. Proton transfer mediator for boosting the current density of biomass electrooxidation to the ampere level. Energy Environ. Sci. 17, 1603–1611 (2024).
Wang, Z., Wang, Z., Liu, W., Xiao, W. & Lou, X. W. D. Amorphous CoSnO3@C nanoboxes with superior lithium storage capability. Energy Environ. Sci. 6, 87–91 (2013).
Zhou, L. et al. A restricted dynamic surface self-reconstruction toward high-performance of direct seawater oxidation. Nat. Commun. 15, 2481 (2024).
Dong, J. et al. A robust & weak-nucleophilicity electrocatalyst with an inert response for chlorine ion oxidation in large-current seawater electrolysis. J. Energy Chem. 90, 486–495 (2024).
Li, S. et al. Coordination environment tuning of nickel sites by oxyanions to optimize methanol electro-oxidation activity. Nat. Commun. 13, 2916 (2022).
Wei, L. et al. Amorphous bimetallic oxide–graphene hybrids as bifunctional oxygen electrocatalysts for rechargeable Zn–air batteries. Adv. Mater. 29, 1701410 (2017).
Huang, C., Huang, Y., Liu, C., Yu, Y. & Zhang, B. Integrating hydrogen production with aqueous selective semi‐dehydrogenation of tetrahydroisoquinolines over a Ni2P bifunctional electrode. Angew. Chem. Int. Ed. 131, 12142–12145 (2019).
Lu, L. et al. Tailoring the electron structure and substrate adsorption energy of Ni hydroxide via Co doping to enhance the electrooxidation of biomass-derived chemicals. J. Catal. 424, 1–8 (2023).
Ge, R. et al. Selective electrooxidation of biomass‐derived alcohols to aldehydes in a neutral medium: Promoted water dissociation over a nickel‐oxide‐supported ruthenium single‐atom catalyst. Angew. Chem. Int. Ed. 61, e202200211 (2022).
Zhou, P. et al. Heterogeneous interface enhanced adsorption of organic and hydroxyl for biomass electrooxidation. Adv. Mater. 34, 2204089 (2022).
Shao, L. et al. In situ generation of molybdate‐modulated nickel‐iron oxide electrodes with high corrosion resistance for efficient seawater electrolysis. Adv. Energy Mater. 14, 2303261 (2024).
Yang, H. et al. Sequential phase conversion‐induced phosphides heteronanorod arrays for superior hydrogen evolution performance to Pt in wide pH media. Adv. Mater. 34, 2107548 (2022).
Görlin, M. et al. Oxygen evolution reaction dynamics, faradaic charge efficiency, and the active metal redox states of Ni–Fe oxide water splitting electrocatalysts. J. Am. Chem. Soc. 138, 5603–5614 (2016).
He, Z. et al. Promoting biomass electrooxidation via modulating proton and oxygen anion deintercalation in hydroxide. Nat. Commun. 13, 3777 (2022).
Xia, Z. et al. Vacancy optimized coordination on nickel oxide for selective electrocatalytic oxidation of glycerol. ACS Catal. 14, 1930–1938 (2024).
Qian, Q. et al. Electrochemical biomass upgrading coupled with hydrogen production under industrial‐level current density. Adv. Mater. 35, 2300935 (2023).
Vorontsov, I. I. et al. Capturing and analyzing the excited-state structure of a Cu (I) phenanthroline complex by time-resolved diffraction and theoretical calculations. J. Am. Chem. Soc. 131, 6566–6573 (2009).
Lu, X. et al. Highly efficient electro‐reforming of 5‐hydroxymethylfurfural on vertically oriented nickel nanosheet/carbon hybrid catalysts: structure–function relationships. Angew. Chem. Int. Ed. 133, 14649–14656 (2021).
Chen, L. et al. Microscopic‐level insights into P–O‐induced strong electronic coupling over nickel phosphide with efficient benzyl alcohol electrooxidation. Small 20, 2306410 (2024).
Acknowledgements
This work was partly supported by the National Natural Science Foundation of China (NSFC, No. 22078052, C.Y.), the Fundamental Research Funds for the Central Universities (DUT23LAB612, C.Y.), and the National Key Research and Development Program of China (Grant No. 2022YFB4101600, J.Q.).
Author information
Authors and Affiliations
Contributions
J.Q. and C.Y. supervised and revised the manuscript. L.C. conceived the concept and carried out the experiments. L.C. wrote the paper and conducted the material characterization. L.C. and X.S. performed the DFT calculations studies. L.C., X.S., J.D. and J.M. discussed and reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jing-Li Luo, Chularat Wattanakit and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, L., Yu, C., Song, X. et al. Integrated electrochemical and chemical system for ampere-level production of terephthalic acid alternatives and hydrogen. Nat Commun 15, 8072 (2024). https://doi.org/10.1038/s41467-024-51937-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51937-y
This article is cited by
-
Redesigning electrolysers in coupled electrolysis
Nature Catalysis (2025)
-
Synergistic enhancement of electrochemical alcohol oxidation by combining NiV-layered double hydroxide with an aminoxyl radical
Nature Communications (2025)
-
Selective electrooxidation of 5-hydroxymethylfurfural at pilot scale by engineering a solid polymer electrolyte reactor
Nature Catalysis (2025)
