
Abstract
Neoadjuvant immune checkpoint blockade (ICB) has shown unprecedented activity in mismatch repair deficient (MMRd) colorectal cancers, but its effectiveness in MMRd endometrial cancer (EC) remains unknown. In this investigator-driven, phase I, feasibility study (NCT04262089), 10 women with MMRd EC of any grade, planned for primary surgery, received two cycles of neoadjuvant pembrolizumab (200 mg IV) every three weeks. A pathologic response (primary objective) was observed in 5/10 patients, with 2 patients showing a major pathologic response. No patient achieved a complete pathologic response. A partial radiologic response (secondary objective) was observed in 3/10 patients, 5/10 patients had stable disease and 2/10 patients were non-evaluable on magnetic resonance imaging. All patients completed treatment without severe toxicity (exploratory objective). At median duration of follow-up of 22.5 months, two non-responders experienced disease recurrence. In-depth analysis of the loco-regional and systemic immune response (predefined exploratory objective) showed that monoclonal T cell expansion significantly correlated with treatment response. Tumour-draining lymph nodes displayed clonal overlap with intra-tumoural T cell expansion. All pre-specified endpoints, efficacy in terms of pathologic response as primary endpoint, radiologic response as secondary outcome and safety and tolerability as exploratory endpoint, were reached. Neoadjuvant ICB with pembrolizumab proved safe and induced pathologic, radiologic, and immunologic responses in MMRd EC, warranting further exploration of extended neoadjuvant treatment.
Similar content being viewed by others


Introduction
Endometrial cancer (EC) is the most common gynaecological malignancy in high-income countries, with a rising incidence and mortality1,2. 20-30% of cases demonstrate DNA mismatch repair deficiency (MMRd), due to germline defects in MMR genes MLH1, MSH2, MSH6 or PMS2 (Lynch syndrome) or somatic MLH1 loss, resulting in a higher tumour mutational burden (TMB) in the majority of MMRd EC cases. Currently, these are managed by total hysterectomy and bilateral salpingo-oophorectomy, with adjuvant (chemo)radiotherapy given depending on disease grade and stage. Despite appreciable short- and long-term morbidity of such therapy3, disease recurrence is common, particularly in high-grade tumours. The unprecedented activity of immune checkpoint blockade (ICB) in MMRd tumours in the metastatic setting has motivated its investigation in earlier disease stages. A recent study in colon cancer demonstrated ~95% major pathological responses (MPRs) after only 15 days of treatment with little toxicity, raising possibility of organ-sparing approaches4. However, despite the considerable efficacy of ICB in advanced EC5,6, its activity in localised disease remains unknown. Here, we present results of the phase I PAM trial (NCT04262089), in which patients with resectable EC received 2 cycles of neoadjuvant pembrolizumab monotherapy intravenously (i.v.) every three weeks, before surgical resection and adjuvant therapy where indicated. All patients completed treatment without severe toxicity. A partial radiologic response was observed in 3 out of 10 patients, 5 out of 10 patients had stable disease on Magnetic Resonance Imaging (MRI). A pathologic response was observed in 5 out of 10 patients, with 2 patients showing a major pathologic response.
Results
Patient characteristics
Twelve patients were screened for enrolment between December 2020 and October 2022. Two patients were excluded during screening prior to initiation of treatment. Ten patients met the inclusion criteria and were treated with two cycles of neoadjuvant pembrolizumab 200 mg IV Q3W prior to their standard-of-care (SoC) hysterectomy (Supplementary Fig. 1a,b). This regimen was chosen based on previous neoadjuvant ICB trials and the need to balance benefit with toxicity risk in this population with relatively favourable prognosis. Detailed baseline patient characteristics are shown in Table 1. The median age was 61.5 years (range, 54–71 years). Loss of MLH1 and PMS2 were most common (80%). Two patients had Lynch syndrome. Most tumours were of endometrioid histotype (90%) and high-grade (grade 3 (G3)) (60%). All ten patients completed treatment with pembrolizumab on schedule and were evaluated for safety, efficacy and translational endpoints.
Safety and feasibility
Neoadjuvant pembrolizumab was feasible and could be administered safely (exploratory endpoint), with no ≥grade 3 immune-related adverse events (irAEs). (Table 2). Toxicities were consistent with those expected, including fatigue, myalgia, arthralgia and hypothyroidism (managed medically). One patient developed pleural effusion and ascites. This could be established not to be related to the investigational treatment. Pleural effusion was characterized by lymphocytosis, histiocytes and reactive mesothelial cells, but not with signs of malignancy. This pleural effusion required drainage. Both the pleural effusion and ascites resolved spontaneously. All patients underwent SoC hysterectomy within the predefined eight weeks after diagnosis. Surgically-related serious adverse events were observed in one patient (10%), who developed an abdominal and subcutaneous abscess after surgery. No evidence of a relationship to pembrolizumab treatment was found.
Neoadjuvant pembrolizumab induces pathological and radiological responses
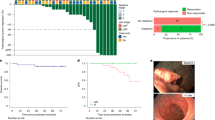
Pathological (primary endpoint) and radiological response (secondary endpoint) were determined according to our predefined criteria for treatment response. Radiological evaluation of treatment response was performed using MRI at baseline and post-treatment immediately prior to surgery. Eight of the ten patients had measurable disease as per RECIST v1.1 at baseline and could be evaluated for radiological tumour response (ORR) (Fig. 1a,c). Of the two patients that were not evaluable for ORR, one patient (PAM-003) had a target lesion that was not measurable on both baseline and post-treatment MRI and one patient (PAM-008) did not have MRI scans due to COVID-19 restrictions. None of the evaluable patients (0%) showed progressive disease and six patients (75%) showed a decrease in the sum of the longest diameter (SLD) of the tumour on MRI after neoadjuvant pembrolizumab treatment (Fig. 1a). By investigator review, the ORR reached 37.5% (95% CI 8.52–75.51%), with all responding patients achieving a partial response (PR).

a Waterfall plots showing the percentage pathologic regression and the percentage radiographic change from baseline in the sum of the target lesion after neoadjuvant administration of 2 cycles of pembrolizumab (n = 10). The first dotted line indicates the cutoff for radiological partial response (PR) according to RECIST 1.1. The second dotted line depicts the threshold for major pathological response (MPR) corresponding to 90% regression. The orange bar colours reflect the pathologic response (dark orange: MPR; light orange: pathological Partial Response (pPR)). The pink bar colours represent the radiologic response (dark pink: partial response (PR); light pink: stable disease (SD)). Pathological features are annotated for each patient and include histological subtype, tumour grade at baseline and in the resection specimen, (pathologic) FIGO stage after treatment, expression of MisMatch Repair (MMR) proteins, and the aetiology of MMR deficiency (MMRd). The numbers above the bars represent the study ID of each patient. *: no pathologic response; **: no pathologic response and radiologic response not measurable and/or not determined due to Covid restrictions. b Swimmer plot depicting per patient the timeline of follow-up, type of surgery, lymph nodes debulking at time of surgery, type of adjuvant therapy, and recurrence / mortality (n = 10). c Representative MRI images of the uterus pre- versus post-treatment with 2 cycles of neoadjuvant pembrolizumab from PAM-004. Similar experiments conducted for 8 patients d Representative H&E images of pre- versus post-treatment biopsy tissue from PAM-004. The pretreatment biopsy shows a grade 3 endometrioid adenocarcinoma. The post-treatment biopsy shows the fibrotic tumour bed with immune-cell infiltration. Similar experiments conducted for all 10 patients. Source data are provided as a source data file. Abbreviations: ICB immune checkpoint blockade, MRI magnetic resonance imaging, MPR major pathological response, pPR partial pathological response, PR partial response, SD stable disease.
All ten patients were evaluated for pathological response (Fig. 1a,d). Five patients (50%) had a pathological response, of whom two patients (20%) had a MPR with 10% residual viable tumour cells. Patients classified as pathological partial responders (pPR) had residual viable tumour varying from 12–50% (Fig. 1a). The tumour regression observed in most patients was characterized by a tumour bed without viable tumour cells consisting of fibroinflammatory changes and in some instances focal aggregations of macrophages, and a mixed distribution (both in the tumour centre and the surrounding areas) of treatment response. Alongside aforementioned characteristics, multinucleated giant cells, individual or in combination with histiocytes, and a band-shaped lymphocyte infiltration at the advancing front were present in the resection specimens of patients with a MPR.
9/10 patients are still alive. One patient passed away due to reasons not related to her treatment or disease. The median follow-up time was 22.5 months (12–32 months) with a median and longest disease-free survival of 20 and 32 months, respectively (Fig. 1b; cut-off date 01-11-2023). Disease recurrence was observed in two non-responders (NR) by imaging and histologically, at 47 and 93 weeks after surgery (Fig. 1b). 3/10 patients received adjuvant therapy, while 7/10 patients underwent follow-up without adjuvant treatment (Table 3, Fig. 1b).
Clinicopathologic predictors and correlates of response
Post-hoc analysis revealed that radiologic and pathologic responses were deeper in patients with pre-operative grade 3 disease, although this did not reach statistical significance (Fig. 2a). Lynch syndrome was observed only in non-responding patients (Fig. 2b). Furthermore, upon comparing mutations in MMR genes with hypermethylation of these genes, a more favourable treatment response was observed in the hypermethylation group (8/10 patients) (Fig. 2c). Two patients from the hypermethylation group exhibited mutations within the MMRd genes, as detected by TruSight Oncology 500 (TSO500) sequencing. Despite a higher tumour mutational burden in patients with MMR-gene mutations, this elevated mutational burden did not subsequently impact the response rate (Fig. 2d). Analysis of common driver mutations using the TSO500 panel did not reveal any significant associations with treatment response (Fig. 2e). Notably, both patients experiencing recurrent disease after study treatment exhibited TP53 mutations.

a Violin plots showing the percentage of pathologic and radiologic tumour regression among patients with pre-operative low-grade tumour (grade 1 and 2) (n = 4) versus patients with a pre-operative high-grade tumour (grade 3) (n = 6). The white dots represent the means of the data, and the error bars indicate the 95% confidence intervals around the means. Statistical significance was assessed using the Mann–Whitney U test. b Violin plots depicting the percentage of pathologic and radiologic tumour regression among patients with MMRd caused by Lynch syndrome (n = 2) versus patients with a sporadic mutation (n = 8). The white dots represent the means of the data, and the error bars indicate the 95% confidence intervals around the means. Statistical significance was assessed using the Mann–Whitney U test. c Violin plots showing the percentage of pathologic and radiologic tumour regression among patients with a mutation in their MMR genes (n = 4) versus patients with hypermethylation of their MMR genes (n = 6). The white dots represent the means of the data, and the error bars indicate the 95% confidence intervals around the means. Statistical significance was assessed using the Mann–Whitney U test. d Dot plot showing the mutations per megabase (Mbase) among patients with a mutation in their MMR genes (n = 4) versus patients with hypermethylation of their MMR genes (n = 6) and showing the mutations per Mbase among patients with a MPR/pPR (n = 5) versus patients with NR (n = 5). e, Analysis of (potentially) pathogenic variants using TSO500, represented in an oncoprint format (n = 10). Mutations are visually differentiated by type, with the frequency of mutations per gene and per patient depicted. Source data are provided as a Source Data file. Abbreviations: G1,2,3 grade, MMRd mismatch repair deficiency, MPR major pathological response, pPR partial pathological response, NR non-responders.
Immunologic predictors and correlates of response
The infiltration of T and B cells was evaluated post-hoc by immunohistochemical analysis of CD3, CD8, and CD19, using the pre-treatment biopsies, post-treatment biopsies and resection specimens (Fig. 3a). Baseline infiltrate was not associated with radiologic response (Fig. 3b), nor with pathologic responses (Fig. 3c). Indeed, most responders were characterized by a baseline immune “desert” phenotype. On-treatment, ICB was associated with increases in both epithelial and stromal T and B cell density in 8 of 10 cases (Figs. 3b,c). However, no association was observed with either radiologic or pathologic responses (Figs. 3b,c, respectively).

a Representative H&E, CD3, CD8, and CD19 staining images of pre- (top) and post-treatment biopsy tissue (bottom) from PAM-004. Similar experiments conducted for all 10 patients. b Pre- to post-treatment changes in CD3+, CD8+, CD19+ densities in patients with a radiologic partial response (PR) (n = 3) and patients with stable disease (SD) on MRI (n = 5). Statistical significance was assessed using a mixed-effects model. c Pre- to post-treatment changes in CD3+, CD8+, CD19+ densities in patients with a pathologic response (pPR and MPR) (n = 5) and patients without a pathologic response (Non-pPR) (n = 5). Statistical significance was assessed using a mixed-effects model. d Heatmap of included patients with data on pathologic and radiologic response, location of immune infiltrate, TLS, and CD3+, CD8+ and CD19+ densities. Clustering of CD3+, CD8+ and CD19+ densities stratified by location was done by hierarchical clustering using Ward’s method. Source data are provided as a Source Data file. MPR major pathological response, pPR partial pathological response, PR partial response, SD stable disease, TLS tertiary lymphoid structure.
Hierarchical clustering of immune infiltrate in the post-treatment tumour, invasive margin and peritumoral regions of the resection specimens similarly revealed no overt association with response (Fig. 3d). Five patients harboured mature tertiary lymphoid structures (TLSs), characterized by a Bcl6+ germinal centre (GC) and CD21+ follicular dendritic cells (FDCs) in the peritumoral myometrium of their resection specimen (Supplementary Fig. 2). Immature TLS, characterized by scattered Bcl6+ cells and a minimal FDC network, and without the characteristic structural organisation of a mature TLS were also present in three patients (Supplementary Fig. 2). TLS did not associate with radiologic or pathologic response (Fig. 3d).
We finally assessed T cell clonality and diversity by analysing the complementary determining region 3 (CDR3) repertoire of the TCR β-chain (TCRβ) in pre- and post-treatment biopsies (Supplementary Data 1). Paired analysis (n = 8) revealed a significant monoclonal expansion in 3/3 radiologic responders and both MPR cases (Fig. 4a; P = 0.016). No monoclonal expansion was observed in partial pathologic responders or non-responders (Fig. 4a). Of note, TCRβ CDR3s were not shared between patients and were largely concordant between post-treatment biopsies and resection specimens (Fig. 4b). Nevertheless, TCRβ heterogeneity was observed between post-treatment biopsies and resection specimens, in line with recent work7.

a Paired dot plot of bulk TCR CDR3 sequencing from pre- and post-treatment biopsy tissues (n = 8). Individual patients are annotated by their pathologic and radiologic response. b Heatmap showing correlation between TCR CDR3 sequences in post-treatment biopsies and resection specimens (n = 6), corresponding to those used for IHC in Fig. 3. c UMAP visualization of 11,870 T cells with identical CDR3 TCRs present in bulk TCR sequencing data from the same patient. Clusters are colour-coded by inferred identity. d Bubble plot showing expression and abundance of key canonical marker genes for T cell clusters. e Bar graph of cell subset fractions within significantly expanded and non-expanded populations as determined by bulk TCR sequencing. f Bar graph of clone sizes within expanded and non-expanded populations from bulk TCR sequencing. g UMAP visualization of 199 significantly expanded CD4 T cells, colour-coded by inferred identity. h Circle plot showing shared full-length TCR sequences between significantly expanded CD4 T cells from different clusters. i UMAP visualization of 3014 CD8 T cells with significantly expanded CDR3 TCRs from pre- vs post-treatment bulk TCR sequencing. Clusters are colour-coded by inferred identity. j Bubble plot of marker gene expression and abundance differentially expressed among CD8 T cell subsets. k Scatter plot showing clonal diversity as a function of clone size for significantly expanded CD8 T cells, colour-coded by most abundant cluster. l Bar graph of cell subset fractions within the most abundant ( > 10 identical TCRs) expanded CD8 T cell clones, with each bar representing an individual clone. m Bar graph showing inferred identity of cycling T cells within individual expanded CD8 T cell clones, with each bar representing cycling T cells from an individual clone. Abbreviations: MPR major pathological response, pPR partial pathological response, PR partial response, SD stable disease. Source data are provided as a Source Data file.
Pathologic responders are characterized by a CD8-driven T cell response
To determine the T cell phenotype associated with anti-PD1 response, we generated post-hoc single-cell combined transcriptome and T cell receptor (TCR) data from 21,765 tumour-infiltrating CD4+ and CD8+ T cells from 3 patients for which single T cells could be retrieved from the tumour: PAM-010 ( ~ 50% pPR), PAM-011 ( ~ 20% pPR) and PAM-007 (NR) (Supplementary Fig. 3).
In total, 11,870 T cells expressed a unique TCRβ sequence also detected by bulk TCRβ sequencing of the corresponding post-treatment biopsy, and were used in subsequent analyses. T cells were broadly distributed among seven main populations (Fig. 4c), including cycling PD1high cells (MKI67, PDCD1), exhausted T (Tex) cells (PDCD1 TOX), activated CD4 T cells (CXCL13) and regulatory T (Treg) cells (FOXP3) (Fig. 4d, Supplementary Fig. 3b, Supplementary Data 2). Similar clusters were observed when analysing all 21,765 T cells (Supplementary Fig. 3c,d). ICB-expanded T cells were enriched for CD8 (exhausted) clusters (Fig. 4e), but only a subtle relationship with overall post-treatment clone size was observed (Fig. 4f). We next analysed both expanded CD4 and CD8 T cells in more detail. The small number of ICB-expanded CD4 T cells consisted largely of Tregs (FOXP3), activated (CXCL13) and exhausted CD4 T cells (CXCL13 LAG3) (Fig. 4g, Supplementary Data 3). CD4 Tex cells were clonally related to activated CD4 T cells (Morisita index 0.357), as were a subset of Tregs (Morisita index 0.097). ICB-expanded CD8 T cells consisted of a precursor exhausted (Tpex) population (IL7R TCF7 XCL18), exhausted T (Tex) cells (NR4A1 NR4A29), precursor (CXCR610) epithelial-infiltrating (ITGAE) T cells (Tep11), a TOX-high cycling (MKI67) Tep subset12, and cytotoxic (NKG7) LAG3 Tep cells (Figs. 4h,4i, Supplementary Data 4, Supplementary Fig. 3e). An atypical population of CD8 T cells was also observed, defined by activation-induced genes JUN13 and IL3214 (Fig. 4i), the latter of which has recently been linked to improved immunity in melanoma15. A subset of IL32 CD8 cells were cycling, as determined by G2M- and S-phase scoring (Supplementary Fig. 3f). We explored whether a potential relationship existed between TCR(β) usage and specific T cell states following ICB. To this end, we used a recently reported clonotype bias metric16. Clonotype bias was minimal (Fig. 4j), concordant with the significant phenotype heterogeneity observed when analysing clones on an individual level (Fig. 4k). In line with recent models, most clones contained at least one cell with a Tpex phenotype (Fig. 4k)17,18. Finally, in 27/29 expanded cycling Tep clones, cycling Tep were derived from pre-Tep or LAG3low Tep cells, suggesting LAG3high Tep cells represent a terminally-differentiated cytotoxic subset (Fig. 4l,m).
Responding T cell clones are disseminated across the draining lymph node network
Stem-like and/or Tpex clones in the tumour-draining lymph nodes (TDLNs) have been linked to the success of ICB in preclinical models of cancer. We therefore performed an exploratory post-hoc analysis on the presence of expanded T cells clones in the TDLN from patients that underwent a SoC lymphadenectomy alone, or in combination with a sentinel lymph node (SLN) biopsy. ICB-expanded clones were observed in the TDLN for MPR cases PAM-004 (50%) and PAM-009 (78%), for pPR cases PAM-010 (22%) and PAM-011 (100%), and for non-responder PAM-007 (17%; Fig. 5a). With the exception of non-responder PAM-007, the largest expanded clone was always observed in the TDLN. Phenotypic analysis using the single-cell dataset revealed that the naïve/Tcm-like cluster (Fig. 5c), which includes expanded CD8 Tpex cells (Fig. 5b), was enriched in intra-tumoural T cell clones also observed within the TDLN (Fig. 5c), with the highest TDLN frequency corresponding to clones only observed in this cluster (Fig. 5d). We speculate these cells may represent bystander T cells not involved in the ICB response.

a Scatter plot of bulk TCR CDR3 sequencing from patients with post-treatment biopsy and TDLN tissue (n = 5). Significantly expanded clones (pre- vs post-treatment bulk TCR sequencing) are highlighted in orange. Dot size represents BH-adjusted P-value for ICB-induced clonal expansion. Patients are annotated by pathological and radiological response. b Projection of significantly expanded CD8 T cells onto the global T cell UMAP, colour-coded by inferred CD8 T cell identity. Non-expanded CD8 and CD4 T cells are shown in grey. c Left: UMAP of 2910 T cells with identical CDR3 TCRs in bulk TCR sequencing from at least one TDLN of the same patient. Clusters are colour-coded by inferred identity. T cell clones not detected in any TDLN are shown in grey. Right: Bar graph showing the fraction of indicated cell subsets within clones detected or not detected in a TDLN by bulk TCR CDR3 sequencing. d UMAP visualization of 2910 T cells with identical CDR3 TCRs in bulk TCR sequencing data from the same patient. Cells from a given T cell clone are colour-coded by their relative frequency in the TDLN. T cell clones not detected in any TDLN are shown in grey. e UMAP of 519 T cells from patient PAM-007 with identical CDR3 TCRs in bulk TCR sequencing. T cell clones are colour-coded by CDR3 sequence. Left: Clones in ‘left’ sentinel TDLN. Middle: Clones in ‘right’ sentinel TDLN. Right: Clones in both TDLNs. f Venn diagram showing overlap of T cell clones in 3 sentinel TDLNs from patient PAM-004. g Venn diagram showing overlap of T cell clones in 4 TDLNs from the Obturator station of patient PAM-004. h Alluvial plot showing distribution of significantly expanded T cell clones from patient PAM-004 across post-treatment biopsy and indicated (sentinel) TDLNs. Source data are provided as a Source Data file. Abbreviations: BH Benjamini–Hochberg, MPR major pathological response, pPR partial pathological response, PR partial response, SD stable disease, TDLN tumour-draining lymph node, SLN sentinel lymph node.
We next took advantage of the multiple lymph nodes removed in some patients to perform additional exploratory analyses on a patient-by-patient basis. Two SLNs were removed from PAM-007, which contained phenotypically distinct T cell clones (Fig. 5e). In addition, Tcm-like clones were observed in both SLNs of this patient. The entire TDLN system from best-responder PAM-004 was also available for analysis. Comparison between 3 different SLN revealed a diverse composition of (overlapping) clones (Fig. 5f). The 4 (SLN and non-SLN) lymph nodes from the obturator station contained a similarly heterogenous constellation of (overlapping) T cell clones (Fig. 5g). Nevertheless, 4 out of the total 8 (50%) expanded T cell clones observed after treatment were found in at least 2 TDLN, with some found in all 6/6 nodes analysed (Fig. 5h). These data suggest that for both highly responsive and non-responsive cases, anti-PD1 ICB-responding T cells disseminate widely across the (tumour-draining) lymph node network.
Heterogeneous MMR expression as response determinant
In a single patient (PAM-004) we observed that despite a MPR, a mismatch repair proficient (MMRp) component remained in her resection specimen, comprising almost the entirety of the residual tumour cells (Fig. 6a). By contrast, only a minimal number of MMRd cells were detected after treatment (Fig. 6a). This residual MMRp area was associated with a reduced immune-cell infiltration compared to the fibrotic tumour bed after treatment (Fig. 6b). As recent work has nevertheless demonstrated effective ICB responses in experimental models of (mixed) MMRp and MMRd tumours4, we analysed post-hoc T cell gene expression in the MMRp regions after treatment. We compared differentially expressed genes in T cells of PAM-004 from before the initiation of neoadjuvant pembrolizumab (i.e., pre-treatment biopsy) and after the administration of neoadjuvant pembrolizumab (i.e., resection specimens) for both the MMRp area and the fibrotic tumour bed (where MMRd cells most likely previously resided).

a Representative H&E image of PAM-004 hysterectomy sample showing MPR after two ICB cycles (n = 1). b CD3 staining on PAM-004 hysterectomy sample. c MLH1 staining on PAM-004 hysterectomy sample showing MMRp (left) and fibrotic tumour bed (FTB) (right) regions (n = 1). d Fluorescent staining of PAM-004 hysterectomy sample: CD3 (red/orange), CD20 (yellow), DNA (green), PanCK (blue) (n = 1). e Spatial transcriptomics of PAM-004 hysterectomy sample with ROIs: ROI005, ROI001, ROI003 in MMRp (left) and ROI006, ROI002, ROI004 in FTB. f,g Images of ROI006 in FTB (upper row) and ROI005 in MMRp (upper row): f CD3 mask for T cells (orange), g PanCK mask for tumour cells (blue). h Representative H&E image of pretreatment biopsy from PAM-004 (n = 1). i Fluorescent staining of PAM-004 pretreatment biopsy: ROIs ROI008, ROI013, ROI007, ROI009. CD3 (red/orange), CD20 (yellow), DNA (green), PanCK (blue) (n = 1). j Image of ROI009 with CD3 mask for T cells (orange). k,l Volcano plots comparing gene signatures in T cell areas (CD3 mask) of pretreatment biopsy vs. hysterectomy sample from FTB or MMRp regions. Blue dots: genes enriched in pretreatment biopsy. Orange dots: genes enriched in FTB or MMRp hysterectomy sample. Differential expression analysis was done using a linear mixed model with fixed effects for condition and random effects for subject-specific variability. P-values adjusted by Benjamini–Hochberg procedure. m Volcano plot comparing gene signatures in T cell areas (CD3 mask) within FTB vs. MMRp regions of hysterectomy sample. Blue dots: genes enriched in FTB region. Differential expression analysis was done using a linear mixed model with fixed effects for condition and random effects for subject-specific variability. P-values adjusted by Benjamini–Hochberg procedure. n Scatter plot of genes expressed in MLH1+ cells within tumour cell areas (PanCK mask) of FTB vs. MMRp region. Source data are provided as a source data file. Abbreviations: FTB fibrotic tumour bed, MMRp mismatch repair proficient. Source data provided as a Source Data file.
T cells from three regions of interest (ROIs) in both areas were selected (Fig. 6a,d–e). The fibrotic tumour bed exhibited minimal Pan-cytokeratin (Pan-CK) staining and a high number of CD3 cells compared to the pre-treatment biopsy (Fig. 6f,g,j, Supplementary Fig. 4d,e). The MMRp areas were characterized by Pan-CK staining comparable to the pre-treatment biopsy and a minimal infiltration of T cells (Fig. 6f,g, Supplementary Fig. 4f,g).
Differential expression analysis revealed an upregulation of genes involved in T cell activation in both the fibrotic tumour bed and MMRp areas, suggesting T cells in the MMRp areas were activated to a similar extent as those found in the fibrotic tumour bed (Fig. 6k–m, Supplementary Fig. 4h). Notably, comparing the transcriptomic profile of Pan-CK+ cells from the MMRp areas to the surviving Pan-CK+ cells in the fibrotic tumour bed revealed a highly conserved signature and no differentially expressed genes (Fig. 6n). Subsequent IHC analysis of MLH1 and PMS2 expression in both areas revealed that the residual Pan-CK+ cells were MMRp (Fig. 6c, Supplementary Fig. 4c). Taken together, these data suggest that the determinants of effective cytolysis against the tumour did not occur at the level of T cell activation in this patient.
Discussion
In the present study, we show that neoadjuvant treatment of resectable MMRd EC with two cycles of pembrolizumab monotherapy leads to a major or partial pathological response in 50% of the patients. Neoadjuvant treatment was safe and feasible without delay of planned surgery and without any grade >2 treatment-related AEs (trAEs). Robust immune activation was observed in almost all patients in both the primary tumour and the TDLN.
Despite the high rate of pathological responses on histologic examination of the primary tumours, the radiological response rate was 37.5%. Two patients showing a pathological response did not demonstrate concomitant radiological shrinkage by imaging. In line with our findings, previous studies with neoadjuvant ICB also indicated a discrepancy between radiological response and the actual degree of pathologic tumour regression4,19,20,21,22. This discrepancy between the radiological and pathological response may be attributed to immune-cell infiltration into the tumour and/or by degradation of tumour tissue by fragmentation rather than by shrinkage that RECIST v1.1 does not account for.
Previous studies with neoadjuvant ICB in early-stage resectable melanoma, colon, lung, and urothelial carcinoma reached pathological response rates varying from 30-100% after one to three cycles of mono- or combination ICB4,19,20,21,22,23,24,25,26. Trials that reported higher rates of pathological responses ( > 75%) were predominantly those that utilized combination ICB regimes comprising anti-PD1 and CTLA-4, most notably the recent NICHE-I and II trials in MMRd colon cancers4,19. Combined with the recent data on neoadjuvant anti-PD1 monotherapy in MMRd rectal cancer, it is likely that differences in response rates between the current study and the NICHE trials are related to the dual treatment regime. Nevertheless, it cannot be excluded that baseline immunological disparities between MMRd tumours at different sites, such as T cell density and IFNγ pathway activity27, predispose to distinct clinical responses to ICB. In addition, the time required to respond in EC is largely unknown. It could be hypothesized that response evaluation within a few weeks after the second cycle of immunotherapy does not offer the opportunity to observe the maximal therapeutic effect.
A notable portion of MMRd EC patients currently undergo adjuvant radio(chemo)therapy, which affects quality of life (QoL). Although adjuvant therapy improves loco-regional control, it does not improve disease-specific survival in MMRd patients28 and there is no significant difference in the effect of radiotherapy versus radiotherapy combined with chemotherapy in patients with MMRd EC29. If extended neoadjuvant ICB can achieve response rates similar to those reported in rectal cancer, neoadjuvant ICI followed by surgery could offer a viable alternative to the standard-of-care. Furthermore, neoadjuvant pembrolizumab may provide a treatment option for patients with comorbidities contraindicating surgery or as an organ-sparing treatment for young women desiring fertility preservation. Future studies will need to evaluate the efficacy and benefits of neoadjuvant pembrolizumab in these populations.
Conclusive data on the efficacy of ICB in patients with Lynch syndrome (LS) compared to sporadic MMRd is lacking. A phase 2 trial in 24 patients with MMRd EC (6 LS and 18 sporadic tumours) suggested higher ORR, progression-free survival (PFS), and overall survival (OS) in LS-associated recurrent MMRd EC compared to sporadic MMRd EC30. However, outcomes were not adjusted for endometrial cancer grade and/or stage. Conversely, analysis of NRG GY018 data following combination treatment with chemotherapy and immunotherapy found no differences in outcomes and responses between methylated and non-methylated MMRd patients31, nor did a retrospective analysis of methylated and non-methylated MLH1 loss in EC patients treated with pembrolizumab32. In our cohort, although small, patients diagnosed with LS-associated MMRd EC did not show higher rates of pathological and radiological response to neoadjuvant ICB, when compared to patients with sporadic MMRd EC. In our cohort, we observed that patients with high-grade (grade 3) disease at baseline, characterized by a tumour consisting of >50% solid architecture at baseline, exhibited a more favourable treatment response than patients with low-grade (grade 1–2) disease, predominantly glandular architecture. These data are in line with NRG GY018, where a trend towards favourable response was observed for grade 3 tumours31. Whether these differences reflect underlying immune biology such as a higher TMB and/or influx of lymphocytes or kinetic differences in response remains to be determined.
The majority of responders in the current trial were characterized by a low baseline density of T and B cells, often referred to as an “immune desert”. Such deserted tumours have generally been characterized as less responsive to ICB33. However, this has not been thoroughly explored within the confines of a single molecularly-stratified tumour type, such as MMRd EC. It could be speculated that genomic response features such as TMB underly the baseline immune differences linked to response in other tumour types. On a relatively homogenous genomic background such as MMRd, these differences may carry different predictive value. It will be interesting explore this hypothesis in larger molecularly-stratified randomized trials, such as the RAINBO-green trial for MMRd EC34.
Despite the low baseline immune infiltration, our data on clonal expansion and TLDN-involvement are consistent with observations that cytotoxic reinvigoration, clonal replacement, and clonal resurgence all operate to drive T cell expansion within distinct timeframes post-ICB35. In particular, we note that robust clinical responses in the current study are paralleled by a single (TDLN-associated) T cell clone that emerged as the dominant occupant of the clonal space after ICB treatment. The TCR dynamics associated with (long-term) responses to ICB have not been fully characterized, and may differ between tumour type. In lung cancer patients, durable response to immune checkpoint blockade correlated with early and sustained expansion of one to two dominant T cell clones36. By contrast, recent work in melanoma found that a higher amount of TCR clonotypes after ICB discriminated responders from non-responders37. In addition to tumour type, clonotype discrepancies may also result from the timing of analysis, analysis of PBMC versus tumour compartment, or the use of mono- versus dual-ICB37. With respect to the latter, our work and that of others38 suggests anti-PD1 monotherapy minimally expands CD4 T cells, which might account for the more restricted clonotype expansion. The lack of CD4 expansion might also account for the absence of mature TLS in the majority of our patients. In the neoadjuvant ICB NICHE trial in MMRd colon cancer, TLS were observed in the majority of cases following combined treatment with anti-PD1 and anti-CTLA-44. As CD4 cells are thought to play a key role in TLS formation and maturation, it is plausible that the lack of CD4 cell expansion observed here and in other studies with anti-PD1 monotherapy is a contributing factor to the lack of TLS38. Altogether, the time window of the study treatment, the use of mono- instead of dual-ICB and the differences in tumour microenvironment (TME) between cancers could contribute to the discrepancies observed.
Our observation that a subset of the ICB-expanded clones was also detected within the TDLNs aligns with previous studies where an overlap between clonotypes of TDLNs and the corresponding tumour was observed39,40. It is interesting to note, however, that not all clones distributed to the entire TDLN network, and substantial heterogeneity was observed, even within a given TLDN station. It is therefore possible that a subset of clones has disseminated systemically to non-TLDN and expanded upon systemic ICB, followed by tumour homing. This might account for the observation that not all expanded clones in the current study where present in the TDLN, and the further absence of mature TLS in the majority of patients suggests that these structures do not form the only reservoir for clones absent from the TDLN as previously proposed41.
Our study is limited by the small cohort and the relatively short duration of post-operative follow-up. To establish more robust conclusions, larger studies combined with a minimum follow-up period of three years are necessary to determine if the observed (M)PRs correlate with improved disease-free and overall survival. Additionally, investigating the potential association between changes in the TME (such as TCR clonality and T cell influx) observed in some of the non-responding patients and improved survival outcomes would be of interest.
In contrast to work in MMRd (colo)rectal cancer, no complete pathologic responses were observed in the current study. The response rate to neoadjuvant ICB in MMRd EC can likely be improved by inclusion of anti-CTLA-4 therapy or extended anti-PD1 monotherapy. Indeed, recent work in MMRd rectal cancer demonstrated a remarkable 100% complete response rate after nine cycles of neoadjuvant dostarlimab monotherapy42. Considering the potential of trAEs with anti-PD1 and anti-CTLA-4 combination therapy, a follow-up study (PAM-II) has been initiated to study the effect of nine cycles of neoadjuvant pembrolizumab monotherapy, followed by standard-of-care treatment.
In conclusion, our findings demonstrate that neoadjuvant pembrolizumab in surgically resectable MMRd EC elicits pathological, radiological and immunological responses. Extended treatment is warranted to determine if complete pathologic responses can be achieved.
Methods
Study design
This single institution investigator-initiated study (Clinicaltrial.gov identifier NCT04262089) was carried out in the Netherlands at the University Medical Centre Groningen (UMCG). The study protocol and all amendments were approved by the independent ethics committee of the UMCG (EudraCT registration number: 2018-001816-31). The study was conducted in accordance with the protocol, the declaration of Helsinki, and the International Conference on Harmonization Guidelines for Good Clinical Practice. All patients provided written informed consent. Funding was provided by the KWF Dutch Cancer Society and MSD.
Patient population
Eligible patients were aged 18 years and older, had histologically confirmed primary diagnosis of MMRd EC at any stage or grade, and were intended to be treated with primary surgery (at least a SoC hysterectomy). EC was diagnosed according to routine pathological workup on the biopsy. Testing for MMRd is part of the routine pathological workup for patients <70 years of age. If patients were >70 years of age, MMRd testing was conducted for study purposes, in addition to standard pathological workup. 9/10 patients underwent abdominal and thoracic CT scans and/or thoracic X-rays as part of the routine diagnostic evaluation for EC prior to study enrolment. Key exclusion criteria were prior therapy with immune checkpoint blockers or other recent systemic anti-cancer therapy, immunosuppressive treatment, active autoimmune disease and other active cancer, immunodeficiency, and pregnancy.
Treatment
Between December 2020 and September 2022, 12 patients were recruited, of whom 10 patients met our inclusion and none of our exclusion criteria (Supplementary Fig. 1b). The first patient was enroled on 14-12-2020 and the last patient was enroled on 01-09-2022.
Patients were treated with neoadjuvant pembrolizumab monotherapy between diagnosis and SoC resection and adjuvant radiotherapy if indicated. Pembrolizumab was administered IV at 200 mg every three weeks for a total of two cycles (Supplementary Fig. 1a). After SoC resection, patients visited the hospital 30 days, three months and six months after they received their last cycle of pembrolizumab to monitor potential long-term effects, including blood collections for safety values.
Study endpoints
Primary endpoints were feasibility and efficacy, which was assessed by pathological response rate in the primary tumour at resection. Feasibility was determined based on any treatment-related complications leading to delays in surgery past the eight weeks after informed consent. Tumour tissue was assessed by two independent experienced pathologists and was assessed for evidence of necrosis, viable residual tumour cells, fibrosis, degree of inflammation, immune infiltration, presence of lymphoid aggregates, presence of mature TLS, mucin degeneration, granulomas, multinucleated giant cells, cholesterol clefts, and the distribution of treatment response. Aligning with the tumour regression grading system of Mandard et al43., the following percentages are used (our adaptation): >90% tumour cells, 50–90% tumour cells, 10–50% tumour cells and MPR was characterized as ≤10% residual tumour cells, and complete responses as no residual tumour cells at all. The Mandard paper does not suggest combination of the middle categories and there is no consensus regarding PR definition post-immunotherapy44,45. Therefore, we deemed it appropriate to combine these categories for neoadjuvant ICB for MMRd EC. The secondary endpoint included ORR according to RECIST v1.1 criteria. Hereto, the longest diameter of the target lesion (tumour) was assessed by Magnetic Resonance Imaging (MRI) at baseline and after administration of two cycles of pembrolizumab, but prior to surgery.
Exploratory endpoints included safety, changes in the tumour and TME, and the predictive value of a post-treatment biopsy for pathological response in the primary tumour. Throughout the study adverse events (AEs) were monitored according to standard Common Terminology Criteria for Adverse Events Version 4 (CTCAE4) criteria. All AEs were followed until they were abated or until a stable situation was reached. Safety was assessed by evaluation of trAEs and serious AEs (SAEs). Changes in the tumour and TME were determined by image-based quantification of immunohistochemical stainings, spatial transcriptomics, TCR sequencing, and single-cell sequencing.
DNA extraction and TruSight Oncology 500 assays
Estimation of the percentage of neoplastic cells and selection of the area for macrodissection was performed by a pathologist. DNA extraction from tumour-rich Formalin-Fixed Paraffin-Embedded (FFPE) material with more than 20% neoplastic cells was carried out using the Maxwell® RSC FFPE Plus DNA Kit (AS1720) on the Maxwell® CSC device according to manufacturer instructions. For DNA library preparation and enrichment, The TruSight Oncology 500 (TSO500) hybrid-based capture assay (Illumina, San Diego, CA, USA) was used according to manufacturer’s instructions. Post-enriched libraries were quantified, pooled, and sequenced using the NovaSeq6000 (illumina) by GenomeScan BV (Leiden, The Netherlands). Data interpretation was performed by a molecular scientist of the pathology department at UMCG using their proprietary pipeline based on TruSight Oncology 500 Local App (Illumina). Tumour mutational burden was reported as mutations per megabase sequenced. Data outputs exported from the TSO500 pipeline (Illumina) were filtered in the categories pathogenic, likely pathogenic and variants of uncertain significance (VUS) using Genoox’s Franklin web-based analysis tool. Based on information from JAX-CKB (https://ckb.jax.org/) and cBioPortal46,47,48, the mutations were further classified. Pathogenic and likely pathogenic variants were subsequently used in the results of this paper.
Immunohistochemical (IHC) analysis
IHC analysis for MMR proteins (MLH1, PMS2, MSH2, and MSH6) was performed on all pre-treatment biopsies as standard of care, in accordance with national guidelines. IHC analysis for CD3, CD8, and CD19 were performed on all pre- and post-treatment biopsies and resection specimens, where available. IHC analysis for Bcl6 and CD21 were performed on all resection specimens. All IHC stainings were carried out on the fully-automated Benchmark ULTRA platform (Roche, Ventana Medical Systems, Oro Valley, AZ, USA), under standardized laboratory conditions, accredited following the ISO15189 quality system. In short, paraffin tissue sections (3 µm) were incubated with antibodies against MLH1 (R.T.U, Ventana, M1), PMS2 (R.T.U, Ventana, A16-4), MSH2 (R.T.U, Ventana, G219-1129), MSH6 (R.T.U, Ventana, SP93), CD3 (R.T.U, Ventana, 2GV6), CD8 (1:20, Dako, C8/144B), CD19 (1:1600, ThermoFisher, LE-CD19), Bcl6 (R.T.U., Ventana, Cl191E/A8) and/or CD21 (R.T.U., Ventana, 2G9). Each slide contained a suitable tissue section, serving as external control.
Machine learning-based IHC quantification
All slides were scanned on a (Philips IntelliSite Ultra Fast Scanner) at 20x and reviewed digitally by two pathologists (DL, VHK) for staining artefacts, folds or signs of necrosis. Regions with artefacts were excluded from further analysis. In each hysterectomy specimen, a tumour invasion front was defined as a 1000 µm band around the tumour invasion front. Further regions annotated were the tumour centre and the peritumoral tissue region. Infiltration density was computed for each area separately. Image analysis was carried out using HALO digital image analysis software v3.6.4134.137 (Indica Labs, Corrales, NM, USA). A deep neural network algorithm was trained using HALO AI to localize and quantify tumour epithelial tissue and stroma regions within each tissue region. Graphical overlays for each tissue class were generated and classification accuracy was confirmed by pathologist review. Results for CD3+, CD8+, CD19+ infiltration were recorded as positive cell infiltration density (cells/mm2) for tumour and stroma compartments separately. TLS were quantified on CD19 stained tissue slides in the peritumoral region by visual pathologist review.
Spatial transcriptomics
Tumour tissue, from the pre-treatment biopsy and the standard-of-care hysterectomy, was processed following the GeoMX DSP slide prep user manual (MAN-10150-01). In brief, FFPE slides were baked in an oven at 60 °C for at least 30 mins. Deparaffinization and rehydration were performed in xylene for 3 × 5 mins, 100% ethanol for 2 times 5 mins, 95% ethanol for 1x5minutes and once in 1× PBS for one minute. Antigen retrieval was performed in 1x Tris EDTA pH 9.0 in a pressure cooker for 15 mins at 100 °C. Thereafter the slides were washed in 1x PBS for 5 mins. To expose tissue RNA targets, the slides were incubated in 1 ug/ml proteinase K in 1x PBS at 37 °C for 15 mins. Slides were washed in 1x PBS for 5 mins, then immediately placed in 10% Neutral Buffered Formalin (Electron Microscopy Sciences, 15741-01) for 5 mins, followed by washing 2 times 5 mins in NBF stop buffer, and subsequently in 1× PBS for 5 mins. Whole Transcriptome Assay (WTA) hybridizations were performed at 4 nM final probe concentration in Buffer R (Nanostring). Slides were dried of excess 1× PBS, set in a hybridization chamber lined with Kimwipes wetted with 2x SSC, and covered with 200 µL hybridization solution individually. After applying HybriSlips, the slides were left to incubate at 37 °C overnight. After incubation, the HybriSlips were removed by dipping the slides in 2× SSC/0.1% Tween20. To remove unbound probes the slides were washed twice in a stringent wash solution consisting of 50% formamide/2× SSC at 37 °C for 25 mins, subsequently washed twice in 2× SSC for 2 mins. Slides were blocked in 200 µL buffer W (Nanostring), placed in a humidity chamber and incubated at room temperature for 30 mins. Morphology marker/antibody solution was prepared with 22 µL SYTO83, 5.5 µL PanCK-488, 5.5 µL CD3-647, 5.5 µL CD20-594, and 143 µL Buffer W for a total volume of 220 µL/slide. Slides were dried of excess Buffer W and the tissue was covered with 200 µL morphology marker/antibody solution. Hereafter, the slides were put in a dark humidity chamber and left to incubate at room temperature for 1 h. After incubation, slides were washed in 2x SSC for 2 times 5 mins and were immediately loaded onto the Nanostring DSP instrument and areas of interest collected into separate wells in the DSP collection plate.
DSP collection plates were dried at room temperature overnight, followed by incubation in a thermocycler at 65 °C for 1 h. After resuspension of the wells with 10 µL of nuclease-free water, the plates were incubated for 10 mins on room temperature and briefly centrifuged. A 96-well PCR plate was prepared by mixing 2 µL PCR mix (Nanostring), 4 µL GeoMx SeqCode primer mix (Nanostring) and 4 µL of the DSP sample, followed by incubation in a thermocycler. The indexed libraries were pooled by combining 4 µL of each PCR well into a 1.5 mL tube and incubated with AMPure XP beads (Beckman Coulter) at a 1.2× bead to sample ratio for 5 mins. Beads were washed twice with 1 ml 80% ethanol, on a magnetic stand and afterwards resuspended in 54 µL elution buffer. 50 µL of the library, was extracted to a new tube with 60 µL of AMPure XP beads (Beckman Coulter). Following magnetic incubation for 5 min and removal of supernatants, the beads were washed twice with 1 mL of 80% ethanol and dried for 1 min before eluted with 16 µL elution buffer. Quality of the library was assessed with Agilent Bioanalyzer for the expected library size of 162 bp. Library quantity was assessed using Qubit (Thermo Fisher). Total target counts per DSP collection plate were 100 counts/ µm2. Each library was diluted to 4–10 nM and combined to react the estimated counts/µm2 per library in the final pool. Data was analysed using differential expression analysis in the GeoMX DSP Analysis Suite (version 3.0.0.111). The 250 most abundantly expressed genes were further analysed using a co-functionality network tool (UMCG, Groningen, The Netherlands)49.
TCR sequencing
TCR sequencing was performed on all pre- and post-treatment biopsies, where available. Genomic DNA (gDNA) from FFPE tumour tissue was extracted using the Cobas kit following manufacturer’s instructions. Briefly, FFPE tissue sections (10 µm) were deparaffined and lysed using Proteinase K and a DNA Tissue Lysis Buffer, followed by the addition of isopropanol. The mixture was centrifuged through a column with a glass fibre filter and washed. gDNA quantity was assessed using Qubit (Thermo Fisher).
TCR sequencing was done using the immunoSEQ hsTCRB kit (Adaptive Biotechnologies), according to manufacturer’s protocol. Each FFPE sample was run in duplicate. In short, in a first multiplex PCR, the CDR3 region was amplified using V- and J-gene specific primers. This PCR product was diluted and subsequently uniquely identified by barcodes during a second PCR. All libraries were equally pooled according to volume, and the superpool was purified using cleanup beads (supplemented in the kit). The molar concentration was determined using the KAPA Library Quantification Kit (Roche). 1.6 pM was loaded onto the NextSeq 500 (Illumina inc., San Diego, CA, USA). A 150 bp mid-output NextSeq reagent kit was used (paired end). The data was analysed using the immunoSEQ analyser version 3.0 (Adaptive Biotechnologies) and R statistical software (version 4.2.2).
Single cell isolation of T cells isolated from resected tumours
Tumour digests were thawed in FCS and resuspended in RPMI + 10% FCS. After 1.5 h incubation at 37 °C/5% CO2, samples were stained and incubated with a CXCR5 PerCP-Cy5.5 antibody for an additional 0.5 h at 37 °C. After a total of 2 h recovery at 37 °C/5% CO2, samples were centrifuged and resuspended in 100 µl PBS + 2% FCS. Samples were incubated for 30-45 mins on ice (in dark) with the following antibodies: CD45 BV605, CD8 APC Cy7, CD4 PE, CD19 BV421, PD1 APC, CD3 PE-Cy7 and the dump channel antibodies CD14 FITC, CD56 FITC and EpCAM FITC. More information on the antibodies used can be found in Supplementary Data 5. 3 µl of each antibody was used per 1x10e6 cells. Samples were washed twice with PBS + 2% FCS and filtered using a 35 µm strainer (Falcon) before sorting on a Beckman Coulter MoFlo Astrios cytometer. UltraComp eBeads (Thermo Fisher Scientific) were used as compensation controls. Before sorting, Propidium Iodide was added to exclude dead cells. Sorting was done in 1.5 mL Low Bind DNA tubes (Eppendorf) in 150 µl PBS 0.04 % BSA. CD45+ CD3+ CD19− CD8+ and CD4+ cells were sorted. Sorted cells were centrifuged and resuspended at a concentration of 727 cells/µl (total 16.5 µl) for PAM-007 or 2121 cells/µl (total 16.5 µl) for PAM-010 and PAM-011, in PBS 0.04% BSA. Subsequent steps were performed according to manufacturer’s protocol (10x genomics): ‘chromium next GEM single-cell 5’ reagents kit v2 (dual index)’. For the cDNA amplification step, 13 cycli were done. For each sample, both the V(D)J library as well as the gene expression (GEX) library was constructed. All cDNA quality control and quantification steps were done with the Qubit 4 Fluorometer (Thermo Fisher) and the Agilent 4200 Tapestation (with D5000 and high sensitivity D5000 screen tapes). For the sequencing, V(D)J and GEX libraries were pooled at a 1:4 molar ratio, with 2 GEX libraries and 2 V(D)J libraries in one pool. 1.5 pM of this superpool was sequenced on a NextSeq 500 (Illumina inc., San Diego, CA, USA). A 150 bp NextSeq high-output reagent kit was used for the sequencing (paired end).
Sequencing read alignments, quality control, cell clustering, and annotation
Reads from single cells isolated using 10x chromium were demultiplexed and aligned to the GRCh38.p12 human reference genome (from 10× Genomics) using Cell Ranger (version 6.0.1; 10x Genomics50). Cell Ranger outputs were loaded into R (version 4.2.2) (Seurat (version 4.3.0) package51) and cells with fewer than 500 genes, more than 4000 genes, or >10% mitochondrial counts were excluded from further analysis. At this stage, TCR libraries were demultiplexed and processed using Cell Ranger. Cell Ranger outputs were loaded into R (scRepertoire(V1.0.2) package52) and contig annotations combined using the ‘combineTCR’ command, removing doublets with >1 TCR with the ‘removeMulti’ command. RNA expression and TCR profiles per cell were combined into a single Seurat object using the ‘combineExpression’ command with cloneCall set to ‘strict’ (utilizing both gene segment and CDR3 sequence). Seurat objects from individual patients were subset to retain only T cells with a single unique TCR that corresponded to a TCR identified using bulk TCR sequencing of tumour material from the same patient (identical CDR3 amino acid sequence) using the ‘SubsetData’ command. Seurat objects from different patients were subsequently integrated using sequentially the ‘NormalizeData’, ‘FindVariableFeatures’, ‘ScaleData’, ‘RunPCA’, ‘FindIntegrationAnchors’ (with rpca as reduction), and ‘IntegrateData’ commands (default settings with dimensions determined empirically using the ‘ElbowPlot’ command). Expression values of the merged Seurat object were converted to z-scores using the ‘ScaleData’ command. Principle components were calculated using the ‘RunPCA’ command. Global clusters of similar cells were detected using the Louvain method for community detection to construct the shared nearest neighbour map and an empirically set resolution using the ‘FindNeighbours’ and ‘FindClusters’ commands.
Clusters were annotated based on canonical expression of markers such as CD8A, CD4, PDCD1 and divided into main subsets (CD8 Tex, CD8 Teff, Tcm-like CD8, CD4 Tex, CD4 Teff, Treg and cycling T cells). Expansion of individual T cell clones was determined based on pre- and post- bulk TCR sequencing as defined above. TDLN-residence of clones was determined based on bulk TDLN TCR sequencing. Expanded CD8 or CD4 T cells were subset using the ‘SubsetData’ command and the above-described procedure (from ‘ScaleData’ onwards) was applied to subset expanded cells. The number of clusters was iteratively defined using canonical markers previously reported to represent meaningful T cell biology (e.g. Tpex, intra-epithelial cells, etc.). If clusters could not be resolved by canonical marker expression, we prepended a representative marker gene or gene signature to their ‘canonical’ identity (e.g. LAG3high T cells, or IL32+ T cells). Differential gene expression between the annotated cell populations was determined using the ‘FindAllMarkers’ command. Cell cycle inference was performed using the ‘CellCycleScoring’ command using the built-in s- and g2m features. Clonotype bias was determined according to Andreatta et al53. using the ‘clonotypeBias’ command. For visualization, dimensional reduction was performed on the total annotated dataset using the ‘RunUMAP’ command. The same number of principle components were used as applied for clustering.
Statistical analysis
Statistical analysis was performed using GraphPad Prism (version 8.4.2). Differences in clinicopathologic characteristics between groups were evaluated by Mann–Whitney U tests (Fig. 2a,b). Differences in immune infiltration between groups were evaluated using a mixed-effects model with Bonferroni correction (Fig. 3). Differences in TCR diversity (Simsons clonality) between groups was assessed using a paired t-test. A P-value of <0.05 was considered to be statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw and processed scRNA-seq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE268903. Genomic data have been deposited in the European Genome-Phenome Archive (EGA) and are accessible through accession number EGAD50000000694. The data are available under restricted access and can be obtained by contacting the Data Access Committee. The clinical data, tumour material and peripheral blood cells generated during, or analysed in, the present study are not publicly available owing to restrictions by privacy laws. Data are held by the coauthors of this article (see also: https://doi.org/10.34760/668fb3edd96e2). Requests for sharing of data and material, including the study protocol, should be addressed to the corresponding author(s) within 15 years of the date of publication of this article and include a scientific proposal. Depending on the specific research proposal, the coauthors will determine when, for how long, for which specific purposes and under which conditions the requested data can be made available, subject to ethical consent and the composition of a material and/or data transfer agreement. Requests for data access will be processed within a 3-month timeframe. The remaining data are available within the Article, Supplementary Information or Source Data file. Source data are provided with this paper.
References
Crosbie, E. J. et al. Seminar Endometrial Cancer. www.thelancet.com vol. 399 (1995).
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Nout, R. A. et al. Five-year quality of life of endometrial cancer patients treated in the randomised Post Operative Radiation Therapy in Endometrial Cancer (PORTEC-2) trial and comparison with norm data. Eur. J. Cancer 48, 1638–1648 (2012).
Chalabi, M. et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat. Med. 2020 26, 566–576 (2020).
O'Malley, D.M. et al. Pembrolizumab in patients with microsatellite instability-high advanced endometrial cancer: results from the KEYNOTE-158 study. J. Clin. Oncol. 40, 752–761 (2022).
Le, D. T. et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (1979) 357, 409–413 (2017).
Kist de Ruijter, L. et al. Whole-body CD8+ T cell visualization before and during cancer immunotherapy: a phase 1/2 trial. Nat. Med 28, 2601–2610 (2022).
Andreatta, M. et al. Interpretation of T cell states from single-cell transcriptomics data using reference atlases. Nat. Commun. 12, 2965 (2021).
Liu, X. et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature 567, 525–529 (2019).
Muthuswamy, R. et al. CXCR6 by increasing retention of memory CD8 + T cells in the ovarian tumor microenvironment promotes immunosurveillance and control of ovarian cancer. J. Immunother. Cancer 9, e003329 (2021).
Workel, H. H. et al. CD103 defines intraepithelial CD8+ PD1+ tumour-infiltrating lymphocytes of prognostic significance in endometrial adenocarcinoma. Eur. J. Cancer 60, 1–11 (2016).
Anadon, C. M. et al. Ovarian cancer immunogenicity is governed by a narrow subset of progenitor tissue-resident memory T cells. Cancer Cell 40, 545–557.e13 (2022).
Lynn, R. C. et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576, 293–300 (2019).
Goda, C. et al. Involvement of IL-32 in activation-induced cell death in T cells. Int Immunol. 18, 233–240 (2006).
Gruber, T. et al. IL-32γ potentiates tumor immunity in melanoma. JCI Insight 5, e138772 (2020).
Andreatta, M. et al. A CD4+ T cell reference map delineates subtype-specific adaptation during acute and chronic viral infections. Elife 11, e76339 (2022).
Miller, B. C. et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 20, 326–336 (2019).
Liu, B. et al. Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti-PD-1 therapy in lung cancer. Nat. Cancer 3, 108–121 (2022).
Cascone, T. et al. Neoadjuvant nivolumab or nivolumab plus ipilimumab in operable non-small cell lung cancer: the phase 2 randomized NEOSTAR trial. Nat. Med 27, 504–514 (2021).
Forde, P. M. et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N. Engl. J. Med. 378, 1976–1986 (2018).
Blank, C. U. et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat. Med 24, 1655–1661 (2018).
Gao, S. et al. Neoadjuvant PD-1 inhibitor (Sintilimab) in NSCLC. J. Thorac. Oncol. 15, 816–826 (2020).
Amaria, R. N. et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat. Med 24, 1649–1654 (2018).
Rozeman, E. A. et al. Identification of the optimal combination dosing schedule of neoadjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma (OpACIN-neo): a multicentre, phase 2, randomised, controlled trial. Lancet Oncol. 20, 948–960 (2019).
Tong, B. C. et al. Perioperative outcomes of pulmonary resection after neoadjuvant pembrolizumab in patients with non–small cell lung cancer. in Journal of Thoracic and Cardiovascular Surgery vol. 163 427–436 (Elsevier Inc., 2022).
Powles, T. et al. Clinical efficacy and biomarker analysis of neoadjuvant atezolizumab in operable urothelial carcinoma in the ABACUS trial. Nat. Med 25, 1706–1714 (2019).
Glaire, M. A. et al. Discordant prognosis of mismatch repair deficiency in colorectal and endometrial cancer reflects variation in antitumour immune response and immune escape. J. Pathol. 257, 340–351 (2022).
Horeweg, N. et al. Molecular classification predicts response to radiotherapy in the randomized PORTEC-1 and portec-2 trials for early-stage endometrioid endometrial cancer. J Clin Oncol 41, 4369–4380 (2023).
Leon-Castillo, A. et al. Molecular classification of the PORTEC-3 Trial for high-risk endometrial cancer: impact on prognosis and benefit from adjuvant therapy. J. Clin. Oncol. 38, 3388–3397 (2020).
Bellone, S. et al. A phase 2 evaluation of pembrolizumab for recurrent Lynch-like versus sporadic endometrial cancers with microsatellite instability. Cancer 128, 1206–1218 (2022).
Eskander, R. N. et al. LBA43 Updated response data and analysis of progression free survival by mechanism of mismatch repair loss in endometrial cancer (EC) patients (pts) treated with pembrolizumab plus carboplatin/paclitaxel (CP) as compared to CP plus placebo (PBO) in the NRG GY018 trial. Ann. Oncol. 34, S1284 (2023).
Toboni, M. D. et al. Differential outcomes and immune checkpoint inhibitor response among endometrial cancer patients with MLH1 hypermethylation versus MLH1 “Lynch-like” mismatch repair gene mutation. Gynecol. Oncol. 177, 132–141 (2023).
Wang, M. M., Coupland, S. E., Aittokallio, T. & Figueiredo, C. R. Resistance to immune checkpoint therapies by tumour-induced T-cell desertification and exclusion: key mechanisms, prognostication and new therapeutic opportunities. Br. J. Cancer 2023 129, 1212–1224 (2023).
Bosse, T. et al. Refining adjuvant treatment in endometrial cancer based on molecular features: the RAINBO clinical trial program. Int. J. Gynec. Cancer 33, ijgc-2022-004039 (2023).
Oliveira, G. & Wu, C. J. Dynamics and specificities of T cells in cancer immunotherapy. Nat. Rev. Cancer 2023 23, 295–316 (2023).
Olugbile, S. O. et al. Sustained oligoclonal T cell expansion correlates with durable response to anti-PD1 therapy. 10.1200/JCO.2017.35.15_suppl.3061 35, 3061–3061 (2017).
Puig-Saus, C. et al. Neoantigen-targeted CD8+ T cell responses with PD-1 blockade therapy. Nature (2023) https://doi.org/10.1038/s41586-023-05787-1.
Franken, A. et al. CD4+ T cell activation distinguishes response to anti-PD-L1+anti-CTLA4 therapy from anti-PD-L1 monotherapy. Immunity 57, 541–558.e7 (2024).
Matsuda, T. et al. TCR sequencing analysis of cancer tissues and tumor draining lymph nodes in colorectal cancer patients. Oncoimmunology 8, e1588085 (2019).
Pai, J. A. et al. Lineage tracing reveals clonal progenitors and long-term persistence of tumor-specific T cells during immune checkpoint blockade. Cancer Cell (2023) https://doi.org/10.1016/j.ccell.2023.03.009.
Jiao, S. et al. Intratumor expanded T cell clones can be non-sentinel lymph node derived in breast cancer revealed by single-cell immune profiling. J. Immunother. Cancer 10, 3325 (2022).
Cercek, A. et al. PD-1 blockade in mismatch repair–deficient, locally advanced rectal cancer. N. Engl. J. Med. 386, 2363–2376 (2022).
Mandard, A.M. et al. Pathologic assessment of tumor regression after preoperative chemoradiotherapy of esophageal carcinoma clinicopathologic correlations. https://doi.org/10.1002/1097-0142.
Tetzlaff, M. T. et al. Pathological assessment of resection specimens after neoadjuvant therapy for metastatic melanoma. Ann. Oncol. 29, 1861–1868 (2018).
Cottrell, T. R. et al. Pathologic features of response to neoadjuvant anti-PD-1 in resected non-small-cell lung carcinoma: a proposal for quantitative immune-related pathologic response criteria (irPRC). Ann. Oncol. 29, 1853–1860 (2018).
Cerami, E. et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal complementary data sources and analysis options. Sci. Signal 6, 1–20 (2013).
de Bruijn, I. et al. Analysis and visualization of longitudinal genomic and clinical data from the AACR project GENIE biopharma collaborative in cBioPortal. Cancer Res (2023) https://doi.org/10.1158/0008-5472.can-23-0816.
Urzúa-Traslaviña, C. G. et al. Improving gene function predictions using independent transcriptional components. Nat. Commun. 12, 1464 (2021).
Zheng, G. X. Y. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
Borcherding, N. & Bormann, N. L. scRepertoire: An R-based toolkit for single-cell immune receptor analysis. F1000Res 9, 47 (2020).
Ryan, N. A. J. et al. Histological and somatic mutational profiles of mismatch repair deficient endometrial tumours of different aetiologies. (2021) https://doi.org/10.3390/cancers.
Acknowledgements
We thank all patients, their families, caregivers and clinical investigators for their participation in the PAM trial. The present trial was funded by the Dutch Cancer Society (KWF) (2021-1 / 13537; HWN) and the UMCG Cancer Research Fund (KRF) (4901157; HWN). Merck Sharp & Dome (MSD), the Netherlands, provided the pembrolizumab. The funding source had no role in the execution of the trial, data analysis and writing of the manuscript.
Author information
Authors and Affiliations
Contributions
A.L.E. and K.B. coordinated the trial procedures, collected and processed patient samples, analysed and interpreted clinical and translational data and wrote the manuscript, with input, edits and approval from all authors. A.V. coordinated trial procedures and collected patient samples. S.P. wrote the study protocol, coordinated trial procedures and collected patient samples. A.V., S.P., and M.R. contributed equally to this manuscript. M.R., N.R., D.S. performed the TCR and single-cell sequencing experiments. N.R., A.P., F.J.H. and P.K. isolated PBMCs and processed tumour tissue. D.L. and V.H.K. developed and performed digital quantification of IHC staining. A.E. developed and performed the TSO500 assays. R.Y., T.R., N.M.L., and R.K. informed patients, executed trial procedures, and collected patient samples. D.C. provided scientific input during manuscript writing and critically reviewed the manuscript. R.W. and F.F. performed bioinformatics analysis of translational data. T.B. and J.B. assessed tumour tissue for pathological response. J.B. performed IHC staining. A.R. conceived the trial and wrote the study protocol. M.J. and A.R. executed screening and study procedures. M.B. and H.W.N. conceived the trial, wrote the study protocol, analysed and interpreted clinical and translational data and supervised the writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
HWN and MB received grants from the Dutch Cancer Society (KWF), the European Research Council (ERC), Health Holland (HH), Mendus, BioNovion, Aduro Biotech, Vicinivax, Genmab and IMMIOS (all paid to the institute); received non-financial support from BioNTech, Surflay Nanotec and Merck Sharp & Dohme; are stock option holders in Sairopa. DC reports to be part of the advisory board for Merck Sharp & Dohme, his immediate family member is an Amgen employee, and received funding from HalioDx, unrelated to the subject of this manuscript. VHK served as an invited speaker on behalf of Sharing Progress in Cancer Care (SPCC) and Indica Lab, reports to be part of the advisory board of Takeda, and has sponsored research agreements with Roche and IAG, all unrelated to the current trial. TB received grants from the Dutch Cancer Society, unrelated to the subject of this manuscript. JB received grants from the Dutch Cancer society and AstraZeneca. MJ reports to be part of the advisory board of Pierre Fabre, AstraZeneca, BMS, and GSK. All other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Eerkens, A.L., Brummel, K., Vledder, A. et al. Neoadjuvant immune checkpoint blockade in women with mismatch repair deficient endometrial cancer: a phase I study. Nat Commun 15, 7695 (2024). https://doi.org/10.1038/s41467-024-52098-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-52098-8
This article is cited by
-
Therapeutic targeting of mismatch repair-deficient cancers
Nature Reviews Clinical Oncology (2025)
-
Epidemiology, pathogenesis, biology and evolving management of MSI-H/dMMR cancers
Nature Reviews Clinical Oncology (2025)
