
Abstract
Powdery mildew, caused by Blumeria graminis f. sp. tritici (Bgt), reduces wheat yields and grain quality, thus posing a significant threat to global food security. Wild relatives of wheat serve as valuable resources for resistance to powdery mildew. Here, the powdery mildew resistance gene Pm6Sl is cloned from the wild wheat species Aegilops longissima. It encodes a nucleotide-binding leucine-rich repeat (NLR) protein featuring a CC-BED module formed by a zinc finger BED (Znf-BED) domain integrated into the coiled-coil (CC) domain. The function of Pm6Sl is validated via mutagenesis, gene silencing, and transgenic assays. In addition, we develop a resistant germplasm harbouring Pm6Sl in a very small segment with no linkage drag along with the diagnostic gene marker pm6sl-1 to facilitate Pm6Sl deployment in wheat breeding programs. The cloning of Pm6Sl, a resistance gene with BED-NLR architecture, will increase our understanding of the molecular mechanisms underlying BED-NLR-mediated resistance to various pathogens.
Similar content being viewed by others

Introduction
Wheat (Triticum aestivum L., 2n = 6x = 42, AABBDD) is a major staple crop worldwide, contributing approximately 20% of the total dietary protein and calories for the global human population1. Powdery mildew poses a substantial threat to global food security by affecting wheat yield and quality2. The identification and deployment of effective resistance genes in wheat varieties is widely recognized as one of the most economical, effective and environmentally sustainable approaches to reduce the impact of wheat powdery mildew. To date, more than 100 alleles of 69 powdery mildew resistance genes (Pm) have been officially designated, of which 19 genes have been cloned3,4,5,6. These genes include 12 (63.16%) Pm genes, namely, Pm1a, Pm2, Pm3b/Pm8/Pm17, Pm5e, Pm12/Pm21, Pm41, Pm55, Pm60 and Pm69, which encode nucleotide-binding leucine-rich repeat (NLR) proteins; one gene encoding an ATP-binding cassette (ABC) transporter (Pm38); and one gene encoding a hexose transporter (Pm46). Recently, several resistance genes containing novel/unusual domains, including Pm24 encoding a tandem kinase protein (TKP), Pm4 encoding a putative chimeric protein of a serine/threonine kinase, and Pm36 encoding a unique tandem kinase with a transmembrane domain, have been reported6. We also reported two powdery mildew resistance genes: Pm13 encodes a mixed lineage kinase domain-like (MLKL) protein that contains an N-terminal MLKL_NTD domain and a C-terminal STK domain7, and Pm57 encodes an unusual TKP protein with putative kinase-pseudokinase domains followed by a von Willebrand factor A (vWA) domain (WTK6b-vWA)8, which sheds light on various plant resistance strategies. However, more effort is needed to understand the diverse array of complex mechanisms that activate plant resistance.
NLRs constitute the largest class of immune receptors in plants and play a pivotal role in the plant defence surveillance system by monitoring pathogen effectors delivered into the plant cells9,10. NLRs share a tripartite domain structure, comprising an N-terminal domain, a central nucleotide-binding (NB) domain, and a C-terminal leucine-rich repeat (LRR) domain. On the basis of their N-terminal domains, NLRs can be further classified into three subclasses: NLRs containing Toll/interleukin-1 receptor (TIR) (TNLs), NLRs with N-terminal CC domains (CNLs), and NLRs with RPW8-like CC (CCR) domains (RNLs)11. The N-terminal domains, CC, CCR, and TIR, are expected to initiate downstream signalling, as the expression of these domains alone often triggers immune responses and hypersensitive response (HR)12. In addition to these typical tripartite domain NLRs, other NLRs with additional integrated domains (NLR-IDs), including those containing WRKY13, kinase14, heavy metal-associated (HMA)15, and zinc finger (Znf) BED domains16, exist. NLRs with Znf-BED domains (BED-NLRs) constitute the second largest NLR-ID group after WRKYs17. To date, six BED-NLRs linked to resistance to plant pathogens have been isolated. These genes include Yr5/Yr7/YrSP, which provide resistance to wheat yellow rust16; Xa1/Xo1, which confer resistance to rice blight and leaf streak18,19; and Rph15, which induces resistance to barley leaf rust20. The Znf-BED domain, which was originally identified in transposases, possesses DNA binding capabilities21,22. It is required for the proper function of BED-NLRs, as demonstrated by mutations in the Znf-BED domain resulting in increased susceptibility16. However, the mechanism by which the Znf-BED domain is involved in the pathogen resistance of plant NLRs remains largely unknown. The identification, cloning and characterization of more resistance genes will enhance our understanding of resistance activation and signalling mechanisms, accelerate the deployment of resistance genes and lead to the engineering of new strategies for effective disease management.
Over the past century, breeders have carried out numerous crosses to expand the genetic diversity of wheat via introgression of resistance genes from its wild relatives. More than 200 (42.83%) of the currently designated 467 resistance genes in cultivated bread wheat originated outside the bread wheat gene pool23. The introgression of chromosome segments from wild relatives of wheat is a well-established strategy for broadening the genetic diversity of disease resistance genes24. Aegilops longissima Schw. et Musch. (2n = 2x = 14, SlSl) is a diploid S-genome member of the Sitopsis section in the Triticeae tribe25, representing a valuable reservoir of genetic diversity for resistance to stem rusts26,27, powdery mildew28,29, Septoria glume blotch (SNB)30, eyespot31, and drought stress tolerance32. Among these genes, Pm13 on chromosome 3Sl 25 and Pm66 on 4Sl 29 were introgressed into wheat. Previously, we identified a powdery mildew resistance gene, designated Pm6Sl, on chromosome 6Sl#3 from Ae. longissima, and transferred it to bread wheat via homoeologous recombination between chromosome 6Sl#3 and its wheat counterparts induced by the ph1b gene33,34, which enhances homoeologous recombinants in common wheat. We mapped Pm6Sl to the long arm of 6Sl#3 in a 42.80 Mb distal interval between the markers Ael58410 and Ael57699 via an F2 population generated by crossing the wheat-Ae. longissima disomic addition line TA7548 containing Pm6Sl with TA3809, a Chinese Spring (CS) ph1b-deletion mutant28.
In this work, we report the cloning of the Pm6Sl gene. The function of Pm6Sl is validated via ethyl methanesulfonate (EMS) mutagenesis, virus-induced gene silencing (VIGS), and transgenic assays. Pm6Sl encodes an NLR with a Znf-BED domain integrated in the coiled-coil (CC) domain and is thus designated the Znf-BED CNL. Additionally, we develop several Pm6Sl stocks containing small alien segments along with a diagnostic gene marker for Pm6Sl. This study documents a resistance gene for elucidating the molecular mechanisms underlying gene-for-gene interactions between wheat and the Bgt pathogen. Moreover, wheat lines with small Pm6Sl-harboured segments and a diagnostic marker for rapid deployment of Pm6Sl are developed to facilitate powdery mildew resistance breeding in wheat.
Results
Pm6Sl is finely mapped in a 210 kb interval on the long arm of 6Sl#3
To fine map Pm6Sl, a secondary Pm6Sl segregation population (F3) was produced by self-pollinating previously developed heterozygous Pm6Sl recombinant plants in a homozygous ph1b background. First, we retrieved the Ael58410 and Ael57699 corresponding genomic sequences from the Ae. longissima cv. TL05 reference genome (Fig. 1a). We then prioritized the low-copy number regions and developed 20 6Sl#3-specific PCR markers (Supplementary Data 1 and 2). Next, we screened 8,000 F3 individuals and identified 105 recombinants lacking either Ael58410 or Ael57699. With the subsequent genotyping of these 105 recombinants via 20 newly developed markers and phenotypic responses to the Bgt isolate E26, we narrowed Pm6Sl to a 0.93 Mb interval between Ael03991 (654.11 Mb) and Ael04335 (655.04 Mb) (Fig. 1b).

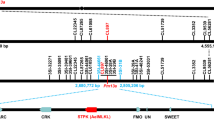
a Schematic diagram of the wheat-Ae. longissima recombined chromosome 6Sl#3. b Resistance assay and marker analysis of 105 6Sl#3 recombinants narrowed Pm6Sl down to a 0.93 Mb interval. c Analysis of an additional five markers and evaluation of powdery mildew resistance in two additional distinct types (U and V) of 6Sl#3 recombinants delimited the Pm6Sl interval to 210 kb. The number of recombinants of each type is shown in brackets; all eight recombinants were resistant to the Bgt isolate E26. d Seven genes discovered within the 210 kb interval, of which only Ae.longissima.TL05.6S01G0714700 (named CNL1) and Ae.longissima.TL05.6S01G0715000 (named CNL2) are annotated to encode resistance proteins.
Next, we developed five additional markers in the 0.93 Mb region and screened eight 6Sl#3 recombinants of two distinct types (U and V) from 800 F4 plants derived from heterozygous Pm6Sl-containing recombinants (T27-VI and T3012) (Fig. 1c). Among these, five U-type recombinants displayed both markers Ael039960 (654.21 Mb) and Ael16698 (654.28 Mb), whereas three V-type plants (T3012I-26, T3012I-29, T3012II-3) carried only the Ael16698 marker. However, all eight recombinants were resistant to Bgt isolate E26. By integrating the Bgt responses of the eight recombinants via five-marker analysis, we further delimited Pm6Sl to a 210 kb region (654.21-654.42 Mb) between Ael39960 and Ael09126 (Fig. 1c and Supplementary Data 1). Annotation of this 210 kb region revealed seven genes, of which only TL05.6S01G0714700 (designated CNL1) and TL05.6S01G0715000 (CNL2) were annotated to encode disease resistance (R) proteins, whereas the remaining five were predicted to encode unknown functional proteins (Fig. 1d and Supplementary Data 3).
Mutagenesis and gene silencing indicated CNL1 to be the best candidate for Pm6Sl
The primers SALF-CNL1 and SALF-CNL2 (Supplementary Data 2) were designed to amplify CNL1 and CNL2 sequences, respectively, from the Pm6Sl donor TA7548 via the use of genomic DNA (gDNA) or cDNA. Sanger sequencing of amplicons and comparison of sequences between gDNA and cDNA revealed that the gDNA sequence of CNL1 from TA7548 is 4496 bp long and contains three exons encoding a protein of 1431 amino acids (Supplementary Data 4). For CNL2, the gDNA sequence from TA7548 is 3262 bp long, which transcribes 2979 bp coding sequences (CDSs) in three exons encoding a protein of 992 amino acids (Supplementary Data 5). In addition, both the CNL1 and CNL2 sequences were absent in CS (Supplementary Fig. 1). Therefore, the CNL1 and CNL2 genes were prioritized as Pm6Sl candidate genes.
To determine the causal gene responsible for Pm6Sl, a mutational screening protocol was employed to identify potential Pm6Sl candidates via 30 independent susceptible mutants generated from EMS-treated TA7548S (Supplementary Fig. 2). Analysis of variation in CNL1 in 30 susceptible mutants revealed mutations either introducing a premature stop codon or leading to nonsynonymous amino acid alterations in CNL1 in 29 mutants, whereas only two mutants (Mut12 and Mut24) presented mutations in CNL2, but they also carried nonsynonymous mutations in CNL1 (Fig. 2a and Supplementary Data 6). Furthermore, we used a barley stripe mosaic virus-induced gene silencing system (BSMV-VIGS) to knock down either CNL1 or CNL2 in TA7548 to verify their function. The results revealed that silencing CNL1 with the BSMV:γCNL1 construct resulted in susceptibility of TA7548 plants to powdery mildew (Fig. 2b), whereas no notable phenotypic changes were observed in the plants treated with the BSMV:γ and BSMV:γPDS (phytoene desaturase gene) controls. Conversely, silencing CNL2 with the BSMV:γCNL2 construct did not change resistance levels compared with those in plants treated with the BSMV:γ or BSMV:γPDS controls (Fig. 2b). These results confirmed that CNL1, rather than CNL2, is the most promising candidate for Pm6Sl.

a EMS-induced mutation sites in CNL1 and CNL2. Two mutant lines, Mut12 and Mut23, carry two mutations in CNL1, and two mutant lines, Mut12 and Mut24, carry nonsynonymous mutations in CNL1 and CNL2. b BSMV-VIGS-mediated functional validation of CNL1 and CNL2. Representative leaves showing the resistance phenotype of TA7548 plants subjected to the silencing of CNL1 (BSMV:γCNL1) or CNL2 (BSMV:γCNL2) through BSMV-VIGS; BSMV:γPDS was used as a control to illustrate the effect of gene silencing, and BSMV:γ was used as the empty vector control. Images show a representative phenotype of the third leaf at 7 days post inoculation (dpi) from seedlings infected with Bgt isolate E26. Scale bars = 0.5 cm. The relative expression of CNL1 and CNL2 in the gene-silenced plants was analysed for silencing efficiency via quantitative real-time PCR (qRT‒PCR). The TaACTIN gene was used for expression normalization. The infected leaves of each plant were mixed as one sample. qRT‒PCR was performed three times for each sample, and each time as a technical replicate. The values are the means of three technical replicates with standard deviation (SD) as error bars. Different lowercase letters above the bars denote significant differences at the p < 0.05 level (one-way ANOVA). CNL1 and CNL2 were efficiently silenced in infected leaves of TA7548 individuals (presented by no. 1, 2, 3), while they were normally expressed in BSMV:γ infected leaves. Source data are provided as a Source Data file.
Transgenic assay verified candidate CNL1 as Pm6Sl
To confirm whether CNL1 is responsible for powdery mildew resistance in plants containing Pm6Sl, the CDS of CNL1 driven by the maize Ubi promoter was introduced into the recipient wheat Fielder, which is susceptible to powdery mildew (Supplementary Fig. 3a). A total of 24 T0 plants were regenerated, of which 20 were CNL1 positive and 4 were negative. These plants self-pollinated and advanced to generation T1 (Fig. 3a). Among the 20 positive T1 lines, L17 and L21 presented CNL1 expression levels comparable to those of T3012II-3, a resistant recombinant with a small 6Sl segment carrying Pm6Sl, whereas the other 18 T1 lines presented significantly higher levels of CNL1 expression (Fig. 3b). An assessment of the resistance of these 20 T1 lines revealed that all the CNL1-positive transgenic plants were resistant to the Bgt isolate E26, whereas the CNL1-negative plants were susceptible (Fig. 3a and Supplementary Fig. 3b). In addition, CNL1-positive T1 plants at the heading stage continued to show resistance to powdery mildew, suggesting that Pm6Sl is capable of conferring all-stage resistance (Fig. 3c).

a Resistance assay of transgenic T1 lines 10 dpi with the Bgt isolate E26, with the receptor Fielder as a susceptible control and recombinant T3012II-3 with a small 6Sl segment carrying Pm6Sl as a resistance control. “+“ indicates the presence of CNL1, whereas “−“ indicates the absence of CNL1. A representative leaf segment phenotype from one plant in each T1 line is shown. Scale bar, 0.5 cm. b qRT‒PCR analysis of CNL1 expression in positive T1 plants and T3012II-3 plants at the seedling stage. Data are mean ± SD from three biological replicates. N.D: not detected. Asterisks (*, **, ***) represent significant differences at the p < 0.05, p < 0.01, and p < 0.001 levels, respectively (two-tailed Student’s t test). Three biological replicates, two leaves from two resistant seedlings per replicate, were carried out for each T1 line. c T1 transgenic plant response to Bgt isolate E26 at the heading stage. Positive plants harbouring CNL1 are resistant to powdery mildew, whereas negative control plants in the same pot are susceptible. Source data are provided as a Source Data file.
CNL1 function was also validated by a transgenic assay using its native promoter and genomic sequence. The ProCNL1:CNL1 construct comprising a 9727 bp genomic fragment including a presumed 4569 bp native promoter, 4496 bp exon and intron regions, and 662 bp terminator of CNL1 was delivered to the Fielder (Supplementary Fig. 4a), generating six independent transgenic T0 seedlings. All plants were positive for CNL1 and exhibited resistance to powdery mildew akin to the resistance control T3012II-3 (Supplementary Fig. 4b). These results indicate that both normal and elevated CNL1 expression can effectively confer resistance to Bgt isolate E26. Taken together, the results of the mutagenesis, VIGS-induced gene silencing, and transgenic assays consistently confirmed that CNL1 is the Pm6Sl gene.
Pm6Sl encodes an NLR protein containing a CC-BED module
To study the structural characteristics of the Pm6Sl protein, we used the AlphaFold2 artificial intelligence-augmented system to generate a 3D model. The results revealed that Pm6Sl encodes an NLR protein that contains a Znf-BED domain in addition to the expected N-terminal CC domain, a central NB-ARC site, and a C-terminal LRR region (Fig. 4a). The CC domain of Pm6Sl comprises four α-helices (α1–4), analogous to that of a conventional CNL protein35; nevertheless, the Znf-BED domain is integrated between its α3 and α4 helices to form an α1-3-BED-α4 chimeric structure that we designated the “CC-BED module” (Fig. 4b). Moreover, the incorporation of amino acid changes in susceptible mutants into the Pm6Sl 3D model revealed that all four domains had mutations leading to amino acid changes: two in the CC-BED module, 11 in the NB-ARC domain, and 18 in the LRR domain (Supplementary Fig. 5a). Interestingly, the amino acid changes in the LRR domain in the mutants were mainly localized on the outer α-helices and disordered coils (Supplementary Fig. 5b), rather than on the continuous β-sheet inside the LRR domain, which is generally considered an important region for effector binding36,37. Loss of resistance as a result of mutations in all domains of Pm6Sl implies the necessity of these domains in Pm6Sl to mediate resistance to wheat powdery mildew.

a 3D model of the Pm6Sl protein predicted via AlphaFold2. b 3D model of the Pm6Sl CC-BED module predicted via AlphaFold2. c TPN-Coomassie blue-stained leaf sections and cell death statistics for infected leaves of T3012II-3 and CS at 48 hpi with Bgt isolate E26. Scale bar, 100 μm. d Leaf segments with DAB-Coomassie blue and H2O2 accumulation cell statistics at 48 hpi with Bgt isolate E26. Brown staining indicates intracellular reactive oxygen species (IntraROS), while blue staining indicates Bgt fungus. Scale bar, 100 μm. Hyp: hyphae. The values are the means with SD as error bars. **p < 0.01, ***p < 0.001 (two-tailed Student’s t test). Source data are provided as a Source Data file.
Pm6Sl is involved in cell death
To explore how Pm6Sl provides resistance to the pathogen, T3012II-3 and CS seedlings were infected with Bgt isolate E26 and stained with trypan blue (TPN). Compared with that in CS seedlings, increased cell death, as shown by deeper staining, was observed in T3012II-3 seedlings, with a proportion of 38.3% cell death at 48 hpi, which was 3.4 times greater than that in CS seedlings (11.3%) (Fig. 4c). Moreover, a diaminobenzidine (DAB) staining assay revealed that the T3012II-3-infected cells accumulated significantly higher and more stable levels of intracellular reactive oxygen species (intraROS) than the CS-infected cells did (Fig. 4d and Supplementary Fig. 6). These results suggested that Pm6Sl may mediate resistance to wheat powdery mildew by promoting cell death.
Previous studies have shown that host cell death in response to effector-triggered immunity is controlled by the CC domain of NLR proteins38. To elucidate the mechanism by which Pm6Sl triggers cell death, a transient assay with Nicotiana benthamiana leaves was performed. The results revealed that only the CC-BED module triggered cell death, whereas neither the full-length Pm6Sl protein nor the other truncated Pm6Sl N-terminal proteins, including α1-3, α1-3-BED, α1-4 (CC) protein or Znf-BED, exhibited this ability (Supplementary Figs. 7 and 5a). In addition, two mutated CC-BED modules, CC-BEDL21F and CC-BEDM172I, which convert TA7548S from resistant to susceptible to powdery mildew, retained the ability to induce cell death in N. benthamiana (Fig. 5b). These findings suggested that the integrity of the CC-BED module is crucial for inducing cell death, at least under the tested conditions.

a Transient expression, TPN staining, and western blot of different truncated forms of the Pm6Sl CC-BED module in N. benthamiana cells. 1–5: schematic diagram illustrating the truncated forms of the Pm6Sl CC-BED module; Empty vector (EV), negative control; Pm21-CC, positive control. b Transient expression, TPN staining, and western blot of the Pm6Sl CC-BED module and two mutants (CC-BEDL21F and CC-BEDM172I) identified in the Pm6Sl CC-BED module through EMS mutagenesis in N. benthamiana cells. 6-7: Schematic diagram depicting two mutants (L21F and M172I) identified within the Pm6Sl CC-BED module via EMS mutagenesis. c Transient expression and TPN staining of the CC-BED proteins Pm6Sl, Yr7, Yr5, Rph15 and Xa1 in N. benthamiana cells. 8–11: Schematic diagram of the CC-BED domains of Yr7, Yr5, Rph15 and Xa1. The total proteins were extracted from the leaves of N. benthamiana at 48 hours after infection, and Ponceau S was employed as a stain for normalizing the total protein content. Following this, equivalent samples were utilized, and the tagged proteins were detected through western blot analysis using an anti-GFP antibody. The black arrows indicate the location of the target protein. The experiment was independently repeated three times with similar results. Source data are provided as a Source Data file.
Structural prediction analysis was also conducted to determine the presence of the N-terminal CC-BED module in the six other identified resistant BED-NLRs, including Yr5/Yr7/YrSP from wheat, Xa1/Xo1 from rice, and Rph15 from barley. The results revealed that all the BED-NLR proteins also had a CC-BED module at their N-terminus, which was comparable to that of Pm6Sl (Supplementary Fig. 8). Nevertheless, transient expression of CC-BED, CC (α1–4), and Znf-BED in N. benthamiana leaf cells revealed that, unlike Pm6Sl, none of the truncated proteins of these six BED-NLRs induced cell death under the tested conditions (Fig. 5c and Supplementary Fig. 9). Therefore, it is proposed that the CC-BED module represents a conserved structure for BED-NLRs, however, the module may function differently in BED-NLR-mediated resistance.
Pm6Sl relationships with NLRs from Gramineae species
To investigate the evolutionary origin of Pm6Sl, we first searched for Pm6Sl homologues in the National Center for Biotechnology Information (NCBI) nonredundant protein sequence database. The results revealed that the sequences of the obtained proteins presented low similarity (<75% identity) to those of Pm6Sl (Supplementary Data 7). A subsequent comparative analysis of published genome sequences revealed that Pm6Sl orthologues were absent on homoeologous Group 6 chromosomes of species related to wheat (Supplementary Fig. 10). Therefore, it is assumed that Pm6Sl may be present exclusively in Ae. longissima, although further research is necessary to substantiate this assumption.
To explore the relationships of Pm6Sl with other known NLR proteins, phylogenetic analysis was performed using Pm6Sl and six other cloned BED-NLR proteins, along with 174 known NLR proteins in the Gramineae family. The results indicated that Pm6Sl was most closely related to Rph15 from barley (Supplementary Fig. 11). However, sequence alignment analysis revealed very low amino acid sequence identity (48.8%) between Rph15 and Pm6Sl (Supplementary Fig. 12). Furthermore, phylogenetic tree analysis of the Znf-BED domains revealed that Pm6Sl is distinct from the Znf-BED domains of the other six cloned BED-NLR proteins, which are highly conserved among proteins annotated in homoeologous group 6 chromosomes of wheat-related species (Supplementary Fig. 13). In contrast, there was 38–45% identity between the ZnF-BED domain of Pm6Sl and those of the six resistant BED-NLR proteins (Supplementary Fig. 14). Therefore, Znf-BED domain integration in Pm6Sl is likely independent of those in the six characterized BED-NLR proteins.
Development of Pm6Sl germplasms and a diagnostic gene marker
During Pm6Sl cloning, multiple recombinant stocks of CS-Ae. longissima containing Pm6Sl were developed, with T3012II-3 encapsulating a 0.21 Mb fragment carrying Pm6Sl (Supplementary Data 1), equivalent to approximately 0.03% of chromosome 6Sl (666.21 Mb) of Ae. longissima cv. TL05. Agronomic traits, such as plant height, number of tillers per plant, spike length, number of spikelets per spike, number of grains per spike, grain length, grain width, and 1000-grain weight, of T3012II-3 were not significantly different from those of CS (the introgression parent), suggesting the absence of deleterious linkage drag associated with Pm6Sl in T3012II-3 plants (Fig. 6a, b). Simultaneously, a gene marker, pm6sl-1, specific to Pm6Sl, was designed (Fig. 6c and Supplementary Data 2). A total of 112 wheat cultivars or advanced breeding lines were evaluated using this marker and Pm6Sl stock lines as controls and displayed no Pm6Sl loci (Supplementary Data 8), indicating its breeding utility as a diagnostic gene marker for Pm6Sl.

a Visual phenotypes of adult plants, spikes and grains of T3012II-3 (Pm6Sl +ve) and CS (Pm6Sl −ve) plants. b Comparing the agronomic traits between T3012II-3 and CS in plant height (n = 10), number of tillers per plant (n = 10), spike length (n = 30), number of spikelets per spike (n = 30), number of grains per spike (n = 30), grain length (n = 30), grain width (n = 30), and 1000-grain weight (n = 30). The data are displayed as box and whisker plots with individual data points. The error bars represent the maximum and minimum values. Centerline, median; box limits, 25th and 75th percentiles. Significant differences were determined by two-tailed Student’s t test. ns: no significant difference. c Electrophoresis patterns of the Pm6Sl gene marker pm6sl-1. M: 2 kb DNA Ladder Marker; 1: CS; 2-4: Pm6Sl stocks TA7548, TA7548S, and T3012II-3; 5–24: common wheat varieties Aikang58, Guomai301, Ningchun4, Zhoumai22, Kenong9204, Nongda399, Jimai22, Fielder, Xiaoyan81, Xiaoyan60, Yannong19, Jing411, Xinong528, Fengchan3, Aiqianniu, Nonglin11, Pingan602, Pingan0518, Pastor and Japper. The PCR amplicons of Pm6Sl are indicated by red arrows. The experiment was independently repeated three times with similar results. Source data are provided as a Source Data file.
To further investigate the value of Pm6Sl in wheat breeding, we performed a resistance spectrum assay using 36 Bgt isolates with different virulence spectra collected from primary wheat-growing provinces of China. Our results revealed that T3012II-3, harbouring Pm6Sl, exhibited immunity or resistance (IT 0–2) against 35 of these Bgt isolates at the seedling stage (Supplementary Data 9). These findings confirmed that Pm6Sl confers resistance to multiple Bgt isolates. In addition, as previously mentioned, Pm6Sl is capable of conferring all-stage resistance, thus reinforcing its significant potential for application in wheat breeding.
Discussion
The wild relatives of wheat harbour a diverse array of resistance genes, many of which have been introgressed into common wheat through the development of wild wheat species amphidiploids, addition or substitution lines, and translocation lines39. However, the process of cloning these alien genes in common wheat via map-based cloning was previously challenging because of the suppression of homologous recombination between wheat and wild species, the limited number of chromosome-specific markers, and the complexity of identifying candidate genes40. In recent years, the exploitation of ph1b deletion mutants33 has enabled increased homologous recombination among wild wheat species, thus accelerating map-based cloning of genes from wild wheat relatives. Further integration of mutagenesis and next-generation sequences has resulted in map-based cloning of many resistance genes, such as Pm2141,42, Fhb743, and Lr4744. Mapping and cloning alien genes from wild relatives in a hexaploid wheat background offers an additional advantage in the form of ready-to-deploy germplasm that contains shortened segments harbouring resistance genes from wild relatives suitable for wheat breeding. In this study, we successfully isolated Pm6Sl from Ae. longissima in common wheat by combining map-based cloning with ph1b-induced homologous recombination and compared the candidate gene sequences of loss-of-function mutants and Pm6Sl donors. Furthermore, we developed several wheat-Ae. longissima recombinant stocks containing small Pm6Sl segments without significant adverse agronomic effects. These outcomes highlight the benefits of our map-based cloning strategy and the development of germplasms with shortened alien segments carrying alien genes that increase wheat resistance.
To date, 19 wheat Pm genes have been cloned, and their encoding proteins can be divided into four classes: typical NLR proteins, kinase fusion proteins (KFPs), ABC transporters, and hexose transporters3,4,5,6. Although 12 of the 19 cloned Pm genes encoded NLR proteins, none of the cloned Pm genes prior to Pm6Sl encoded an NLR protein with an additional integrated domain. In this study, Pm6Sl from Ae. longissima was shown to encode an NLR containing a Znf-BED domain. The cloning of Pm6Sl provides a valuable resistance gene for revealing the molecular mechanism of resistance to powdery mildew in wheat. To date, in addition to Pm6Sl, only six other NLR-IDs possessing a Znf-BED domain conferring resistance to barley or wheat rust, rice blight and leaf streak have been cloned from different crop species. Furthermore, two Znf-BED-containing non-NLRs, YrNAM, which confers resistance to wheat stripe rust, and the NAC transcription factor protein Rph7, which regulates resistance to barley leaf rust, were recently cloned45,46. Pm6Sl is a cloned BED-NLR architecture used to control powdery mildew resistance in wheat. In addition, by 3D structure prediction of Pm6Sl and the other six cloned pathogen-resistant BED-NLR architectures via Alphofold2, we discovered that all these genes had an N-terminal CC domain composed of four helixes (α1-4), and the Znf-BED domain was integrated between α3 and α4 of the CC domain, forming a CC-BED module (Supplementary Fig. 8). In this study, two mutations in the CC-BED domain of Pm6Sl, which convert leucine to phenylalanine and methionine to isoleucine, respectively, were identified (Supplementary Fig. 5a). These two mutations resulted in susceptibility to both Mut28 and Mut29, indicating that CC-BED is necessary for Pm6Sl to mediate resistance.
Zinc finger proteins constitute a superfamily of proteins that play crucial roles in diverse aspects of plant functions. Recent reports highlight the importance of zinc finger domains in host‒pathogen interactions. For example, TaZF, a wheat zinc finger protein, facilitates Pm2a-mediated recognition of AvrPm2, and in particular, TaZF’s zinc finger domain alone is sufficient for interaction with AvrPm247. In addition, Xa1, a rice BED-NLR protein, recognizes the transcription activator-like (TAL) effectors of Xanthomonas oryzae pv oryzae (Xoo), AvrXa7 and Xoo1132, through its BED domain, which forms a complex with these two TAL effectors. Furthermore, the AP2/ERF-type transcription factor OsERF101 may regulate recognition and thus positively regulate Xa1-mediated immunity48. Even so, the BED domain was unlikely the sole domain of a BED-NLR protein determining its recognition specificity to pathogen effectors, as ZnF-BEDs with identical BED domain sequences, such as Yr5 and YrSP16, Xa1 and Xo118, can confer resistance to different isolates of a pathogen or different pathogens. In this study, although the expression of the Pm6Sl CC-BED domain in N. benthamiana triggered a cell death response, the individual CC or BED domains, as well as other N-terminal truncations, failed to elicit this response (Fig. 5a). This further corroborates the hypothesis that the CC and BED domains of a BED-NLR function as an integrated CC-BED module. Additionally, the two mutant CC-BED types (CC-BEDL21F and CC-BEDM172I) maintained an N. benthamiana cell death response (Fig. 5b). These findings suggested that these two mutations resulted in the loss of Pm6Sl resistance, most likely by influencing other processes, such as pathogen recognition, rather than directly affecting the induction of cell death. However, reports on the roles of Znf-BEDs in plant immunity are scarce, necessitating further experimental evidence to elucidate their molecular mechanism.
Furthermore, on the basis of transient expression in N. benthamiana leaves, we observed that the CC-BED modules are structurally conserved in crop-derived BED-NLRs that confer resistance to various pathogens, but the functions of the module most likely vary in different proteins; for example, the CC-BED module of Pm6Sl, unlike the other six BED-NLRs, triggered cell death. In addition, as revealed by sequence alignment and phylogenetic analysis, the sequences of both the Znf-BED domain and CC-BED module of Pm6Sl are distinct from those of six other cloned BED-NLRs (Supplementary Fig. 15) and may result in functional diversification of CC-BED modules. Nevertheless, the roles of CC-BED modules in BED-NLR-mediated resistance to diverse pathogens have yet to be experimentally verified. Pm6Sl provides a new member of the BED-NLR family for understanding the molecular mechanisms of Znf-BED-containing architecture-mediated resistance to diverse pathogens.
In conclusion, we isolated Pm6Sl from Ae. longissima, which was integrated into a common wheat background, via positional cloning with ph1b-induced homoeologous recombination, integrating gene function validation via mutagenesis, gene silencing and transgenic assays. Pm6Sl encodes an NLR architecture with a CC-BED module, which is structurally conserved for crop BED-NLRs with resistance to diverse pathogens, enhancing our understanding of the NLR protein-mediated resistance mechanism. Furthermore, multiple wheat-Ae. longissima recombinant stocks encapsulating short Pm6Sl fragments devoid of detrimental agronomic traits and a diagnostic gene marker, pm6sl-1, were generated to increase the deployment of Pm6Sl in wheat breeding.
Methods
Plant materials
The plant materials used in this study included common wheat CS (TA3808) and the CS ph1b mutant (TA3809), which can induce high-frequency homologous pairing and recombination; CS-Ae. longissima disomic 6Sl#3 addition line TA7548, where a pair of 6Sl#3 chromosomes carrying Pm6Sl from Ae. longissima was added to the CS background; CS-Ae. longissima 6Sl#3[6 A] substitution line TA7548S, where a pair of CS 6 A chromosomes were replaced by 6Sl#3 pairs; CS-Ae. longissima 6Sl#3 recombinants T24, T27 and R43, containing Pm6Sl-harbouring segments; and Fielder, a common wheat variety used as a receptor for wheat genetic transformation (Supplementary Data 8). Among these materials, TA3808, TA3809, TA7548, and TA7548S were obtained from Wheat Genetics Resource Center (WGRC), Kansas State University, USA, and Fielder was obtained from the Shandong Academy of Agricultural Sciences, China. The CS-Ae. longissima 6Sl#3 recombinants were developed from a homozygous ph1b population segregating for 6Sl#3 that was derived from self-pollinated hybrids of TA7548 crossed with TA3809. In addition, 112 common wheat varieties or advanced lines collected from wheat breeders were used to validate the diagnostic value of gene markers for Pm6Sl selection (Supplementary Data 8). N. benthamiana was also used as a transformation receptor for transient expression analysis. All the plant materials were maintained at the Experimental Station of Henan Agricultural University, Zhengzhou, China.
Pathogen inoculation and phenotype identification
Bgt isolate E26 and 35 other genetically divergent isolates collected from various regions of China (Supplementary Data 9) were used to evaluate the powdery mildew resistance spectrum of Pm6Sl, while the isolate E26 was used for all other resistance assays in this study. When the first leaves were fully expanded, the seedlings were inoculated with Bgt isolates and grown in an incubator maintained at 20–22 °C, 16 h light/8 h dark cycle and approximately 95% relative humidity24. At 7–10 post inoculation (dpi), the plant responses to Bgt were scored on a 0 to 4 scale (0, 1, 2, 3, and 4), with ITs 0, 1 and 2 indicating resistance and ITs 3 and 4 indicating susceptibility.
Mutagenesis
In total, 6000 seeds of the CS-Ae. longissima disomic 6Sl#3[6 A] substitution line TA7548S were subjected to EMS treatment to generate loss-of-function mutants. The seeds were exposed to 0.6% (v/v) EMS solution and shaken at 150 rpm for 16 h in darkness at room temperature. Approximately 80 EMS-treated seeds were sown in rows of 2 × 0.3 m in the field. A total of 906 M1 plants were harvested. Thirty M2 seeds from each M1 plant, along with CS (susceptible control) and TA7548S (resistant control) plants, were germinated and cultivated in an incubator at 22–25 °C with 60% humidity and a 16-h light/8-h dark photoperiod. Once the leaves were fully expanded, the seedlings were inoculated with Bgt isolate E26 and grown at 20–22 °C with 95% humidity and 16 h of supplemental light. After 10 dpi, 41 M2 individuals with ITs of 3–4 from 30 M2 lines segregating for powdery mildew resistance were selected for further validation via the 6Sl#3-specific markers Ael69501, Ael67319, Ael63185, and Ael42958 to exclude susceptible plants caused by 6Sl#3 loss or seed impurity. Three M3 seedlings from one of the 41 M2 susceptible plants for each of the three replicates were subsequently reassessed for susceptibility. The gDNA and cDNA of thirty independently susceptible M3 mutant lines derived from different M1 plants were extracted and used to amplify candidate gene sequences to identify mutation sites through Sanger sequencing.
RNA extraction and qRT‒PCR analysis
Fresh leaves obtained from plant material were cryopreserved in liquid nitrogen and used for RNA extraction. TRIzol reagent (Cat. No. R401, Vazyme, Nanjing, China) was used to extract total RNA samples, and 1 µg of total RNA was then subjected to reverse transcription via HiScript III RT SuperMix for qPCR (+ gDNA wiper) (Cat. No. R323, Vazyme, Nanjing, China) following the manufacturer’s instructions. The resulting reverse transcripts were diluted fivefold and used for quantitative real-time RT‒PCR (qRT‒PCR). A 10 μl qRT‒PCR mixture comprising 5 μl of Taq Pro Universal SYBR qPCR Master Mix (Cat. No. Q712, Vazyme, Nanjing, China), 0.2 μl of 10 mM forward and reverse primers, and 4.6 μl of diluted cDNA was used. The PCR amplification started at 95 °C for 30 s, followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. The instrument’s default melting curve acquisition procedure was then utilized. Wheat TaACTIN (TraesCS5B02G124100) was used as an internal reference gene49. qRT‒PCR was performed via an ABI StepOne plus fluorescence quantitative PCR instrument (ABI, USA), and transcription levels were calculated via the comparative CT method50. The primers used to determine Pm6Sl transcript levels are listed in Supplementary Data 2.
Fine mapping of gene Pm6Sl
Molecular markers specific to the 6Sl#3 chromosome were designed based on Ae. longissima cv. TL05 genomic reference. Genomic DNA was obtained via the CTAB method. The 15 µL PCR mixture contained 2.0 µL of template gDNA (100 ng/µL), 1.0 µL of each primer (5.0 µmol/L), 7.5 µL of Taq MasterMix (Cat. No. CW0862; Cowin Biotech Co. Ltd., Beijing, China) and 3.5 µL of ddH2O. PCR amplification was performed using a Touchdown-63 reaction procedure51. Following electrophoresis on a 2.0% agarose gel with 4SGelred nucleic acid dye (Cat. No. A616697, BBI Life Science Corporation, Shanghai, China), the PCR products were visualized using GBOXSYDR4/1494 Syngene Automated Gel Imager (Syngene, Frederick, MD21704, USA) under UV light.
Heterozygous CS-Ae. longissima 6Sl#3 recombinants T24, T27 and R43, in a homozygous ph1b background, were used to generate secondary populations segregating for Pm6Sl. Two rounds of gene mapping were conducted. In the first round, fifty seedlings at a fully extended leaf stage were inoculated with Bgt isolate E26, and resistance was assayed for each of the 6Sl#3 recombinants. The secondary population consisting of seedlings that segregated for powdery mildew resistance was used to generate 6Sl#3 recombinants via the markers Ael58410 and Ael57699, which flank the Pm6Sl-containing interval. Plants lacking either marker were considered 6Sl#3 recombinants and were further analysed via the 20 markers in the candidate gene region (Supplementary Data 2). Fifteen unfolded first-leaf seedlings from each F2 progeny of the 6Sl#3 recombinants were inoculated with Bgt E26 to evaluate the resistance of the recombinants. The recombinant was determined to have a susceptible phenotype if all 15 F2 seedlings were susceptible. On the other hand, if the 15 F2 seedlings segregated for resistance and only resistant seedlings presented with the 6Sl#3-specific marker, the recombinant parent was confirmed to be resistant. Pm6Sl mapping was performed carefully by integrating genotypic and phenotypic data from each 6Sl#3 recombinant between markers that were present in all resistant recombinants but absent in susceptible recombinants. In the second round, the resistant recombinants T27-6 and T3012 (Supplementary Data 1), which were selected in the first round, were used to generate a secondary population segregating for Pm6Sl. 6Sl#3 recombinants were identified via the markers Ael04335 and Ael03991 flanking Pm6Sl identified via first-round mapping. The genotype of each recombinant was determined via five newly designed markers, Ael39960, Ael16698, Ael09126, Ael34119, and Ael17152, within the Pm6Sl mapping interval (Supplementary Data 1 and 2). The responses of each recombinant to Bgt isolate E26 were evaluated to further finely map Pm6Sl via the method described for initial mapping.
Identification and cloning of the candidate genes
Pm6Sl candidate genes were identified by aligning the sequences of Pm6Sl flanking markers with the genomic reference sequences of Ae. longissima cv. TL05. CNL1 and CNL2, two annotated resistance (R) genes in the Pm6Sl interval, were selected as Pm6Sl candidates. PCR primer pairs were designed to isolate the CNL1 and CNL2 sequences from the Ae. longissima cv. TL05 reference genome sequence, with forward and reverse primers located in the 5′ untranslated region (UTR) and 3′ UTR regions, respectively. The genomic DNA sequences of CNL1 and CNL2 were amplified via gDNA templates from TA7548 and CS. The 30 μl PCR mixture contained 15 μl of high-fidelity enzyme 2× Phanta Flash Master Mix (Dye Plus) (Cat. No. P520, Vazyme, Nanjing, China), 2 μl of each primer (5.0 µmol/L), 4 μl of template gDNA (100 ng/µL), and 7 μl of ddH2O. Amplification proceeded at 98 °C for 30 s, followed by 35 cycles of 98 °C for 10 s, 58–60 °C (depending on the primers) for 5 s, 72 °C for 5 s/kb, and a final extension at 72 °C for 1 min. A 5 μl amplification product was electrophoresed on a 1% agarose gel to estimate the product size. The remaining PCR product was purified via the MonPure Gel & PCR Purification Kit (Cat. No. MI17101, Monad Biotech Co., Ltd., Suzhou, China), cloned, and inserted into the TOPO vector (Cat. No. C602, Vazyme, Nanjing, China) and sequenced at Sangon Biotech (Shanghai) Co., Ltd. To amplify cDNAs of CNL1 and CNL2, total RNA isolated from TA7548 and CS was transcribed. The complementary DNA (cDNA) was used as a template to amplify CDSs via the primers SALF-CNL1 and SALF-CNL2, and the same methods were used for gDNA sequence isolation.
Virus-induced gene silencing
BSMV-VIGS was performed to investigate whether candidate genes mediate powdery mildew resistance in TA7548. The sequences of the candidate genes CNL1 and CNL2 were aligned with the reference genome sequences of CS and Ae. longissima cv. TL05 to select targeted fragments with less homology to CS (identity <50% and 100% nucleotide identity <21 nt) for designing VIGS primers (Supplementary Data 2). The 182 bp sequence of CNL1 and the 210 bp sequence of CNL2 were separately amplified from cDNA sequences and reversely cloned and inserted into BSMV:γ vectors via homologous recombination (ClonExpress II One Step Cloning Kit, Cat. No. C112, Vazyme Biotech, Nanjing, China). Each construct of BSMV:γCNL1 and BSMV:γCNL2, together with BSMV:γ (negative control) and BSMV:γPDS (phytoene desaturase genes, positive control), was transcribed in vitro using the mMESSAGE mMACHINE T7 Kit (Cat. No. AM1344, Invitrogen, USA) according to the manufacturer’s protocol. Each of the transcripts was then mixed equally with the transcripts from the BSMV:α and BSMV:β vectors before equal amounts of 2×GKP (50 mM glycine, 30 mM K2HPO4, pH 9.2, 1% w/v bentonite, and 1% w/v celite) were added. After the mixture was applied to the second leaves of TA7548, the seedlings were moistened to maintain 100% humidity at 23 °C in darkness for 24 h before being grown at 20–22 °C with 16 h of supplemental light. At 14 days after viral infection, the third leaf segments were taken for resistance assessment via in vitro culture on agarose medium supplemented with 2.5 mg/L 6-benzylaminopurine (6-BA) and inoculated with Bgt isolate E26. At 7 dpi, the infection type of each in vitro leaf segment was recorded on a 0–4 scale, with ITs of 0–2 rated as resistant and ITs of 3-4 rated as susceptible. The transcript levels of CNL1 and CNL2 were determined via qRT‒PCR via total RNA templates extracted from infected leaves. The primer qPCR-CNL1 was used for CNL1, and the primer qPCR-CNL2 was used for CNL2 and was designed based on non-VIGS targeted sequences. For silencing each candidate gene using BSMV-VIGS, at least 15 plants were challenged by BSMV vector, and the experiments were repeated three times.
Genetic transformation of wheat
The 4296 bp CDS was amplified from the CNL1 cDNA construct based on the XmaI and SpeI restriction sites. This CDS was subsequently cloned downstream of the Ubi promoter in the pWMB110-ProUbi construct, yielding a pWMB110-ProUbi:CNL1 vector for genetic transformation. The resulting pWMB110-ProUbi:CNL1 plasmid was subsequently transformed into Agrobacterium strain EHA105 for genetic transformation of the common wheat cultivar Fielder, generating a total of 24 transgenic T0 plants. For resistance assessment of the transgenic T1 lines derived from self-pollinated T0 plants, the first leaf was harvested from each plant, cultured in vitro on agarose medium containing 2.5 mg/L 6-BA, inoculated with Bgt isolate E26, and kept in an incubator at 20 °C for 16 h light and 18 °C for 8 h in darkness. After resistance was recorded at 10 dpi, each leaf segment was used to isolate gDNA to examine CNL1 via the primer pairs Ubi-F and CNL1-R. The second leaves from two resistant plants of each T1 line were used to examine CNL1 gene expression levels via qRT‒PCR. Three biological replicates, two leaves from two resistant seedlings per replicate, were carried out for each T1 line.
Transgenic wheat plants expressing CNL1 gene driven by its native promoter ProCNL1 were also generated. The genomic DNA of ProCNL1 and CNL1 was amplified from TA7548 using primers CNL1-COM, which incorporated HindIII and SacI restriction sites for cloning. The digested PCR fragment was cloned into the linearized pWMB110 vector by homologous recombination (ClonExpress II One Step Cloning Kit, Cat. No. C112, Vazyme Biotech, Nanjing, China) to yield the ProCNL1:CNL1 construct. Genetic transformation, resistance assessment and qRT-PCR followed the above-mentioned methods, except that one half a single completely expanded new leaf from each transgenic T0 seedling was used for Bgt resistance screening and the other half for CNL1 expression analysis.
Evaluation of agronomic traits
The agronomic traits of CS-Ae. longissima recombinant T3012II-3 containing Pm6Sl in the smallest 6Sl segment and CS were evaluated to determine whether any linkage drag was associated with the Pm6Sl or 6Sl segment. T3012II-3 and CS were planted in experimental fields in Beijing, China. A total of 21 plants for each plant material were planted in a 2 × 0.3 m row. Field management practices, including irrigation, fertilization, and herbicide and pesticide applications, strictly followed local practices. Ten plants were randomly selected from each row to measure plant height (from the ground to the tip of the main spike, excluding awns), tiller number per plant and spike number per plant. Three of the early-tillering spikes from each plant were then measured to determine spike length (the length from the base of the rachis to the uppermost spikelet top excluding awns), number of spikelets per spike, number of grains per spike, grain length, grain width, and one thousand-grain weight. The significant differences in agronomic traits between T3012II-3 and CS were determined for each trait via two-tailed Student’s t test.
H2O2 accumulation and wheat cell death assays
To investigate the differential accumulation of hydrogen peroxide (H2O2) in T3012II-3 cells mediated by Pm6Sl and CS lacking Pm6Sl following pathogen infection, we used DAB staining to observe intracellular reactive oxygen species (intraROS) in infected cells. To do so, leaves infected with Bgt isolate E26 were treated with a 0.1% (w/v) solution of DAB (pH 3.8) at 25 °C in darkness for 12 h. The leaves were then decolorized in a solution of ethanol and acetic acid (3:1, v/v) at room temperature until complete decolorization was achieved. Finally, the decoloured leaves were stained with a 0.6% (w/v) solution of Coomassie brilliant blue for 10 s before being rinsed with water to visualize the Bgt fungal structure for observation.
To quantify cell death levels in infected cells, leaves inoculated with Bgt isolate E26 were obtained from CS and TA7548 at 48 hpi and then immersed in a 0.4% trypan blue solution that was brought to the boiling point for exactly one minute. After immersion, the leaves were bleached for 24 h with chloral hydrate solution containing 2.5 g/ml and then stained with a 0.6% (w/v) Coomassie blue solution for 10 s. The treated leaves were observed under a Nikon ECLIPSE Ni-U microscope. All the experiments were carried out with three biological replicates, each with a single leaf. A minimum of 100 infected cells were assessed per leaf to calculate the proportions of cells exhibiting intraROS and cell death responses52.
Prediction of structural domains and 3D structures
The websites SMART, NCBI and InterPro (http://www.ebi.ac.uk/interpro/search/sequence/) were used to predict Pm6Sl structural domains. The MPI Bioinformatics Toolkit (https://toolkit.tuebingen.mpg.de/tools/deepcoil2) was used to predict the CC domain of Pm6Sl, and the software “ColabFold v1.5.2: AlphaFold2 in using MMseqs2” was used to predict its three-dimensional (3D) construction. Five structural models were predicted in total, and the first-ranked model was selected for structural analysis. The protein sequences and domain compositions of Yr5, Yr7, Xa1, and Rph15 are referenced from published articles. We individually extracted the sequence before the NB-ARC domain of each protein and used Alphafold2 to predict their 3D structures. PyMOL 2.6.0a0 was used to open the 3D structural model and polish and mark the model.
Agrobacterium-mediated transient expression in N. benthamiana
To investigate whether individual or combined domains of Pm6Sl can induce cell death, expression vectors containing various truncated forms of Pm6Sl were generated for transient expression in N. benthamiana. The different truncated Pm6Sl sequences were amplified using primers listed in Supplementary Data 2 and cloned between the double 35S promoter and GFP in the pCambia 1305.1-GFP vector based on SpeI and BamHI restriction sites. This resulted in the construction of pCambia 1305.1-truncated Pm6Sl-GFP vectors, where GFP was fused to the C-terminus of truncated Pm6Sl proteins. The truncated sequences of the α1–4 (CC), Znf-BED, and α1-3-BED-α4 (CC-BED) modules of Yr5, Yr7, Xa1 and Rph15 were synthesized by Sangon Biotech (Shanghai) Co., Ltd., on the basis of published gene sequences. The pCambia 1305.1-truncated Yr7/Yr5/Xa1/Rph15-GFP vectors were constructed using the same method used for Pm6Sl. These vectors were subsequently transformed into Agrobacterium strain GV3101, using the pCambia 1305.1-GFP vector as a negative control and pCambia 1305.1-Pm21CC-GFP causing N. benthamiana cell death as a positive control53. After supplementation with 10 mmol/L MES and 20 μmol/L acetosyringone, the activated Agrobacterium strain was cultured until the OD600 value reached ~1.0. Agrobacterium cells were subsequently collected via centrifugation and resuspended in an injection buffer containing 10 mmol/L MgCl2, 10 mmol/L MES, and 100 μmol/L acetosyringone to adjust the OD600 value in the range of 1.0–1.5. Following a 3-h incubation at room temperature, each Agrobacterium strain containing a distinct plasmid carrying one truncated form was injected into a circular area of 1 cm diameter on one leaf of an N. benthamiana plant at the 4–6-leaf stage. After 3 d of culture at 22 °C under a light source, the phenotype of the injected area was recorded, and TPN staining was conducted to confirm the cell death phenotype54. Proteins transiently expressed in N. benthamiana were detected via western blotting using an anti-GFP antibody (1:5000, QiYan, Beijing, China, No.QYA03914A).
Phylogenetic analysis
Homologues of Pm6Sl were identified via the Homologous function in the TGT System (http://wheat.cau.edu.cn/TGT/index.html)55 and NCBI BLAST. A phylogenetic tree of Pm6Sl was constructed along with 180 other proteins from the Gramineae family used to analyze the CNL network56, as well as six other BED-NLRs with resistance to diverse pathogens, including Rph15, Yr7, Yr5, YrSP, Xa1 and Xo1. For phylogenetic analysis of the Znf-BED domains, Pm6Sl and six other crop BED-NLRs, along with 154 non-NLR and NLR proteins containing Znf-BED domains in the Gramineae family reported by Marchal et al57. were used. For phylogenetic analysis of the Pm6Sl and CC-BED modules, six reported crop BED-NLRs, along with 46 other BED-NLRs in the Gramineae family, were used. Multiple sequence alignment was performed via MAFFT v7.475 L-INS-I (https://mafft.cbrc.jp/alignment/software/) with default parameters. A phylogenetic tree was constructed via the approximate maximum likelihood method via FastTree v2.1.11 (http://www.microbesonline.org/fasttree/). The tree was visualized via iTOL (https://itol.embl.de/)58.
Development and validation of a diagnostic gene marker
A gene marker, pm6sl-1, for Pm6Sl was designed based on genomic DNA sequences of Pm6Sl in comparison with wheat genomic reference sequences collected from WheatOmics 1.0 (http://wheatomics.sdau.edu.cn/)59. PCR amplification was conducted via the primer pair 5′-GCCGAGCTAACAACCTTCCTC-3′/5′-GCATGGGAATTAATGTTGTG-3′ for pm6sl-1, resulting in amplicons of 904 bp. To determine the diagnostic value of the marker, Pm6Sl stocks, including TA7548, TA7548S and CS-Ae. longissima recombinant T3012II-3, along with a selection of 112 common wheat varieties, were used (Supplementary Data 8). The amplification products were separated on 1.0% agarose gels, stained with 4SGelred nucleic acid dye (Cat. No. A616697, BBI Life Science Corporation, Shanghai, China), and detected using a GBOXSYDR4/1494 Syngene Automated Gel Imager (Syngene, Frederick, MD21704, USA) under UV light.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated or analyzed in this study are included in this article and Supplementary Information files as well as the public databases. The detailed sequence data of Pm6Sl are accessible on the NCBI website under accession OR736058. Source data are provided with this paper.
References
Asif, M., Iqbal, M., Randhawa, H. & Spaner, D. Managing and breeding wheat for organic systems, enchaining competitiveness against weeds. Springer, New York (2014).
Savary, S. et al. The global burden of pathogens and pests on major food crops. Nat. Ecol. Evol. 3, 430–439 (2019).
Wang, B. et al. Fighting wheat powdery mildew: from genes to fields. Theor. Appl. Genet. 136, 196 (2023).
Li, Y. et al. Dissection of a rapidly evolving wheat resistance gene cluster by long-read genome sequencing accelerated the cloning of Pm69. Plant Commun. 5, 100646 (2024).
Lu, C. et al. Wheat Pm55 alleles exhibit distinct interactions with an inhibitor to cause different powdery mildew resistance. Nat. Commun. 15, 503 (2024).
Li, M. et al. A membrane associated tandem kinase from wild emmer wheat confers broad-spectrum resistance to powdery mildew. Nat. Commun. 15, 3124 (2024).
Li, H. et al. Wheat powdery mildew resistance gene Pm13 encodes a mixed lineage kinase domain-like protein. Nat. Commun. 15, 2449 (2024).
Zhao, Y. et al. Pm57 from Aegilops searsii encodes a tandem kinase protein and confers wheat powdery mildew resistance. Nat. Commun. 15, 4796 (2024).
Bi, G. & Zhou, J. Regulation of cell death and signaling by pore-forming sesistosomes. Annu. Rev. Phytopathol. 59, 239–263 (2021).
Saur, I. M. L., Panstruga, R. & Schulze-Lefert, P. NOD-like receptor-mediated plant immunity: from structure to cell death. Nat. Rev. Immunol. 21, 305–318 (2021).
Kourelis, J., Sakai, T., Adachi, H. & Kamoun, S. RefPlantNLR is a comprehensive collection of experimentally validated plant disease resistance proteins from the NLR family. PLoS Biol. 19, e3001124 (2021).
Chia, K. S. & Carella, P. Taking the lead: NLR immune receptor N-terminal domains execute plant immune responses. N. Phytol. 240, 496–501 (2023).
Sarris, P. et al. A plant immune receptor detects pathogen effectors that target WRKY transcription factors. Cell 161, 1089–1100 (2015).
Brueggeman, R. et al. The stem rust resistance gene Rpg5 encodes a protein with nucleotide binding site, leucine rich, and protein kinase domains. Proc. Natl. Acad. Sci. USA 105, 14970–14975 (2008).
Maqbool, A. et al. Structural basis of pathogen recognition by an integrated HMA domain in a plant NLR immune receptor. Elife 4, e08709 (2015).
Marchal, C. et al. BED-domain containing immune receptors confer diverse resistance spectra to yellow rust. Nat. Plants 4, 662–668 (2018).
Grund, E., Tremousaygue, D. & Deslandes, L. Plant NLRs with integrated domains: unity makes strength. Plant Physiol. 179, 1227–1235 (2019).
Ji, C. et al. Xa1 allelic R genes activate rice blight resistance suppressed by interfering TAL effectors. Plant Commun. 1, 100087 (2020).
Zhang, B. et al. Multiple alleles encodinga typical NLRs with unique central tandem repeats in rice confer resistance to Xanthomonas oryzae pv. oryzae. Plant Commun. 1, 100088 (2020).
Chen, C. et al. BED domain containing NLR from wild barley confers resistance to leaf rust. Plant Biotechnol. J. 19, 1206–1215 (2021).
Hayward, A., Ghazal, A., Andersson, G., Andersson, L. & Jern, P. ZBED evolution: repeated utilization of DNA transposons as regulators of diverse host functions. PLoS One 8, e59940 (2013).
Aravind, L. The BED finger, a novel DNA-binding domain in chromatin-boundary-element- binding proteins and transposases. Trends Biochem. Sci. 25, 421–423 (2000).
Yu, G. et al. The wheat stem rust resistance gene Sr43 encodes an unusual protein kinase. Nat. Genet. 55, 921–926 (2023).
Wulff, B. B. & Moscou, M. J. Strategies for transferring resistance into wheat: from wide crosses to GM cassettes. Front. Plant Sci. 5, 692 (2014).
Li, H. et al. Development of novel wheat-Aegilops longissima 3Sl translocations conferring powdery mildew resistance and specific molecular markers for chromosome 3Sl. Plant Dis. 105, 2938–2945 (2021).
Huang, S., Steffenson, B. J., Sela, H. & Stinebaugh, K. Resistance of Aegilops longissima to the rusts of wheat. Plant Dis. 102, 1124–1135 (2018).
Anikster, Y., Manisterski, J., Long, D. L. & Leonard, K. J. Resistance to leaf rust, stripe rust, and stemrust in Aegilops spp. in Israel. Plant Dis. 89, 303–308 (2005).
Tian, X. et al. Development and characterization of Triticum aestivum-Aegilops longissima 6Sl recombinants harboring a novel powdery mildew resistance gene Pm6SI. Front. Plant Sci. 13, 918508 (2022).
Li, H. et al. A spontaneous wheat-Aegilops longissima translocation carrying Pm66 confers resistance to powdery mildew. Theor. Appl. Genet. 133, 1149–1159 (2020).
Ecker, R., Dinoor, A. & Cahaner, A. The inheritance of resistance to Septoria glume blotch. Plant Breed. 102, 113–121 (1989).
Sheng, H., See, D. R. & Murray, T. D. Mapping QTL for resistance to eyespot of wheat in Aegilops longissima. Theor. Appl. Genet. 125, 355–366 (2012).
Zhou, J. et al. Identification of drought stress related proteins from 1Sl(1B) chromosome substitution line of wheat variety Chinese Spring. Bot. Stud. 57, 20 (2016).
Sears, E. R. An induced mutant with homoeologous pairing in common wheat. Can. J. Genet. Cytol. 19, 585–593 (1977).
Koo, D., Liu, W., Friebe, B. & Gill, B. S. Homoeologous recombination in the presence of Ph1 gene in wheat. Chromosoma 126, 531–540 (2017).
Wang, J., Han, M. & Liu, Y. Diversity, structure and function of the coiled-coil domains of plant NLR immune receptors. J. Integr. Plant Biol. 63, 283–296 (2021).
Bella, J., Hindle, K. L., McEwan, P. A. & Lovell, S. C. The leucine-rich repeat structure. Cell Mol. Life Sci. 65, 2307–2333 (2008).
Förderer, A., Yu, D., Li, E. & Chai, J. Resistosomes at the interface of pathogens and plants. Curr. Opin. Plant Biol. 67, 102212 (2022).
Wang, J. et al. Reconstitution and structure of a plant NLR resistosome conferring immunity. Science 364, (2019).
Friebe, B., Jiang, J., Raupp, W. J., McIntosh, R. A. & Gill, B. S. Characterization of wheat-alien translocations conferring resistance to diseases and pests: current status. Euphytica 91, 59–87 (1996).
Wang, Y. et al. Mapping stripe rust resistance gene YrZH22 in Chinese wheat cultivar Zhoumai 22 by bulked segregant RNA-Seq (BSR-Seq) and comparative genomics analyses. Theor. Appl. Genet. 130, 2191–2201 (2017).
Xing, L. et al. Pm21 from Haynaldia villosa encodes a CC-NBS-LRR protein conferring powdery mildew resistance in wheat. Mol. Plant 11, 874–878 (2018).
He, H. et al. Pm21, encoding a typical CC-NBS-LRR protein, confers broad-spectrum resistance to wheat powdery mildew disease. Mol. Plant 11, 879–882 (2018).
Wang, H. et al. Horizontal gene transfer of Fhb7 from fungus underlies Fusarium head blight resistance in wheat. Science 368, eaba5435 (2020).
Li, H. et al. Cloning of the wheat leaf rust resistance gene Lr47 introgressed from Aegilops speltoides. Nat. Commun. 14, 6072 (2023).
Ni, F. et al. Sequencing trait associated mutations to clone wheat rust resistance gene YrNAM. Nat. Commun. 14, 4353 (2023).
Chen, C. et al. A pathogen induced putative NAC transcription factor mediates leaf rust resistance in barley. Nat. Commun. 14, 5468 (2023).
Manser, B. et al. Wheat zinc finger protein TaZF interacts with both the powdery mildew AvrPm2 protein and the corresponding wheat Pm2a immune receptor. Plant Commun. 5, 100769 (2024).
Yoshihisa, A. et al. The rice OsERF101 transcription factor regulates the NLR Xa1-mediated immunity induced by perception of TAL effectors. N. Phytol. 236, 1441–1454 (2022).
Athiyannan, N. et al. Long-read genome sequencing of bread wheat facilitates disease resistance gene cloning. Nat. Genet. 54, 227–231 (2022).
Schmittgen, T. D. & Livak, K. J. Analyzing real time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108 (2008).
Liu, W. et al. Homoeologous recombination-based transfer and molecular cytogenetic mapping of powdery mildew resistant gene Pm57 from Aegilops searsii into wheat. Theor. Appl. Genet. 130, 841–848 (2017).
Li, Y. et al. Intracellular reactive oxygen species aided localized cell death contributing to immune responses against wheat powdery mildew pathogen. Phytopathology 113, 884–892 (2023).
Gao, A. et al. Pm21 CC domain activity modulated by intramolecular interactions is implicated in cell death and disease resistance. Mol. Plant Pathol. 21, 975–984 (2020).
Zhu, S. et al. Orthologous genes Pm12 and Pm21 from two wild relatives of wheat show evolutionary conservation but divergent powdery mildew resistance. Plant Commun. 4, 100472 (2023).
Chen, Y. et al. A Collinearity incorporating homology inference strategy for connecting emerging assemblies in the Triticeae Tribe as a pilotpractice in the plant pangenomic era. Mol. Plant 13, 1694–1708 (2020).
Contreras, M. P., Ludke, D., Pai, H., Toghani, A. A. & Kamoun, S. NLR receptors in plant immunity: making sense of the alphabet soup. EMBO Rep. 24, e57495 (2023).
Marchal, C., Haberer, G., Spannagl, M. & Uauy, C. Comparative genomics and functional studies of wheat BED-NLR loci. Genes (Basel) 11, 1460 (2020).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, 293–296 (2021).
Ma, S. et al. WheatOmics: a platform combining multiple omics data to accelerate functional genomics studies in wheat. Mol. Plant 14, 1965–1968 (2021).
Acknowledgements
We are grateful to Prof. Guihua Bai from Kansas State University, Manhattan, KS, USA for amending this manuscript and valuable suggestion. This research was financially supported by the National Natural Science Foundation of China (32372089 to W.X.L., 32401805 to Q.W.L. and 32272070 to H.H.L.), the Major Science and Technology Projects of Henan Province (221100110700 to G.H.Y.), the Natural Science Foundation of Henan (242300420489 to Q.W.L.), the Graduate Education Reform Project of Henan Province (2023SJGLX052Y to Y.Z.) and the China Postdoctoral Science Foundation (2024M750806 to Q.W.L.).
Author information
Authors and Affiliations
Contributions
Q.W.L., Y.S.Z., W.X.L. and Y.Z. designed the study. C.M., X.B.T., Z.J.D., H.H.L., X.X.C., G.H.Y., S.Y.M., L.W.Z., H.F.Y. and S.W.W. performed the research. Y.Z., Z.B.Z., C.L. and S.S. analyzed the data. W.X.L., X.B.T., C.M. and Y.S.Z. wrote the manuscript and S.S., Y.Z., A.Z.C. and B.L. contributed to revising the draft. All authors have read and approved the final manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Brian Steffenson and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, C., Tian, X., Dong, Z. et al. An Aegilops longissima NLR protein with integrated CC-BED module mediates resistance to wheat powdery mildew. Nat Commun 15, 8281 (2024). https://doi.org/10.1038/s41467-024-52670-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52670-2
This article is cited by
-
Identification and characterization of a novel powdery mildew resistance gene PmCWI45575 in wild emmer wheat
BMC Plant Biology (2025)
-
Divergent molecular pathways govern temperature-dependent wheat stem rust resistance genes
Nature Communications (2025)
-
An optimized disease resistance gene cloning workflow for wheat
Nature Communications (2025)
-
Essential role of rice ERF101 in the perception of TAL effectors and immune activation mediated by the CC-BED NLR Xa1
Plant Cell Reports (2025)
-
Fine mapping of a novel powdery mildew resistance gene PmDM8 derived from a cultivated emmer (Triticum dicoccum)
Theoretical and Applied Genetics (2025)
