
Abstract
The challenging synthesis of thermodynamic-unfavored cis-olefins through catalytic cross-coupling reactions requires the synergistic interaction of substrate-activating units and configuration-regulating catalysts. Successfully hitting these two birds with one stone, we herein develop a convenient photoredox access to Z-alkenes from alkynes and light alkanes with a bifunctional iron-catalyzed system possessing both C(sp3)−H activation and configuration-controlling abilities. The protocol exhibits 100% atom utilization, mild conditions, a broad substrate scope, and compatibility with multitudinous functional groups. The detailed reaction mechanism and the origin of geometry regulation are well investigated by experimental and computational studies. Progressively, a catalytic amount of diaryl disulfides is introduced for consecutive photoinduced Z−E isomerization via reversible radical addition and flipping. Big steric hindrance substituents assembled on the disulfide emerge necessity for suppressing double-bond migration. This tandem strategy paves a promising way for stereoselective alkene construction and will bring significant inspiration for the development of transition metal photocatalysis.
Similar content being viewed by others

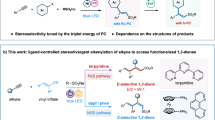
Introduction
The alkene motif is one of the most fundamental and essential organic functional groups for further multiple functionalization and carbon-chain extension, playing an important role in synthetic chemistry, pharmacology, biochemistry, polymer chemistry, and material science1,2,3,4,5,6. Developing a feasible, universal, and atom-economic strategy to construct stereo-defined alkenes has demonstrated great necessity, given the crucial role they play in these fields. Historically, methods including Wittig and Heck-type reactions, olefin metathesis, and hydrogenation and hydro-functionalization of alkynes have been well established for alkene construction7,8,9,10,11,12,13,14,15,16. Nonetheless, preactivated substrates and noble-metal catalysts are usually essential in these cases, and the generated olefins are mostly in the thermodynamic-preferred E-type or an uncertain Z-E mixture17,18,19,20,21,22,23,24,25,26,27. It is of great significance to realize the user-defined regulation of olefin geometry on account of an obvious property difference affected by steric configuration. Among the two configurations, preparation of the thermodynamic-unfavored Z-olefin is still challenging due to the uncontrollability and undetectable intermediates. In recent decades, a restricted number of highlighted works focusing on this issue have been reported. The most typical strategy should be the direct isomerization from E-alkene to Z-isomer by triplet energy transfer induced by an excited photosensitizer under light irradiation, although pre-synthesized internal olefins are needed and the practical types are limited (Fig. 1a)28,29,30,31,32,33. Impressively, two novel approaches for in situ construction of cis-olefins through transition metal-catalyzed cross-coupling hydroalkylation of alkynes were reported respectively by the Hu and the Lalic groups (Fig. 1b)34,35. Later in 2022, Silvi and coworkers pathbreakingly integrated photocatalysis and traditional Wittig-type synthesis into one system to provide geometry-defined alkenes through the photoredox generation of a phosphonium ylide36. Despite satisfactory cis-selectivities, these systems still suffered from the requirements of excess additives (metal reductants or strong bases), high temperatures, and low atom-utilization efficiency resulting from the large leaving groups, posing a pressing demand on developing mild, low-cost, and atom-economic olefination protocols to meet the criteria of green and circular economy.

a Triplet energy transfer-induced alkene isomerization. b Transition metal-catalyzed Z-selective hydroalkylation of alkynes. c Our design plan for a bifunctional iron-catalyzed system. d Graphical abstract for this work of bifunctional iron-catalyzed alkyne Z-selective hydroalkylation and Z-E inversion. PS photosensitizer, LG leaving group, SET single electron transfer, L ligand, C alkyl.
The selective C–H activation strategy, which effectively simplifies the pre-functionalization step and promotes atom-utilization efficiency, has brought major impetus to the landscape of molecular synthesis37,38. Recently, much effort has been made into the challenging C(sp3)−H activation of methane and other light alkanes via photoinduced hydrogen atom transfer (HAT) strategy by Zuo39, Noël40, and many other distinguished groups41,42,43,44,45,46,47,48,49,50,51,52,53,54 including us55,56,57,58. The chlorine radical generated by the photo-driven ligand-metal charge transfer (LMCT) process of a simple Fe−Cl complex is one of the most efficient and widely applied HAT catalysts46,47,48,49,50,51,52,53,54,55,56,57. Meanwhile, iron, as the most abundant transition metal element in the Earth’s crust, has been broadly employed in traditional transition metal catalysis59. Many ingenious iron catalysts in different oxidation states have been developed for a broad range of alkyne transformations including hydroboration, hydrosilylation, hydromagnesiation, hydrocarbonylation, and hydrogenation by Thomas25,26,60,61,62,63,64, Chikkali27,65,66, Beller67,68, Zhu14,69,70, and Kirchner71,72 et al. These original works greatly promote the development of this field. However, iron catalysis has been rarely used as the partner catalytic cycle in photo-metal dual catalysis73. According to our goal of developing a low-cost and atom-economic cis-olefination protocol, integrating the photoinduced C(sp3)−H activation unit and the transition metal-catalyzed configuration-regulated alkenylation strategy into one system seems to be an ideal means. Depending on the works mentioned above and our research experience, a bifunctional iron catalytic system is designed to hit two birds with one stone (Fig. 1c), in which the photo-mediated Fe−Cl catalytic cycle can transform the alkane into a carbon radical (1st bird), and the iron-ligand catalytic section is proposed to control the spatial configuration of the olefination process via steric hindrance effect in either radical trapping-molding route or alkyne preactivation-controllable addition route14,25,26,27,60,61,62,63,64,65,66,67,68,69,70,71,72,74,75,76,77,78 (2nd bird). Furthermore, the Z-E inversion of the achieved cis-olefins with high trans-selectivity is another key point to acquire olefin photosynthesis with a user-defined geometry. Nevertheless, photo-driven isomerization from Z-alkene to E-isomer has been rarely reported due to the inherent contradiction between the energy-absorbing process of the alkene photoexcitation to a triplet state and the energy-releasing process of the thermodynamically spontaneous Z-E transformation28,29,30,31,32,33. A reversible radical addition-elimination strategy instead of the energy transfer protocol appears to be an effective solution while only a few non-catalytic examples with stoichiometric disulfide additives were reported to our knowledge36,79,80. Based on our design plan and considerable efforts, we herein develop a convenient and tandem photoredox access to configuration-defined alkenes from alkynes and light alkanes via radical molding and flipping. A bifunctional iron-catalyzed system possessing both C(sp3)−H activation and configuration-controlling abilities is utilized for direct cis-olefin synthesis, and catalytic-amount diaryl disulfide with big steric hindrance substituents is employed for performing photoinduced Z−E isomerization and suppressing double-bond migration. The strategy exhibits 100% atom utilization, mild conditions, broad substrate scope, and compatibility with multitudinous functional groups.
Results
Condition optimization for alkyne Z-selective hydroalkylation
We began our condition optimization using phenylacetylene (1a) and cyclohexane (2a) as the model substrates (Table 1). The optimized conditions for the amination of cyclohexane established in our previous research55 were tried first, and only a trace amount of product was obtained. (Entry 1), indicating the weaker radical-trapping ability of alkynes than that of azo compounds. Investigation on different metal catalysts that can assist the radical capture (details see Supplementary Table 1) showed that only CuCl and FeCl2•4H2O were effective, and the cooperation of FeCl3 and FeCl2•4H2O afforded the corresponding internal alkene 3a and 4a in a promising yield of 36% with a 66% Z-isomer selectivity (Entry 2). Studies on the catalyst loading scale indicated that increasing the usage of FeCl2•4H2O contributed to the improvement of both the yield and the cis-selectivity, and the loading of FeCl3 could be reduced to 0.001 equivalent without any decrease in the yield and stereoselectivity (Entry 3, details see Supplementary Table 2). The solvent effect was then explored and CH3CN was the best choice (Entries 3–6). To further promote the Z-selectivity, various kinds of ligands were screened (Entries 7–9, details see Supplementary Table 3). The outcomes suggested the chalcogen ligands (L3–L6) to be better candidates than nitrogen-group ligands (L1–L2, L7–L10), and the reaction reached a remarkable 90% yield and 90:10 Z/E ratio with a bi-2-naphthol ligand equipped with two 2,4,6-triisopropylphenyls (Entry 9). Control experiments presented that FeCl3 and light irradiation were both essential for this transformation (Entries 10–11). Extension of the light wavelength to 405 nm would almost terminate the reaction completely (Entry 12).
Substrate scope for alkyne Z-selective hydroalkylation
With the optimized conditions in hand, we first investigated the substrate scope for alkynes (Fig. 2). Many aryl alkynes with diverse substituent groups reacted with cyclohexane smoothly under standard conditions, delivering the alkenylation products in good to excellent yields and Z-selectivities (3a−3ad). A variety of functional groups including methoxyl (3b–3d, 3m–3n), tert-butyl (3e), acetamido (3f), methyl (3g–3i), ethyl (3j), propyl (3k), phenyl (3l), fluoro (3o–3p), chloro (3q–3s), bromo (3t), trifluoromethyl (3u–3v), cyano (3w), carboxyl (3x) and methoxycarbonyl (3y–3z) at different substituted positions, some of which are vital synthons for further derivatization, were introduced to the alkene products, showing the excellent functional group tolerance of this method. It is worth mentioning that, as a classical motif in the combretastatin family and a promising anticancer agent81, the 3,4,5-trimethoxyphenyl ethylenyl group-based alkylation product was successfully constructed with good results (3n). Similarly, 3aa, a promising compound for treating allergic diseases, was prepared through this method with an 82% yield and 93:7 Z/E ratio, for which a poor cis-selectivity was given in traditional Wittig-type synthesis82. Heterocycle-containing alkynes were also examined (3ab−3ad) and exhibited satisfying yields and excellent Z-selectivities (up to over 99:1). Noteworthily, the easily oxidized thioether component, which is always treated as a poison to metal catalysts, was well tolerated in this process with little influence in efficiency (3ac). Silylacetylene is well acknowledged as a remarkable synthon in organic synthetic chemistry, especially in terminal alkyne synthesis. Here with triisopropylsilylacetylene as substrate, a silicon-based olefin (2-cyclohexylvinyl)triisopropylsilane (3ae) was constructed directly by this strategy. Moreover, we managed to expand the scope to an alkyl alkyne but only a moderate yield and poor Z-selectivity were obtained (3af), which revealed that the accessible π-π interaction between aryl alkynes and the bi-2-naphthol ligand might play a positive role for stereoselectivity control. Finally, to demonstrate the practicability, a gram-scale reaction was carried out with 1a and 2a, affording 3.01 g of 3a without obvious loss in performance.

aReaction conditions: alkyne (0.3 mmol), cyclohexane (10 equiv.), FeCl3 (0.001 equiv.), FeCl2•4H2O (0.5 equiv.), L6 (0.5 equiv.), and CH3CN (3 mL) under N2 and irradiation of a 30 W 365 nm LED for 48 h. bFeCl3 (0.1 equiv.). cReacting for 24 h. dReacting for 7 d.
The scope of alkanes was also investigated (Fig. 3). Many liquid and solid alkanes including cyclic (3ag–3ak) and linear (3al–3ap) ones were attempted to react with phenylacetylene, generating the corresponding C(sp3)−H alkenylation products in appreciable yields and geometry-selectivity. Enlarging the size of the cyclic alkanes seems to lead to an insignificant decrease in yields and a slight improvement of Z-selectivities (3ag−3aj). Besides the similar success in stereoselectivities at different positions of one alkane molecule, the regiomeric ratios (r.r.) towards different sites were also studied (3ak−3ar). Amazingly, the summarized characteristics of the regioselectivities were partly distinct from those reported previously83. Except for the stability of obtained carbon radicals (3°/2°/1°) and the quantity of hydrogens with a same chemical environment, the stereo-hindrance effect around the corresponding alkyl radical was also critical in deciding the regiomeric ratio thanks to the crowded capturing process with a linear alkyne as the radical trap56. As a consequence, the regiomeric ratio for 1° and 2° carbon sites increased to a certain extent for 3al to 3aq. In addition, an important component of liquid petroleum gas (LPG), iso-butane, was imported instead of the N2 atmosphere to form the alkenylated product (3aq), and the moderate outcomes might be ascribed to the inferior solubility of the gaseous substrate in acetonitrile. A typical heterocycle-involved compound 3ar was also prepared from one of the most common solvents, tetrahydrofuran, with this method.

aReaction conditions: alkyne (0.3 mmol), alkane (10 equiv., 1 atm for gaseous alkane), FeCl3 (0.001 equiv.), FeCl2•4H2O (0.5 equiv.), L6 (0.5 equiv.), and CH3CN (3 mL) under N2 and irradiation of a 30 W 365 nm LED for 48 h. bFeCl3 (0.1 equiv).
Applications
Olefin is considered to be a unique and key synthon in synthetic chemistry, biochemistry, and pharmacology1,2,3,4,5,6. Consequently, extending the applicable scope to late-stage functionalization (LSF) of drugs and biorelevant compounds will greatly enhance the application value of our strategy (Fig. 4). For this purpose, some representative amino acids and peptides were introduced first, and the derived alkylated alkenes from Cbz-protected proline, phenylalanine, and glycylglycine were efficiently acquired in high cis-selectivities (3as−3au). Furthermore, alkynes from four natural products, including spice-based thymol and borneol (3av, 3aw), terpenoid dehydroabietylamine (3be), and steroid antineoplastic lithocholic acid (3bf), were attempted, and the reactions proceeded with cyclohexane smoothly with good results. Various drugs with diverse functional groups and therapeutic abilities, such as Ibuprofen (antiphlogistic, 3ax), Naproxen (analgesic, 3ay), Acetylsalicylic acid (antipyretic, 3az), Amantadine (antiviral, 3ba), Probenecid (treating gout, 3bb), Desloratadine (treating allergy, 3bc), and Fluoxetine (treating depressive disorder, 3bd) were also involved in this strategy. To our delight, the corresponding hydroalkylation products were all delivered under the universal conditions with satisfactory yields and Z-selectivities, rendering this protocol great potential in LSF and drug discovery.

aReaction conditions: alkyne (0.3 mmol), cyclohexane (10 equiv.), FeCl3 (0.1 equiv.), FeCl2•4H2O (0.5 equiv.), L6 (0.5 equiv.), and CH3CN (3 mL) under N2 and irradiation of a 30 W 365 nm LED for 48 h.
Mechanistic study
As shown in Fig. 5, some mechanistic experiments were carried out to clarify the mechanism of the stereoselective alkenylation. Firstly, a radical trapping experiment using 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) as the radical scavenger was performed under the standard conditions (Fig. 5a). The corresponding adduct 1-(cyclohexyloxy)-2,2,6,6-tetramethylpiperidine between TEMPO and a cyclohexyl radical was clearly observed by Hydrogen Nuclear Magnetic Resonance (1H NMR) and Gas Chromatography-Mass Spectrometry (GC-MS) (Supplementary Figs. 9 and 10). A radical clock experiment was conducted then with (2-ethynylcyclopropyl)benzene (Fig. 5b), and the ring-opening allene 5b was successfully isolated in a 25% yield and confirmed by 1H NMR and GC-MS with the direct olefination product 5a not observed (Supplementary Fig. 12). These two results demonstrated the system to undergo a radical pathway with an alkyl radical involved. The kinetic isotope effect (KIE) towards the C(sp3)–H activation procedure was investigated next by applying 1) 2a/d12-2a, respectively, as the substrate or 2) a 1:1 mixture of 2a and d12-2a as the substrates (Fig. 5c). The KIE values given by kH/kD and PH/PD were calculated as 1.19 and 3.00, which indicated the relative HAT process to be a “product-determining step” but not a “rate-determining step” (Supplementary Figs. 13−15)84. The quantum yield (QY) of the model reaction was measured as 0.018 (details see Supplementary Figs. 16 and 17), suggesting that radical chain processes made little contribution to generating the corresponding olefin products. A similar conclusion was also verified by the light-on-off experiment (Supplementary Fig. 18). The interaction between the Fe(II) catalyst and alkyne substrates was next investigated through the 1H NMR titration experiments. With different equivalents of FeCl2 added to the CH3CN-d3 solution of phenylacetylene, neither obvious chemical shift change of the characteristic peaks of the alkyne nor new peaks were observed, indicating the unobvious complexation between the Fe(II) catalyst and terminal alkynes in this system (Supplementary Fig. 19). To further explore the interaction between the iron catalysts and the ligand, we prepared the complexes L4-FeCl2 and L4-FeCl3 (details shown in SI). The formation of these complexes was confirmed by Ultraviolet-Visible spectroscopy (UV-Vis) and 1H NMR detection (Supplementary Figs. 3–8). Followingly, we respectively used the combinations of FeCl3/L4-FeCl2, L4-FeCl3/FeCl2, and L4-FeCl3/L4-FeCl2 as catalysts to conduct the model reaction under standard conditions (Supplementary Table 8). Compared to entry 7 in Table 1, similar outcomes were obtained with the combinations of FeCl3/L4-FeCl2 and L4-FeCl3/L4-FeCl2. These results indicate that the coordination of the ligand and FeCl2 plays an important role in alkyne transformation and stereoselectivity regulation. Meanwhile, whether the ligand coordinates with FeCl3 has little influence on the yield and selectivity due to the abilities of both FeCl3 and L4-FeCl3 for photoinduced generation of chlorine radicals (demonstrated by another radical trapping experiment shown in Supplementary Fig. 20).

a Radical trapping experiment. b Radical clock experiment. c Kinetic isotope effect experiments with 1a and 2a/d12-2a. d hydrogen source studies with D12-cyclohexane and D2O. e Kinetic profile of alkenylation of cyclohexane.
To gain deep insights into the influence factors of configuration regulation, some control experiments were implemented with the cis-major 3a gained from the model reaction. Initially, a blank control experiment for the isomerization of 3a was explored only under the irradiation of 365 nm LEDs, of which the results presented an obvious trend of an unstopped transformation to E-isomer (Supplementary Fig. 21). Subsequently, 3a instead of the two substrates was put back into the model conditions. As a result, the Z-E inversion efficiency was found to remarkably decrease and the Z/E ratio could reach a constant value at 70:30 after long enough (Supplementary Fig. 22). These outcomes suggested that Z-form olefins should be generated and regulated during the alkene skeleton-establishing process. Further evidence was provided by the following kinetic exploration of the time-tracking experiments. As displayed in Fig. 5e, the initiate cis-selectivity for 3a could attain 93:7. With the extension of reaction time, a steady increase of yield along with a gradual but slight decline of Z-selectivity within 48 h were observed, supporting that the iron-ligand-catalyzed geometry-regulating process was crucial for the Z-isomer formation. Additionally, the hydrogen source of the hydroalkylation reaction was tested with isotope-labeled substrates (Fig. 5d). When d12-cyclohexane was employed instead of cyclohexane, the benzyl hydrogen of the obtained olefin was almost unexchanged with deuterium (Supplementary Fig. 23). With an increasing amount of D2O added to the system, the deuterium-labeling radio of the benzyl hydrogen was rising simultaneously (Supplementary Fig. 24). These phenomena reflected that the protonation of an alkenyl anion from water was more likely to occur than a HAT process between an alkenyl radical and cyclohexane during the final procedure.
To further elucidate the reaction pathway and the detailed origin of the Z/E selectivity, we conducted density functional theory (DFT) calculations with the model substrates and 1,1’-bi-2-naphthol ligand (Fig. 6)85,86. According to the previous research and our experience46,47,48,49,50,51,52,53,54,55,56,57, a chlorine radical as an active HAT catalyst originates from the FeCl3 species through LMCT in the presence of light. Subsequently, it can abstract a hydrogen atom from cyclohexane through transition state ts-HAT to afford a cyclohexyl radical, which is a fast process (details see Supplementary Fig. 25). In our design plan, the configuration is predicted to be controlled through two possible distinct routes (Fig. 1c). For the first radical trapping-molding pathway (Fig. 6a, black route), the cyclohexyl radical underwent a radical addition process to the phenylacetylene 1 at its terminal C site via transition state ts1 with a free energy barrier of 13.8 kcal/mol. A following single electron oxidation between the generated benzyl radical 8 and the quintet FeCl2 complex 4 is supposed to occur in two probable geometry, affording Fe(III) complex 6-Z or 6-E respectively via transition state ts3-Z or ts3-E. The kinetic analysis shows the formation of 6-Z needs to overcome a free energy barrier of 11.0 kcal/mol which is lower than that of 6-E for 1.7 kcal/mol, indicating the formation of 6-Z to be more favorable. Finally, an energy-releasing protolysis process of 6-Z with a proton and a chloride ion leads to the Z-configuration product 9-Z and the regeneration of the photo-sensitive sextet FeCl3 complex 7 for the next LMCT procedure. Further hole-electron analysis of the excited state of 7 was performed (Supplementary Fig. 26). The result shows that the electrons of Cl atoms (blue: hole) will transfer to the Fe atom (green: electron) during the exciting process, demonstrating the ability of 7 for the photoinduced generation of Cl radicals. In contrast, considering that the alkyne as a common π-acid may be pre-activated by the Fe(II) species 4, another alkyne preactivation-controllable addition route was also studied. As presented in the red route of Fig. 6a, the alkyne preactivation is realized through the coordination of alkyne 1 to complex 4, and the subsequent radical addition from the cyclohexyl radical can be regulated by the steric hindrance effect around the iron center. However, this pathway is proposed to be excluded due to the high free energy barrier of ts2. Furthermore, the energy decomposition analysis (EDA) of ts3-Z and ts3-E was performed to reveal the origin of cis-selectivity (details see section 5.1 in supplementary information). As displayed in Fig. 6b, the steric hindrance (ΔΔEsteric = 9.8 kcal/mol) is suggested to be the dominant factor for controlling the hydroalkylation geometry, which faultlessly fits our design.

a Free energy profiles of the Z-configuration alkene generation under iron catalysis. b Energy decomposition analysis of ts3-Z and ts3-E. DFT calculations at the M06/6-311 + G(d,p)-SDD(for Fe)/SMD(acetonitrile)//M06/6-31 G(d,p)-SDD(for Fe)/SMD(acetonitrile) level of theory. The energies are given in kcal/mol.
Depending on our experimental and computational studies, a proposed mechanism is shown in Fig. 7. Inside the catalytic cycle, most of the steps have been proven to be reasonable as we design. It is worth mentioning that since the alkenyl radical 8 is well known as a HAT-active species, a potential radical-chain process that generates undesired nonselective products may also be involved in this system (shown in the bottom half). To avoid this pathway and confirm the configuration control, a 50% loading of FeCl2 and the ligand is applied, which results in enough concentration of species 4 for trapping the radical 8 in time.

Proposed reaction mechanisms for the·bifunctionaliron-catalyzed cycle and the unfavored radical-chain process.
Design of tandem isomerization to E-form
Based on our goal of constructing olefins with a user-defined configuration, we next devoted ourselves to the tandem transformation of the obtained olefins to their E-stereoisomers. As we discussed in the introduction part, a reversible radical addition-elimination strategy instead of the energy transfer protocol appears to be an effective solution to realize the Z-E inversion via radical flipping. According to the previous non-catalytic report with stoichiometric disulfide reagents36,79,80 and our investigation experience, we planned to utilize a catalytic amount of a well-designed diaryl disulfide as a practical arylthiyl radical precursor under light irradiation to accomplish the tandem Z-E conversion and inhibit the potential alkene migration.
Condition optimization for tandem E-alkene synthesis
An iodine radical from I2 photolysis as a well-known intermediate with reversible addition-elimination abilities was attempted first to catalyze the consecutive Z-E inversion process and acquire E-olefins from the crude products of the model reaction (Fig. 8). With the addition of 5 mol% I2 as the catalyst and CH3CN as the solvent to the cis-alkene-major mixture under irradiation of 365 nm LED, a 64% total yield of alkenes and a moderate E-selectivity were observed with a tiny amount of migration byproducts (5a and 6a) appearing. Further screening on diverse solvents demonstrated that n-hexane was able to markedly promote the transformation efficiency along with an undesired increasing yield of byproducts (Supplementary Table 5). Meanwhile, the dosage of I2 could be lowered to 1% without obvious loss in outcomes, but rising the iodine loading to 10% would dramatically damage the desired olefins to some unknown compounds (Supplementary Table 6). The previously used diphenyl disulfide was also tried under similar conditions instead of I2, and as a result, the fantastic trans-selectivity could remain with a reduced amount of migration products. To further inhibit the radical migration process, three isopropyls were assembled on each phenyl ring to amplify the steric hindrance effect. Satisfyingly, the double-bond migration was almost suppressed to only 5% with an up to 96% E-selectivity of the olefin products. Moreover, the reaction results could be further improved by lengthening the wavelength of light, reaching an excellent 2-step yield of alkenes containing 99% trans-isomer with the migration byproducts completely disappearing (Supplementary Table 7).

Reaction conditions: 1) 1a (0.3 mmol), cyclohexane (10 equiv.), FeCl3 (0.001 equiv.), FeCl2•4H2O (0.5 equiv.), L6 (0.5 equiv.), and CH3CN (3 mL) under N2 and irradiation of a 30 W 365 nm LED for 48 h. 2) Catalyst (1 mol%) and solvent (3 mL) under N2 and irradiation of a 30 W LED for 3 h (C1 with 365 nm, 405 nm, 455 nm for 0.5 h, 5 h, and 12 h, respectively). The total yields were the isolated yields after two successive steps. Yields of 3a + 4a or 5a + 6a were determined by 1H NMR. The E-selectivity was detected by 1H NMR or GC-MS.
Substrate scope for 2-step E-alkene synthesis
With our brilliant catalytic system established, we next selected some typical substrates from the scope of the 1st step to test the applicability of the Z-E inversion strategy (Fig. 9, 4a-4ab). Initially, the alkyne substrates carrying electron-donating or electron-withdrawing groups at different positions transformed to the corresponding E-selective hydroalkylation products smoothly in good 2-step yields and superb selectivities (4a-4t). Various cyclic and linear alkane substrates were also employed, affording the C(sp3)−H trans-selective alkenylation products efficiently and stereoselectively (up to >99%) (4u-4y). Furthermore, three derivated alkynes from Naproxen, dehydroabietylamine, and Fluoxetine were picked out and they worked well under the thiyl radical-mediated conditions to deliver the desired E-isomers.

aReaction conditions: 1) 1 (0.3 mmol), alkane (10 equiv.), FeCl3 (0.001 equiv.), FeCl2•4H2O (0.5 equiv.), L6 (0.5 equiv.), and CH3CN (3 mL) under N2 and irradiation of a 30 W 365 nm LED for 24–48 h. 2) C1 (1 mol%) and n-hexane (3 mL) under N2 and irradiation of a 30 W 455 nm LED for 12 h. bIsolated yield after 2 steps. cFeCl3 (0.1 equiv.).
Mechanistic study for Z-E inversion
Considering that the designed disulfide catalyst was the only additive in this system, we directly performed our mechanistic investigation with the generated arylthiyl radical from the photoinduced homolytic S−S bond cleavage based on DFT calculations at the same level above (Supplementary Fig. 28). We started the studies with the model cis-hydroalkylation product 9-Z and a simplified phenylthiyl radical. The sulfur-center radical first undergoes a radical addition to the C=C bond of 9-Z through ts5, generating a benzyl radical species 10. The corresponding free energy barrier of this process is calculated to be 14.1 kcal/mol and 4.1 kcal/mol endothermically in total. In contrast, if the benzenethiol radical attacks 9-Z at its benzyl carbon site, the free energy barrier through transition state ts6 will be 16.1 kcal/mol which is 2.0 kcal/mol higher than that of ts5. Meanwhile, the relative energy of the corresponding product 11 is 9.6 kcal/mol higher than that of 10, suggesting the more unstable nature of 11 compared to 10. Both results proposed the ts6-11 route to be a disfavored pathway. In the preferred intermediate 10, as the π bond of the C=C double bond is already broken, a C-C bond rotation that can also be treated as a radical flipping procedure will occur through transition state ts7 with a 10.6 kcal/mol free energy barrier to give the radical intermediate 12. Finally, the radical elimination of 12 through ts8 provides the rebirth of the phenylthiyl radical and the target E-configuration 13 with a 4.4 kcal/mol energy release.
Discussion
In a nutshell, we have successfully merged the light-alkane C(sp3)−H activation function from the Fe−Cl photocatalysis and the alkene-configuration regulation ability from the iron-ligand catalysis into one bifunctional iron-catalyzed system to realize the low-cost and atom-economic Z-olefin construction protocol through alkyne cis-hydroalkylation. A chlorine radical-mediated HAT process and an iron-ligand-controlled Z-selective radical molding procedure are the two key points responsible for the high reaction yield and stereoselectivity. Furthermore, the Z-E inversion of the achieved cis-olefins is well established via a reversible addition-elimination pathway catalyzed by a well-designed disulfide compound assembled with big steric hindrance substituents for inhibiting double-bond migration. This remarkable synthetic strategy for achieving arylalkyl alkenes with a user-defined geometry exhibits 100% atom utilization, outstanding selectivities, mild conditions, broad substrate scope, and compatibility with multitudinous functional groups. The detailed reaction mechanism and the origin of configuration control were meticulously investigated depending on experimental and computational studies. This tandem strategy paves a promising way for stereoselective alkene construction and will ignite new sparks via the collision of traditional approaches with emerging methods.
Methods
General procedure for C(sp3)–H Z-alkenylation of liquid/solid alkanes
A 10 mL quartz reaction tube was added a magnetic stir bar, alkyne (1 equiv.), alkane substrates (10 equiv.), FeCl3 (0.001 equiv.), FeCl2•4H2O (0.5 equiv.) and ligand (0.5 equiv.) followed by CH3CN under N2 environment. The reaction tube was capped with a greased two-way septum cock. The resulting mixture was degassed and backfilled with N2 five times, then set to stir (600 rpm) and irradiated with a 30 W 365 nm LED (2 cm away, with circulating water to keep the reaction at room temperature for 24 to 48 h). After the reaction ended (monitored by TLC or GCMS), the reaction solution was diluted with DCM, washed with deionized water two times, and the organic layer was concentrated in vacuo. The crude product was purified by silica gel column chromatography using the appropriate solvent system to give the desired product.
General procedure for Z-alkenylation of gaseous alkanes
A 10 mL quartz reaction tube was added a magnetic stir bar, alkyne (1 equiv.), FeCl3 (0.1 equiv.), FeCl2•4H2O (0.5 equiv.), and ligand (0.5 equiv.) followed by CH3CN under N2 environment. The reaction tube was sealed with a greased three-way septum cock. The reaction mixture was degassed by gaseous alkane five times and kept at ambient pressure all the time. Then the quartz tube was set to stir (600 rpm) and irradiated with a 30 W 365 nm LED (2 cm away, with circulating water to keep the reaction at room temperature for 48 h). After the reaction ended, the reaction solution was diluted with DCM, washed with deionized water two times, and the organic layer was concentrated in vacuo. The crude product was purified by silica gel column chromatography using the appropriate solvent system to give the desired product.
General procedure for tandem configuration transformation
The 1st-step reaction was carried out following the general procedures described above. After the reaction ended, the reaction solution was diluted with DCM, washed with deionized water two times, and the organic layer was concentrated in vacuo. The crude product and 1,2-bis(2,4,6-triisopropylphenyl)disulfane (C1, 1 mol%, 1 mg) was dissolved in n-hexane (3 mL) and added into a 10 mL quartz reaction tube equipped with a stir bar. The reaction tube was capped with a greased two-way septum cock. The resulting mixture was degassed and backfilled with N2 three times, then set to stir and irradiated with a 30 W 455 nm LED (2 cm away, with an electric fan to keep the reaction at room temperature for 12 h). If the transformation was suspensive (monitored by GC-MS or 1H NMR), an extra addition of 1 mol% C1 was needed. After the reaction ended, the mixture was purified by silica gel column chromatography using the appropriate solvent system to give the desired trans-product.
Data availability
All data that support the findings of this study are provided within the paper and its supplementary information files, and are also available from the corresponding author upon request. Source data are provided with this paper.
References
Oger, C., Balas, L., Durand, T. & Galano, J. M. Are Alkyne Reductions Chemo-, Regio-, and Stereoselective Enough To Provide Pure (Z)-Olefins in Polyfunctionalized Bioactive Molecules? Chem. Rev. 113, 1313–1350 (2013).
Williams, J. M. J. Preparation of Alkenes: A Practical Approach (Oxford University Press, 1996).
Wille, U. Radical Cascades Initiated by Intermolecular Radical Addition to Alkynes and Related Triple Bond Systems. Chem. Rev. 113, 813–853 (2013).
Liu, R.-Y. & Buchwald, S. L. CuH-Catalyzed Olefin Functionalization: From Hydroamination to Carbonyl Addition. Acc. Chem. Res. 53, 1229–1243 (2020).
Zhang, Z. K., Bera, S., Fan, C. & Hu, X. L. Streamlined Alkylation via Nickel-Hydride-Catalyzed Hydrocarbonation of Alkenes. J. Am. Chem. Soc. 144, 7015–7029 (2022).
Wu, D., Kong, W.-Y., Bao, Y., Zhao, D.-Y., Li, Y.-Q. & Yin, G.-Y. Alkene 1,1-difunctionalizations via organometallic-radical relay. Nat. Catal. 6, 1030–1041 (2023).
Vougioukalakis, G. C. & Grubbs, R. H. Ruthenium-Based Heterocyclic Carbene-Coordinated Olefin Metathesis Catalysts. Chem. Rev. 110, 1746–1787 (2010).
Meek, S. J., O’Brien, R. V., Llaveria, J., Schrock, R. R. & Hoveyda, A. H. Catalytic Z-selective olefin cross-metathesis for natural product synthesis. Nature 471, 461–466 (2011).
Koh, M. J., Khan, R. K. M., Torker, S., Yu, M., Mikus, M. S. & Hoveyda, A. H. High-value alcohols and higher-oxidation-state compounds by catalytic Z-selective cross-metathesis. Nature 517, 181–186 (2015).
Slack, E. D., Gabriel, C. M. & Lipshutz, B. H. A Palladium Nanoparticle–Nanomicelle Combination for the Stereoselective Semihydrogenation of Alkynes in Water at Room Temperature. Angew. Chem. Int. Ed. 53, 14051–14054 (2014).
Garnes-Portolés, F. et al. Regioirregular and catalytic Mizoroki–Heck reactions. Nat. Catal. 4, 293–303 (2021).
Chen, C., Dugan, T. R., Brennessel, W. W., Weix, D. J. & Holland, P. L. Z-Selective Alkene Isomerization by High-Spin Cobalt(II) Complexes. J. Am. Chem. Soc. 136, 945–955 (2014).
Wech, F. & Gellrich, U. In Situ Formation of an Efficient Catalyst for the Semihydrogenation of Alkynes from Imidazolone and BH3. ACS Catal. 12, 5388–5396 (2022).
Hu, M.-Y. et al. Iron-Catalyzed Regiodivergent Alkyne Hydrosilylation. J. Am. Chem. Soc. 142, 16894–16902 (2020).
Li, Y., Liu, D. G., Wan, L., Zhang, J. Y., Lu, X. & Fu, Y. Ligand-Controlled Cobalt-Catalyzed Regiodivergent Alkyne Hydroalkylation. J. Am. Chem. Soc. 144, 13961–13972 (2022).
Corpas, J. et al. One-Metal/Two-Ligand for Dual Activation Tandem Catalysis: Photoinduced Cu-Catalyzed Anti-hydroboration of Alkynes. J. Am. Chem. Soc. 144, 13006–13017 (2022).
Zhu, Y. F. & Wei, Y. Y. Copper catalyzed direct alkenylation of simple alkanes with styrenes. Chem. Sci. 5, 2379–2382 (2014).
Kancherla, R. et al. Oxidative Addition to Palladium(0) Made Easy through Photoexcited-State Metal Catalysis: Experiment and Computation. Angew. Chem. Int. Ed. 58, 3412–3416 (2019).
Hazra, A., Chen, J. & Lalic, G. Stereospecific Synthesis of E‑Alkenes through Anti-Markovnikov Hydroalkylation of Terminal Alkynes. J. Am. Chem. Soc. 141, 12464–12469 (2019).
Cao, H. et al. Photoinduced site-selective alkenylation of alkanes and aldehydes with aryl alkenes. Nat. Commun. 11, 1956–1963 (2020).
Lee, G. S., Kim, D. & Hong, S. H. Pd-catalyzed formal Mizoroki–Heck coupling of unactivated alkyl chlorides. Nat. Commun. 12, 991–1002 (2021).
Nattmann, L., Lutz, S., Ortsack, P., Goddard, R. & Cornella, J. A Highly Reduced Ni−Li−Olefin Complex for Catalytic Kumada−Corriu Cross-Couplings. J. Am. Chem. Soc. 140, 13628–13633 (2018).
Yue, F. Y., Dong, J. Y., Liu, Y. X. & Wang, Q. M. Visible-Light-Mediated Alkenylation of Alkyl Boronic Acids without an External Lewis Base as an Activator. Org. Lett. 23, 2477–2481 (2021).
Nambo, M. et al. Desulfonylative Coupling of Alkylsulfones with gem-Difluoroalkenes by Visible-Light Photoredox Catalysis. ACS Catal. 12, 9526–9532 (2022).
Docherty, J., Peng, J. Y., Dominey, A. & Thomas, S. Activation and discovery of earth-abundant metal catalysts using sodium tert-butoxide. Nat. Chem. 9, 595–600 (2017).
Challinor, A., Calin, M., Nichol, G., Carter, N. & Thomas, S. Amine Activated Iron Catalysis: Air- and Moisture Stable Alkene and Alkyne Hydrofunctionalization. Adv. Synth. Catal. 358, 2404–2409 (2016).
Sen, A. et al. Iron-catalyzed (E)-selective hydrosilylation of alkynes: scope and mechanistic insights. Catal. Sci. Technol. 14, 2752–2760 (2024).
Singh, K., Staig, S. J. & Weaver, J. D. Facile Synthesis of Z-Alkenes via Uphill Catalysis. J. Am. Chem. Soc. 136, 5275–5278 (2014).
Nevesely, T., Wienhold, M., Molloy, J. J. & Gilmour, R. Advances in the E → Z Isomerization of Alkenes Using Small Molecule Photocatalysts. Chem. Rev. 122, 2650–2694 (2022).
Zhu, C., Yue, H.-F., Maity, B., Atodiresei, I., Cavallo, L. & Rueping, M. A multicomponent synthesis of stereodefined olefins via nickel catalysis and single electron/triplet energy transfer. Nat. Catal. 2, 678–687 (2019).
Molloy, J. J., Schäfer, M., Wienhold, M., Morack, T., Daniliuc, C. G. & Gilmour, R. Boron-enabled geometric isomerization of alkenes via selective energy-transfer catalysis. Science 369, 302–306 (2020).
Xu, J.-T., Li, Z.-L., Xu, Y.-M., Shu, X.-M. & Huo, H.-H. Stereodivergent Synthesis of Both Z- and E-Alkenes by Photoinduced, Ni-Catalyzed Enantioselective C(sp3)–H Alkenylation. ACS Catal. 11, 13567–13574 (2021).
Zähringer, T. J. B., Wienhold, M., Gilmour, R. & Kerzig, C. Direct Observation of Triplet States in the Isomerization of Alkenylboronates by Energy Transfer Catalysis. J. Am. Chem. Soc. 145, 21576–21586 (2023).
Cheung, C. W., Zhurkin, F. E. & Hu, X. L. Z‑Selective Olefin Synthesis via Iron-Catalyzed Reductive Coupling of Alkyl Halides with Terminal Arylalkynes. J. Am. Chem. Soc. 137, 4932–4935 (2015).
Lee, M. T., Goodstein, M. B. & Lalic, G. Synthesis of Isomerically Pure (Z)‑Alkenes from Terminal Alkynes and Terminal Alkenes: Silver-Catalyzed Hydroalkylation of Alkynes. J. Am. Chem. Soc. 141, 17086–17091 (2019).
Filippini, D. & Silvi, M. Visible light-driven conjunctive olefination. Nat. Chem. 14, 66–70 (2022).
Arockiam, P. B., Bruneau, C. & Dixneuf, P. H. Ruthenium(II)-Catalyzed C−H Bond Activation and Functionalization. Chem. Rev. 112, 5879–5918 (2012).
Gensch, T., Hopkinson, M. N., Glorius, F. & Wencel-Delord, J. Mild metal-catalyzed C–H activation: examples and concepts. Chem. Soc. Rev. 45, 2900–2936 (2016).
Hu, A., Guo, J.-J., Pan, H. & Zuo, Z. W. Selective functionalization of methane, ethane, and higher alkanes by cerium photocatalysis. Science 361, 668–672 (2018).
Laudadio, G. et al. C(sp3)–H functionalizations of light hydrocarbons using decatungstate photocatalysis in flow. Science 369, 92–96 (2020).
Shields, B. J. & Doyle, A. G. Direct C(sp3)–H Cross Coupling Enabled by Catalytic Generation of Chlorine Radicals. J. Am. Chem. Soc. 138, 12719–12722 (2016).
Heitz, D. R., Tellis, J. C. & Molander, G. A. Photochemical Nickel-Catalyzed C–H Arylation: Synthetic Scope and Mechanistic Investigations. J. Am. Chem. Soc. 138, 12715–12718 (2016).
Treacy, S. M. & Rovis, T. Copper catalyzed C(sp3)–H bond alkylation via photoinduced ligand-to-metal charge transfer. J. Am. Chem. Soc. 143, 2729–2735 (2021).
Birnthaler, D., Narobe, R., Lopez-Berguno, E., Haag, C. & König, B. Synthetic Application of Bismuth LMCT Photocatalysis in Radical Coupling Reactions. ACS Catal. 13, 1125–1132 (2023).
Sang, R., Han, W., Zhang, H., Saunders, C. M., Noble, A. & Aggarwal, V. K. Copper-mediated dehydrogenative C(sp3)–H borylation of alkanes. J. Am. Chem. Soc. 145, 15207–15217 (2023).
Wang, M., Huang, Y. H. & Hu, P. Terminal C(sp3)–H borylation through intermolecular radical sampling. Science 383, 537–544 (2024).
Chinchole, A., Henriquez, M. A., Cortes-Arriagada, D., Cabrera, A. R. & Reiser, O. Iron (III)-light-induced homolysis: A dual photocatalytic approach for the hydroacylation of alkenes using acyl radicals via direct HAT from aldehydes. ACS Catal. 12, 13549–13554 (2022).
Dai, Z. Y., Zhang, S. Q., Hong, X., Wang, P. S. & Gong, L. A practical FeCl3/HCl photocatalyst for versatile aliphatic C–H functionalization. Chem. Catal. 2, 1211–1222 (2022).
Tu, J. L., Hu, A. M., Guo, L. & Xia, W. J. Iron-catalyzed C(sp3)–H borylation, thiolation, and sulfinylation enabled by photoinduced ligand-to-metal charge transfer. J. Am. Chem. Soc. 145, 7600–7611 (2023).
Kang, Y. C., Treacy, S. M. & Rovis, T. Iron-Catalyzed Photoinduced LMCT: A 1 °C-H abstraction enables skeletal rearrangements and C(sp3)-H alkylation. ACS Catal. 11, 7442–7449 (2021).
Zhang, Z. N., Li, X. Y., Zhou, D. Z., Ding, S. J., Wang, M. Y. & Zeng, R. Controllable C−H alkylation of polyethers via iron photocatalysis. J. Am. Chem. Soc. 145, 7612–7620 (2023).
Chang, L. et al. Resurgence and advancement of photochemical hydrogen atom transfer processes in selective alkane functionalizations. Chem. Sci. 14, 6841–6859 (2023).
Yuan, X. Y., Wang, C. C. & Yu, B. Recent advances in FeCl3-photocatalyzed organic reactions via hydrogen-atom transfer. Chin. Chem. Lett. 35, 109517–109525 (2024).
Zhong, P.-F. et al. Photoelectrochemical oxidative C(sp3)-H borylation of unactivated hydrocarbons. Nat. Comm. 14, 6530–6540 (2023).
Jin, Y. H., Zhang, Q. Q., Wang, L. F., Wang, X. Y., Meng, C. G. & Duan, C. Y. Convenient C(sp3)–H bond functionalisation of light alkanes and other compounds by iron photocatalysis. Green. Chem. 23, 6984–6989 (2021).
Jin, Y. H., Wang, L. F., Zhang, Q. Q., Zhang, Y. Q., Liao, Q. & Duan, C. Y. Photo-induced direct alkynylation of methane and other light alkanes by iron catalysis. Green. Chem. 23, 9406–9411 (2021).
Zhang, Q. Q. et al. Iron-catalyzed photoredox functionalization of methane and heavier gaseous alkanes: scope, kinetics, and computational studies. Org. Lett. 24, 1901–1906 (2022).
Zhang, Y. Q., Jin, Y. H., Wang, L. F., Zhang, Q. Q., Meng, C. G. & Duan, C. Y. Selective C(sp3)–H activation of simple alkanes: visible light-induced metal-free synthesis of phenanthridines with H2O2 as a sustainable oxidant. Green. Chem. 23, 6926–6930 (2021).
Shang, R., Ilies, L. & Nakamura, E. Iron-catalyzed C−H bond activation. Chem. Rev. 117, 9086–9139 (2017).
Greenhalgh, M., Jones, A. & Thomas, S. Iron-Catalysed Hydrofunctionalisation of Alkenes and Alkynes. ChemCatChem 7, 190–222 (2015).
Greenhalgh, M., Frank, D. & Thomas, S. Iron-Catalysed Chemo-, Regio-, and Stereoselective Hydrosilylation of Alkenes and Alkynes using a Bench-Stable Iron(II) Pre-Catalyst. Adv. Synth. Catal. 356, 584–590 (2014).
Greenhalgh, M. & Thomas, S. Chemo-, regio-, and stereoselective iron-catalysed hydroboration of alkenes and alkynes. Chem. Commun. 49, 11230–11232 (2013).
Greenhalgh, M. & Thomas, S. Iron-Catalyzed Hydromagnesiation of Olefins. Synlett 24, 531–534 (2013).
Le Bailly, B. & Thomas, S. Iron-catalysed reduction of carbonyls and olefins. RSC Adv. 1, 1435–1445 (2011).
Sen, A., Kuma, R., Tewari, T., Gonnade, R. & Chikkali, S. Iron-Catalyzed Alkoxylation, Dehydrogenative-Polymerization and Tandem Hydrosilylative-Alkoxylation. Chem. Eur. J. 29, e202301375 (2023).
Tewari, T., Kumarab, R. & Chikkali, S. Iron-catalysed highly selective hydroalkoxycarbonylation of alkynes using CO as C1 source. Catal. Sci. Technol. 13, 5549–5555 (2023).
Driller, K., Prateeptongkum, S., Jackstell, R. & Beller, M. A General and Selective Iron-Catalyzed Aminocarbonylation of Alkynes: Synthesis of Acryl- and Cinnamides. Angew. Chem. Int. Ed. 50, 537–541 (2011).
Wienhöfer, G., Westerhaus, F., Jagadeesh, R., Junge, K., Junge, H. & Beller, M. Selective iron-catalyzed transfer hydrogenation of terminal alkynes. Chem. Commun. 48, 4827–4829 (2012).
He, P., Hu, M.-Y., Li, J.-H., Qiao, T.-Z., Lu, Y.-L. & Zhu, S.-F. Spin effect on redox acceleration and regioselectivity in Fe-catalyzed alkyne hydrosilylation. Natl Sci. Rev. 11, nwad324 (2024).
Li, W.-T., Hu, M.-Y., Xiong, J.-W., Zhang, X.-Y. & Zhu, S.-F. Iron-catalysed hydroalumination of internal alkynes. Chem. Sci. 13, 7873–7879 (2022).
Gorgas, N., Alves, L., Stöger, B., Martins, A., Veiros, L. & Kirchner, K. Stable, Yet Highly Reactive Nonclassical Iron(II) Polyhydride Pincer Complexes: Z‑Selective Dimerization and Hydroboration of Terminal Alkynes. J. Am. Chem. Soc. 139, 8130–8133 (2017).
Gorgas, N., Stöger, B., Veiros, L. & Kirchner, K. Iron(II) Bis(acetylide) Complexes as Key Intermediates in the Catalytic Hydrofunctionalization of Terminal Alkynes. ACS Catal. 8, 7973–7982 (2018).
Rao, H., Schmidt, L. C., Bonin, J. & Robert, M. Visible-light-driven methane formation from CO2 with a molecular iron catalyst. Nature 548, 74–77 (2017).
Wen, H. N., Liu, G. X. & Huang, Z. Recent advances in tridentate iron and cobalt complexes for alkene and alkyne hydrofunctionalizations. Coord. Chem. Rev. 386, 138–153 (2019).
Wei, D. & Darcel, C. Organophosphorus and Iron Catalysis: Good Partners for Hydrometalation of Olefins and Alkynes. J. Org. Chem. 85, 14298–14306 (2020).
Liu, J. G. et al. Iron-Catalyzed Regiodivergent Hydrostannation of Alkynes: Intermediacy of Fe(IV)−H versus Fe(II)−Vinylidene. J. Am. Chem. Soc. 143, 409–419 (2021).
Huang, Z. J., Dong, Y. N., Li, Y. D., Makha, M. & Li, Y. H. Enhancing Ligand-free Fe-Catalyzed Aminocarbonylation of Alkynes by ZrF4. ChemCatChem 11, 5236–5240 (2019).
Nakajima, K., Kato, T. & Nishibayashi, Y. Hydroboration of Alkynes Catalyzed by Pyrrolide-Based PNP Pincer−Iron Complexes. Org. Lett. 19, 4323–4326 (2017).
Golub, M. A. The radiation induced cis-trans isomerization of polybutadiene. II. J. Am. Chem. Soc. 81, 54–58 (1959).
Moussebois, C. & Dale, J. A method of cis,trans-isomerisation of non-conjugated olefins without double-bond migration. J. Chem. Soc. C. 1966, 260–264 (1966).
Tron, G. C., Pirali, T., Sorba, G., Pagliai, F., Busacca, S. & Genazzani, A. A. Medicinal Chemistry of Combretastatin A4: Present and Future Directions. J. Med. Chem. 49, 3033–3044 (2006).
Buckle, D. R. (Beecham Group p.l.c., England) Novel Compounds. U.S. Patent US4713486 (1987).
For r.r. comparison with other HAT reagents, see, An, Q. et al. Cerium-catalyzed C–H functionalizations of alkanes utilizing alcohols as hydrogen atom transfer agents. J. Am. Chem. Soc. 142, 6216–6226 (2020).
Simmons, E. M. & Hartwig, J. F. On the interpretation of deuterium kinetic isotope effects in C−H bond functionalizations by transition-metal complexes. Angew. Chem. Int. Ed. 51, 3066–3072 (2012).
Zhao, Y. & Truhlar, D. G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 120, 215–241 (2007).
Rassolov, V. A., Ratner, M. A., Pople, J. A., Redfern, P. C. & Curtiss, L. A. 6-31G* Basis Set for Third-Row Atoms. J. Comput. Chem. 22, 976–984 (2001).
Acknowledgements
We acknowledge the assistance of Dr. Yuming Sun and Dr. Huihui Wan in the DUT Instrumental Analysis Center for their great help in HRMS analysis. We acknowledge the support of the National Natural Science Foundation of China (21901032 for Y.J., 21890381 for C.D., 21820102001 for C.D.) and the Fundamental Research Funds for the Central Universities (DUT21LK13 for Y.J.).
Author information
Authors and Affiliations
Contributions
Y.Z., Y.J., and C.D. conceived and designed the experiments. Y.Z., Z.C., R.C., W.H., H.W., J.C., and Y.C. performed and analyzed the experiments. D.F., S.L., and Y.L. designed and performed the theoretical calculations. All authors discussed the results and contributed to the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Y., Fu, D., Chen, Z. et al. Bifunctional iron-catalyzed alkyne Z-selective hydroalkylation and tandem Z-E inversion via radical molding and flipping. Nat Commun 15, 8619 (2024). https://doi.org/10.1038/s41467-024-53021-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-53021-x
This article is cited by
-
Photo-driven bifunctional iron-catalyzed one-pot assembling of indoles from arylamines and alkanes/carboxylic acids
Nature Communications (2026)
-
Photocatalytic anti-Markovnikov hydro- and haloazidation of alkenes
Nature Communications (2025)
