
Abstract
Obesity and related diseases pose a major health risk, yet current anti-obesity drugs inadequately addressing clinical needs. Here we show AA005, an annonaceous acetogenin mimic, resists obesity induced by high-fat diets and leptin mutations at non-toxic doses, with the alpha subunit of the mitochondrial trifunctional protein (HADHA) as a target identified through proteomics and in vitro validation. Pharmacokinetic analysis shows AA005 enriches in adipose tissue, prompting the creation of adipose-specific Hadha-deficient mice. These mice significantly mitigate diet-induced obesity, echoing AA005’s anti-obesity effects. AA005 treatment and Hadha deletion in adipose tissues increase body temperature and energy expenditure in high-fat diet-fed mice. The beneficial impact of AA005 on obesity mitigation is ineffective without uncoupling protein 1 (UCP1), essential for thermogenesis regulation. Our investigation shows the interaction between AA005 and HADHA in mitochondria, activating the UCP1-mediated thermogenic pathway. This substantiates AA005 as a promising compound for obesity treatment, targeting HADHA specifically.
Similar content being viewed by others


Introduction
The worldwide obesity epidemic represents a critical public health crisis1,2. Globally, more than 2.1 billion individuals are classified as overweight or individuals with obesity, and approximately 3.4 million deaths per year are attributed to obesity-related diseases3. For instance, obesity plays a significant role in the development of debilitating conditions such as type 2 diabetes, cardiovascular disease, hypertension, obstructive sleep apnea, and fatty liver disease. These conditions have a detrimental impact on both the quality and duration of life4,5,6,7. Consequently, the availability of drugs capable of safely reversing obesity would provide substantial benefits for a large population.
Currently, the Food and Drug Administration (FDA) has approved several classes of medications for long-term weight management. These classes primarily consist of single agents, including orlistat, liraglutide, lorcaserin, semaglutide, as well as two combination drugs: phentermine-topiramate and naltrexone-bupropion8,9,10,11,12. These drugs offer moderate weight loss results for patients even that long-term usage of them are associated with multiple side effects13,14,15,16,17. It is necessary to explore new compounds that are both safe and effective for the treatment of obesity, with a particular emphasis on those derived from or inspired by natural plants.
Annonaceae (custard-apple) family encompasses 130 genera and 2300 species18, and their fruits, roots, and leaves, have been widely employed in traditional medicine to enhance blood circulation, alleviate pain, repel insects, and combat malaria19. Products from the Annonaceae family, called annonaceous acetogenins (AAs), are very potent inhibitors of mammalian mitochondria NADH-ubiquinone reductase (Complex I)20. To date, more than 400 members of this compound family have been discovered, with most demonstrating significant cytotoxic and antitumor activities18. In recent years, significant progress has been made in the development of a range of AA mimetics. Among these mimics, AA005 stands out for its remarkable attributes. This particular compound incorporates a lengthy alkyl chain, a terminal γ-lactone ring, and a tetra-hydrogen ring as its principal structural components. Notably, AA005 has displayed exceptional efficacy in combating various forms of cancer, showcasing its potential as a potent antitumor agent21,22. Importantly, AA005 displayed selective antitumor effects with minimal toxicity to normal cells21,22,23,24,25,26. Despite the well-documented antitumor activities and traditional medicinal uses, there are limited studies exploring the other pharmacological effects and mechanisms of the Annonaceae family. Recent research has unveiled the potential anti-diabetic and antioxidant effects of crude leaf extracts from Annonaceae plants27, which motivates further exploration of additional pharmacological activities of the Annonaceae.
Fatty acid β-oxidation is a fundamental energy-yielding metabolic pathway present in organisms ranging from bacteria to humans. The β-oxidation pathway consists of four reactions: acyl-CoA dehydrogenase (ACAD), 2-enoyl-CoA hydratase (ECH), 3-hydroxyacyl-CoA dehydrogenase (HACD), and 3-ketothiolase (KT). The majority of fatty acid β-oxidation takes place within the mitochondria. The initial step is catalyzed by very long-chain acyl-CoA dehydrogenase (VLCAD). The final three steps are primarily facilitated by the mitochondrial trifunctional protein (TFP), a protein complex bound to the inner mitochondrial membrane28. TFP consists of two subunits. The alpha subunit (TFPα or HADHA), encoded by the Hadha gene, encompasses the ECH and HACD activities. The beta subunit (TFPβ or HADHB), encoded by the Hadhb gene, contains the KT activity29. Mutations in these two subunits that result in the loss of TFP function lead to mitochondrial trifunctional protein deficiency (MTPD), an autosomal recessive disorder characterized by a wide range of clinical manifestations, including liver dysfunction, retina abnormality, muscle atrophy, cardiomyopathy, and sudden death30,31. Additionally, deficiency of the long-chain HACD activity in HADHA has been observed in children of women who develop HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome and AFLP (acute fatty liver pregnancy) syndrome32, both of which are life-threatening obstetric conditions. The mice with homozygous deletion of Hadha and Hadhb exhibited characteristics such as low birth weight, neonatal hypoglycemia, and experienced sudden death within 6-36 h after birth33. However, the currently known functions of TFP cannot fully explain all of the aforementioned phenotypes, especially the low birth weight.
In this work, we find that annonaceous acetogenins mimic AA005 targets HADHA, which activates UCP1 as monolysocardiolipin acyltransferase-1 (MLCLAT-1), thereby mitigating obesity.
Results
AA005 resists obesity and its metabolic disorders in diet-induced obese mice
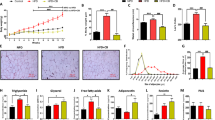
In order to investigate the impact of AA005 (Fig. 1a) on obesity, we conducted experiments involving mice fed a high-fat diet (HFD) for 22 weeks, with daily administration of AA005 or a vehicle as a negative control (Supplementary Fig. 1a). Remarkably, AA005 prevented weight gain induced by the HFD starting from week 3 onwards (Fig. 1b). Importantly, this effect was not accompanied by changes in food intake (Supplementary Fig. 1b), indicating that AA005 did not influence appetite. At the end of the 22nd week, mice receiving AA005 exhibited a reduction in body weight from 52.38 ± 1.90 g to 41.98 ± 1.61 g, corresponding to a 20% weight loss relative to the vehicle-treated mice (Fig. 1b, c). Additionally, the body fat content of AA005-treated mice was significantly decreased compared to vehicle-treated mice (Fig. 1d). Specifically, AA005 prevented fat accumulation in five fat depots, including two subcutaneous depots (inguinal white adipose tissue (iWAT) and anterior-subcutaneous white adipose tissue (asWAT)) and three visceral fat depots (epididymal white adipose tissue (eWAT), retroperitoneal white adipose tissue (rWAT), and mesenteric white adipose tissue (mWAT)) (Fig. 1e). Notably, AA005-treated mice exhibited significantly reduced the area of iWAT and eWAT adipocytes and reduced the size of lipid droplets (LDs) in BAT (Fig. 1f). Consistent with the anti-obesity phenotype, AA005 treatment significantly improved HFD-induced glucose tolerance (Fig. 1j, h) and insulin resistance (Fig. 1i, j) and decreased liver weight, lipid storage and decreased total cholesterol levels compared to vehicle controls after 22 weeks of HFD feeding (Supplementary Fig. 1d–h). Intriguingly, AA005 treatment also resulted in a notable increase in body temperature (Fig. 1k). To further evaluate physical activity and energy expenditure, we performed metabolic cage assays. As expected, energy expenditure (Fig. 1l, m), oxygen consumption (Fig. 1n, o), and carbon dioxide production (Fig. 1p, q), was significantly elevated in AA005-treated diet-induced obese mice. The respiratory quotient (RQ) values of mice treated with AA005 showed no significant difference during the daytime but were reduced at night (Supplementary Fig. 1i).

a Chemical structure of AA005. b Weight gain of male littermates fed HFD without or with AA005 (1.7 mg/kg) was recorded for 22 weeks (n = 9 for each group). c Representative photograph of vehicle and AA005-treated HFD-induced obese mice at experimental endpoint. Scale bar: 1 cm. d Fat weight was measured after 22 weeks of treatment (n = 9). e Fat weight of inguinal white adipose tissue (iWAT), abdominal subcutaneous white adipose tissue (asWAT), epididymal white adipose tissue (eWAT), retroperitoneal white adipose tissue (rWAT), mesenteric white adipose tissue (mWAT), and brown adipose tissue (BAT) in vehicle and AA005-treated obese mice (n = 9). f, Representative images of haematoxylin and eosin (H&E) stained of iWAT, eWAT, and BAT. iWAT, n = 79 (Vehicle) and 163 (AA005) adipocytes per group; eWAT, n = 90 (Vehicle) and 142 (AA005) adipocytes per group; BAT, n = 541 (Vehicle) and 392 (AA005) LDs per group. Scale bar, 50 μm. Glucose tolerance test (GTT) (g), area under the curve (AUC) of GTT (h), insulin tolerance test (ITT) (i), and AUC of ITT (j) in vehicle and AA005-treated obese mice (n = 9). k Body temperature was compared between the two groups (n = 5 per group). Three different biological replicates were tested. Mice at the end of week 21 were performed metabolic cage assays for energy expenditure (l, m), O2 consumption (n, o), and CO2 production (p, q) (n = 5). Data are presented as mean ± SEM. Unpaired, two-tailed Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001, NS indicates not significant. Source data are provided as a Source Data file.
Additionally, we administered vehicle or AA005 to mice maintained on a chow diet (CD) for 22 weeks and observed no significant changes in body weight, food intake, fecal energy excretion, blood glucose, or important physiological activities following long-term chronic AA005 administration, indicating the absence of toxic effects in mice (Supplementary Fig. 2). To assess whether AA005 exhibited a therapeutic effect on weight loss in established HFD-induced obese mice, we administered AA005 treatment to obese mice that had been previously subjected to 10 weeks of high-fat diet induction (Supplementary Fig. 3a). The results revealed that at the end of the 13th week, the weight and fat content of AA005-treated mice were significantly lower than those of the control obese mice (Supplementary Fig. 3b–e). Furthermore, mice treated with AA005 exhibited improved glucose tolerance and insulin resistance, accompanied by an increase in body temperature (Supplementary Fig. 3f–l). The results showed that AA005 at a dose of 8.5 mg/kg could significantly decrease the levels of serum ALT and AST, suggesting that AA005 had no obvious hepatotoxicity in mice (Supplementary Fig. 3m).
AA005 ameliorates obesity independent of leptin
Leptin, an adipocyte-derived hormone, serves as the primary messenger conveying information regarding peripheral energy stores to the central nervous system34,35. It has been proposed that obesity is characterized by leptin resistance or insensitivity36. To investigate whether AA005 acts as a leptin sensitizer, we initially assessed the circulating leptin levels in high fat diet-induced obese mice and leptin-deficient (ob/ob) mice following long-term AA005 treatment. Interestingly, we observed no significant alterations in plasma leptin levels (Fig. 2a, b). To further confirm these findings, we subsequently examined the weight loss effects of AA005 in ob/ob mice (Supplementary Fig. 4).

a Plasma leptin levels in vehicle and AA005-treated mice fed CD or HFD (n = 5). b Plasma leptin levels in wild-type (WT) and ob/ob mice treated with vehicle or AA005 (n = 5). c Body weight of WT and ob/ob male littermates treated with vehicle or AA005 (1.7 mg/kg) (n = 9 for WT mice, n = 8 for ob/ob mice for each group). d The average daily food intake of each mouse was recorded (n = 9 for WT mice, n = 8 for ob/ob mice for each group). e A representative photograph of WT and ob/ob mice treated with vehicle or AA005 at experimental endpoint. Scale bar, 1 cm. f Fat weight of iWAT, asWAT, eWAT, rWAT, mWAT, and BAT in WT and ob/ob mice treated with vehicle or AA005 (n = 9 for WT mice, n = 8 for ob/ob mice). g Fat and lean mass (n = 9 for WT mice, n = 8 for ob/ob mice for each group). h Representative images of H&E stained of iWAT, eWAT, and BAT. iWAT, n = 136 (ob/ob;vehicle) and 221 (ob/ob; AA005) adipocytes per group; eWAT, n = 93 (ob/ob;vehicle) and 165 (ob/ob; AA005) adipocytes per group; BAT, n = 546 (ob/ob;vehicle) and 384 (ob/ob; AA005) LDs per group;. Scale bar, 50 μm. Glucose tolerance test (GTT) (i), area under the curve (AUC) of GTT (j), insulin tolerance test (ITT) (k), and AUC of ITT (l) in vehicle and AA005-treated WT and ob/ob mice (n = 9 for WT mice, n = 8 for ob/ob mice for each group). Data are mean ± SEM. Unpaired, two-tailed Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001, NS indicates not significant. Source data are provided as a Source Data file.
Similar to the observations made in HFD-induced obese mice, the administration of AA005 resulted in a significant reduction in body weight gain in ob/ob mice, amounting to approximately 16% (Fig. 2c, e). Importantly, this effect was not accompanied by changes in food intake (Fig. 2d), ruling out an anorectic effect of AA005 on mice. Furthermore, AA005 treatment led to a decrease in fat mass in ob/ob mice (Fig. 2f). Further analysis revealed that AA005-treated ob/ob mice exhibited smaller cell volumes in inguinal white adipose tissue (iWAT) and epididymal white adipose tissue (eWAT), as well as an increased number of multilocular brown-like adipocytes in brown adipose tissue (BAT) (Fig. 2g). Notably, AA005-treated ob/ob mice exhibited significantly reduced the area of iWAT and eWAT adipocytes and reduced the size of LDs in BAT compared to vehicle-treated ob/ob mice (Fig. 1h). Additionally, AA005 improved glucose tolerance (Fig. 2i, j) and insulin resistance (Fig. 2k, l) in ob/ob mice compared to vehicle-treated ob/ob mice. Collectively, these findings demonstrate that the anti-obesity effect of AA005 is not dependent on the modulation of the leptin signaling pathway.
In our previous study, we demonstrated that high doses of AA005 exhibit anti-tumor effects in tumor cells by inducing the nuclear translocation of Apoptosis-inducing factor (AIF) and subsequent downregulation of mitochondrial complex I expression23,24. Interestingly, other studies have suggested that the specific ablation of AIF in muscle or liver could counteract the development of obesity, insulin resistance, and diabetes in mice37. To investigate whether the anti-obesity effects of low-dose AA005 are mediated through AIF, we examined the nuclear translocation of AIF and the expression of mitochondrial complex subunits in the liver, muscle, and iWAT of AA005-treated obese mice. We observed no significant alterations (Supplementary Fig. 5a–c). Furthermore, the protein levels of AIF and complex I subunit NDUFB6 were assessed in AA005-treated obese mice, but no significant changes were detected compared to the vehicle control mice (Supplementary Fig. 5d, e).
AA005 targets the mitochondrial trifunctional enzyme α subunit (HADHA)
To identify potential targets of AA005 for its anti-obesity effects, we investigated the impact of AA005 on mouse 3T3-L1 cells, a well-established cell model for studying lipid metabolism38. The 3T3-L1 cells were induced to undergo adipogenesis through exposure to a mixture of hormonal stimuli. Upon staining with Oil-Red-O, we observed a significant reduction in lipid droplets in the 300 nM AA005-treated group compared to the control group (Fig. 3a). Importantly, the viability of the cells remained unaffected following treatment with 300 nM AA005 (Supplementary Fig. 6a), thereby excluding any cytotoxic effects of this concentration on 3T3-L1 cells.

a Preadipocyte 3T3-L1 cells were induced to undergo adipogenesis and concomitantly treated with different concentrations of AA005 on day 8. The cells were stained with Oil-Red-O. Scale bar: 10 μm. Three different biological replicates were tested. b Workflow illustrating the use of biotin-tagged AA005. c On day 8 after induction of adipogenesis concomitant treatment with biotin or biotin-AA005, 3T3-L1 cells were stained with Oil-Red-O. Scale bar, 10 μm. Three different biological replicates were tested. d Immunofluorescence imaging showing the localization of biotin-AA005 and HADHA in cells. Mitotracker was used to label mitochondria, and DAPI was used to label nuclei. Three different biological replicates were tested. Scale bar, 10 μm. e Schematic representation of the discovery of interacting proteins of AA005 using chemical proteomics. f Mitochondrial fractions of 3T3-L1 cells were incubated with biotin or biotin-AA005 in the presence or absence of a 5- or 10-fold excess of unlabeled AA005. The proteins were then pulled down with streptavidin-agarose, and western blotting was performed to detect HADHA. Three different biological replicates were tested. g Structural overview of the predicted HADHA–AA005 complex model. The docking result was performed by Swiss-docking method. The protein is shown in cartoon, and AA005 is shown in a stick representation. Residues around the predicted binding pocket were highlighted with a red rectangle. On the right is the zoomed-in view of the predicted HADHA–AA005 interface. Potential key interface residues (V174, L175, L202, T203, R205, T294, Q293, E500, K505, M506, P581, V582, E590, G708, G709, and L708) in HADHA were colored in green and labeled. h Microscale thermophoresis (MST) binding assay determined the Kd value (WT, Kd = 1.1 μΜ; Mut-V174, Kd = 9.6 μΜ; L175, Kd = 20 μΜ; L202, Kd = 6.1 μΜ) for the binding of AA005 toward wild and mutant HADHA. Three different biological replicates were tested. i Immunoprecipitation analysis of the physical interaction between wild and mutant HADHA and biotin-AA005. Three different biological replicates were tested. j On day 8 after induction of adipogenesis, 3T3-L1 cells with normal HADHA expression (3T3-L1-NC) or cells with Hadha knockdown (3T3-L1-siHadha) were stained with Oil-Red-O. Scale bar: 10 μm. Three different biological replicates were tested. Data are mean ± SEM. Source data are provided as a Source Data file.
In the past decade, chemical proteomics has emerged as a powerful method for target identification by combining affinity purification using small molecules as bait and protein identification through mass spectrometry39,40. This approach offers great promise in discovering novel targets due to its unbiased nature41. With this in mind, we designed a chemical probe called biotin-AA005, based on structure-activity relationship information (Fig. 3b). Importantly, biotin-AA005 retained the activity of reducing lipid droplets in 3T3-L1 cells (Fig. 3c). To investigate its subcellular localization, we conducted immunofluorescence using streptavidin-fluorescein isothiocyanate (FITC) and a Mito-Tracker probe, which demonstrated that biotin-AA005 localized within the mitochondria (Fig. 3d). This finding aligns with previous reports indicating that AA005 exerts its effects within the mitochondria of cancer cells42. We isolated subcellular fractions, including mitochondria, nucleus, and cytosol, from 3T3-L1 cells, and employed western blotting to confirm the purity of the isolated fractions (Supplementary Fig. 6b). Subsequently, we incubated lysates of the subcellular fractions with an equivalent dose of either biotin or biotin-AA005, followed by affinity purification using streptavidin-coated beads. The proteins were then separated by SDS-PAGE (Fig. 3e). As demonstrated in Fig. 3e, the proteins enriched by biotin-AA005 were predominantly localized in the mitochondria. To identify the true interacting proteins of AA005 among all mitochondrial proteins in 3T3-L1 cells, we compared the relative abundances of tryptic peptides from each group using label-free spectral count-based quantitation. We identified 39 proteins that were significantly enriched by biotin-AA005 (Supplementary Table 1). To verify the affinity of these proteins to biotin-AA005, we performed western blotting on the six enriched proteins of interest to us. The α subunit of mitochondrial trifunctional protein (HADHA) exhibited the strongest affinity to AA005 (Supplementary Fig. 6c–e).
Notably, affinity purification using biotin-AA005, instead of biotin alone, resulted in the enrichment of HADHA (Fig. 3e, f, Supplementary Fig. 6f). Furthermore, the binding of HADHA by biotin-AA005 could be competitively blocked by a high concentration of AA005 (Fig. 3f), suggesting that both AA005 and biotin-AA005 bind to the same site on HADHA. Consistently, immunofluorescent staining using antibodies against HADHA and streptavidin-fluorescein isothiocyanate (FITC) revealed colocalization of biotin-AA005 with HADHA in mitochondria (Fig. 3d). To gain insights into the protein-small molecule interaction under conditions closer to the in vivo situation, we employed microscale thermophoresis (MST), which is a tool to characterize these interactions in complex biological liquids43.
To assess the binding pocket of AA005 in HADHA, we performed protein-ligand docking to predict the possible binding mode between AA005 and HADHA by Swiss-docking method (Supplementary Fig. 7a). The top five docking pockets were used for in-depth analysis. Amino acid residues that potentially interact with AA005 were identified by Chimera X. In these five poses, V174 and L175 were found to be the most likely amino acid sites to interact with AA005. The optimal docking result revealed a good binding mode and the possible binding sites between AA005 and the binding pocket of HADHA (Fig. 3g). Based on this, we used the Schrödinger-Glide procedure and found that biotin-AA005 could also dock to this pocket without steric hindrance. To confirm the interaction of AA005 with HADHA, 293 T cells overexpressing HADHA were constructed and incubated with AA005 under varying concentrations in the microscale thermophoresis (MST) binding assay (Supplementary Fig. 7a). Microscale thermophoresis (MST) binding assay determined the dissociation constant (Kd) value for the binding of AA005 toward wild and mutant HADHA (WT, Kd = 1.1 μΜ; Mut-V174, Kd = 9.6 μΜ; L175, Kd = 20 μΜ; L202, Kd = 6.1 μΜ). The results showed a notably high affinity of AA005 to wild HADHA (Kd = 1.1 μM) (Fig. 3h), while the Kd values were higher at Val174 and Leu175 (Fig. 3h), suggesting that mutations in these two sites may affect the affinity of HADHA to AA005. Then, we performed immunoprecipitation analysis by using anti-Flag antibody. As shown in Fig. 3i, HADHA-WT and AA005 physically interacted, while the mutation at Val174 and Leu175 blunted the physical interaction between HADHA and AA005 (Fig. 3i, Supplementary Fig. 7c). This result further strengthens our conclusion that HADHA exhibits strong AA005-binding properties at the Val174 and Leu175 sites.
To investigate the functional role of Hadha in lipid metabolism, we utilized two pairs of shRNAs specifically targeting Hadha (siHadha-1 and siHadha-2) to knockdown its expression, while a non-specific scramble shRNA (NC) was used as a negative control (Supplementary Fig. 7d, e). Subsequently, Oil-Red-O staining was performed to assess the impact of Hadha silencing on lipid droplet formation. The results revealed a significant reduction in lipid droplets compared to control cells, indicating that suppressing Hadha expression led to decreased lipid accumulation (Fig. 3j). This phenotype closely resembled the effect observed with AA005 treatment, suggesting that Hadha plays a crucial role in mediating the anti-obesity effect of AA005. Importantly, knockdown of the other top 9 candidate genes, as well as the Hadhb gene encoding the mitochondrial trifunctional protein β subunit, did not result in significant changes in cellular fat content (Supplementary Fig. 7f).
AA005 resists HFD-induced obesity in mice by targeting HADHA
To gain a deeper understanding of the mechanism underlying the anti-obesity effect of AA005, we conducted an analysis of its plasma concentration, half-life, and dynamic tissue distribution in mice. The findings revealed that AA005 attained its peak concentration in the bloodstream approximately 1 h after administration, with its basic metabolism being completed within 24 h (Supplementary Table 2 and Supplementary Fig. 8a). The half-life of AA005 was determined to be approximately 5 h, with a maximum plasma concentration of 10.47 ng/mL (Supplementary Table 3), and no accumulation of this compound was detected in mice. In order to further obtain safety data of AA005 in mice and to eliminate the interference of high-fat diet factors, we administered a single oral gavage of different concentrations of high-dose AA005 to mice on a chow diet for acute toxicity assessment. The mice were continuously observed for one week for phenotype, behavioral status, body weight, and food intake after administration. The results of this experiment indicate that compared with the control group, the mice in all dosed groups maintained a good survival status. Even the mice treated with the highest dose of AA005 (1000 mg/kg) did not exhibit significant adverse reactions (Supplementary Table 4). This suggests that the maximum tolerated dose of a single administration of AA005 is greater than 1000 mg/kg. Remarkably, pharmacokinetic analysis revealed a significant enrichment of AA005 in adipose tissue (Fig. 4a). Additionally, we examined the tissue distribution after 22 weeks of long-term AA005 treatment and obtained similar results (Supplementary Fig. 8b). To elucidate the primary role of Hadha in adipose tissues, we generated adipose-specific Hadha-deficient (HadhaΔAdipo) mice using the CRISPR-Cas9 system. This was achieved by crossing Hadha floxed mice (Hadhafl/fl) with Adiponectin-Cre mice (Supplementary Fig. 8c). The HadhaΔAdipo mice were born healthy, viable, and exhibited normal Mendelian ratios. The efficient deletion of the Hadha allele specifically in adipose tissue was confirmed through Western blotting (Supplementary Fig. 8d).

a Heatmap and histogram depicting the tissue distribution of AA005 in mice at various time points after AA005 administration (n = 3 per group). b Weight gain of male littermates fed a high-fat diet (HFD) in Hadhafl/fl and HadhaΔAdipo (adipose Hadha deficiency) mice was recorded for 18 weeks (n = 6). c Representative photograph of Hadhafl/fl and HadhaΔAdipo mice fed HFD at experimental endpoint. Scale bar, 1 cm. d Fat and lean mass were recorded after 18 weeks treatment (n = 5). e Fat weight of iWAT, asWAT, eWAT, rWAT, mWAT, and BAT in Hadhafl/fl and HadhaΔAdipo mice fed HFD (n = 6). Glucose tolerance test (GTT) (f), and insulin tolerance test (ITT) (g) performed on Hadhafl/fl and HadhaΔAdipo mice fed HFD (n = 6 for each group). h Comparison of body temperature between the two groups (n = 5 per group). Three different biological replicates were tested. Mice at the end of week 17 were performed metabolic cage assays for comparison of 24 h energy expenditure between Hadhafl/fl and HadhaΔAdipo mice fed HFD (i) (n = 5 per group); The adjusted means of energy expenditure in the two groups analysed by ANCOVA. (j) (n = 5); O2 consumption (k, l) and CO2 production (m, n) measured during a 24 h period (k, m) (n = 5); Average values for the light and dark periods, respectively (l, n) (n = 5). o, Weight gain recorded over 20 weeks (n = 6) in male littermates fed HFD with or without AA005 treatment in HadhaΔAdipo mice. Glucose tolerance test (GTT) (p), area under the curve (AUC) of GTT (q), insulin tolerance test (ITT) (r), and AUC of ITT (s) performed on vehicle and AA005-treated HadhaΔAdipo mice (n = 6). Data are presented as mean ± SEM. Unpaired, two-tailed Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001, NS indicates not significant. Source data are provided as a Source Data file.
We proceeded to investigate whether adipose-specific loss of Hadha would confer resistance to obesity in mice (Supplementary Fig. 8e). As anticipated, HadhaΔAdipo mice exhibited a notable resistance to HFD-induced body weight gain (Fig. 4b, c), providing further evidence for the substantial impact of Hadha ablation on weight loss. Analysis of adipose tissue content and morphology confirmed that control mice displayed significant fat deposition following high-fat feeding, whereas HadhaΔAdipo mice demonstrated significantly reduced fat mass, and area of iWAT and eWAT adipocytes and reduced the size of LDs in BAT (Fig. 4d, e and Supplementary Fig. 8f–i). Importantly, HadhaΔAdipo mice exhibited improved glucose tolerance (Fig. 4f and Supplementary Fig. 8j) and insulin resistance (Fig. 4g and Supplementary Fig. 8k). As expected, HadhaΔAdipo mice displayed a phenotype similar to that of AA005-treated mice, including elevated body temperature and substantially increased energy expenditure (Fig. 4h–j). This was further confirmed by the observed increase in O2 consumption (Fig. 4k, l) and CO2 production (Fig. 4m, n). Similar to the results after AA005 treatment, the RQ values of mice with hadha adipose tissue deficiency showed no significant difference during the daytime, but were reduced at night (Supplementary Fig. 8q). Importantly adipose-specific Hadha-deficiency did not affect serum insulin levels, fecal calories, activity levels, and dietary intake in mice (Supplementary Fig. 8l–p). We compared the body weight and fat content of control and HadhaΔAdipo mice fed a control diet (CD) and found no significant changes in HadhaΔAdipo mice compared to wild-type controls (Supplementary Fig. 9a-d). Morphological, glycemic, and energy metabolic analyses, assessing overall vitality of the mice, revealed no significant abnormalities in HadhaΔAdipo mice fed CD for 18 weeks (Supplementary Fig. 9e–q), thus excluding any toxicity associated with adipose-specific deletion of Hadha in mice. Furthermore, we administered AA005 to HadhaΔAdipo mice to evaluate its therapeutic effect on high-fat diet-induced obesity. The results demonstrated that the weight loss effect in HadhaΔAdipo mice remained largely unchanged after AA005 treatment compared to vehicle-treated HadhaΔAdipo mice (Fig. 4o–s and Supplementary Fig. 10). This suggests that adipose Hadha knockout mice no longer respond to AA005 treatment. In summary, these findings indicate that AA005 exerts its anti-obesity effects in mice by targeting HADHA.
AA005 treated mice and Hadha ΔAdipo mice exhibit similar alterations in cardiolipin profiling
Previous studies have highlighted the role of the hetero four polymer complex consisting of HADHA and HADHB in catalyzing mitochondrial long-chain fatty acid β-oxidation44. To investigate how AA005 affects these biological functions of HADHA, we examined whether AA005 influences the endogenous expression of HADHA. Our results demonstrated that AA005 did not impact the expression of HADHA either in vitro or in vivo (Fig. 5a, b). Furthermore, through detailed analysis, we observed that AA005 had no effect on the enzymatic activity of ECH and HACD (Fig. 5c–e), which are the two protein domains responsible for the execution of long-chain fatty acid β-oxidation functions in HADHA. Additionally, AA005 did not influence the enzymatic activity of HADHB (Fig. 5f). These findings suggest that the function and mechanism of AA005 differ from those of inhibitors targeting mitochondrial trifunctional protein complexes, such as trimetazidine45. This result aligns with previous reports indicating that trimetazidine does not induce weight loss in obese mice fed a high-fat diet45.

a The protein expression and quantification analysis of HADHA in 3T3L1 cells after treatment with AA005 for different days (n = 3). b The protein expression and quantification analysis of HADHA in iWAT from vehicle and AA005-treated mice fed CD or HFD (n = 3). c Schematic presentation of HADHA structure and fatty acid β-oxidation pathway. Left: HADHA structure (from PDB code: 5ZQZ) is colored according to domain architecture (N-terminal ECH domain, middle core linker, C-terminal HACD domain). ECH domain (residues 37-333) is colored hot pink; middle core linker (residues 334-349) is colored coral; HACD domain (residues 350-763) is colored cornflower blue. Right: Schematic diagram of fatty acid β-oxidation process. ECH domain catalyzes the second step hydration reaction of FAO, while HACD domain catalyzes the third step dehydrogenation reaction. In 3T3L1 cells, the activities of ECH (d), HACD (e), and KACT (f) enzymes were detected after treatment with AA005 and inducer for 8 days (n = 6 per group). Three different biological replicates were tested. g Structure of HADHA was obtained from human mitochondrial trifunctional protein (TFP) (PDB code: 5ZQZ). MLCLAT-1 structure model was predicted by I-TASSER server and colored pink. The region corresponding to MLCLAT-1 in HADHA was colored light blue, and the extra N-terminal 190 residues were colored green-yellow respectively. h Schematic diagram of how HADHA works to remodel CL. i Relative mRNA levels of Mlclat-1 and Tafazzin genes in BAT of vehicle or AA005-treated HFD-fed mice (n = 5). j The protein expression and quantification analysis of HADHA and MLCLAT-1 in BAT of vehicle or AA005-treated HFD-fed mice (n = 3). k Relative mRNA levels of Mlclat-1 and Tafazzin genes in BAT of Hadhafl/fl and HadhaΔAdipo mice fed HFD. (n = 5). l The protein expression and quantification analysis of HADHA and MLCLAT-1 in BAT of Hadhafl/fl and HadhaΔAdipo mice fed HFD. (n = 3). m Heatmaps of saturation immature cardiolipin (CLSat) (upper) and unsaturation mature cardiolipin (CLUnsat) (lower) species level in BAT. (n = 5 per group, Data are presented as mean ± SEM. Unpaired, two-tailed Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001). Source data are provided as a Source Data file.
Recent studies have highlighted the similarity between some amino acid sequences of HADHA and MLCLAT-1, a protein involved in cardiolipin maturation46. To investigate this further, we conducted a comparison of the amino acid sequences and structures of these two proteins. We found that the C-terminal residues of HADHA were identical to those of MLCLAT-1, spanning 573 residues (Supplementary Fig. 11). Additionally, the three-dimensional structures of the two proteins exhibited remarkable similarity (Fig. 5g). These findings suggest that HADHA likely performs MLCLAT-1-like functions (Fig. 5h). According to previous study47, we tested the effect of AA005 on the expression of Mlclat-1 and tafazzin genes, and the results showed that AA005 had no effect on the expression of Mlclat-1 and tafazzin genes (Fig. 5i). However, compared with the wild type, the Mlclat-1 gene in the BAT tissue of adipose tissue-specific HAHDA knockout mice was significantly reduced (Fig. 5k). More importantly, compared with the control group, AA005 treatment or fat-specific knockout of hadha gene can significantly reduce the expression of MLCLAT-1 protein (Fig. 5j, l). Under normal physiological conditions, MLCLAT-1 is involved in the maturation of cardiolipin48. To evaluate the effects of Hadha deficiency on cardiolipin expression, we extracted brown adipose tissue from both HadhaΔAdipo mice and control mice treated with a high-fat diet (using epididymal white adipose tissue (eWAT), representative of white fat, and inguinal white adipose tissue (iWAT), representative of beige fat, as controls)49. Utilizing real-time fluorescence quantitative PCR, we detected the mRNA levels of BAT-specific marker genes Ucp1 and Cidea, with the results indicating good purity of the brown adipose tissue (Supplementary Fig. 12a). As monolysocardiolipin (MLCL) can serve as a substrate for various enzymes, including MLCLAT-1, to participate in the maturation of cardiolipin50, we also analyzed the content of MLCL. The results showed that, compared to the control group, there was a slight increase in the levels of certain species of MLCL in mouse brown adipose tissue following treatment with AA005 or in the absence of hadha function (Supplementary Fig. 12b). Subsequently, we analyzed the changes in the cardiolipin profile of the mice from different groups using LC-MS/MS methods. The findings revealed that, in comparison with the control mice, the deficiency of Hadha led to an increased expression of immature cardiolipins and a decreased expression of mature cardiolipins among the top 50 most abundant cardiolipins. Interestingly, after treatment with AA005, a similar alteration in cardiolipin expression was observed, consistent with the findings in HadhaΔAdipo mice (Fig. 5m). Cardiolipin is primarily located in the inner mitochondrial membrane and plays a crucial role in maintaining the normal structure and function of mitochondria51. Considering the potential differences in mitochondrial load in the brown adipose tissue of AA005-treated and untreated, as well as wild-type and Hadha-deficient mice, which could affect the changes in cardiolipin profiles among the groups. To assess the differences in mitochondrial content in the brown adipose tissue of mice from each group, the mitochondrial DNA (mtDNA) copies was measured using real-time quantitative PCR. mtDNA, which is usually located at the mitochondrial matrix, encodes several key components of respiratory chain complexes that produce cellular energy through oxidative phosphorylation52,53,54. The results showed that there was no significant difference in the levels of mtDNA in the brown adipose tissue cells among all mouse groups (Supplementary Fig. 12c), suggesting that there was no obvious change in the mitochondrial levels in the brown adipose tissue cells of the mice in each group. To further rule out the impact of mitochondrial levels on the cardiolipin profile, we recalibrated and analyzed the cardiolipin data according to the mtDNA content of each group (Supplementary Fig. 12d). The results obtained are in agreement with the previous findings (Fig. 5m), indicating that the changes in the cardiolipin profile detected in each group of mice are mainly due to AA005 treatment or Hadha deficiency, rather than mitochondrial levels. Considering the key roles of ubiquinol 9 (CoQ9H2) and ubiquinol 10 (CoQ10H2) in the mitochondrial respiratory chain and cellular homeostasis55,56, we measured the levels of CoQ9H2 and CoQ10H2 in equal amounts of mouse brown adipose tissue. The results indicate that, compared to the control group, there was no significant difference in the levels of CoQ9H2 and CoQ10H2 after treatment with AA005 or in the absence of Hadha function (Supplementary Fig. 12e, f). This finding is consistent with our observation of essentially unchanged mtDNA content across the mouse groups, suggesting that treatment with AA005 or the lack of Hadha function in adipose tissue does not affect the normal function of mitochondria. The combined results suggest that AA005 may affect the maturation of cardiolipin by binding to HADHA and inhibiting its MLCLAT-1 enzyme activity.
The Ucp1-mediated thermogenic pathway is activated in AA005-treated mice and Hadha ΔAdipo mice
Cardiolipin alterations have been known to impact adipocyte metabolism57,58. To gain a comprehensive and unbiased understanding of the molecular mechanisms underlying the anti-obesity effects of AA005 and Hadha deletions, we conducted RNA-seq analysis using fresh residual inguinal white adipose tissue (iWAT) from mice treated with AA005, high-fat diet-fed HadhaΔAdipo mice, and untreated control mice. Strikingly, our analysis revealed a high degree of similarity in the gene expression profiles between AA005-treated mice and high-fat diet-fed HadhaΔAdipo mice (Fig. 6a). We then identified differentially expressed genes and transcripts for further analysis. Biological function analysis of these genes revealed the notable activation of Kyoto Encyclopedia of Genes and Genome (KEGG) terms associated with “metabolic process” (Fig. 6b). The heatmap visualization demonstrated that AA005 treatment and Hadha deficiency predominantly upregulated genes involved in the thermogenesis signaling pathway, while downregulating genes related to other pathways such as PPAR gamma targets, fatty acid metabolism, and cholesterol metabolism (Fig. 6c). Interestingly, consistent with the strong upregulation of thermogenesis-associated genes in AA005-receiving mice and high-fat diet-fed HadhaΔAdipo mice, Hadha was identified as a central node in the network interacting with Ucp1, Ucp2, and Ucp3 (Fig. 6d).

a Heatmap generated by RNA-seq analysis displaying differentially regulated genes between AA005-treated and HadhaΔAdipo mice fed HFD in iWAT (n = 5). Red and blue colors indicate upregulated or downregulated genes. b Significant enrichment in KEGG terms. Bars indicate the number or proportions of genes within each functional class, ranked based on protein coverage. c Heatmap showing the mean fold-change of the indicated metabolic processes in vehicle control, AA005-treated and HadhaΔAdipo mice fed HFD. Red and blue colors indicated upregulated or downregulated genes. d Hadha as the central node in the network of gene interactions with thermogenesis pathways. The protein expression and quantification analysis of UCP1, p-PKA, and p-HSL in BAT tissue derived from vehicle- or AA005-treated mice fed HFD for 22 weeks (e), as well as Hadhafl/fl or HadhaΔAdipo mice fed HFD for 18 weeks (f) (n = 3). Actin was used as the loading control. g–l Vehicle- or AA005-treated mice fed CD subjected to cold room stress before analysis. Rectal temperature of vehicle- or AA005-treated mice (4 °C acute cold in (g) programmed cooling to 6 °C chronic cold in (j), n = 5 per group). Representative western blot and quantification of UCP1 and PKA activation in the BAT (acute cold in (i), chronic cold in (l)) (n = 3). Representative images of haematoxylin and eosin (H&E) stained of iWAT and BAT (acute cold in (h), iWAT, n = 248 (vehicle) and 301 (AA005) adipocytes per group; BAT, n = 287 (vehicle) and 193 (AA005) LDs per group, chronic cold in (k), iWAT, n = 100 (vehicle) and 309 (AA005) adipocytes per group; BAT, n = 600 (vehicle) and 478 (AA005) LDs per group; Scale bar, 50 μm. Data are presented as mean ± SEM. Unpaired, two-tailed Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001). Source data are provided as a Source Data file.
To validate the results obtained from RNA-seq analysis, we conducted real-time RT-PCR and Western blot analyses to assess the mRNA and protein levels of specific marker genes associated with the Ucp1-mediated thermogenic pathway. Consistently, we observed a significant increase in the expression of BAT-specific marker Ucp1 in adipose tissue of high-fat diet-fed AA005-receiving mice and HadhaΔAdipo mice. Furthermore, the mRNA levels of other thermogenic genes, Pgc-1 α, Dio2, Cidea, Tfam, Pgc-1 β, and Prdm16, and the protein levels of p-PKA substrate, p-HSL were significantly elevated in both AA005-treated and Hadha-deleted mice (Fig. 6e, f, and Supplementary Fig. 13a–h)
To further investigate the role of AA005 in regulating body temperature defense, we examined the response to acute hypothermia exposure. We found that a substantial enhancement of acute cold tolerance and reduced area of iWAT adipocytes and reduced the size of LDs of BAT in AA005-treated mice (Fig. 6g, h). In accordance, an elevated expression of UCP1 in BAT (Fig. 6i) were observed in AA005-treated mice. AA005-treated mice showed similar effects to chronic hypothermia exposure (Fig. 6j-l). Consistently, in vitro experiments demonstrated that AA005 was capable of inducing increased UCP1 expression and modestly elevated expression of mitochondrial genes, as well as an increased oxygen consumption rate (OCR) in brown adipocytes (Supplementary Fig. 13i-l).
The Ucp1-mediated thermogenic pathway contributes to the anti-obesity effect of AA005
Ucp1−/− mice were subjected to a high-fat diet to induce obesity, with or without AA005 treatment (Fig. 7a, Supplementary Fig. 14). At the end of week 20, it was observed that AA005 did not induce weight loss in Ucp1−/− mice (Fig. 7b, c). Consequently, the effects of AA005 on adipose tissue content, morphology, and blood glucose levels in obese Ucp1−/− mice were no longer observed (Fig. 7d–k). These findings suggest that the Ucp1-mediated thermogenesis pathway is essential for AA005 to exert its anti-obesity pharmacological effects.

a PCR confirmation of Ucp1 allele deletion in Ucp1−/− mice. Three different biological replicates were tested. b Weight gain recorded over 20 weeks in male littermates fed HFD with or without AA005 treatment in Ucp1−/− mice (n = 6 for vehicle-treated Ucp1−/− mice, n = 7 for AA005-treated Ucp1−/− mice for each group). c Representative photograph of vehicle- or AA005-treated Ucp1−/− mice fed HFD at experimental endpoint. Scale bar, 1 cm. d The average food intake of each mouse for 1 week was recorded (n = 6 for vehicle-treated mice, n = 7 for AA005-treated mice). e Fat and lean mass were recorded after 20 weeks treatment (n = 6 for vehicle-treated mice, n = 7 for AA005-treated mice). f Fat weight of iWAT, asWAT, eWAT, rWAT, mWAT, and BAT in Ucp1−/− mice treated with vehicle or AA005 (n = 6 for vehicle-treated mice, n = 7 for AA005-treated mice). Glucose tolerance test (GTT) (g), area under the curve (AUC) of GTT (h), insulin tolerance test (ITT) (i), and AUC of ITT (j) in vehicle and AA005-treated Ucp1−/− mice fed HFD (n = 6 for vehicle-treated mice, n = 7 for AA005-treated mice). k Representative images of haematoxylin and eosin (H&E) stained of iWAT, eWAT, and BAT. iWAT, n = 107 (Vehicle) and 114 (AA005) adipocytes per; eWAT, n = 115 (Vehicle) and 108 (AA005) adipocytes per group; BAT, n = 473 (Vehicle) and 474 (AA005) LDs per group. Scale bar, 50 μm. l AA005 targets HADHA to counteract obesity by inhibiting cardiolipin maturation and enhancing Ucp1-mediated thermogenesis pathway. AA005 demonstrates the ability to induce a brown-like transformation in adipose tissue and enhance energy expenditure in obese mice, thereby potentially exerting a pharmacological effect in combating obesity and improving glucose metabolism. Mechanistically, AA005 selectively enters adipocyte mitochondria and targets the binding site on HADHA, leading to inhibition of cardiolipin maturation in mitochondria. Consequently, AA005 alters the expression profile of cardiolipin. Notably, the presence of immature cardiolipin activates the Ucp1-mediated thermogenic pathway, consequently promoting mitochondrial thermogenesis. Data are presented as mean ± SEM. Unpaired, two-tailed Student’s t test. NS indicates not significant. Source data are provided as a Source Data file.
Discussion
Although annonaceous acetogenins, natural products derived from the Annonaceae family, have demonstrated significant antitumor activity22,23, many other pharmacodynamics and mechanisms remain unclear. In our study, we introduce AA005, a mimic of annonaceous acetogenins, as an anti-obesity compound that selectively targets HADHA, thereby enhancing energy expenditure without affecting food intake or lean mass. Mechanistically, AA005 enters adipocyte mitochondria and binds to HADHA, inhibiting cardiolipin maturation and causing alterations in cardiolipin expression profiles. Consequently, this process activates the Ucp1-mediated thermogenic pathway, leading to heightened mitochondrial thermogenesis. These mechanisms elucidate the observed pharmacological effects of AA005, which include brown-like changes in adipose tissue, increased energy expenditure, weight loss, and improved glucose metabolism in obese mice (Fig. 7l). This study unveils a previously unrecognized pharmacological activity of annonaceous acetogenins. Based on the data presented herein, further exploration and development of AA005 as a potential therapeutic agent for obesity and metabolic diseases are promising.
Obesity is a chronic disease influenced by various factors, including genetics, lifestyle, and imbalanced energy metabolism. Current anti-obesity drugs target specific mechanisms, such as lipase inhibition (e.g., orlistat), incretin modulation (e.g., liraglutide), central nervous system modulation (e.g., lorcaserin), and mitochondrial uncoupling (e.g., 2,4-dinitrophenol). However, long-term use of these drugs often leads to limited weight loss, and their associated side effects cannot be overlooked. For instance, orlistat can cause significant gastrointestinal discomfort and even lead to liver damage and acute pancreatitis59. Liraglutide commonly elicits adverse reactions like nausea, vomiting, and hypoglycemia60. Weight-loss drugs that target the central nervous system are associated with headaches, fatigue, and even potential risks of cancer, mental illness, and cardiovascular disease61. Recently, there has been growing interest in the development of drugs that increase energy expenditure as a promising and appealing strategy against obesity. In our research, multiple lines of evidence support the idea that AA005 exerts its anti-obesity effects primarily by enhancing Ucp1-mediated thermogenesis in adipocytes, which suggests AA005 may serve as an anti-obesity agent via a non-classical target.
Notably, the mitochondrial uncoupling agent DNP has demonstrated favorable effects on obesity in preclinical models. However, its clinical application is hindered by its excessive toxicity. DNP functions as a proton carrier within the human body, facilitating the entry of protons into the inner mitochondrial membrane while bypassing ATP synthase. Consequently, this process reduces cellular efficiency in ATP synthesis. As the concentration of DNP increases, the metabolic rate escalates, leading to uncoupling and the generation of substantial heat, resulting in increased fat consumption. It is important to note that this phenomenon is considered “non-genomic”. DNP induces an elevation in cyclic adenosine monophosphate (cAMP), a second messenger, most likely through the activation of adenylate cyclase. The production of cAMP transitions the non-genomic event into a genomic event, thereby triggering the expression of numerous genes. Consequently, excessive and prolonged usage of DNP may result in uncontrolled fever and even death62,63. In the present study, AA005 emerges as a class of anti-obesity compounds exhibiting a distinctive mechanism of action and several desirable features. Unlike the mitochondrial uncoupling agent DNP, the target of AA005 is the protein HADHA, and protein signaling pathways that play a role in metabolic processes in vivo are relatively mild and controllable. AA005 binds to HADHA, inhibiting the enzymatic activity of mitochondrial cardiolipin maturation. Consequently, this inhibition promotes the activation of UCP1-mediated thermogenic pathways, contributing to resistance against obesity without inducing toxic side effects. In contrast to DNP, although AA005 also increases energy expenditure, long-term administration of AA005 in mice resulted in only a modest increase in body temperature, ~1 °C, as opposed to a sustained and substantial elevation. This outcome can be attributed to the mild nature of mitochondrial uncoupling achieved by AA005 through oral administration, as well as the robust regulatory mechanisms governing body temperature homeostasis in mice. Furthermore, pharmacokinetic studies have revealed that AA005 is predominantly distributed in adipose tissue, limiting its absorption in tissues other than fat. Moreover, AA005 is rapidly metabolized with a half-life of approximately 5 h and does not exhibit significant accumulation in mice. This rapid metabolism serves as an additional safety mechanism, preventing the potential buildup of excessive AA005.
Previous studies have emphasized the involvement of the heterotetrameric complex composed of HADHA and HADHB in facilitating the oxidation of long-chain fatty acids within the mitochondria. Intriguingly, mice with homozygous deletion of Hadha and Hadhb exhibited characteristics such as low birth weight, neonatal hypoglycemia, and experienced sudden death within 6–36 h after birth33. This unforeseen phenomenon cannot be solely accounted for by the established functions of HADHA and HADHB, implying the potential existence of novel biological functions associated with lipid metabolism for these proteins.
In this study, we made an intriguing discovery regarding the amino acid sequences and protein structures of HADHA and MLCLAT-1, the enzyme responsible for cardiolipin maturation. Through lipidomic analysis of brown adipose tissue from different mouse groups, we observed striking similarities in the changes of cardiolipin expression profiles between AA005-treated mice and mice with adipose-specific deletion of Hadha, as depicted in Fig. 5m. These findings suggest that HADHA may possess a function akin to that of MLCLAT-1, playing a crucial role in the anti-obesity effect of AA005 by remodeling cardiolipin. This aligns with the recently reported function of HADHA in cardiomyocytes64.
Furthermore, our investigations demonstrated that AA005 did not affect the enzymatic activity of ECH and HACD, which are protein domains responsible for the functions of long-chain fatty acid oxidation in HADHA. Additionally, AA005 did not impact the enzymatic activity of HADHB. These findings suggest that the function and mechanism of AA005 differ from those of inhibitors targeting the mitochondrial trifunctional protein complex, such as trimetazidine. This result is consistent with a previous report and provides a plausible explanation for why trimetazidine failed to induce weight loss in obese mice fed a high-fat diet45. Recent studies have shed light on the critical role of cardiolipin in promoting and sustaining fat thermogenesis and glucose uptake65,66. The precise mechanisms underlying the remodeling of cardiolipin by AA005 and Hadha warrant further investigation. Notably, while Hadha homozygous deletion in mice was lethal33, adipose-specific deletion of Hadha improved obesity and glucose homeostasis. This suggests that HADHA may serve as a promising target for guiding the development of more specific weight-loss drugs that target adipose tissue HADHA.
Pharmacokinetic studies have shown that AA005 is primarily distributed in adipose tissue, followed by distribution to liver tissue. Treatment with AA005 can ameliorate the fatty liver disease induced by a high-fat diet in obese mice, including reducing the liver weight, hepatic pathology, and serum TC (Supplementary Fig. 1), suggesting that AA005 can promote hepatic fatty acid metabolism. Recently, it has been reported that overexpression of liver HADHA in mice can reverse gluconeogenesis in the liver, protect the body against disruptions in glucagon response, and perform its functions independently of HADHB67. Perhaps AA005 can exert beneficial effects on glucose and lipid metabolism by binding to liver HADHA. In this study, we found that the main mechanism by which AA005 resists obesity is that AA005 enters the mitochondria of adipocytes and binds to HADHA, inhibiting the maturation of cardiolipin, and further activating the Ucp1-mediated thermogenic pathway, leading to heightened mitochondrial thermogenesis. Considering that Ucp1 primarily functions in adipose tissue, if liver-specific Hadha knockout mice can also resist obesity, then the mechanism of resistance to obesity may be more complex and deserves further investigation in the future.
Methods
Animal experiments and ethics statement
This animal study was conducted in accordance with ethical standards and was approved by the Animal Care and Use Committee of the Experimental Animal Research Center at Minhang Hospital affiliated with Fudan University. The animals received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health. This study utilized Six-week-old male C57BL/6 mice, four-week-old male ob/ob mice, and Ucp1−/− mice purchased from Charles River Laboratories. All animals used in this research were housed in a specific pathogen-free (SPF) environment with a 12 h dark/light cycle (lighting from 7:00 to 19:00), constant and appropriate room temperature (22–25 °C), and had free access to food and water. The C57BL/6J mice were fed either a normal diet (D12450B, 10% energy from fat, Research Diets, Brunswick, NJ, USA) or a high fat diet (D12492, 60% energy from fat, Research Diets, Brunswick, NJ, USA) for at least 12 weeks to establish an obesity model. The Hadhafl/fl mouse line was generated by genome editing using CRISPR/Cas9. There were 20 exons in Hadha gene, and exon 3 (71 bp) was selected as flox region. LoxP sites were localized to ~100–200 bp introns upstream and downstream of exon 3 by homologous recombination. The Hadhafl/fl mouse line was generated and had been backcrossed into the genetic background of C57BL/6. Hadhafl/fl mice were intercrossed with Adipoq-Cre (JAX) transgenic mice expressing the Cre recombinase under the control of Adipoq promoter to generate adipocyte-specifc Hadha-knockout mice, and their wild-type littermates were used as a control group throughout the experiments. In metabolic balance studies, mice were placed in metabolic cages (Columbus Instruments) for 48 h to assess their food intake, energy expenditure, O2 consumption, CO2 production, and physical activity. Rectal temperature was measured with a model BAT-12 thermometer (Physitemp Instruments). For adaptive thermogenic analysis, mice were maintained at thermal neutrality, followed by short-term acute cold exposure in a 4 °C cold chamber and observed for 1 day, during which mice had free access to water and food. For metabolic analysis after chronic cold stimulation, mice were placed in a cold chamber and subjected to programmed cooling to 6 °C for up to 8 days with free access to water and food65. Only male mice were used in the experiments. At the end of the experiments, mice were euthanized by inhalation of carbon dioxide (CO2) followed by cervical dislocation as a secondary method of euthanasia, after which the tissues were harvested and collected for biochemical studies.
Chemicals and reagents
Annonaceous acetogenin mimic AA005 was dissolved in dimethylsulfoxide. Antibodies against AIF (#4642), PPARγ (#2443), LaminB1 (#12586), Cox IV (#4844), UCP1 (#72298), Phospho-PKA Substrate (#9624), HSL (#4107), Phospho-HSL (Ser563) (#4139), and β-actin (#3700) were purchased from Cell Signaling Technology (Beverly, MA, USA), NDUFB6 (#16037-1-AP) and HADHA (#ab203114) were purchased from Proteintech Group.
Glucose and insulin tolerance tests
For glucose tolerance testing, mice were fasted for 14 h and injected with glucose (2 g/kg) intraperitoneally. For insulin tolerance testing, mice were fasted for 6 h and injected with recombinant human insulin (0.75 U/kg) (Actrapid, Novo Nordisk, Bagsværd, Denmark) intraperitoneally. We measured mouse blood glucose levels with whole blood from the tail vein using a glucose meter (Ascensia, Contour, Bayer Healthcare, Oslo, Norway).
Immunohistochemical staining
Immunohistochemical (IHC) staining was applied to detect the protein levels of AIF and NDUFB6 in vehicle or AA005-treated mice fed CD or HFD with anti-AIF (#4642, Cell Signaling Technology, Danvers, MA, USA) or anti-NDUFB6 (16037-1-AP, Proteintech Group, Chicago, IL) antibodies. IHC staining was performed according to the manufacturer’s protocol. For immunostaining quantification, the H-score system was obtained by multiplying staining intensity (0 for no staining, 1 for weak staining, 2 for moderate staining, and 3 for intense staining) by the percentage (0–100) of cells showing that intensity.
Morphological analysis
For hematoxylin and eosin (H&E) staining, adipose and liver tissues were isolated, fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned at 5 μm for WAT and liver and 3 μm for BAT. The size of lipid droplets in the representative area was counted by AdipoCount68.
Adipocyte differentiation
Brown preadipocytes were isolated and immortalized as previously described69. Brown preadipocytes were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). The 3T3-L1 cell line (Cat: #ATCC, CL-173TM) is a fibroblast cell line derived from mouse embryos, purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). 3T3-L1 cells were cultured in DMEM (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% bovine growth serum and 1% penicillin-streptomycin, and maintained at 37 °C in a 5% CO2 incubator. To initiate differentiation, a cocktail containing 0.5 mM IBMX, 125 μM indomethacin, and 1 μM dexamethasone was added to confluent preadipocytes maintained in DMEM supplemented with 10% FBS, 20 nM insulin, and 1 nM T3. Cells were switched to differentiation medium (DMEM, 10% FBS, 20 nM insulin, and 1 nM T3) after 2 days. Mitochondrial abundance was observed following MitoTracker staining under a Leica DM IRB microscope.
Oil Red O staining
Differentiated adipocytes were fixed with 4% paraformaldehyde (Sigma-Aldrich, P6148) in PBS and were washed twice with PBS. Completely air-dried at room temperature, cells were then incubated with Oil Red O working solution (Servicebio, G1016) at room temperature for 1 h. Cells were washed three times with PBS before images were acquired for analysis.
Immunofluorescence
3T3-L1 cells were crawled onto cover slides. After treated with 10 μM biotin-AA005 for 4 h, 3T3-L1 cells were incubated in the mitochondrial probe mito-tracker (1 μM)-containing medium for 30 min. Then, cells were fixed with 4% paraformaldehyde and permeabilized with 0.3% Triton X-100 for 10 min. Slides were blocked with 1% bovine serum albumin and incubated with streptavidin-FITC (1:100) overnight at 4 °C. After washing in PBS, the cells were stained with nuclear counterstaining DAPI. Fluorescence signals were detected on a Olympus BX-51 fluorescence microscope (Tokyo, Japan).
Subcellular cell fractionation and mitochondria purification
Cells were separated into different fractions according to previously described methods70,71. In brief, cells were harvested and rinsed with mitochondria isolation buffer (MIB, 0.25 M sucrose and 10 mM Tris-HCl, pH 7.4), resuspended in MIB supplemented with 1% protease inhibitor cocktail, and homogenized using a glass Dounce homogenizer (Kontes, Sigma-Aldrich) with 20 strokes at 4 °C. The homogenate was centrifuged at 1000 × g for 10 min to pellet the nucleus. The supernatant was centrifuged at 15,000 × g for 20 min at 4 °C to pellet the raw mitochondria. Cytoplasmic proteins in the post-mitochondria supernatant were precipitated with chloroform and methanol according to Klotz’s methods72. To further enrich the mitochondria, the pellet of raw mitochondria was resuspended in 36% iodixanol (Sigma-Aldrich) and overlaid with 30% and 10% iodixanol. The gradient was ultracentrifuged (80,000 × g, 3 h) at 4 °C. The purified mitochondria were collected at the interface between 10% and 30% iodixanol and washed twice with MIB. Samples of each fraction were prepared by addition of sample buffer and subjected to immunoblot analysis.
Western blots
Cells were rinsed with ice-cold 1×PBS and lysed in nonreducing buffer containing 100 mM Tris–HCl, pH 6.8, 2% SDS, 50 mM IAA, supplemented with PMSF and cocktail, or reducing buffer containing an additional 100 mM DTT. Cell lysates were separated on a 8–12.5% SDS-polyacrylamide gel, transferred to a nitrocellulose membrane (Bio-Rad, Richmond, CA), blocked with 5% nonfat milk in PBS, and immunoblotted with the indicated antibodies. After incubation with the horseradish peroxidase-linked second antibody (Cell Signaling, Beverly, MA, USA) at room temperature, detection was performed using the SuperSignal West Pico Chemiluminescent Substrate Kit (Pierce, Rockford, IL) according to the manufacturer’s instructions. Uncropped blots and gel images were provided in the Source Data file.
Pull-down and MS analysis of AA005-bound proteins
The mitochondria was isolated from 3T3-L1 cells, and the mitochondrial fractions lysates were incubated with biotin or biotin-AA005 overnight at 4 °C, followed by pull-down with streptavidin-agarose beads, and the bead-bound proteins were separated by SDS-PAGE and visualized by Coomassie blue staining. The protein-containing band in the gel was excised, followed by in-gel digestion and analysis by LC-MS/MS. Pull-down and MS analysis was performed, and the data were analyzed according to our previous report39,40.
Microscale thermophoresis analysis
A human epithelial-like cell line 293 T (Cat: #ATCC, CRL-3216TM) derived from a patient’s kidney was transfected with empty-negative control plasmid pFlag-EGFP-CMV4 (NC), or plasmid pFlag-EGFP-CMV4-HADHA (WT) to overexpress wild-type HADHA protein or HADHA protein containing mutant sites (Mut-Val174; Mut-Leu175; Mut-Leu202) respectively. The five group cell lysates were kept equal of total proteins and AA005 was titrated from 1 × 10−10 M to 1 × 10−3 M. The affinity was determined. The samples were incubated at room temperature for 10 min before the measurements. The binding assay was performed in a buffer with 20 mM Tris, 100 mM NaCl, and 0.02% Tween-20. A NanoTemper Monolith Instrument (NT.115) was used for measuring thermophoresis.
Tissue cardiolipin profiling by LC-MS/MS
Interscapular brown adipose tissue (BAT) was isolated from mice for cardiolipin profiling. A 100 mg sample was taken in a 2 ml EP tube (L207270p, Eppendorf) and then extracted with chloroform methanol (vol/vol, 2:1) and dried in a vacuum. Samples were dissolved in isopropanol for LC-MS/MS (Bionovogene). Chromatographic separation was accomplished in a Thermo Vanquish system equipped with an ACQUITY UPLC BEH C18 (100 × 2.1 mm, 1.7 μm, Waters) column at 50 °C. The temperature of the autosampler was 8 °C. Gradient elution of analytes was carried out with acetonitrile: water = 60:40 (0.1% formic acid +10 mM ammonium formate) and isopropanol: acetonitrile = 90:10 (0.1% formic acid +10 mM ammonium formate) at a flow rate of 0.25 mL/min. Injection of 2 μL of each sample was done after equilibration. An increasing linear gradient of solvent C (v/v) was used as follows: 0–5 min, 70–57% C; 5–5.1 min, 57–50% C; 5.1–14 min, 50–30% C; 14–14.1 min, 30% C; 14.1–21 min, 30–1% C; 21–24 min, 1% C; 24–24.1 min, 1–70% C; 24.1–28 min, 70% C. The ESI-MSn experiments were executed on the Thermo Q Exactive Focus mass spectrometer with the spray voltage of 3.5 kV and −2.5 kV in positive and negative modes, respectively. analyzed and identified using LipidSearch software. 20 μL liquid from each sample was used as quality control (QC).
Tissue distribution study
To investigate the tissue distribution characteristics of AA005 in mice, C57BL/6 mice (n = 3) were given a single or long-term oral dose of 1.7 mg/kg AA005. After 0.25, 1, 2, and 6 h, all tissues, including the plasma, liver, heart, pancreas, kidney, lung, fat, spleen, brain, testis, stomach, and intestine were separated and homogenized, and their AA005 concentrations were detected by UPLC-MS/MS.
RNA-sequencing
Inguinal white adipose tissue (iWAT) of mice were collected with Trizol reagent. The total RNA was processed by NEBNext®Poly(A) mRNA Magnetic Isolation Module to enrich mRNA, and the product RNA was used for construction Library, via KAPA Stranded RNA-Seq Library Prep Kit (Illumina). Sequencing libraries, denatured by 0.1 M NaOH to generate single-stranded DNA, as amplified in situ Illumina cBot (TruSeq SR Cluster Kit v3-cBot-HS (#GD-401-3001, Illumina)). The ends of the generated fragments were used to run 150 Cycles by the Illumina HiSeq 4000 Sequencer. RNA-sequencing was performed three times.
Real-time quantitative RT-PCR
Total RNA from the cell lines were extracted with TRIzol reagent (Invitrogen, Carlsbad, CA) and treated with RNase-free DNase (Promega, Madison, WI, USA). Quantitative real-time PCR was performed using the SYBR® Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA). The primers used are listed as follows:
Ucp1-5′-GGCATTCAGAGGCAAATCAGCT-3′(forward),
Ucp1-5′-CAATGAACACTGCCACACCTC-3′(reverse),
Pgc1α-5′-AGCCGTGACCACTGACAACGAG-3′(forward),
Pgc1α-5′-GCTGCATGGTTCTGAGTGCTAAG-3′(reverse),
Dio2-5′-GATGCTCCCAATTCCAGTGT-3′(forward),
Dio2-5′-TGAACCAAAGTTGACCACCA-3′(reverse),
Pgc1β-5′-TCCTGTAAAAGCCCGGAGTAT-3′(forward),
Pgc1β-5′-GCTCTGGTAGGGGCAGTGA-3′(reverse),
Cidea-5′-GCAGCCTGCAGGAACTTATCAGC-3′(forward),
Cidea-5′-GATCATGAAATGCGTGTTGTCC-3′(reverse),
Tfam-5′-GGAATGTGGAGCGTGCTAAAA-3′(forward),
Tfam-5′-ACAAGACTGATAGACGAGGGG-3′(reverse),
Prdm16-5′-CGGAAGAGCGTGAGTACAAATG-3′(forward),
Prdm16-5′-TCCGTGAACACCTTGACACAGT-3′(reverse),
Arrpp0-5′-GAAACTGCTGCCTCACATCCG-3′(forward),
Arrpp0-5′-GCTGGCACAGTGACCTCACACG-3′(reverse),
Tafazzin-5′-GCAGACATCTGCTTCACCAA-3′(forward),
Tafazzin-5′-TCTGGTAGACGCCATCTCCT-3′(reverse),
Mlclat-1-5′-GAAGTCATCCGAATCCTCCA-3′(forward),
Mlclat-1-5′-TTTCGCTACATCCACACCAA-3′(reverse),
Real-time RT-PCR was performed, and the data were analyzed according to a previous report73.
Determination of mitochondrial DNA copy number by PCR
Total DNA from mouse brown adipocytes was extracted using the SpeeDNA Isolation Kit (ScienCell, Cat #MB6918) according to the manufacturer’s instructions. The mtDNA levels was determined by real-time quantitative PCR using the Relative Mouse Mitochondrial DNA Copy Number Quantification qPCR Assay Kit (ScienCell, Cat #M8938). A segment of mitochondrial genomic was routinely amplified as mtDNA copy number. All samples were quantitated by SYBR® Green staining and diluted in TE buffer to equal concentrations of 2.5 ng/μl. 5 ng of each template was added to the reaction. Amplification of mouse mtDNA segment was performed using the 2X GoldNStart TaqGreen qPCR Master Mix (ScienCell, Cat #MB6018a-1). PCR conditions: (i) 95 °C, 10 min; (ii) 95 °C, 20 s; (iii) 52 °C, 20 s; (iv) 72 °C, 45 s; and repeat steps 2–4 for 32 cycles; (v) hold 20 °C. Quantitation of each amplified product was performed by SYBR® Green staining using Micro Plate Reader.
Mitochondrial DNA content and respiration
Brown preadipocytes were isolated and immortalized as previously described74. Mitochondrial DNA content was measured using qPCR analysis of total DNA isolated from differentiated brown adipocytes. Primers for mitochondrial DNA (mtND1 and mtCox1) are listed as follows: mtND1-5′-CCTATCACCCTTGCCATCAT-3′(forward),
mtND1-5′-GAGGCTGTTGCTTGTGTGAC-3′(reverse),
mtCox1-5′-CTACTATTCGGAGCCTGAGC-3′(forward),
mtCox1-5′-GCATGGGCAGTTACGATAAC-3′(reverse),
Oxygen consumption rate in differentiated brown adipocytes was measured using an Oxygen Meter (Strathkelvin Instruments) with a Mitocell (MT200) mixing chamber. Cells were suspended in 400 μl differentiation medium, and oxygen concentration was recorded for 5 min followed by injections of FCCP (25 μM) oroligomycin (5 μg/ml) to the chamber. Oxygen consumption rate was calculated using software (782 Oxygen System version 4.0) and normalized to protein content.
Statistical analysis
Each experiment was done independently at least 3 times with similar results. Data were analyzed using two-tailed Student’s t test for independent groups. A p value of less than 0.05 was considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq of adipose tissue is publicly available at NCBI site as BioProject: PRJNA1149567. The raw LC-MS/MS data of cardiolipin profiling is publicly available at MetaboLights site as identifier MTBLS11124. All other data that support the findings of this study can be found in the manuscript, figures, and supplementary data or are available from the corresponding authors upon request. Source data are provided with this paper.
References
Collaboration, N. C. D. R. F. Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 387, 1377–1396 (2016).
Collaboration, N. C. D. R. F. Worldwide trends in diabetes since 1980: a pooled analysis of 751 population-based studies with 4.4 million participants. Lancet 387, 1513–1530 (2016).
Ng, M. et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 384, 766–781 (2014).
Bluher, M. Obesity: global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 15, 288–298 (2019).
Lauby-Secretan, B. et al. Body fatness and cancer-viewpoint of the IARC Working Group. N. Engl. J. Med. 375, 794–798 (2016).
Perdomo, C. M., Cohen, R. V., Sumithran, P., Clement, K. & Fruhbeck, G. Contemporary medical, device, and surgical therapies for obesity in adults. Lancet 401, 1116–1130 (2023).
Valenzuela, P. L. et al. Obesity and the risk of cardiometabolic diseases. Nat. Rev. Cardiol. 20, 475–494 (2023).
Weghuber, D. et al. Once-weekly semaglutide in adolescents with obesity. N. Engl. J. Med. 387, 2245–2257 (2022).
Apovian, C. M. et al. Pharmacological management of obesity: an Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 100, 342–362 (2015).
Christou, G. A., Katsiki, N., Blundell, J., Fruhbeck, G. & Kiortsis, D. N. Semaglutide as a promising antiobesity drug. Obes. Rev. 20, 805–815 (2019).
Heymsfield, S. B. & Wadden, T. A. Mechanisms, pathophysiology, and management of obesity. N. Engl. J. Med. 376, 1492 (2017).
Yanovski, S. Z. & Yanovski, J. A. Long-term drug treatment for obesity: a systematic and clinical review. JAMA 311, 74–86 (2014).
Apovian, C. M. et al. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity 21, 935–943 (2013).
Davidson, M. H. et al. Weight control and risk factor reduction in obese subjects treated for 2 years with orlistat: a randomized controlled trial. JAMA 281, 235–242 (1999).
Gadde, K. M. et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial. Lancet 377, 1341–1352 (2011).
Pi-Sunyer, X. et al. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N. Engl. J. Med. 373, 11–22 (2015).
Smith, S. R. et al. Multicenter, placebo-controlled trial of lorcaserin for weight management. N. Engl. J. Med. 363, 245–256 (2010).
Alali, F. Q., Liu, X. X. & McLaughlin, J. L. Annonaceous acetogenins: recent progress. J. Nat. Prod. 62, 504–540 (1999).
Chang, F. R. & Wu, Y. C. Novel cytotoxic annonaceous acetogenins from Annona muricata. J. Nat. Prod. 64, 925–931 (2001).
Degli Esposti, M., Ghelli, A., Ratta, M., Cortes, D. & Estornell, E. Natural substances (acetogenins) from the family Annonaceae are powerful inhibitors of mitochondrial NADH dehydrogenase (Complex I). Biochem. J. 301, 161–167 (1994).
Huang, G. R. et al. Induction of cell death of gastric cancer cells by a modified compound of the annonaceous acetogenin family. Chembiochem 4, 1216–1221 (2003).
Jiang, S. et al. Parallel fragment assembly strategy towards multiple-ether mimicry of anticancer annonaceous acetogenins. Angew. Chem. Int Ed. Engl. 43, 329–334 (2004).
Han, B. et al. Annonaceous acetogenin mimic AA005 suppresses human colon cancer cell growth in vivo through downregulation of Mcl-1. Acta Pharm. Sin. 40, 231–242 (2019).
Han, B. et al. Annonaceous acetogenin mimic AA005 induces cancer cell death via apoptosis inducing factor through a caspase-3-independent mechanism. BMC Cancer 15, 139 (2015).
Liu, H. X., Huang, G. R., Zhang, H. M., Wu, J. R. & Yao, Z. J. Annonaceous acetogenin mimics bearing a terminal lactam and their cytotoxicity against cancer cells. Bioorg. Med. Chem. Lett. 17, 3426–3430 (2007).
Zeng, B. B. et al. Studies on mimicry of naturally occurring annonaceous acetogenins: non-THF analogues leading to remarkable selective cytotoxicity against human tumor cells. Chemistry 9, 282–290 (2003).
Florence, N. T. et al. Antidiabetic and antioxidant effects of Annona muricata (Annonaceae), aqueous extract on streptozotocin-induced diabetic rats. J. Ethnopharmacol. 151, 784–790 (2014).
Eaton, S. et al. The mitochondrial trifunctional protein: centre of a beta-oxidation metabolon? Biochem. Soc. Trans. 28, 177–182 (2000).
Xia, C., Fu, Z., Battaile, K. P. & Kim, J. P. Crystal structure of human mitochondrial trifunctional protein, a fatty acid beta-oxidation metabolon. Proc. Natl Acad. Sci. USA 116, 6069–6074 (2019).
Fletcher, A. L., Pennesi, M. E., Harding, C. O., Weleber, R. G. & Gillingham, M. B. Observations regarding retinopathy in mitochondrial trifunctional protein deficiencies. Mol. Genet. Metab. 106, 18–24 (2012).
Kompare, M. & Rizzo, W. B. Mitochondrial fatty-acid oxidation disorders. Semin Pediatr. Neurol. 15, 140–149 (2008).
Ibdah, J. A., Yang, Z. & Bennett, M. J. Liver disease in pregnancy and fetal fatty acid oxidation defects. Mol. Genet. Metab. 71, 182–189 (2000).
Ibdah, J. A. et al. Lack of mitochondrial trifunctional protein in mice causes neonatal hypoglycemia and sudden death. J. Clin. Investig. 107, 1403–1409 (2001).
Dietrich, M. O. & Horvath, T. L. Hypothalamic control of energy balance: insights into the role of synaptic plasticity. Trends Neurosci. 36, 65–73 (2013).
Halaas, J. L. et al. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proc. Natl Acad. Sci. USA 94, 8878–8883 (1997).
Considine, R. V. et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 334, 292–295 (1996).
Pospisilik, J. A. et al. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell 131, 476–491 (2007).
Shen, S. M. et al. Apoptosis-inducing factor is a target gene of C/EBPalpha and participates in adipocyte differentiation. FEBS Lett. 585, 2307–2312 (2011).
Gao, Y. H. et al. VHL deficiency augments anthracycline sensitivity of clear cell renal cell carcinomas by down-regulating ALDH2. Nat. Commun. 8, 15337 (2017).
Liu, C. X. et al. Adenanthin targets peroxiredoxin I and II to induce differentiation of leukemic cells. Nat. Chem. Biol. 8, 486–493 (2012).
Schenone, M., Dancik, V., Wagner, B. K. & Clemons, P. A. Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 9, 232–240 (2013).
Liu, H. X. et al. A structure-activity guided strategy for fluorescent labeling of annonaceous acetogenin mimetics and their application in cell biology. Chembiochem 8, 172–177 (2007).
Wienken, C. J., Baaske, P., Rothbauer, U., Braun, D. & Duhr, S. Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 1, 100 (2010).
Ishikawa, M., Tsuchiya, D., Oyama, T., Tsunaka, Y. & Morikawa, K. Structural basis for channelling mechanism of a fatty acid beta-oxidation multienzyme complex. EMBO J. 23, 2745–2754 (2004).
Ussher, J. R. et al. Treatment with the 3-ketoacyl-CoA thiolase inhibitor trimetazidine does not exacerbate whole-body insulin resistance in obese mice. J. Pharm. Exp. Ther. 349, 487–496 (2014).
Taylor, W. A. et al. Human trifunctional protein alpha links cardiolipin remodeling to beta-oxidation. PLoS One 7, e48628 (2012).
Mejia, E. M. et al. Expression of human monolysocardiolipin acyltransferase-1 improves mitochondrial function in Barth syndrome lymphoblasts. J. Biol. Chem. 293, 7564–7577 (2018).
Taylor, W. A. & Hatch, G. M. Identification of the human mitochondrial linoleoyl-coenzyme A monolysocardiolipin acyltransferase (MLCL AT-1). J. Biol. Chem. 284, 30360–30371 (2009).
Perdikari, A. et al. BATLAS: deconvoluting brown adipose tissue. Cell Rep. 25, 784–797.e784 (2018).
Duncan, A. L. Monolysocardiolipin (MLCL) interactions with mitochondrial membrane proteins. Biochem Soc. Trans. 48, 993–1004 (2020).
Falabella, M., Vernon, H. J., Hanna, M. G., Claypool, S. M. & Pitceathly, R. D. S. Cardiolipin, mitochondria, and neurological disease. Trends Endocrinol. Metab. 32, 224–237 (2021).
Miao, N. et al. Oxidized mitochondrial DNA induces gasdermin D oligomerization in systemic lupus erythematosus. Nat. Commun. 14, 872 (2023).
Bratic, A. et al. Complementation between polymerase- and exonuclease-deficient mitochondrial DNA polymerase mutants in genomically engineered flies. Nat. Commun. 6, 8808 (2015).
Shu, L. et al. ATAD3B is a mitophagy receptor mediating clearance of oxidative stress-induced damaged mitochondrial DNA. EMBO J. 40, e106283 (2021).
Deshwal, S. et al. Mitochondria regulate intracellular coenzyme Q transport and ferroptotic resistance via STARD7. Nat. Cell Biol. 25, 246–257 (2023).
Ruiz-Jimenez, J., Priego-Capote, F., Mata-Granados, J. M., Quesada, J. M. & Luque de Castro, M. D. Determination of the ubiquinol-10 and ubiquinone-10 (coenzyme Q10) in human serum by liquid chromatography tandem mass spectrometry to evaluate the oxidative stress. J. Chromatogr. A 1175, 242–248 (2007).
Cole, L. K. et al. Impaired cardiolipin biosynthesis prevents hepatic steatosis and diet-induced obesity. Diabetes 65, 3289–3300 (2016).
Li, J. et al. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 12, 154–165 (2010).
Padwal, R. S. & Majumdar, S. R. Drug treatments for obesity: orlistat, sibutramine, and rimonabant. Lancet 369, 71–77 (2007).
Bray, G. A. Obesity: liraglutide-another weapon in the war against obesity? Nat. Rev. Endocrinol. 11, 569–570 (2015).
Gustafson, A., King, C. & Rey, J. A. Lorcaserin (Belviq): a selective serotonin 5-HT2C agonist in the treatment of obesity. P T 38, 525–534 (2013).
Geisler, JG 2,4 Dinitrophenol as medicine. Cells 8, https://doi.org/10.3390/cells8030280 (2019).
Perry, R. J., Zhang, D., Zhang, X. M., Boyer, J. L. & Shulman, G. I. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 347, 1253–1256 (2015).
Miklas, J. W. et al. TFPa/HADHA is required for fatty acid beta-oxidation and cardiolipin re-modeling in human cardiomyocytes. Nat. Commun. 10, 4671 (2019).
Sustarsic, E. G. et al. Cardiolipin synthesis in brown and beige fat mitochondria is essential for systemic energy homeostasis. Cell Metab. 28, 159–174.e111 (2018).
Lee, Y., Willers, C., Kunji, E. R. & Crichton, P. G. Uncoupling protein 1 binds one nucleotide per monomer and is stabilized by tightly bound cardiolipin. Proc. Natl Acad. Sci. USA 112, 6973–6978 (2015).
Pan, A. et al. The mitochondrial beta-oxidation enzyme HADHA restrains hepatic glucagon response by promoting beta-hydroxybutyrate production. Nat. Commun. 13, 386 (2022).
Zhang, Z. et al. The rs1421085 variant within FTO promotes brown fat thermogenesis. Nat. Metab. 5, 1337–1351 (2023).
Klein, J., Fasshauer, M., Klein, H. H., Benito, M. & Kahn, C. R. Novel adipocyte lines from brown fat: a model system for the study of differentiation, energy metabolism, and insulin action. Bioessays 24, 382–388 (2002).
Morand, J. P., Macri, J. & Adeli, K. Proteomic profiling of hepatic endoplasmic reticulum-associated proteins in an animal model of insulin resistance and metabolic dyslipidemia. J. Biol. Chem. 280, 17626–17633 (2005).
Yu, Y. et al. Subcellular proteome analysis of camptothecin analogue NSC606985-treated acute myeloid leukemic cells. J. Proteome Res. 6, 3808–3818 (2007).
Klotz, A. V., Stegeman, J. J. & Walsh, C. An alternative 7-ethoxyresorufin O-deethylase activity assay: a continuous visible spectrophotometric method for measurement of cytochrome P-450 monooxygenase activity. Anal. Biochem 140, 138–145 (1984).
Zhao, K. W. et al. Protein kinase Cdelta mediates retinoic acid and phorbol myristate acetate-induced phospholipid scramblase 1 gene expression: its role in leukemic cell differentiation. Blood 104, 3731–3738 (2004).
Zhao, X. Y., Li, S., Wang, G. X., Yu, Q. & Lin, J. D. A long noncoding RNA transcriptional regulatory circuit drives thermogenic adipocyte differentiation. Mol. Cell 55, 372–382 (2014).
Acknowledgements
This work was financially supported by Fundamental Research Funds for Shanghai Municipal Health Commission (2022YQ052 (B.H.)), Minhang Hospital (2022MHBJ01 (B.H.), 2021MHJC02 (Z.M.L.), 2021MHJC01 (C.Y.K.), 2023MHBJ01 (YQM) and 2024MHBJ02 (H.L.C.)), National Natural Science Foundation of China (82372626 (L.S.W.), 82303578 (Y.Q.M.), 81803601 (B.H.) and 81872245 (L.S.W.)) and Shanghai (SKLGE-2112 (L.S.W.), 20214Y0327 (Z.M.L.), 20214Y0326 (H.L.C.), 20214Y0328 (Y.Q.M.), 23YF1438700 (Y.Q.M.)).
Author information
Authors and Affiliations
Contributions
B.H. and L.S.W. conceived and designed the experiments; B.H., Z.M.L., X.Y.Z., K.L., Y.Q.M., S.L.Z., L.Y.H., C.Y.K., X.P., H.L.C., J.T.H., Z.X.W., J.Q.Y., P.R.C., Z.Y.Z., and X.M.Z. performed the experiments; B.H., Z.J.Y., G.Q.C., and L.S.W. analyzed the data; B.H., Z.M.L., and L.S.W. wrote the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Fong-Fu Hsu, Elena Zambrano, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Han, B., Li, ZM., Zhao, XY. et al. Annonaceous acetogenins mimic AA005 targets mitochondrial trifunctional enzyme alpha subunit to treat obesity in male mice. Nat Commun 15, 9100 (2024). https://doi.org/10.1038/s41467-024-53118-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-53118-3
