
Abstract
Human mitochondrial tRNAs (mt-tRNAs), critical for mitochondrial biogenesis, are frequently associated with pathogenic mutations. These mt-tRNAs have unusual sequence motifs and require post-transcriptional modifications to stabilize their fragile structures. However, whether a modification that stabilizes a wild-type (WT) mt-tRNA would also stabilize its pathogenic variants is unknown. Here we show that the N1-methylation of guanosine at position 9 (m1G9) of mt-Leu(UAA), while stabilizing the WT tRNA, has a destabilizing effect on variants associated with MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes). This differential effect is further demonstrated, as removal of the m1G9 methylation, while damaging to the WT tRNA, is beneficial to the major pathogenic variant, improving the structure and activity of the variant. These results have therapeutic implications, suggesting that the N1-methylation of mt-tRNAs at position 9 is a determinant of pathogenicity and that controlling the methylation level is an important modulator of mt-tRNA-associated diseases.
Similar content being viewed by others

Introduction
Mitochondria are essential eukaryotic organelles that produce most of the cellular ATP molecules. In humans, due to the high demand for aerobic activity and developmental complexity, the mt-DNA has accumulated much higher levels of mutations as compared to the nuclear genome1,2,3,4. Importantly, most of the disease-causing mt-DNA mutations are mapped to mt-tRNAs (MITOMAP, www.mitomap.org)5,6 and are associated with devastating mitochondrial pathologies, underscoring the need for a better understanding of these tRNAs.
The human mt-DNA encodes genes for 22 mt-tRNAs, 2 mt-rRNAs, and 13 mt-proteins7,8. All 22 mt-tRNAs are used in the mt-ribosome machinery7,9 for synthesis of the 13 mt-DNA-encoded proteins, each of which is a core component of the mitochondrial electron transport chain10, emphasizing the importance of each mt-tRNA in its fully functional state. Notably, mt-tRNAs are transcribed with inherent structural fragility, with low sequence complexity, small loop sizes, weak base pairs and mismatches in stem regions, loss of conserved tertiary interactions of the canonical structure, and even deletions of entire domains, contributing to their generally low thermal stability7,11,12. This structural fragility is mitigated in each mt-tRNA by acquiring a set of post-transcriptional modifications13 that are synthesized by nuclear-encoded enzymes, which are also frequently associated with human pathologies14.
The most prominent post-transcriptional modification in mt-tRNAs is the N1-methylation to the purine R (A/G) nucleotide at position 9, which is conserved in 19 of the 22 mt-tRNAs (5 as m1G9 and 14 as m1A9)15, whereas the remaining 3 mt-tRNAs each contains a pyrimidine at position 9. The m1G9/m1A9 methylation is catalyzed by a nuclear-encoded ternary complex that has dual-specificity. This ternary complex consists of MRPP1-2-3 (mitochondrial RNase P proteins 1, 2, and 3) (Fig. S1a), where MRPP1 (TRMT10C) is the methyl transferase that requires the assistance of MRPP2 (SDR5C1, short-chain oxidoreductase/hydroxysteroid 17β-dehydrogenase 10), while the two enzymes assist MRPP3 (PRORP) to perform 5′-processing of all mt-tRNAs16,17,18. As 5′-processing is the initiation step of maturation of all mt-tRNAs18,19, the associated m1G9/m1A9 methylation is likely the first modification that folds each mt-tRNA into a structure suitable for subsequent modifications20. Indeed, while the transcript of human mt-Lys(UUU) (UUU, the anticodon), lacking any modification, adopts an extended and non-functional stem-loop structure, introduction of the m1A9 methylation to the transcript converts it to a structure closely similar to the native-state21,22,23. Notably, this finding was based on the WT sequence of mt-Lys(UUU), but not a pathogenic variant. We do not yet know whether the N1-methylation of purine 9 would have the same, or a different, effect on a pathogenic variant. As many pathogenic variants are unstable24,25, due to mutations that further weaken an already fragile structure, they might benefit from the N1-methylation of purine 9 to strengthen their structures.
Here we address this question directly, using human mt-Leu(UAA) as a model (Fig. 1a), which has m1G9 (Fig. 1b) as shown in an LC-MS analysis15. This mt-tRNA was previously denoted as mt-tRNALeu(UUR), indicating its reading of the leucine codons UUR during protein synthesis. We chose mt-Leu(UAA) as a different sequence framework than mt-Lys(UUU) to more broadly test the importance of the N1-methylation of purine 9. Notably, mt-Leu(UAA) has been examined by structural probing and aminoacylation experiments26,27, and is one of the few human mt-tRNAs possessing all of the conserved features of the canonical tRNA structure (Fig. 1a)28. A previous study showed that, while the transcript of WT mt-Leu(UAA) has a weakly stable structure, sampling between a folding intermediate and the native-state, the native form of the tRNA, possessing all natural modifications, has a more constrained structure that does not sample the intermediate state29. While this work emphasizes the importance of tRNA modifications, it does not point to a specific modification as the major driver of folding. There has also been no study of mt-Leu(UAA) that contains all natural modifications but lacks m1G9, leaving open the question of how m1G9 would contribute to the tRNA folding. Importantly, human mt-Leu(UAA) is most closely associated with MELAS30. Approximately 80% of patients with MELAS have the mt-DNA A3243G substitution that is mapped to mt-Leu(UAA)31,32, representing the most common single-point pathogenic mutation in human populations33. Another 10% of patients with MELAS have the T3271C substitution that is also mapped to mt-Leu(UAA) (Fig. 1a) as the second most common mutation associated with the disease34. The A3243G substitution corresponds to the A14G mutation in mt-Leu(UAA), which disrupts the 8–14 base pair that normally would connect the acceptor stem with the D stem in the canonical tRNA structure. The T3271C substitution corresponds to the U40C mutation in mt-Leu(UAA), which disrupts the 30–40 base pair of the anticodon stem. The two mutations thus represent two distinct structural perturbations of the same mt-tRNA. However, despite the close association of the two mutations with MELAS, the molecular basis of the pathology remains unclear. Specifically, while both variants are deficient in tRNA stability, aminoacylation, and the 5-taurine-methyl modification to the wobble nucleotide U34 (τm5U34)35 (Fig. 1c), the driver of these deficiencies is unknown.

a Sequence and cloverleaf structure of human mt-Leu(UAA) showing the A3243G and T3271C substitutions in blue and the m1G9 and τm5U34 modifications in red, both mapped to the canonical L-shaped tRNA structure. b Chemical structure of m1G, showing the N1-methyl group in red. c Chemical structure of τm5U, showing the taurine-C5-methyl group in red.
To address this question, we functionally separated m1G9 from all other natural modifications in mt-Leu(UAA). We find that, while m1G9 indeed stabilizes the WT mt-Leu(UAA) structure, it has an opposite effect on both the A3243G and T3271C variants, trapping each in an aberrant structure that is sensitive to degradation. Conversely, while removal of m1G9 is damaging to the WT mt-Leu(UAA), it is beneficial to the A3243G variant, improving the stability and functional state of the variant within the mitochondrial milieu. These results suggest a conceptual framework that emphasizes the ability of m1G9 to differentially control and regulate mitochondrial health and disease. This framework suggests that, instead of enhancing m1G9 in mt-Leu(UAA) as a remedy for MELAS, limiting the methylation in variants of mt-Leu(UAA) would have a better outcome. Given the broad conservation of m1G9/m1A9 in human mt-tRNAs, and the high-stoichiometry of the methylation even in their pathogenic variants, this framework is likely generalizable to other mt-tRNA-associated diseases.
Results
The m1G9 methylation in mt-Leu(UAA) is the major driver to the native structure
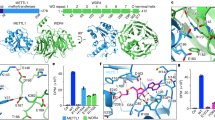
We tested whether m1G9 is the major driver that folds human mt-Leu(UAA) to its native structure. To determine folding, we measured translocation of the tRNA through solid-state nanopores using our label-free single-molecule assay36. This measurement distinguishes different RNA structures by the pore translocation times and ion-current signals37. As a voltage is applied across the small synthetic pore (7–10 nm thickness, ~3 nm diameter), a tRNA molecule is electrophoretically driven to unfold and to translocate through an electrolyte filled pore (Fig. 2a, top), displacing some of the ions as it passes through, thus lowering the open pore current in an event defined by two parameters37. The “current blockade” is the difference between the open pore current and the measured current amplitude, while the “dwell time” is the duration of the molecule within the pore. These two parameters provide structural information on the tRNA passing through the pore, such as the shape, size, and the conformation dynamics in both folded and unfolded states38,39. Using pores of the described dimension, a properly folded tRNA typically resides in the pore longer until it eventually unfolds and passes through the pore with a higher current blockade, whereas an improperly folded tRNA passes through the pore faster with a lower current blockade38,39.

a Top: Electrophoretic unfolding of a tRNA through a solid-state nanopore. Schematics show steps required for tRNA passage through a nanopore with an example of a translocation event. Bottom: Heat-map scatter plots of fractional current blockade as a function of dwell time of the WT human mt-Leu(UAA) in the G9-state (left), in the m1G9-state (middle), and the Bos taurus liver mt-Leu(UAA) in the native-state (right). Statistics for the different dwell time and current blockade values are in Table S1. b A native 12% PAGE separation of the WT from the A3243G variant of human mt-Leu(UAA) in the heteroplasmy series as probed by Northern blot analysis. c A Northern blot analysis of the steady-state level of human mt-Leu(UAA) relative to the 5S rRNA in WT and A3243G cybrids, showing size markers superimposed from a stained gel. Bar graph represents the average ± SD (n = 3). No adjustments were made for multiple comparisons. d A representative primer extension analysis of the m1G9 level on mt-Leu(UAA) from WT and A3243G cybrids, showing the region of interest in a red box and the band due to termination of primer extension by an orange arrow. e A representative primer extension analysis of the m2G10 level in mt-Leu(UAA) in WT and A3243G cybrids. The red box and the orange arrow are as in (d). f A representative primer extension analysis of the m1A58 level in mt-Leu(UAA) in WT and A3243G cybrids. The red box and the orange arrow are as in (d). g Analysis of the τm5U34 level in mt-Leu(UAA) by a CMC-assisted primer extension assay (CMC (+) or buffer (−)) in WT and A3243G cybrids. The τm5U34 level was calculated as the fraction of the band at A35 in the sum of the bands at A35, τm5U34, U33, and C32. Bar graphs are average ± SD (n = 3). Source data are provided as a Source Data file, and p values were obtained from two-sided paired t test (c, g).
To separate m1G9 from all other natural modifications in the folding of the WT mt-Leu(UAA), we prepared two states of the tRNA—the transcript G9-state lacking any modification, and the m1G9-state containing the single m1G9 modification in the transcript-state. Each state was allowed to fold in the presence of Mg2+ and then to pass through nanopores. Measurement of the translocation kinetics showed that, while the transcript G9-state had a lower fractional current blockade (20%) and a shorter dwell time (2 μs) (Fig. 2a, bottom, left), the m1G9-state had a higher fractional current blockade (70%) and a longer dwell time (log102.4 μs) (Fig. 2a, bottom, middle, Table S1). The increase in the current blockade indicates that the single m1G9 methylation transformed the G9-state to a more stable structure that required higher energetics of unfolding during passage through the pore. Notably, the translocation kinetics of the m1G9-state was quantitatively similar to that of the native mt-Leu(UAA) of bovine liver (Fig. 2a, bottom, right, Table S1), possessing all natural modifications40. This similarity, based on the similar sequence and modification profile between bovine and human mt-Leu(UAA) in the native-state (Fig. S1b, c), indicates that the single m1G9 is the major driver that folds human mt-Leu(UAA) into a structure similar to the native structure of the bovine counterpart.
A different structure of the A3243G variant, despite having m1G9
We used a cybrid cell model to compare the WT and the A3243G variant of MELAS. Cybrids are created by intercellular transfer of the mt-DNA of a clinical patient into human r0 cells lacking mt-DNA41,42. Such cybrids have been created for the A3243G variant43,44 and others45,46. A unique strength of cybrids is that they provide direct evidence for the association of a mt-DNA mutation with the disease pathology47, excluding the role of the nuclear genome. We previously generated a series of cybrids, starting with a patient cell line heteroplasmic of the A3243G mutation and isolated clones that ranged in the frequency of the mutation (0, 20, 30, 50, 60, 90, and 100%) (Fig. 2b)48. Using this series of the A3243G variant, we systematically monitored the structural folding of the A3243G variant vs. the WT. Analysis of total RNA isolated from each cybrid line, followed by Northern blot analysis of mt-Leu(UAA) in a non-denaturing gel, showed that the WT and A3243G variant migrated differently (Fig. 2b), indicating a distinct structure of each. Importantly, the two structures distributed relative to each other in a way consistent with the mutation frequency of each cybrid line (Fig. 2b), enabling assignment of the variant to the faster migrating species. The distinct structure of the variant from the WT tRNA was further supported in a Northern blot analysis of total RNA from the two cybrid lines, showing that the abundance of the variant tRNA, after normalization to the 5S rRNA level, was 40% of the WT (Fig. 2c), consistent with a previous analysis35,49. The reduced abundance indicates that the variant tRNA was sensitized to degradation, most likely due to a different structure than the WT. In experiments below, unless otherwise noted, we used the WT (0% mutation) and the A3243G cybrids (100% mutation) of this heteroplasmy series.
Despite having a different structure, the A3243G variant has retained m1G9 in stoichiometry. While the retention of m1G9 was previously documented in an A3243G cybrid line35, we validated this retention in our cybrid line. Due to the limited quantities of mt-Leu(UAA) isolated from cybrids, LC-MS measurement of the methylation was not practical. We thus probed the methylation using a primer extension assay, where the methyl group of m1G9 occurring in the W-C face of the guanosine (Fig. 1b) would interfere with primer extension by a reverse transcriptase (RT). The nature of the interference varied with the chemical environment of the modification and the processivity of the RT. While some RTs would read through m1G9, others would stop and generated an extension product that terminated one nucleotide before the modification. We designed each primer-extension assay by placing the 3′-end of the primer ~2 nucleotides before the modification (Fig. S2, Table S2), and by including the ddNTP that would terminate the extension at the nucleotide after the modification. We then calculated the fraction of the extension product that terminated one nucleotide before the modification relative to the sum of all extension products.
We used the presence of m1G9 in the WT mt-Leu(UAA) as the guide to screen for RTs that would terminate primer extension in response to the modification. We found that the Luna RT was the only one that made a complete stop at m1G9 by extending the 3′-end of a primer from position C11 to m2G10 in a reaction containing dCTP and ddATP (Figs. 2d and S2a). The RT stop at m2G10 indicated that primer extension incorporated a nucleotide opposite m2G10 but that it was terminated by m1G9. Analysis of the termination product relative to the readthrough to U8 showed ~91% of m1G9 in samples isolated from WT cybrids (Fig. 2d), consistent with the LC-MS analysis15. To confirm that the termination was indeed the readout for m1G9, and not for m2G10, we analyzed a series of mt-Leu(UAA) in the transcript-state, lacking m2G10 but containing m1G9 at 0, 25, 50, and 100% (Fig. S2b). The linear correlation between the prepared m1G9 and the measured RT stop (Fig. S2c) indicated that the assay was quantitative with m1G9. In this titration, lacking m2G10, we detected m1G9 at the full stoichiometry when the prepared sample was homogeneous with the methylation, consistent with the level measured in the presence of m2G10 in WT cybrids (91%) (Fig. 2d). Thus, the readout was independent of m2G10 but was responsive to the level of m1G9. Applying this assay to total RNA of A3243G cybrids, we observed an RT stop at a level similar to that of WT cybrids (Fig. 2d), validating that the variant has retained m1G9 at stoichiometry.
We tested whether the variant changed the status of other tRNA modifications due to its different structure than the WT. We focused on modifications that are present in the WT tRNA and quantified a subset of these in the variant using the primer extension assay. At m2G10, immediately on the 3′-side of m1G9, primer extension with SuperScript III (SSIII) identified a similar level of modification in the variant as in the WT (Figs. 2e and S2d, e). At m1A58, adjacent to m1G9 in the tRNA tertiary structure, primer extension with SSIII also identified a similar level of the modification in the variant as in the WT (Figs. 2f and S2f). The τm5U34 modification permits efficient decoding of mt-Leu(UAA) of not only the UUA codon but also the UUG codon for Leu50,51,52,53,54,55. The τm5U34 level is closely correlated with clinical presentation of MELAS, where patients with pathogenic variants deficient of τm5U34 (e.g., A3243G, T3271C) present severe symptoms, while those retaining τm5U34 show few symptoms56. Also, restoration of the level of τm5U34, by over-expression of the mt-MTO1 enzyme that installs taurine to the modification57 or by co-expression of the haplotypic T3290C mutation58, improves mitochondrial respiration. These results demonstrate the close association of this modification with mitochondrial pathology. To measure the level of τm5U34 in cybrids, we performed a CMC (N-cyclohexyl-N′-b-(4-methyl-morpholinium) ethyl-carbodi-imide)-assisted primer extension assay15,55,57, where CMC modified the taurine moiety in total RNA, generating a covalent adduct that was detectable as an RT stop. We observed in the WT mt-Leu(UAA) a strong AMV RT stop at A35, one nucleotide before the modification in the RT reaction, but a much lower level of the stop in the A3243G variant (Fig. 2g, left, Fig. S2g). The τm5U34 level in the variant was estimated as 48% of the WT (Fig. 2g, right), based on the fraction of the extension product that terminated at A35 among all extension products. Thus, of the modifications that we examined for the A3243G variant, τm5U34 was the only one that was deficient, consistent with previous analysis of an A3243G line35,56.
m1G9 is the major driver to the altered structure of the A3243G variant
The distinct gel migration of the A3243G variant, and its loss of abundance relative to the WT (Fig. 2b, c), suggests that it has an altered structure. We tested whether m1G9 was the major driver to the altered structure by isolating the effect of the methylation from other natural modifications in mt-Leu(UAA). We thus compared the structure of the variant relative to the WT in the m1G9-state vs. the native-state, using the SHAPE analysis (selective 2′-hydroxyl acylation analyzed by primer extension)59,60 to probe RNA local backbone flexibility. We employed NAI (2-methyl-nicotinic acid imidazolide)61 as the SHAPE reagent, which formed an adduct at each reactive site, leading to an RT stop during primer extension. The results were analyzed for the differential effect of NAI as the ratio of the chemical reactivity across nucleotide positions of the A3243G variant relative to the WT. A ratio of 1 indicated a similar chemical reactivity of the two, while a ratio different than 1 indicated that the variant had a different reactivity due to structural alterations from the WT.
We first probed the m1G9-state of the A3243G variant vs. the WT to assess the methylation as the single determinant of each tRNA structure. Among variations of the ratio across the tRNA sequence, the most notable was at positions 12-16, 23-24, and 27-28 in the D stem-loop and at positions 34-40 (Fig. 3a, left, Fig. S3a), which are in the region encompassing m1G9, the A14G substitution, and the upper part of the coaxially stacked D-anticodon stem-loop (Fig. 3a, right). We next probed the variant vs. the WT in the native state to assess the effect of m1G9 in the presence of all natural modifications. Cybrids were grown for the WT and the variant, treated with NAI, and total RNA of each was probed by RT stops with a primer specific to mt-Leu(UAA). Across the tRNA sequence, the most prominent differences between the variant and the WT were at positions 13-18, 24-27, and at the wobble U34 (Fig. 3b, left, Fig. S3b), which also mapped to the D-anticodon stem-loop and to the wobble nucleotide U34 (Fig. 3b, right). Thus, the mapping data of the variant vs. the WT were extensively overlapped between the m1G9-state and the native-state (Fig. 3a, b). As m1G9 is the single driver of the WT structure (Fig. 2a), this extensive overlap indicates that m1G9 is also the major driver of the variant structure. Additionally, the mapping data show how the variant differed from the WT, primarily in the D stem-loop, into the upper part of the anticodon stem-loop, and the wobble U34. The mapping difference at U34 is notable, where the variant is deficient of the τm5U34 modification (Fig. 2g), suggesting that the structural differences have prevented the variant from recognition by the enzyme complex mt-MTO1-mt-GTPBP3 that synthesizes τm5U3462.

a SHAPE analysis of the m1G9-state of the A3243G variant relative to the m1G9-state of the WT tRNA using NAI as the chemical probe. Left: Plots represent the NAI reactivity ratio across nucleotides 12–40 in the sequence framework of mt-Leu(UAA), where regions with NAI reactivity above or below the ratio of 1.0 are shown in red boxes. Right: Differential NAI reactivity is mapped in the canonical L-shaped tRNA structure, where the A3243G mutation is shown as a cyan ring, while m1G9 is shown as a yellow ring. b SHAPE analysis of the native-state of the A3243G variant relative to the native-state of the WT tRNA using NAI as the chemical probe. Left: Plots represent the NAI reactivity ratio across nucleotide positions 12–40 in the sequence framework of mt-Leu(UAA), where regions with NAI reactivity above or below the ratio of 1.0 are shown in red boxes. Right: Differential NAI reactivity is mapped in the canonical L-shaped tRNA structure as in (a). c Stability of the WT and A3243G variant in the G9-state and the m1G9-state. Each transcript was 5′-32P-labeled and incubated with a mouse liver mitochondrial lysate over time, where aliquots were analyzed by a 12% PAGE/7 M urea sequencing gel. Plots are the average ± SD (n = 3). The half-life t1/2 of each tRNA was calculated from curve fitting to a linear regression equation y = a + bx, where a was the dependent variable (intensity at time 0 set to 100), b was the slope, y = 50, and x was the half-life t1/2. Source data are provided as a Source Data file.
We then tested whether the variant structure was more sensitive to degradation than the WT in a cellular environment. To directly test m1G9 as the single determinant of tRNA folding, we prepared G9- and m1G9-state of the WT and the A3243G variant, each in the 5′-32P-labeled transcript form, and measured the abundance of the label in mitochondrial lysates isolated from a mouse liver. As each tRNA was directly monitored through the 5′-32P-label, the decrease in abundance reflected the decrease in stability in the mitochondrial lysate. Due to the focus on m1G9 as the single determinant of each tRNA structure, this analysis could not be performed in live cells, where all mt-tRNAs contained the full complement of modifications. Analysis of the label over time showed that, while m1G9 improved the stability of the WT transcript (t1/2 from 52 min of the G9-state to 129 min of the m1G9-state), it decreased the stability of the variant (t1/2 from 114 min of the G9-state to 63 min of the m1G9-state) (Fig. 3c and Fig. S3c). Thus, m1G9 differentially regulates the stability of the WT and the A3243G variant.
The folding kinetics of the A3243G and T3271C variants are impeded by m1G9
To determine how m1G9 guided the A3243G variant to a different and less stable structure than the WT, we compared their folding kinetics. To isolate the methylation from all other natural modifications in mt-Leu(UAA), we prepared the WT and the A3243G variant in the G9-state and the m1G9-state. While the G9-state was made by in vitro transcription, the m1G9-state was made by an assembly approach, in which the 5′-fragment of each tRNA (nucleotides G1 to C17a) was chemically synthesized and was joined by T4 Rnl2 with the 3′-fragment (nucleotides G18 to A76) generated by in vitro transcription (Fig. S4a, b). We monitored the folding kinetics of each using the enzyme mt-leucyl-tRNA synthetase (mt-LeuRS) as the probe29,63 to identify the structure that was recognizable for aminoacylation. Each tRNA was incubated without Mg2+ and was initiated for the aminoacylation reaction by simultaneous addition of mt-LeuRS and Mg2+. Notably, the addition of Mg2+ separated the unfolded tRNA into two populations, one in a meta-stable intermediate state (due to local intra-molecular base parings) that was competent to fold to the chargeable state, whereas the other in a disordered state. As shown previously29, the folding of mt-Leu(UAA) from the meta-stable state to the chargeable-state was slow (kobs = 0.66 min−1), while aminoacylation of the chargeable-state was fast for these enzymes (kobs = 10 − 20 s−1)64,65,66,67,68 (Fig. 4a), indicating that the kinetics of aminoacylation was rate-limited by the kinetics of folding from the meta-stable state to the chargeable-state.

a An illustration of human mt-Leu(UAA) folding from a meta-stable intermediate-state to the chargeable-state for aminoacylation. The rate constants 0.66 min−1 and 10–20 s−1 were from published reports29,64,65,66,67,68, respectively. b The fraction of the charged tRNA as a function of time was monitored for the m1G9-state of the WT tRNA, the G9-state of the WT tRNA, the G9-state of the A3243G variant, and the m1G9-state of the A3243G variant. For each tRNA, the rate constant (k1) and the amplitude of the first phase (amp1) are shown as the average ± SD (n = 3). c The fraction of the charged tRNA as a function of Mg(OAc)2 concentration for the 4 tRNAs described in (b). Each value was the average ± SD (n = 3). d The fraction of the charged tRNA as a function of time was monitored for the m1G9-state of the WT tRNA, the G9-state of the WT tRNA, the G9-state of the T3271C variant, and the m1G9-state of the T3271C variant. For each tRNA, the rate constant (k1) and the amplitude of the first phase (amp1) are shown as the average ± SD (n = 3). e The fraction of the charged tRNA at 8 min as a function of Mg(OAc)2 concentration for the 4 tRNAs described in (d). Each value was the average ± SD (n = 3). Source data are provided as a Source Data file.
We observed biphasic kinetics for all 4 tRNAs, where the initial rapid phase represented aminoacylation of the meta-stable state that was competent to fold to the chargeable state, while the slower phase represented re-arrangement of the disordered species to the meta-stable state after enzyme addition. For the WT transcript, the addition of m1G9 improved the rate constant k1 (2.4 ± 0.2 to 5.4 ± 0.4 min−1) of aminoacylation of the first phase (Fig. 4b), indicating that the methylation facilitated conversion of the meta-stable species to a structure more favorable for charging. The addition of m1G9 also increased the amplitude of the first phase (amp1) (46 ± 14 to 53 ± 4 %), indicating that it increased the fraction of the meta-stable species to more than 50%. In contrast, the addition of m1G9 to the A3243G variant had a negative effect relative to the unmethylated variant. It decreased both k1 (2.4 ± 0.2 to 0.71 ± 0.06 min−1) and amp1 (46 ± 14 to 19 ± 3%) (Fig. 4b), indicating that the methylation led the meta-stable species to a structure less favorable for charging and that it reduced the fraction of the meta-stable species to less than 20%. As the kinetics continued into the second phase, the most notable deficiency of the m1G9-modified A3243G variant was the low level of the aminoacylation amplitude (~25%), indicating that most of the species were kinetically trapped in a non-chargeable structure. Even the unmethylated variant reached a higher amplitude (~55%).
The negative effect of m1G9 on the A3243G variant was also observed in a titration of Mg2+, a diffusible metal ion that can bind discrete folding intermediates69. All 4 tRNAs showed sigmoidal binding kinetics (Fig. 4c), indicating that Mg2+ binding at one site facilitated binding at another site. In each titration series, the mt-tRNA was incubated with Mg2+ for 20 min, followed by aminoacylation for 3 min, providing information on the fraction of the meta-stable intermediate that was competent to fold into the chargeable state. Notably, the m1G9-modified variant exhibited the lowest amplitude (35%) relative to others (60-75%), indicating that it had the lowest fraction that was competent to convert to the chargeable state but the largest fraction that was trapped in a non-competent state. In addition, the m1G9-modified variant required the highest Mg2+ concentration (10.8 ± 0.4 mM) relative to others to transition from the meta-stable state to the chargeable-state. Even the unmethylated variant required a lower concentration of Mg2+ for this transition (6.9 ± 0.2 mM) (Fig. 4c).
We also tested the folding kinetics of the T3271C variant, which contains m1G9 in the native state35 and represents a distinct structural perturbation than the A3243G variant (Fig. 1a). As above, the G9- and m1G9-state of the WT and the T3271C variant were prepared (Fig. S4c). Similar to the A3243G variant, addition of m1G9 to the WT tRNA improved the folding kinetics (k1 from 0.56 ± 0.03 to 0.98 ± 0.1 min−1 and amp1 from 38 ± 0.9 to 40 ± 2 %) (Fig. 4d), indicating that it led the meta-stable state to a more favorable structure for aminoacylation and that it shifted the fraction of the meta-stable state to a higher level. By contrast, addition of m1G9 to the T3271C variant had a negative effect, substantially reducing the fraction of the meta-stable state (amp1 from 35 ± 0.6 to 9.7 ± 0.3%) (Fig. 4d). Also, in the Mg2+ titration analysis, while the unmodified variant showed similar kinetics as the WT tRNA in reaching a high amplitude (35%), the m1G9-modified variant was limited to a much lower amplitude (10%) (Fig. 4e). Thus, in both the A3243G and T3271C variants, m1G9 impeded the folding kinetics, trapping each in a non-chargeable aberrant structure.
Hypomethylation of m1G9 improves the relative stability of the A3243G variant
The negative effect of m1G9 on the A3243G variant raised the possibility that hypomethylation of the variant in a cell model would improve its structure and activity. We thus targeted the methyl transferase gene MRPP1 by siRNAs to induce gene-specific KD of the methylation, as there is currently no small molecular inhibitor of the enzyme. Complete MRPP1-KO was not possible, due to the growth-dependent essentiality of the gene70,71. We screened four siRNAs that targeted different regions of the mRNA (Fig. S5a, Table S3) and found that 48 h post-transfection had a sufficient KD effect of all four siRNAs on both the mRNA and the protein (Fig. S5b, c), while 72 h post-transfection had no further effect on the protein (Fig. S5d). We monitored 48 h post-transfection and found that siRNA1 and siRNA3 were the most effective on the MRPP1 mRNA, reducing the level to 10% and 13% in WT cybrids, respectively, and to 13% and 15% of the variant, relative to the negative control of each cybrid line mediated by a scrambled siRNA (Fig. S5e). These two siRNAs also had the strongest silencing effect on the MRPP1 protein, reducing it to 22% and 36% in whole cell lysates of WT cybrids, respectively, and to 30% and 34% of A3243G cybrids (Fig. S5f). Importantly, using the same set of cells for measurement of MRPP1 mRNA level, we showed by a primer extension analysis that the m1G9 level in mt-Leu(UAA) was reduced to 87% and to 88% in WT treated with siRNA1 and siRNA3 and to 79% and to 80% in the variant (Fig. 5a). The similar effects of the two siRNAs recapitulated their similar effects of targeting, indicating consistency between the two measurements. While modest, these effects were statistically significant. Notably, while 72 h siRNA treatment reduced the m1G9 level further, we avoided this condition as it had a different effect on the WT and variant (73 vs. 42% with siRNA1; 76 vs. 44% with siRNA3, Fig. S5g), which would complicate interpretation of downstream assays. Further, we verified that MRPP1-KD by siRNA targeting also had a global effect on mt-tRNAs. It reduced the m1G9 level of mt-Gln(UUG) and mt-Ile(GAU) (Fig. S5h, i), demonstrating an impact beyond mt-Leu(UAA). It also reduced 5′-processing of the MRPP1-2-3 complex (Fig. S5j). Together, these results validated that siRNA targeting of MRPP1 not only hypomethylated mt-Leu(UAA), but also had an adverse effect on the biogenesis of other mt-tRNAs.

a Left: A primer extension analysis of WT and the A3243G variant of mt-Leu(UAA), using cells validated for MRPP1-KD (Fig. S5e). Neg controls are from cybrids transfected with a scrambled siRNA. The m1G9 level was the fraction of the RT stop at position 10 in the sum of positions 10, 8, and the primer. Right: Relative m1G9 levels to the negative control in bar graphs (blue for WT; red for A3243G). Data are the average ± SD (n = 3). b A Northern analysis of the stability of mt-Leu(UAA), showing size markers superimposed from a stained gel. c Data in (b) in a bar graph, where the steady-state level of mt-Leu(UAA) relative to 5S rRNA was quantified for each and normalized to that of WT in the negative control and d compared to that of the WT in the same targeting condition. Data (c, d) are the average ± SD (n = 3). e Steady-state levels of τm5U34 in a CMC-assisted primer extension analysis of WT and A3243G, showing the fraction of A35 in the sum of A35, τm5U34, U33, and C32 and reported as the level in the CMC (+) lane subtracted from that of the corresponding CMC (−) lane. f Data in (e) in a bar graph, where the τm5U34 level in mt-Leu(UAA) was quantified for each and normalized to that of WT in the negative control and g compared to that of the WT in the same targeting condition. Data (f, g) are the average ± SD (n = 4). h Steady-state levels of charged mt-Leu(UAA) in an acid-urea gel. i Data in (h) in a bar graph, where the steady-state level of mt-Leu(UAA) was quantified for each and normalized to that of WT in the negative control and j compared to that of the WT in the same targeting condition. Data (i, j) are the average ± SD (n = 4). Source data are provided as a Source Data file, and p values were from two-sided paired t test (a, c, d, f, g, i, j).
We next determined whether hypomethylation of mt-Leu(UAA) would improve the activity of the A3243G variant relative to the WT. We first probed the tRNA stability. Total RNA of the WT and A3243G cybrids were isolated and the level of mt-Leu(UAA) in each was quantified and normalized to the 5S rRNA (Fig. 5b). In the negative control with a scrambled siRNA, the stability of the variant was 38% relative to the WT (Fig. 5c), similar to the value reported above (Fig. 2c), whereas upon targeting by siRNA1 and siRNA3, while the stability of the WT decreased from 100% in the control to 65% and 76%, respectively, that of the variant showed no statistically significant change (Fig. 5c and Table S4). However, when we compared the stability of the variant relative to the WT, we observed an increase of the stability of the variant from 38% to 48% and to 45% upon targeting by siRNA1 and siRNA3, respectively, each with statistical significance (Fig. 5d and Table S4).
We next determined whether hypomethylation of m1G9 would increase the level of the τm5U34 modification in the A3243G variant, based on the loss of the modification in the non-targeted state (Fig. 2g). Total RNA of the WT and A3243G cybrids were isolated and the level of τm5U34 in mt-Leu(UAA) of each was probed by the CMC-assisted primer extension assay. While the τm5U34 level of the variant relative to the WT in the negative control was 46% (Fig. 5e, f), similar to that reported above (Fig. 2g), this level changed differentially for the WT and the variant upon targeting. Upon targeting by siRNA1 and siRNA3, while the level of the WT decreased from 100% to 83 and 85%, respectively, that of the variant was unchanged (Fig. 5e, f and Table S5). As a result, hypomethylation increased the τm5U34 level of the variant relative to the WT from 46% to 58% and to 60%, respectively (Fig. 5g and Table S5). Additionally, we evaluated if hypomethylation would increase the aminoacylation level of the variant. Total RNA of WT and A3243G cybrids were isolated in an acid condition to preserve charged aminoacyl-tRNAs (aa-tRNAs) and were then probed for mt-Leu(UAA) in an acid Northern blot analysis. Quantification of the charged level of mt-Leu(UAA) in the negative control showed a decrease in the variant to 83% relative to the WT (Fig. 5h, i), consistent with previous reports35,49. Upon siRNA1 and siRNA3 targeting, while the charged level in the WT decreased from 100% to 78% and 77%, that in the variant was unchanged (Figs. 5h, i and Table S6). As such, hypomethylation increased aminoacylation of the variant relative to the WT from 83% to 108% and to 104%, respectively (Fig. 5j and Table S6). The increases in both τm5U34 and charging in the variant relative to the WT upon targeting suggest an improvement of the variant structure to a state that is better recognized by the respective enzymes.
Hypomethylation of m1G9 in the A3243G variant improves mitochondrial respiration
We then tested whether hypomethylation of m1G9 would improve mitochondrial respiration in the A3243G variant relative to the WT cybrids. Due to the global effect of targeting on all mt-tRNAs, we focused on the early phase of targeting to capture the potential differential effect. We first assessed the effect of targeting by culturing siRNA-treated cells in a medium containing galactose, where oxidation of the non-fermentable sugar to pyruvate was via glycolysis, yielding no net ATP and forcing cells to generate ATP by mitochondrial oxidative phosphorylation. Thus, when cultured in a medium with galactose as the only carbon source, cells with defective mitochondria would not proliferate. We transfected WT and the A3243G cybrids with siRNA1 or siRNA3 in a glucose medium 2 days before the switch to a galactose medium, upon which the proliferation analysis was marked as Day 0 (Fig. 6a). We then monitored cell counts 2 days post-siRNA treatment, where the inhibitory effect is usually most discernible72,73. In WT cybrids, while cells in the negative control continued to grow in the galactose medium for several days, those targeted by siRNA1 or siRNA3 were immediately arrested in growth (Fig. 6b), showing the importance of MRPP1 for cell survival. In A3243G cybrids, by contrast, while cells in the negative control gradually declined in number over time, indicating a mitochondrial deficiency when cultured in galactose, those targeted by siRNA1 or siRNA3 increased in number from Day 0 to Day 1 and then declined (Fig. 6c). Measurement of cell counts relative to WT cybrids in the negative control showed that, while the cell number of WT declined from 100% to 60% and to 58% upon siRNA1 and siRNA3 targeting, respectively, that of the variant increased from 43% to 90% and to 88% upon targeting (Fig. 6d), indicating cell growth. This is remarkable, given that the half-life of the A3243G variant of mt-Leu(UAA) (6 h) is substantially shorter relative to WT (56 h) in a stress-free condition35. Here, in the m1G9-deficiency stress, the temporary gain-of-function of the variant indicates that its improved structure and activity more than enough compensated for its inherently shorter half-life within the one-day period. We confirmed that siRNA1- and siRNA3-targeting was effective even 5 days post-transfection as the MRPP1 protein remained low relative to 2 days post-transfection (Fig. S6a, b). Together, these results showed that hypomethylation of m1G9, while destructive to WT cybrids, was beneficial to A3243G cybrids, enabling growth of the latter in the galactose medium.

a The timeline of the growth curve analysis, showing the start of siRNA treatment 2 days before the switch from glucose to galactose, which was marked as Day 0. b Growth curves in a galactose medium of WT. c Growth curves in a galactose medium of A3243G cybrids in different targeting conditions. The region of interest is boxed in red, and data (b, c) are the average ± SD (n = 3). d Bar graphs showing cell counts (%) relative to WT in the negative control in the region of interest. Each comparison is the average ± SD (n = 3). e Measurement of OCR of cybrids without siRNA targeting, showing an example of the measurement (left), basal OCR of A3243G relative to WT (middle); and ATP-linked respiration of A3243G relative to WT (right). The unit of OCR is pmol/min/μg. Data are the average ± SD (n = 3). f Basal OCR of WT and A3243G upon siRNA1 targeting relative to the respective negative control, showing average ± SD (n = 3). g Basal OCR and ATP-linked respiration upon siRNA1 targeting of cybrids of A3243G relative to the WT in the negative control. h Data in (g) relative to the WT in the same targeting condition, showing average ± SD (n = 3). i Basal OCR of WT and A3243G upon siRNA3 targeting relative to the respective negative control, showing average ± SD (n = 3). j Basal OCR and ATP-linked respiration upon siRNA3 targeting of cybrids of A3243G relative to the WT in the negative control. k Data in (j) relative to the WT in the same targeting condition, showing average ± SD (n = 3). Source data are provided as a Source Data file, and p values were obtained from two-sided paired t test (d, e, g, h, j, k).
We then examined the effect of targeting on mitochondrial respiration. We measured the basal oxygen consumption rate (OCR), followed by addition of oligomycin, an inhibitor of ATP synthase, to measure the ATP-linked respiration. This was followed by addition of the uncoupler FCCP (carbonyl cyanide-p-tri-fluoro-methoxy-phenylhydrazone), which disrupts the mitochondrial membrane potential to reveal the maximal electron transport chain activity. Without targeting, A3243G cybrids had 50% of the basal OCR and 47% of the ATP-linked respiration of the WT (Fig. 6e). Upon siRNA1 targeting, while WT cybrids lowered basal OCR to 85% and the ATP-linked respiration to 87%, the A3243G variant showed no significant change (Fig. 6f, g). When the value of the variant was compared to the WT, the activity of the targeted variant was 63% in OCR and was 62% in ATP-linked respiration relative to the WT, whereas the activity of the untargeted variant was 49% (Fig. 6h), indicating an improvement of the respiration upon targeting. Similarly, with siRNA3 targeting, while WT cybrids lowered basal OCR to 79% and the ATP-linked respiration to 82%, the variant showed no significant change (Fig. 6i, j). When the value of the variant was compared to the WT, the activity of the targeted variant was 69% in OCR and was 65% in ATP-linked respiration relative to the WT, whereas the activity of the untargeted variant was 49% in both (Fig. 6k), indicating an improvement of the respiration upon targeting.
Discussion
Here we address the question of whether m1G9/m1A9, while promoting the stable folding of mt-tRNAs, would have the same, or a different effect, on the folding of pathogenic variants that are associated with mitochondrial pathologies. We use MELAS as the model, focusing on the two most frequent mutations – A3243G and T3271C, both mapped to human mt-Leu(UAA)—to determine the effect of m1G9 in the tRNA. We find that, while m1G9 indeed promotes stable folding of the WT tRNA, it has an opposite effect on the two variants, trapping each in an aberrant structure with reduced activities (Fig. 7). This opposite effect is confirmed in our separate nanopore analysis74, where we developed an ultra-thin nanopore (effective thickness of 1–2.5 nm, diameter of 3 nm) than that used in this study (effective thickness of 7–10 nm, diameter of ~2.7 nm, Fig. 2a). With the ultra-thin nanopore, we measured tRNA translocation through the pore in an electronic condition in which a higher current blockade or a shorter dwell time indicates a more stable structure relative to a less stable structure74. Using the ultra-thin nanopore, we showed that while m1G9 stabilizes the WT transcript, it destabilizes the A3243G variant74, further supporting the notion that the methylation differentially controls the stability of the WT and the variant. Future studies are necessary to elucidate how and why m1G9 destabilizes the variant.

A model for differential contribution of m1G9 to the WT and variants of mt-Leu(UAA), represented by the A3243G and T3271C substitutions (cyan filled circles). While the addition of m1G9 (green filled circle) drives the WT tRNA to a correctly folded structure, it drives the A3243G variant to a mis-folded structure that differs from the correct structure in regions marked by red (closed circle). This model is also supported by the folding kinetics of the T3271C variant, although its structural variation has not been mapped (open circle).
An additional differential effect of m1G9 is shown between the A3243G and T3271C variants (Fig. 4b–e). While the unmethylated-state of the two variants differs from each other in the folding kinetics (e.g., amp1 of the G9-state of A3243G at 47 ± 6% vs. amp1 of the G9-state of T3271C at 35 ± 0.6%, a 1.3-fold difference), the m1G9-state of each amplifies the difference (amp1 of the m1G9-state of A3243G at 19 ± 3% vs. amp1 of the m1G9-state of T3271C at 9.7 ± 0.3%, a 2.0-fold difference) (Fig. 4b, c). This observation indicates that the methylation is an active determinant to accelerate the decline of each variant, even though the decline may proceed in distinct pathways between them.
The differential effect of m1G9 on the WT and the A3243G variant is further supported by analysis of hypomethylation in cell-based assays. In each of these assays, we show that while hypomethylation is damaging to the WT, it is beneficial to the variant. Specifically, while hypomethylation reduces the activity of the WT cybrids, it does not change the activity of the variant, leading to an improvement of the variant relative the WT on a comparative basis (Figs. 5, 6). The lack of a robust positive effect of the variant is due to the competing negative effect of siRNA targeting on all other mt-tRNAs by reducing both the m1G9/m1A9 methylation and the 5′-processing activity (Fig. S5h, i, j). Additionally, it can also be attributed to the compromised state of the A3243G variant, which is under continuous stress related to impairment of mitochondrial translation.
Given the biological complexity of siRNA targeting, we suggest that therapeutics for the A3243G-associated MELAS should focus on only reducing m1G9 in the variant, but not other mt-tRNAs. Also, given the inter-dependence within the MRPP1-2-3 complex, hypomethylation of m1G9 should not affect 5′-processing of the A3243G variant to allow the latter to function on the mt-ribosome. These considerations can be addressed by screening for small molecular inhibitors that target the A3243G variant in a complex with MRPP1-2-3 enzymes. As each variant has a unique sequence and structure, as shown in the distinct kinetic profile of the A3243G vs. T3271C variant, this approach is feasible. Indeed, the cryo-EM structure of the human MRPP1-2-3 complex, bound with a pre-mt-tRNA substrate18, reveals that the subcomplex MRPP1-2 is sufficient to catalyze methylation, which is followed by recruitment of MRPP3 to catalyze 5′-processing. Notably, the recruitment of MRPP3 is using a distinct protein-protein interface of the subcomplex than that for methylation, supporting the notion of separation of 5′-processing from methylation75. Once small-molecular inhibitors are found by screening, they can be optimized based on the cryo-EM structure of the ternary complex with the bound substrate18. Thus, while the approaches of over-expression of mt-LeuRS76,77, or of mt-MTO157, have elevated mitochondrial respiration, these are technically difficult to translate the result from bench to bedside. Instead, small molecular targeting of a tRNA-specific m1G9/m1A9 represents an attractive therapeutic strategy.
The result of this study is likely generalizable to other mt-tRNA-associated diseases. All 22 human mt-tRNAs have been associated with pathogenic mutations, each mapped to a position that would perturb the tRNA structure (MITOMAP). A study of 11 of these mutations, localized in various positions throughout sequences of human mt-Ile(GAU), mt-Met(CAU), and mt-Leu(UAA), showed that all reduced the 5′-processing activity catalyzed by the MRPP1-2-3 complex78. This consistency in the reduction of kinetics indicates that each mutation has altered the respective mt-tRNA to a structure that is compromised in 5′-processing. Additionally, among the pathogenic variants of mt-tRNAs that have been analyzed for post-transcriptional modifications, all have retained the m1G9/m1A9 methylation in high stoichiometry (e.g., A3243G, T3271C, and A8344G)35,79. Combined, these two observations suggest that the destabilizing effect of pathogenic variants is frequently coupled with high stoichiometry of m1G9/m1A9, where the methylation can take an active role to accelerate mis-folding of each variant and sensitize it to degradation, leading to mitochondrial deficiency. We suggest that this accelerated loss of stability is the root cause of mitochondrial pathology that would occur before the loss of aminoacylation or the loss of the τm5U34 modification of the variant.
Methods
Cell culture
WT and cybrid lines with 100% of the A3243G mutation48, were cultured in DMEM (Corning, 10-013-CV) supplemented with 10% FBS (Corning, 35-010-CV), 50 µg/mL uridine and maintained at 37 °C in the presence of 5% CO2. Heteroplasmy series of cybrid lines48, with 0, 20, 30, 50, 60, 90, and 100% of the A3243G mutation were cultured under the same condition. Cells were seeded and grown in a fresh culture medium until the confluency was 75%, in 3–4 days, and were split into a fresh medium.
Preparation of G9- and m1G9-state of tRNA transcripts
Unmodified G9-state of transcript tRNAs, containing the WT sequence, the A3243G mutation, or the T3271C mutation, were prepared by in vitro transcription using in-house T7 RNA polymerase (T7 RNAP). Since the 5′-end of these tRNAs did not contain two consecutive guanosine nucleotides, they were transcribed with an upstream self-cleaving hammerhead ribozyme to generate mature tRNAs with a 5′-hydroxyl group. The m1G9-state of each transcript tRNA was prepared by ligation of a chemically synthesized 18-mer 5′-RNA fragment, encoding nucleotides G1 to C17a, with a 60-mer 3′-RNA fragment prepared by in vitro transcription, encoding nucleotides G18 to A76. By including GMP in the transcription reaction, most of the RNAs contained a 5′-phosphate. Ligation reactions were carried out in the absence of a splint by incubating equimolar amounts of the 18-mer and the 60-mer RNAs with T4 Rnl2 and ATP at 37 °C for 1 h. All transcripts and ligation products were purified by denaturing 12% PAGE.
Nanopore analysis of tRNA folding
All tRNA samples were lyophilized and resuspended in the buffer of 10 mM Tris-HCl, pH 8.0, 1 mM EDTA (TE) to a final concentration of ~200 ng/µL. For each analysis, an aliquot of each tRNA was diluted to 10 ng/µL in 1X TE with 20 mM MgCl2 and incubated at room temperature for ~30 min. Each was then individually run on a nanopore experimental setup, consisting of an insulating SiN membrane (effective thickness of 7–10 nm, diameter of ~2.7 nm) that separated two chambers of electrolyte solution (400 mM KCl in 1X TE). After incubation, samples were individually added to the cis chamber of the setup by replacing the test buffer with sample buffer. A bias voltage of 300 mV was applied across the membrane, driving the negatively charged tRNA molecules in the cis chamber through the nanopore in the membrane and towards the positively charged electrode in the trans chamber. The current through the nanopore was monitored using a high bandwidth current amplifier (Chimera Instruments, New York, NY), which provided a current sampling rate of 4.16 MHz. This current signal was then filtered at 200 kHz and analyzed using custom python data analysis software (https://github.com/rhenley/Pyth-Ion).
The time-series current data was used to analyze the folding efficiency of each tRNA. The transient current spikes resulting from individual molecules of a tRNA translocating the pore were extracted for the fractional current blockade—the ratio of the signal amplitude to the open pore current value. This fractional current blockade number was fit to a simple geometric model for nanopore conductance to extract a rough size value for the molecule translocating through the pore. Additionally, the dwell time (the duration of the signal; defined as the time for the molecule to translocate the pore) was also determined.
Northern blot analysis of a heteroplasmy series of A3243G cybrids
A series of cybrid lines with different levels of heteroplasmy of the A3243G mutation (0, 20, 30, 50, 60, 90, and 100%) were cultured in DMEM with 10% FBS, 50 µg/mL uridine and 5% CO2 at 37 °C until the cell confluency reached 80% in a 10 cm dish. After removal of the media, the cells were lysed on the plate with addition of 1 mL TrizoleTM reagent (Thermo Fisher Scientific, 15596026) and processed according to the manufacturer’s protocol to isolate total RNA from each cell line. Each total RNA sample (4 µg) was mixed with 5 µL of a non-denaturing dye (100 mM Tris-HCl, pH 7.0, 50% glycerol, 0.25% xylene cyanol, and 0.25% bromophenol blue), and loaded onto a 12% non-denaturing PAGE in 1X TBE (90 mM Tris-borate, pH 8.0, 2 mM EDTA) buffer in a mini BioRad apparatus, and run for 30 min at 200 V (at 4 °C, cold room). Quantification of the WT and A3243G variant in each cell line was achieved by Northern blot analysis.
Northern blot gels, either native or denaturing PAGE, were used to quantify the level of mt-Leu(UAA) in each cell sample. Samples for the native PAGE were prepared as above, while those for the denaturing PAGE were prepared with total RNA (4 μg) from WT and 100% A3243G cells, mixed with 5 µL of a denaturing dye (7 M urea, 0.25% xylene cyanol and 0.25% bromophenol blue), pre-heated at 85 °C for 3 min, and run in a 12% PAGE/7 M urea gel in a mini Bio-Rad apparatus in 1X TBE for 30 min at 200 V (room temperature). Each gel, run in the native or denaturing condition, was electroblotted (25 V and 1 mA for 20 min) under semi-dry conditions onto a positively charged Hybond®-N+ hybridization membrane (Millipore Sigma, GERPN203B) using a Trans-Blot® TurboTM Transfer System (Biorad, 1704150). The electroblotted membrane was briefly air dried and the RNA was then crosslinked to the membrane using the option of “optimal crosslink” in a UV crosslinker (Fisher Scientific, FB-UVXL-1000). The crosslinked membrane was incubated in a hybridization buffer (0.9 M NaCl, 90 mM Tris-HCl, pH 7.5, 6 mM EDTA, 0.3% SDS, 1% dry milk) at 37 °C for 1 h. A 5′-32P-oligonucleotide probe (~106 cpm), targeting positions 39-56 of human mt-Leu(UAA) (5ʹ-GAACCTCTGACTGTAAAG-3ʹ), and a separate 5′-32P-oligonucleotide probe (~106 cpm), targeting positions 1-19 of human 5S rRNA (5ʹ-GGGTGGTATGGCCGTAGAC-3ʹ), were added together to the membrane and incubated for 12 h with shaking. Unbound probes were washed off by shaking the membrane twice in 2XSSC buffer (0.3 M NaCl, 30 mM Na-Citrate) for 10–15 min at 37 °C. The membrane was then dried, exposed to an imaging plate overnight, and the tRNA on the membrane was quantified relative to 5S rRNA by a phosphor-imager (Amersham Typhoon model 9500, GE), using ImageJ software v.1.51 (NIH).
Quantification of modifications by primer extension assays
Cells cultured in a 10 cm dish at 80% confluency were lysed on the plate by replacing the media with 1 mL TrizoleTM to isolate total RNA. Primers to monitor and quantify the level of m1G9 (5ʹ-GATTACCGGGCTCTG-3ʹ), m2G10 (5ʹ-GATTACCGGGCTCT-3ʹ) and m1A58 (5ʹ-TGGTGTTAAGAAGAGGA-3ʹ) were synthesized by IDT. Each primer was 5ʹ-labeled with γ-32P-ATP (PerkinElmer) by T4 PNK (polynucleotide kinase, NEB, M0201), purified through a MicroSpinTM G-25 spin column (Cytiva AmershamTM, 27532501), and used at 0.33 pmol to anneal with total RNA from WT (3 µg) and with total RNA (6 µg) of MELAS cells in ddH2O. The annealing mixture was heated at 90 °C, 3 min, and then slowly cooled to room temperature. To monitor the presence of m1G9, Luna WarmStart RT (NEB, M3002) was used for primer extension at 55 °C, 1 h with dCTP (0.5 mM) and ddATP (0.1 mM) in the reaction. To monitor the presence of m2G10, SuperscriptTM III (SSIII) RT (Thermo Fisher Scientific, 18080093) was used for primer extension at 55 °C, 30 min with dGTP (0.5 mM) and ddCTP (0.1 mM) in the reaction. The reaction was terminated by incubation at 70 °C, 15 min. To monitor the presence of m1A58, SSIII RT was used for primer extension at 55 °C, 30 min with dATP (0.5 mM) and ddTTP (0.1 mM) in the reaction. The reaction was terminated by incubation at 70 °C, 15 min. An aliquot of each primer extension reaction (5 µL) was mixed with 5 µL denaturing dye, heated at 85 °C, 5 min, and the products resolved on a 12% denaturing PAGE/7 M Urea in 1X TBE in a 40-cm sequencing gel. The gel was transferred to WhatmanTM filter paper (GE Healthcare Life Sciences, 3030-917), dried, and phosphorimaged after overnight exposure.
Monitoring of τm5U34 was assisted by pre-treating total RNA with CMC (Millipore Sigma, C106402) as described57. Each RNA sample (15 µg/12 µL of TE) was diluted with 24 µL of the TEU buffer (50 mM Tris-HCl, pH 8.3, 4 mM EDTA, 7 M Urea), and mixed with 4 µL of CMC (0.1 M final concentration) freshly dissolved in TEU (the CMC reaction) or with 4 µL of TEU (the mocked reaction). The samples were incubated at 30 °C, 16 h, diluted with 160 µL of a stop buffer (50 mM KOAc, 200 mM KCl, pH 7.0), and ethanol precipitated at –80 °C for at least 1 h. The RNA pellet was washed twice with 500 µL of 70% ethanol to remove excess CMC. The pellet was dissolved in 40 µL of an alkaline buffer (50 mM Na2CO3, 2 mM EDTA, pH 10.4) and incubated at 37 °C, 4 h. Upon dilution to 200 µL with 160 µL of the stop buffer, the RNA was ethanol precipitated as before and dissolved in 10 µL of RNase free water.
The DNA primer (5ʹ-ACCTCTGACTGTAAAG-3ʹ) to detect the CMC-reacted τm5U34 was designed to hybridize to positions 39-54 of the tRNA. It was 5ʹ-labeled with γ-32P-ATP (PerkinElmer), purified through a MicroSpin G-25 column, and used at 0.2 pmol to anneal with the CMC-treated or untreated total RNA (2.5 µg) in 10 µL TE. Annealing was achieved by heating at 80 °C, 2 min, followed by slow cooling to room temperature. AMV RT (NEB, M0277L) was used for primer extension at 42 °C, 1 h, with 1.5 mM each dATP, dTTP, and ddGTP. The reaction was terminated by mixing with denaturing loading dye, heat-cooled at 85 °C, 5 min, and the RT products were separated by denaturing 12% PAGE until bromophenol blue reached the bottom of the gel. The level of RT arrest by CMC-modified taurine was quantified by a phosphor-imager as the band intensity that terminated at position 35 relative to the sum of band intensities at positions 32, 33, 34, and 35. To remove background, the ratio obtained from the mock reaction was subtracted from that of the CMC-treated reaction. Each reported value is the average of three independent replicates.
SHAPE analysis of tRNA structures
SHAPE analysis was based on termination of primer extension at the position of a nucleotide adduct upon interaction with the SHAPE reagent (i.e., NAI). The primer sequence was complementary to tRNA nucleotides 75–56, and each primer extended nucleotide was manually counted according to the band position relative to the primer position in a denaturing PAGE/7 M urea gel. Each termination of primer extension, along with a non-treated control, was then run in parallel to map the position of the termination to the position of the tRNA nucleotide across the sequence framework at a single nucleotide resolution. For analysis of transcripts, 1 µL of 4 µM WT or A3243G variant of mt-Leu(UAA) bearing m1G9 was mixed with 16 µL of RNase-free water and heated at 85–90 °C, 3 min. After adding 2 µL of 10X reaction buffer (200 mM MgCl2, 400 mM HEPES, pH 7.5), the tRNA was allowed to fold for 15 min at room temperature followed at 4 °C, 15 min. The SHAPE reaction was initiated with addition of 1 µL of 1 M 2-methylnicotinic acid imidazolide (NAI) in DMSO (50 mM final concentration) and continued at 4 °C, 15 min. A control reaction was similarly treated with DMSO alone. Both reactions were stopped by adding DTT to 30 mM final concentration. The tRNA was extracted with phenol-chloroform-isoamyl alcohol, pH 5.0, followed by ethanol precipitation. Each pellet was dissolved in 4.5 µL of RNase free water and half was supplemented with 1 µL of a 5ʹ-32P-labeled 21-mer primer (5ʹ-TGGTGTTAAGAAGAGGAATTG-3ʹ; complementary to tRNA positions 56-76) and annealed at 65–70 °C, 2 min, followed by quenching in an ice bath. Primer extension was catalyzed by 50 units of Protoscript II RT (New England Biolabs, M0368) in a 5 µL reaction containing 0.5 mM dNTP, 5 mM DTT, in the 1X Protoscript buffer at room temperature, 5 min, and then 46 °C, 20 min. Reactions were stopped by adding 2.5 µL of denaturing loading dye and heating at 85 °C, 2 min. Aliquots were run through a 12% sequencing gel which was dried and imaged on a phosphor screen after 12–18 h. Bands in the NAI and DMSO lanes were quantified using ImageJ software (NIH). The signal from the DMSO sample was subtracted from the NAI signal to get the value for each NAI reacted band.
For SHAPE analysis of cellular mt-Leu(UAA), WT and A3243G cells grown to 80% confluency were treated with 1 µL of DMSO or 1 µL of 1 M NAI (50 mM final), 15 min. Total RNA was isolated using TrizolTM reagent, 3 µg of which was hybridized to the 5ʹ-32P labeled primer as above. All downstream steps were the same as that used for transcript tRNAs.
Analysis of tRNA stability in mitochondrial lysates
One fresh mouse liver was placed in a beaker to rinse excess blood. Using a razor blade, tissue was thoroughly minced on ice. Minced tissue was transferred to a Potter-Elvehjem homogenization vessel with a Teflon pestle. Homogenization media (30 mL; 250 mM sucrose, 10 mM Tris-HCl, pH 7.4, 0.1 mM EGTA, and 0.5% BSA) was added to the vessel, and the tissue was gently homogenized using 10 passes of a motorized tissue grinder at a speed of 400 rpm while on ice throughout. The homogenized tissue was transferred to a 30 mL plastic centrifuge tube and was spun in an SS-34 rotor at 478 × g at 4 °C, 8 min. The supernatant was transferred to a new tube and was centrifuged at the same speed again. After repeating this step at 7650 × g, the resulting pellet was resuspended in 1 mL of isolation buffer (250 mM sucrose, 10 mM Tris-HCl, pH 7.4, 0.1 mM EGTA), layered with an additional 14 mL of isolation buffer, and spun at 7650 × g at 4 °C, 8 min one more time. The pellet was resuspended in 1 mL of mitochondrial resuspension buffer (MRB) (250 mM mannitol, 5 mM HEPES, pH 7.4, and 0.5 mM EGTA) for further processing.
The crude mitochondrial suspension was layered onto 8 mL of a fresh Percoll medium [255 mM mannitol, 25 mM HEPES, pH 7.4, 1 mM EGTA, 30% Percoll (v/v)] followed by 3 mL of MRB in a 13 mL ultracentrifuge tube (Beckman Instruments, Inc, CA 331372) and spun at 95,000 × g for 45 min in a TH641 rotor. Mitochondria formed a layer near the bottom of the tube which was carefully pipette out and transferred to a new 13 mL ultracentrifuge tube. After 10-fold dilution with MRB the mitochondria were centrifuged at 6300 × g for 10 min. The mitochondrial pellet was again diluted 1:10 with MRB and centrifuged at 6300 × g for 10 min. The purified mitochondrial pellet was resuspended in 200 µL of the mitochondrial lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 0.2% (v/v) Triton X100, 0.3% NP-40, 0.2 mM PMSF, and a protease inhibitor cocktail) and kept on ice for 30 min to allow mitochondria to lyse. The lysed mitochondria were centrifuged at 13,500 × g for 30 min at 4 °C. The mitochondrial lysate supernatant was transferred to an ice-cold 1.5 mL tube and protein concentration was determined by Bradford.
WT and A3243G transcripts with or without m1G9 were used for the in vitro stability assay. These four transcripts were 5ʹ-32P-labeled and purified in a 40 cm sequencing gel. The excised band of each full-length tRNA was extracted overnight in TE, followed by ethanol precipitation. The purified tRNAs had a specific activity of 105 cpm/µL. Each tRNA was incubated with 2 µL of the mitochondrial lysate (50 µg/µL) at 37 °C in a 50 μL reaction containing 5 µL of 5ʹ-32P labeled tRNA (104 cpm/µL) and 20 µL of carrier E. coli tRNA (400 ng/µL) in 30 mM HEPES, pH 7.4, 30 mM NaCl, and 5 mM MgCl2. Aliquots of 5 µL were removed over time into equal volumes of denaturing gel loading dye. After heating at 85 °C, 3–5 min, each reaction was loaded onto a 12% 40 cm sequencing gel and run at 1800 V until bromophenol blue reached the bottom. The dried gel was exposed overnight, read on a phosphor-imager, and analyzed using ImageJ software.
Kinetics of tRNA folding monitored by aminoacylation
Aminoacylation by mt-LeuRS of each transcript tRNA, in the G9-state or the m1G9-state, was performed under single-turnover conditions as described63. Each tRNA was 3′-labeled with 32P by exchange with α-32P ATP in a reaction catalyzed by Bst1 CCA adding enzyme at 60 °C, 30 min. The labeled tRNA was extracted with phenol-chloroform-isoamyl alcohol, spun through a gel filtration cartridge, and ethanol precipitated. Specific activity of the labeled tRNA was 20,000–100,000 cpm/pmole. Prior to aminoacylation, each labeled tRNA (1 pmol) was incubated 20 min at room temperature in 5 µL of 1X reaction buffer (80 mM HEPES, pH 7.0, 5 mM DTT, and 8 mM leucine). Aminoacylation was initiated by adding an equal volume of 2X assay mix (10 µM LeuRS, 20 mM Mg(OAc)2, and 25 mM ATP/Mg in 1X reaction buffer) at room temperature. Aliquots of 1 µL were removed over time into 3 µL of quench solution (50 mM NaCl, 200 mM NaOAc, pH 5.0) and stored at –20 °C until analyzed. Folding as a function of Mg(OAc)2 concentration was investigated by incubating each tRNA for 20 min at room temperature with the indicated concentrations of Mg(OAc)2 in 1X reaction buffer. Aminoacylation was carried out for 3 min at room temperature as described without changing the Mg(OAc)2 concentration. Aminoacylation was determined by treating each quenched aliquot with 6 units of S1 nuclease at 37 °C, 20 min, followed by thin layer chromatography on PEI-cellulose 0.1 M NH4Cl/5% HOAc. The ratio of labeled AMP to leucyl-AMP was determined by phosphor-imaging.
Targeting MRPP1 with siRNAs
Cultured WT and A3243G (100% mutation) lines (60 × 103 cells) were seeded on each well of a 12 well-plate 1 day before transfection to reach a confluency of 40–50% for transfection. The cell number was adjusted based on the plate size to 40–50% at the time of siRNA transfection. The cells were transfected with commercially available, predesigned, chemically synthesized siRNAs targeted to the MRPP1 gene (DharmaconTM, J-020813 09-12) along with a scrambled siRNA (DharmaconTM, D-001810-01) as a negative control at 30 nM final concentration using Lipofectamine RNAiMAX reagent (Invitrogen, 13778150), following the manufacturer’s protocol. After 24 h of transfection, culture media were replaced with fresh DMEM/10% FBS. Cells at 48 h post-transfection were harvested for analysis of the mRNA and protein levels of MRPP1 by qRT-PCR and Western blot, respectively.
The effect of siRNA targeting on the MRPP1 mRNA was evaluated by qRT-PCR analysis. Total RNA was extracted from transfected cells using TRIzolTM according to the manufacturer’s protocol. Genomic DNA was removed from the total RNA by treatment with RQ1 RNase-free DNase (Promega, M6101). cDNA synthesis was carried out with the RevertAid First Strand cDNA synthesis Kit (Thermo Scientific, K1622). qPCR was performed on a QuantStudio-3 qPCR instrument (Thermo Fisher Scientific). For targeting MRPP1, 18 µL of qPCR master mix (10 µL of Power SYBRTM Green PCR Master Mix (Applied BiosystemsTM, 4367659), 7.6 µL of nuclease-free PCR grade water, and 0.4 µL of gene-specific forward and reverse primers (0.2 pmol each)) were combined with 2 µL of the cDNA template into each well of a 96-well reaction plate (MicroAmp® Fast 96-well reaction plate, 0.1 mL, Applied BiosystemsTM, 4344907). qPCR amplification was performed using the following cycling conditions: preincubation 95 °C, 10 min, followed by 45 cycles of 3 steps of amplification with 95 °C (10 s), 48 °C (10 s), and 72 °C (1 min). The transcript level for each gene was determined using a standard curve generated from the control template by plotting the Cq values (y-axis) against the log initial concentration (x-axis). GAPDH was used for normalization between samples. The primer sets used for amplification of MRPP1 and GAPDH were: MRPP1 forward primer: 5ʹ-GAGTACACCCCCTTCTGAAG-3ʹ; MRPP1 reverse primer: 5ʹ-GGGTTTTGAGCTCTTCTTCAG-3ʹ; GAPDH forward primer: 5ʹ- ATGGGCAAGGTGAAGGTG −3ʹ; GAPDH reverse primer: 5ʹ-GTACACCATGTAGTTCAGGTC-3ʹ.
The effect of siRNA targeting on protein expression of MRPP1 was analyzed by Western blot analysis. Transfected cells were washed with ice cold PBS three times and lysed on the plate by adding 1 mL of NP-40 lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40; supplemented with Protease inhibitor (Thermo ScientificTM, A32963)) on an ice bath, 30 min. Supernatant was collected from lysed cells by spinning at 16,000 × g, 20 min, and samples were quantified by Bradford assay (Bio Rad, 5000006) for even gel loading. For MRPP1 and GAPDH detection, 20 µg of protein was run on a 12% SDS-PAGE, and then transferred to a PVDF membrane (Millipore, IPVH00010) by semi-dry blotting using the Trans-Blot® TurboTM Transfer System (BioRad, 1704150). After 1 h blocking with 5% skim milk in TBST (1X TBS, 0.1% Tween-20), the membrane was incubated overnight with anti-MRPP1 antibody (Atlas Antibodies, HPA036671) at 1:1000 dilution or anti-GAPDH antibody (Novus Biologicals, NB615) at 1:2500 dilution at 4 °C with gentle agitation. After three 10 min washes with 1X TBST, the membrane was incubated with anti-rabbit secondary antibody (Cell Signaling, 7074) at 1:2000 dilution for the MRPP1 membrane and anti-mouse secondary antibody (Cell Signaling, 7076S) at 1:5000 dilution for the GAPDH membrane for 1 h at room temperature in 1X TBST, followed by three washes of 10 min each. The membrane was incubated with SuperSignalTM West Pico PLUS chemiluminescent substrate (Thermo Scientific, 34580), 1–2 min, and the signal was detected using the ChemiDocTM MP Imaging System (Bio-Rad, 12003154). Band intensity was quantified using ImageJ software.
Analysis of the 5′-processing activity of the MRPP1-2-3 complex
The 5′-processing activity of the MRPP1-2-3 complex was measured on a precursor form of human mt-Leu(UAA) as described80. The substrate tRNA was prepared by an assembly approach in which the 5ʹ-fragment with a leader sequence (69-mer, G − 10 to A57) of mt-Leu(UAA) was generated by in vitro transcription, while the 3ʹ-fragment (54-mer, A58 to A76) labeled with FAM was synthesized by Integrated DNA Technologies (IDT) (Fig. S5j). The two fragments were ligated by T4 Rnl2 according to the base-pairing property to form the acceptor stem of mt-Leu(UAA)81. To detect the 5′-processing activity of the MRPP1-2-3 complex upon siRNA targeting, a mt-lysate was used prepared from 108 cells of the WT cybrids 48 h post-transfected with siRNA1 as the source of the processing complex. The mt-lysate (1.67 mg/mL) was incubated with the precursor form of the mt-Leu(UAA) at room temperature as described80. As a positive control, a purified MRPP1-2-3 enzyme complex was reconstituted with the molar ratio of 1:2:1 of the three enzymes. The reaction was carried out with 70 nM of the substrate tRNA, 2.28 U/μL of the RNase inhibitor (NEB, M0314L), and 1X reaction buffer (50 mM Tris-HCl, pH 8.3, 50 mM NaCl, 2.5 mM MgCl2 and 1 mM DTT). As a negative control, the substrate tRNA was treated with the reaction buffer without the mt-lysate but containing the RNase inhibitor. For each reaction, an aliquot was collected and quenched at 2 min and 5 min in the denaturing dye containing 9 M urea, 40% formamide, and 0.2% SDS. The 5ʹ-processed product was separated from the precursor tRNA on a 12% PAGE/7 M Urea gel. The fractional conversion of the substrate to the 5ʹ-processed product was quantified by imageJ (NIH) by measuring the processed band over the sum of both the processed and unprocessed bands.
Growth curve analysis in a galactose medium
A 48 well-plate was seeded with 0.5 × 104 cells per well in 200 µL of glucose media. The cells were transfected with MRPP1 siRNA1 and siRNA3 along with a scrambled siRNA as a negative control (final siRNA concentration 30 nM) using Lipofectamine RNAiMAX reagent (Invitrogen, 13778-075). Galactose media were prepared by using DMEM without glucose (Gibco, 11966-025) but supplemented with 10 mM galactose, 2 mM glutamine, 5 mM HEPES, 1 mM sodium pyruvate, and 10% FBS. Cells were washed with PBS after 48 h of transfection and media were changed to galactose media. The cells in each well were counted after trypsinization every 24 h for 3–5 days unless mentioned otherwise.
Acid-Urea PAGE analysis of tRNA aminoacylation levels in cybrids
Total RNA was extracted from cells in an acidic condition (pH 4.5) to maintain aa-tRNA levels82. Cells harvested from a 10 cm culture dish (1 × 106 cells) were mixed at a 1:1 volume ratio with 10% TCA and incubated on ice, 10 min. Cells were spun down at 7600 × g at 4 °C, 5 min, resuspended in an extraction buffer (0.3 M NaOAc, pH 4.5, 10 mM EDTA), and mixed with an equal volume of ice-cold phenol-chloroform-isoamyl alcohol (83:17:3.4), pH 5.0. The mixture was vortexed for 5 cycles of 1 min vortex and 1 min rest on ice and spun at 13,500 × g at 4 °C, 10 min. An equal volume of cold isopropanol was added to the aqueous phase which was incubated at −20 °C, 30 min for precipitation. The nucleic acid pellet was collected after centrifugation at 13,500 × g at 4 °C, 20 min, washed with 70% ethanol in 30% 10 mM NaOAc, pH 4.5, dried, and dissolved in gel-loading buffer (10 mM NaOAc, pH 4.5,1 mM EDTA). Nucleic acid from WT cells (2.5 µg) and from MELAS cells (5 µg) were mixed with 2 volumes of an acid-urea gel loading buffer (0.1 M NaOAc, pH 5.0, 9 M urea, 0.05% bromophenol blue (BPB), and 0.05% xylene cyanol (XC)). An intermediate sized acid-urea gel (14 × 17 × 0.5 mm) was used to resolve charged from uncharged mt-Leu(UAA). Each acid-urea gel was made with 6.5% PAGE/7 M urea in 0.1 M NaOAc, pH 5.0, and run at 250 V for 3 h 45 min at 4 °C. Following electrophoresis, the region between BPB and XC was excised and washed in 1X TBE. Nucleic acid was transferred from the gel to a wetted nitrocellulose membrane in 1X TBE and quantified by Northern blot analysis using the probe described above. The charged fraction was calculated as the area encompassing the charged band divided by the sum of charged and uncharged bands.
Measurement of the oxygen consumption rate and the ATP-linked respiration rate
The oxygen consumption rate in cybrid cell lines was detected using an XFe24 extracellular flux analyzer (Seahorse Bioscience) as described57 with some modifications. Cells that were transfected with MRPP1 siRNAs along with the scrambled siRNA as a negative control were trypsinized and counted 24 h post-transfection. Cells (104 counts) were seeded in 4 wells of a XFe24 cell culture miniplate precoated with collagen (Seahorse Biosciences) and cultured for an additional 24 h to further knockdown MRPP1. After 24 h, the culture medium was replaced with freshly resuspended DMEM (Sigma-Aldrich, D5030), pH 7.4, containing 25 mM glucose and 4 mM L-glutamine. The rates of oxygen consumption were measured under basal conditions and over the course of programmed injections of oligomycin (Sigma-Aldrich, O4876) (final concentration 5 µg/µL), carbonyl cyanide-p-tri-fluoro-methoxy-phenylhydrazone (FCCP) (Sigma-Aldrich, C2920) (final concentration 500 nM), and antimycin A (Sigma-Aldrich, A8674) (final concentration 10 µM), which is an antibiotic that specifically inhibits metabolic reduction of cytochrome C. Basal respiration was determined as the OCR before oligomycin, subtracted from the minimum OCR after the antibiotic treatment. The ATP-linked respiration rate was determined as the OCR before oligomycin subtracted from the minimum OCR after oligomycin. After completion of measurements, the cells were lysed with the RIPA buffer (Sigma-Aldrich, R0278) and protein concentration was measured by Bradford assay to normalize the cell number variation in each well.
Quantification and statistical analysis
Each experiment was performed with at least 3 biological replicates (n ≥ 3), as described in each Figure legend. The statistical significance of biochemical and cell-based assays was calculated by the two-sided paired t test.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are available from the corresponding authors upon request. Source data for the figures and supplementary figures are provided as a Source Data file. Source data are provided with this paper.
References
Nabholz, B., Glemin, S. & Galtier, N. Strong variations of mitochondrial mutation rate across mammals–the longevity hypothesis. Mol. Biol. Evol. 25, 120–130 (2008).
Nabholz, B., Glemin, S. & Galtier, N. The erratic mitochondrial clock: variations of mutation rate, not population size, affect mtDNA diversity across birds and mammals. BMC Evol. Biol. 9, 54 (2009).
Pesole, G., Gissi, C., De Chirico, A. & Saccone, C. Nucleotide substitution rate of mammalian mitochondrial genomes. J. Mol. Evol. 48, 427–434 (1999).
Lavrov, D. V. Key transitions in animal evolution: a mitochondrial DNA perspective. Integr. Comp. Biol. 47, 734–743 (2007).
Lott, M. T. et al. mtDNA variation and analysis using mitomap and mitomaster. Curr. Protoc. Bioinform. 44, 1 23 21–26 (2013).
Ruiz-Pesini, E. et al. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 35, D823–D828 (2007).
Suzuki, T., Nagao, A. & Suzuki, T. Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu. Rev. Genet. 45, 299–329 (2011).
Rackham, O. & Filipovska, A. Organization and expression of the mammalian mitochondrial genome. Nat. Rev. Genet. 23, 606–623 (2022).
Hallberg, B. M. & Larsson, N. G. Making proteins in the powerhouse. Cell Metab. 20, 226–240 (2014).
Anderson, S. et al. Sequence and organization of the human mitochondrial genome. Nature 290, 457–465 (1981).
Salinas-Giege, T., Giege, R. & Giege, P. tRNA biology in mitochondria. Int. J. Mol. Sci. 16, 4518–4559 (2015).
Helm, M. et al. Search for characteristic structural features of mammalian mitochondrial tRNAs. RNA 6, 1356–1379 (2000).
Bohnsack, M. T. & Sloan, K. E. The mitochondrial epitranscriptome: the roles of RNA modifications in mitochondrial translation and human disease. Cell Mol. Life Sci. 75, 241–260 (2018).
Chujo, T. & Tomizawa, K. Human transfer RNA modopathies: diseases caused by aberrations in transfer RNA modifications. FEBS J. 288, 7096–7122 (2021).
Suzuki, T. et al. Complete chemical structures of human mitochondrial tRNAs. Nat. Commun. 11, 4269 (2020).
Holzmann, J. et al. RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 135, 462–474 (2008).
Vilardo, E. et al. A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase-extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res. 40, 11583–11593 (2012).
Bhatta, A., Dienemann, C., Cramer, P. & Hillen, H. S. Structural basis of RNA processing by human mitochondrial RNase P. Nat. Struct. Mol. Biol. 28, 713–723 (2021).
Reinhard, L., Sridhara, S. & Hallberg, B. M. Structure of the nuclease subunit of human mitochondrial RNase P. Nucleic Acids Res. 43, 5664–5672 (2015).
Reinhard, L., Sridhara, S. & Hallberg, B. M. The MRPP1/MRPP2 complex is a tRNA-maturation platform in human mitochondria. Nucleic Acids Res. 45, 12469–12480 (2017).
Helm, M. et al. The presence of modified nucleotides is required for cloverleaf folding of a human mitochondrial tRNA. Nucleic Acids Res. 26, 1636–1643 (1998).
Helm, M., Giege, R. & Florentz, C. A Watson-Crick base-pair-disrupting methyl group (m1A9) is sufficient for cloverleaf folding of human mitochondrial tRNALys. Biochemistry 38, 13338–13346 (1999).
Voigts-Hoffmann, F. et al. A methyl group controls conformational equilibrium in human mitochondrial tRNA(Lys). J. Am. Chem. Soc. 129, 13382–13383 (2007).
Richter, U., McFarland, R., Taylor, R. W. & Pickett, S. J. The molecular pathology of pathogenic mitochondrial tRNA variants. FEBS Lett. 595, 1003–1024 (2021).
Kelley, S. O., Steinberg, S. V. & Schimmel, P. Functional defects of pathogenic human mitochondrial tRNAs related to structural fragility. Nat. Struct. Biol. 7, 862–865 (2000).
Sohm, B. et al. Towards understanding human mitochondrial leucine aminoacylation identity. J. Mol. Biol. 328, 995–1010 (2003).
Sohm, B., Sissler, M., Park, H., King, M. P. & Florentz, C. Recognition of human mitochondrial tRNALeu(UUR) by its cognate leucyl-tRNA synthetase. J. Mol. Biol. 339, 17–29 (2004).
Giege, R., Sissler, M. & Florentz, C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 26, 5017–5035 (1998).
Bhaskaran, H., Taniguchi, T., Suzuki, T., Suzuki, T. & Perona, J. J. Structural dynamics of a mitochondrial tRNA possessing weak thermodynamic stability. Biochemistry 53, 1456–1465 (2014).
Pavlakis, S. G., Phillips, P. C., DiMauro, S., De Vivo, D. C. & Rowland, L. P. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Ann. Neurol. 16, 481–488 (1984).
Goto, Y., Nonaka, I. & Horai, S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 348, 651–653 (1990).
Kobayashi, Y. et al. A point mutation in the mitochondrial tRNA(Leu)(UUR) gene in MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes). Biochem. Biophys. Res. Commun. 173, 816–822 (1990).
Gorman, G. S. et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 77, 753–759 (2015).
Goto, Y., Nonaka, I. & Horai, S. A new mtDNA mutation associated with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS). Biochim. Biophys. Acta 1097, 238–240 (1991).
Yasukawa, T., Suzuki, T., Ueda, T., Ohta, S. & Watanabe, K. Modification defect at anticodon wobble nucleotide of mitochondrial tRNAs(Leu)(UUR) with pathogenic mutations of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. J. Biol. Chem. 275, 4251–4257 (2000).
Henley, R. Y. et al. Electrophoretic deformation of individual transfer RNA molecules reveals their identity. Nano Lett. 16, 138–144 (2016).
Bandarkar, P., Yang, H., Henley, R. Y., Wanunu, M. & Whitford, P. C. How nanopore translocation experiments can measure RNA unfolding. Biophys. J. 118, 1612–1620 (2020).
Henley, R. Y., Carson, S. & Wanunu, M. Studies of RNA sequence and structure using nanopores. Prog. Mol. Biol. Transl. Sci. 139, 73–99 (2016).
Ivankin, A. et al. Label-free optical detection of biomolecular translocation through nanopore arrays. ACS Nano 8, 10774–10781 (2014).
Suzuki, T. & Suzuki, T. A complete landscape of post-transcriptional modifications in mammalian mitochondrial tRNAs. Nucleic Acids Res. 42, 7346–7357 (2014).
Wilkins, H. M., Carl, S. M. & Swerdlow, R. H. Cytoplasmic hybrid (cybrid) cell lines as a practical model for mitochondriopathies. Redox Biol. 2, 619–631 (2014).
King, M. P. & Attardi, G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246, 500–503 (1989).
King, M. P., Koga, Y., Davidson, M. & Schon, E. A. Defects in mitochondrial protein synthesis and respiratory chain activity segregate with the tRNA(Leu(UUR)) mutation associated with mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes. Mol. Cell Biol. 12, 480–490 (1992).
Chomyn, A. et al. MELAS mutation in mtDNA binding site for transcription termination factor causes defects in protein synthesis and in respiration but no change in levels of upstream and downstream mature transcripts. Proc. Natl. Acad. Sci. USA 89, 4221–4225 (1992).
Chomyn, A. et al. In vitro genetic transfer of protein synthesis and respiration defects to mitochondrial DNA-less cells with myopathy-patient mitochondria. Mol. Cell Biol. 11, 2236–2244 (1991).
Hayashi, J. et al. Introduction of disease-related mitochondrial DNA deletions into HeLa cells lacking mitochondrial DNA results in mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 88, 10614–10618 (1991).
Hayashi, J. et al. Functional and morphological abnormalities of mitochondria in human cells containing mitochondrial DNA with pathogenic point mutations in tRNA genes. J. Biol. Chem. 269, 19060–19066 (1994).
Picard, M. et al. Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc. Natl. Acad. Sci. USA 111, E4033–E4042 (2014).
Park, H., Davidson, E. & King, M. P. The pathogenic A3243G mutation in human mitochondrial tRNALeu(UUR) decreases the efficiency of aminoacylation. Biochemistry 42, 958–964 (2003).
Kurata, S. et al. Modified uridines with C5-methylene substituents at the first position of the tRNA anticodon stabilize U.G wobble pairing during decoding. J. Biol. Chem. 283, 18801–18811 (2008).
Morscher, R. J. et al. Mitochondrial translation requires folate-dependent tRNA methylation. Nature 554, 128–132 (2018).
Dunbar, D. R., Moonie, P. A., Zeviani, M. & Holt, I. J. Complex I deficiency is associated with 3243G:C mitochondrial DNA in osteosarcoma cell cybrids. Hum. Mol. Genet 5, 123–129 (1996).
Hayashi, J. et al. Accumulation of mtDNA with a mutation at position 3271 in tRNA(Leu)(UUR) gene introduced from a MELAS patient to HeLa cells lacking mtDNA results in progressive inhibition of mitochondrial respiratory function. Biochem. Biophys. Res. Commun. 197, 1049–1055 (1993).
Chomyn, A., Enriquez, J. A., Micol, V., Fernandez-Silva, P. & Attardi, G. The mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episode syndrome-associated human mitochondrial tRNALeu(UUR) mutation causes aminoacylation deficiency and concomitant reduced association of mRNA with ribosomes. J. Biol. Chem. 275, 19198–19209 (2000).
Kirino, Y. et al. Codon-specific translational defect caused by a wobble modification deficiency in mutant tRNA from a human mitochondrial disease. Proc. Natl. Acad. Sci. USA 101, 15070–15075 (2004).
Kirino, Y., Goto, Y., Campos, Y., Arenas, J. & Suzuki, T. Specific correlation between the wobble modification deficiency in mutant tRNAs and the clinical features of a human mitochondrial disease. Proc. Natl. Acad. Sci. USA 102, 7127–7132 (2005).
Tomoda, E. et al. Restoration of mitochondrial function through activation of hypomodified tRNAs with pathogenic mutations associated with mitochondrial diseases. Nucleic Acids Res. 51, 7563–7579 (2023).
Ueda, S. et al. Mitochondrial haplotype mutation alleviates respiratory defect of MELAS by restoring taurine modification in tRNA with 3243A > G mutation. Nucleic Acids Res. 51, 7480–7495 (2023).
Merino, E. J., Wilkinson, K. A., Coughlan, J. L. & Weeks, K. M. RNA structure analysis at single nucleotide resolution by selective 2’-hydroxyl acylation and primer extension (SHAPE). J. Am. Chem. Soc. 127, 4223–4231 (2005).
Wilkinson, K. A., Merino, E. J. & Weeks, K. M. Selective 2’-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc. 1, 1610–1616 (2006).
Busan, S., Weidmann, C. A., Sengupta, A. & Weeks, K. M. Guidelines for SHAPE reagent choice and detection strategy for RNA structure probing studies. Biochemistry 58, 2655–2664 (2019).
Asano, K. et al. Metabolic and chemical regulation of tRNA modification associated with taurine deficiency and human disease. Nucleic Acids Res. 46, 1565–1583 (2018).
Bhaskaran, H., Rodriguez-Hernandez, A. & Perona, J. J. Kinetics of tRNA folding monitored by aminoacylation. RNA 18, 569–580 (2012).
Zhang, C. M., Perona, J. J., Ryu, K., Francklyn, C. & Hou, Y. M. Distinct kinetic mechanisms of the two classes of Aminoacyl-tRNA synthetases. J. Mol. Biol. 361, 300–311 (2006).
Uter, N. T. & Perona, J. J. Long-range intramolecular signaling in a tRNA synthetase complex revealed by pre-steady-state kinetics. Proc. Natl. Acad. Sci. USA 101, 14396–14401 (2004).
Guth, E., Connolly, S. H., Bovee, M. & Francklyn, C. S. A substrate-assisted concerted mechanism for aminoacylation by a class II aminoacyl-tRNA synthetase. Biochemistry 44, 3785–3794 (2005).
Guth, E. C. & Francklyn, C. S. Kinetic discrimination of tRNA identity by the conserved motif 2 loop of a class II aminoacyl-tRNA synthetase. Mol. Cell 25, 531–542 (2007).
Francklyn, C. S., First, E. A., Perona, J. J. & Hou, Y. M. Methods for kinetic and thermodynamic analysis of aminoacyl-tRNA synthetases. Methods 44, 100–118 (2008).
Brooks, K. M. & Hampel, K. J. A rate-limiting conformational step in the catalytic pathway of the glmS ribozyme. Biochemistry 48, 5669–5678 (2009).
Sen, A. et al. Loss of the mitochondrial protein-only ribonuclease P complex causes aberrant tRNA processing and lethality in Drosophila. Nucleic Acids Res. 44, 6409–6422 (2016).
Rackham, O. et al. Hierarchical RNA processing is required for mitochondrial ribosome assembly. Cell Rep. 16, 1874–1890 (2016).
Qu, Y. et al. Time is a critical factor when evaluating oligonucleotide therapeutics in hERG assays. Nucleic Acid Ther. 33, 132–140 (2023).
Han, H. RNA interference to knock down gene expression. Methods Mol. Biol. 1706, 293–302 (2018).
Namani, S. et al. Unraveling RNA conformation dynamics in mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episode syndrome with solid-state nanopores. ACS Nano 18, 17240–17250 (2024).
Vilardo, E., Toth, U., Hazisllari, E., Hartmann, R. K. & Rossmanith, W. Cleavage kinetics of human mitochondrial RNase P and contribution of its non-nuclease subunits. Nucleic Acids Res. 51, 10536–10550 (2023).
Park, H., Davidson, E. & King, M. P. Overexpressed mitochondrial leucyl-tRNA synthetase suppresses the A3243G mutation in the mitochondrial tRNA(Leu(UUR)) gene. RNA 14, 2407–2416 (2008).
Li, R. & Guan, M. X. Human mitochondrial leucyl-tRNA synthetase corrects mitochondrial dysfunctions due to the tRNALeu(UUR) A3243G mutation, associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like symptoms and diabetes. Mol. Cell Biol. 30, 2147–2154 (2010).
Karasik, A., Wilhelm, C. A., Fierke, C. A. & Koutmos, M. Disease-associated mutations in mitochondrial precursor tRNAs affect binding, m1R9 methylation, and tRNA processing by mtRNase P. RNA 27, 420–432 (2021).
Richter, U. et al. RNA modification landscape of the human mitochondrial tRNA(Lys) regulates protein synthesis. Nat. Commun. 9, 3966 (2018).
Falk, M. J. et al. A novel HSD17B10 mutation impairing the activities of the mitochondrial RNase P complex causes X-linked intractable epilepsy and neurodevelopmental regression. RNA Biol. 13, 477–485 (2016).
Dupasquier, M., Kim, S., Halkidis, K., Gamper, H. & Hou, Y. M. tRNA integrity is a prerequisite for rapid CCA addition: implication for quality control. J. Mol. Biol. 379, 579–588 (2008).
Masuda, I. et al. Loss of N(1)-methylation of G37 in tRNA induces ribosome stalling and reprograms gene expression. Elife 10, e70619 (2021).
Acknowledgements
We thank Piotr Kopinski of the Wallace lab for sharing the heteroplasmy series of A3243G cybrids, Megumi Shigematsu for contributing to the early phase of the project, and all members of the Hou lab for discussion. This work was supported by National Institutes of Health grants R35 GM134931 to Y.M.H., R35 GM149336 to J.A.P., R01 GM123771 to E.S., CA259635 and AG078814 to D.C.W., and by Japan Society for the Promotion of Science (JSPS 18H05272) and Exploratory Research for Advanced Technology (ERATO, JPMJER 2002) from the Japan Science and Technology Agency (JST) to T.S.
Author information
Authors and Affiliations
Contributions
S.M. designed and performed experiments on most of the tRNA-based assays in vitro and in vivo, including those for analysis of primer extension, stability, acid-urea gels, Northern blots, and Western blots with and without targeting by siRNAs, while H.G. and T.C. designed and performed experiments on aminoacylation-based folding assays and on processing assays. S.M. and Y.Y. designed and performed experiments on cell growth assays in galactose. R.Y.H. and M.W. designed and performed experiments on nanopore assays, N.S.L and J.A.P. synthesized m1G9-containing RNA oligonucleotides, T.S. isolated the bovine liver mt-Leu(UAA), and E.S. guided measurements of mitochondrial respiration assays. M.W., T.S., E.S., and D.C.W. provided discussion, while Y.M.H. wrote the manuscript. All authors approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Maharjan, S., Gamper, H., Yamaki, Y. et al. Post-transcriptional methylation of mitochondrial-tRNA differentially contributes to mitochondrial pathology. Nat Commun 15, 9008 (2024). https://doi.org/10.1038/s41467-024-53318-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-53318-x
