
Abstract
TNF inhibitors have been used to treat autoimmune and autoinflammatory diseases. Here we report an unexpected mechanism underlying the therapeutic effects of TNF inhibitors in Behçet’s disease (BD), an autoimmune inflammatory disorder. Using serum metabolomics and peripheral immunocyte transcriptomics, we find that polymorphonuclear neutrophil (PMN) from patients with BD (BD-PMN) has dysregulated mevalonate pathway and subsequently increased farnesyl pyrophosphate (FPP) levels. Mechanistically, FPP induces TRPM2-calcium signaling for neutrophil extracellular trap (NET) and proinflammatory cytokine productions, leading to vascular endothelial inflammation and damage. TNF, but not IL-1β, IL-6, IL-18, or IFN-γ, upregulates TRPM2 expression on BD-PMN, while TNF inhibitors have opposite effects. Results from mice with PMN-specific FPP synthetase or TRPM2 deficiency show reduced experimental vasculitis. Meanwhile, analyses of public datasets correlate increased TRPM2 expressions with the clinical benefits of TNF inhibitors. Our results thus implicate FPP-TRPM2-TNF/NETs feedback loops for inflammation aggravation, and novel insights for TNF inhibitor therapies on BD.
Similar content being viewed by others


Introduction
Different from the well-characterized damage-associated molecular pattern (DAMP), metabolic intermediates, have recently been identified as a novel class of endogenous danger signals that strongly elicit a variety of biological functions1,2,3,4. In response to external stimulations, polymorphonuclear neutrophil (PMN) tend to adopt specific metabolic pathways5, for a tuned purpose to support specialized effector functions such as neutrophil extracellular trap (NET) formation (NETosis)6,7. Hence, considering the short cell lifespan8, PMN is a potentially important source of serum danger signals during the shift of its metabolic patterns9. However, the immunometabolism of PMN in autoimmune and autoinflammatory diseases remains to be investigated.
Corresponding to the clinical features of autoinflammatory and autoimmune diseases, Behçet’s disease (BD) is a chronic systemic vasculitis characterized by recurrent oral or genital ulcers, skin lesions, and involvement of vital organs, such as ocular, cardiovascular, gastrointestinal, and neurological manifestations10. BD is sight-threatening and even life-threatening, imposing considerable financial burdens on society and individuals. BD is a representative type of immune disorder as it exhibits aberrant and excessive activation of both innate and adaptive immunity, which is thus considered as a unique and crucial clinical condition linking both autoimmunity and autoinflammation11. Although the etiology of BD is still unknown, it is well accepted that during the progression of BD, an interplay between endogenous danger signals and PMN is of note12,13,14. In this regard, elevated serum DAMPs such as high mobility group box 1 (HMGB1)15 and S100 calcium-binding protein A12 (S100A12)16 have been reported in BD. These DAMPs promote acute inflammation and recruitment of PMN to vascular lesions17, and thus BD was also recognized as a “neutrophilic vasculitis”18. Moreover, PMN is particularly prone to undergo cell necrosis, mainly in the form of NETosis under inflammatory conditions19,20, which involves the release of various DAMPs, including DNA, histones19, HMGB1, and S100A8, etc., aggravating the activation and inflammation of macrophages19 and vascular endothelium20,21 in BD. As an integrating mechanism, all these accounts for the production of proinflammatory cytokines, including tumor necrosis factor (TNF), interferon-γ (IFN-γ), interleukin 6 (IL-6), and skewed T-helper (Th) 1 and Th17 cell activation22. Thus, BD is ideal for use as a representative disease to investigate the mechanism of action underlying the therapeutic effects of TNF inhibitors in treating autoimmune and autoinflammatory diseases. Farnesyl pyrophosphate (FPP), a key metabolite in the mevalonate (MVA) pathway, plays a crucial role in cholesterol biosynthesis, has been implicated in inflammatory responses, and potentially contributed to autoinflammatory and autoimmune diseases23,24. However, its implications in BD pathogenesis remain largely to be elucidated.
Here in this study, we conduct comprehensive analyses of serum metabolomics and peripheral immune cell transcriptomics, revealing a dysregulated mevalonate pathway in PMN from BD patients (BD-PMN) and subsequently elevated FPP levels. Further investigations demonstrated that FPP promotes BD-PMN hyperactivation via a calcium-TRPM2-dependent pathway. TNF upregulates TRPM2 expression on BD-PMN, while TNF inhibitors have the opposite effect. Our findings highlight the potential pathogenic involvements of FPP in BD and uncover the immunometabolic mechanisms underlying disease progression. These insights provide novel therapeutic implications for TNF inhibitors in BD and potentially other autoimmune and autoinflammatory disorders.
Results
Multi-omic analyses highlight the proinflammatory contributions of MVA-pathway in BD-PMN hyperactivation
To gain an unbiased understanding of immunometabolic profiles in BD, we first integrated multi-omic analyses in our previously published cohorts containing BD patients and the sex- and age-matched HC, including serum metabolomics and lipidomics25, bulk RNA-sequencing of peripheral blood mononuclear cell (PBMC) (GSE19853311), PMN (GSE20586712), and single-cell RNA-sequencing of PBMC (GSE19861611). We found that the steroid biosynthesis pathway is significantly upregulated in BD serum in comparison to that from HC (Fig. 1A, Supplementary Fig. 1A). Multi-omic analyses of individual immune cell populations revealed that this metabolic alteration in serum was mainly displayed in the BD-PMN (Fig. 1B, and Supplementary Fig. 1B). Considering that there are multiple downstream pathways in steroid synthesis, we then conducted an enrichment analysis of each pathway. We found that the cholesterol biosynthesis pathway, but not the synthesis of bile acid and steroid hormone pathways, was significantly increased in BD-PMN in comparison to HC-PMN (Fig. 1C). It shall be noted that although our single-cell sequencing analyses also indicated a slight upregulation of the steroid synthesis pathway in monocytes from BD patients, their cholesterol synthesis pathway was not significantly upregulated as in the case of PMN (Supplementary Figs. 1C, D).

A Lipidomic analysis between treatment-naïve BD and HC serum. B GSEA of steroid biosynthesis pathway in BD and HC peripheral blood immune cells, including bulk RNA-seq of PBMC (GSE198533), PMN (GSE205867), and single-cell RNA-seq of PBMC (GSE198616). C GSEA of steroid synthesis sub-pathways, including bile acids, steroid hormones, vitamin D, and cholesterol synthesis, in BD and HC PMN (GSE205867). D Heatmap showing enzyme expression of cholesterol synthesis pathway in BD PMN RNA-seq (GSE205867). Significantly increased and decreased genes are marked in red and blue, respectively. E Schematic plot showing the intervention of the cholesterol synthesis pathway. Significantly elevated enzymes validated by both BD PMN RNA-seq and qRT-PCR (N = 20) are marked in red. The agonists and inhibitors are marked by green and purple arrows, respectively. N for all experiments were biological replicates. ZGA, zaragozic acid; SQS, squalene synthase.
Next, we examined the expression profiles of crucial enzymes in the cholesterol biosynthesis pathway from transcriptional datasets of BD-PMN and HC-PMN (Fig. 1D), and further validated the transcription levels of these target genes by qRT-PCR in an independent cohort of twenty BD patients (Supplementary Fig. 1E, and Supplementary Table 1). We found that those most differentially expressed enzymes, including ACAT1, MVK, PMVK, and MVD, were concentrated in the upstream pathway of cholesterol biosynthesis, i.e., the MVA pathway (Fig. 1E). These results reveal an upregulated MVA pathway specifically in BD-PMN.
To determine whether the upregulated MVA pathway is involved in the hyperactivation of BD-PMN, we inhibited 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) and farnesyl pyrophosphate synthase (FPPS, also known as farnesyl diphosphate synthase (FDPS)), which control the start and end points of the MVA pathway, via the clinically-available drugs simvastatin and zoledronic acid, respectively (Fig. 1E). Notably, both inhibitors suppressed proinflammatory cytokines in BD-PMN (Fig. 2A, B). Since FPP is the end product of the MVA pathway that is converted by FPPS enzyme from geranyl-pyrophosphate (GPP), we speculated that FPP might be the primary component participating in the MVA pathway-induced BD-PMN hyperactivation. To confirm this hypothesis, we either inhibited FPP-metabolizing enzymes farnesyl-diphosphate farnesyltransferase (FDFT1) (squalene synthase), via zaragozic acid (ZGA) and BPH-652, or activated FDFT1 function via ferroptosis inducing 56 (FIN56)26. The results demonstrated that intracellular accumulation of FPP increased the production of proinflammatory cytokines in BD-PMN (Fig. 2C, D), and vice versa (Fig. 2E), both of which suggested a proinflammatory role of FPP in PMN. Notably, none of the aforementioned inhibitors or agonists had any effect on PMN viability (Supplementary Figs. 2A, B). To conclude, these results suggested a proinflammatory role of the MVA pathway, especially its metabolite FPP, in PMN activation and inflammation.

A–E BD-PMN was pretreated with either simvastatin (1 μM and 10 μM, A) to inhibit HMGCR, zoledronic acid (1 μM and 10 μM, B) to inhibit FDPS, ZGA (7.5 μM and 15 μM, (C) and BPH652 (40 μM and 80 μΜ, (D) to inhibit squalene synthase FDFT1, or FIN56 (1 μM and 5 μM, E) to activate FDFT1, for 16 hours. The expression levels of TNF, IL-6, IL-18, and IL-1β were detected by qRT-PCR (N = 4, for each group). F Relative abundance of FPP in PMN (N = 24) and serum (N = 23) from active BD and HC. G, H Relative abundance of FPP in PMN (N = 15, G) and serum (N = 14, H) from active and remission BD. I, J Correlations between the relative abundance of FPP in BD-PMN (N = 39, (I) and serum (N = 37, J) with BD clinical activity indicators, CRP and ESR. K, L Relative abundance of FPP in PMN (N = 32 vs 7, (K) and serum (N = 23 vs 14, L) of BD with different BDCAF. N for all experiments were biological replicates. Data are presented as mean ± SD; error bars indicate the SD. A two-sided p-value < 0.05 was considered statistically significant, with *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 indicating significant differences. Source data are provided as a Source Data file. One-way ANOVA tests were used in (A–E), with p-value adjusted for multiple comparisons by FDR using the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli. Independent t-tests were used in (F, K, L), Pearson correlation analysis was used in I, J and paired t-tests were used in (G, H). ZGA, zaragozic acid; SQS, squalene synthase; ESR, erythrocyte sedimentation rate; CRP, C-reactive protein; BDCAF, Behçet’s Disease Current Activity Form.
FPP levels were significantly higher in BD than in HC and correlated with BD disease activity
PMN is particularly prone to undergo cell necrosis, mainly in the form of NETosis under inflammatory conditions19,20, which release DAMPs contributing to BD15,16,17,19. We further investigated the abundance of FPP in both serum and total PMN lysis samples by targeted liquid chromatography-mass spectrometry (LC-MS). The relative levels of FPP were elevated in both types of samples from active BD patients compared to those from HC (Fig. 2F). Notably, our longitudinal follow-up data showed a remarkable decrease in both PMN and serum FPP levels after BD patients achieved remission (Fig. 2G, H). More importantly, we performed clinical evaluations and reviewed the medical records of all participants (Table 1). The results showed that FPP levels in BD-PMN were positively correlated with C-reactive protein (CRP), and serum FPP levels in BD were positively correlated with CRP and erythrocyte sedimentation rate (ESR), indicators of BD disease activity (Fig. 2I, J). In addition, PMN and serum FPP levels were markedly higher in patients with a Behçet’s Disease Current Activity Form (BDCAF) greater than 2 compared with those with a BDCAF of 0 to 1 (Fig. 2K, L). We further analyzed receiver-operating characteristic (ROC) curves of serum FPP levels to investigate its diagnostic value and found that serum FPP level could potentially differentiate BD from HC, with an area under the ROC curve (AUC) of 0.7467 (p value = 0.0041, cutoff = 1.400, sensitivity% = 65.22%, specificity% = 82.61%) (Supplementary Fig. 3A). Moreover, we assessed the disease severity of treatment-naïve BD patients according to the well-established BD Disease Severity Score reported by Krause27,28,29. The median disease severity score was 3 (range 2-6) for the BD-PMN cohort and 5 (range 2-7) for the BD serum cohort. Patients with a Disease Severity Score of less than or equal to 2 were characterized as mild BD patients, while those with a score greater than 2 were defined as moderate-severe BD patients. Of note, serum FPP levels were significantly higher in moderate-severe BD than in mild BD (Supplementary Fig. 3B). Remarkably, the serum levels of FPP were notably higher in BD patients with extracardiac vascular involvement (including deep vein thrombosis and aneurysms) than in those with cardiac involvement (Supplementary Fig. 3C). A possible explanation may be that inflammatory indicators are particularly elevated in BD patients with extracardiac vascular involvement. All these results drove us to examine the DAMPs-mediated direct effects of PMN inflammation and necrosis upon extracellular FPP stimulation in vitro.
Enhanced responsiveness of BD-PMN to extracellular FPP promotes PMN hyperactivation and vascular endothelial inflammation and damage
We chemically synthesized FPP (method reported before24), and investigated the expression of proinflammatory cytokines upon stimulation by FPP in PMN, lymphocytes, and monocytes from both active BD patients and HC. Notably, FPP significantly induced the production of proinflammatory cytokines, including TNF, IL-6, IL-18, and IL-1β, in PMN but not in lymphocytes or monocytes, and BD-PMN exhibited a greater response than HC-PMN (Fig. 3A, D). FPP at concentrations of 5 to 10 µg/mL stimulated the production of proinflammatory cytokines in PMN in a dosage-dependent manner (Fig. 3E), with no effect on their viability (Supplementary Fig. 4A, B). When increasing the concentrations, we found that 60 μg/mL FPP induced the death of PMN, monocytes, and lymphocytes, but more cell death and a lower median lethal concentration (LC50) of FPP were only observed in PMN from BD patients compared to HC (Supplementary Fig. 4C), especially in the form of NETosis (Fig. 3F), again with stronger responsiveness in BD-PMN (Fig. 3G, H, and Supplementary Fig. 4D). Our published paper demonstrated a positive correlation between BD serum NETs-derived dsDNA (NETs-dsDNA) levels and CRP19. To further investigate the involvement of FPP levels and PMN activity in the progression of BD, we measured NETs-dsDNA levels in the serum cohort and found a markedly increased NETs-dsDNA in active BD compared to HC (Fig. 3I). Furthermore, our longitudinal follow-up data demonstrated a notable decrease in serum NETs-dsDNA levels in remission BD after appropriate treatment (Fig. 3J). Of noted, the supernatants of BD-PMN stimulated by 60 μg/mL FPP induced a greater inflammation of vascular endothelial cell (VEC) than that of HC, as demonstrated by the upregulation of proinflammatory cytokines and adhesion molecules (Fig. 3K). Immunofluorescence assays showed that FPP did not lead to the mortality of VECs (Supplementary Fig. 5A). Meanwhile, neither FPP alone nor supernatants of unstimulated PMN contributed to the inflammation of VECs (Supplementary Fig. 5B, C). Taken together, FPP specifically triggers PMN hyperactivation and VEC inflammation, with enhanced responsiveness of BD-PMN.

A–D The expression levels of proinflammatory cytokines in 10 μg/mL FPP-stimulated PMN (red), lymphocytes (dark blue), and monocytes (light blue) including TNF, IL-6, IL-18 and IL-1β (N = 5). E Quantitative analysis of proinflammatory cytokines in BD-PMN, after 0, 5, and 10 µg/mL FPP stimulation (N = 4). F Levels of NETs-derived dsDNA (NETs-dsDNA) in the supernatants of FPP-stimulated PMN from BD and HC (N = 5). G, H Representative immune-fluorescence staining (G) and quantification (H) N = 20) of 60 μg/mL FPP-induced NETosis, immunostaining of MPO (red), NE (green) and 4′,6-diamidino-2-phenylindole (DAPI) counterstained nuclei (blue); scale bar = 10 μm. I NETs-dsDNA levels in the serum of active BD and HC (N = 15). J NETs-dsDNA levels in the serum of active BD and remission BD (N = 14) K. The vascular endothelial cell line HMVEC was stimulated with supernatants of 60 μg/mL FPP-pretreated BD and HC PMN, respectively, and the expression level of proinflammatory cytokines, and adhesion-related molecules, including E-selectin, P-selectin and ICAM-1, were assessed (N = 5). N for all experiments were biological replicates. Data are presented as mean ± SD; error bars indicate the SD. A two-sided p-value < 0.05 was considered statistically significant, with *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 indicating significant differences. Source data are provided as a Source Data file. Paired t-tests were used in (J). Independent t-test was used in (I). Two-way ANOVA tests were used in (A–F, H and K), with the p-value adjusted for multiple comparisons by FDR using the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli. NS, normal saline; DAPI, 4’,6-diamidino-2-phenylindole; PMN, polymorphonuclear neutrophil; MPO, myeloperoxidase; NE, neutrophil elastase; dsDNA, double-stranded DNA; ICAM-1, intercellular cell adhesion molecule-1.
Calcium and TRP channels are essential for FPP-induced PMN activation
FPP is known to trigger calcium influx by interacting with transient receptor potential (TRP) channels24,30, which leads to gene transcription and cellular immune responses31. Thus, as critical controls, we examined the calcium influx in PMN by either removing extracellular calcium or blocking the TRP channels with a universal inhibitor, ruthenium red (RR), in the presence of calcium (method reported before32). We found that both of these conditions inhibited the FPP-induced calcium influx (Supplementary Fig. 6A), the production of cytokines (Fig. 4A), and NETosis of PMN (Fig. 4B, D, and Supplementary Fig. 6B), strongly suggesting that calcium and TRP channels are essential for FPP to promote PMN hyperactivation. As a further validation, RR was found to significantly attenuate the effect of FPP on NETosis-induced VEC activation (Fig. 4E). We also confirmed that NETs were essential components in the supernatants of FPP-stimulated PMN to induce VEC inflammation, as fully digesting the NETs-dsDNA via deoxyribonuclease I (DNase I) resulted in significantly decreased expression of proinflammatory cytokines and adhesion molecules in VECs (Fig. 4C, E). Additionally, we demonstrated that TRP channels mediated the enhanced response of BD-PMN to FPP stimulation, as RR more effectively suppressed the activation of BD-PMN in comparison to that of HC-PMN (Fig. 4F, G). Further experiments by flow cytometry revealed a stronger calcium influx of BD-PMN than that of HC-PMN in response to FPP stimulation (Fig. 4H). Together, all these suggest that the mechanism of FPP-induced PMN activation depends on the influx of calcium ions through TRP channels.

A, B The statistical results of proinflammatory cytokines (N = 4, (A) and NETs-dsDNA (N = 6, (B) in FPP-stimulated BD and HC PMN were detected by qRT-PCR and pico-green dsDNA fluorescent probe, respectively, in the presence or absence of RR and calcium. C, D Representative immunofluorescence staining (C) of 60 μg/mL FPP-induced NETosis together with RR or DNase I, as well as its quantification (D) N = 15); scale bar = 10 μm. E Quantitative analysis of RR and DNase I affecting the expression of proinflammatory cytokines and adhesion molecules in VECs (N = 5). F, G Inhibitory efficiency of RR on FPP-induced proinflammatory cytokine expression (N = 4) and NETs-dsDNA release (N = 6) in PMN of BD and HC. H 60 μg/mL FPP- and calcium-induced calcium fluxes in the PMN of BD and HC (N = 3). N for all experiments were biological replicates. Data are presented as mean ± SD; error bars indicate the SD. A two-sided p-value < 0.05 was considered statistically significant, with *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 indicating significant differences. Source data are provided as a Source Data file. Independent t-tests were used in (G). One-way ANOVA tests were used in (A, B and D), and two-way ANOVA tests were used in (F), with the p-value adjusted for multiple comparisons by FDR using the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli. DNase I, Deoxyribonuclease I; RR, Ruthenium Red.
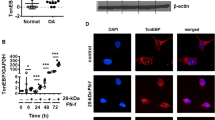
TNF-TRPM2 axis elicits the FPP-induced inflammatory response in PMN
To elucidate the mechanism underlying the hyper-responsiveness nature of BD-PMN in response to FPP stimulation, we conducted a high-throughput transcriptional analysis of RR-sensitive calcium channels including TRP channel, ryanodine receptors, cation channels sperm associated, two pore segment channels and MCU within in our RNA-sequencing database of BD patients. Remarkably elevated expressions of four genes including TRPM2, TRPC1, RYR1, and MCOLN2 were identified (Fig. 5A). Among these, TRPC1 and RYR1 were expressed at extremely low levels in PMN (Supplementary Table 2), and the regulation of cation flux and topology of MCOLN channels was mainly dependent on PH, rather than calcium33. In addition, the other two MCOLN family members, MCOLN1 and MCOLN3, have been reported to be less sensitive to RR34. Consequently, we speculated that the increased FPP responsiveness in BD-PMN was mainly attributed to TRPM2, which was further validated by qRT-PCR (Fig. 5B) and western blot (Fig. 5C). Notably, TRPM2 expression levels were significantly higher in active BD patients than those in remission (Fig. 5D), suggesting a potential involvement of TRPM2 in BD progression. Importantly, silencing TRPM2 on BD-PMN significantly reduced the FPP-induced hyperactivation of BD-PMN, as evidenced by decreased production of proinflammatory cytokines (Fig. 5E) and NETs (Fig. 5F–H, and Supplementary Fig. 6C), reducing the inflammation of VECs (Fig. 5I). As a critical control, the silencing efficiency of TRPM2-specific siRNA was verified at both the transcript (Supplementary Fig. 6D) and protein levels (Supplementary Fig. 6E).

A Differential expression analysis of TRP channels in BD and HC PMN transcriptome sequencing data (GSE205867). Channels with significantly elevated (p < 0.05 and log2FC > 0.5) and decreased expression (p < 0.05 and log2FC > 0.5) are marked in red and blue, respectively. B The quantification of TRPM2 expression on PMN from BD and HC verified by qRT-PCR (N = 9) C Representative and summary of Western blot (WB) analysis of the TRPM2 expression in PMN from BD and HC (N = 6). D The mRNA expression levels of TRPM2 in active BD patients and those in remission (N = 14). E, F The expression of TRPM2 was silenced by TRPM2-specific siRNA, and the levels of proinflammatory cytokines (N = 6, E and NETs-dsDNA (N = 5, F) in FPP-stimulated BD-PMN were analyzed respectively. A scramble siRNA was used as a negative control. G, H Representative immunofluorescence staining diagrams (G) and quantification (N = 15, H) of 60 μg/mL FPP-stimulated PMN after TRPM2-specific siRNA or si-scramble treatment, scale bars =10 µm. I The VECs were stimulated with supernatants from 60 μg/mL FPP-pretreated PMN, after TRPM2-specific or scramble siRNA transduction. The transcriptional expression levels of the above-mentioned proinflammatory cytokines and adhesion molecules in VECs were detected by qRT-PCR (N = 6). N for all experiments were biological replicates. Data are presented as mean ± SD; error bars indicate the SD. A two-sided p-value < 0.05 was considered statistically significant, with *p < 0.05, **p < 0.01, and ***p < 0.001, indicating significant differences. Source data are provided as a Source Data file. Independent t-tests were used in B–D and F, H. Two-way ANOVA tests were used in E and I, with the p-value adjusted for multiple comparisons by FDR using the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli.
To gain further insights into the molecular pathways of FPP-induced PMN activation, we employed a series of inhibitors at graded concentrations to inhibit the downstream molecules activated by second messenger calcium ions, including PKC inhibition with staurosporine (STS), NF-kB inhibition with PDTC, Pyk2 inhibition with PF-5662271, ERK inhibition with PD98059, cPLA2 inhibition with tanshinone I, and calcineurin inhibition with cyclosporin A, respectively (Supplementary Fig. 7A-7F). Notably, only STS effectively reduces FPP-mediated PMN activation, suggesting a PKC-dependent molecular mechanism. Additionally, the critical involvement of PKC molecules was further confirmed by other PKC inhibitors, including VTX27, ruboxistaurin, and Go 6983 (Supplementary Fig. 7G). Thus, all these results indicate that FPP plays a crucial role in the opening of the TRPM2 channel, leading to the facilitated influx of calcium. Consequently, calcium ions act as pivotal second messengers, ultimately resulting in the production of proinflammatory cytokines through the activation of PKC signaling pathway.
Lastly, we investigated the mechanism driving the upregulated TRPM2 levels in BD-PMN than HC-PMN, and in active BD patients than in those in remission. It is interesting to observe that HC-PMN cultured with active BD serum displayed significantly higher TRPM2 expression levels compared to HC serum (Fig. 6A). To identify the serum factors responsible for enhanced TRPM2 levels in BD-PMN, we examined the main proinflammatory cytokines and found elevated TNF, IL-6, and IFN-γ in BD serum than those in HC serum (Fig. 6B). These investigations revealed that only TNF significantly increased TRPM2 expression on PMN (Fig. 6C), but not the other cytokines including IL-6, IL-18, IL-1β, or IFN-γ (Supplementary Figs. 8A, E). Notably, the effective reduction of TRPM2 expression by TNF-neutralizing antibodies further corroborated our conclusion (Fig. 6D). To investigate whether TNF can enhance the TRPM2 expression in PMN in vivo, we examined the expression of TRPM2 in PMN from patients with BD treated with TNF inhibitors (including adalimumab and infliximab). We found that TRPM2 expression was significantly reduced in BD-PMN after a median of 4 months (range 3-6) of TNF inhibitors treatment in vivo (Fig. 6E). In addition, we performed long-term follow-up of these patients upon the treatment of TNF inhibitors with a median term of 10 months (range 7-24), of which 3 patients were lost to follow-up. Consistently, the expression of TRPM2 in BD-PMN was significantly lower at the last visit than at baseline, although no significant difference was observed compared to the level at the mid-term follow-up (Supplementary Fig. 8F). Furthermore, in vitro experiments showed that TNF blockade in BD serum successfully reduced FPP-induced PMN activation (Fig. 6F, G). Therefore, the treatment with TNF inhibitors can reduce FPP-induced PMN activation by downregulating the expression of TRPM2 in BD-PMN.

A The expression levels of TRPM2 in HC-PMN were assayed by qRT-PCR, after incubation with a medium prepared from the serum of active BD and HC (N = 20). B ELISA of proinflammatory cytokines, including TNF, IL-6, and IFN-γ in the serum from HC and BD (N = 16). C The expression levels of TRPM2 in HC-PMN after incubation with medium prepared from 50 ng/mL TNF, IL-6, and IFN-γ (N = 7). D The mRNA expression levels of TRPM2 in HC-PMN incubated before and after TNF-neutralizing antibody addition to active BD serum (N = 20). E The mRNA expression levels of TRPM2 in PMN from BD patients before and after treatment with TNF inhibitors (N = 7). F–G The quantification of proinflammatory cytokines (N = 8, (F) and NETs-dsDNA (N = 8, G in FPP-stimulated BD-PMN after preincubation with BD serum in the presence or absence of TNF-neutralizing antibody. N for all experiments were biological replicates. Data are presented as mean ± SD; error bars indicate the SD. A two-sided p-value < 0.05 was considered statistically significant, with *p < 0.05, **p < 0.01, and ***p < 0.001, indicating significant differences. Independent t-tests were used in (A, D, and G). Paired t-tests were used in E. Source data are provided as a Source Data file. One-way ANOVA tests were used in (B and C) and two-way ANOVA tests were used in F, with the p-value adjusted for multiple comparisons by FDR using the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli.
To explore the generalizability of our findings, we comprehensively analyzed neutrophil RNA-seq or microarray data from publicly available databases for a variety of autoimmune and autoinflammatory diseases, including systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), inflammatory bowel disease (IBD), adult-onset Still’s disease (AOSD), and antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV). These analyses were performed with a focus on the cholesterol synthesis pathway and TRPM2 expression levels. Notably, TRPM2 expression was significantly elevated in patients with IBD, AOSD, AAV, and RA (Supplementary Fig. 9A). In these types of diseases, TNF inhibitors have been widely recommended by guidelines or have potential clinical benefits35,36,37,38,39,40,41,42. In marked contrast, there was no upregulation of TRPM2 expression in patients with SLE (Supplementary Fig. 9A), in the case of which TNF inhibitors are contraindicated for clinical usage43,44,45. Additionally, a remarkable upregulation of the cholesterol synthesis pathway was noted only in IBD patients (Supplementary Fig. 9B). Further analysis of the key enzymes of the cholesterol synthesis pathway in IBD patients revealed that both upstream and downstream enzymes of FPP were elevated (Supplementary Fig. 9C). Notably, PMN hyperactivation is implicated in the pathogenesis of RA and IBD, and TNF inhibitors are guideline-recommended biologics commonly used to treat patients with refractory/severe RA and IBD. We found that serum from patients with active RA and IBD also promoted TRPM2 expression in the HC-PMN (Supplementary Fig. 10A), and further enhanced the response to FPP in the HC-PMN, as evidenced by increased production of proinflammatory cytokines and NETs (Supplementary Fig. 10B, C). Furthermore, the blockade of TNF in RA and IBD sera effectively reduced TRPM2 expression in PMN (Supplementary Figs. 10D–E), thereby decreasing its response to FPP (Supplementary Figs. 10F–I). The above results indicate that the TNF-TRPM2 axis is also potentially implicated in the pathogenesis of other autoimmune and autoinflammatory diseases, thereby further extending the generality of our findings.
The FPP-TRPM2-PMN axis is involved in the mouse model of vasculitis and experimental autoimmune uveitis
To further confirm the critical role of FPP in vascular inflammation and damage, we induced vasculitis in myeloid cell-specific FPP synthetase (FPPS) knockout (FPPSflox/flox LysMcre) mice and FPPSflox/flox mice (control group), according to the previously reported method46. Notably, FPPSflox/flox LysMcre mice demonstrated significantly reduced vascular inflammation and damage compared with control mice, as evidenced by remarkably decreased neutrophil infiltration and vascular permeability of Evans-blue (Fig. 7A–D). In addition, a significantly lower proportion of MPO-positive cells were noted in the skin tissues of FPPSflox/flox LysMcre mice than in control mice, as assayed by both immunohistochemistry and immunofluorescence (Fig. 7E–H). Meanwhile, TRPM2 total knockout (TRPM2-KO) mice also demonstrated significantly reduced vascular damage compared with wild-type control mice, evidenced by reduced Evans-blue permeability (Fig. 7I–K). All these layers of experiments revealed the critical involvement of FPP-TRPM2-PMN-axis in inducing vasculitis.

A Representative images of Evans-blue permeation on the dorsal skin of FPPSflox/flox LysMcre mice and FPPSflox/flox mice taken after induction of vasculitis. Rabbit serum was used as a background control. B Representative microphotographs of paraffin-embedded skin sections from FPPSflox/flox LysMcre mice and FPPSflox/flox mice with H&E staining. Blue arrows indicate neutrophils, and orange arrows indicate erythrocyte extravasation. Scale bar = 25 μm. C Quantification of Evans-blue penetration area in dorsal skin was standardized by the rabbit serum group, after vasculitis induction in FPPSflox/flox LysMcre mice and FPPSflox/flox mice (N = 6, 8 weeks old, 3 female and 3 male). D The number of perivascular neutrophils in HE-stained skin sections observed under a 40x objective microscope (N = 6). E, F Representative and summary of immunohistochemistry staining of MPO in skin tissues of FPPSflox/flox LysMcre mice and FPPSflox/flox mice with induced vasculitis (N = 6). Scare bar = 50 μm. G, H Representative and summary of immunofluorescence staining of MPO and Ly6G in skin tissues of FPPSflox/flox LysMcre mice and FPPSflox/flox mice with induced vasculitis (N = 6). Scare bar = 50 μm. I–K Representative and summary of Evans-blue permeation on the dorsal skin of TRPM2 total knockout mice and control mice taken after induction of vasculitis (N = 4, 8 weeks old, 2 female and 2 male). Quantification of Evans-blue was obtained with Image J (J) and spectrophotometer (K), respectively. N for all experiments were biological replicates. Data are presented as mean ± SD; error bars indicate the SD. A two-sided p-value < 0.05 was considered statistically significant. Source data are provided as a Source Data file. *, p < 0.05. Independent t-tests were used in (C, D, F, H, and J, K).
In addition, after a combined consideration of the clinical features of BD, the phenotypic features of available animal models, and the required experimental conditions, we chose a widely accepted experimental autoimmune uveitis (EAU) model to investigate the involvement of FPP in BD pathogenesis. The EAU model is utilized because ocular inflammation, as characterized by uveitis, is reported to occur in 50–70% of BD patients47. There are also multiple reports confirming the feasibility of inducing EAU in C57BL/6 J mice48,49,50. Of note, PMN hyperactivity has been implicated in EAU, as evidenced by excessive production of NETs51. Based on the aforementioned thoughts and literature studies, we thus induced EAU in either FPPSflox/flox LysMcre or FPPSflox/flox control mice using interphotoreceptor retinoid-binding protein (IRBP) peptide 1-20 according to the previously reported methods48,49,50. Induction of uveitis was assessed by ocular computerized tomography (OCT) and fundoscopy on day 14 (Supplementary Fig. 11A), and a lower incidence of EAU was found in FPPSflox/flox LysMcre mice compared to the control mice (FPPSflox/flox LysMcre mice vs FPPSflox/flox mice: 25% vs. 66.67%). Shedding of CD62L is considered a marker of PMN activation52,53,54. On day 18, peripheral blood and eyes were collected from each type of mice, and a reduced percentage of CD11b+ Ly6G+ cells and higher CD62L expression were observed in the peripheral blood of FPPSflox/flox LysMcre mice compared with FPPSflox/flox mice (Supplementary Fig. 11B, C). Histological assessments were also performed according to previously established criteria55. We found a reduction of PMN infiltration of ocular tissues and a lower pathological score in FPPSflox/flox LysMcre mice compared to the control FPPSflox/flox mice (Supplementary Fig. 11D). These results provide additional support for the involvement of FPP in BD pathogenesis in disease model studies.
Overall, our data suggested the previously unrecognized mechanism by which TNF makes PMN more responsive to FPP by upregulating TRPM2 expression, which, together with the pro-TNF- and NETs-producing effects of FPP, causes positive proinflammatory feedback loops in BD (Fig. 8). This mechanism may also contribute to other autoimmune and autoinflammatory diseases.

Aberrantly elevated metabolite FPP in BD patients promotes PMN hyperactivation via a calcium-TRPM2-dependent pathway, evidenced by the excessive production of proinflammatory cytokines, including TNF, and NETs, which exacerbates vascular endothelium inflammation and damage. In addition, excessive levels of TNF in BD serum triggered the upregulation of TRPM2, which further induced hypersensitivity to FPP in BD-PMN. All these led to significantly increased serum levels of NETs and proinflammatory cytokines, including TNF, ultimately resulting in proinflammatory positive feedback loops in BD. Created in BioRender. Ji, Z. (2024) BioRender.com/d22k224.
Discussion
Hyperactivation and infiltration of PMN are the predominant contributors to recurrent episodes of acute inflammation in BD14,20. Through integrated analyses, we revealed that the MVA pathway metabolite FPP promoted BD-PMN hyperactivation in a calcium-TRPM2-dependent manner, ultimately exacerbating vascular endothelial inflammation. This report also highlighted that the excessive levels of TNF, but not IL-6, IL-18, IL-1β, or IFN-γ, in BD serum triggered the upregulation of TRPM2, which further induced hypersensitivity to FPP in BD-PMN. All these led to significantly increased serum levels of NETs and proinflammatory cytokines, including TNF, ultimately resulting in proinflammatory positive feedback loops. Thus, our findings uncover the involvement of FPP in PMN hyperactivation and suggest that targeting FPP could be a potential strategy for treating BD.
The MVA pathway plays a pivotal role in immune homeostasis, aberrant of which contributes to the pathogenesis of autoimmune and autoinflammatory diseases56,57. Several studies have demonstrated that statins, inhibitors of HMGCR, a key enzyme in the initiation of the MVA pathway, are effective in alleviating the clinical progression of patients with rheumatic diseases, including RA58,59,60, SLE61, Kawasaki’s disease62, and BD62,63. In addition, simvastatin has been demonstrated to reduce neutrophil inflammation and NETosis in a mouse model of severe asthma, thereby alleviating lung inflammation and airway hyper-responsiveness64. As none of the BD patients in our current cohort had a history of hyperlipidemia or statin usage, which made it challenging to find sufficient subjects for further studies. Moreover, metabolites of the MVA pathway have been identified to enhance trained immunity in monocytes and macrophages65, function as a potent antigen for γδT cells, increasing the secretion of proinflammatory cytokines, such as TNF66, and as a vital checkpoint to maintain T-reg functional and lineage stability57. Consequently, suppressive metabolites of the MVA pathway are considered pivotal for the anti-inflammatory, anti-oxidant, and vascular repair properties of stains67. Here, by integrated analysis of serum metabolism and the peripheral immune cell transcriptome, our study is the first to highlight the proinflammatory contributions of the MVA pathway in BD. Interestingly, endogenous accumulation of FPP, an end product of the MVA pathway, mediated by ZGA treatment induces NRF2-mediated oxidative stress responses in keratinocytes68. Excessive extracellular FPP has been implicated in promoting epithelial eotaxin-3 production69, and even triggering cell necrosis as DAMP24. By inhibiting a series of key enzymes, our study proposed for the first time that FPP is a key metabolite in the MVA pathway that contributes to the activation of BD-PMN, and confirmed the promotional effect of FPP on vascular and ocular inflammation and damage using FPPSflox/flox LysMcre mice.
Mechanistically, it has been proposed that FPP can regulate c-fos-directed DNA binding by reducing phosphorylation events of p38 and ERK, which ultimately affects Th1 cell differentiation70. However, the mechanism by which FPP exerts danger signals is to activate the TRPM2 and TRPV3 channels, whereas the latter is mainly expressed in keratinocytes, not immune cells24,30. Although TRPM2-mediated calcium influx has been reported to be responsible for 250 μM H2O2-induced chemokine production in monocytes71, neither monocytes nor lymphocytes were observed to respond to 5 to 10 µg/mL FPP stimulation in our study. Additionally, the response of monocytes and lymphocytes to FPP-induced cell death was comparable in BD and HC. However, BD-PMN responded significantly higher to FPP than HC, suggesting that PMN, but not monocytes in the peripheral blood, is the primary cell in which FPP contributes to the pathogenesis of BD. Our study is the first to propose a TRPM2-calcium-dependent proinflammation mechanism of action of FPP in PMN. Notably, FPP specifically promoted PMN to produce more proinflammatory cytokines such as TNF, which in turn upregulated PMN responsiveness to FPP. Moreover, FPP at the site of inflammation accelerated NETosis, leading to further release of FPP, and exacerbating the proinflammatory microenvironment in BD. The TRPM2-dependent effect on vascular inflammation and damage was also confirmed using TRPM2-KO mice. Taken together, the generation of two types of proinflammatory feedback centering on FPP highlights the theoretical rationale for developing metabolically-targeted drugs for BD but also complements the therapeutic mechanism of TNF inhibitors on BD from an immunometabolism perspective.
Our study has some limitations. First, we only focused on the effects of FPP on immune cells in peripheral blood, and further study of BD lesions would improve our understanding of the role of FPP in BD pathogenesis. Second, due to the lack of specific antibodies against FPP, we merely measured FPP levels in BD serum and PMN using targeted LC-MS. Specific antibodies are worth developing in the future for better quantification and localization of FPP in BD lesions, and for exploring the pathogenesis of BD. Third, this study provides initial insights into the involvement of FPP with BD disease activity and severity. Further multi-center, larger long-term follow-up studies will be valuable in clarifying the significance of FPP in BD pathogenesis.
In summary, our study revealed the proinflammatory implications of FPP in fueling PMN hyperactivation, thus broadening the understanding of PMN hyperactivation-associated diseases from an immunometabolism perspective. It also highlights a novel therapeutic mechanism of TNF inhibitors in BD and potentially other autoimmune and autoinflammatory diseases.
Methods
Patient enrollment
Participants were recruited from the Peking Union Medical College Hospital (PUMCH) between November 2020 and February 2021. To perform the qRT-PCR validation of differentially expressed MVA pathway key enzymes analyzed by GSEA, 20 BD and 20 HC participants were recruited between November 2020 and September 2022 from PUMCH. Furthermore, a cohort of active BD patients was recruited between February 2021 and October 2023 and followed up until April 2024. A total of 39 BD-PMN samples and 37 BD sera were collected for targeted mass spectrometry analysis of FPP. These included 24 active BD-PMN samples and 15 remission BD-PMN samples. Additionally, 23 active BD serum samples and 14 remission BD serum samples were collected. All BD patients met the International Criteria for BD (ICBD), and their disease activity was assessed using ESR, CRP, and the BD Current Activity Form 2006 (BDCAF 2006)72. IBD serum samples were collected from SRRSH IBD Biobank in China (SRRSH-IBC).
The PUMCH Ethical Committee approved this study (I-23PJ1123; I-24PJ0600), and all subjects provided written informed consent.
PBMC and PMN isolation
Blood samples were collected from active BD patients and age- and gender-matched HCs with no personal or family history of autoimmune diseases for mass spectrum analysis. PMN, monocytes, and lymphocytes were isolated using Ficoll-Hypaque density gradient centrifugation according to the manufacturer’s instructions at 500 x g for 20 minutes at 24 °C. The PMN layer was harvested and red blood cell (RBC) lysis was performed using red blood cell lysis buffer (RBC lysis buffer, 10×, BioLegend) according to the manufacturer’s protocol. After washing with PBS, PMN was resuspended in RPMI-1640 growth medium (Gibco, USA), and their purity was confirmed by flow cytometry. Monocytes were isolated from the PBMC layer using CD14 magnetic beads (Miltenyi, Germany, 130-050-201), and lymphocytes were defined as the remaining cells in PBMC after monocyte removal. The cells were washed with PBS and resuspended for further experiments.
Cell culture and transfection
Human microvascular endothelial cell line HMVEC was gifted from Qiong Wu lab in Tsinghua University. The cell line was cultured in DMEM supplemented with 10% FBS, 1% non-essential amino acids, and 1% penicillin/streptomycin solution in a 5% CO2 incubator at 37 °C. To silence the expression of TRPM2, siRNAs targeting TRPM2 were transfected into the cells according to the manufacturer’s instructions.
Mice
Adult C57BL/6 J mice were purchased from GemPharmatech Co., Ltd (Strain NO. N000013) in the present study. Mice were maintained in separately ventilated cages in a specific pathogen-free (SPF) facility, in a room with standard ambient temperature and humidity, and the animals had unrestricted access to food (Xietong Organism, 1010001) and water, with a 12-hour light/dark cycle. TRPM2-KO mice were generated with 4 nucleotide deletion mutation ACTT (ACGAGCAACACTTGGAGGT → ACGAGCAACGGAGGT) in exon 5 by CRISPR-Cas9 technique in C57BL/6 J background and were then backcrossed with wildtype C57BL/6 J for at least three generations before further functional experiments. Myeloid cell-specific FPP synthase (FPPS) knockout mice (FPPSflox/floxLysMcre) and FPPSflox/flox mice were gifted from the laboratory of Prof. Yonghui Zhang26 at Tsinghua University. Mice were euthanized by carbon dioxide (CO2) asphyxiation inhalation, and cervical dislocation was performed as a secondary euthanasia procedure, and then the tissues were isolated. All animal experiments were approved by the Animal Research Ethics Committee and were carried out in accordance with the guidelines of the Laboratory Animal Research Center of Tsinghua University, with an assurance identification number: 15-LWL3 and 19-LWL1 by the Institutional Animal Care and Use Committee (IACUC) of Tsinghua University.
Inhibitors
Simvastatin (1 μM, 10 μM), zoledronic acid (1 μM, 10 μM), FIN56 (1 μM, 5 μM), zaragozic acid (ZGA) (7.5 μM, 15 μM), and BPH-652 (40 μM, 80 μM) were purchased from MedChemExpress (MCE). All the inhibitors used in this study for PKC screening including staurosporine (STS) (1 nM, 5 nM, 10 nM), PDTC (20 μM, 100 μM, 500 μM), PF-5662271 (5 nM, 10 nM, 20 nM), PD98059 (1 μM, 5 μM, 25 μM), tanshinone I (1 μM, 5 μM, 25 μM), cyclosporin A (1 nM, 5 nM, 25 nM), VTX27 (10 nM), ruboxistaurin (10 nM), and Go 6983 (10 nM). These inhibitors were purchased from Tsinghua University active screening platform.
RNA isolation and qPCR
Total RNA was extracted using an RNA-Quick Purification Kit (ES science, China, #RN001) according to the standard protocol. RNA was reverse transcribed into cDNA using an M5 Super plus qPCR RT kit with gDNA remover (Mei5 Biotechnology, China, MF166-plus-01). Quantitative PCR (qPCR) was performed using 2X M5 HiPer SYBR Premix EsTaq (with Tli RNaseH) (Mei5 Biotechnology, China, MF787-02). All primer sequences were presented in Supplementary Table 3.
FPP stimulation assay
Cells were washed twice with PBS and resuspended with normal saline to a final concentration of 106/mL. For the peripheral blood immune cells activation assay, the final concentrations of 0, 5, and 10 μg/mL FPP were incubated with cells for 10 minutes, followed by adding 2 mM CaCl2 for 30 minutes, then cells were collected for follow-up assays. For VEC activation, PMN was incubated with 60 μg/mL FPP for 10 minutes, followed by adding 2 mM CaCl2 for 30 minutes, the supernatant was collected and added to VECs at 80% density for 4 hours, the supernatant was removed and VECs were harvested for subsequent assays. For all the experiments involving inhibitors, 30 minutes were allowed for the inhibitors to fully interact with cells before adding FPP.
Mass spectrum
For cell samples, equal numbers of PMN from HCs and BD were lysed using a solution of methanol and ammonium hydroxide (7:3, v/v). After sonication and deproteinization, samples were centrifuged, and the resulting supernatant was dried and dissolved in acetonitrile and water (1:1, v/v) before injection into the LC-MS/MS system.
For serum samples, 300 μL of serum from HCs and BDs was diluted with 900 μL of 2% formic acid aqueous solution, vortexed, and loaded into solid phase extraction (SPE) cartridges pre-treated with 2% formic acid aqueous solution. After washing, elution was performed using NH4OH:2-propanol:1-hexane (1:7:12, v/v/v). The eluted samples were evaporated and dissolved in acetonitrile and water (1:1, v/v) before injection into the LC-MS/MS system.
Qtrap 6500+ (AB Sciex, USA) coupled with H-Class UHPLC (Waters, USA) for FPP analysis. The “relative abundance” of FPP was determined based on the chromatographic area of the FPP signal. The chromatographic area is directly proportional to the concentration of FPP in the sample. Therefore, by assessing the “relative abundance” of FPP extracted from equal serum or neutrophils, we can determine whether FPP is up- or down-regulated.
Flow cytometry
For cell death measurement after FPP stimulation by flow cytometry, cells were treated as indicated above, and the PI (MCE, USA, HY-D0815, 20 μg/mL) signal was measured after 30 minutes of incubation. PI-positive cells were defined as dead cells.
Peripheral blood was collected from the experimental autoimmune uveitis (EAU)-induced mouse model on day 18. After washing and lysis of red blood cells, surface staining was performed for the neutrophil-specific marker Ly6G (1:100, MultiScience Biotech, China, clone: RB6-8C5, 70-F21LY6G03-25) and the activation indicators CD11b (1:100, Biolegend, USA, clone: M1/70, 101205) and CD62L (1:100, Biolegend, USA, clone: W18021D, 161204). Expression of these surface markers was measured by percentage of positive cells and mean fluorescence intensity (MFI).
The stained cells were analyzed with a BD FACS Aria II and FlowJo Software (Tree Star).
Double-stranded DNA (dsDNA) Quantification
After isolating PMN from HC and BD, 6 × 105 cells were either stimulated with 0, 10, 30, or 60 μg/mL FPP for 10 mins, followed by 2 mM CaCl2 for 30 mins at room temperature in 24-well plates. The resulting supernatant was collected after centrifugation at 500 × g for 10 mins. Double-stranded DNA (dsDNA) in the supernatants was quantified using a Quant-iT Pico-Green dsDNA assay kit (Invitrogen, USA, P7589) by incubating the samples with PicoGreen for 5 mins at room temperature and measuring the fluorescence emission intensity at 520 nm after excitation at 485 nm.
Immunofluorescence and Immunohistochemical Staining
After isolating PMN from HC and BD, 6 × 105 cells were seeded on poly-L-lysine-coated coverslips in 24-well plates and incubated with 60 μg/mL FPP for 10 mins, followed by 2 mM CaCl2, with or without 50 U/mL Dnase I for 60 mins. Cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, and blocked with 2% BSA. Rabbit monoclonal to myeloperoxidase (MPO) (1:500, Abcam, UK, ab208670) were utilized as primary antibodies.
The paraffin-embedded skin sections of FPPSflox/floxLysMcre and FPPSflox/flox mice were stained with neutrophil-specific Ly6G (1:500, CST, USA, Clone: 1A8, 88876S) and MPO (1:500, Abcam, UK, Clone: EPR20257, ab208670) antibodies. YF®594 Goat Anti-Rabbit IgG (1:1000, UElandy, China, Y6107L) were utilized as secondary antibodies. DAPI was used to stain the nuclei.
Confocal microscopy was used to capture images, and the percentage of PMN undergoing NETosis was calculated as the number of cells showing NETosis divided by the total number of cells, multiplied by 100%.
For immunohistochemical staining, paraffin-embedded skin sections of FPPSflox/flox LysMcre and FPPSflox/flox mice were stained with MPO antibody (1:1000, Abcam, UK, Clone: EPR20257, ab208670) according to standard procedures and quantified by Image J software.
Enzyme-linked immunosorbent assay (ELISA)
Levels of proinflammatory cytokines, including TNF, IL-6, and IFN-γ, in serum from BD patients and HC were measured using commercially available ELISA kits (human TNF ELISA Kit, MultiScience Biotech, China, EK182-96; human IL-6 precoated ELISA Kit, Dakewei Biotech, China, 1110602; human IFN-γ ELISA Kit, MultiScience Biotech, China, EK180-96), following the manufacturer’s protocol. Levels of MPO-DNA complex, a specific marker for NETs, in cell supernatant were measured by a commercially available human MPO-DNA complex ELISA Kit (MEIMIAN, China, MM-2467H1) according to the manufacturer’s instructions.
Western blot
Neutrophils were lysed on ice using RIPA buffer (Huaxingbio, China) supplemented with a protease inhibitor and phosphatase inhibitor cocktail (Thermo Scientific, USA, 78446). The protein concentration of lysates was determined using the Pierce™ BCA Protein Assay Kits (Thermo Scientific, USA, 78446) according to the manufacturer’s instructions. Cell lysates were subjected to 10% SDS–PAGE, and transferred onto polyvinylidene fluoride (PVDF) membranes (Bio-Rad, USA, 1620177). The membranes were blocked and then incubated with anti-TRPM2 (1:500, SABbiotech, Clone: MGC133383, 22689) and anti-β-actin (1:1000, abcam, UK, Clone: 13E5, ab8227) overnight at 4 °C, respectively. The membranes were washed and incubated with anti-rabbit IgG-HRP for 1 hour at room temperature. Blots were developed with chemiluminescence and detected by Tanon-5200 (Bio-Tanon, China). Gray value analysis was done by Image J (v.1.50 g, NIH) software.
Public data collection and analysis
Metabolomics data in BD was obtained from the National Genomics Data Center (NGDC) database (OMIX007402 and OMIX007403, https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA030253))25. Gene Set Enrichment Analysis (GSEA) was performed to identify significantly changed metabolic pathways in BD serum with the threshold p-value < 0.05 and normalized enrichment score (NES) | > 1, analyzed by R package MetaboAnalystR.
Bulk RNA sequencing (Bulk RNA-seq) data of BD PBMC and PMN were obtained in the GEO dataset GSE198533 and GSE205867 from our published research, respectively11,12. The fold change, p-value, and adjusted p were calculated by R package DESeq2. Heatmap by R package ComplexHeatmap was employed to demonstrate genes from cholesterol biosynthesis pathway in PMN bulk RNA-seq, normalized by rlog method in DESeq2. Genes with p < 0.01, padj < 0.1, and |log2Fold Change | >0.5 were considered significantly different.
Single-cell RNA-sequencing (scRNA-seq) of PBMC was obtained in the GEO dataset GSE19861611 from our published research. The fold change, p-value, and adjusted p were calculated by Function FindMarkers in the R package Seurat. Subgroups of immunocytes including T cells, B cells, monocytes, NK cells, and dendritic cells were subset according to original research.
GSEA of metabolic pathways in RNA-seq data was performed using the R package ClusterProfiler. Gene sets included KEGG_STEROID_BIOSYNTHESIS, GOBP_VITAMIN_D_BIOSYNTHETIC_PROCESS, WP_CHOLESTEROL_
BIOSYNTHESIS_PATHWAY, KEGG_PRIMARY_BILE_ACID_BIOSYNTHESIS, and KEGG_STEROID_HORMONE_BIOSYNTHESIS from Molecular Signatures Database (MSigDB).
A gene set of calcium channels was obtained from the HUGO Gene Nomenclature Committee (HGNC), including Cation channels sperm associated (CATSPER), Inositol 1,4,5-triphosphate receptors (ITPR), Ryanodine receptors (RYR), and Two pore segment channels (TPCN). We also investigated Mitochondrial Calcium Uniporter (MCU). Genes with p < 0.01, padj <0.1, and |log2Fold Change | >0.5 were considered significantly different.
Induction and evaluation of vasculitis in mice
The FPPSflox/floxLysMcre mice and FPPSflox/flox mice (N = 6, 8 weeks old, 3 female and 3 male), TRPM2 knockout (KO) mice and wild-type (WT) mice (N = 4, 8 weeks old, 2 female and 2 male) were injected by intraperitoneal with 100 μL of PBS containing 2% bovine serum albumin (BSA) (Solarbio, China, PC0001) and 1% Evans blue (MACKLIN, E808783), respectively. Then, the mice were intradermally injected with 40 μL of rabbit anti-BSA antibody (Solarbio, China, SA263). Rabbit serum (Solarbio, China, T8570) was used as a control. 4 hours after injection, the mice were euthanized and dorsal skin samples were collected and digitally photographed. Evans-blue dye was extracted from the dorsal skin of TRPM2 KO mice and WT mice in a vasculitis model, and then dissolved in 50% trichloroacetic acid, the absorbance of each group was then measured at 620 nm by spectrophotometry. Additionally, skin samples were fixed in 10% neutral buffered formalin and stained with hematoxylin for routine microscopic examination. The severity of vasculitis was assessed by Image J analysis of the area of Evans blue permeation area and manual counting of perivascular neutrophils in HE-stained sections under a 40x objective.
Induction and evaluation of EAU in mice
The FPPSflox/flox LysMcre mice and FPPSflox/flox mice (N = 6, 8 weeks old, female) were injected subcutaneously with an emulsion consisting of 200 μg of retinal antigen interphotoreceptor retinoid-binding protein (IRBP) peptide 1–20 (GPTHLFQPSLVLDMAKVLLD, Sangon, China) and complete Freund’s adjuvant (sigma, USA, F5881) containing Mycobacterium tuberculosis strain H37Ra (BD Biosciences, USA, 231141) in a 1:1 volume ratio. Intraperitoneal injections of 250 μg Pertussis toxin (list lab, USA, #180) were given on day 0 and day 2 postimmunization. The induction of uveitis was assessed by ocular computerized tomography (OCT) and fundoscopy on day 14 after immunization. On day 18, peripheral blood and eyes were collected from each type of mice. Neutrophil activation was measured by flow cytometry as indicated above. Histological assessments of eyes were performed according to standard procedures and previously established criteria55.
Statistics
Experiments were repeated at least three times, with one representative dataset shown. Data are presented as mean ± standard deviation (SD), median + quantile, or percentage. The Kolmogorov-Smirnov test was used to test for the normality of data distribution. Student’s t-test and non-parametric test were used for comparisons between two groups, while paired t-test was used for comparisons before and after treatment. One-way ANOVA and two-way ANOVA tests were used with p-value adjusted for multiple comparisons by FDR using the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli. Correlations were calculated using Pearson correlation analysis. Statistical analyses were performed using SPSS V22.0 (IBM, USA) and GraphPad Prism V6.01 (GraphPad Software Inc, USA). A two-sided p-value < 0.05 was considered statistically significant, with *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 indicating significant differences.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the data supporting the findings of this study are present in the article and the supplementary information files, or can be obtained from the corresponding author upon reasonable request. Source data are provided with this paper.
Change history
03 January 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41467-024-55702-z
References
Hotamisligil, G. S. Inflammation, metaflammation and immunometabolic disorders. Nature 542, 177–185 (2017).
Hotamisligil, G. S. Foundations of immunometabolism and implications for metabolic health and disease. Immunity 47, 406–420 (2017).
Wang, X., Wang, Y., Antony, V., Sun, H. & Liang, G. Metabolism-associated molecular patterns (MAMPs). Trends Endocrinol. Metab.: TEM 31, 712–724 (2020).
Kang, N. et al. Metabolite-derived damage-associated molecular patterns in immunological diseases. FEBS J. 291, 2051–2067 (2023).
Jeon, J. H., Hong, C. W., Kim, E. Y. & Lee, J. M. Current understanding on the metabolism of neutrophils. Immune Netw. 20, e46 (2020).
Alarcón, P. et al. d(-) Lactic acid-induced adhesion of bovine neutrophils onto endothelial cells is dependent on neutrophils extracellular traps formation and CD11b expression. Front. Immunol. 8, 975 (2017).
Azevedo, E. P. et al. A metabolic shift toward pentose phosphate pathway is necessary for amyloid fibril- and phorbol 12-myristate 13-acetate-induced neutrophil extracellular trap (NET) formation. J. Biol. Chem. 290, 22174–22183 (2015).
Lahoz-Beneytez, J. et al. Human neutrophil kinetics: modeling of stable isotope labeling data supports short blood neutrophil half-lives. Blood 127, 3431–3438 (2016).
Kumar, S. & Dikshit, M. Metabolic insight of neutrophils in health and disease. Front. Immunol. 10, 2099 (2019).
Greco, A. et al. Behçet’s disease: new insights into pathophysiology, clinical features and treatment options. Autoimmun. Rev. 17, 567–575 (2018).
Zheng, W. et al. Single-cell analyses highlight the proinflammatory contribution of C1q-high monocytes to Behçet’s disease. Proc. Natl Acad. Sci. USA 119, e2204289119 (2022).
Yu, X. et al. Transcriptional analysis of neutrophils from patients with Behçet’s disease reveals activation and chemotaxis of neutrophils. Clin. Immunol. (Orlando, Fla.) 245, 109161 (2022).
Emmi, G. et al. Behçet’s syndrome as a model of thrombo-inflammation: the role of neutrophils. Front. Immunol. 10, 1085 (2019).
Safi, R. et al. Neutrophils contribute to vasculitis by increased release of neutrophil extracellular traps in Behçet’s disease. J. Dermatological Sci. 92, 143–150 (2018).
Ahn, J. K., Cha, H. S., Bae, E. K., Lee, J. & Koh, E. M. Extracellular high-mobility group box 1 is increased in patients with Behçet’s disease with intestinal involvement. J. Korean Med. Sci. 26, 697–700, (2011).
Han, E. C. et al. Expression of pro-inflammatory protein S100A12 (EN-RAGE) in Behçet’s disease and its association with disease activity: a pilot study. Ann. Dermatol. 23, 313–320, (2011).
Pittman, K. & Kubes, P. Damage-associated molecular patterns control neutrophil recruitment. J. Innate Immun. 5, 315–323 (2013).
Hayasaki, N. et al. Neutrophilic phlebitis is characteristic of intestinal Behçet’s disease and simple ulcer syndrome. Histopathology 45, 377–383 (2004).
Li, L. et al. Neutrophil extracellular traps promote aberrant macrophages activation in Behçet’s disease. Front. Immunol. 11, 590622 (2020).
Le Joncour, A. et al. Critical role of neutrophil extracellular traps (NETs) in patients with Behcet’s disease. Ann. Rheum. Dis. 78, 1274–1282 (2019).
Bettiol, A. et al. Vascular Behçet syndrome: from pathogenesis to treatment. Nat. Rev. Rheumatol. 19, 111–126 (2023).
Zhou, Z. Y., Chen, S. L., Shen, N. & Lu, Y. Cytokines and Behcet’s disease. Autoimmun. Rev. 11, 699–704 (2012).
Okin, D. & Medzhitov, R. The effect of sustained inflammation on hepatic mevalonate pathway results in hyperglycemia. Cell 165, 343–356 (2016).
Chen, J. et al. Farnesyl pyrophosphate is a new danger signal inducing acute cell death. PLoS Biol. 19, e3001134 (2021).
Zheng, W. et al. Metabolomic alterations associated with Behçet’s disease. Arthritis Res. Ther. 20, 214 (2018).
Xia, Y. et al. The mevalonate pathway is a druggable target for vaccine adjuvant discovery. Cell 175, 1059–1073.e1021 (2018).
Cheng, L. et al. Proteomics landscape mapping of organ-resolved behçet’s disease using in-depth plasma proteomics for identifying hyaluronic binding protein 2 expression associated with vascular involvement. Arthritis Rheumatol. (Hoboken, N. J.) 75, 424–437 (2023).
Krause, I. et al. Behçet’s disease in Israel: the influence of ethnic origin on disease expression and severity. J. Rheumatol. 28, 1033–1036 (2001).
Krause, I., Molad, Y. & Weinberger, A. Association of HLA-B5 with CLINICAL EXPRESSION AND SEVERITY OF BEHCET’S DISEASe in Israel. J. Clin. rheumatology: practical Rep. Rheum. Musculoskelet. Dis. 5, 137–140 (1999).
Bang, S., Yoo, S., Yang, T. J., Cho, H. & Hwang, S. W. Farnesyl pyrophosphate is a novel pain-producing molecule via specific activation of TRPV3. J. Biol. Chem. 285, 19362–19371 (2010).
Berridge, M. J., Bootman, M. D. & Roderick, H. L. Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529 (2003).
Neuberger, A., Nadezhdin, K. D. & Sobolevsky, A. I. Structural mechanisms of TRPV6 inhibition by ruthenium red and econazole. Nat. Commun. 12, 6284 (2021).
Lev, S. et al. Constitutive activity of the human TRPML2 channel induces cell degeneration. J. Biol. Chem. 285, 2771–2782 (2010).
Venkatachalam, K., Wong, C. O. & Zhu, M. X. The role of TRPMLs in endolysosomal trafficking and function. Cell Calcium 58, 48–56 (2015).
Kerschbaumer, A. et al. Efficacy of pharmacological treatment in rheumatoid arthritis: a systematic literature research informing the 2019 update of the EULAR recommendations for management of rheumatoid arthritis. Ann. Rheum. Dis. 79, 744–759 (2020).
Smolen, J. S. et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2022 update. Ann. Rheum. Dis. 82, 3–18 (2023).
Chan, H. C. & Ng, S. C. Emerging biologics in inflammatory bowel disease. J. Gastroenterol. 52, 141–150 (2017).
Chebli, J. M. et al. A guide to prepare patients with inflammatory bowel diseases for anti-TNF-α therapy. Med. Sci. Monit.: Int. Med. J. Exp. Clin. Res. 20, 487–498 (2014).
Sfriso, P., Bindoli, S. & Galozzi, P. Adult-onset still’s disease: molecular pathophysiology and therapeutic advances. Drugs 78, 1187–1195 (2018).
Macovei, L. A. et al. Adult-onset still’s disease-a complex disease, a challenging treatment. Int. J. Mol. Sci. 23, 12810 (2022).
Lee, R. W. & D’Cruz, D. P. Novel therapies for anti-neutrophil cytoplasmic antibody-associated vasculitis. Drugs 68, 747–770 (2008).
McAdoo, S. P. & Pusey, C. D. Is there a role for TNFα blockade in ANCA-associated vasculitis and glomerulonephritis? Nephrol., Dialysis, Transplant.:. Publ. Eur. Dialysis Transpl. Assoc. - Eur. Ren. Assoc. 32, i80–i88 (2017).
Katz, U. & Zandman-Goddard, G. Drug-induced lupus: an update. Autoimmun. Rev. 10, 46–50 (2010).
Williams, E. L., Gadola, S. & Edwards, C. J. Anti-TNF-induced lupus. Rheumatol. (Oxf., Engl.) 48, 716–720 (2009).
Ghorbaninezhad, F. et al. Tumor necrosis factor‑α in systemic lupus erythematosus: Structure, function and therapeutic implications (Review). Int. J. Mol. Med. 49, 43 (2022).
Jiang, D., De Vries, J. C., Muschhammer, J., Sindrilaru, A. & Scharffetter-Kochanek, K. Mouse Model of Immune Complex-mediated Vasculitis in Dorsal Skin and Assessment of the Neutrophil-mediated Tissue Damage. Bio-Protoc. 7, e2660 (2017).
Tang, C. et al. Surgical treatment of Behcet’s disease with severe aortic regurgitation. Front. Cardiovascular Med. 10, 1290615 (2023).
Nian, H. et al. Activated gammadelta T cells promote the activation of uveitogenic T cells and exacerbate EAU development. Investigative Ophthalmol. Vis. Sci. 52, 5920–5927 (2011).
Peng, Y. et al. Minimally activated CD8 autoreactive T cells specific for IRBP express a high level of Foxp3 and are functionally suppressive. Investigative Ophthalmol. Vis. Sci. 48, 2178–2184 (2007).
Tajiri, N. et al. The protective function of invariant natural killer T cells in the relapse of experimental autoimmune uveoretinitis. Exp. Eye Res. 203, 108406 (2021).
Shu, Q. et al. IL-8 triggers neutrophil extracellular trap formation through an nicotinamide adenine dinucleotide phosphate oxidase- and mitogen-activated protein kinase pathway-dependent mechanism in uveitis. Investigative Ophthalmol. Vis. Sci. 64, 19 (2023).
McCreedy, D. A. et al. Spleen tyrosine kinase facilitates neutrophil activation and worsens long-term neurologic deficits after spinal cord injury. J. Neuroinflammation 18, 302 (2021).
Morrison, P. J., Suhrkamp, I., Gerdes, S. & Mrowietz, U. Oral dimethyl fumarate induces changes within the peripheral neutrophil compartment of patients with psoriasis that are linked with skin improvement. Br. J. Dermatol. 185, 605–615 (2021).
Yu, P. F. et al. TNFα-activated mesenchymal stromal cells promote breast cancer metastasis by recruiting CXCR2(+) neutrophils. Oncogene 36, 482–490 (2017).
Thurau, S. R., Chan, C. C., Nussenblatt, R. B. & Caspi, R. R. Oral tolerance in a murine model of relapsing experimental autoimmune uveoretinitis (EAU): induction of protective tolerance in primed animals. Clin. Exp. Immunol. 109, 370–376 (1997).
Haas, D. & Hoffmann, G. F. Mevalonate kinase deficiencies: from mevalonic aciduria to hyperimmunoglobulinemia D syndrome. Orphanet J. Rare Dis. 1, 13 (2006).
Timilshina, M. et al. Activation of mevalonate pathway via LKB1 Is essential for stability of T(reg) cells. Cell Rep. 27, 2948–2961.e2947 (2019).
Chodick, G. et al. Persistence with statins and onset of rheumatoid arthritis: a population-based cohort study. PLoS Med. 7, e1000336 (2010).
Nagashima, T., Okazaki, H., Yudoh, K., Matsuno, H. & Minota, S. Apoptosis of rheumatoid synovial cells by statins through the blocking of protein geranylgeranylation: a potential therapeutic approach to rheumatoid arthritis. Arthritis Rheum. 54, 579–586 (2006).
Kim, J. Y. et al. Atorvastatin inhibits osteoclastogenesis by decreasing the expression of RANKL in the synoviocytes of rheumatoid arthritis. Arthritis Res. Ther. 14, R187 (2012).
Amuro, H. et al. Statins, inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme a reductase, function as inhibitors of cellular and molecular components involved in type I interferon production. Arthritis Rheum. 62, 2073–2085 (2010).
Tremoulet, A. H. The role of statins in inflammatory vasculitides. Autoimmunity 48, 177–180 (2015).
Inanc, M. T. et al. Effects of atorvastatin and lisinopril on endothelial dysfunction in patients with Behçet’s disease. Echocardiogr. (Mt. Kisco, N. Y.) 27, 997–1003 (2010).
Chen, Y. R. et al. Simvastatin reduces NETosis to attenuate severe asthma by inhibiting PAD4 expression. Oxid. Med. Cell. Longev. 2023, 1493684 (2023).
Bekkering, S. et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell 172, 135–146.e139 (2018).
Bendersky, A. et al. Cellular interactions of synovial fluid γδ T cells in juvenile idiopathic arthritis. J. Immunol. (Baltim., Md.: 1950) 188, 4349–4359 (2012).
Greenwood, J., Steinman, L. & Zamvil, S. S. Statin therapy and autoimmune disease: from protein prenylation to immunomodulation. Nat. Rev. Immunol. 6, 358–370 (2006).
Pastar, I. et al. Skin metabolite, farnesyl pyrophosphate, regulates epidermal response to inflammation, oxidative stress, and migration. J. Cell. Physiol. 231, 2452–2463 (2016).
Bratt, J. M. et al. Farnesyltransferase inhibition exacerbates eosinophilic inflammation and airway hyperreactivity in mice with experimental asthma: the complex roles of ras GTPase and farnesylpyrophosphate in type 2 allergic inflammation. J. Immunol. (Baltim., Md.: 1950) 200, 3840–3856 (2018).
Dunn, S. E. et al. Isoprenoids determine Th1/Th2 fate in pathogenic T cells, providing a mechanism of modulation of autoimmunity by atorvastatin. J. Exp. Med. 203, 401–412 (2006).
Yamamoto, S. et al. TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 14, 738–747 (2008).
The International Criteria for Behçet’s Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J. Eur. Acad. Dermatol. Venereol. 28, 338–347 (2014).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (82371822-W.Z., 82171800-W.Z., 82302036-N.K., 32141004-W.L., 32430035-W.L.). This study was also supported by the Natural Science Foundation of Beijing (7232124-W.Z., Z230014-W.L.), Fundamental Research Funds for the Central Universities (3332023113-M.Z. and 3332023125-Z.W.), National High Level Hospital Clinical Research Funding (2022-PUMCH-C-008-W.Z.), CAMS Innovation Fund for Medical Sciences (2023-I2M-C&T-B-049-W.Z.), Tsinghua University Spring Breeze Fund, Shenzhen Medical Research Fund (B2402012-W.L. and C2404002-W.L.), and grants from Ministry of Science and Technology of China (2021YFC2300500-W.L. and 2021YFC2302403-C.L.). We thank the health professional staff from the Department of Rheumatology and Clinical Immunology, Peking Union Medical College Hospital, and appreciate for the participation of all the patients and healthy volunteers in this study.
Author information
Authors and Affiliations
Contributions
W.Z. and W.L. conceptualized and designed the project and supervised the project. X.Y. conducted the bioinformatics analysis with the help of M.Z. and Y.L. M.Z. and N.K. performed the experiments and verified the underlying data reported in the manuscript with the help of X.Z., Q.D., Y.Z., and C.Z. X.M. provided FPP and Q.Z. provided inhibitors and agonists of the MVA pathway. M.Z., Z.W., X.W, Y.L., Q.C., R.L., X.B., H.Y., L.Z., and J.L. participated in the patient's enrollments, sample collection, and clinical analysis, with the help from J.J., R.G., X.M., and C.L. M.Z. and N.K. drafted the manuscript. W.Z., W.L., H.C., and Y.Z. critically reviewed the manuscript and provided valuable input. All authors read and approved the manuscript. M.Z., N.K., and X.Y. are co-first authors, and the order of the co-first authors was determined by workload.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yacine Boulaftali, Ricardo Silvestre and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, M., Kang, N., Yu, X. et al. TNF inhibitors target a mevalonate metabolite/TRPM2/calcium signaling axis in neutrophils to dampen vasculitis in Behçet’s disease. Nat Commun 15, 9261 (2024). https://doi.org/10.1038/s41467-024-53528-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-53528-3
