
Abstract
Developing efficient electrocatalysts for acidic electrosynthesis of hydrogen peroxide (H2O2) holds considerable significance, while the selectivity and stability of most materials are compromised under acidic conditions. Herein, we demonstrate that constructing amorphous platinum–selenium (Pt–Se) shells on crystalline Pt cores can manipulate the oxygen reduction reaction (ORR) pathway to efficiently catalyze the electrosynthesis of H2O2 in acids. The Se2‒Pt nanoparticles, with optimized shell thickness, exhibit over 95% selectivity for H2O2 production, while suppressing its decomposition. In flow cell reactor, Se2‒Pt nanoparticles maintain current density of 250 mA cm−2 for 400 h, yielding a H2O2 concentration of 113.2 g L−1 with productivity of 4160.3 mmol gcat−1 h−1 for effective organic dye degradation. The constructed amorphous Pt–Se shell leads to desirable O2 adsorption mode for increased selectivity and induces strain for optimized OOH* binding, accelerating the reaction kinetics. This selenization approach is generalizable to other noble metals for tuning 2e‒ ORR pathway.
Similar content being viewed by others


Introduction
Hydrogen peroxide (H2O2) is a valuable chemical for a variety of industrial applications, such as paper bleaching, wastewater treatment, medical disinfection, and fine chemical synthesis1,2,3,4. Currently, H2O2 is almost manufactured through a large-scale indirect anthraquinone route, which, however, involves multiple complex processes and generates vast organic wastes5,6. An appealing alternative is the electrochemical method through two-electron oxygen reduction reaction (2e‒ ORR) that can be operated under mild conditions and coupled with renewable energy sources7,8. Over the past decades, electrosynthesis of H2O2 in alkaline conditions has been widely explored with the use of heterogeneous catalysts as the electrode9,10,11,12,13. However, the produced H2O2 in alkali is unstable and tends to decompose rapidly14,15,16, thus extra stabilizers (e.g., acids and halides) are usually introduced to delay the self-decomposition for long-term usage17. In comparison, H2O2 in acidic media is more stable and exhibits stronger oxidation ability, which is highly desired for many applications18. Nevertheless, most catalysts are unstable and even dissolvable in acids, which seriously hinders its further development19,20.
Compared with the carbon- and 3d transition metal-based catalysts, platinum-group metal nanomaterials such as Pt represent the most potential catalysts for the production of H2O2 in acid because of the relatively appropriate electronic configuration and adequate structural stability under acidic media21,22. Nevertheless, molecular O2 on the surface of pure noble metals is inclined to be reduce into H2O via a 4e− pathway rather than the 2e‒ pathway into H2O223,24. It is therefore of great significance to regulate the structure of noble metals to maximize the selectivity for the reaction. Engineering the surface structures of catalysts can regulate the performance of typical electrochemical processes25,26, while its importance to H2O2 electrosynthesis has been largely unexplored. Hence, manipulating the reduction pathway on Pt nanomaterials via structure engineering provides a feasible route for designing efficient 2e‒ ORR catalysts for acidic conditions. In addition to the point of catalyst design, the discrepancy of techniques between the rotating ring-disk electrode (RRDE) and continuous flow reactors also dramatically influences the H2O2 selectivity. Different from the rapid transportation of H2O2 in RRDE technique, the additional transport factors must be considered in real devices because of the slow diffusion rate of H2O2 from the catalyst and gas diffusion layer to the output stream27. Consequently, the generated H2O2 tends to discompose in the presence of catalysts via electrochemical reduction reaction or disproportionation reaction, which is unfavorable for the accumulation of H2O228,29. However, much less attention has been paid to this problem at present. Therefore, designing active, durable, and selective electrocatalysts for consecutive production of H2O2 in acids has remained elusive due to the challenges of long-term operation under harsh conditions and inhibition of side reactions.
Herein, through selenization induced straining and phasing we demonstrate the successful construction of a series of core-shell structures consisting of crystalline Pt core and amorphous Pt‒Se shells with tunable thicknesses. The core-shell nanoparticles can boost the 2e− reduction of O2 toward H2O2 in acidic media, achieving pronounced enhancements in H2O2 selectivity, production rate, and inhibition of H2O2 decomposition. The catalyst with optimized shell thickness (Se2–Pt NPs) shows high H2O2 selectivity over 95% in different acidic electrolytes and significantly inhibited electrochemical reduction reaction and disproportionation reaction. When employed in a flow cell reactor, the Se2–Pt NPs present long-term stability (400 h) at 250 mA cm−2 for continuous electrosynthesis of H2O2, yielding a high H2O2 production rate of 4160.3 mmol gcat−1 h−1. The ultimate concentration of H2O2 achieves 113.2 g L−1 for efficient removal of organic dyes as well. Detailed experimental evidence reveals that the formation of amorphous Pt‒Se shell leads to the transformation of isolated Pt sites that adsorb O2 in an “end-on” way, which is desirable for the preservation of O‒O bond. Meanwhile, the strain generated by mismatch between the crystalline core and amorphous shell adjusts the electronic structures of the surface shell with weakened binding strength toward *OOH, thus finally making it more conducive to the production of H2O2. Significantly, the selenization induced straining and phasing is general and readily applicable to other noble metals for tuning ORR with 2e‒ pathway. This work provides an avenue to overcome the bottleneck of electrosynthesis of H2O2 in acidic media.
Results
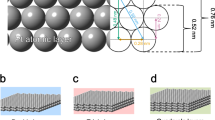
The core-shell nanoparticles consisting of crystalline Pt core and amorphous Pt‒Se shells with controllable thicknesses were prepared via a two-step post-modification method (Fig. 1a). Concretely, the Pt NPs were used as growing seeds, and the introduction of Se can achieve the construction of amorphous Pt‒Se shell on the surface. It should be noteworthy that the structural mismatch at the interface further induces the strain interaction (Fig. 1a). The thickness of the amorphous shell can be finely tuned by varying the introduced amount of Se (Sex–Pt NPs, x represents the extent of selenization), thereby enabling continuous modulation over the surface structure (Fig. 1b). A series of characterizations were then conducted to exhibit the structural evolution of Se‒Pt NPs with various shell thicknesses. As shown in the X-ray diffraction (XRD) patterns, it can be clearly observed a continuous attenuation of peak intensity at (111) peak (~ 39°) from Pt NPs to Se‒Pt NPs with the deepening of selenization degree, indicating that the crystallinity of the NPs continues to decrease as the increasement of amorphous Pt‒Se shell thickness (Fig. 1c)30. Correspondingly, results from the scanning electron microscopy energy-dispersive X-ray spectroscopy (SEM-EDX) suggest a continuously increasing trend of the Se element (Fig. 1d), which is consistent with results from the inductively coupled plasma atomic emission spectrometry (ICP-AES) (Table S1). To clarify the surface species of the core-shell structure, low-energy ion scattering (LEIS) that only probes the outmost atomic layer was conducted31,32. It can be observed from the LEIS spectrum that the Se2‒Pt NPs show an additional presence of Se signal compared to that of Pt NPs, demonstrating that the amorphous shell on the surface is composed of Pt and Se elements (Fig. 1e). Moreover, X-ray absorption fine structure spectroscopy (XAFS) was conducted to confirm the structural evolution of the core-shell structures. As shown in Fig. 1f, compared to the pure Pt NPs, the core-shell catalysts with thicker amorphous shells exhibit a continuously decreased peak intensity. Combined with the fitting results, the Se‒Pt NPs with thicker amorphous shells exhibit decreased coordination numbers of Pt‒Pt bonds but increased Pt‒Se bonds, demonstrating the deepening of selenization degree (Supplementary Fig. 1 and Table S2). In addition, the negative shift of peak position in the wavelet transform counter plots indicates more Pt‒Se coordination because of the smaller atomic numbers of coordinated Se atoms, which supports the coordination environment evolution of Pt atoms in Se‒Pt NPs (Fig. 1g)33.

a Schematic illustration of the synthetic process of Se2‒Pt NPs and corresponding structural evolution on the surface. b Surface structures of Se1‒Pt NPs and Se3‒Pt NPs. c XRD patterns, (d) SEM-EDX spectra, (e) LEIS spectra for the surface species, (f) Pt K-edge EXAFS in R space, and (g) wavelet transforms of Pt NPs and Se‒Pt NPs with different shell thicknesses.
The structures of Se‒Pt NPs with tunable shell thicknesses were further investigated by transmission electron microscopy (TEM). Firstly, the corresponding mappings demonstrate the gradual deepening of the spatial distribution of Se element into the core of Pt element, proving the evolutions of Pt‒Se shell thicknesses (Fig. 2a–d). As revealed by the TEM images at different selenization stages (Fig. 2e–h and Supplementary Figs. 2–5), the selenization process progresses from outside to inside and it begins with the doping of Se atoms on the surface of Pt core in the initial stage (Fig. 2f and Supplementary Fig. 3). As the reaction progresses, a thin layer of amorphous Pt‒Se shell around the Pt core is generated and the thickness of amorphous shell is gradually getting thicker (Fig. 2g and Supplementary Fig. 4). Finally, core-shell structured nanomaterials consisting of a well-defined crystalline core and amorphous shell can be obtained (Fig. 2h and Supplementary Fig. 5). The electrochemical behaviors of nanomaterials via cyclic voltammogram (CV) technique further demonstrate the different selenization stages of nanomaterials. As shown in Fig. 2m, the CV curve of Pt NPs exhibits typical hydrogen adsorption/desorption peaks between 0.05 ~ 0.40 V (vs. RHE)34, whereas the Se‒Pt NPs show a continuously decreased hydrogen region until a flat profile with deeper selenization degree. Therefore, it can be reasonably inferred that the Pt atoms in the surface will gradually transform into isolated sites with the selenization progressing until the formation of an amorphous shell, and further selenization will lead to the insertion of Se atoms into deeper Pt lattices, being consistent with the TEM results (Fig. 2e–h). Besides the amorphization of crystal structure, the insertion of Se into the Pt lattice can also expand the volume of the corresponding region, inducing the strain interaction at the interface (Fig. 1a, b)35. The strain mapping by geometric phase analysis shows that the color intensity, which represents the degree of strain, varies very little throughout the Pt NPs, indicating that the strain within the structure is negligible (Fig. 2i and Supplementary Fig. 6). As the amorphous shell gradually forms, strain is generated and gradually increases (Fig. 2j, k and Supplementary Figs. 7, 8), and reaches the maximum value until the formation of an excessively thick amorphous shell (insets in Fig. 2l and Supplementary Fig. 9)36. Correspondingly, the intensity profiles of the Pt lattice near the interface exhibit a gradual positive shift (Fig. 2n), indicating the existence of tensile strain for the crystalline Pt core. The lattice spacings along the Pt (111) are measured to be 2.52 Å, 2.58 Å, 2.68 Å, and 2.69 Å at samples after different selenization degrees, which correspond to the lattice tensions of about 2.3%, 6.3%, and 6.7%, respectively. The intensity profiles of the Pt lattice parallel to the (200) plane also exhibit a positive shift, indicating the existence of tensile strain (Supplementary Figs. 10, 11). On the contrary, the amorphous shell is under compressive strain (Fig. 1a, b), and the compression strain decreases with the increase of shell thickness due to the limitation of the influence range of strain effect36,37. Furthermore, the influence of the strain on the electronic structures of the surface Pt sites was studied using X-ray photoelectron spectroscopy (XPS) and surface valence band spectra (Supplementary Fig. 12). It can be found that the d-band centers of Pt are highly sensitive to the lattice strain, shifting negatively as the generation of strain and positively with the weakening of strain effect (Fig. 2o), demonstrating the successful regulation of the surface structures38,39.

a–d HAADF-STEM-EDS elemental mappings, (e–h) HRTEM images, and (i–l) corresponding strain mappings of (a, e, i) Pt NPs, (b, f, j) Se1‒Pt NPs, (c, g, k) Se2‒Pt NPs, and (d, h, l) Se3‒Pt NPs. m CV curves at 50 mV s−1 in N2-saturated 0.1 M HClO4, n Intensity profiles taken from the corresponding region in (e–h), and (o) surface valence band photoemission spectra of Pt NPs and Se‒Pt NPs with different shell thicknesses. The shaded area in Fig. 2m is the hydrogen adsorption/desorption peaks.
The 2e‒ ORR performance of Se‒Pt NPs with different shell thicknesses was explored in O2-saturated acids through the RRDE measurement (see Materials and methods for details, Supplementary Figs. 13–15). As displayed in Fig. 3a, the recorded linear scan voltammogram (LSV) curves exhibit the currents for O2 reduction measured on the disk electrode (solid lines) and the currents for H2O2 oxidation detected on the Pt ring electrode (dashed lines) in 0.1 M HClO4. Accordingly, the H2O2 selectivity (%) of Se2‒Pt NPs is > 95% over a wide range of potential (0.0 V ‒ 0.6 V vs. RHE), indicating a highly active and selective 2e− pathway with the transferred number of electrons approaching ~2.0 (Fig. 3b and Supplementary Fig. 16). In contrast, the Se1‒Pt NPs with less selenization degree display a relatively dominant 4e− pathway (H2O2% < 36%, n ~ 3.5), and the thicker ones, although showing the characteristics of 2e− pathway (H2O2% > 70%, n ~ 2.6), exhibit significantly larger overpotentials and lower ring currents (Fig. 3a, b and Supplementary Fig. 16), revealing the correlation between the 2e‒ ORR performance and the thickness of amorphous shell. Importantly, the Se2‒Pt NPs can also exhibit high selectivity toward 2e− ORR in various acidic electrolytes, including 0.1 M H2SO4 (≈ 95.6%), 0.5 M HClO4 (≈ 96.1%), and 0.5 M H2SO4 (≈ 96.3%), which are superior to most of the reported catalysts in a wider potential range (Fig. 3c, Supplementary Fig. 17, and Table S4).

a RRDE voltammograms at 1600 rpm in O2-saturated 0.1 M HClO4 with the ring current (dashed lines) and disk current (solid lines) for C support, Pt NPs, and Se‒Pt NPs with different shell thicknesses (without iR corrected) (scan rate: 10 mV s−1, loading amount: 3 μgPt; O2 flow rate: 10 mL min−1; temperature: 25 °C). b Values of the selectivity (%) calculated from the RRDE polarization curves. The shaded area represents the error bar derived from three independent measurements. c The comparison of the selectivity values of Se2‒Pt NPs in different electrolytes and the reported electrocatalysts. d Schematic illustration of an ideal catalyst surface that inhibit the disproportionation reaction and electrochemical reduction reaction. e electrochemical reduction reaction studied by following a similar method for ORR test but using a degassed 0.1 M HClO4 containing 10 mM H2O2 and (f) disproportionation reaction measured by adding 10 mg catalysts into 10 mM H2O2 aqueous solution at room temperature. g The photo taken after adding 10 mg Pt NPs and Se2‒Pt NPs into 10 mM H2O2 aqueous solution at room temperature.
Besides the high performance tested in the RRDE experiment, it is noteworthy that, in a practical flow cell reactor, the residence time of generated H2O2 on the catalyst surface would be much longer than that of the RRDE measurement, leading to the consecutive decomposition of H2O2 on the catalyst (disproportionation reaction and electrochemical reduction reaction, Table S3)28,29. Hence, for the practical application of H2O2 electrosynthesis, both the favourable 2e− pathway and the supersession of decomposition reactions are essential for an ideal catalyst (Fig. 3d). We then systemically studied the activities of the as-prepared catalysts toward disproportionation reaction and electrochemical reduction reaction. Based on the results of significantly lower H2O2 reduction currents in electrochemical reduction reaction (Fig. 3e) and inhibited H2O2 decomposition in disproportionation reaction (Fig. 3f, g), it can be concluded that the construction of amorphous shell can significantly suppress the thermal and electrochemical H2O2 decomposition reactions. In addition, the stability of Se2‒Pt NPs in RRDE measurement was also studied. As shown in Supplementary Figs. 18, 19, the activity and stability of Se2‒Pt NPs exhibit no obvious decays during the long-term stability test (~ 15 h) in 0.1 M HClO4, indicating superior recyclability and stability of the catalyst for 2e‒ ORR performance. According to the Pourbaix diagram for Se-H2O system (Supplementary Fig. 20), it can be observed that Se predominantly exists in the form of elemental solid or high-valent oxide solid within the potential range associated with the 2-electron ORR, suggesting that Se element in the amorphous shell can theoretically maintain stable presence during the reaction. Furthermore, the structural characterizations of the spent Se2‒Pt NPs further confirm the excellent corrosion resistance of the core-shell structure under harsh conditions (Supplementary Figs. 21–23).
Inspired by the promising catalytic performance and suppressed H2O2 decomposition reactions of Se2‒Pt NPs, we further incorporated the catalyst into practical flow cell reactor to continuously produce H2O2 in acids at high current density and volume (Fig. 4a). Figure 4b and Supplementary Fig. 24 show the typical components and parts of a flow cell device with the Pt mesh as anode electrode, Se2‒Pt NPs coated gas diffusion layer as cathode electrode, and Nafion 211 membrane for proton transport and elimination of gas crossover. The operational conditions (such as working temperature, liquid flow rate, and electrolyte environment and volume) for electrosynthesis of H2O2 in flow cell reactor are summarized in the insets of Fig. 4c. The polarization curve collected at a high current density of 250 mA cm-2 under a continuous O2 flow exhibits a steady state, indicating the potential of Se2‒Pt NPs for practical application at operational conditions (Fig. 4c). Furthermore, it can be observed from Fig. 4d that the corresponding H2O2 concentration gradually increases as the time progressing, and the calculated Faradaic efficiency (FE) maintains high level with ~90% along the process (Supplementary Fig. 25). After operating at 250 mA cm−2 for 400 h, the accumulated H2O2 concentration reaches 3328.2 mmol L−1 and corresponding FE remains at 89.2%, which are well-placed among previously reported catalysts in acidic media (Table S5) and also suggesting possibility in practical applications3,7,40. As a proof of concept, the degradation experiments using Se2‒Pt NPs as the electrode were performed. Figure 4e–g show that the degradation of organic pollutants including methylene blue, malachite green, and rhodamine B solutions is achieved, demonstrating the applicability in organic wastewater treatment.

a Photograph of the three-phase flow cell reactor system and (b) schematic diagram of the three-phase flow cell reactor. Rflow cell = 1.9 ± 0.1 Ω. c The V-t curve of Se2‒Pt NPs tested in flow cell reactor with current density of 250 mA cm−2 in 0.1 M HClO4. Insets are the photograph of cathode electrolyte and the working conditions, respectively. (mass loading: 0.2 mgPt cm−2; O2 flow rate: 10 mL min−1; temperature: 25 °C) (d) H2O2 production and corresponding Faradaic efficiency of H2O2 of Se2‒Pt NPs tested in the flow cell reactor with the increase of time. The UV-vis spectra and photographs of (e) methylene blue (MB), (f) malachite green (MG), and (g) rhodamine B (RB) solutions before and after degradation.
To investigate the correlation between the 2e‒ ORR performance and the thickness of amorphous shell, detailed characterizations and discussions have been provided to understand the mechanism. As discussed above (Fig. 2m), Pt atoms on the surface will exhibited as isolated sites with the insertion of Se atoms34,41,42. This transformation of the Pt sites leads to the change of O2 adsorption mode from “side-on” to “end-on”, which can hinder the cleavage of O‒O bond such that H2O2 can be preferentially formed (Supplementary Fig. 26)43,44. However, the aforementioned results partly explain the reason why Se1‒Pt NPs show a relatively dominant 4e‒ pathway while the thicker amorphous shell (Se2‒Pt NPs and Se3‒Pt NPs) prefer a 2e‒ pathway. On the basis of formed isolated Pt sites, it should be noteworthy that the thicker amorphous shell causes relatively poor 2e‒ ORR performance (Fig. 3a, b). An ideal catalyst for 2e‒ ORR should not only suppress the O‒O bond cleavage in *OOH (the key intermediate during ORR) but also possess suitable binding strength toward *OOH7,45. For pure Pt nanomaterials, which bind the *OOH intermediate relatively too strongly, it is therefore essential to lower the binding strength for the subsequent reactions. With the formation of appropriate shell thickness (Se2‒Pt NPs), the d-band center of surface Pt sites shifts negatively because of the compressive strain, indicating the reduced binding strength toward intermediates (Fig. 1a)35, which is beneficial for the production of H2O2 with respect to the pure Pt. In comparison, the surface active sites on Se3‒Pt NPs with an excessively thick shell thickness are under weaker strain effect (Fig. 1b), leading to the positive shift of the d-band center and enhancing the binding strength with the intermediate36,37.
The density functional theory (DFT) calculation was then conducted to gain deeper understanding of the influence of selenization degree on the catalytic performance and selectivity. Systematic DFT calculations have demonstrated that the incorporation of Se atoms induces surface amorphization of Pt NPs, with a thicker amorphous Pt‒Se shell formed as the amount of Se introduced increases (Supplementary Fig. 27). The structural evolution agrees well with the results observed in the experiment. The calculated total density of states (TDOS) and projected density of states (PDOS), as depicted in Fig. 5a, b, reveal the metallic nature of all four catalysts with excellent electronic conductivity, a crucial factor for facilitating rapid charge transfer and promoting the ORR. Furthermore, the orbital overlap and hybridization between catalysts and intermediates demonstrate the effective charge transfer and formation of bonding interactions. The free energy diagrams of ORR on four contrastive catalysts at selected potentials are shown in Supplementary Fig. 28 and Tables S6, S7. It can be inferred that both Pt NP and Se1‒Pt NPs exhibit a 4e− pathway, as *O*OH tends to dissociate due to the presence of continuous active sites. Conversely, Se2‒Pt NPs and Se3‒Pt NPs with increased Se content display isolated Pt sites on their surface. Consequently, O2 is adsorbed in end-on mode where the O‒O bond is less prone to breakage, resulting in the formation of H2O2. In addition, from the perspective of reaction kinetics, the CI-NEB method is used to search for the transition states for the key elementary step (*OOH → H2O2/*O + H2O). As a benchmark, the continuous Pt/Au sites in pure Pt/Au surface were demonstrated to follow the 4e‒ pathway via the structural optimization and kinetic barrier analysis (Supplementary Fig. 29 and Table S8). Based on the calculated results in Fig. 5c and Supplementary Figs. 28–30, it can be inferred that the reaction barriers for 2e‒(4e‒) pathway on Se2‒Pt NPs and Se3‒Pt NPs are determined to be 0.42 (0.91) eV and 0.79 (2.06) eV, respectively. The barriers of the 2e‒ pathway are significantly lower than those of the 4e‒ pathway in both catalysts, indicating that the former is the predominant process. Furthermore, the barrier for the 2e‒ pathway on Se2‒Pt NPs is even smaller compared to that on Se3‒Pt NPs, thereby demonstrating superior catalytic performance of Se2‒Pt NPs in H2O2 production.

a, b Density of states of OOH adsorbed on Pt, Se1‒Pt, Se2‒Pt, and Se3‒Pt. c The 2e−/4e− ORR path and corresponding energy barriers for Se2-Pt and Se3-Pt. d The planar-average-potential along with differential charge density at the interface between amorphous Pt–Se shell and crystalline Pt core with the adsorption of OOH intermediate (cyan and yellow represent charge depletion and accumulation, respectively). The planar-average-potential along with differential charge density distribution on (e) OOH intermediate, and (f) active-site Pt atoms with OOH intermediate. g The d-band centers of the active-site Pt atoms in various catalysts. h Schematic illustration of the reaction mechanism on the surface of Pt NPs and Se2‒Pt NPs.
More importantly, in order to gain insights into the strain effect caused by selenization on the activity, we performed in-depth electronic structure analysis on the catalysts, intermediates, and active sites from different perspectives by using different characterization tools. The charge density distribution on different objects studied at the interface between amorphous Pt‒Se shell and crystalline Pt core is illustrated in Fig. 5d–f and Supplementary Fig. 31. Specifically, the optimal state of charge transfer between the interface and the active-site Pt atoms is achieved when an appropriately thick amorphous shell is formed, which plays a significant role in activating oxygen-containing intermediates. However, as the thickness of the Pt‒Se amorphous shell increases, the influence of interface charge transfer gradually diminishes. We then further calculated the d-band centers of the active-site Pt atoms to elucidate the impact of the various interface on the surface-active sites (Fig. 5g). It can be concluded that the d-band center of the active-site Pt atoms follows the order εd(Pt) > εd(Se1‒Pt) > εd(Se3‒Pt) > εd(Se2‒Pt), highlighting the crucial influence of amorphous shell thickness, which aligns with experimental observations. Therefore, with the increase of selenium content, amorphous Pt‒Se shells are formed and the corresponding strain effects are generated. The appropriate shell thickness induces optimized charge transfer and d-band center of the surface Pt sites owing to the strongest interaction between the amorphous shell and crystalline core. In a nutshell, with the detailed experimental evidence and analyses, it can be concluded that the active and selective 2e‒ ORR performance of Se2‒Pt NPs is derived from the construction of amorphous Pt‒Se shell with appropriate thickness, which leads to the formation of isolated Pt sites and downshift of d-band center, thereby changing the O2 adsorption mode and decreasing the binding strength toward *OOH (Fig. 5h).
To explore the universality of selenization induced straining and phasing in improving the 2e‒ ORR performance, we then extended other nanomaterials and investigated their electrocatalytic performance under the same catalytic conditions. As shown in Fig. 6a and Supplementary Fig. 32, in comparison with Pd NPs, the selenated Pd NPs exhibit the characteristics of the formation of amorphous shell, indicating the successful construction of the structure consisting of crystalline core and amorphous shell (Se‒Pd NPs). Interestingly, the Se‒Pd NPs show a dominant 2e‒ reduction pathway with significantly improved ring currents compared to that of Pd NPs (Fig. 6b). Furthermore, this strategy is general and can be even extended to commercial catalysts (commercial Pt/C and commercial Pd/C) (Fig. 6a and Supplementary Fig. 33). Figure 6c and Supplementary Fig. 34 demonstrate that the construction of amorphous shell can significantly modulate the reduction pathway of commercial catalysts. Specifically, the Se‒Pd NPs show remarkably enhancement of H2O2% over 80% between 0.15 and 0.47 V (vs. RHE) compared to Pd NPs with ~0%, and the Se‒Commercial Pt/C and Se‒Commercial Pd/C also exhibit obviously improvement in H2O2% form ~0% to ~55% and ~7% to ~67% (Fig. 6d and Supplementary Fig. 35a). Moreover, based on the statistical results (Fig. 6e and Supplementary Fig. 35b), it can be inferred that the construction of core-shell structure transforms the nanomaterials from a primary 4e‒ reaction pathway catalyst to a catalyst dominated by 2e‒ pathway, demonstrating its regulation toward ORR pathway and the universality of selenization induced straining and phasing.

a XRD patterns of different catalysts before and after selenization. Insets are the corresponding surface structural evolution before and after selenization. b, c RRDE voltammograms in O2-saturated 0.1 M HClO4 for catalysts before and after selenization of (b) Pd NPs and Se‒Pd NPs, (c) commercial Pt/C and Se‒commercial Pt/C (without iR corrected). d Comparison of the selectivity (%) and (e) average transferred electrons (n) calculated from the RRDE polarization curves of catalysts before and after selenization. The shaded area represents the error bar derived from three independent measurements.
Discussion
In summary, we have demonstrated that the strategy of selenization induced straining and phasing on noble metal nanomaterials can enhance the reduction of O2 toward H2O2 with high activity and selectivity in acidic media. The optimized Se2‒Pt NPs composed of crystalline Pt core and amorphous Pt‒Se shell exhibit pronounced enhancements in H2O2 selectivity, production rate, and inhibition of side reactions. Impressively, Se2‒Pt NPs exhibit high H2O2 selectivity over 95% under various acidic electrolytes and significantly inhibited electrochemical reduction reaction and disproportionation reaction. Significantly, when employed in a flow cell reactor, Se2‒Pt NPs present long-term stability (400 h) under 250 mA cm-2 to reach high H2O2 concentration of 113.2 g L−1 with production rate of 4160.3 mmol gcat−1 h−1. With the detailed experimental proofs, it can be concluded that the formation of amorphous Pt‒Se shell transforms Pt atoms into isolated sites, which adsorbs O2 in the desirable “end-on” way, and the strain generated between the crystalline core and amorphous shell leads to the surface sites with weakened binding strength toward *OOH, thus finally making it more conducive to the production of H2O2. Moreover, the selenization induced straining and phasing is general and readily applicable to other noble metals for tuning ORR with 2e‒ pathway. This universality of selenization induced straining and phasing in improving electrocatalytic activity is of great significance for designing highly efficient and selective metal-based nanomaterials toward acidic H2O2 electrosynthesis and beyond in sustainable energy applications.
Methods
Chemicals
Platinum (ІІ) acetylacetonate (Pt(acac)2, 99%), palladium (ІІ) acetylacetonate (Pd(acac)2, 99%), ascorbic acid (AA, C6H8O6, reagent grade, 99.0%), Nafion (5%) and commercial Pd/C (10 wt% Pd) were purchased from Sigma-Aldrich. Polyvinylpyrrolidone (PVP, K58) was purchased from Energy Chemistry. Selenous acid (H2SeO3, ≥95.0%), ethylene glycol (EG, C2H6O2, ≥99.7%), ethanol (C2H6O, analytical reagent, ≥99.7%), acetone (C3H6O, analytical reagent, ≥99.5%), isopropanol (C3H8O, analytical reagent, ≥99.7%), trichloromethane (CHCl3, analytical reagent, ≥99.0%), perchloric acid (HClO4, analytical reagent, 70.0 to 72.0%), and sulfuric acid (H2SO4, analytical reagent, 95.0 to 98.0%) were purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). Commercial Pt/C (20 wt% Pt) was purchased from Johnson Matthey (JM) Corporation. All the chemicals were used as received without further purification. The water (18 MΩ cm–1) used in all experiments was prepared by passing through an ultra-pure purification system (Aqua Solutions).
Preparation of Pt NPs and Pd NPs
In a typical synthesis of Pt NPs, 9.8 mg Pt(acac)2, 36.0 mg AA, 20.0 mg PVP, and 10.0 mL EG were added into a glass vial (volume: 30 mL). The mixture in the capped vial was ultrasonicated for 1 h. After that, the homogeneous mixture was then heated from room temperature to 180 °C and maintained at 180 °C for 3 h in an oil bath. After cooling to room temperature, the products were washed with ethanol/acetone mixture and then collected by centrifugation. The synthetic procedures for Pd NPs are the same as that of Pt NPs except for changing 9.8 mg Pt(acac)2 with 7.6 mg Pd(acac)2.
Preparation of Se‒Pt NPs with different shell thicknesses
In a typical synthesis of Se‒Pt NPs with different shell thicknesses, a certain amount of H2SeO3 is dispersed in EG to form a solution with an H2SeO3 concentration of 2 mg/mL, and then 65 μL, 320 μL, and 800 μL solution were introduced into the above Pt NPs solution that has been cooled down. The above mixture was heated from room temperature to 140 °C and maintained at 140 °C for 2 h in an oil bath for the preparation of Se1‒Pt NPs, Se2‒Pt NPs, and Se3‒Pt NPs, respectively. After cooling to room temperature, the products were washed with ethanol/acetone mixture and then collected by centrifugation.
Preparation of Se‒Pd NPs, Se‒commercial Pt/C, and Se‒commercial Pd/C
In a typical synthesis of the Se‒Pd NPs, 320 μL of the EG solution (H2SeO3 concentration: 2 mg/mL) was added into the above Pd NPs solution that has been cooled down. The mixture was then heated from room temperature to 140 °C and maintained at 140 °C for 2 h in an oil bath. After cooling to room temperature, the products were washed with ethanol/acetone mixture and then collected by centrifugation. The synthetic procedures for Se‒commercial Pt/C and Se‒commercial Pd/C are the same as that of Se‒Pd NPs.
Preparation of supported catalysts
9.8 mg carbon black (VXC-72) and 3 mL trichloromethane were added into a 30 mL vial. After that, as-prepared nanomaterials dispersed in 2 mL ethanol/ trichloromethane (v:v = 1:1) solution were mixed with the above mixture under ultrasonicated for 1 h. Afterwards, the products were washed with acetone via centrifugation and then dried under ambient conditions.
Preparation of acidic electrolytes
In the preparation of 0.1 M and 0.5 M HClO4 solution, 3.95 mL and 19.75 mL HClO4 were diluted to a volume of 0.5 L, respectively. For 0.1 M and 0.5 M H2SO4 solution, 2.72 mL and 13.60 mL were diluted to a volume of 0.5 L, respectively. The obtained acidic electrolytes were placed in a dry, shady, and well-ventilated place for further utilization.
Characterization
Transmission electron microscopy (TEM) was conducted on a JEM-1400 transmission electron microscope at an accelerating voltage of 100 kV. High-resolution TEM (HRTEM) and high-angle annular dark-field scanning TEM (HAADF-STEM) were conducted on FEI Tecnai F30 transmission electron microscope at an accelerating voltage of 300 kV. Low-resolution EDX was performed on a scanning electron microscope (SEM, ZEISS Sigma 300). X-ray diffraction (XRD) patterns were collected on the Ultima-IV diffractometer with a Cu Kα X-ray source (λ = 1.540598 Å). X-ray photoelectron spectroscopy (XPS) was carried out on a Thermo Scientific XPS Spectrometer (ESCALAB 250 XI). The carbon peak at 284.8 eV was used as a reference to correct for charging effects. The loading amount of Pt was determined by the inductively coupled plasma atomic emission spectroscopy (ICAP 7000, ThermoFisher, USA). Low-energy ion scattering (LEIS) was performed on Qtac-100 LEISS-XPS. X-ray absorption spectra (XAS) data at Pt L-edge were collected at the TPS-44A beamline of the National Synchrotron Radiation Research Center (NSRRC, Hsinchu, Taiwan). Data were processed according to standard procedures using the Demeter program package (Version 0.9.24)46.
Strain analysis around the boundary of crystalline core and amorphous shell
The geometric phase analysis method is used for quantifying displacements and strain fields at atomic resolution, which is based on the reciprocal space of the HRTEM image and ER-C plugin in DigitalMicrograph software. The diffraction spots we choose were (0—22) and (20—2) along the face-centered cubic zone [111] axis to obtain the strain distribution via the εxx field.
Electrochemical measurements
The ORR measurements were performed by using a rotating ring-disk electrode (RRDE; Pine Research Instrumentation; disk diameter, 5.61 mm, disk area, 0.2475 cm2; ring inner diameter, 6.25 mm, ring outer diameter, 7.92 mm, ring area, 0.1866 cm2) connected to the installation of rotating electrode speed control (Pine Research Instrumentation, model: AFMSRCE) and CHI660 electrochemical station (Chenhua, Shanghai). The raw test data was acquired directly and further processed in the subsequent charting process. The catalysts were dispersed in the mixture containing isopropanol and Nafion (5%) (v:v = 1:0.005) to form the homogeneous catalyst ink by sonicating for 30 min. The concentration of Pd was controlled to be 0.6 mgPd/Pt mL−1 based on ICP-AES measurement. 5 μL catalysts ink was dropped on the glassy carbon disk of RRDE and the corresponding mass of Pt was estimated to be 3 μg. A saturated calomel electrode (SCE) was used as the reference electrode, and a graphite rod was used as the counter electrode. The activity and selectivity for electrosynthesis of H2O2 were conducted in O2-saturated acidic solutions (0.1 M HClO4, 0.1 M H2SO4, 0.5 M HClO4, 0.5 M H2SO4) at the scan rate of 10 mV s−1 from 0.0 to 1.0 V (vs. RHE) with a rotation rate of 1600 rpm for independent measurements three times (without iR corrected). In the ORR polarization curves, the current densities were normalized to the geometric area of the electrode area. The potential applied to the Pt ring of RRDE is 1.2 V (vs RHE) during the entire testing process to oxidize the produced H2O2 from the disk electrode. The H2O2 selectivity of the catalyst on RRDE was calculated based on the current of the disk electrode and ring electrode (Eq. 1).
Where Ir, Id, and N represent the ring current, the disk current, and the collection efficiency (0.330 after calibration), respectively. The number of transferred electrons (n) at the disk electrode during the oxygen reduction process was calculated with (Eq. 2)
Electrosynthesis of H2O2 in practical conditions was evaluated using a flow cell reactor. The resistance value was measured to be 1.9 ± 0.1 Ω through iR compensation in CHI660 electrochemical station (Chenhua, Shanghai). The Se2–Pt NPs catalyst (1 mg, 20 wt% Pt) were homogeneously mixed with 1000 µL ethanol and 30 µL of 5 wt.% Nafion by ultrasonicating for 0.5 h. Afterward, the obtained catalyst ink was pipetted onto the carbon paper (1 × 1 cm2, Pt loading: 0.2 mgPt cm−2) and dried under ambient conditions as the working electrode in the flow cell device. A saturated calomel electrode (SCE) and a platinum mesh were used as reference electrode and counter electrode, respectively. The Nafion 211 membrane (thickness: 183 μm, size: 1.5 × 1.5 cm2) was used to separate the chambers. The Nafion 211 membrane was first treated in a 5% H2O2 solution at 80 °C for 1 h, followed by a soaking in deionized water for 0.5 h. Then, it was treated in a 5% dilute H2SO4 solution at 80 °C for 1 h, followed by another soaking in deionized water with 0.5 h for further use. The cathode and anode compartments were filled with 0.1 M HClO4 (500 mL). After introducing the continuous O2 flow into the reactor, the production rate and stability of the catalyst were tested at a constant current of 250 mA cm−2. H2O2 concentration was quantified by cerium sulfate titration based colorimetric method9, and the amount of H2O2 can be calculated as half of the consumed Ce4+ (1 Ce4+ ~ 1/2 H2O2). The concentrations of Ce4+ before and after the reaction were determined by ultraviolet-visible spectroscopy at a wavelength of 316 nm (Eq. 3).
The 1.0 mmol L−1 Ce4+ standard solutions were obtained by dissolving 33.2 mg Ce(SO4)2 in 100 mL 0.5 M H2SO4. The produced H2O2 can be acquired through mixing the sample solution with the standard Ce4+ solution for calculating the consumed Ce4+. The produced H2O2, Faradaic efficiency (FE), and H2O2 concentration were calculated as follows (Eqs. 4–6):
Where cH2O2 is the molarity of generated H2O2, cCe4+,before is the molarity of the Ce4+ standard solution before test, cCe4+,after is the molarity of the Ce4+ solution after test, Vconsumed Ce4+ is the volume of the standard Ce4+ solution, Vremoved electrolyte is the volume of the removed electrolyte for test, Vtotal electrolyte is the total electrolyte volume, and Q is the amount of charge during the test.
DFT calculations
Spin-polarized density functional theory (DFT) computations are performed for the present systems (pure Pt and different degrees of selenization Pt‒Se core-shell nanoparticles) based on Vienna ab initio simulation package (VASP)47. The generalized gradient approximation (GGA) in the form of Perdew-Burke-Ernzerhof (PBE) functional is employed for the exchange correlation interactions between electrons48. The projector-augmented wave (PAW) method is used to describe the interactions between ions and electrons49. A dispersion correction using the DFT-D3 method is used to describe the van der Waals (vdW) interactions50. The construction of the structural model for calculation is as follows. First, a 2 × 2 × 2 supercell is constructed from the face-centered cubic platinum, then a 6-layer slab model Pt (111) (96 atoms) is established with a vacuum layer of about 15 Å to avoid the interactions between adjacent layers. Through relatively reducing Pt atoms and increasing Se atoms in the first few layers (one to four) of 6-layer slab model Pt(111), the Pt‒Se core-shell structural models with different degrees of selenization (Se1‒Pt, Se2‒Pt, and Se3‒Pt) corresponding to different experimental samples can be obtained. All the structures are fully relaxed. The plane wave cutoff energy is set to 450 eV. The convergence thresholds in all structure relaxation calculations are 0.02 eV/Å and 10−5 eV for the force and energy, respectively. The k-points are set to 3 × 3 × 1 and 6 × 6 × 1 with Γ-centered Monkhorst-Pack grids for geometry optimization and electronic structure calculations (DOS, charge, etc), respectively. The Bader charge and charge density difference are employed to obtain the information of charge transfer, charge redistribution and analyze the bonding interactions between the intermediates and active sites, the isosurface level of the charge plots is set to 0.0026 e/Å3.
The Gibbs free energy change (ΔG) for each elementary step is calculated by the following equation based on the computational hydrogen electrode (CHE) model51 (Eq. 7):
where ΔE is the energy change based on DFT calculations, ΔEZPE is the zero-point energy difference. TΔS (T = 298.15 K) and ∫CpdT are the contribution of entropy and enthalpy, respectively. ΔGU = neU, where n is the number of electrons involved in reaction and U is the applied electrode potential. ΔGpH represent the free energy correction of pH. The elementary step with the maximum free energy change is defined as the potential-determining step (PDS) of the reaction (ΔGPDS).
In order to obtain kinetic information of the reaction and competitive selectivity between different reaction pathways, climbing-image nudged elastic band (CI-NEB)52 method is used to identify the transition state and estimate the kinetic barrier in the reaction pathway. Six images are interpolated between the initial state and the final state to determine the minimum energy barrier. The minimum energy path was optimized using the force-based conjugate-gradient method until the total energy and force threshold are less than 1 × 10−5 eV and 0.05 eV/Å, respectively. The original data of computational models are provided in Supplementary data 1.
Data availability
All data supporting the findings of this study are available in the main text or Supplementary Information. Source data are provided with this paper.
References
Agarwal, N. et al. Aqueous Au-Pd colloids catalyze selective CH4 oxidation to CH3OH with O2 under mild conditions. Science 358, 223–227 (2017).
Hage, R. & Lienke, A. Applications of transition‐metal catalysts to textile and wood‐pulp bleaching. Angew. Chem. Int. Ed. 45, 206–222 (2006).
Brillas, E., Sirés, I. & Oturan, M. A. Electro-Fenton process and related electrochemical technologies based on Fenton’s reaction chemistry. Chem. Rev. 109, 6570–6631 (2009).
Perry, S. C. et al. Electrochemical synthesis of hydrogen peroxide from water and oxygen. Nat. Rev. Chem. 3, 442–458 (2019).
Yi, Y., Wang, L., Li, G. & Guo, H. A review on research progress in the direct synthesis of hydrogen peroxide from hydrogen and oxygen: noble-metal catalytic method, fuel-cell method and plasma method. Catal. Sci. Technol. 6, 1593–1610 (2016).
Campos‐Martin, J. M., Blanco‐Brieva, G. & Fierro, J. L. Hydrogen peroxide synthesis: an outlook beyond the anthraquinone process. Angew. Chem. Int. Ed. 45, 6962–6984 (2006).
Yang, S. et al. Toward the decentralized electrochemical production of H2O2: a focus on the catalysis. ACS Catal. 8, 4064–4081 (2018).
Zhang, X., Xia, Y., Xia, C. & Wang, H. Insights into practical-scale electrochemical H2O2 synthesis. Trends Chem. 2, 942–953 (2020).
Lu, Z. et al. High-efficiency oxygen reduction to hydrogen peroxide catalysed by oxidized carbon materials. Nat. Catal. 1, 156–162 (2018).
Kim, H. W. et al. Efficient hydrogen peroxide generation using reduced graphene oxide-based oxygen reduction electrocatalysts. Nat. Catal. 1, 282–290 (2018).
Jung, E. et al. Atomic-level tuning of Co–N–C catalyst for high-performance electrochemical H2O2 production. Nat. Mater. 19, 436–442 (2020).
Jiang, K. et al. Highly selective oxygen reduction to hydrogen peroxide on transition metal single atom coordination. Nat. Commun. 10, 3997 (2019).
Zheng, Z., Ng, Y. H., Wang, D. W. & Amal, R. Epitaxial growth of Au–Pt–Ni nanorods for direct high selectivity H2O2 production. Adv. Mater. 28, 9949–9955 (2016).
Qiang, Z., Chang, J.-H. & Huang, C.-P. Electrochemical generation of hydrogen peroxide from dissolved oxygen in acidic solutions. Water Res 36, 85–94 (2002).
Duke, F. R. & Haas, T. W. The homogeneous base-catalyzed decomposition of hydrogen peroxide1. J. Phys. Chem. 65, 304–306 (1961).
Roberts, J. L. Jr., Morrison, M. M. & Sawyer, D. T. Base-induced generation of superoxide ion and hydroxyl radical from hydrogen peroxide. J. Am. Chem. Soc. 100, 329–330 (1978).
Othmer, D. F. & Kirk, R. E. Kirk-Othmer encyclopedia of chemical technology. Wiley (2004).
Zhou, W. et al. Selective H2O2 electrosynthesis by O-doped and transition-metal-O-doped carbon cathodes via O2 electroreduction: A critical review. Chem. Eng. J 410, 128368 (2021).
Zheng, Y. R. et al. Black phosphorous mediates surface charge redistribution of CoSe2 for electrochemical H2O2 production in acidic electrolytes. Adv. Mater. 34, 2205414 (2022).
Zhang, X. L. et al. Strongly coupled cobalt diselenide monolayers for selective electrocatalytic oxygen reduction to H2O2 under acidic conditions. Angew. Chem. Int. Ed. 60, 26922–26931 (2021).
Zhang, X. et al. Optimizing the Pd sites in pure metallic aerogels for efficient electrocatalytic H2O2 production. Adv. Mater. 35, 2211512 (2023).
Kodama, K., Nagai, T., Kuwaki, A., Jinnouchi, R. & Morimoto, Y. Challenges in applying highly active Pt-based nanostructured catalysts for oxygen reduction reactions to fuel cell vehicles. Nat. Nanotechnol. 16, 140–147 (2021).
She, Z. W. et al. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 355, eaad4998 (2017).
Huang, L. et al. Advanced platinum-based oxygen reduction electrocatalysts for fuel cells. Acc. Chem. Res. 54, 311–322 (2021).
Wang, P. et al. Phase and structure engineering of copper tin heterostructures for efficient electrochemical carbon dioxide reduction. Nat. Commun. 9, 4933 (2018).
Yao, Q., Yu, Z., Li, L. & Huang, X. Strain and Surface Engineering of Multicomponent Metallic Nanomaterials with Unconventional Phases. Chem. Rev. 123, 9676–9717 (2023).
Choi, C. H. et al. Hydrogen peroxide synthesis via enhanced two-electron oxygen reduction pathway on carbon-coated Pt surface. J. Phys. Chem. C 118, 30063–30070 (2014).
Bianchi, G., Mazza, F. & Mussini, T. Catalytic decomposition of acid hydrogen peroxide solutions on platinum, iridium, palladium and gold surfaces. Electrochim. Acta 7, 457–473 (1962).
Katsounaros, I. et al. Hydrogen peroxide electrochemistry on platinum: towards understanding the oxygen reduction reaction mechanism. Phys. Chem. Chem. Phys. 14, 7384–7391 (2012).
Cheng, H. et al. Surface anion promotes Pt electrocatalysts with high CO tolerance in fuel-cell performance. J. Am. Chem. Soc. 144, 22018–22025 (2022).
Chen, G. et al. Interfacial effects in iron-nickel hydroxide–platinum nanoparticles enhance catalytic oxidation. Science 344, 495–499 (2014).
Zhan, C. et al. Medium/High‐Entropy Amalgamated Core/Shell Nanoplate Achieves Efficient Formic Acid Catalysis for Direct Formic Acid Fuel. Cell. Angew. Chem. Int. Ed. 62, e202213783 (2023).
Yu, Z. et al. Phase-controlled synthesis of Pd–Se nanocrystals for phase-dependent oxygen reduction catalysis. Nano Lett. 21, 3805–3812 (2021).
Siahrostami, S. et al. Enabling direct H2O2 production through rational electrocatalyst design. Nat. Mater. 12, 1137–1143 (2013).
He, T. et al. Mastering the surface strain of platinum catalysts for efficient electrocatalysis. Nature 598, 76–81 (2021).
Smith, A. M., Mohs, A. M. & Nie, S. Tuning the optical and electronic properties of colloidal nanocrystals by lattice strain. Nat. Nanotechnol. 4, 56–63 (2009).
Zhao, J., Chen, B. & Wang, F. Shedding light on the role of misfit strain in controlling core–shell nanocrystals. Adv. Mater. 32, 2004142 (2020).
Hammer, B. & Nørskov, J. K. Theoretical surface science and catalysis—calculations and concepts. In: Advances in catalysis. Elsevier (2000).
Kitchin, J. R., Nørskov, J. K., Barteau, M. A. & Chen, J. Role of strain and ligand effects in the modification of the electronic and chemical properties of bimetallic surfaces. Phys. Rev. Lett. 93, 156801 (2004).
Tang, C. et al. Tailoring acidic oxygen reduction selectivity on single-atom catalysts via modification of first and second coordination spheres. J. Am. Chem. Soc. 143, 7819–7827 (2021).
Yu, Z. et al. Low‐Coordinated Pd Site within Amorphous Palladium Selenide for Active, Selective, and Stable H2O2 Electrosynthesis. Adv. Mater. 35, 2208101 (2023).
Verdaguer-Casadevall, A. et al. Trends in the electrochemical synthesis of H2O2: enhancing activity and selectivity by electrocatalytic site engineering. Nano Lett 14, 1603–1608 (2014).
Jiang, K., Zhao, J. & Wang, H. Catalyst design for electrochemical oxygen reduction toward hydrogen peroxide. Adv. Funct. Mater. 30, 2003321 (2020).
Yang, S., Kim, J., Tak, Y. J., Soon, A. & Lee, H. Single‐atom catalyst of platinum supported on titanium nitride for selective electrochemical reactions. Angew. Chem. Int. Ed. 55, 2058–2062 (2016).
Jung, E. et al. Recent advances in electrochemical oxygen reduction to H2O2: catalyst and cell design. ACS Energy Lett 5, 1881–1892 (2020).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541 (2005).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Peterson, A. A., Abild-Pedersen, F., Studt, F., Rossmeisl, J. & Nørskov, J. K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 3, 1311–1315 (2010).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Acknowledgements
This work was financially supported by the National Key R&D Program of China (2022YFA1504500), the National Natural Science Foundation of China (22025108, U21A20327, 22121001), the start-up fundings from Xiamen University and the Postdoctoral Fellowship Program of CPSF under Grant Number GZC20240895. We acknowledge support from the Max Planck-POSTECH-Hsinchu Center for Complex Phase Materials.
Author information
Authors and Affiliations
Contributions
X.H. conceived and supervised the research. X.H. and Z.Y. designed the experiments. X.H., Z.Y., and Q.Y. performed most of the experiments and data analysis. X.H., Z.Y., Q.Y., and T.H. participated in various aspects of the experiments and discussions. Z.Y., L.Z., and F.X. collected the TEM data. H.D. and L.Y. performed the DFT simulations. Z.H. W.H. and C.P. collected the XAS data. X.H. and Z.Y. wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ziyun Wang and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yu, Z., Deng, H., Yao, Q. et al. Selective and durable H2O2 electrosynthesis catalyst in acid by selenization induced straining and phasing. Nat Commun 15, 9346 (2024). https://doi.org/10.1038/s41467-024-53607-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-53607-5
This article is cited by
-
Carbon-based electrocatalysts for selective two-electron oxygen reduction
Catal (2026)
-
Membrane electrode assembly for hydrogen peroxide electrosynthesis
Nature Reviews Clean Technology (2025)
-
Microbial-vulcanized organic-inorganic dual-modulated cobalt hydroxide for oxygen evolution reaction
Nature Communications (2025)
-
Oxygen functionalization of carbon quantum dots enables efficient acidic hydrogen peroxide electrosynthesis
Nature Communications (2025)
