
Abstract
Few aerobic hyperthermophilic microorganisms degrade polysaccharides. Here, we describe the genome-enabled enrichment and optical tweezer-based isolation of an aerobic polysaccharide-degrading hyperthermophile, Fervidibacter sacchari, previously ascribed to candidate phylum Fervidibacteria. F. sacchari uses polysaccharides and monosaccharides for growth at 65–87.5 °C and expresses 191 carbohydrate-active enzymes (CAZymes) according to RNA-Seq and proteomics, including 31 with unusual glycoside hydrolase domains (GH109, GH177, GH179). Fluorescence in-situ hybridization and nanoscale secondary ion mass spectrometry confirmed rapid assimilation of 13C-starch in spring sediments. Purified GHs were optimally active at 80–100 °C on ten different polysaccharides. Finally, we propose reassigning Fervidibacteria as a class within phylum Armatimonadota, along with 18 other species, and show that a high number and diversity of CAZymes is a hallmark of the phylum, in both aerobic and anaerobic lineages. Our study establishes Fervidibacteria as hyperthermophilic polysaccharide degraders in terrestrial geothermal springs and suggests a broad role for Armatimonadota in polysaccharide catabolism.
Similar content being viewed by others


Introduction
Polysaccharides are the most abundant biological macromolecules on Earth and their structural and energy storage roles extend to all domains of life1. Their innate recalcitrance derives from their structural complexity and low solubility, and their degradation into oligomers and monomers determines whether they are buried or recycled. Polysaccharide catabolism relies on the concerted efforts of diverse carbohydrate-active enzymes (CAZymes), among which glycoside hydrolases (GHs) play a pivotal role by hydrolyzing glycosidic bonds and forming a sugar hemiacetal or hemiketal along with the corresponding aglycon.
In high-temperature biotopes such as terrestrial geothermal springs, polysaccharides can be produced in situ by microorganisms or delivered as allochthonous material. Regardless of source, they can be degraded by thermophilic (45 < Topt < 80 °C) and hyperthermophilic (Topt ≥ 80 °C) bacteria and archaea2 among which most are fermenters, despite the energetic favorability of aerobic metabolism and the dominance of aerobic respiration among extant prokaryotes3,4,5. A better understanding of polysaccharide metabolism by thermophiles stands to improve our knowledge of high-temperature ecosystems, and may provide new enzymes and paradigms for the biofuels industry, which is rate-limited by the depolymerization of lignocellulose wastes into fermentable sugars6.
While there are substantial gaps in our understanding of polysaccharide metabolism at high temperatures, even larger gaps limit our understanding of prokaryotic diversity. For example, uncultivated taxa account for ~85% of the phylogenetic diversity of prokaryotes7 and only 46 of 161 bacterial phyla in the Genome Taxonomy Database (GTDB) have at least one pure culture8. These deep lineages can be studied to some extent without cultivation through meta-omics approaches and targeted experiments such as fluorescence in situ hybridization (FISH) coupled with nanoscale secondary ion mass spectrometry (nanoSIMS)9 or bioorthogonal non-canonical amino acid tagging (BONCAT)10. Nevertheless, the isolation of members of these lineages is a grand challenge in microbiology that would improve our understanding of the scope and scale of prokaryotic biology.
Here, we focus on a yet-uncultivated bacterium that was predicted to be both thermophilic and saccharolytic - Candidatus Fervidibacter sacchari. Evidence for abundant populations of close relatives of Ca. F. sacchari - originally called OctSpA1-106 - was first obtained by rRNA gene surveys and FISH in silica-depositing, alkaline springs in Yellowstone National Park11. Based on phylogenetic analysis of both 16S and 23S rRNA genes, the authors suggested that OctSpA1-106 was the deepest diverging branch in the Bacteria. Five single cell-amplified genomes (SAGs) assigned to the OctSpA1-106 lineage were later obtained from Great Boiling Spring (GBS), NV, as part of a large single-cell genomics study12. The SAGs represented two genera that were predicted to catabolize polysaccharides based on a large number of GHs, thus inspiring the name Ca. F. sacchari for the more abundant genus. A phylogenetic analysis based on conserved markers confirmed that OctSpA1-106 branches deeply within the Bacteria and the name Candidatus Fervidibacteria was proposed. More recently, BONCAT experiments with sediments from a Yellowstone hot spring showed that Ca. Fervidibacteria were highly abundant and active, yet they were not enriched after cellulose, cellobiose, or glucose addition, and their activity decreased following cellobiose addition, calling into question whether they really are cellulolytic or not10.
In this study, we describe the genome-guided isolation of Fervidibacter sacchari and show that it is indeed cellulolytic. We suggest it has a unique niche in circumneutral terrestrial springs as an aerobic, hyperthermophilic polysaccharide degradation specialist. We further reclassify Fervidibacteria as members of the phylum Armatimonadota and suggest that both aerobic and anaerobic polysaccharide catabolism are hallmarks of the phylum based on a high number and diversity of GHs, especially the GH177 and GH179 families.
Results
Genome-guided isolation of Ca. Fervidibacter sacchari
Despite being detected in some terrestrial hot spring sediments like GBS (Fig. 1a), Ca. Fervidibacteria are often missed in 16S rRNA gene surveys and elsewhere due to primer mismatches13,14,15. However, using shotgun metagenomics, we show here that Ca. F. sacchari is the most abundant species in 85 °C clay-rich GBS sediments (Fig. 1b), and identify abundant Ca. F. sacchari cells in GBS sediments by FISH (Fig. 1c). Due to the high abundance of Fervidibacteria in GBS and other terrestrial hot springs, we reasoned that Fervidibacteria are ecologically important in this biome and targeted them for cultivation.

a Great Boiling Spring in Gerlach, NV. b Abundance of metagenomic reads from 85 °C sediments mapping to F. sacchari and other abundant prokaryotes. c Visualization of F. sacchari in 85 °C sediments by FISH using a Cy-3 label. The image is representative of >20 fields of view examined. d Enrichment of F. sacchari in lab cultures with biomass substrates, casamino acids, or volatile fatty acids; media composition and additional results are shown in Supplementary Figs. 1 and 2 (n = 1). e FISH of 6-FAM labeled F. sacchari in locust bean gum lab enrichment containing >99.5% F. sacchari. The image is representative of >20 fields of view examined. f cryo-electron tomogram of pre-divisional F. sacchari cells. The image is representative of ~20 cells examined. A single color legend for taxa is used for (b, d). Source data are provided as a Source Data file.
Initially, Ca. F. sacchari was enriched in the lab from ~85 °C GBS sediments as part of a broad screen to cultivate novel thermophiles, and one of the lab cultures was used for genome-guided cultivation. The broad screen consisted of GBS medium with 22 different combinations of carbon sources, H2 and O2 concentrations, and other terminal electron acceptors (Supplementary Fig. 1). Ca. F. sacchari enrichment was apparent under oxic (10% headspace O2) and microoxic (2% O2) conditions with either 0.005% (mass vol-1) lignin (culture G-4), 0.5 mL of an aqueous extract from GBS photosynthetic mats (culture G-12), an equimolar mixture (1 mM) of the fatty acids butyrate, propionate, and acetate (culture G-2), or the fatty acid mixture plus 0.02% casamino acids (culture G-7) (Fig. 1d and Supplementary Fig. 1). Since those enrichments were not maintained, a fifth culture containing 0.02% casamino acids (culture G-10; Fig. 1d) with a lower abundance of Ca. F. sacchari was used for genome-guided cultivation of Ca. F. sacchari.
Given the enrichment of Ca. F. sacchari under aerobic conditions with biomass substrates and its genome-predicted aerobic and saccharolytic metabolism12,16, we designed lab enrichments derived from the G-10 culture that varied in O2 concentration, incubation time, and genome-predicted polysaccharide substrates, and evaluated results using quantitative PCR (qPCR) and FISH. Initial experiments using 0.02% casamino acids showed no difference in Ca. F. sacchari 16S rRNA gene abundance at 7 days with 5-21% headspace O2 (vol:vol, where 21% O2 refers to a full air atmosphere), and no growth was observed under anaerobic conditions (Supplementary Fig. 2). Further experiments using 10% O2 showed that Ca. F. sacchari reached maximum abundance within 4 days and decreased only slightly after 21 days (Supplementary Fig. 3). Finally, we screened for growth on polysaccharides that were predicted substrates based on GH families identified in Ca. F. sacchari SAGs by dbCAN317. In particular, carboxymethyl cellulose, locust bean gum, xylan, and xyloglucan were chosen as substrates based on the presence of predicted α- and β-mannosidases (GH2 and GH38), β-glucosidases (GH1 and GH3), endo-1,4-β-xylanases (GH10), α-L-fucosidases (GH29 and GH95), and α-N-arabinofuranosidases (GH51) (Supplementary Note 1).
Given the fast growth rate of Ca. F. sacchari relative to other microbes in the G-10 culture, we relied on 4-day transfers (1:50 dilution) in the carboxymethyl cellulose, locust bean gum, xylan, and xyloglucan cultures to dilute slow-growing species. After five months of continuous growth and transfers, Ca. F. sacchari growth was observed on all four polysaccharides (Supplementary Fig. 4), with locust bean gum cultures reaching >99.5% purity (Fig. 1e). To try to obtain a clonal culture (i.e., isolate), we plated a locust bean gum culture onto media solidified with gellan gum (Serva, Heidelberg), with carbon sources and phosphate autoclaved separately to avoid Maillard reactions and phosphate-catalyzed production of reactive oxygen species18; however, no colonies were observed. Instead, a pure culture was obtained from the locust bean gum culture following three rounds of dilution-to-extinction and manual isolation of a single cell using optical tweezers and a sterile polydimethylsiloxane microfluidic chip mounted on an inverted microscope. The genome from the resulting isolate, strain PD1, has 98% average nucleotide identity (ANI) to the medium-quality SAG co-assembly originally designated Ca. F. sacchari12. This strain is proposed as the nomenclatural type under the International Code of Nomenclature of Prokaryotes (ICNP) and is hereafter referred to as Fervidibacter sacchari PD1T (Table 1).
Experiments with strain PD1T were consistent with predictions from the genome and results from enrichment cultures. Strain PD1T grew optimally at 5-10% O2 (Supplementary Fig. 5) and was catalase-positive and oxidase-negative. Weak growth was also observed on xyloglucan under fermentative conditions (Supplementary Fig. 6). The temperature range for growth was 65 to 87.5 °C with an optimum of ≥80 °C, and a minimum generation time of 14.0 ± 0.9 h (Supplementary Fig. 7). Strain PD1T grew on 12 different polysaccharides, three sources of lignocellulose, and seven different 5- and 6-carbon monosaccharides as sole carbon sources (Fig. 2a). Microcrystalline cellulose (90 μm) was also used as a substrate when cultures were supplied with 0.002% (mass vol-1) yeast extract (Supplementary Fig. 8). Strain PD1T also grew on 0.02% casamino acids as the sole carbon source, although growth was lower than on most polysaccharides and monosaccharides (Fig. 2a). Together, these results confirm that F. sacchari is an aerobic and hyperthermophilic polysaccharide-degrading specialist and among the most thermophilic bacteria known19 (Supplementary Data 1).

a F. sacchari growth on carbohydrates, lignocellulosic biomass, and casamino acids versus no-carbon source control (n = 3 experimental replicates; Welch’s one-tailed t-test, *p < 0.05; **p < 0.001). Bars represent the mean. Error bars represent 95% confidence intervals. Sugar mix, 0.005% m v-1 each of D-glucose/D-ribose/D-xylose; AFEX, ammonia fiber expansion. No corrections were applied for multiple statistical tests because each growth experiment had its own negative controls. b Rank-abundance plot of putative GHs based on mean mRNA abundance following growth on eight different polysaccharides (n = 3 experimental replicates). GHs present in the culture G-10 metaproteome, multi-domain GHs, and expressed GHs are highlighted. c NMDS based on F. sacchari RNA-Seq data with PD1T grown on eight different polysaccharides (n = 3 experimental replicates; global one-way ANOSIM p < 0.001). d NMDS based on only CAZymes from the same RNA-Seq data (n = 3 experimental replicates; global one-way ANOSIM p < 0.001). Source data for the figure panels and p values for panel a are provided as Source Data files.
The isolate was imaged with light microscopy and cryo-electron tomography, revealing ovoid- to rod-shaped cells that were 0.9–1.3 µm wide and 1.6–3.6 µm long (Fig. 1c, e, f). Many cells remained attached following division and were Gram-stain-negative. Cryo-EM confirmed the diderm cell envelope structure, including an 8.7 ± 1.3 nm cytoplasmic membrane, a 55.5 ± 3.5 nm periplasm containing a ~12.5 nm peptidoglycan layer, and a 12.1 ± 1.3 nm outer membrane, despite phylogenetic evidence that F. sacchari belongs to the Terrabacteria (see below). Further descriptive information is in Supplementary Note 2.
F. sacchari expresses a high number and diversity of CAZymes
To further probe the metabolism of F. sacchari and mechanisms underlying polysaccharide degradation, the PD1T genome was co-assembled using Illumina and Oxford Nanopore data, resulting in a 3,952,382 bp circular chromosome and a 36,404 bp circular episome. Consistent with its aerobic respiration phenotype, the genome encoded an electron transport chain, including cytochrome c oxidase and the a-subunit of the high-affinity cytochrome bd ubiquinol oxidase (CydA), but CydB was absent. The PD1T CydA homolog belongs to a rare clade that lacks a quinol binding site and instead has a heme c binding motif and likely operates without CydB4. Casamino acid utilization is consistent with the presence of various peptidases, branched-chain amino acid permease, an OPT superfamily oligopeptide transporter, and dicarboxylate/amino acid:cation symporter. Genes encoding NiFe hydrogenase catalytic site synthesis and maturation proteins (hypACDEF) and a Group 3 d NiFe hydrogenase were annotated20, suggesting H2 production as an electron sink for NAD+ regeneration20,21. Additional genes for fermentation included L-lactate dehydrogenase, pyruvate:ferredoxin oxidoreductase, pyruvate formate lyase activating enzyme, acetyl-CoA synthetase, DNA-binding sugar fermentation stimulation protein A (sfsA), and fermentation-respiration switch protein (frsA). KEGG module completion and additional interpretations are in Supplementary Data 2 and Supplementary Note 1.
As originally noted for Ca. F. sacchari SAGs12, the defining signature of the PD1T genome is an extraordinary number and diversity of CAZymes. Based on conservative annotations17, the PD1T genome encodes 191 CAZymes, including 117 genes with GH domains spanning 49 GH families (Supplementary Data 3). This places F. sacchari among prokaryotes with the highest number and diversity of GHs17, with more GHs and GH families than other saccharolytic thermophiles such as Kallotenue papyrolyticum (78 GHs, 37 families), Dictyoglomus turgidum DSM6724 (62 GHs, 35 families), Caldicellulosiruptor bescii DSM6725 (61 GHs, 34 families), Thermotoga maritima MSB8 (54 GHs, 31 families), Saccharolobus solfataricus (31 GHs, 17 families), Geobacillus sp. WSUCF1 (28 GHs, 18 families), Pyrococcus furiosus DSM 3638 (20 GHs, 10 families), and Sulfolobus acidocaldarius (12 GHs, 7 families) (Supplementary Data 1). All of these CAZymes were expressed during growth on eight different polysaccharides as determined by RNA-Seq (191 CAZymes; Fig. 2b–d and Supplementary Data 4), or five different polysaccharides as determined by proteomics (168 CAZymes; Supplementary Fig. 9 and Supplementary Data 5). Together these expression data account for 3579 genes, or 97.4% of PD1T genes.
Among the most highly expressed enzymes in both transcriptomes and proteomes were many of the 31 PD1T enzymes with annotated GH109, 177, or 179 domains (Fig. 2b). These GH families are poorly characterized and biochemically unique. Only two enzymes from these families – both GH109s - have been previously biochemically and structurally characterized, and both proceed through anionic transition states with elimination and reduction rather than oxocarbenium ion-like transition states used by most GHs22. These enzymes use NAD+ as a cofactor and are the only known GHs with activity on both α and β glycosidic bonds as N-acetylgalactosaminidases23,24. Specifically, the only characterized GH179, encoded by Tannerella, is an exo-α-sialidase or α-N-acetylneuraminidase25, and the only characterized GH177, derived from a metagenome fragment from a Sulfolobaceae species, showed NAD+-dependent β-N-acetylglucosaminidase and β-glucosidase activities up to 85 °C26. Based on computed structure models (Supplementary Fig. 10) and protein alignments (Supplementary Data 6), representative PD1T GH109, GH177, and GH179 enzymes also have long, narrow Rossmann fold NAD+ binding pockets and large surface-facing, seven-stranded, antiparallel β-sheets likely involved in dimerization (Supplementary Note 3). However, phylogenetic analysis of F. sacchari PD1T enzymes and all other enzymes in these families from the CAZy Database revealed several lineages of PD1T enzymes that were distant from characterized enzymes (Supplementary Fig. 11). Eleven of these PD1T enzymes have TAT signal peptides, suggesting extracellular polysaccharide metabolism (Supplementary Data 3). The high abundance of these enzymes in PD1T, their conserved Rossmann folds, and their high expression levels is surprising because many hyperthermophilic enzymes replace NAD+ cofactors with functionally equivalent non-heme iron proteins27. However, we were not able to demonstrate activity of any of the 31 PD1T GH109, 177, or 179 enzymes when they were heterologously expressed in E. coli (see below and Supplementary Note 3).
Other CAZymes in the top quartile of RNA-Seq or proteome datasets included 30 different GH families and subfamilies, including five of the 13 multidomain CAZymes: CBM38 + GH116, CBM67 + CBM67 + GH78, PL22 + PL22_2, CBM67 + GH78, and GH148 + CBM4 + GH148 (Fig. 2b). Several of these multidomain CAZymes have motifs with possible roles in biofilm formation or adhesion (e.g., Ig domains28), carbohydrate binding (e.g., sugar-binding jelly roll fold domains29), or protein scaffolding (e.g., tetratricopeptide motifs30) (Supplementary Fig. 12). These domains suggest adhesion of some GHs to polysaccharides; however, cohesion and dockerin domains necessary for cellulosomes31 were absent.
Although several CAZymes were highly expressed under all cultivation conditions, most were differentially expressed, and both transcriptomes and proteomes were different when considering all transcripts/proteins or only CAZymes (Fig. 2, Supplementary Fig. 9, and Supplementary Data 4 and 5). The most distinct transcriptomes and proteomes according to PERMANOVA effect size (with each treatment compared to all others) were chondroitin sulfate (F = 3.91, transcriptome), gellan gum (F = 2.64 and F = 2.94, respectively), and xanthan gum (F = 3.88 and F = 2.77, respectively). Responses to these substrates may reflect modifications of the polysaccharide rather than the component sugars or glycosidic bonds. Chondroitin sulfate is a sulfated glycosaminoglycan, and gellan gum and xanthan gum are acetylated, and acetylated and pyruvylated, respectively. Acetylated and pyruvylated sugars are known components of the cell surfaces of thermophiles32, which may be natural substrates for F. sacchari. Additional interpretations from expression data are included in Supplementary Note 4.
In situ polysaccharide degradation and CAZyme expression
Although the preferred carbon sources of strain PD1T are polysaccharides, the sources of polysaccharides in 80 to 85 °C sediments that could support such a high abundance of F. sacchari are not obvious, given that well-developed photosynthetic mats form below 73 °C in hot springs33 and the dearth of plant material in GBS and most high-temperature springs. To determine whether F. sacchari can readily metabolize polysaccharides or other substrates in situ, we measured the assimilation of 13C atoms from potential substrates in fresh GBS sediment slurries using FISH-nanoSIMS and used the assimilation of 15N atoms from ammonia as a proxy for growth (Fig. 3a, b and Supplementary Fig. 13). These experiments revealed the O2-dependent uptake of 13C atoms from labeled starch and amino acids and concomitant uptake of ammonium within four hours. 13C atoms from starch were not assimilated by non-F. sacchari cells as a whole within four hours, suggesting direct assimilation by F. sacchari. No uptake of 13C atoms from bicarbonate was detected and very little uptake from a fatty acid mixture was detected after 24 h.

a Isotopic enrichments in F. sacchari identified by FISH (green bars) and other cells (gray bars). Each point reflects the percent of cellular 13C in a single cell following stable isotope probing with 13C-labeled substrates (n = number of cells within a single labeling experiment). Boxes represent 25th and 75th percentiles and central mark is the median. Vertical black lines show standard deviations. Asterisks show significance versus unlabeled controls (Wilcoxon rank-sum, *p < 0.05; **p < 0.001). b FISH showing F. sacchari cells (6-FAM, dotted circles), N content mapping (upper right), and nanoSIMS ion ratio images reflecting 13C assimilation (lower left) and 15N assimilation from 15N-ammonium (lower right) in representative F. sacchari cells and other cells (no 6-FAM label). Images shown are representative of 20 F. sacchari cells imaged after labeling with 13C- starch and 15N-ammonium for 24 h. Source data for both panels and p values for panel a are provided as a Source Data file.
To complement FISH-nanoSIMS experiments, we mapped peptides derived from culture G-10, which was fed casamino acids as the carbon source, to the PD1T genome. Among the 254 proteins detected were 35 CAZymes, including three of the multidomain proteins and single-domain enzymes belonging to 18 different GH families (Fig. 2b and Supplementary Data 7). Several of these CAZymes were also among the most highly expressed in PD1T pure cultures, demonstrated by both transcriptomes and proteomes, suggesting a common set of highly expressed CAZymes, even in culture G-10 where peptides were the primary carbon source. The G-10 metaproteome also included peptides that mapped to a nearly complete glycolytic pathway and TCA cycle. Although glycolysis and the TCA cycle could be used for both carbohydrate and amino acid catabolism, the expression of many CAZymes in culture G-10 and the much better growth of PD1T on carbohydrates over peptides suggests sugars may be important for F. sacchari metabolism even in the absence of added sugars. We suggest that F. sacchari may be nourished by polysaccharides produced by other hyperthermophiles in culture G-10 and, by extension, in natural springs as well. Possible sources of sugars that could be produced by hyperthermophiles include glycoprotein S-layers, lipopolysaccharides, or exopolysaccharides32,34,35. Some other F. sacchari proteins detected in culture G-10 suggest peptide catabolism, including M6, M28, S9, S16, S41A, and S55 family peptidases, M20/M25/M40 family metallo-hydrolase, trypsin-like peptidase domain-containing protein, oligogalacturonate lyase family protein, and OPT superfamily oligopeptide transporter, or acetate metabolism, such as acetyl-CoA synthase (AcdAB).
Together, the lab cultivation experiments, FISH-nanoSIMS, and metaproteomics results suggest F. sacchari can metabolize both autochthonous microbial polysaccharides and allochthonous plant polysaccharides, in addition to proteins and acetate. Protein and acetate metabolism are consistent with aspartate and acetate assimilation by Fervidibacter in Gongxiaoshe Hot Spring, China36. The saccharolytic activity of F. sacchari superficially contrasts with BONCAT experiments conducted in a Yellowstone hot spring, which showed a decrease in translation among highly abundant members of the genus Fervidibacter following cellobiose addition10. However, cellobiose and cellotriose are known to inhibit cellulase biosynthesis and activity of saccharolytic, fermentative thermophiles37,38. This has been suggested to be an evolutionary and/or physiological response to tight energy budgets associated with anaerobic cellulolysis, since ATP-dependent transport of oligosaccharides provides more sugar monomers per ATP than monomers39,40. A similar process may explain the inhibition of Fervidibacter in the BONCAT experiments, albeit under oxic conditions. The BONCAT experiments also showed decreased Fervidibacter translation in anaerobic incubations, consistent with the preferred aerobic metabolism of PD1T and O2-dependent activity in our FISH-nanoSIMS experiments.
GHs are hyperthermophilic and have diverse substrates
To investigate the roles of individual GHs in polysaccharide metabolism, we expressed 107 putative PD1T GHs in E. coli (Supplementary Data 8) and screened them for activity against 14 polysaccharides and three lignocellulose sources (Fig. 4a). Nine enzymes showed activity in these screens on agarose (Fsa16295Glu), carboxymethyl curdlan (Fsa16295Glu), carboxymethyl cellulose (Fsa01040Cel), chondroitin sulfate (Fsa07975Nga), β-glucan (Fsa16295Glu, Fsa01040Cel, Fsa11540Glu, Fsa02490Xyn, Fsa04715Cel), galactan (Fsa01670Gal), gellan gum (Fsa11540Glu, Fsa04715Cel), laminarin (Fsa16295Glu), Miscanthus (Fsa02490Xyn), xylan (Fsa02490Cel, Fsa01040Cel, Fsa15405Xyn), and xyloglucan (Fsa04715Cel) (Fig. 4a). These enzymes were classified by dbCAN3 and/or Prokka into GH3 (Fsa15405Xyn), GH5 (Fsa01040Cel), GH10 (Fsa02490Xyn), GH35 (Fsa05935Bga), GH50 (Fsa16295Glu and Fsa11540Glu), GH123 (Fsa07975Nga), GH147 (Fsa01670Gal) and GH148 + CBM4 + GH148 (Fsa04715Cel). Four of these enzymes that were tested were optimally active at 80–100 °C (Fig. 4b–e). Fsa16295Glu, Fsa11540Glu, Fsa07975Nga, and Fsa01670Gal are the first known hyperthermophilic enzymes in families GH50, GH123, and GH147. Several of these GHs were among the most highly expressed GHs in pure cultures and culture G-10 (Fig. 2, Supplementary Fig. 9, and Supplementary Data 4 and 5). However, despite the abundance of annotated GH109, 177, and 179 enzymes in F. sacchari, none were shown to be active (Supplementary Note 3). Collectively, the breadth of substrates and hyperthermophily of these GHs are consistent with the hyperthermophilic polysaccharide-degrading lifestyle of F. sacchari.

a Activity of F. sacchari GHs expressed in E. coli and screened against 15 polysaccharides. Red color represents activity compared with negative controls containing pET21b without a GH gene (n = 1). Asterisks show activities confirmed after repeating in triplicate (n = 3 experimental replicates; unpaired student’s t test p < 0.05). No adjustments were made for multiple statistical tests because significance was confirmed in triplicate separately versus the empty-vector control. b–e Temperature optima for selected GHs, tested after one hour of incubation compared to negative controls (gray circles). Boxes represent 25th and 75th percentiles and central mark is the mean. Shading shows optimal enzyme activity (n = 3 experimental replicates; ANOVA with post-hoc Tukey’s HSD p < 0.05). Source data and p-values are provided as a Source Data file.
F. sacchari belongs to phylum Armatimonadota
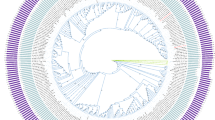
To better understand the ecology and evolution of F. sacchari and its relatives, we first required a better understanding of how F. sacchari is related to other microorganisms. To do this, we initially conducted maximum-likelihood analyses using the PD1T 16S rRNA gene, which placed PD1T into a monophyletic clade with Thermotogota, Candidatus Bipolaricaulota, and UBP14 (Supplementary Fig. 14). This result adds to conflicting results on the deep evolution of Fervidibacteria, which were originally placed at the root of the domain Bacteria11, later designated a candidate phylum12, or most recently assigned to phylum Armatimonadota within GTDB8. To resolve these conflicts, we gathered 64 metagenome-assembled genomes (MAGs) and SAGs from geothermal springs in the U.S. Great Basin, Yellowstone, the Canadian Rockies, Antarctica, Malaysia, and China, along with all high-quality Armatimonadota genomes in GTDB, a representative of each class in the domain Bacteria, and a representative of each order in the Terrabacteria (Supplementary Data 9 and 10). Maximum-likelihood analysis of three marker gene sets – bac120, bcgTree, and UBCG2 – indicated strong support for the monophyly of these genomes and placed them as a deep lineage within phylum Armatimonadota, consistent with the GTDB (Fig. 5 and Supplementary Fig. 15). We note that F. sacchari shares some phenotypic properties with all five Armatimonadota isolates described to date, including aerobic and saccharolytic metabolism, high abundance and diversity of GH109, 177, and 179 domain enzymes, low salt tolerance, and circumneutral pH for growth (Supplementary Data 11; Supplementary Note 2), and other shared properties predicted from representative Armatimonadota genomes, including simple chemosensory systems and moderately complex signaling networks, both suggestive of restricted ecological niches (Supplementary Note 1; Supplementary Figs. 16–18; Supplementary Data 12–17). Taxonomy was then assigned to Fervidibacteria genomes according to patterns of monophyly, relative evolutionary divergence8, average amino acid identity (AAI)41, and ANI42 (Fig. 5 and Supplementary Data 18). These genomes were thus placed into one class, one order, two families, three genera, and 19 species, with each species being restricted to one continent, and the largest genus, Fervidibacter, comprised of eight species in North America and five species in Asia. The wide distribution of two Fervidibacteria genera but limited distribution of their component species is consistent with fast dispersal rates of most bacteria43, although mechanisms of intercontinental dispersal of terrestrial thermophiles are poorly understood. We herein propose a taxonomy and nomenclature for Fervidibacteria within the phylum Armatimonadota by using a combination of the ICNP (Table 1) and SeqCode (Supplementary Data 19), with only high-quality MAGs encoding complete 16S rRNA genes as nomenclatural types44 (Supplementary Note 5).

a Average nucleotide identity (ANI) and average amino acid identity (AAI) among nomenclatural types for each Fervidibacteria species. The same color scheme used in a is used for taxa indicated with numbers. A full ANI and AAI matrix for all medium- and high-quality genomes is shown in Supplementary Data 18. b A maximum-likelihood phylogeny based on the concatenated, partitioned sequence alignment of 120 conserved bacterial marker sequences (bac120), with appropriate evolutionary models for each partition. Filled circles represent supported nodes at the species level and above based on >90% support from 1000 pseudoreplicates. Species are indicated with different colors, with species within the same genus indicated in different shades of the same color. Taxon names in bold and indicated with a star are the proposed nomenclatural types. Gray sequence IDs represent MAGs below quality standards for the SeqCode. c Phylogeny of Fervidibacteria species showing glycoside hydrolase data for each species based on the mean abundance of each category in high-quality genomes assigned to each species. The presence of terminal oxidase subunits for aerobic respiration and hydrogenases are shown in the matrix, with gene gain/loss events numbered. d Key of terminal oxidases and hydrogenases gained and lost; dashed circles show partial gains. Phylogenetic trees generated with different marker gene sets are shown in Supplementary Fig. 15 and an expanded ancestral state reconstruction is shown in Supplementary Fig. 19. Source data are provided as a Source Data file.
Evolution of polysaccharide catabolism in Armatimonadota
With the phylogeny and taxonomy of the Fervidibacteria in place, we examined common features of Fervidibacteria and Armatimonadota and constructed ancestral character states, focusing on polysaccharide catabolism. This revealed that the high number and diversity of GHs encoded by PD1T are typical of both Fervidibacteria and Armatimonadota, which ranged from 51 to 349 total GHs and 25-108 GH families (Fig. 5c and Supplementary Figs. 19 and 20, Supplementary Note 6). Putative GHs with GH177 and 179 domains were also prevalent across the phylogeny and can be considered defining features of the phylum. Despite conservation of polysaccharide catabolism, and the aerobic metabolism of all cultured Armatimonadota, the analysis revealed that anaerobic metabolism was ancestral within the phylum, with a progressive gain of both high-affinity (i.e., CydAB) and low-affinity (i.e., CcoNOP/CoxABC) terminal oxidases through horizontal gene transfer in several lineages. The class Fervidibacteria exemplified this pattern, with a gain of multiple copies of CoxABC in the common ancestor of the family Fervidibacteraceae (node 4) and the subsequent loss of those genes in Fervidibacter sinensis (node 1). In contrast, the family Thermosaccharophagaceae never gained aerobic terminal oxidases, but instead gained a group 1a respiratory H2-uptake [NiFeSe] hydrogenase, suggesting hydrogenotrophy45, and a group 4b formate-respiring, H2-evolving [NiFe] hydrogenase46 (node 8), yet these MAGs lacked formate dehydrogenase (FdhAB). The Thermosaccharophagaceae MAGs encoded the core of the methyl branch of the Wood-Ljungdahl pathway to convert formate to acetyl-CoA but lacked enzymes for the ATP- and acetate-forming reactions, which require acetyl-coA ligase (AcdAB) or the combination of phosphate acetyltransferase (Pta) and acetate kinase (AckA), differentiating them from mostly anaerobic marine sediment Armatimonadota recently classified into the novel classes “Zipacnadia” and “Hebobacteria”47.
Discussion
The exploration of microbial diversity via metagenomics and single-cell genomics has improved our perspective of biodiversity; however, as championed by Koch48, our ability to understand individual species is greatly improved by synergistic studies of pure cultures and their behavior within complex systems. By combining predictions based on single-cell genomes and MAGs, enrichment cultures, and isolation via optical tweezers, we obtained the first pure culture of a major lineage of bacteria that is globally distributed and locally abundant in terrestrial hot springs. Studies of the F. sacchari pure culture, its behavior in enrichment cultures and in situ, and related genomes from three continents confirmed predictions that Fervidibacter is saccharolytic and suggest an ancient and conserved role in polysaccharide metabolism throughout the class Fervidibacteria. Although other hyperthermophilic polysaccharide-degrading prokaryotes are known, most are strict anaerobes that are present at low abundance in situ but can bloom in response to added plant polysaccharides15,49,50. Fervidibacter is unusual based on its high abundance in mineral sediments and its aerobic and hyperthermophilic metabolism. The high abundance of an aerobic hyperthermophilic polysaccharide degrader is surprising given the dearth of plant-derived polysaccharides in most high-temperature springs, the low solubility of O2 at high temperatures, and the reducing chemistry of most hydrothermal fluids51.
Saccharolobus solfataricus and Sulfolobus acidocaldarius are also aerobic hyperthermophiles that degrade polysaccharides such as cellulose, xyloglucan, and starch52,53,54, although they encode only 31 and 12 annotated GHs, using conservative annotations in dbCAN3. They can also be abundant in terrestrial springs, although their ecological niche is distinct from Fervidibacteria because they are restricted to vapor-dominated acid-sulfate springs. Aerobic polysaccharide metabolism imparts substantial differences compared to better-studied fermentative, saccharolytic hyperthermophiles. For example, coupling sugar catabolism with the reduction of O2 translates into ~10 times more ATP per hexose as compared to a mixed acid fermenter (~30 ATP vs 3 ATP, respectively)55 and would not require co-located hydrogenotrophs as electron sinks. Also, energy-limited anaerobes typically anchor CAZymes to the cell wall, which conserves expensive enzymes but limits the physical range of substrate acquisition56,57,58. Alternatively, aerobes typically secrete soluble cellulolytic enzymes into the local environment, which enables acquisition of substrate further from the cell but also results in the loss of some enzymes. This tradeoff is competitive due to higher-yielding energy metabolism of the aerobe56,57,58. Overall, the energetic advantages of aerobic polysaccharide metabolism may contribute to the abundance of Fervidibacter in situ, particularly in springs limited by low concentrations and fluxes of inorganic electron donors13,59,60.
This study also suggests polysaccharide catabolism is prevalent and ancient across the phylum Armatimonadota, and we suggest polysaccharide catabolism is a defining feature of the phylum. We also show that GH177 and 179 enzymes are prevalent throughout the phylum, although none of these Armatimonadota enzymes have been biochemically characterized to date. A better understanding of their substrates and functions may improve our understanding of this poorly characterized phylum and improve their culturability.
Methods
Enrichment, isolation, and pure cultures
Initially, 22 different enrichment cultures (G-1 to G-22, where ‘G’ refers to GBS) were prepared with varying carbon sources, terminal electron acceptors, and headspace gases (i.e., H2 and O2 concentrations), inoculated with ~80 °C sediments, and monitored for microbial growth. The initial enrichment medium was based on the “spring water medium” described in ref. 9. Briefly, 0.5X GBS salts synthetic medium61 was combined 1:1 with GBS water passed through a 0.22 µm Sterivex polyethersulfone filter (Millipore Sigma). GBS salts synthetic medium contains, per liter of 18.2 MΩ⋅cm water: 3 g NaCl, 0.3 g Na2SO4, 0.15 g KCl, 0.015 g CaCl2·2H2O, 0.123 g MgSO4·7H2O, 0.02 g NH4Cl, and 5 mL of a trace mineral solution62. The trace mineral solution contained per liter: 2.5 g Na2-EDTA, 2.2 g KOH, 1.1 g FeSO4·7H2O, 0.029 g ZnSO4·7H2O, 0.098 g MnCl2·4H2O, 0.031 g H3BO3, 0.024 g CoCl2·6H2O, 0.017 g CuCl2·2H2O, and 0.024 g NaMoO4·2H2O and was adjusted to pH 6.5 with HCl. Following combination, the medium was sparged with N2 gas for one hour and 50 mL aliquoted into acid-washed 165-mL borosilicate glass serum bottles in an anaerobic chamber.
The bottles were sealed with butyl rubber stoppers and aluminum crimps, the headspace was exchanged with N2 by three cycles of vacuuming to 25 mmHg and repressurizing to 15 psi with N2, and the medium bottles were sterilized by autoclaving. Prior to inoculation, the following were added at the indicated final concentrations from 0.2 µm-filtered, anoxic stocks: 0.002% m v-1 yeast extract, 0.1 mM Na2HPO4, 0.2 mM NaHCO3, 0.16X strength Wolfe’s vitamins63, 2 mM HEPES buffered to pH 7.5, and corresponding substrate as described in Supplementary Fig. 1. Filter-sterilized air was added by syringe to the headspace at described volume. The medium was inoculated with a sediment slurry collected from the top ~1 cm of sediment at the “A” site of GBS14 (40°39’41.3”N, 119°21’58.1”W; 85 °C, pH 7.1 at time of collection) on July 2, 2016, transferred to the laboratory at ambient temperature, and incubated at described temperature the following day. A stable mixed culture was maintained over two years by transferring 1:100 dilutions to fresh media every 3–4 weeks for the first four transfers, and then every 3 weeks thereafter. Culture composition was monitored by 16S rRNA gene amplicon sequencing using the Illumina MiSeq platform64 at initial sampling and two subsequent transfers (Supplementary Fig. 1). Sequences were taxonomically identified using BLASTN against the nt_prok database65 or using SINA Search and Classify66, and common contaminants were removed67. Stable mixed cultures of select groups were maintained over several years by transferring 1:100 v v-1 dilutions to fresh media every 3–4 weeks.
To target Fervidibacter enrichment with culture G-10 as the inoculum, the “spring water medium” was used and the following were varied: (i) O2 concentrations (0, 5, 10, 15, and 21% v v-1 headspace, with 0.02% m v-1 casamino acids and 7, 14, and 21 days of incubation time); (ii) incubation times (2, 4, 6, 14, and 21 days, with 0.02% m v-1 casamino acids and 10% v v-1 O2 headspace); and (iii) carbon sources in place of the casamino acids (0.2% m v-1 carboxymethyl cellulose, 0.02% m v-1 casamino acids, 0.02% m v-1 casamino acids with 10% v v-1 H2 headspace, 1% m v-1 formate, 0.2% m v-1 myo-inositol, 0.2% m v-1 locust bean gum, sugar mix of 0.025% m v-1 each D-glucose/D-mannose/D-xylose, 0.2% m v-1 xylan, and 0.05% m v-1 xyloglucan, with 10% v v-1 O2 headspace (unless otherwise stated) and 4 and 7 days of incubation time. For each enrichment and time point, cells from four mL of culture were pelleted and preserved at −80 °C. DNA was extracted using the FastDNA Spin Kit for Soil (MP Biomedicals). Fervidibacter abundance in each condition per culture volume was monitored by quantitative PCR using the 16S rRNA gene primers OctSp_For1 (5’-GCTCAACGGGCAGAGGTCG-3’) and OctSp_Rev2 (5’-ACGGGGAGGGTCGARACTC-3’). These primers amplify a ~239 bp product and are specific to, and nearly comprehensive for, the genus Fervidibacter, with >1 mismatch only to F. canadensis, F. japonicum, and Fervidibacter sp. 6. Linearized plasmid containing the near-full length Ca. F. sacchari 16S rRNA gene was used as a standard. Quantitative PCR was performed in 96-well plates in a CFX96 Real-Time PCR Detection System (Bio-Rad) using PerfeCTa SYBR Green SuperMix (Quantabio). The cycling conditions were as follows: initial denaturation at 95 °C for 3 min followed by 36 cycles of denaturation (95 °C for 30 s), annealing (60.5 °C for 60 s), and extension (72 °C for 45 s), and finally 72 °C for 60 s and 55 °C for 60 s, followed by a melt curve analysis from 55 to 95 °C to check for primer specificity.
Cells were sub-cultured from July to December 2018 under the best conditions (10% O2 headspace; four days incubation time; and casamino acids, carboxymethyl cellulose, locust bean gum, xyloglucan, or xylan as sole carbon sources). After confirming the majority of the culture was Fervidibacter cells by FISH (described below) and 16S rRNA gene amplicon sequencing, four rounds of dilution-to-extinction of the locust bean gum culture were performed. Using the highly Fervidibacter-enriched culture with locust bean gum as the sole carbon source (Fig. 1e), cell sorting using optical tweezers and a sterile polydimethylsiloxane microfluidic chip mounted on a Nikon Eclipse Ti-U microscope was performed using a 100 mW, 976 nm laser to obtain ten single cells into sterile chambers68,69 and the cells were flushed into syringes using GBS salts synthetic medium. Each cell was used to inoculate “spring water medium” containing either xyloglucan or locust bean gum. Of the ten cultures, only a single xyloglucan culture resulted in growth, yielding cells of a single morphotype designated strain PD1T.
For pure culture experiments, Fervidibacter medium was used to grow cells of F. sacchari PD1 T. Fervidibacter medium consists of GBS salts synthetic medium as described above with the following additions at the indicated final concentrations: (i) 0.02% m v−1 xyloglucan as the electron donor and carbon source; (ii) a trace element solution containing 100 µM Na2SeO4, 10 µM Na2WO4·2H2O, 1 µM Na2HAsO4·7H2O, 100 nM Ni(NH4)2(SO4)2·6H2O, and 10 nM VOSO4; (iii) vitamins supplied as 1X Wolfe’s vitamins neutralized with 100 µM sodium phosphate, which also served as the source of phosphorous63; and (iv) 2 mM HEPES buffer, pH 7.5 (at 80 °C). Air was added to the N2 headspace to 50% m v-1 final concentration so that the O2 concentration was 10.5%. Cells were routinely grown at 80 °C with 100 rpm of orbital shaking for 4–7 days.
Cryo-electron microscopy
Cell morphology was observed using cells grown on starch at 80 °C after 4–5 days of cultivation. Cells were observed by phase-contrast microscopy with a Zeiss Axioplan 2 imaging microscope or by cryo-electron microscopy (2D, cryo-EM) and cryo-electron tomography (3D, cryo-ET). Briefly, cells were grown to late-exponential phase using Fervidibacter medium (described above) with xyloglucan replaced by soluble starch. Cultures were run through a 5-µm polyethersulfone filter to remove minerals and other insoluble matter. Three µL aliquots were applied to glow-discharged R2/200 mesh copper Quantifoil grids, pre-blotted for 60 s, then blotted for 2 s. Grids were pre-blotted and blotted using an automated Leica EM GP plunge freezer set to 21 °C and 95% humidity and plunge-frozen in liquid ethane. Cells were imaged using a 120-kV Talos L120C transmission electron microscope housed at the Netherlands Centre for Electron Nanoscopy.
Genome sequencing and assembly
DNA was isolated from strain PD1T by using a CTAB procedure (https://jgi.doe.gov/wp-content/uploads/2014/02/JGI-Bacterial-DNA-isolation-CTAB-Protocol-2012.pdf). De novo genome assembly required three separate sequencing runs using: (i) the Illumina MiniSeq platform with 2 × 150 bp paired-end reads at The Sequencing Center (Fort Collins, CO); (ii) the Illumina NovaSeq S4 platform at the Joint Genome Institute (JGI); and (iii) an Oxford Nanopore MinION Mk1B device using a FLO-MIN106 flow cell, using sequencing libraries prepared with the Native Barcoding Expansion (EXP-NBD104) and Ligation Sequencing (SQK-LSK109) kits using Nanopore protocol version NBE_9065_v109_revAH_14Aug2019. Raw reads from the three sequencing runs were combined in a hybrid assembly with SPAdes v3.15.470 using the --nanopore option for the uncorrected long reads and both sets of paired reads generated from Illumina technologies. The final genome sequence was annotated using the NCBI Prokaryotic Genome Annotation Pipeline71. CAZymes were additionally annotated using the dbCAN3 meta server and HMMdb v12.0 and conservatively requiring CAZymes to be identified by at least two tools (HMMER vs. dbCAN, DIAMOND vs. CAZy, and/or HMMER vs. dbCAN-sub)17.
Metagenome sequencing, assembly, and binning
For the G-10 lab enrichment culture, extracted DNA was sequenced using the Illumina MiSeq platform (2×250 bp) and Oxford Nanopore Technology. Briefly, for long-read sequencing, high-molecular weight DNA was extracted using the JGI bacterial CTAB extraction protocol (https://jgi.doe.gov/user-programs/pmo-overview/protocols-sample-preparation-information/jgi-bacterial-dna-isolation-ctab-protocol-2012/), with modifications consisting of three freeze-thaw cycles following lysozyme treatment, in a dry ice/ethanol bath and a water bath at 65 °C for three minutes per cycle. RNase A (Promega, Madison, WI, USA, catalog number: A7973) was used for denaturation of ribonucleic acids, and an additional extraction step following RNAse treatment with chloroform/isoamyl was done. The sequencing library was prepared with the EXP-NBD104 Native Barcoding Expansion Kit (Oxford Nanopore Technologies, Oxford, UK) and the SQK-LSK109 Ligation Sequencing Kit, following the manufacturer’s instructions and sequenced with a MinION Mk1B device (Oxford Nanopore Technologies, Oxford, UK) with a FLO-MIN106 flow cell. Sequence reads for the G-10 culture were then assembled by first assembling all long-read data using metaFLYE v. 2.8.272 using the --nana-raw and --meta options. The obtained assembly was then used as untrusted contigs for assembly of the Illumina reads using metaSPAdes v. 3.12.173.
A total of 27 Fervidibacteria MAGs were available in public databases based on previous studies of geothermal springs in the U.S. Great Basin7,12,15,74, Tengchong, China7, Yellowstone National Park7,75, a Japanese Gold Mine76, SKY Hot Spring, Malaysia77, and Mt. Erebus, Antarctica7. Five others from GBS, Octopus Spring, Gongxiaoshe, and Jinze Spring were derived from existing assemblies of publicly available metagenomes that were binned as part of this study using either MetaWatt v1.778 or ESOM79 or a combination of the two, as previously described80.
The largest source of Fervidibacteria MAGs (36 total) was metagenomes from geothermal springs from Tengchong, China. For these metagenomes, extracted DNA was sequenced on the Illumina HiSeq4000 platform, in a paired-end configuration at Beijing Novogene Bioinformatics Technology Co., Ltd (Beijing, China). Four steps were performed to obtain the clean reads: (i) adapter-contaminated reads were eliminated; (ii) duplicated reads generated by PCR amplification were deleted; (iii) reads with a significant excess of “N” (≥10% of the read) were removed; and (iv) reads with quality score less than 20 at the 3′ end were trimmed. The high-quality reads from each sample were de novo assembled using SPAdes (version 3.9.0)70 individually with the following parameters: -k 33, 55, 77, 99, 111 --meta. Reads were mapped to scaffolds using BBMap (version 38.85; http://sourceforge.net/projects/bbmap/). Genome binning was conducted on scaffolds with length >2.5 kbp using MetaBAT281,82. All MAGs were subjected to quality checks with CheckM v1.0.12 and CheckM2 v1.0.183. Ribosomal RNA genes and tRNA genes were identified with barrnap v0.9 and tRNAscan-SE v2.0.1184, respectively.
Taxonomy was assigned to reads from the 85 °C GBS sediment metagenome (Fig. 1b) by using Kaiju85. Briefly, reads were searched against NCBI NR database and then the unclassified reads were subsequently searched against a custom reference database of high-quality representative genomes comprised of MAGs or isolates from GBS. MAGs were published previously or were assembled and binned as described above (unclassified Desulfurococcales (RAST 6666666.879388), unclassified Geoarchaeales (RAST 6666666.752844), Calditenuis sp. (RAST 3071572.3), Wolframiiraptor gerlachensis (GCA_021323375.2). Isolate genomes included F. sacchari PD1T, (GCA_030520105.1), Pyrobaculum sp. (RAST 2276.22), and Thermoflexus hugenholtzii JAD2T (GCF_900187885.1).
Differential transcriptomics analysis
Cells of F. sacchari were grown in Fervidibacter medium with either 0.05% m v-1 chondroitin sulfate (Alfa Aesar), 0.05% m v-1 β-glucan from oat (Megazyme), 0.05% m v-1 gellan gum (Serva), 0.05% m v-1 locust bean gum (Spectrum), 0.04% m v-1 starch from potato (J.T. Baker), 0.05% m v-1 xanthan gum (Sigma), 0.02% m v-1 xyloglucan from tamarind (CarboMer), or 0.02% m v-1 ammonia fiber expansion-pretreated corn stover (DuPont) as sole carbon sources. Each culture was used as inoculum for same-condition media (1:50 dilution) over at least three transfers. Following the last transfer, cultures were grown in triplicate under each condition to mid-exponential phase. Cultures were rapidly cooled in a dry ice/ethanol bath and pelleted at 5000 × g for eight minutes at 4 °C using a Beckman Coulter J2-HS centrifuge. Supernatant was removed and cell pellets were quickly frozen and stored at −80 °C until RNA extraction. RNA was isolated using the RNeasy Mini Kit (Qiagen) using the enzymatic lysis procedure and on-column DNase treatment. Quantity was measured with a Qubit 3.0 fluorometer, and quality was measured with an Agilent 2100 Bioanalyzer and NanoDrop 1000. Samples with sufficient biomass (≥1 µg total RNA) and RIN (6.5 or higher) were sent to the JGI for library preparation, sequencing, and mapping. An input of 100 ng of total RNA was used to deplete rRNA using the QIAseq FastSelect 5S/16S/23S Kit (Qiagen). Using TruSeq stranded mRNA kit (Illumina), the 300–400 bp heat-fragmented RNA was reverse transcribed to create the first strand of cDNA with random hexamers and SuperScript II Reverse Transcriptase (Thermo Fisher Scientific) followed by second strand synthesis. The double stranded cDNA fragments were treated with A-tailing, ligation with JGI’s unique dual indexed adapters (IDT) and enriched using 10 cycles of PCR. The prepared libraries were quantified using KAPA Biosystems’ next-generation sequencing library qPCR kit and run on a Roche LightCycler 480 real-time PCR instrument. Sequencing of the flowcell was performed on the Illumina NovaSeq sequencer using NovaSeq XP V1.5 reagent kits, S4 flowcell, following a 2 × 151 indexed run recipe. Quality-filtered reads were mapped to the PD1T reference genome using STAR aligner 2.7.10b_alpha_23-06-0986 with the maximum intron size parameter set to 1. Transcripts were mapped to coding sequences from the GCF_030520105.1 assembly using FeatureCounts 2.0.087. Differential expression was calculated using DESeq2 1.40.288. Loci wherein fewer than 10 transcripts were mapped across the entire dataset were omitted and counts were transformed using a regularized log2 transformation. For the sake of ordination, the log2-transformed transcript counts were variance-scaled, and the minimum value added so that all counts were non-negative. The transcriptomes were ordinated using the metaMDS function from the R package vegan 2.6.489, according to Euclidean distance calculated from the positive, scaled feature counts.
Differential proteomics analysis
Cells of F. sacchari were grown in Fervidibacter medium with either 0.05% m v-1 β-glucan from oat (Megazyme), 0.05% m v-1 gellan gum (Serva), 0.05% m v-1 locust bean gum (Spectrum), 0.04% m v-1 starch from potato (J.T. Baker), or 0.02% m v-1 xyloglucan from tamarind (CarboMer) as a sole carbon source. Each culture was used as inoculum for same-condition media (1:50 dilution) over at least three transfers. Cultures were then grown in triplicate under each condition. Following growth of the last transfer to early stationary phase, culture was cooled quickly in a dry ice/ethanol bath and pelleted at 6000 × g for 15 min at 4 °C using a Beckman Coulter J2-HS centrifuge. Cell pellets and culture supernatant were separated into different centrifuge tubes and stored at −80 °C until processing. Soluble cellular proteins were collected from the pellet using the B-PER Complete Protein Extraction Reagent (ThermoFisher). Soluble secreted proteins were collected from the supernatant by methanol/chloroform precipitation as previously described90. The mass of each sample and fraction was measured using the EZQ Protein Quantitation Kit (ThermoFisher). Both cellular and secreted fractions of each replicate were combined 1:1 by mass (19 µg each) at the Mick Hitchcock, Ph.D. Nevada Proteomics Center (Reno, NV, USA). Protein extracts were digested with a trypsin/Lys-C protease mixture using the EasyPep MS Sample Prep Kit (Thermo Scientific). Samples were analyzed using an UltiMate 3000 RSLCnano system (Thermo Scientific). Peptides were trapped prior to separation on a 300 µm i.d. x 5 mm C18 PepMap 100 trap (Thermo Scientific) for 5 min at 10 µL min-1. Separation was performed on a 50 cm uPac C18 nano-LC column (PharmaFluidics) on an EasySpray source (Thermo Scientific) fitted with a 30 µm ID stainless steel emitter (PepSep) at 350 nL min-1 using a gradient from 1 to 45% for 60 min. Data-independent analysis (DIA) was performed using an Eclipse Tribrid Orbitrap mass spectrometer (Thermo Scientific). A chromatographic library was generated using the Spectronaut v.16.5.221115 (Biognosys) search engine Pulsar to generate hybrid (DIA and data-dependent analysis (DDA)) spectra using an aliquot from each sample to form a pool of peptides. The library was generated using a combination of six gas phase fractions (GPF) and full scan DDA of the biological sample pool. GPF acquisition used 4 m/z precursor isolation windows in a staggered pattern at a resolution of 60,000. Automatic gain control (AGC) target was set to custom with a normalized target of 1000%, maximum injection time set to dynamic with a minimum of nine points across the peak, and normalized collision energy set to 33 using higher-energy collision dissociation. Expression data were normalized using Spectronaut v.16.5.221115 (Biognosys). Differentially expressed proteins were identified using R package proDA 1.14.091. For ordination, normalized protein counts were variance-scaled, and the minimum value added so that all counts were non-negative. The proteomes were ordinated using the metaMDS function from the R package vegan 2.6.489, according to Euclidean distance calculated from the positive, scaled protein counts.
Stable isotope labeling
Stable isotope labeling was performed on-site at GBS in July 2017. GBS sediment from the “C” site14 was added to pre-prepared 60-mL amber serum vials containing 8 mL of autoclaved GBS water with an air (aerobic) or N2 (anaerobic) headspace, generating 10 mL microcosms. GBS water was sparged with N2 prior to sterilization for anaerobic incubations. Various individual 13C-labeled substrates (Cambridge Isotope Laboratories) were then added from concentrated, anaerobic stocks to the microcosms at 0.001% (m v-1) final concentration: algal amino acids, algal starch, bicarbonate, or an equal-mass mixture of acetate, propionate and butyrate; a control with no label added was also performed and did not yield enriched cells. Additionally, 0.5 mM 15N-NH4Cl was added to all microcosms. Sealed microcosms with labeled substrates were incubated at 75 °C for 4 or 24 h. After incubation, microcosms were cooled quickly in ice water, shaken vigorously for 30 s, and poured into 15 mL conical tubes. Tubes were centrifuged for 30 s at 11 × g in a clinical centrifuge to pellet large sediment particles. Supernatant was then centrifuged for 5 min at 16,100 × g to pellet cells. Pellets were pooled in 0.25 mL of 1×PBS, and 0.5 mL of freshly prepared 3% paraformaldehyde (PFA) was added and mixed. Samples were fixed for 1 h on ice. After fixation, tubes were centrifuged for 5 min at 16,100 × g, and pellets were washed twice with 1×PBS, resuspended in 200 µL of 50% ethanol, and stored on ice (in the field) and then at −20 °C in the lab. In the lab, cells were further separated from sediments by using Nycodenz density cushion centrifugation. Cells harvested from the aqueous-Nycodenz interface were harvested by centrifugation for 5 min at 16,100 × g, washed twice in water, and resuspended in 50% ethanol.
FISH
Detection of F. sacchari cells was performed with FISH using the cyanine-3 (cy-3) or 6-carboxymethylfluorescein (6-FAM) probe Ferv_891, which is specific to, but not universal for, Fervidibacteria 16S rRNA: 5’ GCGCTTATTGGTTTCCCTGCG (Integrated DNA Technologies). It was designed based on multiple sequence alignment of 16S rRNA genes of Fervidibacteria and other bacteria and checked using the Probe Match function of the Ribosomal Database Project92 and checked for specificity using SILVA’s Testprobe 3.0 tool93. Cells from laboratory enrichments were fixed using a 1% PFA solution for 30 min, followed by storage in 50% v v-1 ethanol at −20 °C until use. Cells from GBS sediment microcosms to be used for nanoSIMS were fixed using 1% PFA for 1 h and stored identically. Fixed cells were spotted and air dried on either 12-well slides or custom synthesized 14-well indium tin oxide (ITO)-coated slides rather than using filters, followed by a series of ethanol dehydration steps. After drying, hybridization was performed for at least 8 h at 46 °C using a solution containing 20% formamide, 0.9 M NaCl, 20 mM Tris (pH 8), and 0.01% SDS, followed by washing at 48 °C using a solution containing 0.225 M NaCl, 5 mM EDTA, 20 mM Tris (pH 8), and 0.01% SDS. Cells were counterstained with 1 µg mL-1 DAPI solution and mounted with a 1:4 Vectashield/Citifuor mixture. Cells from the laboratory enrichments were visualized with an Axio Imager.M2 (Zeiss) using ZEN software. Isotopically labeled cells were visualized with a Leica DM5500B microscope running MetaMorph software with a 100× magnification dry immersion objective. Fluorescence and brightfield images were collected at diverse locations with FISH-positive cells. The X-Y and fiducial locations were noted to enable navigation in the nanoSIMS.
NanoSIMS
ITO-coated well slides were analyzed on a CAMECA NanoSIMS 50 at Lawrence Livermore National Laboratory. Fiducial locations and locations of the FISH-positive cells were found with a charge-coupled device camera using X-Y coordinates and the real time imaging unit. The primary Cs+ ion beam was set to 1.5 pA, corresponding to an approximately 100 nm diameter beam diameter at 16 keV. Rastering was performed over 20 × 20 μm analysis areas with a dwell time of 1 ms pixel–1 for 19–30 scans (cycles) and generated images containing 256 × 256 pixels. Sputtering equilibrium at each analysis area was achieved with an initial beam current of 90 pA to a depth of ~10 nm. After tuning the secondary ion mass spectrometer for mass resolving power of ~7000, secondary electron images and quantitative secondary ion images were simultaneously collected for 12C2–, 13C12C-, 12C14N-, and 12C15N- on individual electron multipliers in pulse counting mode. All nanoSIMS datasets were initially processed using L’Image (http://limagesoftware.net, August 2021) to perform deadtime and image shift correction of ion image data before creating 13C12C/12C2 and 12C14N/12C15N ratio images, which reflected the level of 13C and 15N incorporation into biomass. Regions of interest for isotopic ratio quantification were drawn manually around each cell. A cell was considered isotopically enriched, and therefore anabolically active, if the isotopic composition exceeded three times the standard deviation of the mean isotopic composition of unlabeled (non-Fervidibacteria) cells.
Metaproteomics
The G1-10 culture containing F. sacchari was grown in “spring water medium” as described above with 0.02 % m v-1 casamino acids and 10% v v-1 O2 headspace for three weeks at 80 °C. Cells from 50 mL of culture were pelleted by centrifugation at 10 min at 9300 × g and 4 °C in 50 mL conical tubes and stored at −80 °C prior to protein extraction based on a method by Pan and Banfield94. Cell pellets were resuspended in 1.2 mL of SDS lysis buffer (5% SDS, 50 mM dithiothreitol, and 50 mM Tris-HCl pH 8), transferred to a Lysis Matrix E tube used for DNA extractions (MP Biomedicals soil DNA isolation kit), and processed in a homogenizer three times for 30 s each at 4.5 m s-1. Tubes were then incubated in a simmering water bath for 15 min and centrifuged for 10 min at 16,000 × g. Proteins in the resulting supernatant were precipitated by mixing at a 1:5 ratio with acetone and incubation at −20 °C overnight. Precipitated proteins were pelleted by centrifugation for 10 min at 4 °C and 16,000 × g, washed once with 90% acetone, and then stored at −20 °C with 20 µL 90% acetone remaining prior to shipping to the UC Davis proteomics center. Protein concentrates were dried and then the protein pellets were solubilized in 50 µL SDS solubilization buffer (5% SDS, 50 mM TEAB, pH 7.55) and subjected to clean-up, reduction, alkylation, and tryptic proteolysis via filter-aided sample preparation on suspension-trap (ProtiFi) devices. Data-independent analysis (DIA) was performed via ultra-high-performance liquid chromatography (UHPLC) (nanoElute) coupled to a trapped ion mobility-time of flight (timsTOF) Pro operated in parallel accumulation-serial fragmentation mode. Liquid chromatography was performed on a UHPLC nano-flow Easy nLC (Bruker Baltonics), on a PepSep 150 mm × 25 cm C18 column (PepSep, Denmark; 1.5 μm particle size; 100 Å pores), with a 45-minute gradient. This was directly on-line with hybrid timsTOF Pro (Bruker Daltonics, Bremen, Germany) with a modified nano-electrospray ion source (CaptiveSpray, Bruker Daltonics). The acquisition scheme consisted of four 25 m/z precursor windows per 100 ms TIMS scan, 400–1200 m/z. Sixteen TIMS scans, creating 64 total windows, layered the doubly and triply charged peptides. Raw files were processed with Spectronaut v. 18 (Biognosys, Zurich, Switzerland) using DirectDIA analysis mode. Mass tolerance/accuracy for precursor and fragment identification was set to default settings. Peptides were matched against a custom database of high-quality genomes that were derived from the G-10 culture metagenomes, assembled and binned as described above (unclassified Desulfurococcales (RAST 6666666.879388), unclassified Calditenuis (RAST 3071572.3), or unclassified Geoarchaeales (RAST 6666666.752844), or from isolates from GBS (F. sacchari PD1T, (GCA_030520105.1), Pyrobaculum sp. (RAST 2276.22), and Thermoflexus hugenholtzii JAD2T (GCF_900187885.1)), along with a database of 112 common laboratory contaminants. A maximum of two missing cleavages were allowed, the required minimum peptide sequence length was 7 amino acids, and the peptide mass was limited to a maximum of 4600 Da. Carbamidomethylation of cysteine residues was set as a fixed modification, and methionine oxidation and acetylation of protein N-termini as variable modifications. A decoy false discovery rate (FDR) at less than 1% for peptide spectrum matches and protein group identifications was used for spectra filtering (Spectronaut default). Decoy database hits, proteins identified as potential contaminants, and proteins identified exclusively by one site modification were excluded from further analysis. F. sacchari proteins were annotated using Eggnog-mapper v2.1.995 and the NCBI Prokaryotic Genome Annotation Pipeline71.
GH gene synthesis, expression, purification, and activity
F. sacchari genes encoding putative GHs were codon optimized with an E. coli codon usage table and the balanced strategy using BOOST96, synthesized, and assembled into pET21b (Novagen, Madison, WI) with NEBuilder HiFi Assembly kit (New England Biolabs, Ipswitch, MA) with an N-terminal His6-tag, a GB1 solubility tag, and a TEV protease cleavage site. Plasmids were transformed into E. coli T7 Express cells (New England Biolabs, Ipswich, MA), which were incubated in LB broth with 100 µg mL-1 ampicillin at 37 °C with shaking at 225 rpm until reaching an OD600 of 0.6–0.8 at which time isopropylthio-β-galactoside (0.5 mM) was added followed by overnight incubation at 37 °C with shaking at 150 rpm. Cells were centrifuged at 16,100 × g, resuspended in lysis buffer (50 mM Tris-HCl, 100 mM NaCl, pH 7.0), lysed by sonication for 40 s with 40% cycle and level 4 output with a Branson 450 Sonifier (Branson Ultrasonics, Brookfield, CT), and clarified by centrifugation at 16,100 × g. Enzymes were purified by heating the soluble fraction to 80 °C for 30 min, followed by centrifugation at 16,100 × g to remove denatured host proteins in the pellet. Expression, purity, and solubility were assessed using 7.5% SDS-PAGE. All enzymes expressed well; however, 31 were only visible in the insoluble fraction. Those enzymes with little or no enzyme visible in the soluble fraction were still tested and two showed enzymatic activity (Fsa05935Bga and Fsa04715Cel).
To identify active GHs for further study, soluble enzymes were tested for activity as single replicates using substrates used by F. sacchari, each at 0.5% m v-1: chondroitin sulfate (Alfa Aesar), colloidal chitin97 (Beantown Chemicals), brown algae fucoidan (BestVite), lupin galactan (Megazyme), gellan gum (Serva), karaya gum (Sigma), locust bean gum (Spectrum), xanthan gum (Sigma), birch wood xylan (Sigma), tamarind xyloglucan (CarboMer), oat β-glucan (Megazyme), oyster glycogen (TCI), potato starch (J.T. Baker), and ammonia fiber expansion (AFEX)-pretreated corn stover, Miscanthus, and sugarcane bagasse (DuPont). Substrates and enzymes or a sham protein preparation from an empty vector control were mixed 1:1 in microplate wells (final volume 40 µL), incubated overnight at 80 °C, and reducing sugars were quantified using a 3,5-dinitrosalicylic acid (DNS) assay98 against a standard curve generated using glucose at 0, 5, 10, 15, 20, and 25 mM.
The initial activity screen revealed four active GHs: Fsa01040Cel, Fsa02490Xyn, Fsa15405Xyn, and Fsa16295Glu. Fsa01040Cel and Fsa02490Xyn were further purified using immobilized metal affinity chromatography by loading soluble fractions onto HisPur™ Ni-NTA Spin Columns (Thermo Scientific, Rockford, IL, USA) pre-equilibrated with 20 mM sodium phosphate buffer containing 300 mM sodium chloride and 10 mM imidazole. The columns were then washed with 20 mM sodium phosphate buffer containing 300 mM sodium chloride and 25 mM imidazole, and the enzymes were eluted using 20 mM sodium phosphate buffer containing 300 mM sodium chloride and 250 mM imidazole. To remove imidazole, the eluted fractions were buffer exchanged into lysis buffer using Pierce™ Protein Concentrators with 10 K molecular weight cut-off (Thermo Scientific, Rockford, IL, USA). Protein concentrations were determined by a Pierce™ BCA Protein Assay Kit (Thermo Scientific, Rockford, IL, USA). The initial activities were verified in triplicate, and additional substrates were tested based on known activities from each enzyme’s GH family: carboxymethyl cellulose (Spectrum Chemical, New Brunswick, NJ, USA; 0.5% m v-1) (Fsa01040Cel, GH5); beechwood xylan (Megazyme, Bray, Ireland; 0.5% m v-1) (Fsa02490Xyn, GH10); agarose (VWR Chemicals, Solon, OH, USA; 0.5% m v-1). and carboxymethyl curdlan (Megazyme, Bray, Ireland; 0.5% m v-1) and Laminarin (TCI, Portland, OR, USA; 0.5% m v-1) (Fsa16295Glu, GH50). The temperature ranges of Fsa01040Cel, Fsa02490Xyn, Fsa15405Xyn, and Fsa16295Glu were determined using optimal substrates with enzymes or empty vector controls mixed 1:1, incubated overnight at 4, 20, 30, 40, 50, 60, 70, 80, 90, 95, 100, 110, and 120 °C. Reducing sugars were quantified as above. Temperature optima were determined using one-way ANOVA and post-hoc Tukey’s Honestly Significant Difference tests.
Phylogenomic analysis and ancestral state reconstruction
For phylogenomic analyses, taxon selection for Fervidibacteria consisted of all MAGs and SAGs included in public databases and our own data that were identified as belonging to the GTDB class HRBIN17 (i.e., class Fervidibacteria) using GTDB-Tk v. 2.3.299 with the RS214 database. To place the lineage assigned HRBIN17 into context within the domain Bacteria, taxon selection entailed inclusion of only high-quality genomes, with a representative of each class in the domain bacteria, a representative of each order in the Terrabacteria, and all high-quality genomes in the Armatimonadota. This resulted in a taxon set of 731 taxa, of which 65 belonged to HRBIN17. To assess congruence among phylogenomic trees constructed with different marker sets, all genomes were analyzed with the GTDB-Tk v. 2.3.2, bcgTree v1.2.0100, and UBCG2101 pipelines. Individual marker alignments were extracted and subjected to model testing for identifying the appropriate evolutionary model with ProtTest v3.4.2102 and concatenation as implemented in FASconCAT-G v3.4.2103. This produced three concatenated, partitioned data matrices, with the appropriate evolutionary model assigned to each partition (i.e., marker) within the data matrices (Supplementary Data 20–22). These concatenated data matrices were then subjected to maximum-likelihood analysis with IQ-Tree v. 1.6.8104 using independent model parameter estimation for each partition and allowing for independent evolutionary rates105 (-spp, and branch support was inferred using Shimodaira-Hasegawa approximate likelihood ratio tests106 (SH-aLRT) and ultrafast bootstrapping107 from 1000 replicates each. For confirming the taxonomic placement of the lineage HRBIN17 as a class within the Armatimonadota, trees were rooted with all other bacteria, and positioning of the lineage within the Armatimonadota was assessed. All trees were visualized in iTOL v6.8.1108 and edited in Adobe Illustrator vCS6.
For inferring evolutionary events of genes within the class, presence/absence profiles consisting of consensus counts for all KEGG orthology terms, GHs, and hydrogenases were constructed for all species in the Fervidibacteria, and one representative per order and genomes of isolates in the Armatimonadota, represented by high-completeness, low-contamination genomes. For this, these genomes were subjected to gene calling with Prodigal v. 2.6.2109 and annotated with eggNOG-mapper v. 2.1.995. CAZymes were predicted for these genomes with dbCAN317, and genes identified as hydrogenases were subjected to hydrogenase classification using HydDB20. A cladogram inferred from a Bac120 tree constructed as above, with only these high-completeness and low-contamination genomes was used as the evolutionary hypothesis against which the presence/absence profiles were interrogated. Evolutionary events were predicted through rate optimization and posterior probabilities in COUNT110 and included gene gains and losses. For visualizing these events, a cladogram of the species tree was annotated with node numbers with a summary of events depicted at each node for traits of interest. The full COUNT session file is available as Supplementary Data 23.
Data availability
The complete F. sacchari PD1T genome and MAGs serving as nomenclatural types are available in GenBank under accession numbers listed in Supplementary Data 10, and all other genomes were submitted to eLMSG and/or IMG under accession numbers listed in Supplementary Data 10. Metagenomic reads from which SeqCode nomenclatural types were derived are available in the Sequence Read Archive (SRA) under the SRA accessions listed in Supplementary Data 10. Raw files for 16S rRNA gene sequencing from the G1-G22 enrichments are available in the SRA under BioProject accession number PRJNA1086027. Raw and processed RNA-Seq data are available in the NCBI GEO database under accession number GSE249938 https://www.ncbi.nlm.nih.gov/search/all/?term=GSE249938). The proteomics (MSV000094290; https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=c1fe9f9b8bec4663b307a0548cb5aae8) and metaproteomics (MSV000093641; https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=c4115bc81d584c77a217ef174c8fa6ce) data are available from the Center for Computational Mass Spectrometry’s MassIVE repository via the ProteomeXchange Consortium. Source data are provided with this paper.
Change history
29 April 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41467-025-59415-9
References
Seidi, F. et al. Crystalline polysaccharides: a review. Carbohydr. Polym. 275, 118624 (2022).
Blumer-Schuette, S. E., Kataeva, I., Westpheling, J., Adams, M. W. & Kelly, R. M. Extremely thermophilic microorganisms for biomass conversion: status and prospects. Curr. Opin. Biotechnol. 19, 210–217 (2008).
Brochier-Armanet, C., Talla, E. & Gribaldo, S. The multiple evolutionary histories of dioxygen reductases: implications for the origin and evolution of aerobic respiration. Mol. Biol. Evol. 26, 285–297 (2009).
Murali, R., Gennis, R. B. & Hemp, J. Evolution of the cytochrome bd oxygen reductase superfamily and the function of CydAA’ in Archaea. ISME J. 15, 3534–3548 (2021).
Murali, R., Hemp, J. & Gennis, R. B. Evolution of quinol oxidation within the heme‑copper oxidoreductase superfamily. Biochim. Biophys. Acta (BBA) - Bioenerg. 1863, 148907 (2022).
Lynd, L. R. et al. How biotech can transform biofuels. Nat. Biotechnol. 26, 169–172 (2008).
Nayfach, S. et al. A genomic catalog of Earth’s microbiomes. Nat. Biotechnol. 39, 499–509 (2021).
Parks, D. H. et al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 50, D785–D794 (2022).
Buessecker, S. et al. An essential role for tungsten in the ecology and evolution of a previously uncultivated lineage of anaerobic, thermophilic Archaea. Nat. Commun. 13, 3773 (2022).
Reichart, N. J. et al. Activity-based cell sorting reveals responses of uncultured archaea and bacteria to substrate amendment. ISME J. 14, 2851–2861 (2020).
Blank, C. E., Cady, S. L. & Pace, N. R. Microbial composition of near-boiling silica-depositing thermal springs throughout Yellowstone National Park. Appl. Environ. Microbiol. 68, 5123–5135 (2002).
Rinke, C. et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–437 (2013).
Costa, K. C. et al. Microbiology and geochemistry of Great Boiling and Mud hot springs in the United States Great Basin. Extremophiles 13, 447–459 (2009).
Cole, J. K. et al. Sediment microbial communities in Great Boiling Spring are controlled by temperature and distinct from water communities. ISME J. 7, 718–729 (2013).
Peacock, J. P. et al. Pyrosequencing reveals high-temperature cellulolytic microbial consortia in Great Boiling Spring after in situ lignocellulose enrichment. PLoS ONE 8, e59927 (2013).
Reichart, N. J., Bowers, R. M., Woyke, T. & Hatzenpichler, R. High potential for biomass-degrading enzymes revealed by hot spring metagenomics. Front. Microbiol. 12, 668238 (2021).
Zheng, J. et al. dbCAN3: automated carbohydrate-active enzyme and substrate annotation. Nucleic Acids Res. 51, W115–W121 (2023).
Kato, S. et al. Isolation of previously uncultured slow-growing bacteria by using a simple modification in the preparation of agar media. Appl. Environ. Microbiol. 84, e00807–e00818 (2018).
Hedlund, B. P., Thomas, S. C., Dodsworth, J. A. & Zhang, C. L. Life in high-temperature environments. in Manual of environmental microbiology (eds. Yates, M. V., Nakatsu, C. H., Miller, R. V. & Pillai, S. D.) 4.3.4-1-4.3.4-15 (ASM Press, 2015).
Søndergaard, D., Pedersen, C. N. S. & Greening, C. HydDB: a web tool for hydrogenase classification and analysis. Sci. Rep. 6, 34212 (2016).
Peters, J. W. et al. FeFe]- and [NiFe]-hydrogenase diversity, mechanism, and maturation. Biochim. Biophys. Acta (BBA) - Mol. Cell Res. 1853, 1350–1369 (2015).
Yip, V. L. Y. et al. An unusual mechanism of glycoside hydrolysis involving redox and elimination steps by a family 4 β-glycosidase from Thermotoga maritima. J. Am. Chem. Soc. 126, 8354–8355 (2004).
Liu, Q. P. et al. Bacterial glycosidases for the production of universal red blood cells. Nat. Biotechnol. 25, 454–464 (2007).
Teze, D. et al. The catalytic acid–base in GH109 resides in a conserved GGHGG loop and allows for comparable α-retaining and β-inverting activity in an N-acetylgalactosaminidase from Akkermansia muciniphila. ACS Catal. 10, 3809–3819 (2020).
Ishikura, H., Arakawa, S., Nakajima, T., Tsuchida, N. & Ishikawa, I. Cloning of the Tannerella forsythensis (Bacteroides forsythus) siaHI gene and purification of the sialidase enzyme. J. Med. Microbiol. 52, 1101–1107 (2003).
Strazzulli, A. et al. Discovery of hyperstable carbohydrate‐active enzymes through metagenomics of extreme environments. FEBS J. 287, 1116–1137 (2020).
Daniel, R. M. & Danson, M. J. Did primitive microorganisms use nonhem iron proteins in place of NAD/P? J. Mol. Evol. 40, 559–563 (1995).
Bodelón, G., Palomino, C., Fernández & L, Á. Immunoglobulin domains in Escherichia coli and other enterobacteria: from pathogenesis to applications in antibody technologies. FEMS Microbiol. Rev. 37, 204–250 (2013).
Pires, V. M. R. et al. The crystal structure of the family 6 carbohydrate binding module from Cellvibrio mixtus endoglucanase 5A in complex with oligosaccharides reveals two distinct binding sites with different ligand specificities. J. Biol. Chem. 279, 21560–21568 (2004).
Zeytuni, N. & Zarivach, R. Structural and functional discussion of the tetra-trico-peptide repeat, a protein interaction module. Structure 20, 397–405 (2012).
Artzi, L., Bayer, E. A. & Moraïs, S. Cellulosomes: bacterial nanomachines for dismantling plant polysaccharides. Nat. Rev. Microbiol. 15, 83–95 (2017).
Cava, F., De Pedro, M. A., Schwarz, H., Henne, A. & Berenguer, J. Binding to pyruvylated compounds as an ancestral mechanism to anchor the outer envelope in primitive bacteria. Mol. Microbiol. 52, 677–690 (2004).
Boyd, E. S., Fecteau, K. M., Havig, J. R., Shock, E. L. & Peters, J. W. Modeling the habitat range of phototrophs in Yellowstone National Park: toward the development of a comprehensive fitness landscape. Front. Microbiol. 3, 221 (2012).
Rodrigues-Oliveira, T., Belmok, A., Vasconcellos, D., Schuster, B. & Kyaw, C. M. Archaeal S-layers: overview and current state of the art. Front. Microbiol. 8, 2597 (2017).
Goswami, R., Bandyopadhyay, B. & Sadhukhan, S. Thermophilic bacterial exopolysaccharides: from bio-physicochemical characterization to biotechnological applications. in Advances in environmental engineering and green technologies (eds. Gunjal, A. B., Thombre, R. & Parray, J. A.) 334–361 (IGI Global, 2022).
Lai, D. et al. Resource partitioning and amino acid assimilation in a terrestrial geothermal spring. ISME J. 17, 2112–2122 (2023).
Conway, J. M. et al. Functional analysis of the glucan degradation locus in Caldicellulosiruptor bescii reveals essential roles of component glycoside hydrolases in plant biomass deconstruction. Appl. Environ. Microbiol. 83, e01828–17 (2017).
Yang, S.-J. et al. Efficient degradation of lignocellulosic plant biomass, without pretreatment, by the thermophilic anaerobe “Anaerocellum thermophilum” DSM 6725. Appl. Environ. Microbiol. 75, 4762–4769 (2009).
Zhang, Y.-H. P. & Lynd, L. R. Cellulose utilization by Clostridium thermocellum: bioenergetics and hydrolysis product assimilation. Proc. Natl Acad. Sci. USA 102, 7321–7325 (2005).
Ng, T. K. & Zeikus, J. G. Differential metabolism of cellobiose and glucose by Clostridium thermocellum and Clostridium thermohydrosulfuricum. J. Bacteriol. 150, 1391–1399 (1982).
Konstantinidis, K. T. & Tiedje, J. M. Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 187, 6258–6264 (2005).
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T. & Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 5114 (2018).
Louca, S. The rates of global bacterial and archaeal dispersal. ISME J. 16, 159–167 (2022).
Hedlund, B. P. et al. SeqCode: a nomenclatural code for prokaryotes described from sequence data. Nat. Microbiol. 7, 1702–1708 (2022).
Caffrey, S. M. et al. Function of periplasmic hydrogenases in the sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. J. Bacteriol. 189, 6159–6167 (2007).
Schut, G. J., Boyd, E. S., Peters, J. W. & Adams, M. W. W. The modular respiratory complexes involved in hydrogen and sulfur metabolism by heterotrophic hyperthermophilic archaea and their evolutionary implications. FEMS Microbiol. Rev. 37, 182–203 (2013).
Carlton, J. D. et al. Expansion of Armatimonadota through marine sediment sequencing describes two classes with unique ecological roles. ISME Commun. 3, 64 (2023).
Koch, R. Die Ätiologie der Milzbrand-Krankheit, begründet auf die entwicklungsgeschichte des Bacillus Anthracis (1876). in Robert Koch 19–43 (Springer Berlin Heidelberg, 2018).
Doud, D. F. R. et al. Function-driven single-cell genomics uncovers cellulose-degrading bacteria from the rare biosphere. ISME J. 14, 659–675 (2020).
Vishnivetskaya, T. A. et al. Community analysis of plant biomass-degrading microorganisms from Obsidian Pool, Yellowstone National Park. Microb. Ecol. 69, 333–345 (2015).
Kelly, R. M. & Adams, M. W. W. Metabolism in hyperthermophilic microorganisms. Antonie Van. Leeuwenhoek 66, 247–270 (1994).
Girfoglio, M., Rossi, M. & Cannio, R. Cellulose degradation by Sulfolobus solfataricus requires a cell-anchored endo-β-1-4-glucanase. J. Bacteriol. 194, 5091–5100 (2012).
Quehenberger, J., Shen, L., Albers, S.-V., Siebers, B. & Spadiut, O. Sulfolobus – a potential key organism in future biotechnology. Front. Microbiol. 8, 2474 (2017).
Trincone, A. et al. Enzymatic synthesis and hydrolysis of xylogluco-oligosaccharides using the first archaeal α-xylosidase from Sulfolobus solfataricus. Extremophiles 5, 277–282 (2001).
Doran, P. M. Bioprocess Engineering Principles (Elsevier, 2013).
Anderson, I. et al. Genomics of aerobic cellulose utilization systems in Actinobacteria. PLoS ONE 7, e39331 (2012).
Schwarz, W. H. The cellulosome and cellulose degradation by anaerobic bacteria. Appl. Microbiol. Biotechnol. 56, 634–649 (2001).
Wilson, D. B. Evidence for a novel mechanism of microbial cellulose degradation. Cellulose 16, 723–727 (2009).
Dodsworth, J. A., McDonald, A. I. & Hedlund, B. P. Calculation of total free energy yield as an alternative approach for predicting the importance of potential chemolithotrophic reactions in geothermal springs. FEMS Microbiol. Ecol. 81, 446–454 (2012).
Murphy, C. N., Dodsworth, J. A., Babbitt, A. B. & Hedlund, B. P. Community microrespirometry and molecular analyses reveal a diverse energy economy in Great Boiling Spring and Sandy’s Spring West in the U.S. Great Basin. Appl. Environ. Microbiol. 79, 3306–3310 (2013).
Dodsworth, J. A. et al. Thermoflexus hugenholtzii gen. nov., sp. nov., a thermophilic, microaerophilic, filamentous bacterium representing a novel class in the Chloroflexi, Thermoflexia classis nov., and description of Thermoflexaceae fam. nov. and Thermoflexales ord. nov. Int. J. Syst. Evol. Microbiol. 64, 2119–2127 (2014).
Hanada, S., Hiraishi, A., Shimada, K. & Matsuura, K. Chloroflexus aggregans sp. nov., a filamentous phototrophic bacterium which forms dense cell aggregates by active gliding movement. Int. J. Syst. Bacteriol. 45, 676–681 (1995).
Wolin, E. A., Wolin, M. J. & Wolfe, R. S. Formation of methane by bacterial extracts. J. Biol. Chem. 238, 2882–2886 (1963).
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K. & Schloss, P. D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120 (2013).
Zhang, Z., Schwartz, S., Wagner, L. & Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 7, 203–214 (2000).
Pruesse, E., Peplies, J. & Glöckner, F. O. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829 (2012).
Salter, S. J. et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12, 87 (2014).
Landry, Z. C., Giovanonni, S. J., Quake, S. R. & Blainey, P. C. Optofluidic cell selection from complex microbial communities for single-genome analysis. Methods Enzymol. 531, 61–90 (2013).
Cole, J. K. et al. Kallotenue papyrolyticum gen. nov., sp. nov., a cellulolytic and filamentous thermophile that represents a novel lineage (Kallotenuales ord. nov., Kallotenuaceae fam. nov.) within the class Chloroflexia. Int. J. Syst. Evol. Microbiol. 63, 4675–4682 (2013).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Tatusova, T. et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624 (2016).
Kolmogorov, M. et al. metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat. Methods 17, 1103–1110 (2020).
Nurk, S., Meleshko, D., Korobeynikov, A. & Pevzner, P. A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834 (2017).
Thomas, S. C. et al. Position-specific metabolic probing and metagenomics of microbial communities reveal conserved central carbon metabolic network activities at high temperatures. Front. Microbiol. 10, 1427 (2019).
Fernandes-Martins, M. C. et al. Ecological dichotomies arise in microbial communities due to mixing of deep hydrothermal waters and atmospheric gas in a circumneutral hot spring. Appl. Environ. Microbiol. 87, e01598–21 (2021).
Kato, S. et al. Long-term cultivation and metagenomics reveal ecophysiology of previously uncultivated thermophiles involved in biogeochemical nitrogen cycle. Microbes Environ. 33, 107–110 (2018).
Liew, K. J. et al. Thermophiles and carbohydrate-active enzymes (CAZymes) in biofilm microbial consortia that decompose lignocellulosic plant litters at high temperatures. Sci. Rep. 12, 2850 (2022).
Strous, M., Kraft, B., Bisdorf, R. & Tegetmeyer, H. E. The binning of metagenomic contigs for microbial physiology of mixed cultures. Front. Microbiol. 3, 410 (2012).
Dick, G. J. et al. Community-wide analysis of microbial genome sequence signatures. Genome Biol. 10, R85 (2009).
Becraft, E. D. et al. Single-cell-genomics-facilitated read binning of candidate phylum EM19 genomes from geothermal spring metagenomes. Appl. Environ. Microbiol. 82, 992–1003 (2016).
Hua, Z.-S. et al. Ecological roles of dominant and rare prokaryotes in acid mine drainage revealed by metagenomics and metatranscriptomics. ISME J. 9, 1280–1294 (2015).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019).
Chklovski, A., Parks, D. H., Woodcroft, B. J. & Tyson, G. W. CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat. Methods 20, 1203–1212 (2023).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096 (2021).
Menzel, P., Ng, K. L. & Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 7, 11257 (2016).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 14, 927–930 (2003).
Wessel, D. & Flügge, U. I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 138, 141–143 (1984).
Ahlmann-Eltze, C. & Anders, S. proDA: probabilistic dropout analysis for identifying differentially abundant proteins in label-free mass spectrometry. Preprint at https://www.biorxiv.org/content/10.1101/661496v2 (2020).
Cole, J. R. et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–D642 (2014).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2012).
Pan, C. & Banfield, J. F. Quantitative metaproteomics: functional insights into microbial communities. in Environmental microbiology (eds. Paulsen, I. T. & Holmes, A. J.) 231–240 (Humana Press, 2014).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Oberortner, E., Cheng, J.-F., Hillson, N. J. & Deutsch, S. Streamlining the design-to-build transition with build-optimization software tools. ACS Synth. Biol. 6, 485–496 (2017).
Hsu, S. C. & Lockwood, J. L. Powdered chitin agar as a selective medium for enumeration of Actinomycetes in water and soil. Appl. Microbiol. 29, 422–426 (1975).
Kim, J. J. et al. Effective microwell plate-based screening method for microbes producing cellulase and xylanase and its application. J. Microbiol. Biotechnol. 24, 1559–1565 (2014).
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk v2: memory friendly classification with the genome taxonomy database. Bioinformatics 38, 5315–5316 (2022).
Ankenbrand, M. J. & Keller, A. bcgTree: automatized phylogenetic tree building from bacterial core genomes. Genome 59, 783–791 (2016).
Kim, J., Na, S.-I., Kim, D. & Chun, J. UBCG2: up-to-date bacterial core genes and pipeline for phylogenomic analysis. J. Microbiol. 59, 609–615 (2021).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165 (2011).
Kück, P. & Longo, G. C. FASconCAT-G: extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Front. Zool. 11, 81 (2014).
Nguyen, L.-T., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Chernomor, O., Von Haeseler, A. & Minh, B. Q. Terrace aware data structure for phylogenomic inference from supermatrices. Syst. Biol. 65, 997–1008 (2016).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Hoang, D. T., Chernomor, O., Von Haeseler, A., Minh, B. Q. & Vinh, L. S. UFBoot2: improving the ultrafast bootstrap pproximation. Mol. Biol. Evol. 35, 518–522 (2018).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11, 119 (2010).
Csűös, M. Count: evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics 26, 1910–1912 (2010).
Acknowledgements
We thank Dave and Sandy Jamieson for access to GBS and we acknowledge that GBS is on historical lands inhabited by the Cui Ui Ticutta (Cui-ui eaters) band of Northern Paiutes. We acknowledge the staff of the Yunnan Tengchong Volcano and Spa Tourist Attraction Development Corporation, the operators of Gongxiaoshe Resort and Hotel, Jinze Resort and Hotel, and the people of Diantan, China, for generous permission to collect samples for metagenomics. We thank Ramunas Stepanauskas for obtaining support from JGI for the MDM2 project, which generated a few Fervidibacteria MAGs, and Senthil Murugapiran for help binning some of the Fervidibacteria MAGs. We thank high school teachers Kathleen Diver, Heather Garcia, Juan Salgado, and Chuong Vu for assistance with isotope labeling experiments at GBS. Part of this work was carried out at Lawrence Livermore National Laboratory (LLNL) under Contract DE-AC52-07NA2734 (X.M., J.J., P.W., J.P.-R.). Funding was also provided by the U.S. National Science Foundation (DEB 1557042, N.O.N., D.L., J.A.D., S.B., D.M., X.M., J.P.-R., M.P., B.P.H.), NASA (80NNSC17KO548, N.O.N., S.B., A.D., J.J., J.A.D., S.B., D.M., X.M., J.P.-R., M.P., B.P.H.), Nevada NASA Space Grant (80NSSC20M0043, N.O.N., J.K.C., A.M.C., N.T., S.S., B.P.H.), and the National Natural Science Foundation of China (Nos. 92251302, 32370011, and 32200010, L.L., M.-M.L., J.-Y.J., Z.-H.L. W.-J.L.). The Mick Hitchcock Ph.D. Nevada Proteomics Center is supported by a grant from the National Institute of General Medical Sciences (GM103440 and GM104944) from the National Institutes of Health (N.O.N., J.K.C., B.P.H). The work (proposal: https://doi.org/10.46936/10.25585/60001337, 10.46936/10.25585/60001099) conducted by the U.S. Department of Energy Joint Genome Institute (https://ror.org/04xm1d337), a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy operated under Contract No. DE-AC02-05CH11231 (N.O.N., D.L., J.A.D., M.P., B.P.H.). Additional funds were supplied by the CSUSB OSR 2016 Summer Research Program (J.D., C.G., L.G, M.N.).
Author information
Authors and Affiliations
Contributions
B.P.H., N.O.N., J.A.D., X.M., S.B., and M.P. conceived of the study. J.D., C.G., L.G., M.N., D.M., and J.A.D. conducted original cultivation and 16S rRNA gene screens. N.O.N. conducted genome-enabled lab enrichments and monitored cultures by qPCR and FISH. N.O.N. conducted all other cultivation experiments. D.M. and J.A.D. isolated the strain using optical tweezers. A.R.M. and A.B. conducted cryo-EM and led structural interpretations. M.P., N.O.N., E.E.-F., T.W., and N.S. contributed to DNA isolation, genome sequencing, assembly, and annotation. R.P.C. helped with interpretations related to polysaccharide metabolism and bioenergetics. C.O.S., G.G., and D.L. analyzed transcriptomics and proteomics data. J.A.D., A.E.D., and S.B. performed FISH on short-term incubations with GBS sediments. X.M., J.J., P.K.W., J.P.-R., and D.L. analyzed samples with nanoSIMS and nanoSIMS data analysis/interpretation. J.-F.C., I.K.B., J.K.C, N.T., A.C., S.G.B., S.S., R.M., and M.H. contributed to GH annotation, DNA synthesis, expression, and activity measurements. J.-Y.J., L.L., M.M.L., and W.-J.L. were involved in metagenomic sampling, sequencing, assembly, annotation, and binning. Z.H.L., F.L., B.G., X.S., and S.Z. led on annotations and interpretations related to secretion systems, pili, signal transduction, and motility. M.P. and D.L. performed phylogenomic analyses, and M.P. performed ACS reconstructions. M.H.S. helped with project management and coordination. B.P.H. and N.O.N. wrote the manuscript and all authors contributed to the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This research included local researchers as full authors, when possible, to recognize both logistical and intellectual contributions. No potential or listed authors were discriminated against on the basis of gender, race, ethnicity or any other factors not related to scientific contributions
Peer review
Peer review information
Nature Communications thanks Sara Blumer-Schuette and Kian Mau Goh for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nou, N.O., Covington, J.K., Lai, D. et al. Genome-guided isolation of the hyperthermophilic aerobe Fervidibacter sacchari reveals conserved polysaccharide metabolism in the Armatimonadota. Nat Commun 15, 9534 (2024). https://doi.org/10.1038/s41467-024-53784-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-53784-3
This article is cited by
-
Branched-chain amino acid specialization drove diversification within Calditenuaceae (Caldarchaeia) and enables their cultivation
Nature Communications (2026)
-
Ecological distribution and environmental preference of the deep-branching archaeal lineage in high-temperature hot springs of Central Kyushu, Japan
Extremophiles (2026)
-
Energetic convergence drives metabolic adaptation in lirima chilean high-altitude hydrothermal system
Communications Earth & Environment (2025)
