
Abstract
Palladium-catalyzed directed C − H functionalization/cyclization is an effective approach for synthesizing nitrogen heterocycles. Imine, known for its ease of installation/removal, has been extensively used in the C–H activation of aldehydes, ketones, and alkylamines. Nevertheless, it has been rarely explored in the C(sp2)–H activation of aryl amines because of the generation of a strained four-membered palladacycle. Herein, an imine directed palladium catalyzed C(sp2)–H functionalization of aryl amines assisted by vinylacetic acid is established, providing access to a variety of γ-lactone fused tetrahydroquinolines under mild reaction conditions. The methodology demonstrates broad substrate scope and good functional group tolerance, representing notable advancement in organic synthesis. Mechanistic experiments are performed to clarify how the C(sp2)–H activation occurs, indicating the crucial role of vinylacetic acid. DFT calculations supports the observations, elucidating the strained four-membered ring C–H activation barrier is overcome via coordination and hydrogen bond interaction of vinylacetic acid.
Similar content being viewed by others
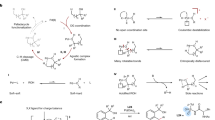
Introduction
Nitrogen containing heterocycles are important organic compounds that are widely found in natural products1,2, organic materials3,4, pharmaceuticals, and other bioactive molecules5,6,7,8. Therefore, the development of methods for the efficient synthesis of nitrogen heterocycles is of great interest and has been long sought9,10,11,12,13. Catalytic C(sp2)−H bond functionalization/cyclization of aryl amines under palladium catalyzed conditions has emerged as one of the most efficient methods for the selective construction of C–C and C–X bonds to deliver different types of nitrogen containing molecules due to its high synthetic efficiency. A wide range of effective methods have been extensively developed to achieve C(sp2)−H bond functionalization/cyclization of aryl amines by promoting cyclometallation to form thermodynamically stable five- or six-membered palladacycle intermediates14,15,16. Cyclometallation usually involves the installation of directing groups, such as sulfonamide17,18,19,20, amide21,22,23,24,25, azo26 and pyridine27,28. On the other hand, imine, a directing group that can be conveniently prepared from a diverse range of amines and aldehydes or ketones, has been extensively employed in the C − H bond functionalization of aldehydes29,30,31,32,33,34,35,36 and ketones30,37,38,39. By contrast, the utilization of imine as a directing group in the C(sp2)−H bond functionalization of aryl amines has been relatively unexplored (Fig. 1a).

a Directing groups applied in ortho-C(sp2)-H activation of amines. b Regioselectivity of imine directed ortho-C(sp2)-H activation; c Our hypothesis of reversing the regioselectivity of imine directed ortho-C(sp2)-H activation.
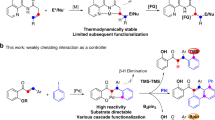
Typically, when an N-benzylideneaniline containing an imine directing group with two ortho-C(sp2)−H bonds at different distances of the imine group is subjected to the C − H activation reaction conditions, a five-membered palladacycle intermediate is more preferable. This intermediate then reacts with other reagents, affording the corresponding cyclization products or the functionalization of aryl aldehydes29,40,41. On the contrary, endeavors attempting to activate the other C(sp2)−H bond require the generation of a high strain four-membered palladacycle42,43, which is thermodynamically less favorable compared to the more stable five-membered palladacycle (Fig. 1b). As a result, it is difficult to achieve ortho-C(sp2)−H activation of aryl amines applying imine as a directing group. We wondered whether the selectivity could be reversed by using a suitable strategy, if so, a different strategy for the C(sp2)−H bond functionalization of anilines may be established, providing a different way to access nitrogen heterocycles.
Based on our focus and interest in vinylacetic acid44,45,46, we envision that the use of vinylacetic acid may make it possible to achieve the imine directed C(sp2)−H activation at the aniline side. Since the vinylacetic acid contains two coordinating groups, the alkenyl group can coordinate with the palladium and the carboxyl group can form a hydrogen bond interaction with the nitrogen atom of the imine. Consequently, the coordination between imine and palladium will be broken, leading to a larger cyclic intermediate and avoiding the high ring strain. Besides, the preference for generating a suitable cyclic intermediate may also help to inhibit the C(sp2)−H activation on the benzaldehyde side36,47. Herein, we report our investigation into this imine directed palladium catalyzed ortho-C(sp2)–H activation (Fig. 1c). The imine is generated in-situ via aryl amines and aldehydes, which then assists ortho-C(sp2)–H activation in conjunction with vinylacetic acid. Subsequent insertion of vinylacetic acid and concerted annulation afford the corresponding product bearing a γ-lactone fused tetrahydroquinoline skeleton, which can be identified in several natural products and potential drugs as a privileged structural scaffold.
Results
Screening of reaction conditions
At the outset of our studies, we employed aniline and benzaldehyde as imine sources, reacting with vinylacetic acid under palladium catalyst conditions to optimize the reaction conditions (Table 1). To our delight, the corresponding product could be obtained in 53% yield with 12.0:1 diastereoselectivity in the presence of Pd(TFA)2 and 2,9-dimethyl-10-phenanthroline in MeCN under 35 °C for 12 h (entry 1). To further improve the yield, we screened different palladium catalysts, and found that palladium catalysts with halogen atoms such as PdCl2, would totally inhibit the reaction (entry 2). Other kinds of palladium catalysts, for instance, Pd(OAc)2, Pd(PPh3)4 and Pd(dba)2, could only afford a trace amount of the corresponding products (entries 3-5). Solvent screening revealed that the use of 1,2- dichloroethane or toluene as solvent gave neither a better result than MeCN nor a better diastereoselectivity (entries 6-7). The reaction worked well with protic solvents like HFIP and AcOH, significantly improving the yield of 4a, albeit with only medium diastereoselectivity (entries 8-9). Employing DMF as solvent would severely inhibit the reaction, whilst the use of DMSO as solvent can result in a medium yield with high diastereoselectivity (entries 10-11). In order to maintain a high diastereoselectivity, we tried to enhance the yield of 4a with DMSO as solvent (See Supplementary Information section 3 for more details). Finally, the corresponding product was obtained in 80% yield with diastereoselectivity keeping higher than 20.0:1 under the following reaction conditions: Pd(TFA)2 (5 mol%), 2,6-dimethoxy-1,4-benzoquinone (1.0 equiv.), DMSO (0.5 M), at 35 °C under air for 24 h (entry 12). Control experiments revealed that the reaction could not proceed without Pd catalyst or oxidants and DMSO did not serve as an oxidant (entries 13-14).
Mechanistic investigations
In order to find out how this reaction occurred, a series of control experiments have been conducted. Initially, we have considered whether the tetrahydroquinoline skeleton was formed via Povarov reaction. And it could be excluded after the intermediate control experiments (see Supplementary Information section 6.1). We then considered the possibility of directed C(sp2)–H activation. As shown in Fig. 2a, when an equal amounts of aniline and benzaldehyde were subjected to a stoichiometric amount of Pd(TFA)2 in DMSO at 35 °C for 12 h, a stable palladacycle complex was isolated by trapping the C − H insertion intermediate with triphenylphosphine. The structure of this complex was confirmed by X-ray crystallography, showing that the C(sp2)–H activation took place at the benzaldehyde side, rather than the aniline side. Additionally, when d5-2a was subjected to the standard reaction conditions, d5-4a was isolated as the sole product without any loss of deuterium atoms (Fig. 2b). These two experiments state that the C(sp2)–H activation of benzaldehyde is inhibited under the standard reaction conditions and the vinylacetic acid may play a crucial role in the C(sp2)–H activation step. With d7-1a as substrate, the corresponding product d4-4a was isolated with all unreacted deuterium atoms remaining, which indicates that the C(sp2)–H activation step is irreversible (Fig. 2c). The intramolecular KIE experiment of aniline provided a KIE value of 1.17, suggesting that the C(sp2)–H activation is not involved in the turnover limiting step (Fig. 2d). In order to illustrate the E/Z configuration of the Heck type intermediate, substrate (Z)-γ-d1-3 was subjected to the standard reaction conditions, affording the corresponding product d-4a in 49% yield, with 0.78 deuterium atoms at the γ position and 0.12 deuterium atoms at the β position (Fig. 2e). As shown in Fig. 2g, the (Z)-alkene intermediate could be generated via β-H elimination, which soon proceeded concerted annulation to afford 4a with most deuterium atoms remaining at the γ position. The (E)-alkene intermediate could also be obtained through β-D elimination. While the (E)-alkene intermediate could not perform concerted annulation, it underwent re-insertion of the Pd−D species to generate the (Z)-alkene intermediate, leading to the migration of the deuterium atom from γ position to the β position. When γ-d2-3 was subjected to the standard reaction conditions, the corresponding product was isolated in 54% yield, with 0.78 deuterium migrating from the γ position to the β position (Fig. 2e). According to the (Z)-γ-d1-3 experiment, the migration of 0.78 deuterium atom from the γ position to the β position indicated that the generation of (E)-alkene intermediate may be more preferable, and subsequent re-insertion of the Pd−D species and β-H elimination afforded the (Z)-alkene intermediate, leading to the target product with additional deuterium atoms at the β position. The (Z)-γ-d1-3 experiment indicated that the generation of (Z)-alkene intermediate was easier, which we speculated was caused by the energy barrier difference. (See Supplementary Information Sections 6.6 and 6.7 for more details).

a Attempt to capture the intermediate. b Deuterium labelled experiment using d5-2a. c Deuterium labelled experiment using d7-1a. d Intramolecular KIE experiment of 1a. e Deuterium labelled experiment using (Z)-γ-d1-3. f Deuterium labelled experiment using γ-d2-3. g Possible outcomes for the “Heck reaction” step.
After obtaining the experimental details of this reaction, density functional theory (DFT) calculations were carried out to seek more details about how the C(sp2)–H activation occurred under the assistance of vinylacetic acid and the origin of high diastereoselectivity. Detailed DFT results are shown in Figs. 3 and 4. During the reaction conditions optimization, we have observed that both the yield and the diastereoselectivity of the reaction improved a lot with DMSO as solvent. Combing with the previous researches that sulfoxides can be used as ligands in many palladium-catalyzed reactions48,49,50,51,52,53, we considered that DMSO not only served as solvent, but also acted as a ligand coordinating with palladium. Additionally, the use of 2,6-DMBQ as an oxidant improved the yield, which indicated that 2,6-DMBQ may also act as a ligand to promote the reaction54,55,56. The interaction between each component was investigated by UV-Vis spectrophotometry, which showed that 2,6-DMBQ did not coordinate with the Pd catalyst (see Supplementary Information Section 6.8).

a Gibbs free energy of C(sp2)–H activation at each side without vinyl acetic acid. b Gibbs free energy of C(sp2)–H activation at each side with vinyl acetic acid. VAA vinyl acetic acid.

a Gibbs free energy profile of the whole reaction pathway. b Origination of diastereoselectivity.
According to the control experiments, the C(sp2)–H activation of benzaldehyde moiety rather than C(sp2)–H activation of aniline moiety was observed in the absence of vinylacetic acid (Fig. 2a). However, the selectivity is exactly reversed when vinylacetic acid was added to the system. We have noticed that Hartwig and co-workers reported a palladium-catalyzed β‑C(sp3)−H activation of primary alkylamines through a four-membered palladacycle intermediate stabilized by a bidentate directing group and an intramolecular hydrogen bond between the oxygen of the directing group and the hydroxyl group of the ligating acetic acid42. We speculated that a similar hydrogen bond interaction may be included in our reaction to realize the C(sp2)–H activation of aryl amines. To explore this interesting regioselectivity difference of C–H activation, DFT studies were conducted. As shown in Fig. 3, in the absence of vinylacetic acid, the palladium-catalyzed C(sp2)–H activation of the in-situ formed imine substrate would proceed through a conventional concerted metalation-deprotonation mechanism, as shown in transition state TS1’ or TS1’-iso in Fig. 3. The transition state of C(sp2)–H activation of the benzaldehyde moiety (TS1’-iso) is more stable than that of the C(sp2)–H activation of the aniline moiety (TS1’) by 13.4 kcal/mol, consistent with the control experiment where the cyclometallation occurs at the benzaldehyde moiety. The relative instability of TS1’ is attributed to the strained four-membered ring transition state structure. Conversely, when vinylacetic acid participated in the C(sp2)–H activation, the transition state of C(sp2)–H activation of the aniline moiety (TS1) is more stable than that of the C(sp2)–H activation of the benzaldehyde moiety (TS1-iso) by 2.2 kcal/mol. This result explains our experimental observation of the unusual aniline regioselectivity. In TS1 and TS1-iso, one vinylacetic acid molecule coordinates with the Pd center, and the other acts as a proton shuttle to deprotonate the C(sp2)–H bond to be activated. Note that, in this C(sp2)–H activation mode, one vinylacetic acid molecule coordinates with the Pd center, and the other interacts with the imine nitrogen via hydrogen-bond interaction, preventing the coordination between the nitrogen atom and the Pd center, thus avoiding the formation of four-membered ring transition state with significant stress in conventional C(sp2)–H activation mode. In comparison with TS1, the higher free energy of TS1-iso may be attributed to the large twisting force reflected by the larger degree of dihedral angle C–C–C–H, and the stronger repulsion force between the two H atoms due to the shorter H ∙ ∙ ∙ H distance in the nine-membered ring. Another case in which the coordinated vinylacetic acid acts as a transient directing group through H-bonding interaction was considered, but the relative free energy of the transition state is 12.2 kcal/mol (see Supplementary Information Section 7.1).
A more comprehensive process is shown in Fig. 4a. Initially, the imine and vinylacetic acid molecules coordinate with the Pd center to form a more stable intermediate IM1. Then, the vinylacetic acid assisted C(sp2)–H activation of aniline moiety occurs via transition state TS1 (ΔG≠ = 21.5 kcal/mol, IM1 → TS1). Subsequently, a DMSO coordinates with the Pd center to exchange the CF3COO⁻ anion, generating intermediate IM3. After that, olefin insertion of the coordinated vinyl acetic acid takes place through transition state TS2 (ΔG≠ = 26.8 kcal/mol, IM1 → TS2). The generated intermediate IM4 can undergo a facile β-H elimination via TS3 (ΔG≠ = 11.8 kcal/mol, IM1 → TS3), and intermediate IM5 is formed by the exchange between CF3COO− and CF3COOH. We also considered the possibility of CF3COO⁻ coordinated species in comparison to the DMSO coordinated species in the olefin insertion and β-H elimination processes (See Supplementary Information section 7.2 for more details). We also calculated the formation of the (E)-alkene intermediate for the β-H elimination step (See Supplementary Information section 7.3 for more details). The free energy of the transition state of the (E)-alkene intermediate is lower than that of the (Z)-alkene intermediate, indicating that (E)-alkene intermediate would be more easily formed via β-H elimination. This is accordance with our deuterium labelled experiment. The relative higher energy of IM5” compared to TS3” indicating that this β-H elimination stage is reversible, the reverse insertion reaction leads to the formation of the Z isomer which could further convert to the final product. Next, IM5 converts to intermediate IM6 by the driving force of oxidation. We calculated this oxidation step by oxygen and benzoquinone, respectively, and found that the oxidizing power of oxygen is 18.1 kcal/mol, which is stronger than that of benzoquinone (See Supplementary Information section 7.4 for more details). Considering the importance of the benzoquinone and the oxygen in the reaction, it implies that is probably driven by the driving force of the oxygen, with benzoquinone as the possible oxidative media. From IM6, an inner-sphere stepwise mechanism involving trans-oxypalladation and subsequent insertion of imine was considered but was less plausible due to the relatively higher free energy barrier (See Supplementary Information section 7.5 for more details), which is in accordance with our previous study44. Alternatively, IM6 could isomerize to intermediate IM6’. Remarkably, from IM6’, DFT studies suggest an outer-sphere concerted annulation process via transition state TS4 to furnish the five- and six-fused rings in a concerted step (ΔG≠ = 28.8 kcal/mol, IM6’ → TS4). The isomer of this outer-sphere concerted annulation transition state was also considered (TS4’). It could be found that TS4’ is less stable than TS4 by 3.9 kcal/mol due to the steric effect between the forming five-member ring and –CH3 group of DMSO in TS4’ (Fig. 4b), which nicely explains the high diastereoselectivity of the annulation product (See the supporting information section 7.6). To explore the coordination mode of DMSO, the analogous Pd(S-DMSO)(O-DMSO) and Pd(S-DMSO)(S-DMSO) species of IM6’, TS4, TS4’, and IM7 were also carefully considered, and their free energies are calculated to be higher than those of IM6’, TS4, TS4’, and IM7 (See the supporting information section 7.7 for more details), indicating that the coordination ability of the O atom of DMSO is stronger than that of the S atom in this system. Finally, the product 4a is generated from a proton transfer step. Overall, this Pd-catalyzed ortho-C(sp2)–H activation/cyclization probably proceeds via C–H activation, olefin insertion, β-H elimination, oxidation, and an outer-sphere concerted annulation (Fig. 5). According to the theoretical calculations and considering the KIE experiment, the concerted annulation is more likely to be the rate-determining step.

2,6-DMHQ: 2,6-Dimethoxyhydroquinone.
Substrate scopes
Having fully grasped this unusual C(sp2)–H activation process, we decided to demonstrate the substrate range of the reaction by testing a variety of aromatic amines and aldehydes. As outlined in Fig. 6, a wide range of aryl amines with diverse electronic and steric properties readily participated in this C(sp2)–H activation cyclization reaction, affording the γ-lactone fused tetrahydroquinoline products in good to excellent yields. Both electron-rich and electron-deficient functional groups on the phenyl ring at para-, meta-, and ortho-positions could be well tolerated, giving the corresponding products in moderate to high yields (4a-4y). Notably, the installation of functional groups with high reactivities such as iodine (4c), pinacol boronate ester (4 h), amide (4i), esters (4j-4k), ketone (4 l), azo (4 m) and a hydroxyl group (4n) could be well tolerated, converting to the products in moderate to high yields, which shows useful features with respect to further synthetic manipulations. When meta-substituted anilines were subjected to the standard conditions, the target products (4o-4r) were also obtained with high yields and complete regioselectivity. As for the ortho-substituted anilines, the yields of the corresponding products were slightly lower (4s-4w), possibly due to the increasing steric hindrance. The 3,5-disubstituted aryl amines also gave the corresponding products in excellent yields (4x-4y). Polycyclic aryl amines were also successfully used under the optimized reaction conditions, further enhancing the utility of this method (4z-4aj). 2-Naphthylamine, 5,6,7,8-tetrahydro-1-naphthylamine, and 5-aminoindan were compatible substrates, delivering the desired products in 35-69% yields(4z-4ab). Incorporating oxygen atom into the 5-aminoindan led to the decline in yields (4ac, 4ad), while the 2,2-difluoro-5-aminobenzodioxole and 1,4-benzodioxan-6-amine could participate in the reaction well, with the desire product yield of 73% and 60%, respectively. Other heterobicyclic aryl amines, such as 5-aminobenzimidazolone and 1-benzothiophen-5-amine were also suitable substrates, converting to the desired product 4ag and 4ah in high yields. Ketone type bicyclic aryl amines could also be transformed into the target products in good yields (4ai, 4aj). The relative configuration of 4a was characterized by single crystal X-ray diffraction.

aReaction conditions: 1 (0.25 mmol), 2a (1.5 equiv.), 3 (1.5 equiv.), Pd(TFA)2 (5 mol%), 2,6-DMBQ (1.0 equiv.), DMSO (0.5 M), at 35 °C under air for 24 h. Yield and dr (4a:4a’) ratio were determined by 1H NMR of the crude product with MeNO2 as internal standard. b1a (0.25 mmol), 2 (1.5 equiv.), 3 (1.5 equiv.), Pd(TFA)2 (5 mol%), 2,9-dmphen (7.5 mol%), AcOH (0.5 M), at 35 °C under air for 12 h. cTBHP (5.5 M in decane, 1.0 equiv.) was added.
This method is also compatible with aryl aldehydes containing different electronic and steric properties, and the results are presented in Fig. 7. The aryl aldehydes bearing electron-neutral, electron-rich, or electron-poor substituents were all tolerated (4ak-4bl). Similarly, sensitive functional groups including iodine (4an, 4az), aldehyde (4as), ketone (4at), and alkene (4ar) at the phenyl ring can also proceed smoothly. Impressively, the aryl aldehyde containing a methylthio group, a strongly chelating functional group, could also provide the corresponding product with good yields (4aw). The meta-substituents have little effect on the product yields (4ay-4ba). Whilst the steric hindrance of ortho-substituents greatly affects the yield, only substituents with little steric hindrance like methyl group and chlorine were tolerated, giving rise to the corresponding products in moderate yields (4bb-4bc). Heterocyclic aldehydes such as thiophene−2-carbaldehyde and thiophene-3-carbaldehyde also provided the corresponding products in medium yields (4bd, 4be). Multi-substituted aryl aldehydes could also transform to the desired products in moderate to high yields (4bf−4bl). Aryl aldehydes with a hydroxyl group, like syringaldehyde and vanillin were also suitable for this reaction, converting to the desired products 4bf and 4bg in 48% and 60% yields, respectively. Polycyclic aldehydes, were also capable of taking part in this process, leading to the corresponding tetrahydroquinoline derivatives 4bh-4bl in 26-79% yields. Unfortunately, the alkyl aldehydes and alkenyl aldehydes were not compatible with this reaction system, which may be caused by the instability of the corresponding imines under the standard reaction conditions (4bm-4bn).

aReaction conditions: 1 (0.25 mmol), 2a (1.5 equiv.), 3 (1.5 equiv.), Pd(TFA)2 (5 mol%), 2,6-DMBQ (1.0 equiv.), DMSO (0.5 M), at 35 °C under air for 24 h. Yield refers to isolated yield. dr (4’:4”) ratio was determined by 1H NMR of the crude product with MeNO2 as internal standard. b1a (0.25 mmol), 2 (1.5 equiv.), 3 (1.5 equiv.), Pd(TFA)2 (5 mol%), 2,9-dmphen (7.5 mol%), AcOH (0.5 mL), at 35 °C under air for 12 h. cTBHP (5.5 M in decane, 1.0 equiv.) was added.
Synthetic applications
We also utilized this reaction in the presence of biologically interesting molecules and natural products (Fig. 8a). This strategy worked for the aryl amines and aldehydes derived from a series of drugs and natural products, delivering the target molecules with good efficiency and diastereoselectivity. Aminoglutethimide, an aromatase inhibitor used for breast cancer continuum57,58, could be transferred to the target products 4bo in excellent yield and decent diastereoselectivity. Similarly, starting from the intermediates of Simendan and Lapatinib, the corresponding products 4 bp and 4bq could also be obtained in moderate to excellent yields. L-Borneol and L-menthol derived aryl amines smoothly converted into the desired products 4br and 4bs in excellent yields with medium diastereoselectivity. Aryl amine derived from a protected D-galactose moiety also gave the corresponding product 4bt in 71% yield and 9.9:1 diastereoselectivity. The more complicated natural product podophyllotoxin can be tolerated, delivering the corresponding product 4bu with 50% yield. Apart from the aryl amine derivatives, the aldehydes derived from pharmaceutical molecules and natural products worked well. Aryl aldehydes derived from S-ibuprofen and gemfibrozil participated in the reaction smoothly, affording the corresponding products 4bv and 4bw in excellent yields and decent diastereoselectivity. The catalytic system also allowed probenecid-derived aryl aldehyde to be a competent partner for the reaction, providing facile access to 4bx in good efficiency. The oleic acid containing an internal alkene moiety was also tolerated, generating the desired product 4by in high yield, while the naproxen derivative also worked well, giving 4bz in moderate yield and better diastereoselectivity. The process is also amenable to gram-scale procedure, with 4a being produced on 5.0 mmol scale in 74% yield and 4c on 5.0 mmol scale in 53% yield. Treating 4a with DIBAL-H under −78 °C would give a hemiacetal 8 in high yield although low diastereoselectivity (dr = 3.7:1). To further demonstrate the practicality of this reaction, iodinated product 4c was chosen as a coupling partner for more functionalized five-membered fused tetrahydroquinoline skeletons. With (triisopropylsilyl)acetylene as a coupling partner, the Sonogashira coupling could deliver the alkynylated product 4br in 78% yield. Besides, by performing Stille coupling with tributyl(vinyl)tin, the alkenylated product 4bs was acquired in 69% yield. The Ullmann type C–N bond coupling with carbazole was also well tolerated, producing the corresponding products 4bt and 63% yield (Fig. 8b).

a Late-stage functionalization of pharmaceutical molecules or natural products; b Synthetic applications. Reaction conditions: (a) 1a, 2a, 3, Pd(TFA)2, 2,9-dmphen, AcOH, at 35 °C under air for 12 h. (b) 1c, 2a, 3, Pd(TFA)2, 2,6-DMBQ, DMSO, at 35 °C under air for 24 h. c 4a, DIBAL-H (diisobutylaluminium hydride), CH2Cl2, at −78 °C under N2 for 1 h. d 4c, (triisopropylsilyl)acetylene, Pd(PPh3)2Cl2, CuI, Et3N, at 25 °C under N2 for 12 h. e 4c, tributyl(vinyl)tin, Pd(PPh3)4, toluene, at 120 °C under N2 for 12 h. f 4c, carbazole, CuI, 1,2-diaminocyclohexane, K3PO4, 1,4-dioxane at 90 °C under N2 for 24 h.
Discussion
In conclusion, we have realized a palladium catalyzed regioselective ortho-C(sp2)–H activation of aniline, using the strategy of vinylacetic acid assisted coordination and hydrogen-bonding to overcome the four-membered ring constraint in conventional C–H activation. This palladium catalyzed ortho-C(sp2)–H activation of aryl amines directed by the imine group undergoes alkene insertion and concerted annulation to give γ-lactone fused tetrahydroquinoline skeletons. A broad range of aryl amines and aryl aldehydes could react with vinylacetic acid to provide the products with high yield and excellent diastereoselectivity under mild reaction conditions. Mechanistic investigations and DFT calculations reveal the crucial roles of vinylacetic acid in coordination and H-bonding, which leads to a vinylacetic acid assisted C–H activation via an eight-membered ring transition state, leading to the unusual aniline C–H bond selectivity. We hope these observations will serve as inspiration for the further development of mild and general catalytic systems for the ortho-C(sp2)–H activation of aryl amines or other C–H activation reactions.
Methods
General procedure for the C(sp2)-H activation/cyclization
Procedure A: To a 25 mL dried test tube equipped with a magnetic stir bar was added Pd(TFA)2 (4.2 mg, 5 mol%), 2,6-DMBQ (42.0 mg, 1.0 equiv.), substituted aniline 1 (0.25 mmol), substituted aldehydes 2 (1.5 equiv.), vinylacetic acid 3 (32 μL, 1.5 equiv.) and DMSO (0.5 mL). The mixture was stirred in a pre-heated 35 °C heating block for 24 h under open-air. Until completion, the mixture was diluted with aqueous saturated Na2CO3 and extracted with EtOAc. The organic layer was collected, dried over anhydrous MgSO4 and passed through a short pad of Celite. After removal of solvent under reduced pressure, the residue was analyzed by 1H NMR to recorded dr ratio in CDCl3 with CH3NO2 as internal standard. The crude product was purified by column chromatography on silica gel using PE/EtOAc as eluent to provide the analytically pure product.
Procedure B: To a 25 mL dried test tube equipped with a magnetic stir bar was added Pd(TFA)2 (4.2 mg, 5 mol%), 2,9-dmphen (3.9 mg, 7.5 mol%), substituted aniline 1 (0.25 mmol), substituted aldehydes 2 (1.5 equiv.), vinylacetic acid 3 (32 μL, 1.5 equiv.) and AcOH (0.5 mL). The mixture was stirred in a pre-heated 35 °C heating block for 12 h under open-air. Until completion, the mixture was diluted with aqueous saturated Na2CO3 and extracted with EtOAc. The organic layer was collected, dried over anhydrous MgSO4 and passed through a short pad of Celite. After removal of solvent under reduced pressure, the residue was analyzed by 1H NMR to recorded dr ratio in CDCl3 with CH3NO2 as internal standard. The crude product was purified by column chromatography on silica gel using PE/EtOAc as eluent to provide the analytically pure product.
Data availability
The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Center (CCDC) under deposition numbers CCDC 2311079 (5) and 2311080 (4a). Copies of the data can be obtained free of charge via www.ccdc.cam.ac.uk/getstructures. All other data supporting the findings of the study, including experimental procedures, compound characterization and NMR spectra are available within the article and its Supplementary Information, or from the corresponding author upon request. Cartesian coordinates of the optimized structures by DFT calculations and UV-Vis data sheets are provided within the Source data file. Source data are provided with this paper.
References
Muthukrishnan, I., Sridharan, V. & Menéndez, J. C. Progress in the chemistry of tetrahydroquinolines. Chem. Rev. 119, 5057–5191 (2019).
Adejoke, H. T., Louis, H., Amusan, O. O. & Apebende, G. A review on classes, extraction, purification and pharmaceutical importance of plants alkaloid. J. Med. Chem. Sci. 2, 130–139 (2019).
Zhou, L., Lokman Hossain, M. & Xiao, T. Synthesis of N-containing heterocyclic compounds using visible-light photoredox catalysis. Chem. Rec. 16, 319–334 (2016).
Bhambri, H., Khullar, S., Sakshi & Mandal, S. K. Nitrogen-rich covalent organic frameworks: A promising class of sensory materials. Mater. Adv. 3, 19–124 (2022).
Chauhan, M. & Kumar, R. A comprehensive review on bioactive fused heterocycles as purine-utilizing enzymes inhibitors. Med. Chem. Res. 24, 2259–2282 (2015).
Tandon, R., Singh, I., Luxami, V., Tandon, N. & Paul, K. Recent advances and developments of in vitro evaluation of heterocyclic moieties on cancer cell lines. Chem. Rec. 19, 362–393 (2019).
Kerru, N., Gummidi, L., Maddila, S., Gangu, K. K. & Jonnalagadda, S. B. A review on recent advances in nitrogen-containing molecules and their biological applications. Molecules 25, 1909 (2020).
Mermer, A., Keles, T. & Sirin, Y. Recent studies of nitrogen containing heterocyclic compounds as novel antiviral agents: A review. Bioorg. Chem. 114, 105076–105124 (2021).
Wu, X. F., Neumann, H. & Beller, M. Synthesis of heterocycles via palladium-catalyzed carbonylations. Chem. Rev. 113, 1–35 (2013).
Gulevich, A. V., Dudnik, A. S., Chernyak, N. & Gevorgyan, V. Transition metal-mediated synthesis of monocyclic aromatic heterocycles. Chem. Rev. 113, 3084–3213 (2013).
He, J., Wasa, M., Chan, K. S. L., Shao, Q. & Yu, J. Q. Palladium-catalyzed transformations of alkyl C−H bonds. Chem. Rev. 117, 8754–8786 (2017).
Teng, S. & Zhou, J. S. Metal-catalyzed asymmetric heteroarylation of alkenes: Diverse activation mechanisms. Chem. Soc. Rev. 51, 1592–1607 (2022).
Liu, C. X. et al. Rhodium-catalyzed asymmetric C−H functionalization reactions. Chem. Rev. 123, 10079–10134 (2023).
Dupont, J., Consorti, C. S. & Spencer, J. The potential of palladacycles: More than just precatalysts. Chem. Rev. 105, 2527–2571 (2005).
Sambiagio, C. et al. A comprehensive overview of directing groups applied in metal-catalysed C−H functionalisation chemistry. Chem. Soc. Rev. 47, 6603–6743 (2018).
Mingo, M. M., Rodríguez, N., Arrayás, R. G. & Carretero, J. C. Remote C(sp3)–H functionalizationviacatalytic cyclometallation: Beyond five-membered ring metallacycle intermediates. Org. Chem. Front. 8, 4914–4946 (2021).
Laha, J. K., Jethava, K. P. & Dayal, N. Palladium-catalyzed intramolecular oxidative coupling involving double C(sp2)–H bonds for the synthesis of annulated biaryl sultams. J. Org. Chem. 79, 8010–8019 (2014).
Bai, L., Wang, Y., Ge, Y., Liu, J. & Luan, X. Diastereoselective synthesis of dibenzo[b,d]azepines by Pd(II)-catalyzed [5+2] annulation of o-arylanilines with dienes. Org. Lett. 19, 1734–1737 (2017).
Youn, S. W., Ko, T. Y. & Jang, Y. H. Palladium-catalyzed regioselective synthesis of 3-arylindoles from N-Ts-anilines and styrenes. Angew. Chem. Int. Ed. 56, 6636–6640 (2017).
Youn, S. W., Kim, Y. H. & Jo, Y. H. Palladium-catalyzed regioselective synthesis of 1-hydroxycarbazoles under aerobic conditions. Adv. Synth. Catal. 361, 462–468 (2019).
Houlden, C. E. et al. Distinct reactivity of Pd(OTs)2: The intermolecular Pd(II)-catalyzed 1,2-carboamination of dienes. J. Am. Chem. Soc. 130, 10066–10067 (2008).
Gao, Y., Huang, Y., Wu, W., Huang, K. & Jiang, H. Pd-catalyzed C–H activation/oxidative cyclization of acetanilide with norbornene: Concise access to functionalized indolines. Chem. Commun. 50, 8370–8373 (2014).
Manna, M. K., Hossian, A. & Jana, R. Merging C–H activation and alkene difunctionalization at room temperature: A palladium-catalyzed divergent synthesis of indoles and indolines. Org. Lett. 17, 672–675 (2015).
Chen, Y. et al. Palladium-catalyzed isoquinoline synthesis by tandem C–H allylation and oxidative cyclization of benzylamines with allyl acetate. Org. Lett. 23, 4209–4213 (2021).
He, Q. & Chatani, N. Palladium-catalyzed site-selective [3+2] annulation via benzylic and meta C–H bond activation. Angew. Chem. Int. Ed. 60, 5189–5192 (2021).
Jayakumar, J., Vedarethinam, G., Hsiao, H. C., Sun, S. Y. & Chuang, S. C. Cascade one-pot synthesis of orange-red-fluorescent polycyclic cinnolino[2,3-f]phenanthridin-9-ium salts by palladium(II)-catalyzed C–H bond activation of 2-azobiaryl compounds and alkenes. Angew. Chem. Int. Ed. 59, 689–694 (2020).
Chen, J. et al. Palladium@cerium(IV) oxide-catalyzed oxidative synthesis of N-(2-pyridyl)indoles via C–H activation reaction. Adv. Synth. Catal. 356, 2955–2959 (2014).
Jie, L. et al. Synthesis of 2-arylindoles through Pd(II)-catalyzed cyclization of anilines with vinyl azides. J. Org. Chem. 83, 10974–10984 (2018).
Tredwell, M. J. et al. Palladium(II)-catalyzed C–H bond arylation of electron-deficient arenes at room temperature. Angew. Chem. Int. Ed. 50, 1076–1079 (2011).
Zhang, F. L., Hong, K., Li, T. J., Park, H. & Yu, J. Q. Organic chemistry. Functionalization of C(sp3)–H bonds using a transient directing group. Science 351, 252–256 (2016).
Liu, X. H. et al. Diverse ortho- C(sp2)–H functionalization of benzaldehydes using transient directing groups. J. Am. Chem. Soc. 139, 888–896 (2017).
Yao, Q. J., Zhang, S., Zhan, B. B. & Shi, B. F. Atroposelective synthesis of axially chiral biaryls by palladium-catalyzed asymmetric C–H olefination enabled by a transient chiral auxiliary. Angew. Chem. Int. Ed. 56, 6617–6621 (2017).
Chen, X. Y. & Sorensen, E. J. Pd-catalyzed, ortho C–H methylation and fluorination of benzaldehydes using orthanilic acids as transient directing groups. J. Am. Chem. Soc. 140, 2789–2792 (2018).
Liao, G. et al. Synthesis of chiral aldehyde catalysts by Pd-catalyzed atroposelective C–H naphthylation. Angew. Chem. Int. Ed. 58, 11464–11468 (2019).
Song, H. et al. Synthesis of axially chiral styrenes through Pd-catalyzed asymmetric C–H olefination enabled by an amino amide transient directing group. Angew. Chem. Int. Ed. 59, 6576–6580 (2020).
Li, Y. H., Ouyang, Y., Chekshin, N. & Yu, J. Q. Pd(II)-catalyzed site-selective β- and γ- C(sp3)–H arylation of primary aldehydes controlled by transient directing groups. J. Am. Chem. Soc. 144, 4727–4733 (2022).
Hong, K., Park, H. & Yu, J. Q. Methylene C(sp3)–H arylation of aliphatic ketones using a transient directing group. ACS Catal. 7, 6938–6941 (2017).
Xu, J. et al. Pd-catalyzed direct ortho-C–H arylation of aromatic ketones enabled by a transient directing group. Org. Lett. 19, 1562–1565 (2017).
Pan, L., Yang, K., Li, G. & Ge, H. Palladium-catalyzed site-selective arylation of aliphatic ketones enabled by a transient ligand. Chem. Commun. 54, 2759–2762 (2018).
Wang, Z., Zhu, F., Li, Y. & Wu, X. F. Palladium-catalyzed carbonylative synthesis of 3-methyleneisoindolin-1-ones from ketimines with hexacarbonylmolybdenum(0) as the carbon monoxide source. ChemCatChem 9, 94–98 (2017).
Shi, R., Liao, F., Niu, H. & Lei, A. Selective formation of phthalimides from amines, aldehydes and co by pd-catalyzed oxidative C–H aminocarbonylation. Org. Chem. Front. 5, 1957–1961 (2018).
Su, B. et al. Palladium-catalyzed oxidation of β- C(sp3)–H bonds of primary alkylamines through a rare four-membered palladacycle intermediate. J. Am. Chem. Soc. 142, 7912–7919 (2020).
Do, D. C. H. & Huynh, H. V. Controlled access to four- and six-membered palladacycles via modifying donor abilities of beta-ketiminato ligands (“NacAcs”). Inorg. Chem. 61, 20087–20094 (2022).
Liu, C. et al. Palladium-catalyzed cascade cyclization for the synthesis of fused benzo-aza-oxa-[5-6-5] tetracycles. Angew. Chem. Int. Ed. 61, e202215020 (2022).
Liu, C. et al. Palladium-catalyzed 1,1-oxamidation and 1,1-diamination of unactivated alkenyl carbonyl compounds. Org. Lett. 25, 2701–2706 (2023).
Liu, C. et al. Access to amino lactones through palladium-catalyzed oxyamination with aromatic amines as the nitrogen source. ACS Catal. 13, 11339–11344 (2023).
Chen, Y. Q. et al. Overcoming the limitations of γ- and δ- C–H arylation of amines through ligand development. J. Am. Chem. Soc. 140, 17884–17894 (2018).
Chen, M. S. & White, M. C. A sulfoxide-promoted, catalytic method for the regioselective synthesis of allylic acetates from monosubstituted olefins via C–H oxidation. J. Am. Chem. Soc. 126, 1346–1347 (2004).
Stang, E. M. & White, M. C. Molecular complexity via C–H activation: A dehydrogenative Diels-Alder reaction. J. Am. Chem. Soc. 133, 14892–14895 (2011).
Diao, T., White, P., Guzei, I. & Stahl, S. S. Characterization of DMSO coordination to palladium(II) in solution and insights into the aerobic oxidation catalyst, Pd(DMSO)2(TFA)2. Inorg. Chem. 51, 11898–11909 (2012).
Trost, B. M. & Rao, M. Development of chiral sulfoxide ligands for asymmetric catalysis. Angew. Chem. Int. Ed. 54, 5026–5043 (2015).
Hsiao, P. Y. et al. Synthesis and mechanistic investigation of bipyrazolo[1,5-a]pyridines via palladium-catalyzed cross-dehydrogenative coupling of pyrazolo[1,5-a]pyridines. J. Org. Chem. 87, 9851–9863 (2022).
Yuan, C. H., Wang, X. X. & Jiao, L. Ligand-enabled palladium(II)-catalyzed enantioselective β- C(sp3)–H arylation of aliphatic tertiary amides. Angew. Chem. Int. Ed. 62, e202300854 (2023).
Hull, K. L. & Sanford, M. S. Mechanism of benzoquinone-promoted palladium-catalyzed oxidative cross-coupling reactions. J. Am. Chem. Soc. 131, 9651–9653 (2009).
Chen, S. Y. et al. Palladium-mediated C(sp3)−H bond activation of N-methyl-N-(pyridin-2-yl)benzamide: Direct arylation/alkylation and mechanistic investigation. J. Org. Chem. 88, 8441–8453 (2023).
Nong, Z. S., Chen, X. R., Wang, P. S., Hong, X. & Gong, L. Z. Enantioconvergent palladium-catalyzed alkylation of tertiary allylic C−H bonds. Angew. Chem. Int. Ed. 62, e202312547 (2023).
Lønning, P. E. The potency and clinical efficacy of aromatase inhibitors across the breast cancer continuum. Ann. Oncol. 22, 503–514 (2011).
Osmaniye, D. et al. Design, synthesis, molecular docking and molecular dynamics studies of novel triazolothiadiazine derivatives containing furan or thiophene rings as anticancer agents. Bioorg. Chem. 122, 105709–105721 (2022).
Acknowledgements
We would like to thank the State Key Laboratory of Pulp and Paper Engineering (2022PY01), the National Natural Science Foundation of China (22231002 and 21871095), and the Key-Area Research and Development Program of Guangdong Province (2020B010188001) for their financial support.
Author information
Authors and Affiliations
Contributions
H.J. designed the project; H.J. and W.W. supervised the work; X.T., J.W. and J.L. performed the experiments, mechanistic studies, and analyzed the data; Z.K., Y.J. and Z.Y. performed the DFT calculations; All authors performed data analysis and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Mengchun Ye and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tan, X., Jing, Y., Wu, J. et al. Palladium catalyzed ortho-C(sp2)–H activation/cyclization of aryl amines assisted by imine and vinylacetic acid. Nat Commun 15, 9877 (2024). https://doi.org/10.1038/s41467-024-54018-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-54018-2
