
Abstract
Identifying individuals at high risk of chronic diseases via easily measured biomarkers could enhance efforts to prevent avoidable illness and death. Using ’omic data can stratify risk for many diseases simultaneously from a single measurement that captures multiple molecular predictors of risk. Here we present nuclear magnetic resonance metabolomics in blood samples from 700,217 participants in three national biobanks. We built metabolomic scores that identify high-risk groups for diseases that cause the most morbidity in high-income countries and show consistent cross-biobank replication of the relative risk of disease for these groups. We show that these metabolomic scores are more strongly associated with disease onset than polygenic scores for most of these diseases. In a subset of 18,709 individuals with metabolomic biomarkers measured at two time points we show that people whose scores change have different risk of disease, suggesting that repeat measurements capture changes both to health status and disease risk possibly due to treatment, lifestyle changes or other factors. Lastly, we assessed the incremental predictive value of metabolomic scores over existing clinical risk scores for multiple diseases and found modest improvements in discrimination for several diseases whose clinical utility, while promising, remains to be determined.
Similar content being viewed by others
Introduction
Identifying individuals at elevated risk of disease can help guide the use of preventative interventions. For example, in the UK the multi-variable QRISK score is used to identify individuals at high risk of cardiovascular disease who should adjust their lifestyle or begin taking cholesterol-lowering or blood pressure-reducing medicine1. This concept of combining multiple measurements or risk factors into a single score has been extended to the use of ‘omic data, such as polygenic scores (PGS)2,3. By adding up the contribution of different genetic variants associated with different diseases, PGS can identify individuals at elevated risk for multiple diseases4 with one measurement (e.g., a GWAS array or genome sequence), and offer complementary information to traditional risk factors5,6. Metabolomic scores, based on adding up the contributions of multiple biomarkers measured from a blood sample, for example via nuclear magnetic resonance spectroscopy7,8,9, have also been shown to predict many common diseases10,11 including cardiovascular disease, type 2 diabetes12, and all-cause mortality13. Furthermore, since the metabolomic scores may change in response to lifestyle and treatment (in contrast to PGS), they can also track changes in people’s risk profiles. A few studies have suggested complementary value for genetics and metabolomics in cardiovascular disease and type 2 diabetes14,15, but the combined use of these ‘omics-based risk predictors has not yet been evaluated at scale.
Here, we generated nuclear magnetic resonance metabolomic biomarker data in blood samples from apparently healthy individuals from three national biobanks with follow-up data on clinical outcomes. We trained risk prediction scores for the 12 leading causes of disability-adjusted life years (DALYs) in high-income countries. We investigated the relative performance of these metabolomic scores, PGS and clinical scores in different diseases and time scales. We replicated the performance of metabolomic scores across the three biobanks in the study and assessed the value of multiple metabolomic time points in two of the biobanks.
Results
Metabolomic risk prediction across top sources of morbidity
Building metabolomic scores
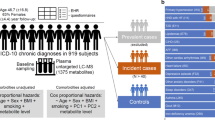
We measured metabolomic biomarkers via nuclear magnetic resonance spectroscopy in blood samples provided at the time of enrollment from 700,217 participants in the UK Biobank, Estonian Biobank (EBB), or Finnish THL Biobank, all with linked comprehensive clinical data (Table 1, Supplementary Data 1). An overview of the study design is shown in Fig. S1. All three biobanks contain adults from Northern European countries, with varying ascertainment, recruitment years, age ranges, and procedures for extracting outcomes from electronic health records (Methods, Fig. S2).
We analyzed 12 diseases causing the most morbidity in the WHO European region in 2019 (excluding falls and back pain, Fig. 1, Table 1, Supplementary Data 2), which cause more than one-third of all DALYs. We trained Cox proportional hazards models to predict incidence of each of these diseases in half of the UK Biobank. We included age and sex in all models as fixed covariates and allowed the model to select (via Lasso with tenfold cross-validation) from among 36 metabolomic biomarkers that have been validated in Europe for use in an in vitro diagnostic medical device (Methods). For all but two of the diseases studied, more than half of the biomarkers were included in the scores: 17 for Alzheimer’s disease, 18 for intracerebral hemorrhage, 21 for colon cancer, 24 for lung cancer, 26 for vascular and other dementias, 27 for stroke, 28 for alcoholic liver disease, 29 for chronic obstructive pulmonary disease (COPD), 30 for liver cirrhosis, 31 for myocardial infarction, 33 for diabetes, and 35 for depression (model coefficients for each biomarker in each score are shown in Supplementary Data 3 and Fig. S3). We evaluated the performance of these scores in the other half of the UK Biobank, as well as the Estonian and Finnish THL biobanks. As we quantify the biomarkers in absolute concentration units (e.g., mmol/l), we can directly use the variable coefficients estimated in the UK Biobank to calculate scores in the other two datasets, without normalizing the biomarkers within each study separately. This is distinct from common practice in other ‘omics analyses, where within cohort normalization is essential16,17. Figure S4 shows that we obtain highly similar results with these normalization steps, but we here present results without them to better mimic predicting a new individual’s risk without additional information (e.g., batch corrections, or cohort means and variances).

A Observed incidence of the 12 diseases divided into one percent bins of the metabolomic score. The observed incidence is shown as a sample size weighted mean of the 4-year incidence in the three biobank cohorts (n = 481,678). Red shading shows the top 10% of the metabolomic score (adjusted for age and sex). Horizontal dashed line shows the population prevalence. B Four-year hazard ratios of metabolomic scores (adjusted for age and sex) comparing the highest 10% to the remaining 90% of the study population for 12 diseases (n = 481,678). The dots represent point estimates (Cox regression estimates for individual cohorts, fixed inverse-variance weighted mean for meta-analysis) and the horizontal error bars denote 95% confidence intervals of the hazard ratio. Source data are provided as a Source Data file.
Baseline age and sex minimally adjusted metabolomic scores and incident disease
We stratified the three test sets into one percent bins of the metabolomic score distribution and meta-analyzed the four-year incidence rates for each disease (Fig. 1A). The risk of incident disease increased with increasing levels of the metabolomic score across all the diseases. As has been observed previously4,10, these curves follow a quantile-logistic function, which rises super-exponentially in the tails, making it possible to identify subsets of individuals that are at much higher risk than average. This effect is especially dramatic for the scores that most strongly predict disease, including type 2 diabetes and liver diseases.
Figure 1B shows the performance of the scores by comparing the relative risk of incident disease in the 10% of individuals with the highest metabolomic scores (high-risk group, red shaded area, Fig. 1A) to the remaining population. Again, to avoid needing within-cohort scaling factors or thresholds, we used the top 10% boundary from our training data to define this group in the other half of the UK Biobank and the other two biobanks. This means the proportion of individuals in the high-risk group varies across the three biobanks (Supplementary Data 4), but this high-risk group nonetheless had consistently increased risk across diseases (Fig. 1B). Only depression, alcoholic liver disease, lung cancer, and COPD showed significant meta-analysis heterogeneity (Cochran’s Q, p < 0.004 to account for multiple testing). The UK Biobank test set had the highest point estimate of effect size in only 4 of 12 diseases, demonstrating that the scores are capturing generalizable risk factors, rather than overfitting to the UK Biobank. The meta-analysis of the three test sets included hazard ratios of ~10 for two types of liver disease and diabetes, ~4 for COPD and lung cancer, and ~2.5 for myocardial infarction, stroke and vascular dementia, and was statistically significant (fixed-effect meta-analysis Z score test, p < 0.004 adjusted for multiple testing) for all diseases (Fig. 1B). The pattern of association is similar when considering hazard ratios per standard deviation in a continuous model (Fig. S4), and population-wide discrimination, as measured by area under the receiver-operating characteristic curve (AUC), shows consistent, though variable, improvement when adding metabolomic scores to age and sex (Supplementary Data 5).
Sensitivity and subgroup analyses of minimally adjusted scores
Having demonstrated that it is possible to construct metabolomic scores that are replicably associated with risk of these diseases, we next sought to use the diverse data available in these biobanks to investigate further properties of metabolomic scores.
First, we assessed the performance of scores using all 249 metabolomic biomarkers we measured, rather than the 36 clinically validated biomarkers described above. Only diabetes and COPD showed consistently improved performance using the extended metabolomics (Fig. S5), likely because many of the biomarkers are correlated, and our clinically validated subset captures a large fraction of the total available information in most cases. In some cases, such as Alzheimer’s and the liver diseases in the EBB, the extended metabolomic scores do not replicate as well as the simpler scores. Taken together, these results suggest some diseases may benefit from additional molecular measurements, but care must be taken that they do not capture cohort-specific effects which are less transferrable.
Second, we sought to understand the extent to which our scores are driven by well-established behavioral risk factors for some of these diseases, in particular tobacco smoking and alcohol consumption. Both lung disease scores show attenuated, but still strong, association when conditioning on pack-years of smoking, suggesting that while they are partly driven by this behavior, they capture additional information beyond the self-reported variables (Fig. S6). The performance of our lung cancer score is reduced to almost zero in never smokers, whereas our COPD score still has significant prediction in that group. Liver cirrhosis is equally well predicted across ever and never drinkers, and virtually unaffected by an adjustment for daily alcohol units. The adjusted alcoholic liver disease prediction is somewhat reduced but remains very strong (Fig. S6).
Third, as we were limited to somewhat coarse ICD-10 based definitions of the diseases we were studying, we examined whether broader or narrower definitions might change our results by investigating cardiovascular disease more closely. The score trained on the narrow outcome of myocardial infarction had a 0.96 correlation with one trained more broadly on ischemic heart disease. This suggests that for deriving the scores the definition of the disease endpoint is not very sensitive, likely because the underlying risk factors are broadly shared. When testing the scores for association in the test datasets, both show a gradient of increasing effect size for more severe outcomes, from unstable angina to first myocardial infarction to subsequent myocardial infarction (Fig. S7), suggesting that the scores may be strongest at predicting severe outcomes.
Comparison of hazard ratios for incident disease among metabolomic, genetic, and combined scores
We next compared the performance of metabolomic scores to PGS, which have received widespread attention for risk stratification to aid prevention4,18,19. We calculated PGS using variant weights from the PGS Catalog (Supplementary Data 2), only using scores that were built from genome-wide association study (GWAS) data that did not include the biobanks studied here. We again trained models in half the UK Biobank always including age and sex, and using Lasso to select from: (i) only the external PGS, (ii) among the metabolomic biomarkers (as above), or (iii) among both the PGS and the metabolomic biomarkers.
PGS were available for 10 of our diseases, and, as expected, the top 10% high-risk groups were at significantly higher risk than the remaining 90% (Fig. 2A). However, the hazard ratio of being in the genetic high-risk group was less than the metabolomic high-risk group in all diseases except colorectal cancer. In most cases, the best performing model included both genetic and metabolomic scores, suggesting that these two data types capture at least partially complementary information. A formal interaction test between metabolomic and genetic scores found a significant effect only for type 2 diabetes (Supplementary Data 6), and the small confidence intervals demonstrated that genetic and metabolomic risk is primarily additive on the log hazard ratio scale. For six diseases we could also calculate PGS in the EBB, which replicated the results in the UK Biobank (Fig. S8).

A Ten-year hazard ratios of metabolomic, polygenic or combined scores comparing the highest risk decile to the remaining study population (UK Biobank test set, n = 242,492). Dots represent Cox regression estimates and horizontal error bars denote 95% confidence intervals of the hazard ratio. B Risk of disease incidence after blood sampling for high genetics risk group stratified by their metabolomic score and for average genetic risk group. Shaded region denotes 95% confidence interval. C Hazard ratios for highest decile of metabolomic or polygenic scores stratified by time to event. Dots and vertical error bars denote Cox regression estimates and 95% confidence intervals per bin, shaded region is 95% confidence interval for a generalized survival model allowing a time-varying effect using natural splines with 2 knots. All scores adjusted for age and sex (n = 242,492). Source data are provided as a Source Data file.
We stratified individuals in the genetic high-risk group by whether they were also in the metabolomic high-risk group (Fig. 2B). Individuals at high risk by both PRS and metabolomics scores are indeed at very elevated risk, but genetically predisposed individuals not in the high metabolomic risk group have risk similar to (or in some cases less than) those not in the genetically predisposed group. This is likely because current PGS capture less than half of the genetic risk for these diseases, and that unexplained heritability, combined with lifestyle and environmental history, is partially reflected in the metabolomic score. The metabolomic and genetic scores also have different patterns of correlation between different diseases (Fig. S9). As has been previously shown, the PGS for different diseases tend to be largely uncorrelated19, whereas the different metabolomic scores are nearly all correlated with each other, reflecting multi-morbidity10,13. Combining the two types of information can yield both improved performance and specificity of risk stratification.
While the three biobanks are dominated by individuals of European ancestry, we did compare the transferability of the metabolomic scores and PGS for 8 endpoints with at least 35 events in multiple ancestries in the UK Biobank (Fig. S10). The metabolomic scores remained significantly predictive across disease-ancestry combinations, though often with weaker effect size estimates than in the European ancestry group. As has been previously shown, the effect sizes of PGS were also attenuated in non-European ancestries, and because they perform worse than metabolomics in Europeans, the estimate was statistically significant in only 3 out of 19 non-European comparisons. For the metabolomic scores, 12 out of 19 were significant. This shows how more diverse datasets will be essential not just to produce transferrable polygenic risk scores, but more generally to produce multi-omic scores that are as widely useful as possible.
The longer follow-up time in the UK Biobank also allowed us to compare short-term and long-term prediction from these scores. As expected, since PGS are fixed throughout life, their hazard ratios remained constant over follow-up time (Fig. 2C). The relationship between hazard ratio for the metabolomic score and time to event varied by disease: diabetes, lung cancer, vascular dementia and alcoholic liver disease scores provide stronger stratification of near-term risk, but for most diseases the metabolomic scores were stable over time, like PGS.
Incident disease risk among participants with two metabolic scores
We generated metabolomic profiles at a second-time point from blood samples donated by 18,709 UK Biobank participants who returned for a repeat visit approximately four and a half years after they initially enrolled in the study (median time difference 4.4 years, mean 4.3 years, range 2.1–6.9 years). The correlations of the scores range from 0.42 for Alzheimer’s disease to 0.71 for diabetes and fall in the middle of the range of correlations for individual biomarkers (e.g., amino acids ~0.3, HDL cholesterol ~0.8) (Supplementary Data 7, Fig. S11).
For eight diseases (myocardial infarction, ischemic stroke, diabetes, COPD, depression, colorectal cancer, lung cancer, and vascular and other dementias) at least 100 events occurred within 10 years of the repeat visit, so we fitted a joint risk model with baseline and follow-up metabolomic score measurements. For diabetes (Cox regression, baseline HRb = 2.52, 95% CI = 2.24–2.83, pb = 2.45 × 10−54, follow-up HRf = 1.57, 95% CI = 1.39–1.77, pf = 1.10 × 10−13) and COPD (HRb = 1.52, 95% CI = 1.33–1.72, pb = 2.2 × 10−10, HRf = 1.31, 95% CI = 1.31–1.70, pf = 1.12 × 10−9) both time points were significantly associated with 10-year risk; for all other diseases except vascular and other dementias the hazard ratio point estimates were all consistently positive but were not significant due to weaker prediction from the scores and smaller sample size. This suggests that both a person’s current metabolomic score value, as well as previously measured score values, contribute information about risk of disease onset.
To further explore this idea, we considered individuals in the top 10% high-risk groups at the first time point and compared the subset of that group who remained in the high-risk group at the follow-up time point to those who had left it. For diabetes, leaving the high-risk group showed a significant reduction in risk (Cox regression, HR = 2.58, 95% CI = 1.74–3.84, p = 2.7 × 10−6), after adjusting for baseline score (Fig. 3). For lung cancer and COPD, risks were reduced fivefold (HR = 4.96, 95% CI = 1.61-15-23, p = 5.2 × 10−3) and 1.9-fold (HR = 1.92, 95% CI = 1.21–3.06, p = 5.8 × 10−3) respectively, but estimates were no longer significant after multiple testing correction. We replicated this analysis in 5038 individuals from the EBB for whom we also profiled a second timepoint from blood samples donated approximately five years after the baseline survey. We observed the same effect for type 2 diabetes, which was the only disease for which we had sufficient cases to test (HR = 4.4, p = 0.002).

Maroon lines show those who were in the high-risk group at both time points ~5 years apart, green lines show those who were in the high-risk group at enrollment but had left it by the second time point, orange lines show those who were in the low risk group at baseline and moved to the high risk group at follow-up, and black lines show those who were in the low-risk group at both time points. Data from 18,709 UK Biobank participants with metabolomic scores from two time points. Shaded areas are 95% confidence intervals, derived from the standard error of the cumulative hazard. Source data are provided as a Source Data file.
While we do not know what caused individual metabolomic scores to change between time points in these observational cohorts, we can assess what differences in lifestyle factors are associated with changes in metabolomic scores. For example, obese individuals who stayed in the high-risk group for diabetes gained an average 0.18 units of body mass index (BMI), but those who changed from high to low risk lost an average of 0.81 units of BMI (difference of 0.99, 95% CI 0.78–1.20, linear regression t(df = 1348) = 9.22, p = 1.08 × 10−19). Among self-reported smokers who were in the high-risk group for COPD at the first time point, 64% of those who continued smoking remained at high risk, compared to just 40% of those who reported quitting between the two time points (Fisher’s exact test OR = 2.72, 95% CI = 2.47–6.68, p = 0.0055). However, these explained only a few percent of the observed metabolomic score changes, demonstrating that the scores integrate a wider range of information than questionnaires.
Comparing multi-omics to existing clinical risk scores
We next compared our multi-omics predictions to published clinical risk scores both in terms of hazard ratios for the top decile and population-wide AUC. For all our diseases except Alzheimer’s we identified scores that are recommended for use either by the National Health Service in England and Wales (NHS) or by professional bodies in the UK, EU, or USA, and calculated them as accurately as possible using available variables in the UK Biobank (Methods). These scores vary in the types of variables they include (e.g., QRISK for cardiovascular disease includes several blood and anthropometric measurements, whereas PHQ2 for depression is based solely on two self-reported questions). The multi-omics scores perform significantly better for 10-year risk prediction of myocardial infarction, the two liver diseases, and colon cancer, while the clinical scores perform significantly better for lung cancer, diabetes, and depression, and the remaining four either have no difference, or inconsistent results between AUC and hazard ratio of the top decile (Fig. 4A, Table 2). For all diseases except intracerebral hemorrhage (which is not well predicted beyond age and sex by any score we tested) a combined clinical+multi-omic score has significantly higher AUC than the clinical score, with increases ranging from 0.006 for lung cancer to 0.118 for alcoholic liver disease (Table 2). These results were also consistent for four years of follow-up (Supplementary Data 8, Fig. S12).

A Ten-year hazard ratios of models with clinical variables compared to those variables plus the best ‘omic data (either just metabolomics or metabolomics plus PGS from Fig. 2, n = 241,750). Dots represent Cox regression estimates and horizontal error bars denote 95% confidence intervals of the hazard ratio. B For each disease, the calibration of three-year observed event rates are shown by 10 equally sized deciles of absolute risk predicted by a metabolomic score adjusted for age and sex (n = 415,592). Dots represent means and vertical lines represent 95% confidence intervals of the observed event rate. Calibration slopes and intercepts were derived from a logistic regression of the observed risk on the predicted risk. Source data are provided as a Source Data file.
To illustrate how the multi-omics score could augment the most widely used risk screening tool in the UK, we further focused on comparing QRISK to QRISK+multi-omics in individuals not using statins at baseline, which approximates the eligible group for a common use of these scores in prioritizing patients for statin treatment. The AUCs improve by 0.029 for myocardial infarction and 0.008 for stroke, which are equivalent to the whole-population values in Table 2 (i.e., there is no interaction between these scores and statin usage). Considering net reclassification index, the continuous changes (i.e., net fraction of individuals whose risk score moves in the right direction) are substantial: 0.21 myocardial infarction events (NRI+) and 0.31 for MI non-events (NRI–) and 0.02 and 0.18 for stroke. Considered categorically (i.e., moving in the right direction between high and low-risk groups) they are NRI+ = 0.09 and NRI– = −0.01 for MI and NRI+ = 0.02 and NRI– = 0.00 for stroke.
Finally, we compared our high-risk groups to the remainder of the population using the Frailty Index20,21 as a surrogate for an overall impression of the health of an individual. The high-risk group has slightly higher frailty index values (Fig. S13) and generally different clinical characteristics (Supplementary Data 9).
Calibration across studies from different countries
Risk prediction models should be evaluated based on both discrimination and calibration22. We therefore tested the calibration of our metabolomic scores by plotting observed event rates against predicted absolute event rates per decile in all three biobanks (Fig. 4B). We estimated calibration slopes and intercepts by fitting a logistic regression model of observed risk on predicted risk, without any study-specific processing or normalization, mimicking real-world patient usage (Methods). For the main calibration analysis, we included diseases with >200 events over 3 years, as recommended by earlier studies23,24. Calibration results for the remaining diseases are shown in Fig. S14.
Overall, the metabolomic scores demonstrated good calibration. In the UK Biobank test set the calibration slopes ranged from 0.95 to 1.24 across diseases, as expected since the models were trained in the other half of this biobank. In the EBB, the calibration slopes ranged from 0.76 to 1.16, except for depression at 0.42. This difference is likely a result of diagnostic differences in depression in different countries, as well as how those diagnoses are encoded in electronic records. In the Finnish THL Biobank the slopes were 1.03 (ischemic stroke), 1.20 (myocardial infarction) and 1.21 (diabetes), though the absolute rates varied considerably, likely reflecting different rates of these diseases in the earlier recruitment waves of these cohorts.
Discussion
We have shown that metabolomic scores can identify individuals at increased risk across a range of diseases, consistent with a previous report that analyzed a subset of the data described here10. We have replicated these findings in two additional national biobanks and demonstrated consistent performance in three countries which have varying sample collection types (e.g., plasma vs. serum), enrollment criteria, fasting protocols, and electronic health record systems. As the biomarkers that constitute these scores are measured in absolute units, the scores can be computed without cohort-specific rescaling, which could aid in clinical translation of the scores. More than 1 in 4 individuals in these biobanks are in the high-risk group for at least one of the cardiovascular diseases, lung diseases, liver diseases or diabetes, where the high-risk group has at least 2.5-fold increase in risk (likely an underestimate of population levels, due to healthy volunteer bias).
Our direct comparison, and combination, of metabolomic and polygenic risk factors suggests the value of multi-omic scores. When good predictors exist from both ‘omics data types (e.g., myocardial infarction, diabetes, colorectal cancer) the scores are complementary and together provide an improved combination of predictive accuracy and specificity. Combining these forms of information may also be useful in maximizing the predictive accuracy while avoiding the perceived determinism25 of fixed genetic predictions: our data suggests both that genetic factors contribute stable long-term predictive information and that these can be outweighed, particularly in the shorter term, by detectable differences in metabolomic risk profile (driven, in part, by factors modifiable by lifestyle and treatment changes). Our analysis of follow-up samples underscores this: the scores show an intermediate level of stability five years apart, meaning they provide long-term risk prediction and can also track measurable change in risk in response to lifestyle changes or treatment. The observational nature of this dataset, as well potential survivor bias in the individuals who participated in the repeat sampling, limit our ability to make causal inferences about associations between changes in lifestyle factors and changes in scores. Future studies that explicitly measure metabolomics before and after an intervention are needed to fully explore how metabolic risk scores track changes in modifiable risk.
For 11 of the diseases we studied, we compared to existing scores used for risk prediction or screening. We identified clear examples where multi-omic scores alone outperformed these clinical scores, including for myocardial infarction and liver diseases. In other diseases the clinical scores perform better, often for understandable reasons. For instance, for type 2 diabetes the QDiabetes score we used includes Hb1Ac, the gold-standard diagnostic biomarker for the disease. For lung cancer, the clinical score directly includes the vital causal risk factor of smoking, and yet the multi-omic score still provides significant additional information: if we restrict to current smokers, the multi-omic score outperforms the clinical score for lung cancer (hazard ratio of 2.2 vs 1.8). Multi-omic scores provide statistically significant improvements when added on top of existing scores in all but one disease. Future work will be needed to quantify the extent to which these statistically significant improvements in prediction, when used to guide health interventions, could translate into improvements in population health, as has been recently studied in the specific case of cardiovascular disease26. Furthermore, it will be important to consider whether and how multi-omic data fit in the diverse clinical contexts represented by these scores. QRISK3 and QDiabetes are mainly tools for identifying high-risk individuals who would benefit from primary prevention efforts, COPD-PS, PHQ2, AUDIT and Fatty Liver Index (FLI) are screening tools for prioritizing further investigation in individuals who may have undiagnosed disease, and QCancer and Liverpool Lung Project (LLP) scores are used for both primary prevention and to aid early detection. In addition to accuracy, our results also allow us to test calibration across populations, which is vital to future applied use. While imperfect, the calibration of our scores is comparable to widely used tools like the pooled cohort equations for cardiovascular risk when compared, for example between the US and Canada27.
Our study has several limitations which we could only partially mitigate. First, although biobanks are powerful for studying many diseases simultaneously, they typically have less detailed and well-curated phenotypes than disease-specific clinical cohorts. For example, we use endpoint definitions based on broad categories of ICD-10 codes. In the case of cardiovascular disease, we have shown using more fine-grained coding does not substantially alter our conclusions, but clinically focused collections will help bring additional resolution. Furthermore, biobanks are known to have healthy volunteer bias, which can affect comparisons between these results and true rates of disease in the overall population. Second, we here defined those groups as the top 10% of each disease’s risk score, but in practice different cut points may be appropriate for different diseases (e.g., in liver disease, risk is strongly concentrated in the top 1%), or cut-offs may not be necessary at all if the goal is to deliver improved prediction across the full range of baseline risk in the population. Third, these three biobanks are nearly all of European ancestry, and while we showed some promising results in the non-European ancestry subset of UK Biobank, analyses of more diverse cohorts will be essential. Finally, in comparing against clinical screening there are certain variables not measured in these biobanks that preclude a complete comparison, including of some widely used low-cost tests (e.g., fecal occult blood samples in colon cancer) and more intensive screening tools (e.g., CT screening for lung cancer or colonoscopies for colon cancer).
Many healthcare systems desire a more personalized and preventative model of disease management, including longitudinal monitoring of risk, early detection of disease, and active, patient-centered management of risk factors. For this reason, it will be valuable to understand how classical risk factors can be supplemented or replaced with newer predictors including metabolomics and genetics, and how and why these risk predictions change over time. We believe our results, and the extremely large dataset that underlies them, are an important part of this puzzle. Future work will need to focus on how we may incorporate additional new predictors to stratify disease risk further (e.g., recent work on proteomics has shown promise28), as well as how this next generation of prediction algorithms, described here for research use, can be validated, made actionable and delivered to patients.
Methods
Study populations and endpoint definitions
We used data from a total of 700,217 individuals from three biobanks, UK Biobank (N = 477,078), EBB (N = 190,785) and Finnish THL Biobank (N = 32,354). Figure S1 shows an overview of our study design and Table 1 shows summaries of participant characteristics.
The UK Biobank is a longitudinal biomedical study of approximately half a million participants between 38 and 71 years old from the United Kingdom29. Participant recruitment was conducted on a volunteer basis and took place between 2006 and 2010. Initial data were collected in 22 different assessment centers throughout Scotland, England, and Wales. Data collection includes elaborate genotype, environmental and lifestyle data. Blood samples were drawn at baseline for all participants, with an average of four hours since the last meal, i.e., generally non-fasting. Nuclear magnetic resonance (NMR) metabolomic biomarkers (Nightingale Health, quantification library 2020) were measured from EDTA plasma samples (100 μL, aliquot 3) during 2019–2023 for the entire cohort. In addition, plasma samples were measured by NMR metabolomics from ~20,000 participants who underwent a repeat-visit assessment on average five years after the baseline visit. The NMR protocol is known to perform well on blind duplicate samples in the UK Biobank8. Follow-up data include a wide range of electronic health-related records, including disease incidence, hospital admissions, primary care, and death records, which are presently still regularly updated. The UK Biobank study was approved by the North West Multi-Centre Research Ethics Committee. This research was conducted using the UK Biobank Resource under Application Number 30418.
The EBB is a curated population-based biobank of Estonia, comprising a cohort of ~210,000 volunteers30. The participants constitute about 20% of the adult Estonian population and the cohort is approximately representative of the nation in terms of age, sex, and geographic dispersion. The enrollment was conducted between 2002 and 2022. A network comprising general practitioners and various medical personnel from private practices, hospitals, and recruitment offices of the Estonian Genome Center was established for participant recruitment, as well as for collection of samples and health data. After recruitment, participants were asked to fill out detailed questionnaires, which encompassed personal information, genealogical data, educational and occupational history, as well as lifestyle habits. Blood samples were generally collected non-fasting31. NMR metabolomic measurements were conducted on EDTA plasma samples (100 μL) for all biobank participants. The EBB database undergoes regular synchronization with several national registries and hospital databases, along with the national health insurance fund’s database that houses comprehensive treatment and service bill information. Disease events are codified in compliance with the ICD-10 standards and medication usage is categorized as per the Anatomical Therapeutic Chemical classification, both with current follow-up data available until the end of 2021. The Estonian Committee on Bioethics and Human Research approved the study. Data was accessed with research approval number 1.1-12/2770.
The Finnish THL biobank data consist of five population cohorts (National FINRISK Studies 1997, 2002, 2007, 2012 and Health 2000 Survey) collected in study specific years between 1997 and 201232,33. Each of the five cohorts is an independent random sample drawn of unique individuals aged 25–98 (25–74 in FINRISK, 30 and over in Health 2000). Recruitment was conducted via invitation-only in multiple urban and rural areas across Finland to be representative of the nation (participation rate 60–70%). The baseline surveys included a wide range of health-related questionnaire and biological measures, including a non-fasting blood sample (median 5 h since last meal) from ~85% of all participants enrolled. NMR metabolomic data were measured from all participants with blood samples available. In contrast to UK and EBBs, NMR metabolomics measurements were done on serum samples (350 μL)8,32. Information on disease outcomes were linked from national hospital discharge registries and reimbursement records with follow-up until 2017 (4 to 19 years of follow-up). The THL biobank cohorts were approved by the Coordinating Ethical Committee of the Helsinki and Uusimaa Hospital District, Finland. Data was accessed with research application number BB2016_86.
In our main analysis, we included 12 diseases that are the top causes of disease-adjusted life years (DALYs) in the European region in 2019 according to the WHO, except falls and back pain34: myocardial infarction, ischemic stroke, intracerebral hemorrhage, lung cancer, type 2 diabetes, COPD, Alzheimer disease, vascular and other dementias, depressive disorders, alcoholic liver disease, cirrhosis of the liver, and colon and rectum cancers. Some WHO groupings were coarse (e.g., all dementias together), so we used narrower definitions in those cases to create more biologically homogenous outcomes aligned with common disease definitions used for PGS development. Instead of using ischemic heart disease and diabetes mellitus, we specified to myocardial infarction and type 2 diabetes, stroke was split into ischemic stroke and intracerebral hemorrhage, Alzheimer’s disease was separated from vascular and other dementias, and alcoholic liver disease was separated from cirrhosis of the liver (Supplementary Data 2). We also included a model for the coarser ischemic heart disease (ICD-10 codes I20-25) endpoint, which we compared to the myocardial infarction model in a supplementary analysis.
Disease incidence was defined based on the first occurrence of ICD-10 codes listed in Supplementary Data 2. In the UK Biobank, we based disease incidence on primary care data (only available in ~45% of UK Biobank participants), hospital inpatient data, death register records, cancer registry data, and self-reports at baseline. In the EBB, we based disease incidence on self-reports at baseline, E-Health, North Estonia Medical Center, Tartu University Hospital, death registry records and cancer registry data. The Estonian Health Insurance Fund was excluded as a source because it appeared that diagnoses exclusively from that source were less severe. In the Finnish THL Biobank, we based disease incidence on the nation-wide hospital discharge registry (HILMO), cause-of-death registry, and for certain diseases, medication reimbursement registry. We considered disease cases occurring after the blood draw at study baseline as incidence cases. Cases that occurred prior to the baseline blood draw were considered prevalent cases and were excluded from the analysis in a disease-specific manner.
Analyses in this paper were all carried out conditional on age and sex. Sex was defined differently in the different cohorts: in UK Biobank, sex at recruitment is taken from the patient’s medical record but can subsequently be edited by the participant if they choose. For the Finnish THL biobank, sex was taken from the population registries held by the Digital and Population Data Services Agency. In the EBB, sex was extracted from the Estonian National Identity number which is created based on the sex recorded in the Estonian Birth Registry.
Written informed consent was obtained from all participants. Participants were not offered compensation for participating in this study.
Metabolomic biomarker profiling
Lipid and metabolite biomarkers were quantified from 757,927 blood samples by high-throughput NMR metabolomics (Nightingale Health Plc). This number includes 23,080 blinded duplicate samples from the UK Biobank for quality control purposes. Additionally, each 96 well-plate contained two internal control samples. We used the sample handling and measurement protocol established and validated for the first phase of metabolomics in UK Biobank8. The measurement protocol was similar for EBB and Finnish THL Biobank32. Briefly, EDTA samples of at least 90 µL were plated onto 96-well plates at UK Biobank laboratory (Stockport, UK) and shipped on dry ice in batches of 5000–20,000 samples to Nightingale Health Laboratories in Finland. Samples were provided to the Nightingale Health lab using randomly assigned pseudonymous sample IDs and were analysed in three tranches blinded to the clinical phenotype of the sample, with UK Biobank only revealing the linkage from pseudonymous sample IDs to true sample IDs after data generated for that tranche was complete. Samples were thawed overnight at +4 °C, mixed and centrifuged, transferred to NMR tubes and mixed in 1:1 ratio with a phosphate buffer (75 mM Na2HPO4 in 80%/20% H2O/D2O, pH 7.4, including also 0.08% sodium 3-(trimethylsilyl) propionate-2,2,3,3-d4 and 0.04% sodium azide). The samples were profiled using a total of nine 500 MHz spectrometers (Bruker AVANCE IIIHD). Two NMR spectra are recorded (a presaturated proton spectrum and a Carr–Purcell–Meiboom–Gill T2-relaxation-filtered spectrum) and proprietary software is used for biomarker quantification in absolute units (Nightingale Health, quantification library 2020)7,8. This provides 249 biomarker measures in a single assay (168 absolute and 81 ratio measures), including routine lipids, lipoprotein profiling of 14 size subclasses, fatty acids, and various low-molecular weight metabolites, such as amino acids, ketones, and glycolysis metabolites as well as two inflammatory protein measures, albumin, and glycoprotein acetyls. For risk model training we used 36 clinically validated biomarkers with CE mark in the NMR metabolomics assay to facilitate rapid translation and clinical applications for model training (Total cholesterol, VLDL cholesterol, Clinical LDL cholesterol, HDL cholesterol, Total triglycerides, Apolipoprotein B, Apolipoprotein A1, Ratio of apolipoprotein B to apolipoprotein A1, Total fatty acids, Omega-3 fatty acids, Omega-6 fatty acids, Polyunsaturated fatty acids, Monounsaturated fatty acids, Saturated fatty acids, Docosahexaenoic acid, Ratio of omega-3 fatty acids to total fatty acids, Ratio of omega-6 fatty acids to total fatty acids, Ratio of polyunsaturated fatty acids to total fatty acids, Ratio of monounsaturated fatty acids to total fatty acids, Ratio of saturated fatty acids to total fatty acids, Ratio of docosahexaenoic acid to total fatty acids, Ratio of polyunsaturated fatty acids to monounsaturated fatty acids, Ratio of omega-6 fatty acids to omega-3 fatty acids, Alanine, Glycine, Histidine, Total concentration of branched-chain amino acids, Isoleucine, Leucine, Valine, Phenylalanine, Tyrosine, Glucose, Creatinine, Albumin, Glycoprotein acetyls)8.
To account for potential glucose degradation prior to plasma sample preparation, we used an estimate of physiological glucose concentration based on observed glucose and lactate as input for the risk models. Further, to correct for spectrometer differences in alanine concentration, the single metabolite most impacted by technical variation35, we shifted mean alanine concentrations observed within each spectrometer in each biobank to the mean and standard deviation of a master spectrometer. The average biomarker detection rate was >99% across the plasma samples. Further details on the individual biomarker measures are provided in the UK Biobank data resource.
Genotype data and polygenic scores
For this study, genotype data was available for UK Biobank and EBB, but not for THL Biobank. The UK Biobank participant have been genotyped on Applied Biosystems UK Biobank Axiom Array and UK BiLEVE Axiom Array, measuring over 800,000 variants and imputed using the Haplotype Reference Consortium and UK10K and 1000 Genomes reference panels outside of this study29. EBB participants have been genotyped with genome-wide chip arrays and further imputed with a population-specific imputation panel consisting of high-coverage (30-fold) whole-genome sequence data from 2244 individuals and over 16 million high-quality genetic variants36.
For 10 of the 12 diseases, we used an existing PGS from the PGS Catalog37 that was developed using GWAS summary statistics that did not include the UK Biobank in their discovery cohort (Supplementary Data 2). These PGS were computed for UK Biobank participants as the weighted sum of risk alleles using imputed genotype data. We were also able to compute six of these PGS in EBB, though we note that a small number (~8000) of EBB samples were included in the GWAS underlying the PGS of diabetes and myocardial infarction. For UK Biobank, we estimated participant’s genetic ancestry with respect to the five superpopulations of the 1000 Genomes Project38 using principal component analysis projection and a random forest classifier, and scaled PGS with respect to their estimated ancestry. For EBB we assumed more homogenous genetic background and scaled PGS within the cohort.
Clinical scores
A disease-specific clinical risk score was chosen for each endpoint to act as a comparator for benchmarking our risk scores. The risk scores were chosen such that they could be fully or partially calculated in UK Biobank participants using data available at baseline, and were taken from national risk assessment, screening and diagnosis guidance from NHS England and Wales where available, and from recommendations from other government or professional bodies where not available. Two risk scores were modified to account for data not available in UK Biobank at baseline (AUDIT-C and COPD-PS), and one was modified to remove a question that showed informative missingness in our dataset (LLPV2). We did not identify a widely used clinical risk score for Alzheimer’s disease, so did not carry out benchmarking for this endpoint. Justifications for the choice of risk scores and details of their implementation are given in the Supplementary Methods document, and details, including code lists, of how each variable is derived from UK Biobank data is given in Supplementary Data 10. We defined high-risk individuals on the basis of the clinical scores as those who were in the top decile of risk, after adjusting for age and sex, in the training set. For some clinical scores (AUDIT-C, COPD-PS and PHQ2) it was not possible to define a top decile of risk, as these scores take discrete values and therefore have exact ties that result in participants being on the border of high and low risk; in these cases we randomly assigned individuals with a borderline risk to the high risk or low category with fixed per-score probabilities, with these probabilities chosen to ensure that 10% of individuals were assigned as high risk in the training set. During model fitting variables were transformed to ensure a close to normal distribution (log transformation for FLI, logit transformation for QRISK3, QDiabetes, QCancer and LLPv2). We additionally assessed the performance of the two lung and two liver models in the context of smoking and drinking behavior. Lung cancer and COPD HR for the top decile versus the bottom 90% were evaluated separately for ever and never smokers, and adjusted for pack years of smoking. Liver cirrhosis and alcoholic liver disease HR were evaluated separately for ever and never drinkers, and adjusted for daily units of alcohol consumption. We also computed correlations between the disease scores, pack years and alcohol units.
Statistical analyses
Prior to epidemiological data analyses, we set any value of the metabolomic biomarkers or PGS to missing if the value was more ±4 standard deviations away from the mean. Samples with any missing information (metabolomic biomarkers, PGS or clinical variables) required for score training or prediction were excluded separately for each analysis. Considering the 36 CE-marked metabolomic biomarkers, 11,676 samples (2.4%) had at least one biomarker missing, and 16,614 samples (3.5%) were excluded due to outlier filtering. Additional analyses comparing performance within excluded samples showed that exclusions had little effect on the results (Fig. S15). We did not filter on genetic ancestry or ethnicity. Metabolomic biomarker measures were log1p-transformed, and all continuous variables were Z-normalized to have a mean of zero and a standard deviation of one in the training set. The means and standard deviations of the training set were subsequently used to scale the metabolomic biomarkers in the testing and replication cohorts.
We assigned all participants with a repeat measurement to the testing set (to increase sample size for that analysis), and then split the remaining UK Biobank data in half for training and testing, resulting in a total of 241,246 individuals to train the risk scores. We used 10 years of follow-up and Cox proportional hazards regression modeling with least absolute shrinkage and selection operator (Lasso) and tenfold cross-validation using the R package hdnom. We favored the parsimonious Lasso models for interpretability after observing no consistent pattern of better-performing prediction between Lasso and Elastic Net (Fig. S16). For each disease, we trained nine models, which all contained (1) age and sex, and additionally included (2) metabolomic measures, (3) a disease-specific PGS, (4) metabolomic measures, and a disease-specific PGS, (5) a disease-specific clinical score, (6) a disease-specific clinical score and PGS, (7) a disease-specific clinical score and metabolomic measures, (8) a disease-specific clinical score, PGS and metabolomic measures, and (9) an extended set of metabolomic biomarkers. Age and sex were not penalized to ensure they were selected for each model and appropriately weighted. The extended set of metabolomic biomarkers contained all 249 available absolute and ratio measures. For the genetic ancestry analyses, we trained another set of models identical to the description above but leaving all participants with non-European genetic ancestry out from the training set to maximize the size of this group in the testing set.
We computed risk scores (for research use only) as the weighted sum of variables selected during training for each model. The variables that were selected for each model during training and their respective coefficients can be found in Supplementary Data 3. In computing the risk scores, we excluded the sex and age coefficients, which for a combined metabolomic and PGS model for example, effectively results in risk scores comprised of the weighted sum of 1–36 metabolomic measures and a PGS, which have been adjusted for age and sex. We evaluated correlations of the scores by computing Pearson correlation coefficients between different endpoints by model type, between duplicate measurements and between baseline and repeat assessments.
We tested risk model performance in all three biobanks by assessing the association of these age- and sex-adjusted scores with disease incidence within the first 4 years (as the EBB has a large number of samples limited to this amount of follow-up) after the blood draw using Cox proportional hazards models and examining the hazard ratios (HR) with 95% confidence intervals between individuals in the top decile of the score and individuals at the bottom 90%. Fixed effect meta-analysis of estimated hazard ratios across the three biobanks was carried out with the R package meta. We calculated the limits for the top deciles for each model in the training set once and subsequently used these to determine top decile classification in validation and replication cohorts (Supplementary Data 4). We assessed HR for 4 and 10 years of follow-up in the UK Biobank for PGS (also including results from the EBB) and disease-specific clinical risk score comparisons. We additionally stratified HR by the source of report of disease incidence (Fig. S17) and whether the individual had primary care data available (Fig. S18). In addition, we computed HR per one standard deviation increments in the age- and sex-adjusted scores using Cox proportional hazards models. We compared these results to another version in which biomarker scaling and decile limits were not determined by the training set, but calculated within each separate cohort (Fig. S4). For statistical significance, we considered p-values < 0.004 corresponding to a 95% confidence level Bonferroni corrected for 12 diseases39,40. We estimated Kaplan–Meier curves stratified by PGS and metabolomic score deciles for 10 years of follow-up using R package survival41 for individuals in the top decile of the PGS score and the bottom 90% of the metabolomic score, the top decile of both the PGS and metabolomic scores, and the bottom 90% of the PGS score. We used the cox.zph function to examine the proportionality of hazards assumption and Schoenfeld residual plots, which revealed that the HR was not constant over the 10-year follow-up time for some disease endpoints. Therefore, we assessed the continuous hazard over the HR in strata across the follow-up time for the metabolomics and PGS models. Using the R package rstpm242 we built a generalized survival model with natural splines using 2 knots to allow for a time-varying effect, and additionally computed the hazard ratio for 1-year strata using the survSplit option in the survival R package41. We also tested for interaction effects between the metabolomic scores and PGS. Area under receiver operating curves (AUC) were estimated utilizing absolute risks using the R package pROC43. Net reclassification improvements (NRI) were computed comparing metabolomic and PGS scores to disease-specific clinical scores. Both continuous and categorical scores were calculated, with categorical scores using limits of top 10% of the scores as cut-off thresholds. The R package nricens44 was utilized in NRI computation.
For analyses examining two time points, we had 18,709 UKB participants with metabolomic scores calculated from both baseline and a repeat visit which took place after 2 to 7 years. We considered the eight diseases that had at least 100 cases within 10 years after the repeat visit (349 cases for COPD, 214 for colon cancer, 288 for depression, 439 for diabetes, 303 for myocardial infarction and 225 ischemic stroke). We fitted Cox proportional hazards models for disease events 10 years after the repeat visit with age- and sex-adjusted metabolomic scores at baseline and repeat visit. Disease events between baseline and repeat visit were excluded. To assesses the risk changes between the baseline and repeat-visit, we categorized participants into three groups: those who stayed in the highest decile of metabolomic score at both time-points, those were in the highest decile of metabolomic score at baseline but left the high-risk category at the repeat visit, and those in the bottom 90% of the metabolomic score at baseline. We tested for a difference between the two groups (stayers vs leavers) using Cox regression, including the baseline metabolomic score as a covariate to control for the fact that stayers had, on average, a higher baseline score than leavers. Additionally, we analyzed 5038 participants with two separate blood metabolomics measurement on average five years apart from EBB as above in the case of diabetes, since this was the only disease with sufficient events to assess replication.
We assessed clinical characteristics of high-risk individuals, defined as those with a metabolomics model score in the highest decile of at least one of the seven best-performing models: alcoholic liver disease, COPD, cirrhosis of the liver, ischemic stroke, lung cancer, myocardial infarction, and type 2 diabetes. In addition to basic clinical characteristics, we evaluated the frailty index, a measure to quantify aging and health, between high- and low-risk individuals. We calculated the frailty index based on the method described by Williams et al. 20, using 49 self-reported disease outcomes in UK Biobank participants. Participants with at least 10 missing items were excluded. We tested the difference in mean for clinical characteristics between high- and low-risk individuals using a two-sided t-test for continuous variables and a Chi-squared test for categorical variables.
To evaluate calibration of the metabolomic scores, we estimated observed and predicted incidence rates in all three biobanks over 3 years of follow-up. We chose to censor at three years to obtain complete and comparable follow-up for as many samples as possible. We estimated calibration slopes and intercepts by fitting logistic regression of individual diseases status (observed risk) on predicted risk23,24. We performed all statistical analyses and modeling in R version 4.3.245.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
For reasons of patient confidentiality and to ensure research is carried out in accordance with the terms of consent the data were collected under, data from the three biobanks are available under controlled access. The UK Biobank data are available for approved researchers through the UK Biobank data-access protocol (https://www.ukbiobank.ac.uk/enable-your-research/apply-for-access). The data from the first ~280,000 UK Biobank participants included in the Data Showcase https://biobank.ndph.ox.ac.uk/showcase/label.cgi?id=220, and full dataset is available on the Resesarch Analysis Platform https://www.ukbiobank.ac.uk/enable-your-research/research-analysis-platform. The average number of weeks from application submission to data release is 15 weeks for UK Biobank. Data from Estonia Biobank can be accessed through a research application to Institute of Genomics of the University of Tartu (https://genomics.ut.ee/en/content/estonian-biobank). Data from FINRISK and Health 2000 cohorts can be accessed through a research application to THL Biobank (https://thl.fi/en/web/thl-biobank). Source data for Figs. 1–4 are provided with this paper. Source data are provided with this paper.
Code availability
Code to reproduce the figures in this paper is available at: https://github.com/NightingaleHealth/ukb-nightingale-omics-prediction/.
References
Hippisley-Cox, J., Coupland, C. & Brindle, P. Development and validation of QRISK3 risk prediction algorithms to estimate future risk of cardiovascular disease: prospective cohort study. BMJ 357, j2099 (2017).
Lambert, S. A., Abraham, G. & Inouye, M. Towards clinical utility of polygenic risk scores. Hum. Mol. Genet. 28, R133–R142 (2019).
Lewis, C. M. & Vassos, E. Polygenic risk scores: from research tools to clinical instruments. Genome Med. 12, 44 (2020).
Khera, A. V. et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224 (2018).
Mars, N. et al. Polygenic and clinical risk scores and their impact on age at onset and prediction of cardiometabolic diseases and common cancers. Nat. Med. 26, 549–557 (2020).
Inouye, M. et al. Genomic risk prediction of coronary artery disease in 480,000 adults. J. Am. Coll. Cardiol. 72, 1883–1893 (2018).
Soininen, P., Kangas, A. J., Würtz, P., Suna, T. & Ala-Korpela, M. Quantitative serum nuclear magnetic resonance metabolomics in cardiovascular epidemiology and genetics. Circulation: Cardiovasc. Genet. 8, 192–206 (2015).
Julkunen, H. et al. Atlas of plasma NMR biomarkers for health and disease in 118,461 individuals from the UK Biobank. Nat. Commun. 14, 604 (2023).
Würtz, P. et al. Quantitative serum nuclear magnetic resonance metabolomics in large-scale epidemiology: a primer on -omic technologies. Am. J. Epidemiol. 186, 1084–1096 (2017).
Buergel, T. et al. Metabolomic profiles predict individual multidisease outcomes. Nat. Med. 28, 2309–2320 (2022).
Pietzner, M. et al. Plasma metabolites to profile pathways in noncommunicable disease multimorbidity. Nat. Med. 27, 471–479 (2021).
Morze, J. et al. Metabolomics and Type 2 Diabetes Risk: An Updated Systematic Review and Meta-analysis of Prospective Cohort Studies. Diab. Care 45, 1013–1024 (2022).
Deelen, J. et al. A metabolic profile of all-cause mortality risk identified in an observational study of 44,168 individuals. Nat. Commun. 10, 3346 (2019).
Lauber, C. et al. Lipidomic risk scores are independent of polygenic risk scores and can predict incidence of diabetes and cardiovascular disease in a large population cohort. PLoS Biol. 20, e3001561 (2022).
Walford, G. A. et al. Metabolite traits and genetic risk provide complementary information for the prediction of future type 2 diabetes. Diab. Care 37, 2508–2514 (2014).
Unterhuber, M. et al. Proteomics-enabled deep learning machine algorithms can enhance prediction of mortality. J. Am. Coll. Cardiol. 78, 1621–1631 (2021).
Godbole, S. et al. A metabolomic severity score for airflow obstruction and emphysema. Metabolites 12, 368 (2022).
Riveros-Mckay, F. et al. Integrated polygenic tool substantially enhances coronary artery disease prediction. Circ. Genom. Precis. Med. 14, e003304 (2021).
Thompson, D. J. et al. A systematic evaluation of the performance and properties of the UK Biobank Polygenic Risk Score (PRS) Release. PLoS One 19, e0307270 (2024).
Williams, D. M., Jylhävä, J., Pedersen, N. L. & Hägg, S. A frailty index for UK biobank participants. J. Gerontol. Ser. A 74, 582–587 (2019).
Mak, J. K. L. et al. Unraveling the metabolic underpinnings of frailty using multicohort observational and Mendelian randomization analyses. Aging Cell e13868 https://doi.org/10.1111/acel.13868 (2023).
Alba, A. C. et al. Discrimination and calibration of clinical prediction models: users’ guides to the medical literature. JAMA 318, 1377–1384 (2017).
Calster, B. V. et al. A calibration hierarchy for risk models was defined: from utopia to empirical data. J. Clin. Epidemiol. 74, 167–176 (2016).
Collins, G. S., Ogundimu, E. O. & Altman, D. G. Sample size considerations for the external validation of a multivariable prognostic model: a resampling study. Stat. Med. 35, 214–226 (2016).
Kullo, I. J. et al. Polygenic scores in biomedical research. Nat. Rev. Genet. 23, 524–532 (2022).
Ritchie, S. C. et al. Cardiovascular risk prediction using metabolomic biomarkers and polygenic risk scores: A cohort study and modelling analyses. Preprint at https://www.medrxiv.org/content/10.1101/2023.10.31.23297859v1 (2023).
Ko, D. T. et al. Calibration and discrimination of the Framingham risk score and the pooled cohort equations. CMAJ 192, E442–E449 (2020).
Gadd, D. A. et al. Blood protein assessment of leading incident diseases and mortality in the UK Biobank. Nat Aging 4, 939–948 (2024).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Leitsalu, L. et al. Cohort profile: Estonian Biobank of the Estonian Genome Center, University of Tartu. Int. J. Epidemiol. 44, 1137–1147 (2015).
Fischer, K. et al. Biomarker profiling by nuclear magnetic resonance spectroscopy for the prediction of all-cause mortality: an observational study of 17,345 persons. PLoS Med. 11, e1001606 (2014).
Tikkanen, E. et al. Metabolic biomarker discovery for risk of peripheral artery disease compared with coronary artery disease: lipoprotein and metabolite profiling of 31 657 individuals from 5 prospective cohorts. J. Am. Heart Assoc. 10, e021995 (2021).
Borodulin, K. et al. Cohort profile: the National FINRISK Study. Int. J. Epidemiol. 47, 696–696i (2018).
World Health Organization. Global health estimates: Leading causes of DALYs. https://www.who.int/data/gho/data/themes/mortality-and-global-health-estimates/global-health-estimates-leading-causes-of-dalys.
Ritchie, S. C. et al. Quality control and removal of technical variation of NMR metabolic biomarker data in ~120,000 UK Biobank participants. Sci. Data 10, 64 (2023).
Mitt, M. et al. Improved imputation accuracy of rare and low-frequency variants using population-specific high-coverage WGS-based imputation reference panel. Eur. J. Hum. Genet. 25, 869–876 (2017).
Lambert, S. A. et al. The Polygenic Score Catalog: new functionality and tools to enable FAIR research. Preprint at https://www.medrxiv.org/content/10.1101/2024.05.29.24307783v1 (2024).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Neyman, J. & Pearson, E. S. On the use and interpretation of certain test criteria for purposes of statistical inference: part I. Biometrika 20A, 175–240 (1928).
Dunn, O. J. Multiple comparisons among means. J. Am. Stat. Assoc. 56, 52–64 (1961).
Therneau T. A Package for Survival Analysis in R. R package version 3.7-0, https://CRAN.R-project.org/package=survival (2024).
Liu, X.-R., Pawitan, Y. & Clements, M. Parametric and penalized generalized survival models. Stat. Methods Med. Res. 27, 1531–1546 (2018).
Robin, X. et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinforma. 12, 77 (2011).
Inoue, E. nricens: NRI for risk prediction models with time to event and binary response dat. https://doi.org/10.32614/CRAN.package.nricens
R Core Team. R: The R project for statistical computing. R foundation for statistical computing, https://www.R-project.org/ (Vienna, Austria, 2022).
Acknowledgements
We acknowledge the lab and spectrometry teams at Nightingale Health for their role in generating the metabolomic data on these cohorts. We are grateful to UK Biobank (Project 30418), Estonian Biobank, and THL Biobank (project BB2016_86) for access to data to undertake this study. We thank all biobank participants for their generous contribution to generating this resource for the scientific community. We acknowledge the Estonian Biobank Research Team of Mari Nelis, Georgi Hudjasov, Reedik Mägi, Andres Metspalu and Lili Milani. Additionally, we thank Kristi Läll, Erik Abner and Kelli Lehto for their help with Estonian Biobank phenotype data. The work was funded by Nightingale Health Plc. Estonian Biobank was supported by Estonian Research Council grant PRG1291. TT was supported by Estonian Research Council grant PSG809.
Author information
Authors and Affiliations
Consortia
Contributions
Conceptualization: J.C.B., T.E., H.J., P.W. Data Curation: H.J., N.K., S.K., S.N.L., K.S. Formal Analysis: L.J.-D., H.J., N.K., S.K., S.N.L., K.S. Methodology: H.J. Investigation: J.C.B., L.J.-D., H.J., K.H., N.K., S.K., A.K., S.N.L., V.M., K.N., K.S., M.S., P.S., M.T., P.W. Resources: T.E., P.J., T.J., J.K., A.L., M.P., V.S., T.T. Supervision: J.C.B., H.J., P.W. Visualization: L.J.-D., H.J., S.K., S.N.L., K.S. Writing – original draft: J.C.B. Writing – review & editing: J.C.B., L.J.-D., H.J., S.K., S.N.L., N.K., K.S., P.W.
Corresponding author
Ethics declarations
Competing interests
JCB, LJD, HJ, AK, NK, SK, HK, SL, VK, KN, KS, MS, PS, MT, and PW are employees of and hold shares or stock options in, Nightingale Health. The remaining authors declare no conflict of interest.
Peer review
Peer review information
Nature Communications thanks Themistocles Assimes, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nightingale Health Biobank Collaborative Group. Metabolomic and genomic prediction of common diseases in 700,217 participants in three national biobanks. Nat Commun 15, 10092 (2024). https://doi.org/10.1038/s41467-024-54357-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54357-0
This article is cited by
-
AI-based multiomics profiling reveals complementary omics contributions to personalized prediction of cardiovascular disease
Nature Communications (2026)
-
Metabolic characteristics of benign and malignant pulmonary nodules and establishment of invasive lung adenocarcinoma model by high-resolution mass spectrometry
BMC Cancer (2025)
-
Interpretable machine learning leverages proteomics to improve cardiovascular disease risk prediction and biomarker identification
Communications Medicine (2025)
-
Mapping the plasma metabolome to human health and disease in 274,241 adults
Nature Metabolism (2025)
-
Pathway level metabolomics analysis identifies carbon metabolism as a key factor of incident hypertension in the Estonian Biobank
Scientific Reports (2025)
