
Abstract
Here we conduct a study involving 12 individuals with retinal dystrophy, neurological impairment, and skeletal abnormalities, with special focus on GPATCH11, a lesser-known G-patch domain-containing protein, regulator of RNA metabolism. To elucidate its role, we study fibroblasts from unaffected individuals and patients carrying the recurring c.328+1 G > T mutation, which specifically removes the main part of the G-patch domain while preserving the other domains. Additionally, we generate a mouse model replicating the patients’ phenotypic defects, including retinal dystrophy and behavioral abnormalities. Our results reveal a subcellular localization of GPATCH11 characterized by a diffuse presence in the nucleoplasm, as well as centrosomal localization, suggesting potential functions in RNA and cilia metabolism. Transcriptomic analysis performed on mouse retina detect dysregulation in both gene expression and splicing activity, impacting key processes such as photoreceptor light responses, RNA regulation, and primary cilia-associated metabolism. Proteomic analysis of mouse retina confirms the roles GPATCH11 plays in RNA processing, splicing, and transcription regulation, while also suggesting additional functions in synaptic plasticity and nuclear stress response. Our research provides insights into the diverse roles of GPATCH11 and identifies that the mutations affecting this protein are responsible for a recently characterized described syndrome.
Similar content being viewed by others


Introduction
The spliceosome is a large ribonucleoprotein (RNP) complex that allows the processing of pre-mRNA from DNA transcription into functional and mature mRNA. Comprising five core small nuclear ribonucleoprotein particles (snRNPs) and numerous splicing factors, the spliceosome requires precise coordination for proper assembly, activation, and regulation to ensure accurate RNA splicing and cellular function1. Spliceosomopathies refer to rare diseases that result from dysfunction of the spliceosome. Despite the ubiquity of the spliceosome, spliceosomopathies tend to affect specific tissues, notably impacting the survival of retinal rod photoreceptors, craniofacial development, or haematopoiesis1.
G-patch domain-containing (GPATCH) proteins form a distinct group characterized by a glycine-rich motif and multiple RNA-binding motifs2,3. Even though they share some common features, GPATCH proteins exhibit considerable variations in size, domain composition, and cellular localization2. Through their protein-protein and protein-nucleic acid interactions, they contribute significantly to RNA metabolism by regulating the remodeling of RNAs and ribonucleoprotein complexes by the DEAH-box RNA helicases with which they associate2,4,5,6. In humans, 23 GPATCH proteins have been identified2. Of these, a dozen have been reported to be involved in pre-mRNA splicing and/or transcription regulation, while others participate in ribosome biogenesis, RNA export, rRNA, and snoRNA maturation2,5,6.
In line with the multifaceted functions of GPATCH proteins, several phenotypic traits have been linked to variations in GPATCH-coding genes, but only variants in RBM107 and SON8 have been associated with very severe multisystem developmental syndromes that may share similarities with spliceosomopathies but are notably more complex in their manifestations7,8.
GPATCH11, also known as coiled-coil domain-containing 75 (CCDC75) and centromere protein Y (CENP-Y), belongs to the lesser explored group of GPATCH proteins. While GPATCH11 is localized in the nucleus, its specific role in RNA metabolism has not yet been demonstrated.
By the analysis of whole exome and whole genome sequencing (WES and WGS) data obtained from affected individuals from six unrelated families, we found compelling evidence showing that biallelic GPATCH11 variants are responsible for a syndrome characterized by early-onset-severe retinal degeneration, neurological symptoms, and abnormal craniofacial features.
To elucidate the function of GPATCH11, we used fibroblasts from patients carrying c.328+1G>T, a recurrent mutation shared by affected individuals from four out of the six described families. This variant specifically removes a part of the G-patch domain while preserving other domains of this protein. Additionally, we generated a mouse model replicating the patients’ phenotypic defects, which presents behavioral abnormalities and retinal dystrophy. Our results revealed a subcellular localization characterized by a diffuse presence in the nucleoplasm and centrosomal localization, indicating potential roles in RNA and cilia metabolism. Transcriptomic analysis of the mouse retina identified dysregulation in both gene expression and splicing activity, affecting crucial processes such as photoreceptor light responses, RNA regulation, and primary cilia-associated metabolism. Proteomic analysis of mouse retina confirmed its involvement in RNA processing, splicing, and transcription regulation, while also suggesting additional roles in synaptic plasticity and nuclear stress responses. These findings highlight the essential functions of GPATCH11 in nuclear and centrosomal activities related to synaptic plasticity, function, and maintenance, particularly under stress conditions. They underscore its importance in regulating gene expression and supporting retinal, neurological, and skeletal functions.
Results
Biallelic GPATCH11 mutations cause retinal dystrophy, neurodevelopmental delay and skeletal anomalies
The families were initially referred to a genetic consultation for distinctive presenting symptoms, namely, severe visual dysfunction from birth (Families 1 and 6), intellectual disability (Family 2), intellectual disability with encephalopathy (Family 3), intellectual disability with retinitis pigmentosa (RP, Family 4) and a complex medical history including failure to thrive in infancy, recurrent febrile illness and abnormal skull shape (Family 5). A review of clinical files, re-examination and/or disease progression revealed common symptoms of RP, variable intellectual disability, and dysmorphic features (Supplementary Data 1).
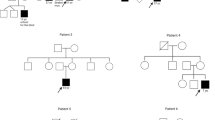
Family 1’s index patient (P2; Fig. 1a) and her elder sister (P1) have experienced severe visual deficiencies since infancy, characterized by nystagmus and flat electroretinograms (ERG). Childhood eye exams revealed poor vision, pigmentary deposits, macular atrophy, and retinal thinning (Supplementary Fig. 1). P1 had high bilateral hypermetropia, while P2 had low unilateral hypermetropia. They also exhibited neurodevelopmental anomalies, including language delays, agitation, hyperactivity, and frustration, despite having normal cerebral magnetic resonance imaging (MRI). Recently, P1, now aged 13, developed seizures. Both presented with facial dysmorphic features such as enophthalmos, a short philtrum, large incisors, and diastema (Fig. 1b). Their paternal uncles (P3 and P4), born to a first-cousin marriage, also showed severe visual impairment and neurodevelopmental delays, including poor language skills, mild ataxia, and general movement disorganization. The older uncle passed away at 16 during a fever episode (Supplementary Data 1).

a Pedigrees of six analyzed families, including segregation analysis of GPATCH11 variants: M1: c.328+1G>T (p.Gly97_Thr110del); M2: c.454C>T (p.Arg152*); M3: c.449+1G>C (p.Arg150ArgFs*1); M4: c.393C>G (p.Tyr131*); +: wild-type allele; P: patient. P1 and P2 are affected individuals whose fibroblasts were analyzed. b Clinical images of affected individuals P1, P2, P5, P8, P9, P11, P12. c Color fundus photographs and fundus autofluorescence images of P10, RE: right eye; LE: left eye. d GPATCH11 diagram indicating the positions of c.328+1G>T, c.454C>T, c.449+1G>C, and c.393C>G variants relative to NM_174931.4 transcript. e Representative chromatograms of gDNA sequences from affected individuals show variants: c.328+1G>T (M1) homozygously (as in P1-P4, P8, P9, P12), c.454C>T (M2) in exon 6 heterozygously (as detected in compound heterozygosity with M1 in P5-P7), c.449+1G>C (M3) homozygously (P10), and c.393C>G in exon 5 in homozygosity (P11). f Chromatograms of cDNA sequences reveal a shorter transcript (r.287-328del) in P1 and P2 due to M1, absence of transcripts (r.0) in P5 (M2), and two distinct transcripts (r.449+128 and r.449+48) in P10 caused by M3. Electrophoresis of PCR products from cDNA from P1 and control individual (C1) shows exclusion of exon 4 (42 base pairs, bp), leading to creation of a shorter aberrant transcript. Electrophoresis of RT-PCR products from leukocytes of control (C3) and P10, and schematic representation showing two aberrant transcripts present for P10 and absent in C3, corresponding to the retention of full and partial intron 5. Blots are representative of three independent experiments. g Western blot analysis of protein extracts using anti-GPATCH11 antibody targeting residues 111-192 showing detection of shorter protein isoform (GPATCH11-Δex4 (36 KDa)) in patients’ fibroblasts (P1, P2) as compared to the controls (GPATCH11-WT (37 KDa)) in C1 and C2. The abundance of GPATCH11 products in fibroblasts from affected individuals is comparable to control samples. The statistical significance was estimated using one-way ANOVA with a post-hoc Tukey test. Bars represent means ± SEM from three experimental replicates (five technical replicates). n.s. not significant. Source data are provided as a Source Data file.
The eldest affected sibling of Family 2 (P5; Fig. 1a) was initially seen for intellectual disability, speech delays, psychomotor retardation, and dyspraxia, with normal electroencephalogram (EEG) and cerebral MRI. At the age of 4 years, she developed visual symptoms, with examination revealing reduced visual acuity (20/50 in each eye), peripheral pigmentary deposits, mild foveolar hypoplasia, low hypermetropia, and flat ERG. She was seen by a dysmorphologist who noted enophthalmos, a short philtrum, large incisors, and diastema (Fig. 1b). Her younger brother (P6) had better visual acuity (20/25 in each eye) with a fundus with peripheral pigmentary deposits and milder neurological symptoms with normal language acquisition. The youngest sibling (P7) had no overt visual problems but exhibited psychomotor retardation and will be monitored for evaluation over time (Supplementary Data 1).
The older of the two affected sisters in Family 3 (P8; Fig. 1a) displayed short stature, psychomotor delay, encephalopathy, seizures, lack of muscular coordination in early childhood and developed diabetes at age 20. Her visual status is unknown. She passed away at 30 due to a thalamic stroke. Her younger affected sister (P9) exhibited similar clinical features, along with pigmentary deposits at the fundus, reduced caliber of retinal vessels, and optic atrophy consistent with advanced RP. She presented with facial dysmorphic features similar to those observed in affected individuals from Families 1 (P1, P2) and 2 (P5), including enophthalmos, a short philtrum, large incisors, and diastema (Fig. 1b, Supplementary Data 1).
The affected individual in Family 4 (P10, Fig. 1a–c) has had night blindness since childhood and was diagnosed with RP at age 20 (Supplementary Fig. 1). Cognitive deficits and developmental delays were observed at age 3, along with dysmorphic features including macrocephaly, large external ears, small pre-auricular tubercle on the right side, small and deep-set eyes, long prominent nose with long columella, hypoplastic nasal alae, and short 5th toes (Supplementary Data 1).
The proband in Family 5 (P11, Fig.1a) was referred to a geneticist at 1.5 years of age, due to failure to thrive, which later improved, recurrent febrile illnesses suggestive of immunodeficiency, and an abnormal skull shape caused by bony protuberances. Cerebral MRI revealed fluid-filled spaces around the optic nerves, indicative of dural ectasia, but treatment with acetazolamide did not yield significant changes and was subsequently discontinued. At 4.5 years, an ophthalmic examination revealed retinal pigmentary deposits and poor ERG responses under photopic and scotopic conditions suggesting early-onset severe retinal dystrophy (Supplementary Fig. 1). At 7.5 years, he had speech and motor delays, learning difficulties, and showed abnormalities on EEG, requiring treatment with sulthiame. Physical examination noted relative macrocephaly (head circumference above the 97th percentile, height at the 10th percentile, weight at the 25th percentile), frontal bossing treated by osteotomy, down-slanting palpebral fissures, bulbous nose, receding midface, and café au lait macules (Fig. 1b).
The affected child in Family 6 (P12; Fig. 1a) exhibited severe visual impairment with nystagmus at 5 months. Ophthalmic evaluation showed poor vision (hand movements), abnormal macula, and dull retina, hemeralopia leading to a diagnosis of Leber congenital amaurosis. He had feeding difficulties, delayed language acquisition, psychomotor delay, concentration and comprehension issues, and severe sleep disorder. Neuropsychological assessment revealed severe intellectual disability and autism, with a normal cerebral MRI. He also had short stature (head circumference -1 SD, height -2.3 SD, weight -2.7 SD) and dysmorphic features, including enophthalmos, hypertelorism, prominent nasal bridge, anteverted nares, flattened cupid’s bow, thick lower lip, small ear lobes, and a single transverse palmar crease (Fig. 1b, Supplementary Data 1).
Next-generation sequencing (NGS) performed in these six independent families enabled the identification of four different variants in GPATCH11. Analysis of WES or WGS datasets revealed the GPATCH11 canonical splice-site variant NM_174931.4: c.328+1G>T (M1) in homozygosity in affected individuals from consanguineous Families 1, 3, and 6, and in compound heterozygosity with the nonsense variant c.454C>T, (p.Arg152*) (M2) in those from nonconsanguineous Family 2 (Fig. 1a, d, e). Haplotype reconstruction using exome datasets from three families (Families 1, 2, and 3), all of whom originate from the Maghreb (Tunisia, Algeria/Morocco, and Algeria, respectively), identified a short (1.27 Mb) common haplotype encompassing the splice-site variant c.328+1G>T (Supplementary Table 1), supporting an ancient founder effect. In the index patients from consanguineous Families 4 and 5, from Portugal and Bedouins of Israel respectively, WES data revealed homozygosity for the splice-site variant c.449+1G>C (M3) and the nonsense variant c.393C>G (p.Tyr131*) (M4) in GPATCH11 (Fig. 1a, d, e). Consistent with autosomal recessive disease transmission, the healthy parents of affected individuals in the six families carried respective variants in heterozygosity (Fig. 1a).
The variants c.328+1G>T and c.454C>T are present 18 and 8 times, respectively, in the heterozygous state in gnomAD v4.09, while the variants c.449+1G>C and c.393C>G are absent from this database. In silico predictions reveal that the c.328+1G>T and c.449+1G>C substitutions disrupt the consensus donor splice sites of exons 4 and 5, respectively, with donor loss scores of 0.95 and 1, according to Splice AI10. Reverse transcription polymerase chain reaction (RT-PCR) analysis of cDNA obtained from fibroblasts from patients P1 and P2, homozygous for M1, revealed that the c.328+1G>T alteration induces in-frame skipping of exon 4 (r.287_328del; p.Gly97_Thr110del, Fig. 1f). Similar analysis performed on cDNA from lymphocytes from patient P10, homozygous for M3, revealed that the c.449+1G>C substitution results in both partial and complete inclusion of intron 5 of GPATCH11 (r.449+128_449+48+ r.449+11g>c, Fig. 1f).
The analysis of blood RNA from the affected subject of Family 2 (P5) revealed an apparent homozygosity for the variant c.328+1G>T, indicating nonsense-mediated mRNA decay (NMD) of the mRNA transcribed from the trans allele carrying the c.454C>T (r.0, p.Arg152*) nonsense variant (Fig. 1f). Collectively these data suggest that the c.328+1G>T variant in GPATCH11 results in a coding mRNA that is 42 base pairs shorter than the normal 792 base pairs sequence, whereas the c.449+1G>C and c.454C>T variants are likely to trigger NMD, resulting in the absence of GPATCH11 protein. No sample was available for RNA analysis of patient P11, who carries the nonsense c.393C>G (p.Tyr131*) variant in homozygosity.
Cells from individuals carrying the c.328+1G>T variant produce a mutant GPATCH11 protein lacking the internal portion of the G-patch domain
The skipping of GPATCH11 exon 4 in the mRNA, resulting from the c.328+1G>T variant is expected to abolish 14 amino acids (aa 96–109) from the protein. These residues bridge the helix and loop braces of the G-patch domain encoded by adjacent exons2. Western blot analysis, utilizing an antibody specific to a sequence downstream of the G-patch domain, revealed a distinctive band of reduced molecular weight in fibroblasts from patients P1 and P2, who harbor the c.328+1G>T variant in homozygosity, compared to controls. The observed shift in size was consistent with the loss of 14 amino acids. The abundance of the mutant protein in patient cells did not significantly differ from the wildtype counterpart in controls (Fig. 1g).
GPATCH11 displays nuclear and centrosomal, but not centromeric, distribution
GPATCH11, primarily known as a nuclear protein, may have additional localizations at the centrosome and kinetochore, suggested by its alternative names, CCDC75 and CENP-Y11.
To explore GPATCH11 nuclear localization, we conducted confocal microscopy analysis on immunostained control fibroblasts fixed with methanol and paraformaldehyde (PFA). Our results revealed a diffuse presence in the nucleoplasm alongside centrosomal localization (Fig. 2a). Notably, centrosomal (basal body) staining was reduced in PFA-fixed cells compared to those fixed with methanol (Supplementary Fig. 2a).

a Nuclear and centrosomal localization. Immunostaining of GPATCH11 (magenta), SC-35, and H3K9me3 (yellow). Magenta arrows show GPATCH11 at the centrosome. Scale bars, 10 µm. b Centrosomal localization of GPATCH11. Immunostaining of GPATCH11 (magenta), Centrin3 (centrioles), Rootletin (linker fibers), and acetylated α-Tubulin (cilia) in cyan. Serum free culture was used to promote ciliation. Cyan arrows show centrioles, linker fibers, or cilia, respectively. Scale bars, 2 µm. c Immunostaining of GPATCH11 (magenta), ACA (yellow), CENP-E (cyan) at different phases of the mitosis. Magenta arrows show GPATCH11 at the centrosome. Scale bar, 10 µm. DAPI is used to label the nucleus (blue). All the pictures are representative of three independent experiments.
For a comprehensive characterization of the protein subcellular distributions, fibroblasts underwent immunostaining for GPATCH11 along with markers for specific cellular compartments, including SC-35 (speckles) and H3K9me3 (heterochromatin) for nucleoplasmic analysis, and acetylated-α-Tubulin (cilium axoneme), CETN3 (centrosome), and Rootletin (centrosome linker) for centrosomal examination, as well as ACA (centromere) and CENP-E (outer kinetochore) for kinetochore analysis. Confocal analysis for nucleoplasmic localization unveiled GPATCH11 proximity to speckles and heterochromatin (Fig. 2a), while Stimulated-emission-depletion (STED) microscopy revealed its localization in the centrosome linker region, associated with Rootletin (Fig. 2b). Further investigation for centrosomal and centromeric localization using confocal microscopy of immunostained cells at various mitotic stages pinpointed GPATCH11 exclusively to the centrosome, with no localization observed at the kinetochore or centromere11 (Fig. 2c).
GPATCH11 subcellular distribution and centrosome-mediated ciliogenesis remain unaffected by the alteration of G-Patch and CCDC domains
In a parallel study, we investigated the potential impact of the loss of the central part of the G-patch domain on the protein localizations in fibroblasts from siblings P1 and P2, who carry the c.328+1G>T mutation in homozygosity in the same conditions as in control cells. Analysis of protein subcellular distribution and abundance using machine learning with Ilastik12 showed no discernible difference in nucleoplasmic localization compared to control fibroblasts (Supplementary Fig. 2a, b). Furthermore, given that CCDC domains are commonly found in centrosomal proteins13,14, we investigated whether the CCDC domain of GPATCH11 influences its subcellular distribution in hTERT-RPE1 cell lines with a homozygous in-frame deletion in exon 3 coding sequence, which we generated using CRISPR‒Cas9. Confirmation was obtained through RT‒PCR and Western blot analysis, revealing shortened mRNA and protein, respectively (Supplementary Fig. 3a, b). Subcellular localization analysis in these cells showed that the absence of the CCDC domain did not alter the centrosomal or nucleoplasmic distribution compared to the wild-type in control hTERT-RPE1 cells (Supplementary Fig. 3c).
Collectively, these findings suggest that GPATCH11 exhibits a nucleo-centrosomal localization pattern, mirroring the distribution observed for centrosome-associated spliceosome components15,16. This alignment strongly indicates that GPATCH11 likely functions as one of these components. Additionally, our results indicate that neither the G-patch domain nor the CCDC domain influence its subcellular localization.
Centrosome-associated spliceosome components have been reported as regulators of ciliogenesis16. Therefore, we aimed to investigate whether the G-patch or CCDC domains of GPATCH11 play a role in cilia formation. To achieve this, fibroblasts from homozygous P1 and P2 individuals, as well as hTERT-RPE1 cells expressing the GPATCH11 protein lacking the CCDC domain, were cultured in a serum-free medium to promote cilia formation and the primary cilium basal body and axoneme were immunostained using Pericentrin and acetylated α-Tubulin antibodies, respectively. The analysis revealed that the abundance of ciliated cells and the mean axonemal length were not significantly different from those detected in controls (Supplementary Fig. 3d, e; Supplementary Fig. 4a, b). This suggests that neither the deletion of the CCDC domain nor the alterations in G-patch domain has an impact on ciliation.
The Sonic Hedgehog (SHH) pathway is a crucial developmental signaling pathway in vertebrates, closely linked to primary cilia17. This pathway includes the ligand SHH, the receptor patched (PTCH), and the activator smoothened (SMO) receptors. When SHH binds to PTCH1, it releases and activates SMO, which then activates GLI transcription factors and PTCH1 itself, which undergoes degradation upon binding with SHH. We investigated the impact of the G-patch domain alteration on primary cilium-dependent SHH signaling in serum-starved fibroblasts from P1 and P2 patients by exposing the cells to smoothened agonist (SAG) and measuring PTCH1, GLI1, and GLI2 expression with RT-qPCR. The expression of PTCH1, GLI1, and GLI2 in P1 and P2 cells was comparable to control counterparts, indicating normal primary cilium-dependent SHH signaling despite G-patch domain alteration (Supplementary Fig. 4c). hTERT-RPE1 cells expressing the GPATCH11 protein lacking the CCDC domain were not considered in this study.
Together, these studies suggest that neither the structure nor the function of the primary cilium is impacted by alterations in the G-patch or CCDC domains.
snRNAs expression is unchanged by the disruption of the G-patch domain
Given that the G-patch domain is suggested to play a specific role in RNA processing and that the nucleo-centrosomal distribution pattern of GPATCH11 implies it might be a centrosome-associated spliceosome component, we decided to analyze whether the loss of the central part of the G-patch domain affects small nuclear RNAs (snRNAs). To achieve this, snRNAs from fibroblasts P1 and P2, as well as from control fibroblasts, were recovered by exclusion chromatography and isolated on acrylamide gel chromatography prior to processing by RNA sequencing, revealing a comparable abundance of U1, U2, U4, U5, and U6 snRNAs. (Supplementary Fig. 5a). Semiquantitative RT-qPCR analysis of snRNA abundance from total RNA extracts was used as an alternative method and confirmed these results (Supplementary Fig. 5b).
Mice expressing the GPATCH11 protein analogous to the human mutant variant display early-onset and severe retinal degeneration and memory impairment
Mice carrying a homozygous genomic deletion of exon 5 (Gpatch11Δ5/Δ5) were viable and developed normally, exhibiting normal appearance, weight, and daily behavior. However, males were completely infertile and had smaller, empty testes (Supplementary Fig. 6), while females retained their normal fertility. The absence of exon 5 in the mRNA and the presence of a shortened protein product in the retina of Gpatch11Δ5/Δ5 mice were verified by RT-PCR, Sanger sequencing, and Western blot analysis, respectively (Fig. 3a–c). The abundance of the mutant protein in Gpatch11Δ5/Δ5 mouse retina did not differ significantly from the wild-type protein in control littermates according to Western blot analyses (Fig. 3d).

a Diagram depicting the human GPATCH11 gene and the murine Gpatch11 gene. The schematic illustrates a CRISPR/Cas9-mediated exon 5 deletion from the mouse Gpatch11 locus (human exon 4 corresponds to murine exon 5). b Representative chromatogram displaying wild-type (WT) and homozygous mutant (Gpatch11Δ5/Δ5) mice cDNA sequences amplified from retina samples using forward and reverse primers in exons 3, and 8, respectively. c Detection and (d) quantification of GPATCH11 wild-type and homozygous mutant isoforms relative to γ-Tubulin by Western blot analysis of retina protein extracts. The significance of variations among samples was estimated using the Two-tailed Student’s t-test. Bars indicate means ± SEM calculated from ≤6 mice per genotype. n.s. not significant. Source data are provided as a Source Data file.
Considering the retinal defects in human patients, we conducted a longitudinal study of retinal function and anatomy in Gpatch11Δ5/Δ5 mice and control wild-type littermates using ERG and histology analysis. Gpatch11Δ5/Δ5 pups displayed normally developed and layered retinas at the age of 15 days (1 day from the eye-opening), with normal rod-specific but moderately reduced cone-specific ERG responses to light stimulations (Fig. 4a, b; Supplementary Fig. 7a, b). As the mice aged, both rod and cone ERG responses declined progressively, accompanied by a reduction in the thickness of the photoreceptor nuclear layer and a decrease in the intensity of immunostaining signals from rod and cone opsins, indicating gradual loss of both photoreceptor types (Supplementary Fig. 7b, c). By the age of 3 months, approximately half of the photoreceptor nuclei were lost. The remaining rod and cone photoreceptors exhibited highly shortened and disorganized outer segments with severely depleted opsin content, resulting in highly altered ERG responses (Supplementary Fig. 7c). At the age of 6 months, ERG responses were flat, and photoreceptor nuclei were almost completely lost (Fig. 4a, b; Supplementary Fig. 7a, b).

a Cone- and rod-specific ERG responses to light in wild-type (WT, black) and Gpatch11Δ5/Δ5 (red) mice at post-natal days 15, 21, 30, 60, 90, 120, 150, and 180. Significance of the difference in a-wave and B-wave amplitudes between age-matched mutant Gpatch11Δ5/Δ5 and wild-type mice was determined through a post hoc Sidak test following a two-way ANOVA. Bars show means ± SEM from nine mice per genotype. n.s. not significant. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. b Spider graph presenting outer nuclear layer (ONL) thickness in wild-type (WT, black) and Gpatch11Δ5/Δ5 (red) mice at 15, 60, 120, and 180 days. The ONL thickness of age-matched mutant Gpatch11Δ5/Δ5 and wild-type mice were compared using a two-way ANOVA with a post hoc Sidak test. Bars represent means ± SEM from three mice per genotype. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. c Immunostaining of retina sections with anti-GPATCH11 (magenta) antibody. Nuclei are stained with DAPI (blue). Scale bars, 50 µm. d Quantification of GPATCH11 protein expression in the ONL, inner nuclear layer (INL), and ganglion cell layer (GCL) in wild-type (WT, black) and mutant Gpatch11Δ5/Δ5 (red) mice. The mean intensity of GPATCH11 of age-matched mutant Gpatch11Δ5/Δ5 and wild-type mice was compared using a two-way ANOVA with a post-hoc Sidak test. Bars represent means ± SEM from three biological replicates. n.s. not significant. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. Source data are provided as a Source Data file.
The presence of neurological impairment in the six families involved in this study compelled us to investigate the presence of neurological deficits in 1-month-old Gpatch11Δ5/Δ5 mice (opsin content in photoreceptors and ERG responses consistent with vision in the mouse model) and their wild-type littermates. The mice were subjected to novel object recognition (NOR), contextual fear conditioning (CFC), Morris water maze (MWM) tests, and the open field test (OF) to assess their episodic memory, associative memory, spatial memory, and anxiety-like behavior, respectively. Both, male and female Gpatch11Δ5/Δ5 mice demonstrated an inability to recognize novel objects in NOR experiments (Fig. 5a), as well as significantly shorter context-elicited freezing times during the training and testing phases in CFC experiments (Fig. 5b). These findings collectively support defective episodic and associative memory. In contrast, their anxiety-like behavior and their spatial memory did not significantly differ from those of controls (Supplementary Fig. 8a, b).

a Episodic memory assessment based on the Novel Object Recognition (NOR). Preference and Discrimination Indexes measurements. Behavioral data were estimated to be statistically significant when p ≤ 0.05 by One-tailed Student’s t-test. b Associative memory assessment using the Contextual Fear Conditioning (CFC). Percentage of freezing time index measured during training and testing phases, respectively. Behavioral data were estimated to be statistically significant when p ≤ 0.05 by Two-tailed Student’s t-test. Bars represent means ± SEM from 20 mice per genotype. ****p ≤ 0.0001. Boxplots show 25–75 percentiles and median; whiskers represent min and max values. c Immunostaining of floating brain sections of wildtype (WT) and Gpatch11Δ5/Δ5 with anti-GPATCH11 (magenta) and anti-CTIP2 (cyan) antibodies with a zoom on hippocampus. DAPI is used to label the cell nucleus (blue). Scale bars, 1 mm and 200 µm, respectively. Source data are provided as a Source Data file.
Given the memory deficit associated with Gpatch11 alteration, we investigated the expression of GPATCH11 in the brain, with a particular focus on the hippocampus, which is crucial for memory functions18. We conducted immunohistochemistry on floating brain sections from 1-month-old Gpatch11Δ5/Δ5 and wild-type mice using antibodies specific to GPATCH11 and CTIP2, a marker known for its distinct hippocampal staining19. We observed strong nuclear GPATCH11 staining in the hippocampus and throughout the rest of the brain in both Gpatch11Δ5/Δ5 and wild-type mice. The prominent expression in the hippocampus, along with observed memory deficits in Gpatch11Δ5/Δ5 mice, suggests a role for GPATCH11 in hippocampal functions (Fig. 5c).
Thus, exploring the retina and behavior in the Gpatch11Δ5/Δ5 mouse model revealed the presence of key symptoms resembling those described in patients.
Gpatch11 mRNA and protein are detected in all retinal nuclear layers, with the accumulation of mutant protein
The retinal expression of Gpatch11 mRNA was examined by RNAscope in eye sections from mutant and wild-type littermates using a probe that targets the wild-type and mutant mRNA isoforms described for Gpatch11. Wild-type mRNA in wild-type mice and the mutant counterpart in Gpatch11Δ5/Δ5 littermates were detected and quantified with no significant difference in all retinal layers (Supplementary Fig. 9a, b).
Immunostaining of the wild-type and mutant protein isoforms in retinal sections using the GPATCH11 antibody showed the presence of the protein in all retinal nuclei (Fig. 4c). Of note, in photoreceptor nuclei the protein displayed a perinuclear distribution, contrasting with the nuclear localization observed in other retinal cells. This inverted pattern in photoreceptor nuclei has been previously ascribed to a peculiar nuclear architecture in nocturnal mammalian photoreceptors20.
Interestingly, quantification of wild-type and mutant mRNA and of protein isoforms over a 6-month period revealed similar mRNA abundance but higher degree of GPATCH11 immunostaining in Gpatch11Δ5/Δ5 mice compared to their wild-type littermates, which suggests an accumulation of the mutant protein (Supplementary Fig. 9a, b; Fig. 4c, d). Because protein degradation is often modulated by ubiquitination or other post-translational modifications, we investigated the latter through the proteomic Nextprot database (NX_Q8N954-1). The database did not identify any modification of the 96–109 amino-acid deleted sequence, whose loss could account for altered protein degradation.
Overall, our data show that the human mutant variant in mice produces a shorter GPATCH11 protein which occurs in accumulated form in all the retinal layers.
Transcriptomic analysis of mutant and wild-type mice retina reveals the role of GPATCH11 in expression and splicing of genes related to visual perception
In order to determine whether the loss of the internal portion of the G-patch domain of GPATCH11 affects gene expression and/or splicing, we compared the transcriptomes of retinas from 15-day-old Gpatch11Δ5/Δ5 mice with that of their wild-type littermates. This resulted in the identification of 160 differentially expressed genes (DEGs) (Fig. 6a). A Gene Ontology (GO) enrichment analysis indicated the enrichment of genes related to photoreceptors, the non-motile cilium, and the response to light stimuli (phototransduction cascade and visual signal propagation) in the Gpatch11Δ5/Δ5 mice compared to their wild-type littermates (Fig. 6b, c, Table 1). Circular visualization based on the calculated z scores indicated that most of the pathways, for which heatmaps are shown, were enriched in downregulated DEGs in particular (Fig. 6b, c). Neither inflammation nor cell death pathways were dysregulated upon the loss of the internal portion of the G-patch domain, supporting the indication that the transcriptome changes at day 15 were primarily due to alterations in Gpatch11.

a Volcano plot displaying differentially expressed genes between Gpatch11Δ5/Δ5 and wild-type (WT) mice. The x-axis represents the difference in gene expression fold change (FC) on a log2 scale, while the y-axis indicates the false discovery rate (FDR) adjusted significance on a log10 scale. Genes significantly upregulated are shown in orange, while downregulated genes are in blue. b Circular visualization depicting selected Gene Ontology (GO) enriched pathways. Up- (red dots) and downregulated genes (blue dots) within each GO pathway are plotted based on logFC. Z score bars indicate whether an entire biological process is more likely to be increased or decreased based on its constituent genes. c Heatmaps from Metascape displaying the GO pathways of interest. Blue represents low expression, while orange represents high expression. The raw data is accessible via BioStudies identifier S-BSST1157.
The differentially expressed isoform analysis identified 299 splicing events—mainly reduced skipped exons (SEs) and increased mutually exclusive exons—in 178 genes (Fig. 7a). Differentially spliced genes (DSGs) included genes related to the photoreceptor response to light (phototransduction and synaptic transmission of the visual message), the photoreceptor connecting the cilium assembly, protein homeostasis, mitochondria, nucleic acid (chromatin, DNA and RNA) binding, and the regulation of RNA splicing (Table 2). Most of these genes showed no differences in expression in the DEG analysis, suggesting that splicing events were not responsible for the changes in expression (Fig. 7b). Specifically, the intersection of DEGs and DSGs identified only 12 genes: Arr3, Asap3, Cabp4, Ccnl2, Dhrs3, Lbhd1, Mpp4, Pex5l, Pitpnm3, Tulp1, Unc13b, and Vtn, all of which, except Asap3, were downregulated upon loss of the internal portion of the G-patch domain (Fig. 7b, c). The splicing events in Asap3, Cabp4, Lbhd1, Pitpnm3, Tulp1, and Vtn are expected to introduce premature termination codons in the mRNA and trigger NMD. In contrast, the analysis of the splicing events in Arr3, which is reported as an example, suggests that mis-splicing is not the main driver of expression dysregulation in this gene (Fig. 7d–f), as well as in Ccnl2, Dhrs3, Mpp4, Pex5l, and Unc13b (Supplementary Table 3).

a Chart generated from rMATS analysis, illustrating that the majority of splicing events correspond to skipped exons. A5SS and A3SS represent alternative 5′ and 3′ splice sites, MXE indicates mutually exclusive exons, RI stands for retained introns, and SE denotes skipped exons. b Venn diagrams comparing differentially expressed genes (DEGs) and differentially spliced genes (DSGs) in Gpatch11Δ5/Δ5 as compared to wild-type (WT) retina samples, with a total of 12 dysregulated and mis-spliced transcripts. c Heatmaps depicting the expression levels of the 12 dysregulated and mis-spliced transcripts. Blue indicates low expression, while orange represents high expression. d Sashimi plots illustrating the alternate splicing event for Arr3 in retina samples from Gpatch11Δ5/Δ5 (red) and wild-type (WT, blue) mice. Orange highlights the alternative splicing events with numbers indicating the junction read count for each event. The raw data can be accessed via BioStudies and the identifier S-BSST1157. e Electrophoresis of Arr3 cDNA and (f) Western blot analysis and ARR3 protein relative to β-Catenin in wild-type (WT) and Gpatch11Δ5/Δ5 retina samples. Blots are representative of three independent experiments. Source data are provided as a Source Data file.
Proteomic study of total lysate from mutant and wild-type mice retina indicates the role of GPATCH11 in gene regulation, cellular dynamics, synapse function and visual perception
A mass spectrometry (MS) analysis of total-retina lysates from 21-day-old Gpatch11Δ5/Δ5 mice and wild-type littermates detected 150 proteins in the Gpatch11Δ5/Δ5 mice with either decreased (n = 63/150) or increased (n = 87/150) abundance. The downregulated proteins are involved in several of the pathways identified by transcriptome analysis, namely, vision perception, synaptic function, mitochondria, cytoskeleton, DNA, and RNA binding and splicing (Supplementary Fig. 10a). Nine of these proteins are encoded by downregulated DEGs, namely, Arr3, Cabp4, Eml3, Lhbd1, Nxnl2, Mpp4, Pex5l, Smad11, and Tulp1 (Supplementary Fig. 10b, c). Among them, mutations in TULP1 and CABP4 have been previously associated with human retinal diseases: Leber congenital amausosis (LCA15)21, and cone-rod synaptic disorder (CRSD)22, respectively, and Arr3 encodes cone arrestin, a key player in the light-dark adaptation of cones23. Among the 54 remaining proteins in this group, 12/54 are involved in RNA processing, and 8/12 are involved in splicing and are present in spliceosomal complexes: SNRPA (in U1), DDX17 and FUS (in Complex A), DDX42 and DNAJC8 (in U2), FAM32A (in Complex C), HNRNPM and HNRNPH3 (in hnRNP) (Supplementary Fig. 10a). 14/54 genes are involved in synaptic functions and are expressed in the neuronal cells or in the retina. They play various roles in neurotransmitter release, synapse formation and stabilization, and calcium signaling, 10/14 of which are essential for the normal functioning of the retina.
Among the 87 upregulated proteins in Gpatch11Δ5/Δ5 mice, 3 are encoded by genes with increased mRNA abundance (e.g., Fgf2, Apobec2, Txnl4a; Supplementary Fig. 10c). The remaining 84 proteins are encoded by genes whose RNA expression was similar in Gpatch11Δ5/Δ5 mice and wild-type littermates. GPATCH11 was included in the list of these 84 upregulated proteins, consistent with immunostaining studies, showing an accumulation of the mutant product in the retinas of Gpatch11Δ5/Δ5 mice (Supplementary Fig. 10d).
The GO enrichment analysis showed that RNA-binding proteins made up more than half of the overrepresented proteins, including PRPF39 (in U1), CHERP, a G-patch domain-containing protein that is known to play a role in pre-mRNA splicing; SF3B2; SF3B3 and SF3B4 (in U2); PRPF3 (in U4/U6); DDX23; PRPF6; and TXNL4 (in U5); PRPF40A and RBM39 (in Complex A); DHX16 (in 1st Step); SRFS6 (in SR); and LSM14A (in LSm) (Supplementary Fig. 10a)24. Several down- and upregulated spliceosomal proteins are reported to be involved in nonretinal (TXNL425, SF3B426, and FUS27) or retina-specific (PRPF328 and PRPF629) spliceosomopathies1.
Overall, these findings support the role of GPATCH11 in the splicing and transcriptional regulation of genes pertinent to retinal function, and suggest that it may also influence other processes by dysregulating genes associated with nonretinal spliceosomopathies.
Proteomic analysis of wild-type and mutant retina protein immunoprecipitated by GPATCH11 suggests functions in synaptic plasticity and nuclear stress response
We employed MS to identify GPATCH11 protein partners in the retina and to evaluate how alterations in the G-patch domain affect its binding profile. This was achieved by isolating proteins using a GPATCH11 antibody from the retinas of 21-day-old Gpatch11Δ5/Δ5 mice and their wild-type littermates. The MS analysis identified 50 proteins associated with GPATCH11. Among these, 30 were found in both wild-type and mutant retinas, while 16 were uniquely enriched in the mutant retina and 4 in the wild-type retina (Supplementary Data 2 and Supplementary Fig. 11). Notably, approximately 23% (12 proteins) of these interactors are involved in RNA metabolism, highlighting GPATCH11’s likely role in RNA processing, splicing, and transcription regulation. Key proteins in this context include DDX23, PRPF31, and PPIL1 for RNA processing and splicing; IRF2BPL, ZFP287, and FGF2 for transcriptional regulation; SAP18 for enhancing transcriptional repression; and others such as NMNAT1, SLX9 (FAM207A), and RRBP1 involved in ribosomal RNA transcription as well as PIH1D1 involved in small nucleolar ribonucleoproteins (snoRNPs) assembly. Additionally, H2AFV contributes to histone and nucleosome functions, further emphasizing GPATCH11’s association with nucleic acid metabolism (Supplementary Data 2). The majority of GPATCH11-interacting proteins are known or presumed to play roles in synaptogenesis, synapse architecture, neuronal morphology and synaptic plasticity (ADD1, ATP8A1, BAI3, DLG3, DLG4, DLGAP1, DLGAP3, DLGAP4, FGF2, IQSEC1, IQSEC2), neurotransmission (BTBD11, FLOT2, GRIN1, KCTD8, KCTD16, SLC1A3, SLC6A9), neuronal development and maintenance (IRF2BPL, PPIL1) and neuroprotection (NMNAT1), with several implicated in neurological and neurosensorial diseases (CHMP1A, CHMP2B, DLG3, DLG4, GRIN1, HSPB1, IQSEC1, IQSEC2, IRF2BPL, NMNAT1, PPIL1, PRPF31, SLC1A3, SLC6A9, USP48) (Supplementary Data 2).
It is interesting to note that 3 of the associated proteins play roles in responding to stress and damage. HSPB1, a molecular chaperone, relocates from the cytoplasm to nuclear splicing speckles in response to stress such as heat shock and oxidative stress30. FGF2, known for its role in cell proliferation and differentiation, contributes to cellular responses to stress and injury by controlling gene expression in the nucleus, enhancing cell survival, and promoting tissue repair31. Additionally, NMNAT1 supports stress response mechanisms in neurons32.
Consistent with the increased immunostaining of GPATCH11 in the retinas of mutant mice compared to wild-type counterparts, MS analysis revealed that GPATCH11 displayed higher abundance in mutant retinas. Interestingly, FGF2 (p < 10e–5), HSPB1 (p < 10e–3), and PSMB4 (p < 10e–3) also showed increased levels in mutant retinas (Fig. 8).

a Volcano plot displaying differentially expressed proteins between WT and Gpatch11Δ5/Δ5 mice retina. The x-axis represents the difference in protein expression on a log2 scale, while the y-axis indicates the p-value. The significance of variations among samples was estimated using the Two-sided Student’s t-test. b Violin plots representing the abundance of the FGF2, HSPB1, and PSMB4 proteins. In blue: abundance in Gpatch11Δ5/Δ5 mice retina; in yellow: abundance in WT mice retina; in green: abundance with IgG immunoprecipitation. Each dot represents data for one mice. Black dots represent imputed data. The raw data is accessible via ProteomeXchange with the identifier PXD051363.
These observations underscore the critical role of GPATCH11’s nuclear and centrosomal functions in synaptic plasticity, function, and maintenance, particularly under stress conditions.
Discussion
In recent years, GPATCH proteins have emerged as pivotal regulators of RNA metabolism, assuming diverse roles that encompass pre-mRNA splicing and transcriptional regulation. The bulk of our knowledge about GPATCH protein functions has been derived from in-vitro studies conducted in yeast and human cell models2,3. Surprisingly, the study of rare genetic diseases has made only minimal contributions to our understanding of GPATCH protein functions, and to date, RBM107and SON8 are the two GPATCH genes with mutations demonstrated to cause human disease.
In this report, we present compelling evidence supporting the notion that biallelic mutations in GPATCH11, one of the lesser-known members of this family2, underlie a syndromic disease prevalent in six families. The families initially sought genetic consultation due to distinct and diversely presenting symptoms, reflecting the variable severity of retinal disease. Notably, retinal impairment was the primary symptom in Family 1, while intellectual disabilities took precedence in the other pedigrees. Moreover, various clinical features, including seizures, short stature, and diabetes, were associated with this retinal disease. It is worth mentioning that these symptoms were reported in the eldest individuals available for examination in the series; evaluations of the youngest affected subjects are still pending. Further to these findings, additional symptoms linked with GPATCH11 mutations are possible. This is highlighted by the sudden death of two unrelated individuals concurrent with a fever episode/a stroke in their teenage years/young adulthood, which could indicate defective responses to pathogens. This points to the need for specific investigations into immune responses among individuals carrying GPATCH11 disease-causing variations.
The presence of dysmorphic features in patients provides evidence that GPATCH11 serves essential functions, both, during early tissue development and in the subsequent maintenance of proper function. While both ciliopathies and spliceosomopathies exhibit features involving the retina, the brain, and the skeletal system, some distinct differences have emerged, particularly in the context of male sterility in mouse models. To our knowledge, thus far, human male sterility has been associated with ciliopathies but not with spliceosomopathies. Considering the severe phenotype affecting male gonads in the mice, it would also be worthwhile to examine the fertility of the affected male individuals in Families 1, 2, 4, and 6.
Genotype-phenotype correlation analyses revealed no significant difference in disease presentation among the following groups: individuals homozygous for the splice site variant (Families 1, 3, and 6), those with a homozygous nonsense mutation (Family 5), subjects with a nonsense mutation in trans with the splice site variant (Family 2), and the individual with a homozygous frameshifting splice site mutation (Family 4). Although the frameshift is predicted to lead to the absence of the protein, the possibility of partial bypassing of protein degradation through self-correcting mechanisms cannot be excluded. To validate the effect of the predicted frameshift splice site mutation, as well as that of the homozygous nonsense mutation, an analysis of the protein derived from the cells of patients P10 and P11 would be valuable.
While investigating the subcellular localization of GPATCH11 in human fibroblasts, we made intriguing observations about the nuclear distribution of the protein, notably that it seems to be proximal to nuclear speckles and show as well, a diffuse distribution in the nucleoplasm, a pattern reported for several spliceosome components15,16.
Additionally, our analyses of the retina of mice expressing a GPATCH11 protein analogous to the human mutant variant -which faithfully recapitulated the retinal disease and the neurological abnormalities associated with human GPATCH11 mutations- unveiled highly significant splicing abnormalities. Interestingly, we also detected a dysregulation of gene expression, which, like splicing defects, impacts mainly photoreceptor light responses. Since we analyze retinas, it is obvious that we mostly find defects and enrichment in retinal genes, and specifically in photoreceptor/phototransduction-specific genes. This dysregulation is known to trigger photoreceptor cell death which, in turn, leads to vision loss33. Reduction of mRNA abundance could be a direct consequence of splicing aberrations which lead to the degradation of mRNA isoforms containing premature termination codons, or it could be an indirect result of splicing dysregulation. However, we identified certain genes, such as Arr3, which exhibited reduced mRNA levels and abnormal splicing without any alterations to the reading frame. This finding suggests that, akin to other GPATCH proteins like SON and ZGPAT2, GPATCH11 is involved in both pre-mRNA splicing and transcriptional regulation. Further, consistent with a role of GPATCH11 in pre-mRNA splicing and transcriptional regulation, proteomic analysis of retina protein immunoprecipitated by GPATCH11 identified among its partners a dozen proteins know to be involved in RNA metabolism and splicing, including the U4/U6-specific protein PRPF3134. Moreover, it is interesting to note that mutations in this protein as well as the U5 proteins PRPF629, PRPF328, and PRPF435, which are all components of the U4/U6.U5 tri-snRNP complex, have all been linked to retinal dystrophy1. Notably, the U4 and U5 snRNAs participate with the U6 snRNA to build a complex known as the U4/U6.U5 tri-snRNP36. The expression of snRNA was assessed by RNA-seq and RT-qPCR, in fibroblasts from patients’ homozygotes for the c.328+1G>T mutation, but we found no measurable anomaly.
Furthermore, consistent with a centrosomal localization of GPATCH11, in addition to protein partners involved in RNA metabolism, immunoprecipitation identified proteins implicated in cytokinesis, cell division, and cilia metabolism. Interestingly, immunocytochemistry in serum-starved human fibroblasts revealed GPATCH11’s localization at the centrosomal linker, correlating with its interaction with Rootletin (CROCC) which also localizes there (Fig. 2b). Given Rootletin’s role in organizing the cytoskeleton and anchoring centrosomes to the nucleus, this finding suggests that GPATCH11 may influence centrosomal dynamics and functions associated with Rootletin.
The nuclear and centrosomal/cilia distribution of GPATCH11 aligns with recent findings describing it as a characteristic feature of spliceosome proteins known as centrosome-associated spliceosome components emerging as crucial players in ciliogenesis and tissue specification. These findings have been reshaping our understanding of RNA metabolism within the cell16. In recent years, the conventional belief that pre-mRNA splicing exclusively takes place within the nucleus has faced challenges. This is primarily due to reports of centrosome/cilia enrichment of both non-coding and coding RNA, along with dedicated translation machinery. Furthermore, the localization of spliceosomal components to these cellular regions further questioned this traditional view. This centrosome-associated RNA machinery is hypothesized to facilitate the nucleocytoplasmic shuttling of RNA between the spliceosome and the cytoplasm16. The precise function of RNA-processing complexes at the centrosome remains an ongoing field of investigation. One possibility is that cell type-specific splicing factors assemble on RNA and congregate around the ciliary basal body, thereby enhancing ciliogenesis and the cellular response to stimuli. A similar concept can be seen in the local translation of nuclearly encoded mitochondrial mRNAs, which facilitates the rapid on-site supply of mitochondrial membrane proteins37. The existence of a close relationship between the spliceosome and the centrosome is evident in the striking phenotypic similarities between splicing and centrosome-related genetic disorders, particularly retinal degeneration, and brain and craniofacial developmental defects observed in individuals carrying GPATCH11 mutations.
The question whether the presence of GPATCH11 at the cilia basal body contributes to cilia-related functions remains yet unanswered. Our investigation into cilia formation and SHH signaling in fibroblasts from patients did not uncover any abnormalities. However, it is essential to consider that this lack of findings may be attributed to the specific cell type that was placed under examination.
Lending support to this perspective is the noteworthy dysregulation in the expression of primary cilium proteins in the retina of mutant mice. These proteins are integral components of the photoreceptor connecting cilium and outer segments, and their dysregulation has been widely reported to contribute to the observed retinal phenotype. Among them, TULP1 has been found to be associated with LCA1521. This suggests that, while our fibroblasts analysis may not have brought to apparent abnormalities, the impact of GPATCH11 mutations on ciliary functions may manifest differently in other cell types or tissues, emphasizing the need for further investigations to comprehensively unravel the intricacies of GPATCH11’s role in cilia-related processes.
Interestingly, the retina of the mutant mice developed and layered normally, responding appropriately to light at eye-opening, and degenerated only later with light exposure. These findings suggest that GPATCH11 plays a crucial role in the function and maintenance of photoreceptor cells but not in the initial development of the retina, similar to many other genes associated with LCA and RP. The variability in the onset and severity of retinal disease in affected individuals from our cohort further supports this hypothesis. Immunoprecipitation analysis using the GPATCH11 antibody identified protein partners involved in the stress response, including the non-syndromic and syndromic LCA-causal protein NMNAT138,39, which has demonstrated a crucial role in neuroprotection against light-induced damage. This suggests that GPATCH11 could have a role in regulating the expression of genes with neuroprotective functions.
Furthermore, we observed an accumulation of GPATCH11 in the retina of mutant mice. This event might result from the loss of residues essential for its degradation, or it could be an indirect consequence of a broader dysregulation40. This accumulation led in turn to the massive dysregulation of RNA-binding protein complexes, most of which themselves also showed increased accumulation. Several of these complexes are known to be involved in the spliceosome, and mutations in their components have been linked to nonretinal (TXNL4A25) or retina-specific (PRPF328, PRPF629) spliceosomopathies.
The origin of the neurological anomalies remains ambiguous. It is unclear whether they are due to developmental issues or emerge later in life, especially given that the neurological problems in the affected sisters of Family 1 appear relatively late in childhood. However, it is important to highlight that the majority of the protein partners identified by GPATCH11 immunoprecipitation play roles in synaptogenesis, synapse architecture, neurotransmission, and overall synaptic plasticity. Interestingly, we failed to detect GPATCH11 in the plexiform layers of the retina, which contain the synapses of retinal neurons.
Altogether, our results raise intriguing questions about the broader functions of GPATCH11 in RNA homeostasis, extending beyond specific roles in pre-mRNA splicing and ciliary functions. Could an investigation of GPATCH11’s involvement in these broader RNA-related processes explain its association with a more diverse spectrum of clinical phenotypes compared to traditional spliceosomopathies and ciliopathies? To gain deeper insights into these questions, further research is clearly warranted.
In summary, our research uncovers a complex syndrome resulting from GPATCH11 mutations, characterized by overlapping clinical features of spliceosomopathies and ciliopathies.
Furthermore, our investigation indicates that GPATCH11 plays a pivotal role in pre-mRNA splicing and in gene transcription regulation. These collective findings shed light on the intricate and diverse roles of GPATCH11 in RNA metabolism, potentially extending to cilia metabolism, underscoring its significance in various physiological processes. They provide valuable insights into the molecular underpinnings of the clinical features associated with GPATCH11 mutations.
Methods
Families
This study involved twelve affected subjects, including six females and six males. They belonged to six different families with varying consanguinity and diverse origins. Family 1 exhibited early-onset retinal dystrophy with neurodevelopmental delay, and it consisted of an inbred Tunisian pedigree with multiple consanguineous connections. It included four affected individuals, two sisters, and two paternal uncles, one of whom had passed away. Family 2 presented cases of intellectual disability and included three affected individuals, a sibling, and their two unrelated parents from Morocco and Algeria. Family 3 included two affected sisters born to consanguineous parents from Algeria, and these cases had been previously reported41. Family 4 consisted of a single affected individual from Portugal. Initially, this individual was referred for mild developmental delay and later for retinitis pigmentosa at around 20 years old. Family 5 included a single affected individual, the first child of parents of Bedouin ancestry who are first cousins. This individual was initially referred to genetics at 1,5 years old due to a complex medical history with failure to thrive since the age of 10 months old. Family 6 comprises a single affected individual from Tunisia, with no reported consanguinity, but the parents originate from the same city.
All individuals provided their consent in line with the principles stipulated by the Declaration of Helsinki for participation in the study which received approval from the institutional review boards: Comité de Protection des Personnes Ile de France II (Families 1 and 2), the Reference Center for Congenital Abnormalities and Malformative Syndromes and Orphonomix Units for Genetic Testing (Family 3), Comissão de Ética para a Saúde do Instituto de Oftalmologia, Cantonal Committee of Canton Vaud for Research Activities on Human Subjects, as well as Ethikkommission Nordwest- und Zentralschweiz (Family 4), Rabin Medical Center (RMC) institutional review board committee (Family 5), Comité de Protection des Personnes Méditerrannée (Family 6). Genomic DNA was extracted from peripheral blood using standard procedures. Consents for identifiable images were obtained for patients P1, P2, P8, P9, P11, and P12.
GPATCH11 variant identification
GPATCH11 variants were identified through a two-step process: initially, variants in the known genes associated with inherited retinal dystrophies (IRD) were excluded via the analysis of a curated IRD panel. Then, WES in a research setting was performed for Families 1 and 4, WES in a diagnostic setting for Families 2 and 3, and whole genome sequencing (WGS) for Family 6. In Family 5, genetic testing that included chromosomal microarray analysis and WES was carried out before the diagnosis of retinal dystrophy was made. Following the observation of retinal changes, an IRD panel was analyzed, followed by a research reanalysis of the WES data. The WES of P1, P2, P4 from Families 1 and F2:I-1, F2:I-2, and P5 from Family 2 was sequenced as described in ref. 42. WES and variant filtration in Family 3 have been previously documented41. For Family 4, WES was conducted at CeGaT GmbH in Tübingen, Germany. Here, sequencing libraries were generated using the Twist Human Core Exome Plus Kit (Twist Bioscience) following the manufacturer’s protocol. Paired-end sequencing on a Novaseq 6000 produced 100-base sequences. Variant filtration and prioritization were performed using an in-house pipeline43. Variants in GPATCH11 were identified by selecting rare sequence changes (minor allelic frequency <1%) with the highest scores of pathogenicity and consistent with a recessive model of inheritance. For Family 5, trio WES (CentoXome Gold®) was carried out at Centogene, Rostock, Germany. Regions of interest from fragmented DNA were enriched using Agilent’s SureSelect Human All Exon V6 kit (Agilent Technologies). The generated library was sequenced using an Ilumina platform with an average coverage depth of ×100 (97% covered at 10× or more). Centogene’s in-house bioinformatics pipeline was applied for base calling, alignment to genome assembly, primary filtration of low-quality variants, and variant annotation. Recently, the research analysis of this data was carried out at the Raphael Recanati Genetics Institute (Petah Tikva, Israel) using Emedgene’s platform (Emedgene Technologies, Mazor, Israel) as previously described in ref. 44, focusing on rare, homozygous variants. The WGS of individuals F6:I-1, F6:I-2 and P12 was conducted as follows: Genome sequencing was performed at the AURAGEN laboratory (LBMMS Auragen, Auvergne Rhône-Alpes) following the recommendations of the PFMG. Whole-blood extracted genomic DNA was sequenced according to standard procedures for a PCR-Free genome on a NovaSeq6000 instrument (Illumina). Sequencing data were aligned to the GRCh38p13 full assembly using bwa 0.7+. Variants were called by several algorithms, including GATK4+, Bcftools1.10+, Manta1.6+, CNVnator0.4+, and annotated using the variant effect predictor. Detected variants were prioritized using in-house procedures. Further details are available upon request on the website of the AURAGEN platform (https://www.auragen.fr). Variants have been annotated using the GRCh38 reference genome and GenBank transcript (NM_174931.4).
Cell culture
A skin biopsy was obtained from the P1 and P2 probands and four controls (C1–4). Primary fibroblasts were isolated and cultured, as described in ref. 45. hTERT-RPE1 cells were maintained as described previously for the primary fibroblasts, but in DMEM/F-12, supplemented with 10% Fetal Bovine Serum (FBS) and 1% streptomycin/penicillin.
Analysis of c.328+1G>T, c.454C>T and c.449+1G>C variants on mRNA
Genomic DNA and total RNA were extracted from trypsinized cells using the Quickextract DNA and RNeasy Mini Kits according to the manufacturers’ protocols (Lucigen and Qiagen, respectively). RNA from peripheral blood white cells of patient P5 (carrying the c.328+1G>T and c.454C>T variants in compound heterozygosity) and P10 (carrying the c.449+1G>C in homozygosity) was prepared using the PAXgene Blood RNA Kit (50) v2 protocol (Qiagen) or Tempus Spin RNA Isolation kit (Applied Biosystems). All samples were DNase treated by the Rnase-free Dnase set (Qiagen). First-stranded cDNA synthesis was performed from total RNA (500 ng) as described in ref. 45 or by using the MultiScribe Reverse Transcriptase from the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems), according to the manufacturer’s instructions.
For analysis, gDNA (50 ng) and cDNA (5 µL of a 1:25 dilution in nuclease-free water) were PCR amplified with 1 µM of primers specific to GPATCH11, (Eurofins, Supplementary Table 2) and subjected to a sequencing reaction using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) on a 350xL Genetic Analyzer ABI and analyzed using the Sequencing Analysis v5.2 sequencing software.
Quantitative analysis of U1, U2, U4, U5, and U6 snRNA abundance and SHH signaling in patient and control fibroblasts
snRNA from P1, P2, and control fibroblasts were lysed using TRIzol and extracted with the Direct-zol RNA MiniPrep Plus (Qiagen and Zymo Research, respectively). For RT-qPCR analysis, cDNAs were amplified using specific primers designed from the sequences of U1, U2, U4, U5, U6 snRNA, and HPRT1 and RPLP0 housekeeping genes. All primers have been previously reported46,47, except for the U5 snRNA specific primers (Eurofins): Forward 5’-CTCTGGTTCTCTTCAGATCGC-3’, and Reverse 5’-CTCAAAAAATTGGGTTAAGACTCAG-3’, and U6 snRNA specific primers: Forward 5’-GCTTCGGCAGCACATATACTAAAAT-3’, and Reverse 5’-CGCTTCACGAATTTGCGTGTCAT-3’. cDNAs (5 µL) were subjected to real-time PCR amplification as described in ref. 45. The expression levels of U1, U2, U4, U5, and U6 snRNAs were normalized by using the relation ΔΔCq relative to HPRT1 and RPLP0.
Patient and control fibroblasts were subjected to a 48-h period of serum starvation. Subsequently, they were either exposed to a smoothened agonist (SAG, 100 nM) or left untreated as a negative control for a duration of 24 h. RNA was separately extracted for each condition and then converted into cDNA, following previously described procedures. The cDNAs were then amplified using specific primers, designed based on the sequences of GLI1, GLI2, and PTCH1, along with the housekeeping genes GUSB and HPRT147.
Absence of amplification when using mRNA (non-RT) and water (W) as templates was controlled in each run (Cq values non-RT > 30 and W = undetermined). The quantitative data are the means ± SEM of three independent experiments and these are presented as ratio among values for individual mRNAs.
Transcriptomic analysis of small nuclear RNA
snRNA sequencing libraries were prepared as follows. First, RNA in the snRNA size range (100–300 nt) was isolated from total RNA on 10% denaturing urea polyacrylamide gels; 4 µg total RNA was heat denatured for 2 min at 80 °C followed by cooling on ice and gel migration. The 100–300 nt size range was cut out, the gel was crushed, and RNA was eluted in 300 µL 0.3 M NaCl during 2 h at room temperature. Then, the RNA was ethanol precipitated and resuspended in 20 µL water. Proper size selection was checked on an Agilent Bioanalyzer chip. Then, 10 µL of the RNA was treated with antarctic phosphatase to dephosphorylate RNAs other than the capped snRNAs by incubating for 30 min at 37 °C and 2 min enzyme inactivation at 80 °C. RNA was purified using AMPure beads (2× volume) and resuspended in 10 µL water. Next, snRNA were decapped with the RppH enzyme in 1× Thermopol buffer (20 mM Tris pH 8.8, 10 mM (NH4)2SO4, 10 mM KCl, 2 mM MgSO4, 0.1% Triton) for 1 h at 37 °C, followed by 5 min incubation at 65 °C. The RNA was again purified using AMPure beads (2× volume) and resuspended in 10 µL water. Then, 6 µL RNA was used for NGS library preparation using the NEBNext small RNA preparation kit following the manufacturer’s instructions and 12 cycles of PCR amplification. Libraries were purified with AMPure beads (1.3× volume), quality checked using the Agilent Bioanalyzer, and sequenced using an Illumina NextSeq2000 instrument.
Quality control of raw Illumina reads was performed using FastQC v0.12.1. Read adapters, low-quality bases (with a Phred score cutoff of 20), and short reads (less than 10 bp) were trimmed using Cutadapt v3.548.Trimmed reads were aligned to the human reference genome (GRCh38.p14) or the mouse reference genome (GRCm39) using STAR v2.7.8a49. The default STAR settings were adjusted with the parameters “--outFilterScoreMinOverLread 0.3 --outFilterMatchNminOverLread 0.3 --outFilterMultimapNmax 100 --winAnchorMultimapNmax 100” to enhance the mappability of snRNA reads. Only primary alignments in proper pairs were retained for downstream analyses using Samtools v1.1450 with the parameters “view -f 3 -F 256”. Annotations for U1, U2, U4, U5 and U6 snRNAs were obtained from RNAcentral v24 for human (taxonomy 9606) and mouse (taxonomy 10090). Overlapping snRNAs were merged using the merge command in Bedtools51 v2.30.0, resulting in 206, 274, 134, and 50 loci for U1, U2, U4, U5 and U6 snRNAs, respectively. Bedtools coverage was utilized to count the number of aligned reads per locus. Counts for various snRNA variants were summed and normalized to the library size, yielding reads per million mapped reads (RPM). These RPM values were used for data visualization and for statistical analysis, including Welch’s t-test, performed in R.
Western blot analysis
Cell proteins were prepared as described in ref. 45 and run in an Any kD Mini-PROTEAN TGX Stain-Free Gel (Bio-Rad). The membrane was incubated overnight at 4 °C with the polyclonal rabbit anti-CCDC75 (anti-GPATCH11) primary antibody raised against the antigenic sequence, encompassing amino acids 111-192 of the human protein (ab122451, Abcam) (1/4000). The goat anti-rabbit IgG-HRP was used as secondary antibody (Invitrogen; 1/4000). Immunoblots were revealed and analyzed as reported in ref. 45. GPATCH11 abundance was compared to ß-Actin (ab8226, ABCAM). The quantitative data are the means ± SEM of three independent protein extractions.
15-day-old Gpatch11Δ5/Δ5 and wild-type mice were sacrificed by cervical dislocation and enucleated. The retina was lysed with RIPA-PIC (Thermo Fisher Scientific) and proceeded further for Western blot analysis as described previously, but to compare GPATCH11 abundance across samples, the membranes were secondarily incubated with mouse anti-γ-Tubulin (T6557-2ML, MERK) and goat anti-rabbit IgG-HRP primary and secondary antibodies, respectively. The abundance of GPATCH11 relative to γ-Tubulin was estimated.
Immunocytochemistry analysis
Fibroblasts and ΔCCDC hTERT RPE1 were seeded at 2 × 105 cells/well on glass coverslips in 12-well or in 6-well plates Cells were then fixed with PFA (4%) or methanol (100%) and blocked with bovine serum albumin (BSA) 3% and triton 0.1% in phosphate buffered saline (PBS). GPATCH11 was immunostained overnight at 4 °C using the polyclonal rabbit anti-GPATCH11 (1:400; ab122451, Abcam) and co-immunostained with the monoclonal mouse anti-SC-35 (1:2000, S4045, Sigma-Aldrich), anti-H3K9me3 (1:500, 61013, Active Motif), and anti-CENP-E (1:250, ab5093, Abcam) and the human anti-ACA (1:500, ImmunoVision, HCT-0100) antibodies. The primary antibodies were labeled for 1 h at room temperature using the Alexa 555-conjugated donkey anti-rabbit (1:1000; Life Technologies), the Alexa 488-conjugated donkey anti-mouse (1:1000; Life Technologies) and the Dylight 649 rabbit anti-human (1:1000; Abnova) secondary antibodies. Nuclei were stained using DAPI (Invitrogen). The images were recorded from the Spinning Disk Zeiss microscope (Zeiss) using a 40x/1.3 Oil objective. For the analysis of GPATCH11 immunostaining, we first used a machine learning with ilastik12 (v1.3.3post3) for GPATCH11 and Cellpose 2.052 nucleus detection. Then, means of the intensities of nuclear and cytoplasmic GPATCH11 staining were measured with Fiji macro37. Image figures were made through FigureJ53.
Serum-starved cells were fixed in methanol (100%) and immunostained using the polyclonal rabbit anti-GPATCH11 (1:400; ab122451, Abcam) antibody and the monoclonal mouse anti-acetylated α-Tubulin (1:1000, clone6-11b-1, Sigma-Aldrich; axonemal staining), anti-Rootletin (1:200, sc-374056, Santa Cruz Biotechnology; centrosome linker), anti-CETN3 (1:200, H00001070-MO1, Abnova; mother centriole), primary antibodies and ATTO 550-conjugated goat anti-rabbit (1:700; Sigma-Aldrich), and Alexa 514-conjugated goat anti-mouse (1:700; Life Technologies) secondary antibodies, respectively. Nuclei were stained using DAPI (Invitrogen). The images were scanned with the Confocal Leica SP8 gSTED (Leica) and the final images were analyzed with Fiji54. Ciliary axonemes and basal bodies were stained using mouse monoclonal anti-acetylated α-Tubulin (clone6-11b-1, Sigma Aldrich; 1:2000) and rabbit polyclonal anti-Pericentrin (1:5000, ab4448, Abcam) primary antibodies overnight at 4 °C, and Alexa 488-conjugated donkey anti-mouse (1:1000; Life Technologies) and Alexa 555-conjugated donkey anti-rabbit (1:1000; Life Technologies) secondary antibodies for 1 h at room temperature, respectively. Images were recorded from Spinning Disk Zeiss microscope (Zeiss) using a 40×/1.3 Oil objective. The mean numbers of ciliated cells were calculated from >100 cells from the four individual controls, each, in three independent experiments. Cilia lengths were measured from the same immunofluorescent images. Mean values were calculated from >100 cells in three independent experiments for each cell line.
Generation and analysis of genome-edited hTERT-RPE1
To establish a cell line expressing a GPATCH11 protein with its CCDC domain removed while keeping the other domains intact, we employed the CRISPR-Cas9 genome editing strategy, as previously outlined in ref. 55. In brief, we designed two guide RNAs (sgRNAs, Integrated DNA Technologies) with specific targeting for the in-frame GPATCH11 exon 3 using the CRISPOR software, which encodes the CCDC domain. These sgRNAs, namely Guide 5’ (5’-TGCCTTAGCATTGGCAATCCTGG-3’) and Guide 3’ (5’-GAACAAGAAAGACGTGACATTGG-3’). The selection was based on their on-target and off-target scores, accessible at (http://crispor.tefor.net/).
The CRISPR-Cas9/sgRNA RNP complex was created by annealing a crRNA XT recognition domain with an ATTO 550-tagged tracrRNA transactivator domain (Integrated DNA Technologies) and assembling with the S. pyogenes HiFi Cas9 Nuclease V3 nuclease (Integrated DNA Technologies), following the manufacturer’s protocol. These RNP complexes were mixed with hTERT-RPE1 cells and introduced into a 16-well reaction cuvette using the 4D-Nucleofector System (Lonza). The cells were nucleofected utilizing program CA137 on the 4D-Nucleofector System. After 24 h, cells displaying ATTO 550 fluorescence were sorted via flow cytometry (BD FACS ARIA II SORP, BD Biosciences) and subsequently placed into 96-well plates for single-cell selection. Cell clones were propagated on 96-well plates in Dulbecco’s Modified Eagle’s Medium (DMEM F-12), supplemented with 10% FBS (fetal bovine serum) and 1% streptomycin/penicillin (Thermo Fisher Scientific). Upon cell passage, a portion of the cells was used for DNA extraction and PCR amplification, employing intronic primers flanking exon 3 (Eurofins, Supplementary Table 2). Total RNA and protein were extracted as described previously.
Generation of Gpatch11 Δ5/Δ5 mouse model
Gpatch11Δ5/Δ5 mouse model was generated at the laboratory of Animal experimentation and Transgenesis facility (LEAT) of the institute Imagine, using CRISPR-Cas9 methodology, that was employed to delete Gpatch11 exon 5, which corresponds to exon 4 in humans (Fig. 4a). Two sgRNA (Integrated DNA Technologies) guides were designed: sgRNA_DEL1 (5’-ATACCATCTCCTAAAAAAGG-3’) and sgRNA_DEL2 (5’-TTCGGGTGATATCAATTATA-3’) from the mouse Gpatch11 intron 4 and intron 5 sequences, respectively, using the CRISPOR software. Newborn mice were genotyped using genomic DNA through PCR amplification, followed by Sanger sequencing employing appropriate primers (Eurofins, Supplementary Table 2). All animal procedures were conducted with the approval from the French Ministry of Research, in accordance with the French Animal Care and Use Committee of Paris Descartes University (APAFIS#31460), and in compliance with the ethical principles within the LEAT Facility of Imagine Institute.
Electroretinographic analysis of mice model
Gpatch11Δ5/Δ5 and wild-type mice aged 15 days to 6 months underwent retinal function analyses via electroretinography (ERG) using the Celeris (Diagnosys LLC) apparatus, as described in ref. 56.
Longitudinal analysis of retina and testis histology
Eyes from mutant and wild-type mice, aged 15 days to 6 months, were collected, fixed in 4% PFA overnight at 4 °C, embedded, and sectioned as described in ref. 56. Three mice from each group were included in the analysis.
Testis from mutant and wild-type mice, aged 21 days, were collected and then fixed in 4% PFA overnight at 4 °C and proceed subsequently as described above.
Longitudinal analysis of Gpatch11 mRNA and protein expression in the retina
RNAscope analysis was conducted on eye sections obtained as previously described. Specifically designed RNAscope probes, manufactured by Advanced Cell Diagnostics, were employed to target Gpatch11 mRNA. The image capture was performed using a Spinning Disk Zeiss microscope equipped with a 63×/1.4 Oil objective. Fiji54 was used to prepare images for RNAscope quantification. The quantification was then carried out using Icy57 (v2.4.3.0) with the spot detector plugin. Image figures were generated using FigureJ53.
For the immunohistochemistry analysis, deparaffined eye sections were incubated in trisodium citrate (10 mM, pH 6) and Tween (0.05%) for 30 min at 95 °C to achieve antigen retrieval. Subsequently, the sections were blocked for 1 h with blocking solution containing 5% BSA in PBS. The primary antibody rabbit anti-GPATCH11 (1:100, ab122451, Abcam) was prepared in blocking solution and eye sections were further incubated overnight at 4 °C in a humidifying chamber. Sections were washed three times with PBS and incubated for 1 h with Alexa 555-conjugated donkey anti-rabbit (IgG, 1:200, Life Technologies, Thermo Fisher Scientific). DAPI was used to label nuclei; sections were mounted with Fluoromount medium (Sigma) under glass coverslips. Images were recorded on a Spinning Disk Zeiss microscope (Zeiss) using a 63×/1.4 Oil objective. For the analysis, we first used machine learning for the detection of GPATCH11 and for the tissue detection we used Ilastik12 (v1.3.3post3). Then we measured the mean intensity of GPATCH11 with a Fiji54 macro. Image figures were made through FigureJ53.
Murine sections were also immunostained using polyclonal rabbit anti-Blue Opsin (1:100, PA5-78371, Invitrogen), anti-Green Opsin (1:1000, OSR00222W, Invitrogen) and monoclonal mouse anti-Rhodopsin (1:500, NBP1-48334, Novus) primary antibody, as well as Alexa 555-conjugated donkey anti-rabbit and 633-conjugated goat anti-mouse (1:200, Life Technologies, Thermo Fisher Scientific).
Behavioral characterization of mice model
All mice of 1 month old were under constant environmental enrichment and were handled for 3–5 days for at least 3 min before the beginning of the behavioral tasks. For each test, mice were transported a short distance from the holding mouse facility to the testing room in their home cages. The tests were performed by an experimentalists blind to the genotypes or treatment of the mice under study. The NOR paradigm, the 3-foot shock CFC, the OF, and MWM were performed as reported in ref. 58.
Brain harvesting and dissection, and Immunohistochemistry on floating sections
Brain from intracardially perfused Gpatch11Δ5/Δ5 and wild-type mice aged 1 month, were collected and fixed as described in ref. 58. Embedded brains were sectioned on a cryostat at 50 μm thickness. Immunohistochemistry on brain slices was performed as described in ref. 58 with rabbit anti-GPATCH11 (1:500; ab122451, Abcam) and monoclonal rat anti-CTIP2 (1:1000, ab18465, Abcam).
Transcriptomic analysis of mutant and wild-type mouse retina
Total RNA was extracted from retinal tissue using the RNeasy Mini Kit (Qiagen). The concentration and purity of the total RNA were assessed using an Agilent 2100 Bioanalyzer.
For RNA-Seq analysis, retinas from both wild-type (n = 5) and Gpatch11Δ5/Δ5 (n = 5) mice at 15 days were used. RNA-Seq libraries were constructed from 1 μg of total RNA using the TrueSeq Stranded mRNA Sample Prep Kit (Illumina) and paired-end sequencing with 100 base pair read length was performed on a Novaseq 6000 Illumina sequencer. Pass-filtered reads were aligned to the Ensembl genome assembly GRCm39.108 using STAR 2.7.10b.
Differential expression analysis was conducted using featureCounts59 to generate a count table of gene features. For gene-level analysis, the edgeR package was employed for normalization, differential expression analysis, and the computation of TPM (transcripts per million) values60. DEGs were filtered based on a fold change greater or <2, a FDR lower than 0.05, and a minimum expression of five TPM.
Alternative splicing analysis was performed using rMATS61, allowing for the assessment of differential exon usage for various event types (e.g., SEs, alternative 5’ and 3’ splice sites, retained introns, and mutually exclusive exons). rMATS was run with default settings using sorted BAM files produced by STAR (two-pass). The identified splicing changes were filtered based on a minimum count of 2 in at least one sample, an FDR of <0.05, and a change in the inclusion-level difference of more than 20%.
A comprehensive analysis of gene lists, including enrichment in biological pathways, and gene annotations from both differential expression and alternative splicing analysis, was conducted using the GO classification system. Metascape was utilized for this purpose62. Data mining and graphical representations of pathway data were performed using R packages, including GOplot63. The raw data have been deposited in BioStudies with the identifier S-BSST1157.
The downregulation of Arr3 in Gpatch11Δ5/Δ5 retinas, compared to their wild-type counterparts, was confirmed through RT-PCR using specific primers Arr3_forward (Eurofins): 5’- CAAGATTGCAGTTGTCCAGA-3’ and Arr3_reverse: 5’-TGGCTGGCAGACCTCCT-3’, following previously described procedures.
TTP-Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS)
The retina was lysed 1 h on 100 µl of RIPA-PIC (Thermo Fisher Scientific) and sonicated before dosage. 1 mg of protein were subjected to immunoprecipitation using SurebeadsTM protein A/G, accordingly to the protocol (Biorad). We subjected retina lysates (50 µg) and immunoprecipation extract (1 mg) (n = 4 for each genotype) to digestion using S-Trap™ micro spin columns (Protifi, Huntington, USA). The eluted peptides were vacuum-dried and then reconstituted in a 125 µl solution containing acetonitrile (2%) and formic acid (0.1%) in HPLC-grade water before being prepared for MS analysis.
Input samples (1 µL) were injected into a nanoelute HPLC system (Bruker Daltonics, Germany) coupled to a timsTOF Pro mass spectrometer (Bruker Daltonics, Germany). High-performance liquid chromatography (HPLC) separation was performed at a flow rate of 250 nL/min using a packed emitter column (C18, 25 cm × 75 μm, 1.6 μm; Ion Optics, Australia) with a 70-min gradient elution profile: from 2% to 13% solvent B over 41 min, followed by 13% to 20% over 23 min, then 20% to 30% over 5 min, 30% to 85% for 5 min, and finally, 85% for 5 min to wash the column.
Mass-spectrometric data were acquired using the parallel accumulation serial fragmentation (PASEF) acquisition method in data-dependent acquisition (DDA) mode. Measurements were conducted within the m/z range of 100 to 1700 Th (Thomson). The ion mobility values ranged from 0.75 to 1.25 V s/cm² (1/k0). The total cycle time was set to 1.17 s, and 10 PASEF MS/MS scans were performed.
Data processing LC-MS/MS acquisition
The MS data files were processed using the MaxQuant software version 2.1.3.0 and searched against the Mus musculus database from SwissProt and TrEMBL (release 11/2021, containing 88,132 entries) using the Andromeda search engine. We configured the search with an initial mass deviation of 10 ppm for parent masses and 40 ppm for fragment ions. The minimum peptide length was set to seven amino acids, and we enforced strict specificity for trypsin cleavage, allowing up to two missed cleavage sites.
Carbamidomethylation was specified as a fixed modification, while oxidation (Met) and N-terminal acetylation were set as variable modifications. We also enabled ‘match between runs’. For label-free quantification (LFQ), the minimum ratio count was set to 1. FDRs were controlled at both the protein and peptide levels, set to 1%. Scores were calculated within MaxQuant as previously described64.
To ensure data quality, we removed reverse and common contaminant hits from the MaxQuant output. Protein quantification was achieved using the MaxQuant label-free algorithm with LFQ intensities64,65. We analyzed four independent lysate replicates for both wild-type and homozygote Gpatch11Δ5/Δ5 mutant samples using Perseus software (version 1.6.15.0), which is freely available at www.perseus-framework.org66.
For statistical comparisons, we divided the data into two groups, each consisting of up to four biological replicates. We filtered the data to retain only proteins with at least three valid values in at least one group. Missing data points were imputed by generating a Gaussian distribution of random numbers with a standard deviation of 33% relative to the standard deviation of the measured values, along with a 1.8 standard deviation downshift of the mean to simulate the distribution of low signal values.
The statistical analysis was performed using a t-test with an FDR threshold of <0.05 and S0 = 0.01. Volcano plots of proteins were generated in Perseus using logarithmized LFQ intensities with an FDR threshold of <0.05 and S0 = 0.01. Furthermore, statistical and bioinformatic analysis were performed with MassDynamics software available at https://massdynamics.com. The raw data from the lysates have been deposited at the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD041849 [http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID = PXD041849] (Lysat) and PXD051363 (Immunoprecipitation).
Statistical analysis
Statistical parameters including the exact sample size (n), post hoc tests, and statistical significance are reported in every figure and figure legend. Data were estimated to be statistically significant when p ≤ 0.05 by Student’s t-test, one or two-way ANOVA. Statistical analyses were performed using GraphPad PRISM (v6) (GraphPad Software Inc., La Jolla, CA, USA). All values are shown as mean ± SEM. Source data are provided as a Source data file.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RNA-seq data generated during the current study are available in BioStudies and are publicly available under accession number S-BSST1157. snRNAs data are available in ArrayExpress (https://www.ebi.ac.uk/biostudies/arrayexpress) with the accession code: E-MTAB-14501. The mass spectrometry proteomics data have been deposited to the in ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD041849 and PXD051363, respectively. Clinical datasets generated during and/or analyzed during the current study are available in the ClinVar repository, https://www.ncbi.nlm.nih.gov/clinvar/submitters/507510 with the submission ID SUB13926389. Source data are provided with this paper.
References
Griffin, C. & Saint-Jeannet, J.-P. Spliceosomopathies: diseases and mechanisms. Dev. Dyn. Publ. Am. Assoc. Anat. 249, 1038–1046 (2020).
Bohnsack, K. E., Ficner, R., Bohnsack, M. T. & Jonas, S. Regulation of DEAH-box RNA helicases by G-patch proteins. Biol. Chem. 402, 561–579 (2021).
Robert-Paganin, J., Réty, S. & Leulliot, N. Regulation of DEAH/RHA helicases by G-patch proteins. BioMed. Res. Int. 2015, 931857 (2015).
Studer, M. K., Ivanović, L., Weber, M. E., Marti, S. & Jonas, S. Structural basis for DEAH-helicase activation by G-patch proteins. Proc. Natl. Acad. Sci. USA 117, 7159–7170 (2020).
Abdelhaleem, M. Helicases: an Overview. in Helicases (ed. Abdelhaleem, M. M.) 587 1–12 (Humana Press, 2009).
Sloan, K. E. & Bohnsack, M. T. Unravelling the mechanisms of RNA helicase regulation. Trends Biochem. Sci. 43, 237–250 (2018).
Johnston, J. J. et al. Massively parallel sequencing of exons on the X chromosome identifies RBM10 as the gene that causes a syndromic form of cleft palate. Am. J. Hum. Genet. 86, 743–748 (2010).
Wynn, S. L. et al. Organization and conservation of the GART/SON/DONSON locus in mouse and human genomes. Genomics 68, 57–62 (2000).
Gudmundsson, S. et al. Variant interpretation using population databases: lessons from gnomAD. Hum. Mutat. 43, 1012–1030 (2022).
de Sainte Agathe, J.-M. et al. SpliceAI-visual: a free online tool to improve SpliceAI splicing variant interpretation. Hum. Genom. 17, 7 (2023).
Ohta, S. et al. The protein composition of mitotic chromosomes determined using multiclassifier combinatorial proteomics. Cell 142, 810–821 (2010).
Berg, S. et al. Ilastik: interactive machine learning for (bio)image analysis. Nat. Methods 16, 1226–1232 (2019).
Jerber, J. et al. The coiled-coil domain containing protein CCDC151 is required for the function of IFT-dependent motile cilia in animals. Hum. Mol. Genet. 23, 563–577 (2014).
Becker-Heck, A. et al. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat. Genet. 43, 79–84 (2011).
Spector, D. L. & Lamond, A. I. Nuclear speckles. Cold Spring Harb. Perspect. Biol. 3, a000646 (2011).
Busselez, J., Uzbekov, R. E., Franco, B. & Pancione, M. New insights into the centrosome-associated spliceosome components as regulators of ciliogenesis and tissue identity. Wiley Interdiscip. Rev. RNA 14, e1776 (2023).
Gigante, E. D. & Caspary, T. Signaling in the primary cilium through the lens of the Hedgehog pathway. Wiley Interdiscip. Rev. Dev. Biol. 9, e377 (2020).
Bird, C. M. & Burgess, N. The hippocampus and memory: insights from spatial processing. Nat. Rev. Neurosci. 9, 182–194 (2008).
Leid, M. et al. CTIP1 and CTIP2 are differentially expressed during mouse embryogenesis. Gene Expr. Patterns GEP 4, 733–739 (2004).
Solovei, I. et al. Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell 137, 356–368 (2009).
Hanein, S. et al. Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis. Hum. Mutat. 23, 306–317 (2004).
Zeitz, C. et al. Mutations in CABP4, the gene encoding the Ca2+-binding protein 4, cause autosomal recessive night blindness. Am. J. Hum. Genet. 79, 657–667 (2006).
Zhang, H. et al. Light-dependent redistribution of visual arrestins and transducin subunits in mice with defective phototransduction. Mol. Vis. 9, 231–237 (2003).
Hegele, A. et al. Dynamic protein-protein interaction wiring of the human spliceosome. Mol. Cell 45, 567–580 (2012).
Wieczorek, D. et al. Compound heterozygosity of low-frequency promoter deletions and rare loss-of-function mutations in TXNL4A causes Burn-McKeown syndrome. Am. J. Hum. Genet. 95, 698–707 (2014).
Bernier, F. P. et al. Haploinsufficiency of SF3B4, a component of the pre-mRNA spliceosomal complex, causes Nager syndrome. Am. J. Hum. Genet. 90, 925–933 (2012).
Hewitt, C. et al. Novel FUS/TLS mutations and pathology in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 67, 455–461 (2010).
Chakarova, C. F. et al. Mutations in HPRP3, a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa. Hum. Mol. Genet. 11, 87–92 (2002).
Tanackovic, G. et al. A missense mutation in PRPF6 causes impairment of pre-mRNA splicing and autosomal-dominant retinitis pigmentosa. Am. J. Hum. Genet. 88, 643–649 (2011).
Bryantsev, A. L., Chechenova, M. B. & Shelden, E. A. Recruitment of phosphorylated small heat shock protein Hsp27 to nuclear speckles without stress. Exp. Cell Res. 313, 195–209 (2007).
Maddaluno, L., Urwyler, C. & Werner, S. Fibroblast growth factors: key players in regeneration and tissue repair. Development 144, 4047–4060 (2017).
Sasaki, Y., Margolin, Z., Borgo, B., Havranek, J. J. & Milbrandt, J. Characterization of leber congenital amaurosis-associated NMNAT1 mutants. J. Biol. Chem. 290, 17228–17238 (2015).
Rodrigues, A. et al. Modeling PRPF31 retinitis pigmentosa using retinal pigment epithelium and organoids combined with gene augmentation rescue. NPJ Regen. Med. 7, 39 (2022).
Deery, E. C. et al. Disease mechanism for retinitis pigmentosa (RP11) caused by mutations in the splicing factor gene PRPF31. Hum. Mol. Genet. 11, 3209–3219 (2002).
Chen, X. et al. PRPF4 mutations cause autosomal dominant retinitis pigmentosa. Hum. Mol. Genet. 23, 2926–2939 (2014).
Burke, J. E., Butcher, S. E. & Brow, D. A. Spliceosome assembly in the absence of stable U4/U6 RNA pairing. RNA 21, 923–934 (2015).
Golani-Armon, A. & Arava, Y. Localization of nuclear-encoded mRNAs to mitochondria outer surface. Biochem. Mosc. 81, 1038–1043 (2016).
Perrault, I. et al. Mutations in NMNAT1 cause Leber congenital amaurosis with early-onset severe macular and optic atrophy. Nat. Genet. 44, 975–977 (2012).
Bedoni, N. et al. An Alu-mediated duplication in NMNAT1, involved in NAD biosynthesis, causes a novel syndrome, SHILCA, affecting multiple tissues and organs. Hum. Mol. Genet. 29, 2250–2260 (2020).
Awwad, S. W., Darawshe, M. M., Machour, F. E., Arman, I. & Ayoub, N. Recruitment of RBM6 to DNA double-strand breaks fosters homologous recombination repair. Mol. Cell. Biol. 43, 130–142 (2023).
Bruel, A.-L. et al. Increased diagnostic and new genes identification outcome using research reanalysis of singleton exome sequencing. Eur. J. Hum. Genet. EJHG 27, 1519–1531 (2019).
Gerber, S. et al. Autosomal recessive pathogenic MSTO1 variants in hereditary optic atrophy. EMBO Mol. Med. 15, e16090 (2023).
Peter, V. G. et al. The first genetic landscape of inherited retinal dystrophies in Portuguese patients identifies recurrent homozygous mutations as a frequent cause of pathogenesis. PNAS Nexus 2, pgad043 (2023).
Basel-Salmon, L. et al. Improved diagnostics by exome sequencing following raw data reevaluation by clinical geneticists involved in the medical care of the individuals tested. Genet. Med. J. Am. Coll. Med. Genet. 21, 1443–1451 (2019).
Barny, I. et al. Basal exon skipping and nonsense-associated altered splicing allows bypassing complete CEP290 loss-of-function in individuals with unusually mild retinal disease. Hum. Mol. Genet. 27, 2689–2702 (2018).
Egloff, S. et al. The 7SK snRNP associates with the little elongation complex to promote snRNA gene expression. EMBO J. 36, 934–948 (2017).
Perrault, I. et al. IFT81, encoding an IFT-B core protein, as a very rare cause of a ciliopathy phenotype. J. Med. Genet. 52, 657–665 (2015).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10 (2011).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Li, H. et al. The sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Pachitariu, M. & Stringer, C. Cellpose 2.0: how to train your own model. Nat. Methods 19, 1634–1641 (2022).
Mutterer, J. & Zinck, E. Quick-and-clean article figures with FigureJ. J. Microsc. 252, 89–91 (2013).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013).
De Malglaive, F. et al. Pharmacological cAMP stimulation via prostaglandin receptors rescues ciliary defects in CEP290-deficient human and mouse models. Preprint at https://doi.org/10.1101/2023.10.06.561156 (2023).
de Chaumont, F. et al. Icy: an open bioimage informatics platform for extended reproducible research. Nat. Methods 9, 690–696 (2012).
Riva, M. et al. Aberrant survival of hippocampal Cajal-Retzius cells leads to memory deficits, gamma rhythmopathies and susceptibility to seizures in adult mice. Nat. Commun. 14, 1531 (2023).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinform. Oxf. Engl. 26, 139–140 (2010).
Shen, S. et al. MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Res. 40, e61 (2012).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019).
Walter, W., Sánchez-Cabo, F. & Ricote, M. GOplot: an R package for visually combining expression data with functional analysis. Bioinforma. Oxf. Engl. 31, 2912–2914 (2015).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Luber, C. A. et al. Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity 32, 279–289 (2010).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016).
Acknowledgements
This work was supported by grants from Retina France; Fondation JED Belgique; Fondation Visio and MSDAVENIR (DEVO-DECODE program) to J.M.R. and I.P., Fédération des Aveugles et Amblyopes de France, PhD international Institut Imagine to A.Z. and by grant # 310030_204285s from the Swiss National Science Foundation to C.R. J.M.R. is member of the European Reference Network for Rare Eye Diseases (ERN-EYE), which is co-funded by the Health Program of the European Union under the Framework Partnership Agreement n°739534. The authors thank the Transgenesis, Neurobehavioral and Metabolism, Necker Bioimage Analysis, Cell Imaging, Histology (Sofian Ameur, Damien Conrozier, and Sophie Berissi), Cytometry, Genomic, and Proteomic platforms for valuable discussions and help. Special thanks go to Nicolas Cagnard for the submission to BioStudies. We acknowledge the sequencing and bioinformatics expertise of the I2BC High-throughput sequencing facility, supported by France Génomique (funded by the French National Program “Investissement d’Avenir” ANR-10-INBS-09. The authors would like to acknowledge all patients and their families for participating in this study. The authors are also grateful to Virginie G. Peter and Raquel Rodriguez for their help throughout the study and to Sitta Föhr for careful proofreading of the manuscript. This research was made possible through access to the data generated by the France Genomic Medicine Plan 2025 and the authors thank Dr. Marine Lebrun for her help in the genome analysis.
Author information
Authors and Affiliations
Contributions
A.Z. performed in vitro and in vivo experiments, analyzed the data, and wrote the paper. P.D. and L.F-T. performed creation of animal models. J.A., C.G., I.A., M.P.R., N.B., S.B., A-L. B., C.C-F., N.P-S., D.A., N.A.B, A.B-Z, D. Z., J.K., C.S., L.C.S., N.O., M.P., S.S., M.S., E.S., K.Z-K., and A.B.S. collected clinical data. S.M. performed behavioral analysis of mice. N.G. created analytic tools. J.R. analyzed data from RNASeq. E.L.V.D performed snRNASeq. R.S-L. and G.D. analyzed data from snRNASeq. I.C.G. and V.J. analyzed data from mass spectrometry. S.B. collected skin biopsies from patients. I.P. analyzed exome data from Family 1. J.A. analyzed exome data from Family 2. C.T.-R. and P.K. analyzed exome data from Family 3. K.K., M.Q., and C.R. analyzed exome data from Family 4. L.B.S analyzed exome data from Family 5. M.P. analyzed exome data from Family 6. J.-M.R. and I.P. supervised the research, designed the experiments, and wrote the paper. All authors discussed the results and participated in manuscript preparation and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Stephen Tsang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zanetti, A., Dujardin, G., Fares-Taie, L. et al. GPATCH11 variants cause mis-splicing and early-onset retinal dystrophy with neurological impairment. Nat Commun 15, 10096 (2024). https://doi.org/10.1038/s41467-024-54549-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54549-8
