
Abstract
With advanced mass spectrometry (MS)-based proteomics, genome-scale proteome coverage can be achieved from bulk tissues. However, such bulk measurement lacks spatial resolution and obscures tissue heterogeneity, precluding proteome mapping of tissue microenvironment. Here we report an integrated wet collection of single microscale tissue voxels and Surfactant-assisted One-Pot voxel processing method termed wcSOP for robust label-free single voxel proteomics. wcSOP capitalizes on buffer droplet-assisted wet collection of single voxels dissected by LCM to the tube cap and SOP voxel processing in the same collection cap. This method enables reproducible, label-free quantification of approximately 900 and 4600 proteins for single voxels at 20 µm × 20 µm × 10 µm (~1 cell region) and 200 µm × 200 µm × 10 µm (~100 cell region) from fresh frozen human spleen tissue, respectively. It can reveal spatially resolved protein signatures and region-specific signaling pathways. Furthermore, wcSOP-MS is demonstrated to be broadly applicable for OCT-embedded and FFPE human archived tissues as well as for small-scale 2D proteome mapping of tissues at high spatial resolutions. wcSOP-MS may pave the way for routine robust single voxel proteomics and spatial proteomics.
Similar content being viewed by others

Introduction
The human body comprises trillions of cells with hundreds of different cell types, which are spatially organized along with their extracellular matrix to form tissues and organs with diverse functionalities1,2. A tissue is an ensemble of certain types of cells spatially located at distinct anatomical regions to execute region-specific functions3. There is significant heterogeneity across different anatomical regions within an organ tissue4,5,6. Therefore, spatial proteomics enabling precise, comprehensive, proteomic mapping of human tissues at high spatial resolution is crucial for phenotypic characterization of tissue heterogeneity and microenvironment within spatial context to improve our understanding of tissue biology and how unique cell types contribute to heterogeneous tissues7.
Antibody-based immunoassays such as immunohistochemistry (IHC)8,9, immunofluorescence (IF)9, CODEX (codetection by indexing)10,11, MIBI (multiplexed ion beam imaging)12,13, and mass cytometry14 are primarily used for spatial targeted proteomics mapping of human tissues at single-cell or near single-cell resolution15. However, they require a priori knowledge which limits the ability for discovery studies and only can open a small window into complex tissue microenvironments due to their inherent limitations (e.g., low multiplex with up to 60 protein markers, and long lead time and difficulties in generating high-quality antibodies for spatial proteomics imaging). Unlike antibody-based targeted proteomics, mass spectrometry (MS)-based global proteomics enables reliable simultaneous detection and quantification of >10,000 proteins from bulk tissue samples with a large number of mixed populations of cells (e.g., ≥10 million of cells ≈1 mg of tissues)16,17,18,19. However, such bulk measurement averages out cell-type specific tissue heterogeneity particularly when cells of interest only account for a small portion of the cell population. Thus, they cannot provide any insights into tissue heterogeneity and microenvironment within spatial context which are necessary for understanding functional spatial biology.
Laser capture microdissection (LCM) is routinely used for microscopic isolation and capture of subpopulations of cells, individual cells, or regions of interest in a complex heterogeneous tissue section20,21,22,23,24,25. In the past two decades, a surge of papers have reported using LCM for tissue spatial molecular profiling24. LCM collection combined with advanced MS-based proteomics, LCM-MS, allows for region- and cell type-resolved proteome characterization of human tissues23,25. Currently, the combined LCM and cap-assisted methods are most widely used due to their cost-effective, easy implementation and an easy-to-use tissue voxel collection and processing workflow21,23,26. For these methods, LCM-dissected tissue voxels are collected to the tube cap. The collected voxels or tissue voxel lysates on the cap are then transferred to the tube, and processed in the tube. For example, Davis et al. performed a series of voxel sample preparation optimizations and developed a sensitive cap-assisted SP3 method for enabling identification of ~1500 proteins from as little as 100 Purkinje cells dissected by LCM with the total area of 60,000 µm2 at a thickness of 10 µm27. Drummond et al. developed a wet cap-assisted localized proteomics method for quantitative cell type-specific proteomics in Alzheimer’s disease (AD) research28,29. 1000 s of neurons, amyloid plaques, or neurofibrillary tangles (NFTs) (the total area of 0.5–2.5 mm2) from AD patient brain tissues dissected by LCM were collected to the tube cap prefilled with distilled water and then transferred to the tube by centrifugation for sample processing28,30,31,32. ~500 and ~900 proteins were identified from dissected neurons (NFTs) and amyloid plaques, respectively. Comparative analysis of neighboring non-diseased tissue voxels allows pathway and network enrichment analysis for discovery of potential therapeutic targets and biomarkers for AD. However, current cap-assisted methods are more suitable for collection and processing of 100–1000s of pooled tissue voxels or large tissue regions of interest. They generally lack robustness and reproducibility for effective single voxel proteomics primarily due to significant sample loss during transferring and processing of single tissue voxels at small sizes (e.g., 400–40,000 µm2 equivalent to the area of 1–100 cell region).
When combined with subcellular resolution imaging, semi-automated LCM, and an ultrasensitive proteomics platform, Deep Visualization Proteomics (DVP) allows proteome profiling of as little as 100 cells from image-guided selection to reveal cell type-specific proteome and spatially resolved proteome changes at different disease states33. Very recently, with further improved sample preparation and LC-MS workflow single-cell DVP termed scDVP has been developed for deep proteome characterization of murine hepatocytes at single-cell spatial resolution34,35. But broad implementation of the 384-well plate-based DVP method for spatial proteome mapping of tissues remains to be demonstrated.
The ability for robust proteomic analysis of single microscale voxels (i.e., single voxel proteomics) is essential for high spatial resolution proteome mapping of human tissues and comprehensive characterization of tissue microenvironment. Furthermore, single voxel proteomics is crucial for analysis of few cells of interest within the complex tissue microenvironment (e.g., few tumor cells in biopsy tissue at the early cancer stage) with great potential of moving towards precision medicine. Two chip-assisted methods, LCM-nanoPOTS36,37,38 and LCM-microPOTS39, were recently reported for enabling proteome profiling of single microscale tissue voxels. LCM-nanoPOTS, when combined with an advanced LC-MS platform, allows for identification and quantification of >2000 proteins from single voxels at 10,000 µm2 with a thickness of 10 µm and generation of 2D proteome images at 100 µm spatial resolution across mouse uterine tissue sections37. However, the chip-assisted methods require specifically designed nanowell or microwell chips, and thus they are not easily adoptable for broad benchtop applications.
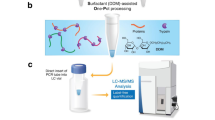
To alleviate the shortcomings of existing spatial proteomics methods, we have recently developed a broadly adoptable, robust, cap-assisted method for quantitative label-free single voxel proteomics. This method capitalizes on both wet collection and Surfactant-assisted One-Pot processing of single voxels termed wcSOP on the tube cap prefilled with a cocktail of buffer containing DDM surfactant and protease (Fig. 1a). Unlike SOP for collection and processing of single cells in the PCR tube, wcSOP uses the prefilled PCR tube cap to collect and process single tissue voxels. It takes advantages of the full compatibility of the PCR tube cap with LCM instruments for robust collection of single voxels with easy microscope inspection and the tight seal with a dome-shaped PCR tube cap to avoid evaporation for effective processing of single voxels in the tube cap. wcSOP shares the same one-pot concept as DVP and nanoPOTS with collection and processing of single voxels in the same pot (i.e., PCR tube cap for wcSOP, the plate well for DVP, and chip nanowell for nanoPOTS). But all these one-pot methods are essentially different because each pot has their unique physical dimensions that require different voxel collection and processing procedures. Furthermore, wcSOP has similar performance as other methods, but unlike DVP and nanoPOTS it is easier to implement and operate at low cost and without the need of a highly skilled person.

a There are three steps for wcSOP-MS: wet cap collection, SOP processing, and LC-MS/MS analysis. A tissue slide mounted on PEN membrane is cut by laser and a single tissue voxel is catapulted to a dome-shaped PCR tube cap prefilled with a cocktail of buffer containing DDM surfactant for tissue lysis and a mixture of Lys-C and trypsin protease for protein digestion. Single voxels are incubated at 75 °C for 1 h (tissue lysis and protein denaturation) and 37 °C for overnight (protein digestion) on the cap at a hanging droplet position to have full interaction between the voxel and the cocktail buffer. After voxel processing, the digested peptides are transferred to the bottom of the tube by centrifugation at 2000 × g for 5 min. Prior to LC-MS analysis, the cap of the PCR tube is removed, and the tube is inserted into a sample vial to avoid sample transfer loss. Single voxels are analyzed by using a standard LC-MS platform for quantitative proteomic analysis. The freely-available open-source MaxQuant and DIA-NN software tools are used for DDA and DIA label-free quantitation, respectively. b Comparison of the number of identified protein groups (No. of protein groups) between DDA and DIA for single spleen tissue voxels at a size of 200 µm × 200 µm × 10 µm (n = 3 biological replicates; data are presented as mean values ± SD). c Venn diagram of protein overlap between DDA and DIA for single spleen tissue voxels at a size of 200 µm × 200 µm × 10 µm. d Proteome dynamic range for DDA and DIA quantification of single spleen tissue voxels at a size of 200 µm × 200 µm × 10 µm. e Comparison of the coefficient of variations (CVs) in protein abundance between DDA and DIA for analysis of single spleen tissue voxels at a size of 200 µm × 200 µm × 10 µm with 4 biological replicates. Source data are provided as a Source Data file.
In this work we show that with its one-pot voxel collection and processing, wcSOP greatly reduces sample transfer and surface adsorption losses, thus significantly improving detection sensitivity for MS analysis of single voxels. A commonly and easily accessible Q Exactive Plus or HF MS platform was used for the development of wcSOP-MS and its application demonstration. wcSOP-MS was demonstrated to enable reliable label-free quantification of ~900 and ~4600 proteins from single voxels at 20 µm × 20 µm (400 µm2) and 200 µm × 200 µm (40,000 µm2) at a thickness of 10 µm with standard LC-MS platforms, respectively. We further evaluated its broad applicability to different types of human tissues (fresh frozen healthy spleen, OCT-embedded breast tumor, and FFPE AD brain) for single voxel proteomics analysis. wcSOP-MS can identify spatially resolved proteome changes and enriched pathways for different types of tissues at high spatial resolutions. These results demonstrate great potential of wcSOP-MS for broad applications in the biomedical research.
Results
Systematic evaluation of cap-assisted methods for tissue voxel collection and processing
The combined LCM and various cap-assisted methods are most widely used for convenient spatial proteome profiling of subpopulations of cells, image-guided regions of interest, and tissue microenvironment from heterogenous tissues due to their cost-effective, easy implementation without the need of any special devices or a highly skilled person21,23,26,40,41,42. Commercially available adhesive tube caps or standard PCR tube caps are used for collection and processing of tissue voxels dissected by LCM. However, current cap-assisted methods are more suitable for collection and processing of 100–1000s of pooled tissue voxels with the total area of ~0.5–2.5 mm2 at relatively large processing volumes28,30,31,32. They generally lack robustness and reproducibility for high-resolution single voxel proteomics because there is a technical challenge in effective transferring and processing of single tissue voxels at small sizes (e.g., 400–40,000 µm2 equivalent to the area of 1–100 cell region). Single voxel proteomics is crucial for precise spatial characterization of tissue heterogeneity and generation of high spatial resolution tissue maps.
To address this issue, we have systematically evaluated and optimized cap-assisted methods for single voxel proteomics. Single spleen tissue voxels at the size of 40,000 µm2 (200 µm × 200 µm) with a thickness of 10 µm were collected into the tube caps, processed by our previously developed SOP, and analyzed by data dependent acquisition (DDA) MS analysis. There are two types of cap-assisted collection methods: dry collection with adhesive polymer attached to the cap and wet collection with prefilled water or buffer on the cap.
We first evaluated a conventional dry cap-assisted collection and processing method for single voxel proteomics (Supplementary Fig. 1). Tissue lysis occurs on the cap, and protein denaturation and digestion are performed in the tube (Supplementary Fig. 1a). Different lysis buffer volumes on the cap were evaluated, and high irreproducibility was observed with protein identification from 715 to 1432 (Supplementary Fig. 2a). This irreproducibility is most likely due to incomplete tissue lysis and/or significant adsorption loss to adhesive polymer. We next evaluated all tissue lysis, denaturation, and digestion on the cap (i.e., “all-in-one” dry cap-assisted SOP termed dcSOP) with the processing volume of 10 µL (the maximal volume for the adhesive cap) (Supplementary Fig. 1b). The selection of 10 µL processing volume is to fully cover a single tissue voxel since it may be collected at the edge of the cap. The technical reproducibility was greatly improved with 1724 proteins and CV < 10% (Supplementary Fig. 2b). However, there are two drawbacks for dcSOP, adhesive polymer detached from the cap during centrifugation and difficulty in tightly sealing the adhesive tube cap. They can significantly reduce its robustness in sample processing and injection for single voxel proteomics analysis.
In parallel, we revisited a conventional wet cap-assisted method used for spatial proteomics. Prior to voxel collection, 25 µL buffer was added to the dome-shaped PCR tube cap with the maximal volume of ~25 µL to form the droplet with the largest surface area at a diameter of ~5.4 mm. The large droplet ensures precise, convenient collection of a single microscale voxel with ~100% success rate. After collection, the PCR tube was centrifuged at 2000 × g to transfer the voxel from the cap to the bottom of the tube (Supplementary Fig. 3). The collected single voxel was then processed in the tube using SOP. To maximize sample recovery and processing reproducibility, we have optimized a series of SOP conditions, including different types of buffers, different buffer volumes, and different temperatures for tissue lysis and protein denaturation (Supplementary Tables 1, 2). Unfortunately, low reproducibility was observed no matter what conditions were used. With the demonstrated performance of SOP-MS for robust single-cell proteomics, we inferred that there is an issue for transferring the collected voxel from the cap to the bottom of the well. This was confirmed by close voxel tracing using microscope during the transferring step. The collected single voxels were found almost everywhere, such as on the cap, on the top, the middle, and the low side tube wall, and at the bottom of the tube, resulting in low processing reproducibility for single voxel proteomics. This can be attributed to much stronger electrostatic interaction between the tissue voxel and the plastic tube wall or cap than the ultracentrifugation force for single microscale voxels.
With this technical challenge in transferring single voxels to the well bottom as well as our experience in dcSOP, we turned to pursue a different wet cap-assisted SOP method for both collection and processing of single voxels on the cap. The wet cap-assisted SOP method termed wcSOP capitalizes super tight sealing of the dome-shaped PCR tube cap, low-bind PCR tube cap, and precise collection of single voxels. We evaluated three different types of wcSOP methods (i.e., 3-step, 2-step, and “all-in-one” 1-step wcSOP) (Supplementary Fig. 4) based on the number of steps for the addition of three types of buffers (50 mM TEABC, 0.024% DDM, and Lys-C/trypsin) to the cap for voxel collection, tissue lysis, and protein digestion. For example, 3-step wcSOP involves the first step to add 50 mM TEABC for voxel collection, the second step to add 0.024% DDM for tissue lysis, and the third step to add Lys-C/trypsin for protein digestion (Supplementary Fig. 4a). After the buffer addition, PCR tube was in the normal position for easy operation with the collected voxel hanging on the cap.
With the use of 25 µL buffer for collection of single voxels, the total processing volume is ~22 µL due to partial water evaporation/condensation to the tube wall (Supplementary Fig. 5). The voxel processing volume is in the range of 5–50 µL previously used for SOP processing of 1–100 human cells43,44. All the three wcSOP methods enabled for identification of >10,000 unique peptides and >2100 protein groups with high reproducibility from single human spleen voxels at the size of 40,000 µm2 (200 µm × 200 µm) (Supplementary Fig. 6). However, as the voxel sample processing steps reduced, the number of identified protein groups (unique peptides) was increased to ~2200 protein groups (~11,100 unique peptides) for “all-in-one” 1-step wcSOP. Consequently, the total area for the extracted ion chromatogram (XIC) was also increased from 3-step to “all-in-one” 1-step wcSOP methods.
To further improve the performance of wcSOP-MS, we have further evaluated different processing conditions of wcSOP including voxel processing positions (hanging and sitting in the PCR tube cap) and the addition of the boosting trypsin enzyme (Supplementary Fig. 7). Different from the recent report with significant improvement in the proteome coverage for the nanowell-based hanging droplet processing45, similar performance between the hanging and sitting position for voxel processing was observed for wcSOP-MS with only slight improvement in the proteome coverage for the hanging position (~7% of protein groups higher than the sitting-droplet position (Supplementary Fig. 7)). This can be attributed to interaction surface area between the collected voxels and the buffer. For wcSOP the collected voxels are fully soaked into the buffer all the time no matter which position is used for voxel processing, while for nanoPOTS after drying the voxel collection solvent DMSO there is contact surface area between the dried voxels and the chip well that cannot be accessed by the buffer45. Surprisingly, the addition of the boosting enzyme has no effect on the proteome coverage (Supplementary Fig. 7a), but it can reduce the percentage of miscleaved peptides from 21% to 13% for wcSOP-MS. Therefore, the voxel hanging position and no boosting enzyme were included in the wcSOP workflow. With all the evaluation results as well as easy convenient operation, “all-in-one” wcSOP with voxel hanging was selected as the optimal cap-assisted voxel collection and processing for single voxel proteomics analysis (Fig. 1a).
Comparison of DIA with DDA for sensitive “all-in-one” wcSOP-MS
To improve proteome coverage of “all-in-one” wcSOP-MS for single voxel proteomics, we have systematically compared label free data-independent acquisition (DIA) with DDA for analysis of different sizes of tissue voxels derived from fresh frozen human spleen tissue. Tissue voxels at the sizes of 20 µm × 20 µm (400 µm2), 50 µm × 50 µm (2500 µm2), 100 µm × 100 µm (10,000 µm2), and 200 µm × 200 µm (40,000 µm2), which are equivalent to 1 cell region, 6 cell region, 25 cell region, and 100 cell region, respectively, were collected (Supplementary Fig. 8) and processed using wcSOP followed by DIA or DDA MS analysis with 4 biological replicates per condition. For DDA MS analysis with (without) the match-between-run (MBR) function, 344 ± 99 (138 ± 53), 622 ± 172 (336 ± 111), 1542 ± 39 (1036 ± 59) and 2051 ± 18 (1708 ± 168) protein groups were identified from single voxels at the sizes of 400 µm2, 2500 µm2, 10,000 µm2 and 40,000 µm2, respectively, while DIA MS analysis with (without) MBR allows for identification of 881 ± 51 (556 ± 16), 1959 ± 227 (1282 ± 136), 4061 ± 385 (3312 ± 496), 4509 ± 40 (3832 ± 223) protein groups (Fig. 1b and Supplementary Fig. 9). It is not surprising that MBR has greatly improved the number of identified protein groups and mitigated the missing value issue especially for smaller voxels with 36% and 57% for DIA and DDA MS analysis of voxels at the size of 400 µm2, respectively (Supplementary Fig. 9a). While DIA covers almost all of proteins identified by DDA, protein abundance distribution shows that DDA detected only those in high abundant region with lower quantitation reproducibility (Fig. 1c, d). This suggests that DIA MS analysis can provide higher proteome coverage with ~2.5–3-fold more protein groups than DDA MS analysis. High reproducibility with lower CV% was observed for DIA MS analysis (Fig. 1e).
To avoid potential evaluation bias caused by the heterogeneity of biological replicates with individual voxels from different locations, we have further evaluated the comparison in proteome coverage between DDA and DIA using technical replicates from tissue voxel digests. Two large tissue voxels at the size of 500,000 µm2 from human spleen red pulp (RP) and white pulp (WP) regions were collected and processed by wcSOP, respectively. The voxel digests from each region were used to generate voxel technical replicates with the sample amount equivalent to single voxels at 200 µm × 200 µm (4 technical replicates per condition). The outperformance of DIA over DDA for small tissue voxels was further confirmed with >2-fold higher proteome coverage with high quantitation reproducibility (Supplementary Fig. 10). For those technical replicates, DDA and DIA have median CVs of ~9% and ~5%, respectively (Supplementary Fig. 10d), while for biological replicates higher median CVs were observed with 31% for DDA and 17% for DIA (Fig. 1e). Unless otherwise noted, DIA MS analysis with MBR was used for later voxel data generation to evaluate wcSOP-MS performance on different types of tissues.
Evaluation of wcSOP-MS performance using fresh frozen human spleen tissue
Fresh (snap) frozen tissue is often the first choice for MS-based proteomics analysis as well as for evaluation of method or platform performance because of its higher protein yield and less sample processing steps than other types of tissues. Different sizes of tissue voxels from fresh frozen spleen tissue were selected to evaluate wcSOP-MS performance. The spleen is the largest secondary lymphoid organ which contains highly organized and mainly compartmentalized structure of the red pulp (RP) through which the blood is continuously filtered to remove old or damaged red blood cells and the WP responsible for immune surveillance against pathogens46. The spleen immune system plays a significant role in innate and adaptive immunity47. Thus, the two distinct regions (RP and WP) having different cell types are an ideal model for evaluation of wcSOP-MS performance on fresh frozen tissues.
Spleen tissue voxels at the sizes of 400 µm2, 2500 µm2, 10,000 µm2, and 40,000 µm2 from both RP and WP regions were collected and processed by wcSOP followed by DIA MS analysis. wcSOP-MS allows for identification of 709 ± 300, 2252 ± 42, 3630 ± 208 and 4673 ± 40 protein groups from single WP voxels at the sizes of 400 µm2, 2500 µm2, 10,000 µm2 and 40,000 µm2, respectively (Fig. 2a). Similarly, 881 ± 51, 1959 ± 227, 4061 ± 385, 4509 ± 40 protein groups can be identified from single RP voxels at the sizes of 400 µm2, 2500 µm2, 10,000 µm2 and 40,000 µm2, respectively (Fig. 2a). >3000 and >30,000 unique peptides were identified from voxels at the sizes of 400 µm2 and 40,000 µm2, respectively (Fig. 2a). This strongly suggests high reproducibility of wcSOP-MS for label free proteome profiling of small tissue voxels.

a The number of identified unique peptides and protein groups at different sizes of single voxels (n = 4 biological replicates for 200 µm and n = 3 biological replicates for the other sizes; data are presented as mean values ± SD). b Proteome dynamic range from DIA quantification of single voxels at different sizes against the largest voxel size of 40,000 µm2. c PCA analysis of the commonly expressed protein abundance between single voxels from the red and white pulp regions at different sizes. d Volcano plots between single voxels from the red and white pulp regions at different sizes (DEP%: the percentage of the number of differentially expressed over total proteins). The number of down/up regulated protein groups from the red pulp region: 2/45 for 400 µm2, 815/373 for 2500 µm2, 1151/1347 for 10,000 µm2, and 2447/1361 for 40,000 µm2. DEPs are displayed with blue and red dots for upregulated proteins in white and red pulp regions, respectively (two-sided t-test, FDR < 0.05, s0 = 0.1, n = 4 biological replicates for 200 µm and n = 3 biological replicates for the other sizes). e Correlation analysis of the protein abundance ratio of the red over white pulp regions for differentially expressed proteins (DEPs) from small single voxels with the largest single voxels at 40,000 µm2. Single voxels at 400 µm2 (20 µm × 20 µm equivalent to 1 cell region), 2500 µm2 (50 µm × 50 µm equivalent to 6 cell region), 10,000 µm2 (100 µm × 100 µm equivalent to 25 cell region) and 40,000 µm2 (200 µm × 200 µm equivalent to 100 cell region) with a thickness of 10 µm were collected, processed, and analyzed using wcSOP-DIA MS with 4 biological replicates per condition. Source data are provided as a Source Data file.
For the largest voxels at 40,000 µm2 with the deepest proteome coverage, ~6 orders of magnitude dynamic range was observed, reflecting high detection sensitivity of wcSOP-MS even though a commonly accessible MS platform was used. As expected, most proteins identified from voxels at 400 µm2 are highly abundant (Fig. 2b). As the voxel size increased to 2500 µm2, moderate abundance proteins were detected, and for voxels at 10,000 µm2 low abundance proteins can be identified (Fig. 2b). Principal component analysis (PCA) of the WP and RP regions has shown distinct separation of the two regions based on protein expression alone even for ultrasmall voxels at 2500 µm2 (Fig. 2c). With increased voxel sizes the separation was gradually increased due to the increased number of differentially expressed proteins (i.e., signatures or biomarkers) (Fig. 2c). Statistical analysis has revealed 47, 1189, 2499, and 3814 protein markers with significantly differential expression between the WP and RP regions for 400 µm2, 2500 µm2, 10,000 µm2, and 40,000 µm2, respectively (Fig. 2d). Interestingly, with the voxel size increasing, the percentage of differentially expressed proteins (DEPs) (adjusted p value of <0.05) was gradually increased, reflecting large difference in proteome between RP and WP regions (Fig. 2d). To evaluate quantitation precision, we performed Pearson correlation analysis by comparing the ratio of the RP over WP abundance for DEPs at smaller voxel sizes with the largest voxel size of 40,000 µm2. High Pearson correlation coefficients of 0.88 and 0.94 were observed for voxel sizes at 2500 µm2 and 10,000 µm2, respectively. However, for the smallest voxels at the size of 400 µm2 with only 29 overlapping DEPs (Supplementary Fig. 11b), there is positive correlation but with a moderate correlation coefficient (Fig. 2e). This can be attributed to unreliable protein quantitation due to the lack of sufficient sensitivity for ultrasmall tissue voxels. In addition, we have compared our spleen tissue voxel data with one recent bulk study for in-depth proteome map of the human body including spleen tissue. ~90% of proteins from spleen tissue voxels were observed in the spleen proteome with the total ~11,000 proteins48. Among these overlapped proteins 274 proteins are spleen tissue specific proteins, which accounts for 81% of all spleen tissue specific proteins48. All these results demonstrate the reproducibility, quantitation dynamic range, and quantitation accuracy of wcSOP-MS for robust single voxel proteomics analysis.
Single voxel proteomics for revealing region-specific markers and signaling pathways
Using wcSOP-DIA for single voxel proteomics analysis, 1189, 2499, and 3814 of protein signatures (biomarkers) can be identified for enabling differentiation of two distinct RP and WP regions for tissue voxel sizes at 2500 µm2, 10,000 µm2, and 40,000 µm2, respectively (Fig. 2d). They include many cell surface CD protein markers (Fig. 3a), and some CD markers are known to be specific to certain cell types within the two regions (e.g., B cell markers: CD20, CD22; T cell markers: CD4, CD8A; macrophage marker: CD163). CD20, CD22, and CD44 were upregulated in the WP region, whereas CD8A, CD4, CD47, and CD163 upregulated in the RP region (Fig. 3a and Supplementary Data 1). This observation is consistent with region-specific cell type distribution and function difference between the RP and WP regions.

a Differentially expressed CD protein markers between the red and white pulp regions (two-sided t-test, FDR < 0.05, s0 = 0.1, n = 4 biological replicates). b Enriched signaling pathways between the red and white pulp regions (FDR < 0.05). c Antibody-based CODEX image of human spleen tissue slide to validate label-free DIA MS quantitation: CD8 (yellow), CD20 (red), CD4 (green), and CD163 (cyan). Prior to image data generation, CODEX imaging was extensively optimized with multiple combinations of antibodies on human spleen tissue slides to ensure high reproducibility. Source data are provided as a Source Data file.
RP is a loose spongy tissue with chords of reticular cells located between venous sinuses that primarily contain erythrocytes, lymphocytes, macrophages, granulocytes, and plasma cells. Unique to human spleen red pulp, CD8A lines these sinuses, termed littoral cells49. Senescent erythrocytes are destroyed by macrophages with hemoglobin:haptoglobin complexes being loaded onto CD163 positive macrophages for metabolic processing. Thus, CD8A, haptoglobin and CD163 were observed to have higher expression levels in the RP regions (Fig. 3a). By contrast, as a B cell specific membrane protein and an attractive target for therapeutic antibodies50, CD20 protein has higher expression level in the WP region. All these results demonstrate that single voxel proteomics can precisely reveal functionally important proteins across different regions at high spatial resolutions.
We have further performed KEGG pathway analysis of DEPs between the RP and WP regions. Several signaling pathways were enriched and closely related to the WP or RP functions (Supplementary Fig. 12). One example is the B-cell receptor (BCR) signaling pathway, which is known to be crucial for normal B cell development and adaptive immunity as well as for B-cell malignancies51. B cell activation begins with initiation of signaling pathways (e.g., NFAT, NF-κB, and MAPK) and endocytosis of the BCR-antigen complex52. Our single voxel proteomics analysis can cover 56 BCR pathway proteins among the total 78 proteins with 30 proteins upregulated and 8 proteins downregulated in the WP region (Fig. 3b). One important upregulated protein in the BCR pathway is spleen tyrosine kinase (SYK), which is known to have a crucial role in adaptive immune receptor signaling53. SYK was a cytosolic non-receptor protein tyrosine kinase recognized as a critical element in the BCR signaling pathway and its phosphorylation activates downstream signaling pathways. Several oral Syk inhibitors are being assessed in clinical trials for multiple malignancies54. This suggests that with its deep proteome coverage wcSOP-based single voxel proteomics can provide high spatial resolution mapping of signaling pathways and improve our understanding of tissue microenvironment.
Validation of label-free MS quantification using CODEX imaging
As mentioned above, many cell surface CD protein markers can be detected and quantified from human spleen tissue voxels using wcSOP-MS. To further validate label-free DIA MS quantitation, we have performed an antibody-based multiplexed CODetection by indexing (CODEX) approach for imaging four representative region-specific protein markers, CD20, CD4, CD8A, and CD163. CODEX imaging of human spleen tissue has shown that CD20 is highly expressed in the WP region, whereas CD4, CD8A, and CD163 have higher expression levels in the red pulp region (Fig. 3c). The results from CODEX imaging are consistent with the data from label-free DIA MS quantification (Figs. 2d and 3a), which confirmed reliable single voxel proteomics analysis with wcSOP-MS.
Application of wcSOP-MS for 2D proteome mapping of human spleen tissues
To further demonstrate its robustness for single voxel proteomics, we next applied wcSOP-MS for 2D proteome mapping of human spleen tissues at 100 µm spatial resolution. A total of 42 consecutive voxels covering one WP and its surrounding RP region were dissected by LCM in the grid mode and collected into PCR tube caps (Fig. 4a). In total ~6000 protein groups were detected and quantified across the 42 voxels with an average of ~3500 protein groups and ~16,000 precursors per voxel (Fig. 4b). Region-specific proteins were observed to have distinct expression patterns in the spleen tissue maps. For example, a B-cell marker CD20 was detected only in the WP region but barely detected in the adjacent RP regions (Fig. 4c). By contrast, a macrophage marker CD163, known to play a critical role in red blood cell clearance, was primarily detected in the RP regions (Fig. 4c). Global PCA analysis of all the 42 voxels was performed based on the abundance of 2837 proteins quantified in at least 70% of the voxels. There are two distinct clusters corresponding to WP and RP regions (Fig. 4d). Without guidance by staining or microscopic image, PCA analysis enabled to precisely annotate 12 and 24 voxels into WP and RP regions, respectively, and the other 6 voxels as transition between WP and RP regions. Comparative analysis of protein abundance from annotated WP with RP voxels has shown that region-specific immune related pathways were enriched (Fig. 4e), reflecting the power of spatial proteome mapping. We have further generated the pathway maps based on the abundance of proteins associated with the pathways (Fig. 4f). All these results demonstrate the robustness of wcSOP-MS for spatial proteome mapping of tissues.

a Tissue sections before, during, and after LCM collection. b The number of quantified protein groups for each voxel across the mapping region. c The normalized protein expression across the mapping region for region-specific protein markers. d Principal component analysis (PCA) of all the tissue voxels based on protein expression. Data point colors: Blue, red and green correspond to white pulp (WP), red pulp (RP) and the mixed regions from the histology view, respectively. e Signaling pathway enrichment based on differentially expressed proteins between WP and RP regions (FRD < 0.05). f 2D maps of the enriched immune signaling pathways using the average abundance of proteins involved in these pathways. In total 42 voxels at the size of 100 µm × 100 µm × 10 µm were collected to cover one WP and adjacent RP regions, and then processed by wcSOP followed by DIA-MS analysis. Source data are provided as a Source Data file.
Evaluation of wcSOP-MS applicability to OCT-embedded breast cancer tissue
With demonstrated performance of wcSOP-MS in fresh frozen healthy human spleen tissue, we next evaluated its applicability to OCT (optimal cutting temperature)-embedded triple negative breast cancer tissue. As one common method for perseveration of fresh frozen tissues, OCT embedding enables preserving molecular structure in a regular shaped block without drying for easy cutting into thin slices. However, signals from polymers polyvinyl alcohol and polyethylene glycol which account for over 14% of OCT ingredients can severely interfere with MS analysis, causing strong ion suppression in peptide detection55. Thus, prior to wcSOP-MS analysis we evaluated different tissue slice washing conditions and the optimal condition (Methods) was determined and then used for analysis of voxels from OCT-embedded triple negative breast cancer (TNBC) tumor tissue.
Breast cancer is the second leading cause of cancer death in women in the United States56. TNBC is an aggressive heterogenous subtype of breast cancer with genetic abnormalities associated with high rates of local recurrence and a propensity for distant metastases. It accounts for ~12% of all breast cancers lacking treatment regimes57. A TNBC tissue block was cryosectioned to generate a series of slices with a 10 µm thickness. Hematoxylin and eosin (H&E) staining was used to identify tumor and adjacent normal regions (Fig. 5a). Voxels at the size of 100 µm × 100 µm from the two regions were collected and processed using wcSOP followed by DIA MS analysis. Large variations in the number of identified proteins were observed for these voxels (Supplementary Table 3), suggesting high intratissue heterogeneity. Voxels with high proteome coverage (9 voxels with >1800 protein groups for tumor region and 3 voxels with >1000 protein groups for normal region) were selected for comparative proteomic analysis because they have high protein content and are presumably more representative of their regions. An average of 2199 ± 192 and 1613 ± 369 protein groups were identified and quantified from tumor and adjacent normal regions, respectively (Fig. 5b). The proteome coverage from breast tissue voxels is lower than that from human spleen tissue voxels probably due to high fat composition in breast tissue and/or certain levels of ion suppression from OCT ingredients. To evaluate whether the identified proteins can be used to differentiate the two regions, we performed PCA. The tumor and adjacent normal regions were clearly segregated based on the protein expression alone with biological replicates from the same regions being clustered together (Fig. 5c). To identify protein signatures distinguishing the two regions, analysis of variance (ANOVA) was performed with a volcano plot of DEPs (Fig. 5d), revealing ~1000 proteins to be significantly different with p < 0.05. Among the ~1350 proteins, 1220 and 132 proteins were highly expressed in tumor and adjacent normal regions, respectively (Supplementary Data 2).

a The images of TNBC tumor and adjacent normal tissues using H&E staining. H&E staining was performed on multiple TNBC slides to ensure high reproducibility and the H&E stained slide with the highest cell counts was selected for wcSOP-MS analysis. b The number of identified protein groups from tumor and normal regions (9 biological replicates for tumor region with >1800 protein groups per voxel and 3 biological replicates for normal region with >1000 protein groups per voxel; data are presented as mean values ± SD). c PCA analysis of the commonly expressed protein abundance between single voxels from tumor and normal regions. d Volcano plots between single voxels from tumor and normal regions. DEPs are displayed with blue and red dots for upregulated proteins in normal and tumor regions, respectively (two-sided t-test, FDR < 0.05, s0 = 0.1, n = 3 biological replicates for normal region and n = 9 biological replicates for tumor region). e Differential expression of known breast cancer protein markers (CD14, CD44, PLXNB2, EGFR) between the tumor and normal regions (n = 3 and 9 biological replicates for normal and tumor regions, respectively). The p value is estimated by Welch’s t-test (two-sided). For unequal sample sizes (3 normal versus 9 tumor biological replicates), the box plots show floating bars from minima to maxima along with solid lines for medians. f Immunohistochemistry (IHC) images of normal and tumor regions, stained for CD14, CD44, PLXNB2, and EGFR. Scale bar = 50 µm. Source data are provided as a Source Data file.
To further validate label-free DIA quantification of OCT-embedded tissue voxels, we performed antibody-based IHC analysis of four representative proteins CD44, EGFR, PLXNB2 and CD14 upregulated in the tumor region (Fig. 5e). Selection of the four proteins was based on literature reports that they are closely associated with breast cancer. For example, the CD44 expression level in breast cancer was correlated with both poor and favorable outcomes58. EGFR overexpression in breast cancer is associated with large tumor size, poor differentiation, and poor clinical outcomes. EGFR is more frequently overexpressed in aggressive TNBC59,60. Plexin-B2 (PLXNB2) was reported to be upregulated in breast cancer with a role of promoting tumor growth and invasion61. Results from IHC staining (Fig. 5f) are in agreement with the data from label-free DIA quantification (Fig. 5e and Supplementary Fig. 13), which confirmed reliable label-free quantification of OCT-embedded tissue voxels with wcSOP-MS. All these results demonstrate the applicability of wcSOP-MS for proteome profiling of OCT-embedded human tissues at high spatial resolution.
Evaluation of wcSOP-MS applicability to FFPE AD patient brain tissue
To further evaluate its broad utility, we applied wcSOP-MS to human FFPE (formalin fixed paraffin embedded) tissue for single voxel proteomics analysis. FFPE preservation of tissues is routinely used for archived samples in clinical settings because of the low-cost sample storage. FFPE tissues can be stored at room temperature for decades, and thus there are many large repositories of FFPE tissues available for retrospective studies with associated patient clinical data. As an initial demonstration, FFPE tissue from AD patient was selected because it is of particular interest to study amyloid plaque microenvironment (e.g., amyloid plaque region (APR) and adjacent nonplaque region (NPR)) for improved understanding of molecular mechanisms underlying AD pathogenesis.
Thioflavidin staining of amyloid plaques was used to identify APR and adjacent NPR for voxel generation with LCM (Fig. 6a). Using wcSOP-MS, 2836 ± 323 and 3055 ± 58 protein groups were identified from APR and NPR voxels (5 pooled voxels at 100 µm × 100 µm × 10 µm for each APR or NPR) with 4 biological replicates per region, respectively (Fig. 6b). When compared to one recent study of amyloid plaque proteome in which ~2250 proteins were identified from ~750 pooled FFPE amyloid plaques (the total volume of 2 mm2 × 8 µm)32, wcSOP-MS has higher sample processing efficiency and detection sensitivity because ~160-fold less protein amount was used for spatial proteomics analysis. Among the quantified proteins many proteins are known to be associated with AD (e.g., APP, tau protein, APOE4, and GFAP). PCA analysis has shown that the two regions, APR and NPR were clearly separated with the 4 replicates from the same region being clustered together (Fig. 6c). To identify protein signatures distinguishing APR and NPR, we performed ANOVA analysis with a volcano plot of DEPs (Fig. 6d), revealing 183 proteins to be significantly different with p < 0.05. Among the 183 proteins, 110 and 73 proteins were more highly expressed in APR and NPR, respectively (Supplementary Data 2). We further performed KEGG signaling pathway analysis of DEPs, resulting in enrichment of AD associated pathways, such as extracellular matrices, focal adhesion and PI3-Akt signaling62 for upregulated proteins in APR and cAMP signaling63, GABAergic synapse64 and retrograde endocannabinoid signaling65 for downregulated proteins in APR (Fig. 6e). Protein network analysis suggests that all these pathways highly interact with each other through AD associated proteins APP, APOE4 and MAPT (Fig. 6f). Box plots showed differential expression profiles between APR and NPR for several selected proteins such as COL6A2, TNC and ITGB6 upregulated in APR and NDUFS6, PRKACB and GABRB3 downregulated in APR (Fig. 6g).

a Thioflavidin staining to identify amyloid plaques for single voxel collection. In the image of thioflavin staining the amyloid plaque containing region framed with square shape was defined as “APR” and the adjacent non-plaque region was defined as “NPR”. b The number of identified protein groups for 5 × single voxels from APR and NPR (APR: amyloid plaque region; NPR: non-plaque region) with an average of 2805 and 3024 protein groups and median CVs of 11% and 2% for APR and NPR, respectively (n = 4 pooled biological replicates for APR and NPR; data are presented as mean values ± SD). c PCA analysis of the commonly expressed protein abundance for 5 × single voxels between APR and NPR. d Volcano plots between 5 × single voxels from APR and NPR (p < 0.01 and ≥2-fold change). DEPs are displayed with blue and red dots for upregulated proteins in NPR and APR, respectively (two-sided t-test, FDR < 0.05, s0 = 0.1, n = 4 pooled biological replicates for NPR and APR). e The enriched signaling pathways between APR and NPR via KEGG pathway analysis (FDR < 0.05). f Network analysis of differentially expressed proteins. Except the middle AD associated proteins APP, APOE4, and MAPT, the network proteins at the left and the right sides were upregulated in NPR and APR, respectively. g Examples of 6 differentially expressed protein signatures between APR and NPR (n = 4 pooled biological replicates for APR and NPR). The p value is estimated by Welch’s t-test (two-sided). The box plots define the range of the data (whiskers), 25th and 75th percentiles (box), and medians (solid line). Outliers are plotted as individual dots outside the whiskers. Source data are provided as a Source Data file.
We also performed wcSOP-MS analysis of single voxels at 200 µm × 200 µm from APR and NPR regions (4 biological replicates per region). ~3000 protein groups were identified from each voxel with clear separation between APR and NPR regions (Supplementary Fig. 14), which is consistent with the results from 5 pooled voxels at the size of 100 µm × 100 µm. However, there are much less DEPs between APR and NPR when compared to the pooled voxels at 100 µm spatial resolution (Supplementary Fig. 14). A total of 97 proteins were found to be differentially expressed between APR and NPR with 93 and 4 more highly expressed in APR and NPR, respectively. This can be attributed to much lower percentage of APR for voxels containing APR at 200 µm than 100 µm spatial resolutions. Based on an average area of ~3000 μm2 per amyloid plaque30 and one amyloid plaque per voxel, the estimated percentages of APR over the whole voxel regions are ~7.5% for single voxels at 200 µm × 200 µm and ~30% for the pooled voxels at 100 µm × 100 µm. This result reflects the importance of high spatial resolution in proteome-wide mapping of human tissues. All these results demonstrate that wcSOP-MS can be applied to voxels from FFPE tissues. Further optimization of wcSOP-MS is needed to improve proteome coverage for proteomic analysis of voxels from FFPE tissues.
Discussion
Recent advances in sample preparation and MS instrumentation make MS-based single cell proteomics feasible43,66,67,68,69,70, but the lack of spatial context limits its use for high-resolution comprehensive characterization of tissue microenvironment to improve our understandings of spatial biology. MS-based spatial proteomics has been broadly used for analysis of 100 s of subpopulations of cells and voxels from regions of interest21,23,26. For example, Deep Visual Proteomics (DVP), a spatial technology which combines imaging, cell segmentation, LCM and MS into a single workflow33, was recently developed for reliable identification of cellular phenotypes through pooling the enough number of LCM-dissected cells with similar morphological features and staining patterns. However, proteome profiling of single voxels at high spatial resolution is still challenging primarily due to the need for precise collection and effective processing of single voxels for proteomic analysis. So far there are only three spatial proteomics methods reported for single voxel proteomics analysis36,39,71 but all of them use specialized devices for collection and processing of single voxels. To address this challenge, we have developed a convenient robust “all-in-one” wcSOP-MS method for sensitive label-free single voxel proteomics, where single voxels are collected and processed on the easily accessible PCR tube cap.
wcSOP is built on the same “all-in-one” concept as SOP. The major difference between wcSOP and SOP is that for wcSOP collection and processing of single voxels occur on the PCR tube cap, while for SOP the PCR tube is used for collection and processing of single cells. Another major difference is the collection and processing volume with ~25 µL for wcSOP and ~5 µL for SOP. The selection of ~25 µL for wcSOP is to fully cover the dome-shaped PCR tube cap to form a large droplet that can provide the largest surface area for robust collection of single voxels with ~100% success rate. In addition, unlike conventional tube cap-based methods with voxel collection on the PCR tube cap and processing in the PCR tube and with multiple steps for processing and transfer, “all-in-one” wcSOP on the PCR tube cap is one-pot collection and processing, resulting in greatly improved robustness and reproducibility for single voxel proteomics.
The performance of wcSOP-MS (e.g., robustness, sensitivity, reproducibility, and quantitation accuracy) was demonstrated by label-free MS analysis of small tissue voxels from 400 µm2 (20 µm × 20 µm equivalent to 1 cell region) to 40,000 µm2 (200 µm × 200 µm equivalent to 100 cell region) at a thickness of 10 µm. Using a commonly accessible Q Exactive plus platform, wcSOP-MS can identify ~900, ~2100, ~3800 and ~4600 protein groups from single voxels at the sizes of 400, 2500, 10,000, and 40,000 µm2, respectively. The number of identified protein groups from wcSOP is comparable to that from the specialized devices (e.g., nanoPOTS38 and DVP33,34,35) (Supplementary Table 4), suggesting high efficiency single voxel collection and processing. For example, using nanoPOTS-MS for label free single voxel proteomics, ~1300, ~2000, and ~2800 protein groups were identified from single mouse tissue voxels at the sizes of 2500, 10,000, and 40,000 µm2, respectively38. Using more advanced MS platforms, the number of identified protein groups from wcSOP-MS could be significantly increased even with a shorter active gradient time, which was supported by our recent comparative analysis of mouse brain tissue voxels (Supplementary Fig. 15).
Intrigued by two recent reports for identification of ~90 and ~500 phosphopeptides from single cells using direct MS analysis without phospho-enrichment72,73, we also performed database searching for phosphopeptides from our direct DIA-MS data for single spleen tissue voxels at different sizes. Similar to the findings from the two recent reports, >400 and ~1800 phosphopeptides were seemly identified from single voxels at 20 µm × 20 µm (400 µm2, ~1 cell region) and 200 µm × 200 µm (40,000 µm2, ~100 cell region), respectively (Supplementary Fig. 16a, b). Pairwise comparison analysis has indicated that >50% of phosphopeptides have a significant abundance difference between the red and WP regions of human spleen tissue (Supplementary Fig. 16c). However, the overlap of phosphopeptides between direct DIA-MS analysis of single voxels and IMAC-MS analysis of bulk spleen tissue slides is low, with only 15 and 36 phosphopeptides for the voxel sizes at single-cell and 100-cell levels, respectively (Supplementary Fig. 17). As IMAC-based phospho-enrichment followed by MS analysis is considered as gold standard for phosphoproteomic analysis74,75, this suggests that phosphoproteomics analysis from direct DIA-MS data is not reliable, and more stringent criteria need to be implemented in the database searching. Such unreliable identification would not be surprising due to: (1) the low stoichiometry of protein phosphorylation (~1% of phosphorylation in the entire protein amount or ~1% of phosphopeptides over the total peptides76), (2) ion suppression by abundant unmodified peptides, and (3) lower ionization efficiency (2 negative charges on a phosphate group) than unmodified counterparts75,77,78.
When compared to other voxel collection devices (e.g., standard 384 well plate with a diameter of ~3.1 mm or nanoPOTS chip with a diameter of ~1.2 mm36,37,38), the dome-shaped PCR tube cap has a much wider diameter of ~5.4 mm, suggesting that the tube cap has much larger collection surface area to ensure robust precise collection of single voxels. By contrast, higher LCM precision is required for voxel collection using the 384 well plate or the nanoPOTS chip. Furthermore, due to the tight sealing for the dome-shaped PCR tube cap, wcSOP can be broadly applied to single voxels from different types of tissues (fresh frozen, OCT-embedded, and FFPE) with operation at a wide range of temperatures. wcSOP-MS enabled to spatially resolve different regions at the single voxel level with identification of 100 s of protein signatures (biomarkers) and revealing signaling pathways closely linked to region-specific biology. Most importantly, unlike currently available label-free single voxel proteomics methods that need specific devices and are difficult to access by research community, wcSOP-MS can be easily adopted for broad applications because commercially available low-cost dome-shaped PCR tube caps are used for voxel collection, and it is simple to operate only by heating at 75 °C for 1 h and 37 °C for overnight without other additional steps. Besides for single voxel proteomics, wcSOP-MS can also be applied for reproducible analysis of small numbers of voxels with high sample recovery, which could greatly improve the robustness of the conventional cap-assisted methods for spatial proteomics. Therefore, wcSOP-MS represents a significant advancement for label-free MS-based single voxel proteomics.
With its demonstration for label-free MS analysis, wcSOP-MS can be equally used for other types of single voxel proteomics analysis (e.g., targeted spatial proteomics and TMT-based sample multiplexing). Future developments will focus on improvements in detection sensitivity and sample throughput for rapid deep proteome profiling of single tissue voxels. Enhancing detection sensitivity could be achieved by effective integration of ultralow-flow LC or capillary electrophoresis (CE) and a high-efficiency ion source/ion transmission interface with the most advanced MS platform. In contrast to our currently used nanoLC-Q Exactive plus MS platform significant improvement in proteome coverage for single voxel proteomics is expected, which was strongly supported by 2–3-fold improvement in the single-cell proteome coverage from a newly released Orbitrap Astral MS platform. Sample throughput could be increased by replacement of single tube cap with parallel eight tube caps for tissue voxel collection and processing (Supplementary Fig. 18). Our initial evaluation is highly promising with 8-fold improvement in wcSOP. Sample throughput could also be increased by using ultrafast high-resolution ion mobility-based gas-phase separation (e.g., SLIM79) to replace current slow liquid-phase (LC or CE) separation, and effective integration of liquid- and gas-phase separations (e.g., SLIM79 or FAIMS80) for greatly reducing separation time but without trading off separation resolution. Alternatively, sample multiplexing with barcoding (e.g., mTRAQ or dimethyl labeling for multiplex DIA34,69,81 and TMT-based BASIL for isobaric labeling68,82,83) and implementation of a multiple LC column system84,85 can also be considered to increase sample throughput. All these improvements could lead to more powerful wcSOP-MS for high spatial resolution proteome mapping of human tissues.
In summary, we report an easily implementable wcSOP-MS method that capitalizes on wet cap collection of single microscale voxels and one-pot voxel processing for robust, sensitive, label-free single voxel proteomics. With its convenient features (e.g., low-cost PCR tube cap and surfactant) and broad applicability to different types of tissues as well as its being easy to use, wcSOP-MS can be readily implemented in any MS laboratory where LCM and MS are available. Its quantitation accuracy was validated with traditional antibody-based imaging methods. wcSOP-MS may open an avenue for routine single voxel proteomics and spatial proteomics.
Methods
Reagents
Triethylammonium bicarbonate (TEABC), trifluoroacetic acid (TFA), formic acid (FA), acetonitrile (ACN), and n-Dodecyl β-D-maltoside (DDM), thioflavin, hematoxylin and eosin were obtained from Sigma (St. Louis, MO). Ethanol was purchased from Decon Labs, Inc. (King of Prussia, PA). Water was processed using a Millipore Milli-Q system (Bedford, MA). Promega trypsin gold was purchased from Promega Corporation (Madison, WI), and Lys-C was purchased from FUJIFILM Wako (Richmond, VA).
Human tissue block collection
The fresh (snap) frozen human spleen tissue was collected from an organ donor under informed consent of the Human Biomolecular Atlas Program of the National Institutes of Health Common Fund, which received ethical approval, and the study protocol (IRB201600029) was approved by the University of Florida Institutional Review Board. The OCT (optimal cutting temperature)-embedded human breast tissue from one triple negative breast cancer patient was collected under guidelines from the Institutional Review Board at Northwestern University (IRB protocol STU00203283) in compliance with NIH human subject study guidelines. All patients have provided written informed consent. The FFPE (formalin fixed paraffin-embedded) brain tissue block from the temporal region of one AD patient was purchased from AMSBio (Abingdon, UK). Both fresh frozen and OCT-embedded tissue blocks were stored at −80 °C freezer, while the FFPE AD tissue block was stored at 4 °C freezer until further processing.
Cryosectioning of fresh frozen, OCT-embedded, FFPE tissue blocks
The fresh frozen spleen tissue block was mounted on a cryomicrotome chuck by freezing a small droplet of water and then sectioned using a blade temperature of −16 °C and specimen temperature of −23 °C. Ten micrometer-thick sections were thaw-mounted onto polyethylene naphthalate (PEN) membrane slides (Zeiss) for subsequent LCM analysis. Cryosectioning was performed using CryoStar NX70 from Epredia. The tissue section was washed and dehydrated by immersing slides in the gradient of ethanol solutions for 30 s for each change (70% ethanol, 95% ethanol, and 100% ethanol, respectively). After the final ethanol wash, slides were naturally dried in a fume hood, vacuum sealed, stored in a slide holder (5-slide per set), and then stored in a −80 °C freezer for further LCM experiment.
Similarly, OCT-embedded triple negative breast cancer tissue block was cryosectioned to 10-µm thick sections using a blade temperature −35 °C and specimen temperature of −25 °C then thaw and mounted onto PEN membrane slides. The OCT compound was removed from the tissue sections by immersing slides into 50% ethanol for 30 s followed by a gradient of ethanol solutions (70%, 96% and 100%) for 30 s each change to fix the tissue sections. After the final ethanol wash, slides were naturally dried in a fume hood, stored in a slide holder (5-slide per set), and then stored in a −80 °C freezer for further LCM experiment.
FFPE AD tissue block was sectioned by microtome into 10-µm thick sections. Sections were temporarily floated on 42 °C deionized water to remove the wrinkles and then picked onto PEN membrane slides. Sections were dried vertically on the iron platform, and then remove the paraffin by immersing slides into a gradient of ethanol solutions (70%, 96%, and 100%) for 30 s each change and xylene for 30 s. After the final xylene wash, slides were naturally dried in a fume hood, stored in a slide holder (5-slide per set), and then stored in a −80 °C freezer for further LCM experiment.
Laser capture microdissection (LCM) of tissue sections
Prior to LCM experiments, for wet collection a dome-shaped cap of PCR tube (0.2 ml of PCR individual tubes with domed cap, GeneMate) was prepopulated with a 25 µL buffer droplet, and for dry collection a PCR tube cap with adhesive polymer membrane was used. LCM was performed on a PALM MicroBeam system (Carl Zeiss MicroImaging, Munich, Germany). Voxelation of the tissue section was achieved by selecting the area on the tissue using PalmRobo software, followed by tissue cutting and catapulting. Tissue sections containing two distinct regions (red vs white pulps for human spleen tissue, tumor vs adjacent normal regions for triple negative breast cancer tissue, and amyloid plaque vs surrounding normal regions for AD patient tissue) were cut at an energy level of 63 and with an iteration cycle of 1 to completely separate tissue voxels at different sizes with a thickness of 10 µm. The “CenterRoboLPC” function with an energy level of delta 20 and a focus level of delta 5 was used to catapult tissue voxels into the cap. The “CapCheck” function was activated to confirm successful sample collection from tissue sections to adhesive polymer membrane or buffer droplet. After tissue voxel collection, the collected tissue voxels were either processed directly or stored at −80 °C until use.
Tissue section staining
Prior to LCM, H&E staining of breast tissue sections were performed to assist selection of regions of interest. The detailed procedure was as follows: 70% ethanol (30 s), distilled water (30 s), hematoxylin (30 s), distilled water (30 s), 70% ethanol (30 s), eosin (1 min), 95% ethanol (1 min), 95% ethanol (1 min), 100% ethanol (2 min), 100% ethanol (2 min) and xylene (3 min).
Thioflavin staining of amyloid plaques in AD patient brain tissue section was performed as follows: 100% ethanol (1 min), 95% ethanol (1 min), 80% ethanol (1 min), 70% ethanol (1 min), distilled water (5 s), 1% Thioflavin S (1 min), distilled water (1 min), 70% ethanol (1 min), 80% ethanol (1 min), 95% ethanol (1 min), 100% ethanol (1 min), 100% ethanol (1 min) and xylene (1 min).
Single tissue voxel lysis, denaturation, and digestion
For “all-in-one” dry cap-assisted SOP termed dcSOP, after single voxel collection 10 µL of a cocktail of buffer containing 0.1% DDM in 50 mM TEABC, 20 ng of Lys-C, and 80 ng of trypsin was added to the adhesive cap with the enzyme concentrations of 2 ng/µL for Lys-C and 8 ng/µL for trypsin. Unlike the dome-shaped PCR tube, tightly sealing the adhesive cap to the PCR tube requires using thermoplastic parafilm to warp the tube at multiple times. The inverted tube was incubated at 75 °C for 60 min for tissue lysis and protein denaturation followed by incubation at 37 °C for overnight (∼16 h) for protein digestion. The voxel digests were then centrifuged at 2000 × g for 5 min and transferred down to the bottom of the PCR tube. 1 µL of a mixture of boosting enzyme containing 10 ng/µL Lys-C and 40 ng/µL trypsin was added to the tube for further digestion for 5 h. After digestion, 3 µL of 5% TFA was added to the PCR tube to stop the enzyme reaction. The PCR tube was then inserted into the LC vial with the removal of the cap and the voxel digests were either directly analyzed or stored at −20 °C for later LC-MS analysis.
For “all-in-one” wet cap-assisted SOP termed wcSOP, prior to single voxel collection 25 µL of a cocktail of buffer containing 0.024% DDM, 0.8 ng/µL Lys-C, and 3.2 ng/µL trypsin in 50 mM TEABC was added to the dome-shaped PCR tube cap. After single voxel collection, the PCR tube was incubated at 75 °C for 60 min for tissue lysis and protein denaturation followed by incubation at 37 °C for overnight (∼16 h) for protein digestion in a hanging droplet position (i.e., the normal position of the PCR tube). The voxel digests were then centrifuged at 2000 × g for 5 min and transferred down to the bottom of the PCR tube. 1 µL of a mixture of boosting enzyme containing 10 ng/µL Lys-C and 40 ng/µL trypsin was added to the tube for further digestion for 5 h. After digestion, 3 µL of 5% TFA was added to the PCR tube to stop the enzyme reaction. The sample digests were partially dried to reduce the volume down to ~20 µL (lower than the LC sample loop) using a SpeedVac concentrator. The PCR tube was then inserted into the LC vial with the removal of the cap and the voxel digests were either directly analyzed or stored at −20 °C for later LC-MS analysis. The detailed workflows for other methods were presented as Supplementary Information.
Immunohistochemistry (IHC)
Immunohistochemical studies were performed on 10 µm frozen sections (OCT-removed) of breast tumor and normal tissues on charged slides by using the Leica Bond-Max Autostainer. The following steps were employed by using Leica Bond Polymer Refine Detection Kit (DS9800) with first Peroxide Block for 5 min, followed by use of antibodies for 15 min [Antibodies used in this study: EGFR, Santa Cruz (sc373746) 1:250; CD14, Sigma Prestige (cat#AMAB90897) 1:250; PlexinB2, Proteintech (cat#10602-1-AP) 1:800; CD44, Dako (cat#M7082) 1:100]. Post Primary and Polymer steps were applied for 8 min, DAB Refine mixes for 10 min, followed by Hematoxylin for 5 min. All slides were rehydrated through alcohol and xylene, mounted, and cover slipped. Stained slides were scanned using digital slide scanner (Nanozoomer 2.0-HT, Hamamatsu Photonics), which mainly converts stained slides at high speed into high-definition digital images. Standard-size (26 mm × 76 mm) slides, with fully automatic operation mode, were scanned with 20 × (0.46 µm/pixel). Scanned images were then viewed with NDP.view2 next-generation viewer software for further pathologic review and quantitative analysis.
CODEX staining and imaging
Barcoded antibody staining of tissue sections mounted on cover slips was performed using a commercially available CODEX Staining Kit according to the manufacturer’s instructions (https://www.protocols.io/view/hubmap-tmc-uf-validation-of-custom-conjugated-anti-81wgbpmjnvpk/v1). Raw images were processed and stitched using CODEX Processor software including cycle alignment, drift compensation, background subtraction, and cell segmentation. Image analysis was performed with the Akoya Multiplex Analysis Viewer in Fiji with KNN/FLANN clustering, gating and spatial network mapping.
LC-MS/MS analysis
Data-dependent acquisition (DDA) on Q Exactive Orbitrap MS
The single tissue voxel digests were analyzed using a commonly available Q Exactive Plus or Q Exactive HF Orbitrap MS (Thermo Scientific, San Jose, CA). Thermo Scientific™ Xcalibur™ software was used to collect MS data. The standard LC system consisted of a PAL autosampler (CTC ANALYTICS AG, Zwingen, Switzerland), two Cheminert six-port injection valves (Valco Instruments, Houston, USA), a binary nanoUPLC pump (Dionex UltiMate NCP-3200RS, Thermo Scientific), and an HPLC sample loading pump (1200 Series, Agilent, Santa Clara, USA). Both SPE precolumn (150 µm i.d., 4 cm length) and LC column (75 µm i.d., 25-cm Self-Pack PicoFrit column, New Objective, Woburn, USA) were slurry-packed with 3-µm C18 packing material (300-Å pore size) (Phenomenex, Terrence, USA). Sample was fully injected into a 20 µL loop and loaded onto the SPE column using Buffer A (0.1% FA in water) at a flow rate of 5 µL/min for 20 min. The concentrated sample was separated at a flow rate of 200 nL/min and a 75 min gradient of 8-35% Buffer B (0.1% FA in ACN). The LC column was washed using 80% Buffer B for 10 min and equilibrated using 2% Buffer B for 20 min. Q Exactive Plus Orbitrap MS (Thermo Scientific) was used to analyze the separated peptides. A 2.2 kV high voltage was applied at the ionization source to generate electrospray and ionize peptides. The ion transfer capillary was heated to 250 °C to desolvate droplets. The DDA mode was employed to automatically trigger the precursor scan and the MS/MS scans. Precursors were scanned at a resolution of 35,000, an AGC target of 3E6, a maximum ion trap time of 100 ms. Top-10 precursors were isolated and fragmented with an isolation window of 2 m/z, an AGC target of 5E5, a maximum ion injection time of 500 ms for 400 μm2, 300 ms for 2500 μm2, 200 ms for 10,000 μm2, and 100 ms for 40,000 μm2 tissue size were used and fragmented by high energy collision with an energy level of 32%. A dynamic exclusion of 40 s was used to minimize repeated sequencing. MS/MS spectra were scanned at a resolution of 17,500.
Data-independent acquisition (DIA) on Q Exactive Orbitrap MS
For DIA analysis, the MS/MS scan was performed in the HCD mode with the following parameters. Precursor ions from 400 to 900 m/z were scanned at a resolution of 35,000 for MS1 with maximum ion injection time of 125 ms and an AGC target of 1E6. A fixed precursor isolation window of 25 m/z with 1 Da overlap was used to acquire DIA MS/MS data at a resolution of 35,000, maximum ion injection time of 125 ms, and an AGC target of 1E6. The isolated ions were fragmented with HCD at 30% level with a loop count of 20 and the fixed first mass at 200 for fragment ions on Q Exactive Plus Orbitrap MS (Thermo Scientific). For DIA analysis on Q Exactive HF Orbitrap MS (Thermo Scientific), similar parameters were used except the resolutions at 60,000 for MS1 and 30,000 for MS/MS and the maximum ion injection time for MS/MS at 90 ms due to high resolution, fast scan speed for Q Exactive HF MS instrument.
Data-independent acquisition (DIA) on Exploris 480 Orbitrap MS
For DIA analysis, the MS/MS scan was performed in the HCD mode with the following parameters. Precursor ions from 400 to 900 m/z were scanned at a resolution of 120,000 for MS1 with maximum ion injection time of 125 ms and an AGC target of 1E6. A fixed precursor isolation window of 25 m/z with 1 Da overlap was used to acquire DIA MS/MS data at a resolution of 30,000, maximum ion injection time set to auto, and an AGC target of 1E6. The isolated ions were fragmented with HCD at 30% level with a loop count of 20 and the fixed first mass at 200 for fragment ions on Exploris 480 Orbitrap MS (Thermo Scientific).
Data analysis
For DDA data, all the MS raw files were processed with MaxQaunt86 (Version 1.6.2.10) and MS/MS spectra were searched against a human UniProt database (fasta file Reviewed sequence, January 2022, along with common contaminants and the total 20, 420 entries) in default parameters (tryptic peptides with 0-2 missed cleavage sites; 10 ppm of parent ion tolerance; 0.06 Da of fragment ion mass tolerance) with the activated match-between-run (MBR) function. Only acetylation (protein N-term), and oxidation (M) were set as variable modifications for global proteome analysis. The final reports were then generated (PSM and protein) and filtered at each level of 1% FDR.
For DIA data, all the MS raw files from the same tissue type were processed in a batch mode and searched together using DIA-NN87 (Version 1.8.1) against UniProt fasta of human proteome (2022-06-15-reviewed with contaminants) in default parameters with several modifications. In silico digest was activated with cleavage at K, R residues to keep a maximum number of missed cleavages set to 2. Peptide length was set 7–30 with a precursor m/z range of 400–900 and charge state +2 to +4. Variable modification was set to a maximum of 3 including mass delta 15.9949 for oxidation at M and mass delta 42.0106 for protein N-terminal acetylation. Fragment m/z range was set 200–1800. Precursor ion generation was activated for both FASTA digest and deep learning-based spectra. Protein inference was set to gene level with single-pass mode neural network classifier, and RT-dependent cross-run normalization, MBR activation, and 1%FDR report filtering.
Protein quantification was performed by using the label-free quantitation (LFQ) function. Contaminants were removed from the protein groups.txt file prior to use for downstream statistical analysis. Among the 12 most common contaminant proteins, except trypsin and albumin with top ranks in protein abundance the other contaminant proteins have moderate or low abundance (Supplementary Fig. 19), suggesting limited impact on the number of identified protein groups. Biological functions and signaling pathways were analyzed by using DAVID Bioinformatics Resources (Version 6.8)88 and Peruses (Version 1.6.2.1)89, and protein-protein association network analysis was performed by the latest version of STRING (Version 11.0)90.
Statistics and reproducibility
Downstream data analysis was performed using Perseus (Version 1.6.2.1)89. At least three biological or technical replicates were used to evaluate reproducibility for wcSOP-MS. No data exclusion was performed, and no randomization or blinding methods were used in data analysis. After label-free quantification with MaxQuant MBR or DIA-NN, the extracted ion chromatogram (XIC) areas of the identified protein groups were log2 transformed, and then normalized by the median value of each column. The non-supervised PCA analysis was used to generate PCA plot. We further used Student t test to prioritize significantly differentiated proteins for the heatmap generation. The extracted data were further processed and visualized using GraphPad Prisim 8, SigmaPlot 14, and Microsoft Excel 2017.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RAW MS data and the identified results from MaxQuant and DIA-NN were deposited in Japan ProteOme STandard Repository (jPOST: https://repository.jpostdb.org/)91 and can be accessed with dataset identifier JPST002191 for jPOST and PXD045043 for ProteomeXchange. Source data are provided in this paper. Source data are provided with this paper.
References
Regev, A. et al. The human cell atlas. eLife 6, e27041 (2017).
Hu, B. C. The human body at cellular resolution: the NIH Human Biomolecular Atlas Program. Nature 574, 187–192 (2019).
Mass, E., Nimmerjahn, F., Kierdorf, K. & Schlitzer, A. Tissue-specific macrophages: how they develop and choreograph tissue biology. Nat. Rev. Immunol. 23, 563–579 (2023).
Uhlen, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Jain, S. et al. Advances and prospects for the Human BioMolecular Atlas Program (HuBMAP). Nat. Cell Biol. 25, 1089–1100 (2023).
Elmentaite, R., Dominguez Conde, C., Yang, L. & Teichmann, S. A. Single-cell atlases: shared and tissue-specific cell types across human organs. Nat. Rev. Genet. 23, 395–410 (2022).
Lundberg, E. & Borner, G. H. H. Spatial proteomics: a powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 20, 285–302 (2019).
Stack, E. C., Wang, C., Roman, K. A. & Hoyt, C. C. Multiplexed immunohistochemistry, imaging, and quantitation: a review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods 70, 46–58 (2014).
Maric, D. et al. Whole-brain tissue mapping toolkit using large-scale highly multiplexed immunofluorescence imaging and deep neural networks. Nat. Commun. 12, 1550 (2021).
Black, S. et al. CODEX multiplexed tissue imaging with DNA-conjugated antibodies. Nat. Protoc. 16, 3802–3835 (2021).
Hickey, J. W. et al. Organization of the human intestine at single-cell resolution. Nature 619, 572–584 (2023).
Angelo, M. et al. Multiplexed ion beam imaging of human breast tumors. Nat. Med. 20, 436–442 (2014).
Greenbaum, S. et al. A spatially resolved timeline of the human maternal-fetal interface. Nature 619, 595–605 (2023).
Giesen, C. et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 11, 417–422 (2014).
Hickey, J. W. et al. Spatial mapping of protein composition and tissue organization: a primer for multiplexed antibody-based imaging. Nat. Methods 19, 284–295 (2022).
Chen, Y. J. et al. Proteogenomics of non-smoking lung cancer in East Asia delineates molecular signatures of pathogenesis and progression. Cell 182, 226–244 (2020).
Mertins, P. et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62 (2016).
Dou, Y. et al. Proteogenomic characterization of endometrial carcinoma. Cell 180, 729–748 (2020).
Vasaikar, S. et al. Proteogenomic analysis of human colon cancer reveals new therapeutic opportunities. Cell 177, 1035–1049 (2019).
Espina, V. et al. Laser-capture microdissection. Nat. Protoc. 1, 586–603 (2006).
Drummond, E. & Wisniewski, T. The use of localized proteomics to identify the drivers of Alzheimer’s disease pathogenesis. Neural Regen. Res. 12, 912–913 (2017).
Bevilacqua, C. & Ducos, B. Laser microdissection: a powerful tool for genomics at cell level. Mol. Asp. Med. 59, 5–27 (2018).
Mao, Y., Wang, X., Huang, P. & Tian, R. Spatial proteomics for understanding the tissue microenvironment. Analyst 146, 3777–3798 (2021).
Liotta, L. A. et al. Laser Capture Proteomics: spatial tissue molecular profiling from the bench to personalized medicine. Expert Rev. Proteom. 18, 845–861 (2021).
Mund, A., Brunner, A. D. & Mann, M. Unbiased spatial proteomics with single-cell resolution in tissues. Mol. Cell 82, 2335–2349 (2022).
Buczak, K. et al. Spatially resolved analysis of FFPE tissue proteomes by quantitative mass spectrometry. Nat. Protoc. 15, 2956–2979 (2020).
Davis, S., Scott, C., Ansorge, O. & Fischer, R. Development of a sensitive, scalable method for spatial, cell-type-resolved proteomics of the human brain. J. Proteome Res 18, 1787–1795 (2019).
Drummond, E. S., Nayak, S., Ueberheide, B. & Wisniewski, T. Proteomic analysis of neurons microdissected from formalin-fixed, paraffin-embedded Alzheimer’s disease brain tissue. Sci. Rep. 5, 15456 (2015).
Drummond, E., Nayak, S., Pires, G., Ueberheide, B. & Wisniewski, T. Isolation of amyloid plaques and neurofibrillary tangles from archived Alzheimer’s disease tissue using laser-capture microdissection for downstream proteomics. Methods Mol. Biol. 1723, 319–334 (2018).
Drummond, E. et al. Proteomic differences in amyloid plaques in rapidly progressive and sporadic Alzheimer’s disease. Acta Neuropathol. 133, 933–954 (2017).
Drummond, E. et al. Phosphorylated tau interactome in the human Alzheimer’s disease brain. Brain 143, 2803–2817 (2020).
Drummond, E. et al. The amyloid plaque proteome in early onset Alzheimer’s disease and Down syndrome. Acta Neuropathol. Commun. 10, 53 (2022).
Mund, A. et al. Deep Visual Proteomics defines single-cell identity and heterogeneity. Nat. Biotechnol. 40, 1231–1240 (2022).
Rosenberger, F. A. et al. Spatial single-cell mass spectrometry defines zonation of the hepatocyte proteome. Nat. Methods 20, 1530–1536 (2023).
Makhmut, A. et al. A framework for ultra-low-input spatial tissue proteomics. Cell Syst. 14, 1002–1014 (2023).
Zhu, Y. et al. Spatially resolved proteome mapping of laser capture microdissected tissue with automated sample transfer to nanodroplets. Mol. Cell Proteom. 17, 1864–1874 (2018).
Piehowski, P. D. et al. Automated mass spectrometry imaging of over 2000 proteins from tissue sections at 100-mum spatial resolution. Nat. Commun. 11, 8 (2020).
Nwosu, A. J. et al. In-depth mass spectrometry-based proteomics of formalin-fixed, paraffin-embedded tissues with a spatial resolution of 50–200 μm. J. Proteome Res. 21, 2237–2245 (2022).
Gosline, S. J. C. et al. Proteome mapping of the human pancreatic islet microenvironment reveals endocrine-exocrine signaling sphere of influence. Mol. Cell Proteom. 22, 100592 (2023).
Xu, R. et al. Spatial-resolution cell type proteome profiling of cancer tissue by fully integrated proteomics technology. Anal. Chem. 90, 5879–5886 (2018).
Clair, G. et al. Spatially-resolved proteomics: rapid quantitative analysis of laser capture microdissected alveolar tissue samples. Sci. Rep. 6, 39223 (2016).
Herrera, J. A. et al. Laser capture microdissection coupled mass spectrometry (LCM-MS) for spatially resolved analysis of formalin-fixed and stained human lung tissues. Clin. Proteom. 17, 24 (2020).
Tsai, C. F. et al. Surfactant-assisted one-pot sample preparation for label-free single-cell proteomics. Commun. Biol. 4, 265 (2021).
Martin, K. et al. Facile one-pot nanoproteomics for label-free proteome profiling of 50-1000 mammalian cells. J. Proteome Res 20, 4452–4461 (2021).
Kwon, Y. et al. Hanging drop sample preparation improves sensitivity of spatial proteomics. Lab Chip 22, 2869–2877 (2022).
Hermida, M. D., de Melo, C. V. B., Lima, I. D. S., Oliveira, G. G. S. & Dos-Santos, W. L. C. Histological disorganization of spleen compartments and severe visceral leishmaniasis. Front. Cell Infect. Microbiol. 8, 394 (2018).
Mebius, R. E. & Kraal, G. Structure and function of the spleen. Nat. Rev. Immunol. 5, 606–616 (2005).
Jiang, L. et al. A quantitative proteome map of the human body. Cell 183, 269–283 (2020).
Ogembo, J. G. et al. SIRPalpha/CD172a and FHOD1 are unique markers of littoral cells, a recently evolved major cell population of red pulp of human spleen. J. Immunol. 188, 4496–4505 (2012).
Klasener, K. et al. CD20 as a gatekeeper of the resting state of human B cells. Proc. Natl. Acad. Sci. USA 118, e2021342118 (2021).
Burger, J. A. & Wiestner, A. Targeting B cell receptor signalling in cancer: preclinical and clinical advances. Nat. Rev. Cancer 18, 148–167 (2018).
Wen, Y. et al. The regulators of BCR signaling during B cell activation. Blood Sci. 1, 119–129 (2019).
Mocsai, A., Ruland, J. & Tybulewicz, V. L. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat. Rev. Immunol. 10, 387–402 (2010).
Liu, D. & Mamorska-Dyga, A. Syk inhibitors in clinical development for hematological malignancies. J. Hematol. Oncol. 10, 145 (2017).
Weston, L. A. & Hummon, A. B. Comparative LC-MS/MS analysis of optimal cutting temperature (OCT) compound removal for the study of mammalian proteomes. Analyst 138, 6380–6384 (2013).
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 72, 7–33 (2022).
Kumar, P. & Aggarwal, R. An overview of triple-negative breast cancer. Arch. Gynecol. Obstet. 293, 247–269 (2016).
Louderbough, J. M. & Schroeder, J. A. Understanding the dual nature of CD44 in breast cancer progression. Mol. Cancer Res. 9, 1573–1586 (2011).
Burness, M. L., Grushko, T. A. & Olopade, O. I. Epidermal growth factor receptor in triple-negative and basal-like breast cancer: promising clinical target or only a marker? Cancer J. 16, 23–32 (2010).
Jiang, W., Wang, X., Zhang, C., Xue, L. & Yang, L. Expression and clinical significance of MAPK and EGFR in triple-negative breast cancer. Oncol. Lett. 19, 1842–1848 (2020).
Gurrapu, S., Pupo, E., Franzolin, G., Lanzetti, L. & Tamagnone, L. Sema4C/PlexinB2 signaling controls breast cancer cell growth, hormonal dependence and tumorigenic potential. Cell Death Differ. 25, 1259–1275 (2018).
Kumar, M. & Bansal, N. Implications of phosphoinositide 3-kinase-Akt (PI3K-Akt) pathway in the pathogenesis of Alzheimer’s disease. Mol. Neurobiol. 59, 354–385 (2022).
Li, H. et al. cAMP/PKA signaling pathway contributes to neuronal apoptosis via regulating IDE expression in a mixed model of type 2 diabetes and Alzheimer’s disease. J. Cell Biochem 119, 1616–1626 (2018).
Jimenez-Balado, J. & Eich, T. S. GABAergic dysfunction, neural network hyperactivity and memory impairments in human aging and Alzheimer’s disease. Semin Cell Dev. Biol. 116, 146–159 (2021).
Mulder, J. et al. Molecular reorganization of endocannabinoid signalling in Alzheimer’s disease. Brain 134, 1041–1060 (2011).
Matzinger, M., Muller, E., Durnberger, G., Pichler, P. & Mechtler, K. Robust and easy-to-use one-pot workflow for label-free single-cell proteomics. Anal. Chem. 95, 4435–4445 (2023).
Brunner, A. D. et al. Ultra-high sensitivity mass spectrometry quantifies single-cell proteome changes upon perturbation. Mol. Syst. Bio 18, e10798 (2022).
Dou, M. et al. High-throughput single cell proteomics enabled by multiplex isobaric labeling in a nanodroplet sample preparation platform. Anal. Chem. 91, 13119–13127 (2019).
Thielert, M. et al. Robust dimethyl-based multiplex-DIA doubles single-cell proteome depth via a reference channel. Mol. Syst. Biol. 19, e11503 (2023).
Cong, Y. Z. et al. Ultrasensitive single-cell proteomics workflow identifies >1000 protein groups per mammalian cell. Chem. Sci. 12, 1001–1006 (2021).
Ma, M. et al. In-depth mapping of protein localizations in whole tissue by micro-scaffold assisted spatial proteomics (MASP). Nat. Commun. 13, 7736 (2022).
Li, Y. Y. et al. An integrated strategy for mass spectrometry-based multiomics analysis of single cells. Anal. Chem. 93, 14059–14067 (2021).
Orsburn, B. C., Yuan, Y. T. & Bumpus, N. N. Insights into protein post-translational modification landscapes of individual human cells by trapped ion mobility time-of-flight mass spectrometry. Nat. Commun. 13, 7246 (2022).
Riley, N. M. & Coon, J. J. Phosphoproteomics in the age of rapid and deep proteome profiling. Anal. Chem. 88, 74–94 (2016).
Polat, A. N. & Ozlu, N. Towards single-cell LC-MS phosphoproteomics. Analyst 139, 4733–4749 (2014).
Reinders, J. & Sickmann, A. State-of-the-art in phosphoproteomics. Proteomics 5, 4052–4061 (2005).
Steen, H., Jebanathirajah, J. A., Rush, J., Morrice, N. & Kirschner, M. W. Phosphorylation analysis by mass spectrometry: myths, facts, and the consequences for qualitative and quantitative measurements. Mol. Cell Proteom. 5, 172–181 (2006).
Solari, F. A., Dell’Aica, M., Sickmann, A. & Zahedi, R. P. Why phosphoproteomics is still a challenge. Mol. Biosyst. 11, 1487–1493 (2015).
Ibrahim, Y. M. et al. New frontiers for mass spectrometry based upon structures for lossless ion manipulations. Analyst 142, 1010–1021 (2017).
Hebert, A. S. et al. Comprehensive single-shot proteomics with FAIMS on a hybrid orbitrap mass spectrometer. Anal. Chem. 90, 9529–9537 (2018).
Derks, J. et al. Increasing the throughput of sensitive proteomics by plexDIA. Nat. Biotechnol. 41, 50–59 (2023).
Tsai, C. F. et al. An improved boosting to amplify signal with isobaric labeling (iBASIL) strategy for precise quantitative single-cell proteomics. Mol. Cell Proteom. 19, 828–838 (2020).
Tsai, C. F. et al. A streamlined tandem tip-based workflow for sensitive nanoscale phosphoproteomics. Commun. Biol. 6, 70 (2023).
Livesay, E. A. et al. Fully automated four-column capillary LC-MS system for maximizing throughput in proteomic analyses. Anal. Chem. 80, 294–302 (2008).
Ye, Z. et al. High-throughput and scalable single cell proteomics identifies over 5000 proteins per cell. bioRxiv https://doi.org/10.1101/2023.11.27.568953 (2023).
Tyanova, S., Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 (2016).
Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44 (2020).
Huang da, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016).
Szklarczyk, D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019).
Watanabe, Y., Yoshizawa, A. C., Ishihama, Y. & Okuda, S. The jPOST repository as a public data repository for shotgun proteomics. Methods Mol. Biol. 2259, 309–322 (2021).
Acknowledgements
This work was supported by a UH3CA256967 grant (to T.S.) from the National Institutes of Health (NIH) Common Fund, Human Biomolecular Atlas Program (HuBMAP) grant, NIH RF1MH128885 (to T.S.), NIH R01GM139858 (to T.S.), P41GM103493 (to R.D.S.), NCI Early EDRN Interagency Agreement ACN20007-001 (to T.L.), NIH R01CA245699 (to H.L.), and ACS127951-RSG-15-025-01-CSM (to H.L.). PNNL is a multi-program national laboratory operated by Battelle for the Department of Energy (DOE) under Contract DE-AC05-76RLO 1830. A portion of this research was performed using EMSL, a national scientific user facility sponsored by the DOE’s Office of Biological and Environmental Research and located at PNNL and the Pathology Core at Northwestern University. We thank M.J. Gaffrey and T.-T. Lin for generous assistance with experiments and S.A. King for assistance with graphics.
Author information
Authors and Affiliations
Contributions
T.S. conceived and designed the study. R.B.K., C.-F.T. and T.S. conceptualized wcSOP-MS and designed the experiments. R.B.K., M.V., Z.X., R.K.C. and W.B.C. performed cryosection and LCM experiments. R.B.K., M.V. and Z.X. performed all proteomics sample preparation. R.B.K., R.Z., D.J.O. R.J.M. and C.-F.T performed all proteomics experiments and data analysis. D.S., J.V.M., D.B.G. and H.L. provided triple negative breast cancer tumor tissues and conducted IHC experiments. B.M.B. and C.H.W. provided healthy human spleen tissues and conducted CODEX experiments. T.Z., P.D.P., T.L., R.D.S., H.L. and C.H.W. provided input on the experimental design, data presentation, and manuscript preparation. R.B.K. and T.S. wrote the manuscript with input from all other authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Nature Communications thanks Florian Rosenberger, Jun Qu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kitata, R.B., Velickovic, M., Xu, Z. et al. Robust collection and processing for label-free single voxel proteomics. Nat Commun 16, 547 (2025). https://doi.org/10.1038/s41467-024-54643-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54643-x
This article is cited by
-
Mass spectrometry-based human spatial omics: fundamentals, innovations, and applications
Journal of Biomedical Science (2026)
