
Abstract
Depression-anxiety comorbidity is commonly attributed to the occurrence of specific symptoms bridging the two disorders. However, the significant heterogeneity of most bridging symptoms presents challenges for psychopathological interpretation and clinical applicability. Here, we conceptually established a common bridging factor (cb factor) to characterize a general structure of these bridging symptoms, analogous to the general psychopathological p factor. We identified a cb factor from 12 bridging symptoms in depression-anxiety comorbidity network. Moreover, this cb factor could be predicted using edge-centric connectomes with robust generalizability, and was characterized by connectome patterns in attention and frontoparietal networks. In an independent twin cohort, we found that these patterns were moderately heritable, and identified their genetic connectome-transcriptional markers that were associated with the neurobiological enrichment of vasculature and cerebellar development, particularly during late-childhood-to-young-adulthood periods. Our findings revealed a general factor of bridging symptoms and its neurobiological architectures, which enriched neurogenetic understanding of depression-anxiety comorbidity.
Similar content being viewed by others


Introduction
Comorbidity between depression and anxiety has been demonstrated to be more common than that of other pairs of psychiatric disorders. Over 50% of patients with depression or anxiety disorder were diagnosed with lifetime depression-anxiety comorbidity1,2, and their treatment outcomes were worse compared to those who diagnosed for either depression or anxiety alone3,4. To clarify the etiological foundation, the comorbidity hypothesis posited that this comorbidity arose from the “bridging symptoms” that actively increased risks of “contagion” from one disorder to another5,6,7. It has been demonstrated that patients diagnosed with anxiety were more likely to experience comorbidity with depression when these “bridging symptoms” present, as opposed to ones who do not exhibit such symptoms8,9. Moreover, deactivating “bridging symptoms” was found to be an effective therapeutic strategy to prevent the concurrence of depression and anxiety disorders8,10. Thus, clinicians have increasingly suggested therapeutically targeting these bridge symptoms to improve treatment outcomes for comorbidity11.
Though there was evidence to support the comorbidity hypothesis, the high heterogeneity of these bridging symptoms in depression-anxiety comorbidity still challenged its psychopathological interpretability and clinical utility. For instance, these depression-anxiety bridging symptoms, such as “psychomotor reaction”, “emotional feelings” and “negative cognition”, were found to be involved in distinct pathological pathways12,13,14. Moreover, these bridging symptoms in depression-anxiety comorbidity were highly inconsistent among existing studies9,12,13,15,16,17,18, with significant discrepancies in identifying bridging symptoms even within one study12. These observed heterogeneities in the depression-anxiety bridging symptoms have been explained by the common cause theory, which posited that such heterogeneous symptoms may originate from a single underlying “common pathogenic factor”15,19. Supporting this point, a general psychopathology factor (i.e., p factor) suggesting a single general cause to derive comorbidity, has been identified as more reliable to characterize psychiatric comorbidities than other multidimensional etiological systems20,21. Despite merits, the sole p factor theory is still challenged as it oversimplifies the inter-symptom structure by merely summarizing the positive inter-correlations across all the symptoms22. Therefore, the p factor showed an unstable structure in explaining comorbidity with highly heterogeneous symptoms, which indicated alternative factors to derive co-concurrences of psychiatric disorders23,24.
Several nosological multi-factorial theories have been established to understand symptomatology structures of psychiatric comorbidities, particularly in the Hierarchical Taxonomy of Psychopathology (HiTOP) and Research Domain Criteria (RDoC) frameworks. The HiTOP proposed a multidimensional diagnostic system embedded within a hierarchical framework, with combinations of subfactors (e.g., internalizing/external problems) to constitute high-order factors for diagnosing comorbidity25,26. However, compared to the single-factor nosological structure, this theory was consistently challenged due to its limited clinical practicability and especially discrepancies in the neurobiological interpretations27,28. To consolidate the theoretical foundation of the multidimensional structure, another nosological system enriched by neurobiological architectures, that was the RDoC, had been established29. Despite its merits in neurobiological interpretability, it is still questioned for its poor theoretical constructs due to its “reductionist” assumptions30,31. Therefore, to address these issues, synthesizing these heterogeneous bridging symptoms into one-factor structure might be one promising pathway and theoretical framework to understand the common neuropsychopathological mechanism to comorbidities11,32,33,34. By combining the common cause theory with the comorbidity hypothesis, we aimed to establish a common bridging component to understand the “bridging factor” in the depression-anxiety comorbidity (referred to as the cb factor) (Fig. 1).

This diagram illustrates the main strength (marked by the “√” button) and key challenge (marked by the “!” button) for the “comorbidity hypothesis” (a) and “common cause hypothesis” (b), respectively. The theoretical hypothesis for the present cb factor was drawn at (c) and was briefly introduced in the box (marked by the “i” button). These icons that used in the present figure are openly available at the web-based software (ICONFINDER, https://www.iconfinder.com/). The “√” icon by Paomedia, titled “Check, sign icon”, used under CC-BY 3.0 license, available at https://www.iconfinder.com/icons/299110/download/png/256. The “!” icon by Tahsin Tahil, titled “Error icon”, used under CC-BY 3.0 license, available at https://www.iconfinder.com/icons/381599/download/png/256. Based on the permission of the CC-BY 3.0 license, this “!” icon is modified by changing its background color to dark yellow and changing the style of the “!” mark in the figure. The “i” icon by Paomedia, titled “Info, sign icon”, used under CC-BY 3.0 license, available at https://www.iconfinder.com/icons/299086/download/png/256. Based on the permission of the CC-BY 3.0 license, this icon is modified by changing the style of this “i” mark.
This conceptualized cb factor may hold the potential to accurately characterize a general structure of bridging symptoms in comorbidities, but the lack of neurobiological evidence limited its interpretability in psychopathology. Connectome, a completed component to describe intrinsic region-to-region functional connections (rFC) in the whole brain, has been broadly demonstrated as a fundamental principle of brain functioning35,36. Research has well-documented that the brain connectome-based features provided robust neurobiological markers to characterize the biotype of depression/anxiety (even in comorbid conditions)37,38,39, especially compared to the regional change in specific regions or plain neural circuits40,41,42. Specifically, the amygdala-modulated downstream rFC-connectomes (e.g., regions of limbic networks) were consistently captured as cross-disorder diagnostic markers for patients who were comorbid with depression and anxiety43,44. Moreover, by synthesizing numerous meta-analytic evidence, the rFC-wise abnormalities in the default mode network and frontoparietal network have been identified as domain-specific biomarkers predicting depression-anxiety comorbidity, showing decreased intra-connections in these brain networks when anxious and depressive symptoms co-occurred45,46. Recently, the edge-centric FC (eFC) has been developed to capture between-rFC communication patterns, which measure the similarity of rFC pairs at each instant time point47,48. The eFC not only surpasses traditional rFC in robustness of phenotyping and in the differential identifiability to individual idiosyncrasies47,48, but also showed superior performance in characterizing intrinsic neural patterns of neuropsychiatric disorders and neurological diseases49,50,51,52. More importantly, compared to rFC, the eFC shifted constructions of brain connectome from between-regions spontaneous synchronization to instantaneous co-fluctuations, thus yielding unique insights into brain high-resolution temporal FC architectures53,54. Therefore, in the present study, we intended to identify the eFC markers associated with the cb factor to probe its neurobiological substrates.
As a crucial intermediate phenotype, the brain connectomes have been reliably demonstrated to be, at least partially, influenced by genetics in deriving clinical phenotypes, such as psychiatric comorbidity55,56,57,58. With the developments of imaging-transcriptomics, neuroimaging-genetic studies hold great potential to elucidate molecular mechanisms and neurobiological architectures underlying heterogeneous psychiatric disorders59,60,61,62. For instance, the connectome changes in the depression were found to be associated with specific protein diversity, interneuron enrichment, and synaptic reorganization that were explained by regional gene expressions58,63. Moreover, previous studies have identified the neurobiological enrichment in the excitatory neuronal systems and synapse functions for concurrent symptoms of depression and other psychiatric disorders, which were attributed to connectome-based alterations correlated with these symptoms64,65,66. Therefore, integrating connectome phenotype with genetic regulations paved a promising way to understand multiscale neurobiological substrates in psychiatric comorbidities, including (but not limited to) their molecular mechanisms and cellular systems62,67,68,69. Here, by probing connectome-genetic markers of this cb factor, we aim to gain more comprehensive insights into the neurogenetic substrates underlying depression-anxiety comorbidity.
In this study, we aimed to conceptually establish a common bridging factor (referred to as the cb factor) to represent the general structure of heterogeneous bridging symptoms in depression-anxiety comorbidity in a large-scale subclinical cohort, which favored theoretically probing and extending comorbidity hypothesis in the network theory (Fig. 2a). Here, we collected the symptoms of depression and anxiety using self-reported questionnaires and identified the cb factor using factor analysis model (see “Methods” section). We hypothesized that the single-factor structure could be optimum in modeling these heterogeneous bridging symptoms. To probe the neurobiological substrates of this conceptualized cb factor, we developed an eFC connectome-based predictive model (eCPM) to examine whether the whole-brain eFC could reliably predict the cb factor (Fig. 2b). From what has been mentioned above, we speculated that the eFC could serve as robust biomarkers of this conceptualized cb factor. Upon confirming this prediction, we employed a multivariate representation similarity analysis (RSA) to delineate what specific eFC markers characterize the cb factor, particularly in limbic, frontoparietal and default mode networks (Fig. 2c). Given the genetic influences on the brain connectome, we finally extended our analysis to capture the eFC-genetic signatures of the cb factor by recruiting an independent twin cohort and incorporating extensive neurocognitive and biological datasets (Fig. 2d).

a The EBICglasso graph-theoretical model was used to identify bridging symptoms in the depression-anxiety inter-symptom network, and the factor analysis was further conducted to identify the common factor to characterize a general structure of these identified bridging symptoms, which was conceptualized as cb factor. b To probe whether this conceptualized cb factor had neural substrates, the edge-centric brain connectome-based feature (i.e., edge-centric functional connectivity, eFC) was calculated as a neural feature for training the eFC connectome-based predictive model (eCPM) to predict the cb factor scores. Here, this model was trained in the discovery sample, and the model performance was evaluated by validating in an independent validation sample and generalization samples. c Once revealing the predictive roles of eFC features, the inter-subject representation similarity analysis (RSA) was further conducted by correlating the eFC pattern (i.e., eFC connectomes at a given eFC) to the behavioral feature (i.e., cb factor scores), to probe how these eFCs characterized the cb factor. d By using the ACE model in an independent twin cohort, these eFCs that identified significant RS to the cb factor, were examined for heritability. Once the heritability was confirmed, the Allen Human Brain Atlas (AHBA) was used to test whether such RS could be predicted by regional gene expression patterns in the partial least square regression (PLS). Finally, if the connectome-transcriptional correlates (i.e., gene expression patterns) were found, these specific gene patterns were annotated by using multiscale normative biological atlases.
Results
Summary of main analytic steps and findings
We included a population-based subclinical cohort (n = 2022) in the present study, with representative sociodemographic conditions in China (e.g., ethnic groups and socioeconomic status) (Fig. 3a, Supplementary Figs. S1–S2, and Supplementary Tables. S1–S4). First, we used an EBICglasso-based (graphic least absolute shrinkage and selection operator with Extended Bayesian Information Criterion) graph-theoretical model for estimating bridge centrality in this depression-anxiety network built from inter-symptom correlations8. Twelve heterogeneous bridging symptoms with high bridge centrality were captured. We identified a statistically reliable common factor representing the general structure of these bridging symptoms, referred to as the cb factor. Second, we used eFC features to construct the eCPM70,71 to probe whether this conceptualized cb factor could be underpinned by eFC connectomes. The results demonstrated that eFCs were generalizable in predicting the cb factor scores. Third, after confirming the predictive roles of eFC connectome, we further carried out multivariate representational similarity analysis (RSA) to delve into what eFC patterns were sensitive to characterize the cb factor, which found that eFCs within the attention and frontoparietal networks outperformed others. Fourth, to clarify the genetic influences, we estimated the heritability of these sensitive eFCs identified from RSA in an independent twin cohort. The findings demonstrated moderate heritability for these eFCs, suggesting the existence of genetic substrates associated with the cb-factor-specific eFCs, which enabled us to capture genetic eFC-transcriptional signatures. Thus, by aligning these sensitive cb-factor-specific eFCs into the Allen Human Brain Atlas (AHBA) and other normative biological atlases, we found the significant eFC-transcriptional markers to the cb factor, which were mainly involved in vasculature systems and cerebellar development, especially in late-childhood-to-young-adulthood periods.

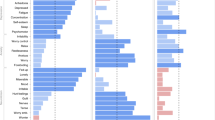
a The geospatial and socioeconomic statistics of this subclinical sample (GGBBP sample recruited from 2019 to 2022) demonstrate the geographic diversity. The scale indicated the number of included subjects after Log transformation. Icons in this panel were generated from the open-access web-based software (ICONFINDER, https://www.iconfinder.com/). The “male gender” icon by Anna Litviniuk, titled “Avatar, male, man icon”, used under Free for commercial use license, available at https://www.iconfinder.com/icons/403019/download/png/512. The “female gender” icon by Anna Litviniuk, titled “Avatar, user, woman icon”, used under Free for commercial use license, available at https://www.iconfinder.com/icons/403023/download/png/512. The “diversity” icon by Dumitriu Robert, titled “Guy, individual, man icon”, used CC-BY 3.0 license, available at https://www.iconfinder.com/icons/3289573/download/png/512. This icon is modified by changing colors and replicating in the figure, as permitted by this license. The “socioeconomic status” icon by Pongsakorn Tan, titled “Banking, business, cash icon”, used under Free for commercial use license, available at https://www.iconfinder.com/icons/4288564/download/png/512. The “family health” icon by Paomedia, titled “House icon”, used CC-BY 3.0 license, available at https://www.iconfinder.com/icons/299061/download/png/512. The “COVID-free” icon by Omeneko, titled “Corona, coronavirus, positive icon”, used CC-BY 3.0 license, available at https://www.iconfinder.com/icons/6217233/download/png/512. This geographic map, along with the compass label, was produced by the open-access software titled “EasyShu (3.61)” (https://www.yuque.com/easyshu/). b Mantel’s test plot was illustrated here (p < 0.001, one-sided Mantel’s test, uncorrected), and each point into the lower triangle indicated the mean values of corresponding items. c We illustrated the centrality of each symptom (item) from the network model in descending order, with the “D” for indicating “depressive symptom” and with the number of this label for indicating the item in this questionnaire (EI Expected Influence). d This showed density with Gaussian kernel function for each symptom by descending order, with each circuit (gray) for indicating the high integrative centrality. Source data are provided as a Source Data file.
Bridging symptoms in depression-anxiety comorbidity network
We found significant correlations for symptoms between depression and anxiety (r = 0.71, p < 0.001, univariate Pearson’s correlation of total scores; r = 0.40, p < 0.001, multivariate Mantel’s correlation of inter-subject across-symptom correlation networks; Fig. 3b), thereby demonstrating the presence of comorbid conditions within this subclinical population. By constructing the graph-theoretical model of the depression-anxiety comorbidity network (see “Methods” section), we identified a series of symptoms showing high bridge centralities, such as “less meaningful life”, “worry” and “less confidence”. Nonetheless, such symptoms varied significantly across distinct bridge centrality metrics (e.g., bridge strength centrality, betweenness, and expected influence centrality) (Fig. 3c, and Supplementary Tables S5–S9). To address the discrepancies arising from these varying metrics, we calculated the normalized Shannon’s entropy (SEnormalized), which quantified the likelihood of each bridging symptom being identified as “bridge node” across these metrics. This analysis identified 12 bridging symptoms with significantly high SE values (all SE > 0.8; Supplementary Table S10), such as “exhaustion” (SEnormalized = 1.0), “meaningless life” (SEnormalized = 1.0), “depressive feeling” (SEnormalized = 0.98) and “psychomotor agitation” (SEnormalized = 0.94) (Fig. 3d). Using the Bootstrapping method and Monte Carlo simulation, we verified the statistical reliability and stability of above findings (Supplementary Figs. S3–S10).
Construction of the conceptualized cb factor
Considering the high heterogeneity of these identified 12 bridging symptoms in the symptomatology, we used factor analysis (see “Methods” section) to identify the factor structure of these symptoms. Results showed that a common factor structure outperformed alternatives (e.g., multi-factor structures) in model fitting (Supplementary Table S5), and explained common variances of these bridging symptoms well (34.0%, p < 0.05; Permutation test at n = 1000). Thus, we conceptualized this one factor as the common bridging factor (cb factor) to characterize the general structure of these bridging symptoms, which was akin to the p factor in psychopathology. In line with the p factor20, we further calculated the cb factor scores by the common factor scores, to quantify a liability to derive general bridging symptoms. Regression models revealed that the cb factor had better goodness-of-fits in predicting the total scores of depression and anxiety symptoms when compared to the individual symptom (s) (Supplementary Table S6). To draw a comparison with the p factor, further regression models were constructed to predict the total scores of depression and anxiety symptoms using the p factor scores and the cb factor, respectively. Results showed that the model with the cb factor significantly outperformed alternatives and presented well-measure invariances (Supplementary Table S7). Overall, we conceptualized a common bridging factor enabling us to characterize the general structure of heterogeneous bridging symptoms in the depression-anxiety comorbidity.
The eFC prediction of the cb factor
Beyond establishing conceptual structure, we further investigated whether this cb factor could be underpinned by neurobiological substrates (see “Methods” section). Next, we developed an eCPM to examine whether the whole-brain eFC connectomes could predict this cb factor (Fig. 4a, b, and Supplementary Table S11). Here, to mitigate data leakage risks72, we used external validations to evaluate the prediction performances of this model, rather in-sample k-fold cross-validation method. We split the original sample into three independent subsamples for model training (one discovery sample, n = 241) and performance evaluation (one external validation sample (n = 240) and one external generalization sample (n = 244)), because these three subsamples were independently curated from three distinct research teams (see “Methods” section). Based on the population characteristics (e.g., ethnic groups, COVID-19 exposure), the remaining participants in the original sample were grouped into three independent generalization samples to rigorously examine model generalizability. We found that the eFC connectomes significantly predicted the cb factor scores in the discovery sample, with models trained on positive eFCs (R2 = 0.23, pperm < 0.01), negative eFCs (R2 = 0.26, pperm < 0.01) or combined ones (R2 = 0.41, pperm < 0.01) (Fig. 4c, Supplementary Fig. S11, and Supplementary Tables S12–S16). In the independent validation sample, we confirmed the predictive powers of this eCPM (all pperm < 0.01) (Fig. 4d). Excepting to the model that trained by the negative eFCs, these findings were generalizable in the independent generalization sample 1 (n = 244, pperm < 0.05) (Fig. 4e). To validate the robustness of generalizability in heterogeneous cohorts, we further tested the eCPM in additional generalization samples. Results showed that this eCPM was still valid in generalization sample 2 (encompassing 29 local ethnic minorities in the Chinese population, n = 133, pperm < 0.05) and sample 3 (the main ethnic group, the Han, n = 237, pperm < 0.05) (Fig. 4f). Considering the potential confounding effects of the COVID-19 exposure, we assessed the generalizability of the eCPM in the generalization sample 4, which was collected after the pandemic. The eCPM also showed significant predictive power on the cb factor in this post-pandemic sample (n = 219, pperm < 0.05) (Fig. 4f). Moreover, we tested the specificity of this eCPM, finding it performed better in predicting cb factor scores than total symptom scores for depression, anxiety, or both (Supplementary Fig. S12). Therefore, our findings support the existence of neurobiological substrates underpinning the conceptualized cb factor by revealing the predictive roles of eFC connectomes.

a We used the open-access Gephi (https://gephi.org/) software to visualize edge-centric connectome. The connectome-based plot and icons were automatically generated by inputting the full-length edge-to-edge matrix into this software. To ensure readability, this connectome density has been threshold to 0.1 and was adjusted by using the Fruchterman-Reingold layout. SMN sensorimotor network, DMN default mode network, VIS visual network, VAN ventral attention network, Cont frontoparietal network. Subscripts embedded in abbreviations of networks (i.e., DMNa, DMNb, DMNc, SMNa, and SMNb) indicated the subnetworks within themselves. b It showed the inter-subject correlations between eFC and the cb factor scores, and brain networks parceled by the Yeo-7 network atlas for improving readability. c To show the trained model performance, we provided scatter plots for the correlation between true cb factor scores and predicted ones (z-scored) within the discovery sample (One-sided Permutation test at n = 5000, uncorrected). The Taylor diagram was drawn to comprehensively evaluate model performance by including models that trained from positive eFCs, negative eFCs, and both of them, respectively. d–f We further displayed edge-centric connectome, along with scatter plots and the Taylor diagram to show the model performance in the external validation and generalization in these independent samples (One-sided Permutation test at n = 5000, uncorrected), respectively. Source data are provided as a Source Data file.
The eFC neural representation patterns of the cb factor
Despite confirming the predictive roles of eFC connectomes to the cb factor as described above, this eCPM fell short in elucidating how these eFC features sensitively characterize the cb factor because it only yielded a selection of eFCs with uncorrected univariate correlations to the cb factor scores72. Thus, we conducted the multivariate representation similarity analysis (RSA) to identify cb-factor-specific eFC characterizations (see “Methods” section, and Supplementary Fig. S13a, b). Results showed statistically significant high representation similarity (RS) in eFCs including the frontal pole, superior frontal cortex, and precuneus (all pperm < 0.05, Benjamini–Hochberg correction; Supplementary Fig. S13c, and Supplementary Table S17). Based on brain network parcellation from Yeo-7 atlas, we found significant RS within the attention, frontoparietal, and default mode networks (all pperm < 0.05, Benjamini–Hochberg correction; Supplementary Fig. S13d, and Supplementary Tables S18–S21). In summary, the eFCs in the attention and frontoparietal networks were sensitive markers for characterizing the neurobiological substrates of the cb factor.
The heritability of the cb-factor-specific eFC markers
Recognizing eFC connectome as a crucial intermediate phenotype, we further probed the heritability of these eFCs exhibiting high RS by building the quantitative ACE (A, additive genetic factor; C, common environment; E, unique environment) model in an independent twin dataset (n = 127 pairs of monozygotic twins, n = 118 pairs of dizygotic twins, see “Methods” section). Results demonstrated a moderate heritability (22.9%, 95% CI: 7.4–37.2) in the optimal best-fitting AE model (Supplementary Fig. S14, and Supplementary Tables S22–S23). Supporting that, we observed a significant within-pair correlation for eFC values in monozygotic twins (intra-class correlation, ICC, r = 0.22, p < 0.0001), but not yet in the dizygotic ones (ICC, r = 0.06, p = 0.28). Thus, the findings suggested the presence of a potential genetic contribution in these cb-factor-specific eFCs.
Connectome-transcriptional markers of the cb factor
Given the confirmation to the heritability of these cb-factor-specific eFCs, we used the normative AHBA (http://human.brain-map.org) to further delve into eFC-transcriptional signatures of this cb factor (see “Methods” section). Therefore, we carried out a partial least squares (PLS) regression model to fit gene expression profiles to the RS values that we calculated in the main sample. The first and second component (s) of PLS (PLS1, PLS2) cumulatively explained 32.4% of the variance in the spatial patterns of gene expressions, showing the anterior-posterior hierarchy (Fig. 5a). Furthermore, results demonstrated significant correlations between neural signatures (i.e., RS values) and gene expression maps (i.e., PLS weighted scores) in both PLS1 and PLS2 (rPLS1 = 0.31, pperm < 0.01; rPLS2 = 0.30, pperm < 0.01) (Fig. 5a), thereby supporting associations between the potential gene expression patterns and the connectome-based markers (i.e., eFCs) that characterize the cb factor.

a We used a partial least squares (PLS) regression model to predict eFC-derived representation similarity by aligning AHBA normative data into Schaefer-100 space (upper panel) and showed the weights for the first and second components (PLS1 and PLS2, bottom panel). Further, the scatter plot was provided to show the linear association of PLS scores (weights) to RS values (p = 0.001, one-sided Permutation test at n = 5000, uncorrected). b The colored table detailed the gene expression patterns for PLS1 (upper panel) and PLS2 (bottom panel), with the threshold for the Z-value > (<) 3.0. The bar plots in the right panel indicated the proportion of the number of genes reaching this statistical threshold from all the candidates. By using this statistical boundary, 27 genes (231 genes) survived from 4177 gene (5823 gene) sets in PLS1, and 44 genes (112 genes) survived from 3994 genes (5989 genes) in PLS2. c We extracted gene expression level for these selected genes from PLS components and illustrated scatter plots for each PLS component that showing the largest correlation strengths (PLS+ for the positive ones and PLS- for negative ones) between this given gene and RS values (z-scored) (p < 0.05, one-sided Permutation test at n = 5000, uncorrected). d The univariate correlations for expression levels of all the genes and RS values (z-scored) were calculated and were presented in this chart in descending order. Source data are provided as a Source Data file.
By examining the statistical significance of gene sets in PLS components, we found that 27 (or 231) genes overexpressed (or under-expressed) with increased (or decreased) RS values (PLS1+, Z > 3.0 or PLS1−, Z < 3.0, p < 0.005; Fig. 5b, c, and Supplementary Tables S24–S25) in the PLS1. Similar results were observed in the PLS2 (Fig. 5b, c, Supplementary Tables S26–S27). We further revealed significant correlations between spatial expression patterns of specific genes and these RS values in both PLS components, such as SCRIB, FTCD, RMND1, and CORO2A (p < 0.05, FDR-corrected; Fig. 5d, and Supplementary Tables S28–S29). Thus, we clarified specific genetic connectome-transcriptional markers to the cb factor enabling us to further probe their neurobiological associations.
Neurobiological enrichment of connectome-transcriptional markers of the cb factor
We used gene expression enrichment analysis to decode biological function-specific annotations of these connectome-transcriptional markers. In the PLS1 component, we capitalized on the Metascape platform that is embedded with the ChatGPT engine, to examine the functional enrichment of these gene sets. We found statistically significant functional enrichment into the biological process (GO) of “blood vessel development” (all p < 5 × 10−6, FDR-corrected; Fig. 6a, Supplementary Fig. S15, and Supplementary Table S30), and mapped their regulations and interactions by identifying the GO network as well as protein-to-protein module (Fig. 6b, c, Supplementary Fig. S16, and Supplementary Table S31). Full results of the PLS2 can be found in Supplementary Table S32, and Supplementary Figs. S17–S18.

a Using the web-based software (i.e., Metascape that was amplified with ChatGPT, https://metascape.org/gp/index.html)124 for the enrichment analysis, we presented the top 20 biological processes/pathways that were enriched from the PLS1 gene set at two-sided q < 0.01 after Benjamini–Hochberg FDR corrections, with the cumulative hypergeometric distribution for estimating corresponding p-values. Once the PLS1 gene list is inputted into this software, these plots or icons would be automatically generated. b Circos plot was illustrated by visualizing the term-to-term connectivity, with edges for showing between-term similarity >0.3 (Two-sided cumulative hypergeometric distribution test, q < 0.05, Benjamini–Hochberg FDR correction). “Count” refers to a number of genes in the PLS1 with membership in the given ontology term provided by Metascape. The “Log10(P)” is the p-value in log base 10, and the “Log10(q)” is the adjusted p-value (i.e., q-value) in log base 10. This plot was generated by Cytoscape embodied into the Metascape tool. c We provided protein-to-protein interaction connectome in this chart, with proteins recolored based on enrichment from this gene list by independent modules detected from the Molecular Complex Detection (MCODE) algorithm. Details for each MCODE can be found in the Supplementary Information. Source data are provided as a Source Data file.
Given the associations of such connectome-transcriptional markers to the GO functional enrichment, we further probed the brain network-specific, tissue-specific, cell type-specific, disease-specific, and neurodevelopment-specific enrichment from these markers. Combining Metascape and SEA (Specific Expression Analysis), the gene set in the PLS1 was significantly enriched across body tissues, particularly in the brain (p < 0.05, FDR-corrected; Fig. 7a, and Supplementary Tables S33–S35). We also observed significant brain network-specific, cell type-specific, and disease-specific enrichment in this PLS component, particularly for enrichment into risks of neurodevelopmental disorders and cerebral metabolic rate of oxygen (CMRO2) (p < 0.05, FDR-corrected; Fig. 7b, c, Supplementary Figs. S19–S20, and Supplementary Tables S36–S44). In conjunction with the BrainSpan atlas, we revealed the specific enrichment of these transcriptional markers in the cerebellar neurodevelopment, especially from late-mid childhood to young adulthood (Fig. 7d). Full results for the PLS2 can be found in Supplementary Tables S45–S47. These findings collectively indicated a cb-factor-connectome-transcriptional pathways, which may enrich understanding of the multiscale neurobiological substrates of cb factor in the depression-anxiety comorbidity.

a We showed the tissue-specific enrichment of this gene set (PLS1) by using both the Metascape124 tool and the Specific Expression Analysis (SEA) database. * indicated the q < 0.05 (two-sided cumulative hypergeometric distribution test, Benjamini–Hochberg FDR correction) that found in the Metascape database in the left panel, while the colors of circles indicated the q-values (Benjamini–Hochberg FDR) in the right panel. The size of the bullseye plots represents the proportion of genes in specific tissues at a given specificity index probability (pSI), which evaluates the level of gene enrichment specificity compared to others, using permutation tests. All the icons within this panel were generated by the web-based plotting platform FigDraw (https://www.figdraw.com/; Unique Copyright Code: YPTUR7d07d). Searching terms in this platform contained each tissue in this panel, including “body”, “brain”, “retina”, “bone”, “heart”, “muscle” and “kidney”, respectively. b Cell type-specific enrichment (left panel) and disease-specific enrichment (right panel) of this gene set (Two-sided cumulative hypergeometric distribution test, q < 0.05, Benjamini–Hochberg FDR correction) were shown. Full names of enriched diseases are detailed below: Down Syndrome (Down synd), Acute pancreatitis (Acute pan), Congenital chromosomal disease (CCD), Complete Trisomy 21 Syndrome (CT21 synd), Middle Cerebral Artery Occlusion (MCAO), Sleep-Apnea-Obstructive (OSA), Cardiomyopathy-Familial-Idiopathic (COP), Endothelial dysfunction (Endo anomaly), Transient Ischemic Attack (TAI), Subarachnoid Hemorrhage (SH), Fatty Liver Disease (FLD), and B-CELL MALIGNANCY-LOW-GRADE (MALIGNANCY). c Bullseye plots, along with q-values, have been illustrated to show the enrichment of the neurodevelopmental periods at different brain areas. d Bullseye plots to show the enrichment of SEA brain regions have been provided though no one reached the statistical significance. Source data are provided as a Source Data file.
Discussion
The present study identified a common factor to represent the general structure of bridging symptoms that were captured in the depression-anxiety comorbidity network, which has been conceptualized as the cb factor. Moreover, our evidence supported the existence of neural substrates of this conceptual cb factor by showing the predictive roles of eFC connectomes on the cb factor. Beyond confirming this prediction, we further elucidated how the cb factor was characterized by the specific representation similarity (RS) patterns of eFC connectomes. Notably, we found that eFCs within the attention and frontoparietal networks are particularly sensitive in characterizing the cb factor. By recruiting an independent twin cohort, we further revealed a moderate heritability of such RS patterns, indicating the potential genetic contributions to these cb-factor-specific eFC connectomes. Thus, we probed into the connectome-transcriptional markers by using AHBA and other normative biological datasets. The findings identified the specific connectome-transcriptional markers of the cb factor and further revealed their functional enrichment in the vasculature and cerebellar development, particularly in late-childhood-to-young-adulthood period. In summary, we established the common bridging factor for comorbidity between depression and anxiety and illuminated multiscale neurobiological substrates of this conceptual factor, which enriched our understanding of the comorbidity hypothesis in psychopathological theory.
Our findings showed that the cb factor could contribute to solving the current challenge to the comorbidity hypothesis, which stemmed from the substantial heterogeneity among bridging symptoms in depression-anxiety comorbidity73,74,75. These bridging symptoms were found to be the mixture of several psychopathological systems, such as psychomotor agitation/retardation13,15,76, negative emotional evaluations9,12,77, and poor physical feelings16,78, which hindered theoretical interpretability. A promising hypothesis posited that, despite high heterogeneity, these bridging symptoms might converge on a common factor that delineated the general structure of a “bridging syndrome”11,75, which was in some way analogous to the p factor in the transdiagnostics. Several lines of evidence reinforced this argument by revealing the single general factor in heterogeneous neuropsychopathological profiles (NP factor) and polygenic risks (PR factor) underlying comorbidities of mood disorders (e.g., depression and anxiety)79,80,81. Thus, establishing a common factor (i.e., cb factor) underlying these heterogeneous bridging symptoms could potentially alleviate the “heterogeneity challenge” in this comorbidity theory. Furthermore, our findings demonstrated that the scores of the conceptual cb factor indeed predicted the severity of comorbid depression and anxiety. As supported by existing evidence, the common factor structure of subclinical self-reported questionnaires has been revealed to outperform in explaining psychopathological networks than individual symptoms or clusters of symptoms in the psychiatric comorbidities20,82,83. Thus, compared to individual heterogeneous bridging symptoms, this cb factor may be a better candidate to predict risks of depression-anxiety comorbidity.
Another notable contribution of the present study was to clarify the neural substrates underpinning this cb factor, with a specific focus on elucidating the role of these eFCs. The eFC connectome reflected whether the communication patterns of pairs between FCs are correlated, depicting complicated hierarchical neural architecture54,84. Compared to the nodal FC, it has been broadly manifested that the eFC outperformed in subject-specific identifiability and predictive robustness for identifying neuropsychiatric and neurological diseases, such as autism, mild cognitive impairment, and migraine50,85,86. Using eFC connectomes, the present study found generalizable neural markers characterizing the cb factor and further clarified the cb-factor-eFC representation similarity in the attention and frontoparietal networks. A robust body of evidence has shown that the nodal FCs both within and between the attention and frontoparietal networks were significantly diminished in patients with comorbidity from depression to anxiety (and vice versa) compared to individuals who have not yet developed comorbid conditions43,87,88. Moreover, existing evidence also indicated that the comorbid conditions between depression and anxiety may result from significant neurocognitive impairments in the selective attention and executive control functions in patients, which were also linked to multiple biological system changes (e.g., symptomatology, genetics, and neural circuits)89,90,91. Thus, the current finding substantiated the neural substrates of depression-anxiety comorbidity into the disruptions of attention and cognitive control systems, especially in changes in the sophisticated edge-centric communication patterns underlying these nodal FCs, which may offer the neurotherapeutic targets to the risks of comorbidity in the subclinical conditions.
The present study further identified specific eFC-transcriptional markers of the cb factor, which probed the biological enrichment shaped by the cb-factor-specific gene expression patterns. We found significant biological enrichment in the vasculature that shared genetic associations with the cb-factor-specific eFCs. Recently, the circulating vascular changes, along with ensuing alternations in brain gene expression and morphology, have been identified to be reliable biomarkers for depression92,93. Emerging studies have further demonstrated the interplay between cardiovascular activities and anxiety disorders, where treatment to cardiovascular diseases has been found to offer therapeutic benefits to anxiety disorders94,95. Such associations had been explained by the well-established vascular hypothesis96: the dysfunctional cardiovascular processes brought about overly neuroinflammatory activities in the brain circuits so as to derive neuropsychiatric symptoms, particularly for those in depression and anxiety97,98. Thus, the bridging factor in the depression-anxiety comorbidity may share the common biological processes in the vasculature, which may imply a potential therapeutic target. Moreover, in the present study, the genetic risks of the cb-factor-specific eFCs were also linked to abnormal cerebellar neurodevelopments. As the internal model hypothesis posited, the cerebellar network constituted an internal system to organize and reorganize emotional reactivity, with the disorganization of this internal system predisposing individuals to mood disorders99,100. This hypothesis has been solidly supported by both animal and human research demonstrating cerebellar anomalies in patients with depression-anxiety comorbidity101,102. Recently, the comorbidity between depression and anxiety has been clarified as the reflection of synaptic plasticity changes in the cerebellum, particularly in adolescence, further implying the roles of cerebellar neurodevelopment in the comorbidity. Compared to early childhood, gene expressions to cerebellar developments, especially in its synaptic plasticity and neural circuit construction, were relatively stable from late childhood to adolescences103,104,105. In adolescence, the dysfunctional changes of gene expressions in the cerebellar development have been demonstrated to increase polygenic risks of neuropsychiatric disorders as well as their comorbidity, such as depression and anxiety106,107,108,109. Thus, we may infer that the biological processes involved in dysfunctional cerebellar development from late-middle childhood to young adulthood may potentially increase the risk of developing comorbidity between depression and anxiety by influencing a common factor underlying these bridging symptoms. These connectome-transcriptional markers advanced the comprehension of how the cb factor in the depression-anxiety comorbidity was supported by multiscale neurobiological underpinnings.
However, several limitations should be warranted. We recruited a large-scale subclinical cohort comprised of adults in young adulthood but did not consider the generalization to clinical patients. The generalization to the clinical population could directly strengthen the clinical utility of this cb factor, especially for early intervention and diagnostics of this comorbidity in the future. Related to this concern, this limitation also restricted the applicability of the current findings to older cohorts. Thus, it is highly worthy to extend the conceptual framework of the cb factor to clinical samples that include older individuals. Given the potential risks of false-positive rates in such brain-wide association analysis, the future clinical cohort study could recruit a large-scale sample for this validation. In addition, the present study established this cb factor using specific self-reported symptom questionnaires for depression and anxiety, but it remained unclear whether this cb factor could be generalizable when established with other measurements (e.g., questionnaires, scales, structural diagnostics). Therefore, further validation of the robustness of establishing the cb factor in different measurements is critically needed. The last aspect of limitations was the moderate strength of evidence. These multiscale neurobiological processes of the cb factor were indirectly inferred by correlating with public normative atlases or annotation datasets, rather than examining direct associations between the cb factor and their neurobiological features. Thus, the evidence strength of these neuroimaging-transcriptomic findings could be enhanced by directly measuring correlations between clinical phenotype (e.g., cb factor scores) and endophenotype (e.g., vascular pressure levels)110,111.
In conclusion, we established a common bridging factor (cb factor) to characterize the general structure of these heterogeneous bridging symptoms in the depression-anxiety comorbidity. By adopting the eCPM and RSA models, we identified neural markers that underpinned this cb factor, showing the crucial roles of eFC connectomes within attention and frontoparietal networks to this comorbidity. In an independent twin cohort sample, we revealed the moderate heritability of these cb-factor-specific eFC connectomes. Thus, by aligning with other normative genetic and neurobiological datasets, we identified specific connectome-transcriptional genetic signatures of the cb factor, which further disentangled complex associations of the cb factor to vasculature and cerebellar developments. Collectively, our study offers novel insights to understand the multiscale neurobiological substrates of bridging factors in depression-anxiety comorbidity, thereby enriching the comorbidity hypothesis in the psychopathological theory.
Methods
Ethical statement
All procedures performed in this study were in accordance with the ethical standards of the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. This study protocol has been approved by the Institutional Review Board (IRB) of Southwest University (Faculty of Psychology) (SWUP-19-20160). Written Informed consents are obtained from all the participants. All the participants are paid for their participation by college credit or monetary rewards.
Participants and neuroimaging data acquisition
We included a large-scale subclinical cohort consisting of 2022 adults, with high sociodemographic diversity (e.g., ethnic groups, geographical distributions, 1326 females; age, ranged from 18 to 29, mean/standard deviation, 19.96/2.01) in the Chinese population (Supplementary Tables S1–S4). The neuroimaging data was not influenced by the potential neurobiological effects of COVID-19, as the data collection occurred either before the onset of the pandemic or afterward from participants with no history of SARS-CoV-2 infection. All the neuroimaging data had been collected from a single site. In the neuroimaging analysis, the whole cohort has been grouped into six independent samples for model training, validation, and generalizations (see below). As covariates of no interests, the self-reported sex, age, self-reported handedness, family incomes, mood states, and mind wondering during scanning are adjusted in statistical analyses.
As for behavioral measurements, Zung’s self-report depression scale (SDS) and trait anxiety inventory (TAI) were used to measure symptom severity (see Supplementary Information). The SDS was one of the most widely-used tools to measure depressive symptoms or activities, with prominently good psychometric merits in the general population112,113. This scale contained 20 items describing the depressive symptoms in daily life, with higher scores for severe symptoms by 5-point Likert-formed style114. Moreover, the trait domain of Spielberger’s STAI (STAI-T) was used to measure one’s anxious symptoms in the present study, given the high reliability in subclinical studies115. This STAI-T also included 20 items to depict anxiety-related effects and feelings, which was widely employed for network analysis116,117,118. Data acquisition and preprocessing for neuroimaging of these participants were all in line with our previously standardized pipelines to this dataset (see Supplementary Information)119,120.
Gaussian graph-theoretical model (GGM) and factor analysis
To capture the bridging symptoms in the depression-depression comorbidity, we carried out the Gaussian graph-theoretical (GGM) model to establish the inter-symptom network. Firstly, we calculated the partial correlations between all the items of SDS (Zung’s self-reported scale) and TAI (trait anxiety inventory) to determine the edges of this inter-symptom network. Secondly, the EBICglasso (graphic least absolute shrinkage and selection operator with Extended Bayesian Information Criterion) algorithm was further used to control the false-positive rates by eliminating connections that might be spurious due to weak correlations within this network (see Supplementary Information)6. Thirdly, the final network adjusted from the EBICglasso algorithm was utilized to estimate bridging centrality for each node by five topological metrics, including bridging strength, bridging betweenness, bridging closeness, 1-step bridging expected influence, and 2-step bridging expected influence (see Supplementary Information). The identification of bridge nodes in network analysis has been found to be inconsistent across studies as researchers arbitrarily chose a single metric from these five centrality measurements to discern bridging symptoms between two distinct disorders11,74,121. To tackle this drawback, we estimated the normalized Shannon’s entropy (SE) value that described the extent to which one node had a relatively higher bridging centrality values than others across all the metrics, irrespective of metric selection. A high SE value indicated a high probability of being identified as a bridging node symptom (see Supplementary Information). Thus, nodes would be identified as “bridging symptoms” once their SE values exceeded 0.8122,123. Network reliability and stability have been validated by the Monte Carlo and Bootstrapping simulations (see Supplementary Information). To capture a common factor characterizing a general structure (conceptualized as “the cb factor”) of these bridging symptoms identified above, we conducted a factor analysis that included all these identified bridging symptoms (items). The common factor that decomposed from these symptoms (items) in this analysis was initially conceptualized as a “general factor”. Then, the factor structure was modified to test the model performance of other alternatives (e.g., bifactor, 3-factor, 4-factor structures), in contrast to this general factor structure (see Supplementary Information). Moreover, the Permutation test with 1000 iterations was conducted to estimate the statistical significance of model fitting of this general factor structure. Finally, as in line with a methodology that was used to define the general psychopathological factor (i.e., p factor)20, this conceptualized cb factor scores were calculated by the common factor scores in this factor analysis.
Edge-centric brain functional connectome (eFC) establishment
The Schaefer-100 atlas was used to parcel cortical areas into 100 regions, and the time series of each region were extracted and z-scored firstly. Then, we obtained “edge time series” by calculating the dot products for time series between nodes within to each pair of these 100 nodes. By doing so, all the 4950 (100 × 99/2) “node pairs” gained their “edge time series”. Moreover, the edge-to-edge connectome was built upon by constructing a 4950 × 4950 eFC matrix from correlating each pair of these 4950 “edge time series” for each participant. Finally, all the 12,248,775 (4950 × 4949/2) unique edge-to-edge functional connectivity (eFC) deriving from 4950 “node pairs” have been extracted as neural feature candidates for each participant (see Supplementary Information).
The eFC connectome-based predictive model (eCPM)
In line with the original CPM, we estimated the inter-subject correlations of each eFC to the cb factor scores and retained eFCs whose correlations reached statistical significance (p < 0.05, uncorrected) as thresholding masks. Here, we generated two thresholding masks: one retaining all the eFCs that positively correlated with the cb factor scores (i.e., positive eFC mask), and another retaining all the eFCs that negatively correlated with the cb factor scores (i.e., negative eFC mask). Moreover, the positive (negative) eFC neural feature to each participant was produced by summing r-values of all the eFC values in positive (negative) eFC mask, respectively. Finally, by using the positive (negative) eFC feature as the independent variable (s), we established the machine-learning models with support vector algorithm to predict the cb factor scores in these independent samples by using MATLAB (MathWorks Inc.) (see Supplementary Information). In the present analysis, the participants in the original sample were divided into six groups beforehand for model training, validation, and generalizations. A total of 724 participants in the original sample were grouped into three independent samples as they were independently recruited from three distinct teams in this data project, including discovery sample 1 (n = 241, used for training this model), validation sample 2 (n = 240, used for validating prediction performance of this trained model) and generalization sample 3 (n = 244, used for testing the generalizability of this trained model). For rigorously examining the generalizability of this eCPM, we generated three independent samples from the remaining participants in the original sample, which were highly heterogeneous compared to the discovery sample that was used to train this model (generalization sample 4, n = 133, derived from 29 ethnic minorities in the Chinese population; generalization sample 5, n = 237, derived from the ethnic majority; generalization sample 6, n = 219, scanned after the COVID-19 pandemic).
Inter-subject representation similarity analysis (IS-RSA)
We deployed the inter-subject representation similarity analysis to capture the correlation between multivariate similar patterns of eFCs and the cb factor, which could interpret what eFCs contributed to the aforementioned prediction. Firstly, each “edge-centric” node was vectorized into a 1 × 4949 “connectome pattern” by including all the eFCs correlated to a given “edge-centric” node (no thresholds to these correlations here). Then, the inter-subject correlations (i.e., r-values) of each “connectome pattern” were calculated, and these 1-r-values were used in the inter-subject correlation matrix to generate a neural representation dissimilarity matrix (RDM) for each “edge-centric” node. Further, we built the behavioral RDM by estimating the Euclidean distance of the cb factor scores across all the pairs of participants. Next, we vectorized all the neural RDMs (and one behavioral RDM) using their upper triangular matrices, excluding diagonal. Each neural RDM was then correlated with the vectorized upper triangular matrix of one behavioral RDM by using Spearman’s rank correlation. By doing so, each correlation reflected the representation similarity (RS) of the neural eFC connectome pattern in a given “edge-centric” node to the cb factor, with a positive (negative) r-value for RS (RDS). Statistical significance for these r-values was set to p < .05 with Benjamini–Hochberg correction to address multiple comparison issues.
Quantitative twin ACE model and the edge-centric connectome-transcriptional signatures
The full model with ACE (A, additive genetic effects; C, common environment; E, unique environment) framework has been established to clarify the heritability of eFC connectome patterns that identified significant RS in the above analysis. Specifically, we decomposed variances of additive genetic effects (A) from the latent factor model for 127 pairs of monozygotic twins and 118 pairs of dizygotic twins (Beijing Twin Study Dataset; see Supplementary Information). Model performances were further compared to these nested submodels dropping out latent factor (s) (e.g., AE or E), in order to determine the optimal model. Quantitative heritability was finally estimated from this optimal model once the statistical significance of Δχ2 was no longer less than 0.5.
Preprocessing of the AHBA dataset followed the standardized pipeline and generated a gene-brain matrix (10,027 genes × 100 parcels) by aligning these gene expression levels into a brain spatial map based on the Schaefer-100 atlas. In addition, we extracted the eFC-cb-factor scores RS vectors (1 RS × 100 parcels) representing the edge-centric neural phenotype of this bridging factor. To capture connectome-transcriptional signatures, we carried out the partial least square (PLS) regression model by fitting the gene-brain matrix (10,027 × 100, independent variables) into this RS vector (1 × 100, dependent variable) (see Supplementary Information). To obviate over-fitting, this PLS regression model has been validated by using the 10-fold cross-validation method. The permutation test (at n = 5000) was used to estimate the statistical significance of each component of this PLS model. Further, the bootstrapping method with n = 5000 was deployed to estimate weights and corresponding statistics (Z-values) for these genes. To balance both Type-I and Type-II errors, the statistical threshold was set to Z > 3 (PLS+) or Z < −3 (PLS-). We finally extracted genes that reached the predefined statistical threshold aforementioned from the first and second components (PLS1 and PLS2), as in line with technical guidelines in such imaging-transcriptomic analysis111.
We capitalized on Gene Annotation by the Macroscale Brain-imaging Association (GAMBA) dataset to estimate associations between these genes that we identified above and brain features, as well as the risks of neurological/psychiatric diseases. These brain features included macroscale brain networks, brain cognitive ontology, cognitive terms, and cortical expansion and metabolism (see Supplementary Information). The statistical significance for each model has been estimated by the ensemble-based (null-brain-gene) model, with Bonferroni–Holm correction.
Finally, we deployed the “Metascape”124 and “Specific Expression Analysis (SEA)” datasets to delineate functional processes that were enriched from gene sets identified in PLS1 and PLS2, respectively. The gene set was used as input for this platform and was further annotated by multiple biological databases (see Supplementary Information). To assess gene enrichment specificity, the specificity index probability (pSI) is used to determine how significantly a gene set is enriched in a particular tissue compared to others across varied thresholds in general background enrichment125,126. The statistical significance of such enrichment for this given gene set was estimated, with Benjamini–Hochberg FDR corrections. The above estimations were implemented by either an online interactive platform or an R package (e.g., pSI).
Data availability
The data generated in this study to reproduce findings/figures have been deposited in the Open Science Framework (OSF) repository under accession code (https://osf.io/nu32z/). The data used for gene expression are available in the Allen Human Brain Atlas (AHBA) database under accession code (https://human.brain-map.org/static/download). The data used for genetic enrichment analysis are available in Metascape databases (https://metascape.org/gp/index.html) and GAMBA databases (http://dutchconnectomelab.nl/GAMBA/), with fully open accesses. The processed data for representation similarity analysis (RSA) are provided in the Supplementary Information. The raw genetic neuroimaging data are available under restricted access for data legal restrictions. Specifically, these genetic neuroimaging data are collected by the GGBBP Data Collection Project, which has not yet completed and is expected to be fully done by May 2028. Thus, as biometric data in an ongoing project, the legal censorship to share them from data regulation authorities is not available at this time, until this project is fully completed. For scientific collaborations or research purposes, any researchers can obtain access to these data by consulting the joint supervision team of GGBBP (PIs: H.Q.H., Q.J., F.T.Y., C.H., and L.X.). Response to reasonable access requests is expected to be done within two weeks. The expiration date to access these data is no more than three months since the data request was approved. Source data are provided with this paper.
Code availability
Codes for estimating gradients and PLS components can be found at the GitHub repository from Xia et al. (https://github.com/mingruixia/MDD_ConnectomeGradient). Codes and software for reproducing eCPM analysis can be accessed at Neurospider (https://neurospider.cn/post/62271/) and Github repository from Faskowitz et al. (https://github.com/brain-networks). Codes for SVM is openly available at https://www.csie.ntu.edu.tw/~cjlin/libsvm/, without access restrictions. Brain maps and plots were built by Surf Ice (https://www.nitrc.org/projects/surfice) and R packages (i.e., ggsegSchaefer, ggplot, ggseg). Self-custom codes have been deposited at Open Science Framework (https://osf.io/nu32z/).
References
Substance Abuse and Mental Health Services Administration. Key Substance Use and Mental Health Indicators in the United States: Results from the 2017 National Survey on Drug Use and Health (HHS Publication No. SMA 18-5068, NSDUH Series H-53) (Center for Behavioral Health Statistics and Quality, Substance Abuse and Mental Health Services Administration, 2018).
Kalin, N. H. The critical relationship between anxiety and depression. Am. J. Psychiatry 177, 365–367 (2020).
Chen, L. C. et al. Antidepressant resistance in adolescents with major depressive disorder: a nationwide longitudinal study. J. Affect. Disord. 262, 293–297 (2020).
Furukawa, T. A. et al. Cognitive-behavioral analysis system of psychotherapy, drug, or their combination for persistent depressive disorder: personalizing the treatment choice using individual participant data network metaregression. Psychother. Psychosom. 87, 140–153 (2018).
Borsboom, D. Psychometric perspectives on diagnostic systems. J. Clin. Psychol. 64, 1089–1108 (2008).
Borsboom, D. & Cramer, A. O. Network analysis: an integrative approach to the structure of psychopathology. Annu. Rev. Clin. Psychol. 9, 91–121 (2013).
Cramer, A. O. et al. Major depression as a complex dynamic system. PLoS ONE 11, e0167490 (2016).
Jones, P. J., Ma, R. & McNally, R. J. Bridge centrality: a network approach to understanding comorbidity. Multivar. Behav. Res. 56, 353–367 (2021).
Konac, D., Young, K. S., Lau, J. & Barker, E. D. Comorbidity between depression and anxiety in adolescents: bridge symptoms and relevance of risk and protective factors. J. Psychopathol. Behav. Assess. 43, 583–596 (2021).
Castro, D. et al. The differential role of central and bridge symptoms in deactivating psychopathological networks. Front. Psychol. 10, 2448 (2019).
Robinaugh, D. J., Hoekstra, R. H. A., Toner, E. R. & Borsboom, D. The network approach to psychopathology: a review of the literature 2008-2018 and an agenda for future research. Psychol. Med. 50, 353–366 (2020).
Groen, R. N. et al. Comorbidity between depression and anxiety: assessing the role of bridge mental states in dynamic psychological networks. BMC Med. 18, 308 (2020).
Wang, Y., Li, Z. & Cao, X. Investigating the network structure and causal relationships among bridge symptoms of comorbid depression and anxiety: a Bayesian network analysis. J. Clin. Psychol. 80, 1271–1285 (2024).
Mihić, L. et al. Comorbidity among depression, anxiety and stress symptoms in naturalistic clinical samples: a cross-cultural network analysis. Clin. Psychol. Psychother. 31, e2927 (2023).
Kaiser, T., Herzog, P., Voderholzer, U. & Brakemeier, E. L. Unraveling the comorbidity of depression and anxiety in a large inpatient sample: network analysis to examine bridge symptoms. Depress. Anxiety 38, 307–317 (2021).
Dobson, E. T. et al. Bridging anxiety and depression: a network approach in anxious adolescents. J. Affect. Disord. 280, 305–314 (2021).
Sánchez Hernández, M. O., Carrasco, M. A. & Holgado-Tello, F. P. Anxiety and depression symptoms in Spanish children and adolescents: an exploration of comorbidity from the network perspective. Child Psychiatry Hum. Dev. 54, 736–749 (2023).
Fan, P., Wang, T. T., Wang, J. W. & Wang, J. Y. Network analysis of comorbid depression and anxiety and their associations with response style among adolescents with subthreshold depression. Curr. Psychol. 43, 8665–8674 (2024).
Frewen, P. A., Schmittmann, V. D., Bringmann, L. F. & Borsboom, D. Perceived causal relations between anxiety, posttraumatic stress and depression: extension to moderation, mediation, and network analysis. Eur. J. Psychotraumatol. 4, 20656 (2013).
Caspi, A. et al. The p factor: one general psychopathology factor in the structure of psychiatric disorders? Clin. Psychol. Sci. 2, 119–137 (2014).
Caspi, A. & Moffitt, T. E. All for one and one for all: mental disorders in one dimension. Am. J. Psychiatry 175, 831–844 (2018).
Watts, A. L., Greene, A. L., Bonifay, W. & Fried, E. I. A critical evaluation of the p-factor literature. Nat. Rev. Psychol. 3, 108–122 (2024).
Borsboom, D., Cramer, A. O. J. & Kalis, A. Brain disorders? Not really: why network structures block reductionism in psychopathology research. Behav. Brain Sci. 42, e2 (2018).
Kendler, K. S., Zachar, P. & Craver, C. What kinds of things are psychiatric disorders? Psychol. Med. 41, 1143–1150 (2011).
Kotov, R. et al. The hierarchical taxonomy of psychopathology (HiTOP): a dimensional alternative to traditional nosologies. J. Abnorm. Psychol. 126, 454–477 (2017).
Kotov, R., Krueger, R. F. & Watson, D. A paradigm shift in psychiatric classification: the hierarchical taxonomy of psychopathology (HiTOP). World Psychiatry 17, 24–25 (2018).
Forbes, M. K. et al. Principles and procedures for revising the hierarchical taxonomy of psychopathology. J. Psychopathol. Clin. Sci. 133, 4–19 (2024).
Conway, C. C., Kotov, R., Krueger, R. F. & Caspi, A. Translating the hierarchical taxonomy of psychopathology (HiTOP) from potential to practice: ten research questions. Am. Psychol. 78, 873–885 (2023).
Cuthbert, B. N. Research domain criteria: toward future psychiatric nosologies. Dialogues Clin. Neurosci. 17, 89–97 (2015).
Bedwell, J. S., Spencer, C. C. & O’donnell, J. P. Promise and challenges with the research domain criteria framework. In Adult Psychopathology and Diagnosis. 8th edn, Ch. 2, 33–44 (Wiley, 2018).
Lilienfeld, S. O. The research domain criteria (RDoC): an analysis of methodological and conceptual challenges. Behav. Res Ther. 62, 129–139 (2014).
Fried, E. I. & Cramer, A. O. J. Moving forward: challenges and directions for psychopathological network theory and methodology. Perspect. Psychol. Sci. 12, 999–1020 (2017).
Fried, E. I. et al. Mental disorders as networks of problems: a review of recent insights. Soc. Psychiatry Psychiatr. Epidemiol. 52, 1–10 (2017).
Blanco, C. et al. The latent structure and comorbidity patterns of generalized anxiety disorder and major depressive disorder: a national study. Depress. Anxiety 31, 214–222 (2014).
Kelly, C., Biswal, B. B., Craddock, R. C., Castellanos, F. X. & Milham, M. P. Characterizing variation in the functional connectome: promise and pitfalls. Trends Cogn. Sci. 16, 181–188 (2012).
Finn, E. S. et al. Functional connectome fingerprinting: identifying individuals using patterns of brain connectivity. Nat. Neurosci. 18, 1664–1671 (2015).
Drysdale, A. T. et al. Resting-state connectivity biomarkers define neurophysiological subtypes of depression. Nat. Med. 23, 28–38 (2017).
Goldstein-Piekarski, A. N. et al. Mapping neural circuit biotypes to symptoms and behavioral dimensions of depression and anxiety. Biol. Psychiatry 91, 561–571 (2022).
Williams, L. M. Precision psychiatry: a neural circuit taxonomy for depression and anxiety. Lancet Psychiatry 3, 472–480 (2016).
Korgaonkar, M. S., Goldstein-Piekarski, A. N., Fornito, A. & Williams, L. M. Intrinsic connectomes are a predictive biomarker of remission in major depressive disorder. Mol. Psychiatry 25, 1537–1549 (2020).
Williams, L. M. Defining biotypes for depression and anxiety based on large-scale circuit dysfunction: a theoretical review of the evidence and future directions for clinical translation. Depress. Anxiety 34, 9–24 (2017).
Bassett, D. S., Zurn, P. & Gold, J. I. On the nature and use of models in network neuroscience. Nat. Rev. Neurosci. 19, 566–578 (2018).
Briley, P. M. et al. Resting-state functional connectivity correlates of anxiety co-morbidity in major depressive disorder. Neurosci. Biobehav. Rev. 138, 104701 (2022).
Pannekoek, J. N. et al. Investigating distinct and common abnormalities of resting-state functional connectivity in depression, anxiety, and their comorbid states. Eur. Neuropsychopharmacol. 25, 1933–1942 (2015).
Brandl, F. et al. Common and specific large-scale brain changes in major depressive disorder, anxiety disorders, and chronic pain: a transdiagnostic multimodal meta-analysis of structural and functional MRI studies. Neuropsychopharmacology 47, 1071–1080 (2022).
Janiri, D. et al. Shared neural phenotypes for mood and anxiety disorders: a meta-analysis of 226 task-related functional imaging studies. JAMA Psychiatry 77, 172–179 (2020).
Faskowitz, J., Esfahlani, F. Z., Jo, Y., Sporns, O. & Betzel, R. F. Edge-centric functional network representations of human cerebral cortex reveal overlapping system-level architecture. Nat. Neurosci. 23, 1644–1654 (2020).
Jo, Y., Faskowitz, J., Esfahlani, F. Z., Sporns, O. & Betzel, R. F. Subject identification using edge-centric functional connectivity. Neuroimage 238, 118204 (2021).
Jo, Y. et al. The diversity and multiplexity of edge communities within and between brain systems. Cell Rep. 37, 110032 (2021).
Yang, B. et al. Edge-centric functional network analyses reveal disrupted network configuration in autism spectrum disorder. J. Affect. Disord. 336, 74–80 (2023).
Idesis, S. et al. Edge-centric analysis of stroke patients: an alternative approach for biomarkers of lesion recovery. Neuroimage Clin. 35, 103055 (2022).
Zamani Esfahlani, F. et al. Edge-centric analysis of time-varying functional brain networks with applications in autism spectrum disorder. Neuroimage 263, 119591 (2022).
Betzel, R. F., Faskowitz, J. & Sporns, O. Living on the edge: network neuroscience beyond nodes. Trends Cogn. Sci. 27, 1068–1084 (2023).
Novelli, L. & Razi, A. A mathematical perspective on edge-centric brain functional connectivity. Nat. Commun. 13, 2693 (2022).
Moreau, C. A. et al. Brain functional connectivity mirrors genetic pleiotropy in psychiatric conditions. Brain 146, 1686–1696 (2023).
Taquet, M. et al. A structural brain network of genetic vulnerability to psychiatric illness. Mol. Psychiatry 26, 2089–2100 (2021).
Romme, I. A., de Reus, M. A., Ophoff, R. A., Kahn, R. S. & van den Heuvel, M. P. Connectome disconnectivity and cortical gene expression in patients with schizophrenia. Biol. Psychiatry 81, 495–502 (2017).
Xia, M. et al. Connectome gradient dysfunction in major depression and its association with gene expression profiles and treatment outcomes. Mol. Psychiatry 27, 1384–1393 (2022).
Martins, D. et al. Imaging transcriptomics: convergent cellular, transcriptomic, and molecular neuroimaging signatures in the healthy adult human brain. Cell Rep. 37, 110173 (2021).
Fornito, A., Arnatkevičiūtė, A. & Fulcher, B. D. Bridging the gap between connectome and transcriptome. Trends Cogn. Sci. 23, 34–50 (2019).
Selvaggi, P., Rizzo, G., Mehta, M. A., Turkheimer, F. E. & Veronese, M. Integration of human whole-brain transcriptome and neuroimaging data: practical considerations of current available methods. J. Neurosci. Methods 355, 109128 (2021).
Buch, A. M. & Liston, C. Dissecting diagnostic heterogeneity in depression by integrating neuroimaging and genetics. Neuropsychopharmacology 46, 156–175 (2021).
Talishinsky, A. et al. Regional gene expression signatures are associated with sex-specific functional connectivity changes in depression. Nat. Commun. 13, 5692 (2022).
Ji, G. J. et al. White matter dysfunction in psychiatric disorders is associated with neurotransmitter and genetic profiles. Nat. Ment. Health 1, 655–666 (2023).
Hettwer, M. D. et al. Coordinated cortical thickness alterations across six neurodevelopmental and psychiatric disorders. Nat. Commun. 13, 6851 (2022).
Ardesch, D. J., Libedinsky, I., Scholtens, L. H., Wei, Y. & van den Heuvel, M. P. Convergence of brain transcriptomic and neuroimaging patterns in schizophrenia, bipolar disorder, autism spectrum disorder, and major depressive disorder. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 8, 630–639 (2023).
Wray, N. R. et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 50, 668–681 (2018).
Flint, J. & Kendler, K. S. The genetics of major depression. Neuron 81, 484–503 (2014).
Howard, D. M. et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 22, 343–352 (2019).
Shen, X. et al. Using connectome-based predictive modeling to predict individual behavior from brain connectivity. Nat. Protoc. 12, 506–518 (2017).
Yarkoni, T. & Westfall, J. Choosing prediction over explanation in psychology: lessons from machine learning. Perspect. Psychol. Sci. 12, 1100–1122 (2017).
Rosenblatt, M. et al. Data leakage inflates prediction performance in connectome-based machine learning models. Nat. Commun. 15, 1829 (2024).
Contreras, A., Nieto, I., Valiente, C., Espinosa, R. & Vazquez, C. The study of psychopathology from the network analysis perspective: a systematic review. Psychother. Psychosom. 88, 71–83 (2019).
McNally, R. J. Network analysis of psychopathology: controversies and challenges. Annu. Rev. Clin. Psychol. 17, 31–53 (2021).
Christensen, A., Garrido, L. & Golino, H. What is bridge centrality? A comment on Jones, Ma, and McNally https://osf.io/preprints/psyarxiv/a8svr (2019).
Ding, F. Q. et al. Network comparison analysis of comorbid depression and anxiety disorder in a large clinical sample before and after treatment. Curr. Psychol. 43, 13267–13278 (2024).
Garabiles, M. R., Lao, C. K., Xiong, Y. & Hall, B. J. Exploring comorbidity between anxiety and depression among migrant Filipino domestic workers: a network approach. J. Affect. Disord. 250, 85–93 (2019).
Ren, L. et al. Network structure of depression and anxiety symptoms in Chinese female nursing students. BMC Psychiatry 21, 279 (2021).
Xie, C. et al. A shared neural basis underlying psychiatric comorbidity. Nat. Med. 29, 1232–1242 (2023).
Neumann, A. et al. Combined polygenic risk scores of different psychiatric traits predict general and specific psychopathology in childhood. J. Child Psychol. Psychiatry 63, 636–645 (2022).
Martin, A. R., Daly, M. J., Robinson, E. B., Hyman, S. E. & Neale, B. M. Predicting polygenic risk of psychiatric disorders. Biol. Psychiatry 86, 97–109 (2019).
Kim, H. & Eaton, N. R. The hierarchical structure of common mental disorders: connecting multiple levels of comorbidity, bifactor models, and predictive validity. J. Abnorm. Psychol. 124, 1064–1078 (2015).
Blanco, C. et al. The space of common psychiatric disorders in adolescents: comorbidity structure and individual latent liabilities. J. Am. Acad. Child Adolesc. Psychiatry 54, 45–52 (2015).
de Reus, M. A., Saenger, V. M., Kahn, R. S. & van den Heuvel, M. P. An edge-centric perspective on the human connectome: link communities in the brain. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369, 20130527 (2014).
Wang, W. P. et al. Edge-centric functional network reveals new spatiotemporal biomarkers of early mild cognitive impairment. Brain‐X 1, e35 (2023).
Dai, W. et al. Abnormal thalamo-cortical interactions in overlapping communities of migraine: an edge functional connectivity study. Ann. Neurol. 94, 1168–1181 (2023).
Beckmann, F. E. et al. Specific alterations of resting-state functional connectivity in the triple network related to comorbid anxiety in major depressive disorder. Eur. J. Neurosci. 59, 1819–1832 (2024).
Crane, N. A. et al. Comorbid anxiety increases cognitive control activation in major depressive disorder. Depress. Anxiety 33, 967–977 (2016).
Keller, A. S., Leikauf, J. E., Holt-Gosselin, B., Staveland, B. R. & Williams, L. M. Paying attention to attention in depression. Transl. Psychiatry 9, 279 (2019).
Jenkins, L. M. et al. Differential engagement of cognitive control regions and subgenual cingulate based upon presence or absence of comorbid anxiety with depression. J. Affect. Disord. 241, 371–380 (2018).
Sindermann, L. et al. Systematic transdiagnostic review of magnetic-resonance imaging results: depression, anxiety disorders and their co-occurrence. J. Psychiatr. Res. 142, 226–239 (2021).
Dion-Albert, L. et al. Vascular and blood-brain barrier-related changes underlie stress responses and resilience in female mice and depression in human tissue. Nat. Commun. 13, 164 (2022).
Burrage, E., Marshall, K. L., Santanam, N. & Chantler, P. D. Cerebrovascular dysfunction with stress and depression. Brain Circ. 4, 43–53 (2018).
Repova, K., Aziriova, S., Krajcirovicova, K. & Simko, F. Cardiovascular therapeutics: a new potential for anxiety treatment? Med. Res. Rev. 42, 1202–1245 (2022).
Tully, P. J., Harrison, N. J., Cheung, P. & Cosh, S. Anxiety and cardiovascular disease risk: a review. Curr. Cardiol. Rep. 18, 120 (2016).
Hodes, G. E., Kana, V., Menard, C., Merad, M. & Russo, S. J. Neuroimmune mechanisms of depression. Nat. Neurosci. 18, 1386–1393 (2015).
Cohen, B. E., Edmondson, D. & Kronish, I. M. State of the art review: depression, stress, anxiety, and cardiovascular disease. Am. J. Hypertens. 28, 1295–1302 (2015).
Zhang, L., Bao, Y., Tao, S., Zhao, Y. & Liu, M. The association between cardiovascular drugs and depression/anxiety in patients with cardiovascular disease: a meta-analysis. Pharm. Res. 175, 106024 (2022).
Hilber, P. et al. Cooperation of the vestibular and cerebellar networks in anxiety disorders and depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 89, 310–321 (2019).
Hilber, P. The role of the cerebellar and vestibular networks in anxiety disorders and depression: the internal model hypothesis. Cerebellum 21, 791–800 (2022).
Hoppenbrouwers, S. S., Schutter, D. J., Fitzgerald, P. B., Chen, R. & Daskalakis, Z. J. The role of the cerebellum in the pathophysiology and treatment of neuropsychiatric disorders: a review. Brain Res. Rev. 59, 185–200 (2008).
Cheng, B. et al. Social support mediates the influence of cerebellum functional connectivity strength on postpartum depression and postpartum depression with anxiety. Transl. Psychiatry 12, 54 (2022).
Bakken, T. E. et al. A comprehensive transcriptional map of primate brain development. Nature 535, 367–375 (2016).
White, J. J. & Sillitoe, R. V. Development of the cerebellum: from gene expression patterns to circuit maps. Wiley Interdiscip. Rev. Dev. Biol. 2, 149–164 (2013).
Butts, T., Green, M. J. & Wingate, R. J. Development of the cerebellum: simple steps to make a ‘little brain’. Development 141, 4031–4041 (2014).
Zhang, C. et al. Arsenic downregulates gene expression at the postsynaptic density in mouse cerebellum, including genes responsible for long-term potentiation and depression. Toxicol. Lett. 228, 260–269 (2014).
Fatemi, S. H., Folsom, T. D., Rooney, R. J. & Thuras, P. D. Expression of GABAA α2-, β1- and ε-receptors are altered significantly in the lateral cerebellum of subjects with schizophrenia, major depression and bipolar disorder. Transl. Psychiatry 3, e303 (2013).
Huang, M., de Koning, T. J., Tijssen, M. A. J. & Verbeek, D. S. Cross-disease analysis of depression, ataxia and dystonia highlights a role for synaptic plasticity and the cerebellum in the pathophysiology of these comorbid diseases. Biochim. Biophys. Acta Mol. Basis Dis. 1867, 165976 (2021).
Chin, P. W. & Augustine, G. J. The cerebellum and anxiety. Front. Cell. Neurosci. 17, 1130505 (2023).
Mandal, A. S., Gandal, M., Seidlitz, J. & Alexander-Bloch, A. A critical appraisal of imaging transcriptomics. Biol. Psychiatry Glob. Open Sci. 2, 311–313 (2022).
Arnatkeviciute, A., Markello, R. D., Fulcher, B. D., Misic, B. & Fornito, A. Toward best practices for imaging transcriptomics of the human brain. Biol. Psychiatry 93, 391–404 (2023).
Chen, I. H., Lin, C. Y., Zheng, X. & Griffiths, M. D. Assessing mental health for china’s police: psychometric features of the self-rating depression scale and symptom checklist 90-revised. Int. J. Environ. Res. Public Health 17, 2737 (2020).
Lu, J. et al. Prevalence of depressive disorders and treatment in China: a cross-sectional epidemiological study. Lancet Psychiatry 8, 981–990 (2021).
Zung, W. W., Richards, C. B. & Short, M. J. Self-rating depression scale in an outpatient clinic. Further validation of the SDS. Arch. Gen. Psychiatry 13, 508–515 (1965).
Spielberger, C. D. State-trait anxiety inventory for adults (STAI-AD). APA PsycTests https://doi.org/10.1037/t06496-000 (1983).
Balsamo, M. et al. The state-trait anxiety inventory: shadows and lights on its construct validity. J. Psychopathol. Behav. Assess. 35, 475–486 (2013).
Slotta, T., Witthöft, M., Gerlach, A. L. & Pohl, A. The interplay of interoceptive accuracy, facets of interoceptive sensibility, and trait anxiety: a network analysis. Pers. Individ. Dif. 183, 111133 (2021).
Heeren, A., Bernstein, E. E. & McNally, R. J. Deconstructing trait anxiety: a network perspective. Anxiety Stress Coping 31, 262–276 (2018).
Zhang, R., Chen, Z. & Feng, T. The triple psychological and neural bases underlying procrastination: evidence based on a two-year longitudinal study. Neuroimage 283, 120443 (2023).
He, L. et al. Functional connectome prediction of anxiety related to the COVID-19 pandemic. Am. J. Psychiatry 178, 530–540 (2021).
Bringmann, L. F. & Eronen, M. I. Don’t blame the model: reconsidering the network approach to psychopathology. Psychol. Rev. 125, 606–615 (2018).
Wu, S., Fu, Y., Shen, H. & Liu, F. Using ranked weights and Shannon entropy to modify regional sustainable society index. Sustain. Cities Soc. 41, 443–448 (2018).
Busu, C. & Busu, M. Modeling the circular economy processes at the EU level using an evaluation algorithm based on Shannon entropy. Processes 6, 225 (2018).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019).
Dougherty, J. D., Schmidt, E. F., Nakajima, M. & Heintz, N. Analytical approaches to RNA profiling data for the identification of genes enriched in specific cells. Nucleic Acids Res. 38, 4218–4230 (2010).
Xu, X., Wells, A. B., O’Brien, D. R., Nehorai, A. & Dougherty, J. D. Cell type-specific expression analysis to identify putative cellular mechanisms for neurogenetic disorders. J. Neurosci. 34, 1420–1431 (2014).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (32300907, C.Z.Y.; 32271123, F.T.Y.), PLA Talent Program Foundation (2022160258, C.Z.Y.), AMU-RD Scholar Foundation (202211001, C.Z.Y.), Key Projects for Technological Innovation and Application Development in Chongqing (CSTB2022TIADKPX0150, F.T.Y.), Innovation Research 2035 Pilot Plan of Southwest University (SWUPilotPlan006, F.T.Y.) and the National Key Research and Development Program of China (2022YFC2705201, F.T.Y.).
Author information
Authors and Affiliations
Contributions
C.Z.Y., and F.T.Y.: Conceptualization, Methodology, Software, Visualization, and Writing - Original Draft, Project administration and Funding acquisition; L.X.R., X.T., Z.R., L.W., and T.Y.C.: Data Curation, Methodology and Validation; X.L., Z.J.X., X.Z.B., H.Y.Y., and H.B.W.: Data Curation; C.J., and Q.S.Z.: Writing - Review & Editing and Methodology; Z.Y., H.Q.H., Q.J., L.X., C.H., and F.Z.Z.: Project Administration, Validation and Data Curation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yumeng Ju and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, Z., Tang, Y., Liu, X. et al. Edge-centric connectome-genetic markers of bridging factor to comorbidity between depression and anxiety. Nat Commun 15, 10560 (2024). https://doi.org/10.1038/s41467-024-55008-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55008-0
This article is cited by
-
Cognitive and neural mechanisms of mental imagery supporting creative cognition
Communications Biology (2025)
