
Abstract
The 2015–2016 Zika virus (ZIKV) outbreak in the Americas revealed the ability of ZIKV from the Asian lineage to cause birth defects, generically called congenital Zika syndrome (CZS). Notwithstanding the long circulation history of Asian ZIKV, no ZIKV-associated CZS cases were reported prior to the outbreaks in French Polynesia (2013) and Brazil (2015). Whether the sudden emergence of CZS resulted from an evolutionary event of Asian ZIKV has remained unclear. We performed a comparative analysis of the pathogenicity of pre-epidemic and epidemic Asian ZIKV strains in mouse embryonic brains using a female immunocompetent intraplacental infection mouse model. All studied Asian ZIKV strains are neurovirulent, but pre-epidemic strains are consistently more pathogenic in the embryos than their epidemic equivalents. Pathogenicity is not directly linked to viral replication. By contrast, an influx of macrophages/microglial cells is noted in infected fetal brains for both pre-epidemic and epidemic ZIKV strains. Moreover, all tested ZIKV strains trigger an immunological response, whereby the intensity of the response differs between strains, and with epidemic ZIKV strains generally mounting a more attenuated immunostimulatory response. Our study reveals that Asian ZIKV strains evolved towards pathogenic attenuation, potentially resulting in CZS emergence in neonates rather than premature death in utero.
Similar content being viewed by others

Introduction
The Zika virus (ZIKV) was first discovered in 1947 during a yellow fever surveillance study amongst monkeys in the Zika forest. During the 1950’s and 1960’s, ZIKV has circulated in human populations in Africa and the Asian-Pacific regions, as evidenced by the detection of antibodies in humans and its first isolation from a human1, causing only sporadic outbreaks associated with no or only mild symptomatic cases2. As a result, ZIKV was not considered a major health threat. However, between 2015 and 2016, ZIKV caused a massive outbreak in the Americas that was associated with an increased number of newborns with congenital Zika syndrome (CZS)3, which encompasses a wide spectrum of birth defects, including microcephaly (smaller skull size), ventriculomegaly (enlarged ventricles), calcification of subcortical regions, neurologic impairment, ocular anomalies, and joint contractures4,5. The emergence of these unprecedented ZIKV-associated devastating neonatal abnormalities led the World Health Organization (WHO) to declare the 2015-ZIKV outbreak a public health emergency of international concern. Since then, various research groups have explored the underlying pathophysiological mechanisms that may explain the high neurovirulence of contemporary ZIKV strains. ZIKV has been reported to be able to infect various cell types in the brain (in vitro and in vivo), such as neural progenitors6,7,8,9,10, mature neurons6,11, astrocytes12,13,14,15,16, oligodendrocytes17,18,19, and microglia20,21,22,23. ZIKV infection of brain cells can induce various deleterious effects, including cell death7,9,24,25, premature differentiation of neural progenitors26,27,28, and secretion of inflammatory factors20,29,30, which could ultimately lead to the development of a microcephaly-like phenotype. Although these studies provided very important insights into the pathophysiological mechanisms underlying ZIKV-associated brain defects, the question on this precipitous emergence remained unanswered. The sudden increase in cases of CZS were linked to ZIKV from the Asian lineage31, which had evolved from the ancestral African lineage32. Several research groups demonstrated that ZIKV from both the African and Asian lineage is equally capable of infecting the placenta33,34 and brain cells35,36,37. Intriguingly, ZIKV from the African lineage was reported to display a more pathogenic profile than ZIKV from the Asian lineage, making it more likely to be associated with miscarriages and spontaneous abortions than with birth defects35,36,38,39,40. Several studies aimed to identify potential genetic determinants of the sudden emergence of ZIKV-associated brain defects caused by the Asian lineage ZIKV41,42,43,44,45, but these yielded conflicting results35,36,41,43. Moreover, some point mutations that were reported to affect the virulence of a particular ZIKV strain did not result in the same effect in the background of other ZIKV strains46. The explanation for the discrepancies in the reported findings is likely multifactorial, such as the use of different mouse models, inoculums, routes of infection, and ZIKV strains, amongst others, increasing the variability in the neurovirulence findings and complicating direct comparisons across studies.
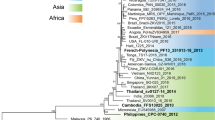
To more comprehensively decipher the relationship between the evolution of Asian lineage ZIKV and the incidence of CZS, we performed a comparative study employing multiple pre-epidemic ZIKV strains (which arose before the 2013-epidemic French-Polynesia strain [PF_2013]), and epidemic ZIKV strains (evolving from PF_2013)32. To recapitulate most accurately first trimester ZIKV infections in humans, which are associated with more severe cerebral pathology in newborns47, we deployed the intraplacental challenge model to infect mouse embryos in utero at a very early stage of embryonic brain development. This infection route better mimics the natural dissemination of ZIKV from the placenta to the fetus (thus, once vertically transmitted), giving the virus access to the developing brain of the embryonic mice. Using this model, we additionally investigated the impact of the S139N amino acid change that was reported to impact fetal brain development41.
Results
Pre-epidemic ZIKV strains cause severe brain defects more consistently than epidemic ZIKV strains
Based on a comparative phylogenetic study by Pettersson and colleagues32, we selected a panel of three pre-epidemic ZIKV (preZIKV) strains (CAM_2010, PHL_2012 and THA_2014) and five epidemic ZIKV (epZIKV) strains (PF_2013, PR_2015, BRA_2015, MEX_2016, and SUR_2016); see Supplementary Table 1 for details. To compare the neurovirulent potential of preZIKV and epZIKV strains, intraplacental (IPL, 5 × 105 PFU/ml) injections were performed at embryonic day 10.5 (E10.5) in immunocompetent SWISS dams (Supplementary Fig. 1), which is a time window corresponding to the first trimester of pregnancy in humans48. As the preZIKV strain CAM_2010 elicited massive fetal death before E18.5 with only 3 harvestable embryos, results obtained with this strain were excluded from all analyses due to the small sample size. Eight days post-infection (pi) at E18.5, we assessed the severity of the brain defects as well as the presence of a microcephaly-like phenotype and ventriculomegaly by measuring the brain weight, the thickness of the cortex, the number of cells present in the cortex (DAPI-positive cells), and the enlargement of the ventricle area. Both preZIKV and epZIKV were capable of inducing brain defects in embryonic mice (Fig. 1). However, brains infected by preZIKV were on average lower in weight than those infected by epZIKV (Fig. 1a), suggesting that preZIKV triggered more severe brain defects. Indeed, although both preZIKV and epZIKV caused a microcephaly-like phenotype, illustrated by a reduction in the cortical length and fewer cells in the cortex compared to mock-infected brains (Fig. 1b, c), preZIKV-infected embryonic brains consistently presented a more severe microcephaly-like phenotype with smaller cortices and fewer cells than epZIKV-infected embryonic brains (Fig. 1a–d). Similarly, ventriculomegaly was observed both in the preZIKV- and epZIKV-infected brains, but with a more significant enlargement of the ventricle area in brains infected by preZIKV (Fig. 1d). Even more striking was that all embryonic brains infected with preZIKV displayed an enlargement of the ventricular region. In contrast, a significant number of brains (35%, n = 24/68; p < 0.0001) infected by epZIKV did not present ventriculomegaly because the size of the ventricular area was comparable or smaller than the maximum ventricular opening observed in mock-infected brains (represented by the dashed line in Fig. 1d). This is for example clearly demonstrated for BRA_2015 (Fig. 1e). This ZIKV strain induced a wide range of ventriculomegaly in the embryonic brain, ranging from almost non-detectable (middle panels Fig. 1e) to severe ventriculomegaly (right panels Fig. 1e), as did all the other epZIKV strains (Supplementary Fig. 2d). A similar trend in variation was observed in brain weight, the thickness of the cortex and the number of cells present in the cortex of brains infected by epZIKV (Supplementary Fig. 2a–c). Thus, embryonic brains infected by epZIKV displayed a broader spectrum of severities, varying from unnoticeable brain defects to an extremely deleterious phenotype, while brains infected by preZIKV exhibited more uniformly severe brain abnormalities.

a–d Measured brain defects in embryonic mouse brains at E18.5 following mock-infection (black, n = 26 for brain weight; n = 12 for the others) or after infection by either pre-epidemic ZIKV strains (blue, n = 28) or epidemic ZIKV strains (purple, n = 68). Embryonic brains were examined morphologically by measuring the brain weight normalized to the head weight (a). Microcephaly-like phenotypes were assessed by measuring the cortical length (b) and the number of DAPI-positive cells (c). Ventriculomegaly was estimated by measuring the ventricle area (d). e Representative images of E18.5 embryonic mouse brains after mock-infection (left, n = 12) or infection with ZIKV BRA_2015 (middle, right, n = 12). Blue, green, red and white indicate DAPI, anti-cleaved caspase3 (ACC3), Zika virus, and Iba1 staining, respectively. The scale bars represent 500 µm and 50 µm. In (a–d), data are presented as mean ± standard deviation. Statistical significance of differences was determined by Brown-Forsythe and Welch ANOVA, followed by Dunnett’s T3 multiple comparisons test for (a, c), or by one-way ANOVA, followed by Kruskal-Wallis’ multiple comparisons test for (b, d). Source data are provided as a Source Data file.
Pre-epidemic ZIKV strains more readily cause subcutaneous edema in embryonic mice
During necropsy of the embryos infected by either preZIKV or epZIKV, subcutaneous edema was observed (Fig. 2a). This is consistent with our previous report, showing subcutaneous edema at E14.5 in 6% and 30% of the embryos infected with PF_2013 or THA_2014, respectively40. Differences in the percentage of embryos displaying subcutaneous edema may reflect differences in pathogenic profile between preZIKV and epZIKV. To assess whether the percentage of infected embryos presenting subcutaneous edema depends on the ZIKV strain used to infect them, we quantified the number of affected embryos for preZIKV and epZIKV at E13.5, E14.5 and E15.5 (Fig. 2b–d). At E13.5, preZIKV tended to cause subcutaneous edema in a higher proportion (18%) of the infected embryos than epZIKV (3%) (Fig. 2b). More specifically, 20% and 17% of the embryos infected with respectively PHL_2012 or THA_2014 displayed subcutaneous edema while it was absent in the embryos infected with any of the epZIKV strains, except for some of the embryos (8%) infected with BRA_2015 (Supplementary Fig. 3b). The trend became even more pronounced at E14.5 with a significantly higher number (44%) of the preZIKV-infected embryos presenting subcutaneous edema compared to the epZIKV-infected embryos (18%) (Fig. 2c). By contrast, a similar proportion of the infected embryos from both groups showed subcutaneous edema at E15.5 (61% versus 43%). (Fig. 2d). Of note, while the proportion of the embryos presenting edema increased from E13.5 to E15.5 for most ZIKV strains, the number remained low for PF_2013 (Supplementary Fig. 3, panel a versus panels b–d). Therefore, results with this strain were omitted from the analysis.

a Picture showing representative E14.5 mouse embryos with mild (arrow left embryo) and severe (arrow right embryo) subcutaneous edema. b Percentage of mouse embryos presenting subcutaneous edema at E13.5 after infection with either pre-epidemic (n = 17 in total) or epidemic (n = 33 in total) ZIKV strains. c, d Percentage of mouse embryos presenting subcutaneous edema at E14.5 (c) or E15.5 (d) after infection with either pre-epidemic (n = 43 and n = 31, respectively, in total) or epidemic (n = 82 and n = 53, respectively, in total) ZIKV strains. Statistical significance of differences was determined by the two-sided Fisher’s exact test (b–d). Source data are provided as a Source Data file.
Asian ZIKV strains have a similar replication profile in embryonic mouse brains
To assess whether the observed differences between preZIKV and epZIKV are associated with differences in viral replication and viral fitness within the embryonic mouse brain, viral RNA levels (by RT-qPCR) and infectious particles (by endpoint titrations) were quantified at different time points pi. However, due to the small size of the embryonic brain on E13.5–E15.5, infectious virus levels were only determined in E18.5 brains.
At E18.5, no significant differences in viral RNA load or infectious virus in the brain were observed between both groups (Fig. 3a, b). One exception is the difference in infectious virus in brains infected with SUR_2016 (epidemic) as compared to those infected with THA_2014 (pre-epidemic), although no significant difference was noted in the viral RNA load for these strains (Supplementary Fig. 4d). Infectious virus titers in brains infected with SUR_2016 were significantly higher than in brains infected with THA_2014 (Supplementary Fig. 4f). Similarly, no significant differences in viral RNA levels were observed between the preZIKV- and epZIKV-infected brains at E13.5 (Fig. 3c, Supplementary Fig. 4a), E14.5 (Fig. 3d) and E15.5 (Fig. 3e), again with one exception: brains infected by PHL_2012 (pre-epidemic) had a higher viral RNA load than those infected by MEX_2016 (epidemic) at E14.5 (Supplementary Fig. 4b). The only significant difference in replication potential at E15.5 was observed between two epZIKV strains, with a significantly higher viral RNA load in brains infected by PR_2015 than in those infected by PF_2013 (Supplementary Fig. 4c). Overall, the preZIKV and epZIKV strains displayed similar replication profiles in embryonic mouse brains with a peak at E15.5 (Fig. 3f), except for PHL_2012 and BRA_2015 which both peaked one day earlier (Supplementary Fig. 4e).

a Infectious virus in brain at E18.5, depicted as log10-transformed 50% tissue culture infectious dose (TCID50) per mg of tissue. b-e Brain viral RNA loads are shown as log10-transformed viral genome copies at various time points: E18.5 (b), E13.5 (c), E14.5 (d), E15.5 (e). f Replication kinetics of the pre-epidemic and epidemic ZIKV strains in embryonic brains at E13.5 to E18.5. Data are summarized data of (b–e). Blue dots/lines represent data of the pre-epidemic ZIKV strains and purple dots/lines represent data of the epidemic ZIKV strains. N numbers are shown in Supplementary Table 2. Data are represented as mean ± standard deviation. Statistical significance of differences was determined by one-way ANOVA, followed by Kruskal-Wallis’ multiple comparisons test. Statistically significant differences are displayed in Supplementary Fig. 4. ns, not significant. Source data are provided as a Source Data file.
To further substantiate that the severity in brain defects is not correlated with the replication competence of the ZIKV strains in the fetal brain, a Spearman r correlation test was performed. However, due to the small size of the fetal brain, brain defect measurements and viral RNA quantification could not be performed on the same sample. Moreover, manipulations of the (severely) affected brains had to be kept to a minimum to avoid loss of integrity of the brain sample. We therefore first assessed whether the ZIKV RNA load in the fetal brain was correlated with that in the heart collected from the same animal. Indeed, a correlation was found between the viral load in the brain and that in the heart, both at E14.4 and E18.5, albeit with a more significant correlation at E18.5 (Supplementary Fig. 5a, b). More interestingly, at E18.5, no significant differences were noted in the viral load in the hearts of the embryos regardless of the severity of the ventriculomegaly in the brains of the same animals (Supplementary Fig. 5c). Finally, we found no correlation between the ZIKV RNA load in the heart and the different brain defect assessments (Supplementary Fig. 5d–f). These results indicate that the viral load in the heart can be used as a proxy for the viral load in the brain, but more importantly, that the severity of the brain phenotype is not correlated with the ZIKV RNA load in the heart and, by extension, the brain.
Asian ZIKV strains display different replication kinetics in human brain cells
We next defined the in vitro neurovirulent profile for each ZIKV strain in the 2 groups using two relevant brain cell types: neuroblastoma cells (SH-SY5Y) and human neural progenitor cells (hNPCs). In neuroblastoma cells, the course of replication was virtually the same for all ZIKV strains, i.e., increasing from d1 to d7 pi (Fig. 4a). However, obvious differences were noted for some ZIKV strains. Among all ZIKV strains, viral RNA levels at endpoint were significantly the highest for BRA_2015. From 3 days post-infection (3 dpi) onwards, PR_2015 replicated to significantly higher viral RNA levels than the other ZIKV strains except BRA_2015 (Fig. 4a). By contrast, PF_2013 replicated less efficiently, resulting in the lowest viral load at 5 and 7 dpi. For all ZIKV strains, the production of viral particles correlated roughly with the viral RNA load, with PR_2015 and BRA_2015 yielding significantly higher virus titers from 5 to 7 dpi than did the other ZIKV strains (Fig. 4b). At earlier time points, virus titers were lower for PR_2015 than for BRA_2015, although levels increased more rapidly from d2-d5 pi for PR_2015, indicative of a higher fitness (also considering the relatively lower viral RNA levels for PR_2015) (Fig. 4b).

a Viral loads in neuroblastoma cells are shown as log10-transformed viral genome copies at different days post-infection (all strains n = 9, except for PF_2013 n = 6). b Infectious virus in neuroblastoma cells is depicted as log10-transformed TCID50 per ml of supernatant at different days post-infection (all strains n = 3). c Viral loads in human neural progenitor cells are shown as log10-transformed viral genome copies at different days post-infection (all strains n = 6, except for SUR_2016 n = 3). d Infectious virus in human neural progenitor cells is depicted as log10-transformed TCID50 per ml of supernatant at different days post-infection (all strains n = 6, except for SUR_2016 n = 3). Blue-colored dots/lines represent data of the two pre-epidemic ZIKV strains and purple-/red-colored dots/lines represent data of the five epidemic ZIKV strains in (a–d). Data are presented as mean values ± standard deviation. Statistical significance of differences was determined by two-way ANOVA followed by Šídák’s multiple comparisons test. Source data are provided as a Source Data file.
When assessing the viral replication kinetics in hNPCs, PR_2015, BRA_2015 and SUR_2016 reached significantly and similarly higher viral RNA levels on 5 to 10 dpi than did the other ZIKV strains. At these time points, viral RNA levels were the highest for SUR_2016 than for the other two ZIKV strains. Similar to the neuroblastoma cells, the lowest viral load in hNPCs was found for PF_2013. In general, all ZIKV strains replicated to peak levels at d9 pi, after which levels slightly decreased at 10 dpi (Fig. 4c). As observed in neuroblastoma cells, infectious virus levels correlated with the viral RNA load in hNPCs with PR_2015, BRA_2015 and SUR_2016 yielding significantly higher virus titers than did the other ZIKV strains (Fig. 4d). Overall, the epidemic ZIKV strains PR_2015 and BRA_2015 exhibited on average slightly superior replication kinetics in the studied human brain cells (illustrated by higher levels of viral RNA and virus titers) than the other ZIKV strains, except for SUR_2016 in hNPCs.
Increased recruitment of microglia and macrophages in embryonic brains infected with Asian ZIKV
We then assessed the number of microglial cells and macrophages in the infected brains that had earlier been analyzed for the presence of brain defects by performing immunostainings with an Iba1 antibody. Both preZIKV- and epZIKV-infected embryonic brains contained significantly higher numbers of microglial cells and macrophages than mock-infected brains, indicating that ZIKV caused a substantial influx of microglia/macrophages (Fig. 5a). To determine whether the increased number of these immune cells in ZIKV-infected brains is correlated with the severity of the brain phenotype, we performed a correlation test with the number of Iba1-positive cells on the one side and the different brain defect measurements (i.e., DAPI-positive cells, cortical length, and the ventricle area) on the other. Indeed, the number of Iba1-positive cells is correlated with the severity of the brain phenotype, with brains displaying a lower number of DAPI-positive cells, smaller cortices and a larger ventriculomegaly having a higher number of Iba1-positive cells (Fig. 5c–e). In conclusion, ZIKV infection of the brain generally resulted in an increase in the number of microglia/macrophages, which is correlated with the severity of the brain phenotype, suggesting a role of these immune cells in ZIKV brain pathogenicity.

a Number of Iba1-positive cells in E18.5 embryonic mouse brain after mock-infection (n = 8) or infection by either pre-epidemic (n = 28) or epidemic (n = 64) ZIKV strains. Data are mean values ± standard deviation. Blue dots represent data of the pre-epidemic ZIKV strains and purple dots represent data of the epidemic ZIKV strains. b Representative images of E18.5 embryonic mouse brains after mock-infection (left pictures) or infection with ZIKV (middle and right pictures). For ZIKV-infected brains, a representative picture of a mild (middle) and severely affected (right) embryonic brain is shown. Blue and green indicate DAPI and Iba1 staining, respectively. Scale bars represent 200 µm and 50 µm. c–e Correlation between the number of Iba1-positive cells at E18.5 and the different brain defect assessments: DAPI-positive cells (c), cortical length (d) and ventricle area (e) of the corresponding embryos after ZIKV infection. Statistical significance in (a) was determined using the Kruskal-Wallis test followed by Dunn’s multiple comparisons test. Correlation in (c–e) was determined using the two-sided Spearman r correlation test. Correlation is indicated by a black solid line with green shaded areas indicating 95% confidence intervals. Source data are provided as a Source Data file.
Asian ZIKV strains differ in inducing immune mediators in embryonic brains
In order to determine whether the ZIKV strains induced different immune responses in the embryonic brain, we evaluated the production of 11 chemokines and cytokines (i.e., CCL3, CCL5, CXCL1, CXCL10, IFN-γ, IL-1β, IL-4, IL-6, IL-10, M-CSF, and TNF-α) at the protein level in embryonic brain lysates (Fig. 6). Measurements were carried out at E14.5 (Fig. 6a) and E18.5 (Fig. 6b) in brains infected with either a preZIKV strain (PHL_2012 or THA_2014) or an epZIKV strain (BRA_2015 or SUR_2016) on E10.5. Generally, the induction of immune mediator production was more pronounced at E18.5 as compared to that in mock-infected brains than at E14.5, as indicated by the significant upregulation of multiple chemokines and cytokines at the later time point, when the infection is already prominent and brain abnormalities are present. Interestingly, although the majority of the cytokines were undetectable or found at similar levels in mock-infected brains at E14.5, the chemokines CCL3, CCL5 and CXCL10 were on average markedly upregulated by ZIKV infection in the embryonic brains at this time point. In more detail, preZIKV strains PHL_2012 and THA_2014 both induced significant increases in the levels of CCL3, CCL5 and CXCL10 as compared to those in the mock-infected brains. Levels of these chemokines were also elevated in BRA_2015-infected brains, albeit only significantly for CCL5, while the immunological stimulation was essentially absent after SUR_2016 infection. Notably, BRA_2015 mounted a highly variable immune response in infected brains, ranging from nearly absent (i.e., comparable to that in mock-infected brains) to very strong (i.e., comparable to that in preZIKV-infected brains), as clearly shown for CCL3, CCL5, CXCL10 and IL-4. In addition, at E14.5, levels of M-CSF were significantly elevated, but only in embryonic brains infected with either preZIKV strain, while an increase in IL-1β production was only measured in brains infected with the preZIKV strain PHL_2012 (not significantly) and the epZIKV strain BRA_2015.

Quantification of eleven chemokine and cytokine levels (i.e., C-C motif chemokine ligand 3 [CCL3], CCL5, C-X-C motif chemokine ligand 1 [CXCL1], CXCL10, Interferon-γ [IFN-γ], Interleukin-1β [IL-1β], IL-4, IL-6, IL-10, Macrophage Colony-Stimulating Factor [M-CSF], and Tumor Necrosis Factor-α [TNF-α]) in embryonic mouse brains after mock-infection or infection with either PHL_2012, THA_2014, BRA_2015 or SUR_2016 at E14.5 (a) or E18.5 (b). Black, dark blue, light blue, red and pink dots represent mock, PHL_2012, THA_2014, BRA_2015 and SUR_2016 data, respectively. N numbers are depicted in Supplementary Table 3. Data are mean values ± standard deviation. Statistical significance was determined using the Kruskal-Wallis test followed by Dunn’s multiple comparisons test, or the Brown-Forsythe and Welch ANOVA test, followed by Dunnett’s T3 multiple comparison test. Source data are provided as a Source Data file.
At E18.5, epZIKV-infected brains still produced elevated levels of CCL3, CCL5 and CXCL10 compared to mock-infected brains; however, this was now accompanied by increased levels of TNF-α (only for PHL_2012), IL-1β (only for PHL_2012), IL-4, IL-6, IFN-γ and M-CSF, demonstrating that preZIKV-infected brains are undergoing a severe inflammatory process at this time point. The immune mediators found in the brain are typical of macrophage-driven immune responses, corroborating our data in Fig. 5. However, it is important to highlight differences between strains at E18.5, with preZIKV PHL_2012 clearly being the most immunostimulatory strain, inducing an upregulation of all immune mediators listed above. This characteristic was largely mirrored by preZIKV THA_2014, except for TNF-α and IL-1β. A large difference in the immunostimulatory properties was noted between the epZIKV strains, being either substantially reduced (BRA_2015) or completely absent (SUR_2016), as was observed at E14.5. Like on E14.5, BRA_2015 induced a highly variable immune response on E18.5, ranging from mild to very strong, as clearly shown for CCL5, CXCL10 and IFN-γ. These data show that preZIKV strains are powerful activators of the immune response in embryonic brains, whereas epZIKV strains are attenuated in terms of chemokine and cytokine induction. Of note, the neutrophil-active chemokine CXCL1 and the immunomodulatory cytokine IL-10 were not significantly upregulated by any ZIKV strain tested, at either time point evaluated. In conclusion, each ZIKV strain tested exhibited a different immunostimulatory and proinflammatory profile, yet with the preZIKV strains eliciting a stronger and more consistent immunological response, while the epZIKV strains displayed a more variable and attenuated immunogenic profile.
Impact of S139N substitution on severity of brain defects
The individual preZIKV and epZIKV strains differ from each other by numerous amino acid changes in their viral genomes. Similarly, different substitutions are present in the genome of ZIKV strains belonging to the same group. However, the only amino acid separating preZIKV from epZIKV is the one at position 139 in the polyprotein (Supplementary Table 1), with a serine (S) shared by preZIKV and an asparagine (N) shared by epZIKV, as was reported previously32. Interestingly, this residue at position 139 was demonstrated to contribute to fetal brain defects41. This prompted us to investigate whether and to what extent this amino acid contributes to the severity of brain abnormalities in embryonic mice using the IPL model. Since brain abnormalities were consistently more severe in (a larger proportion of) embryonic mice infected with preZIKV, the THA_2014 strain was selected for altering the amino acid at position 139. By employing the Infectious Subgenomic Amplicons (ISA) method49, the S was substituted to an N (S139N), resulting in a ZIKV strain referred to as THA_139N. The severity of the brain defects as well as the presence of a microcephaly-like phenotype and ventriculomegaly at E18.5 was assessed as described earlier. Hypothetically, introducing the S139N substitution in the pre-epidemic THA_2014 strain should lead to a microcephaly-like phenotype similar to that induced by epZIKV. Compared with mock-infected embryos, those infected by THA_139N presented pronounced brain defects, as evidenced by a lower brain weight, a microcephaly-like phenotype, and ventriculomegaly (Fig. 7a–e). However, the severity of the brain abnormalities did not differ significantly between embryos infected with THA_139N and those infected with parental THA_2014, indicating that the amino acid at position 139 does not seem to play a decisive role (if any) in the development of (less) severe brain abnormalities induced by ZIKV as shown in Fig. 1.

a–d Measured brain defects in embryonic mouse brains at E18.5 following mock-infection (n = 26 for brain weight; n = 12 for the others) or infection with either THA_2014 (n = 12) or THA_139N (n = 6). Embryonic brains were examined morphologically by measuring the brain weight normalized to the head weight (a). Microcephaly-like phenotypes were assessed by measuring the cortical length (b) and the number of DAPI-positive cells (c). Ventriculomegaly was estimated by measuring the ventricle area (d). e Representative images of E18.5 embryonic mouse brains after mock-infection (left, n = 12) or infection with THA_2014 (middle, n = 12) or THA_139N (right, n = 6). Blue, green, red and white indicate DAPI, anti-cleaved caspase3 (ACC3), Zika virus, and Iba1 staining, respectively. Scale bars represent 500 µm and 50 µm. f Viral loads in neuroblastoma cells, shown as log10-transformed viral genome copies at different days post-infection (THA_2014: n = 6, THA_139N: n = 3). g Infectious virus in neuroblastoma cells, depicted as log10-transformed TCID50 per ml of supernatant at different days post-infection (THA_2014: n = 3, THA_139N: n = 3). h Viral loads in human neural progenitor cells, shown as log10-transformed viral genome copies at different days post-infection (THA_2014: n = 6, THA_139N: n = 3). i Infectious virus in human neural progenitor cells, depicted as log10-transformed TCID50 per ml of supernatant at different days post-infection (THA_2014: n = 6, THA_139N: n = 3). j Number of DAPI-positive cells at day 1 and day 4 after infection of hNPCs (THA_2014: n = 3, THA_139N: n = 3). Data for THA_2014 in (a–d) are the same as those presented in Supplementary Fig. 2. Light blue and orange dots/lines represent THA_2014 and THA_139N data, respectively. Data are mean ± standard deviation in (a–d) and (f–j). Statistical significance of differences was determined by Brown-Forsythe and Welch ANOVA followed by Dunnett’s T3 multiple comparisons test (a–d), or by Two-way ANOVA followed by Šídák’s multiple comparisons test (h, i). Only statistically significant differences are shown. Source data are provided as a Source Data file.
We next investigated whether the S139N substitution affected the viral replication kinetics in neuroblastoma cells and hNPCs. In neuroblastoma cells, the THA_139N variant replicated to a significantly higher viral RNA load at 1 to 5 dpi than the THA_2014 strain (Fig. 7f). Levels of infectious virus were also higher, albeit only significantly at 3 dpi, in neuroblastoma cells infected with THA_139N than in those infected with the parental strain (Fig. 7g). By 7 dpi, the (significant) difference between the two ZIKV strains had disappeared. A similar difference in the replication profile between THA_139N and THA_2014 was noted in hNPCs, i.e., significantly higher viral RNA levels (at 2–9 dpi) and virus titers (at 5 and 7 dpi) for THA_139N than for THA_2014 (Fig. 7h, i). Interestingly, when determining the number of cells, the number of hNPCs infected by THA_139N increased from 1 to 4 dpi, while those infected with THA_2014 decreased from 1 to 4 dpi (Fig. 7j). The difference in the number of hNPCs was significant at 4 dpi. These results suggest that THA_139N may delay virus-induced cytopathic effect in hNPCs.
Discussion
ZIKV strains of the Asian lineage are considered a major public health concern because of outbreaks in several regions around the world, such as Micronesia (2007), French Polynesia (2013), Asia50,51 (e.g., 2012, 2014, 2018), and South and Central America (2014–2016), which also increased in magnitude52,53. Moreover, during the 2015–2016 ZIKV outbreak an increase in the number of newborns with microcephaly and other neurodevelopmental disorders was reported. The detrimental effect on newborns came as a surprise as the virus, which has been circulating for decades, was considered a harmless virus. Various research groups have since tried to decipher the mechanisms that could explain the higher neurovirulence of the Asian lineage ZIKV strains, yielding contradictory results35,36,41,43. This prompted us to comprehensively compare the neurovirulent and pathogenic profile of Asian ZIKV strains belonging to either the pre-epidemic group (ZIKV strains that emerged before PF_2013) or the epidemic group (PF_2013 and all subsequent ZIKV strains) to identify determinant(s) that might have contributed to the proposed gain in neurovirulence and pathogenicity. We demonstrate that the propensity of ZIKV strains of the Asian lineage to cause brain abnormalities, such as a microcephaly-like phenotype and ventriculomegaly, is common to all ZIKV strains that were tested, regardless of their origin (pre-epidemic or epidemic). This indicates that it will be imperative to monitor pregnant women during future ZIKV outbreaks, irrespective of the circulating ZIKV strain. PreZIKV strains, however, were more pathogenic in that they invariably caused more severe brain abnormalities in the embryonic mice. Infections with epZIKV strains, on the other hand, resulted in a greater variation in the severity of brain defects, with brains that did not present major abnormalities and those that were severely affected. This variability in brain defect severity is fully in line with what has been described in clinical reports, i.e., infants presenting varying degrees of adverse outcomes54,55, from undetectable central nervous system malformations56,57 to severe microcephalic phenotypes, and even perinatal mortality55. Thus, our results support the hypothesis that ZIKV evolution along the Asian lineage led to an attenuation of pathogenicity.
The higher pathogenic profile of preZIKV strains as compared to epZIKV strains was also supported by the earlier appearance of subcutaneous edema in a larger proportion of the pre-epidemic ZIKV-infected mouse embryos. Subcutaneous edema has also been described in a human fetus who had developed hydrops fetalis after congenital ZIKV infection58. Hydrops fetalis is characterized by severe swelling (edema) in unborn babies with mortality rates up to 50%59. Although it is extremely difficult to estimate the true incidence of hydrops fetalis caused by congenital ZIKV infections during the various ZIKV outbreaks around the world, the emergence of epidemic ZIKV strains with a reduced pathogenic profile may have contributed to the higher incidence of neurodevelopmental disorders and potentially less cases of fatal hydrops fetalis in infants born during the ZIKV epidemic.
To potentially link the pathogenic profile to the replication fitness, we examined the viral replication kinetics of the preZIKV and epZIKV strain, both in vitro and in vivo. Overall, the infectivity and replication competence of the preZIKV and epZIKV strains were comparable in embryonic brains at all time points tested. Some differences were noted, however not consistently between the (same) preZIKV and epZIKV strains. In neuroblastoma cells and hNPCs, a significantly higher viral load (viral RNA and infectious virus) was found for some viruses, particularly for epidemic BRA_2015 and PR_2015. In addition, the epZIKV strain SUR_2016 also exhibited superior replication kinetics in hNPCs. On the other hand, the strain with the most attenuated replication profile also belonged to the epZIKV strains: PF_2013. More interestingly, BRA_2015 and PF_2013 both caused comparable brain defect severity in embryonic mice, which seems to mirror the observed severity of brain defects in PF_2013- and BRA_2015-infected human fetuses54,60. Taken together, the results indicate that the more pathogenic profile of the preZIKV strains was not associated with an increased viral replication fitness of these strains. Hence, the pathogenicity of ZIKV in the fetal brain appears to be linked to factors other than viral replication. Our results also point out that extrapolation of in vitro findings should be done with caution as critical determinants, such as the immune system, the interplay between the different cell types, and the developmental stage of the embryo, are absent in in vitro cell culture systems.
To explore the potential role of the immune system in ZIKV pathology in the fetal brain, we quantified the number of Iba1-positive cells, representing macrophages/microglial cells, in ZIKV-infected brains. Within the parenchyma, microglial cells represent the major component of the immune system, releasing chemokines and cytokines upon activation. One of the many functions of microglial cells is to exert phagocytotic activities that are essential for clearing pathogens. In our ZIKV mouse model, infection of the embryonic mouse brain by both preZIKV and epZIKV led to elevated numbers of microglia and macrophages in the parenchyma, which correlated with the severity of the brain defects. This indicates that microglial cells are implicated in ZIKV pathogenicity in the fetal brain, irrespective of the status of the infecting ZIKV strain (pre-epidemic/epidemic). This is in line with other studies on abnormalities in the human fetal brain that also reported an increase in infiltrating macrophages61, resident microglial cells, and infection of the microglia20. Future studies will focus on deciphering the (deleterious) role of microglia in ZIKV pathogenicity in the embryonic brain.
Inflammation in the brain can have a dual character. Inflammation is beneficial because it leads to control of viral infection in the brain, likely preventing it from spreading further or increasing in severity. In addition, inflammation also triggers the repair of damaged tissue62. On the other hand, uncontrolled activation of the inflammatory pathways may directly lead to brain damage, as seen in other viral infections and many neurodegenerative diseases63. To obtain a more comprehensive profile of inflammatory mediators in ZIKV-infected fetal brains, we therefore quantified a total of 11 chemokines and cytokines, both at E14.5 and E18.5. ZIKV infection of the embryonic mouse brain resulted in an upregulation of chemokines and cytokines at both time points, with a stronger immune response at the later stage of infection. The elevated immune mediators (i.e., CCL3, CCL5, CXCL10, IFN-γ, IL-1β, IL-6, M-CSF, TNF-α) indicate that anti-inflammatory strategies may be able to restrict the severity of brain abnormalities in embryonic brains. However, important differences were noted in the immunogenic profile of the preZIKV and the epZIKV strains, as well as between the two strains within each of these two groups. While the tested ZIKV strains each presented a unique immunogenic profile, the preZIKV strains triggered a stronger and more consistent immune response than the epZIKV strains that displayed a more variable and attenuated profile, particularly BRA_2015. This indicates that preZIKV strains are powerful activators of the immune response in embryonic brains, whereas epZIKV strains are attenuated in terms of chemokine and cytokine induction. The lower immunogenicity of epZIKV observed in our study is consistent with previous in vivo64 and in vitro65,66 studies. The lower immunostimulatory potential of epZIKV strains could explain the lower pathogenicity of epZIKV in the fetal brain. The differences in the immunogenic profile between preZIKV and epZIKV strains and within each group also indicate that the response of the immune system may occur via different mechanisms for each ZIKV strain, hence caution should be applied when making any generalizations. Notwithstanding these interesting results on the immunogenic profile of preZIKV/epZIKV strains, and the increased influx of Iba1-positive cells, more extensive research is required to further unravel the role of the immune system, and more particularly macrophages/microglia, in ZIKV pathogenicity in the fetal brain.
To investigate whether the S139N substitution could have contributed to the reduced pathogenic profile of the epidemic ZIKV strains, the S139N substitution was introduced into the pre-epidemic THA_2014 strain, resulting in the THA_139N variant. THA_139N replicated more efficiently in vitro than the parental strain. This result is in line with earlier reported results using a modified (pre-epidemic) ZIKV Cambodia strain containing the same S139N substitution. This modified ZIKV strain also exhibited an increased replication profile in mouse neural progenitor cells41. We further showed that the S139N substitution did not alter the in vivo pathogenicity of THA_139N as compared to that of parental THA_2014, corroborating the undetectable differences in the pathogenic profile reported for a modified epZIKV strain (PR_2015 with the N139S substitution)36. Overall, our in vitro and in vivo findings are consistent with earlier reported findings that the S139N substitution seemed to contribute to ZIKV virulence in vitro (i.e., increased viral replication fitness) but did not correlate directly to differences in the in vivo pathogenicity between preZIKV and epZIKV strains. If the S139N substitution was involved at all, it was not the sole genetic determinant responsible for the differences in the clinical outcomes brought about by preZIKV and epZIKV strains. Our results also highlight that to identify genetic determinants underlying the development of ZIKV-associated brain abnormalities in fetuses and newborns in future studies, it is important to design holistic studies that comprehensively examine multiple ZIKV strains in vitro and in vivo.
Studying ZIKV physiopathology during vertical transmission represents a challenge, as ZIKV cannot cross the placental barrier in wildtype (WT) mice, preventing the virus to cause brain defects in the developing embryo. Several mouse models have been developed that circumvent this issue. The most commonly used mouse model to study ZIKV pathophysiology involves intracerebral ZIKV injection in susceptible WT neonatal mice36,41,67. This injection route is also used in another mouse model that uses WT mouse embryos41,68. Although these models have contributed to a better understanding of ZIKV infection of brain cells, they also have their limitations and may not be the most suitable models to study the development of CZS. ZIKV infection occurs at a late stage of cerebral development when performing intracerebral injections on newborn pups, while ZIKV-induced brain abnormalities were found to be mainly associated with congenital infection during the first trimester of pregnancy. Thus, the physiopathology of ZIKV infection in the neonatal brain may differ from that in the embryonic brain. Moreover, intracerebral inoculation largely ignores the potential interactions between ZIKV and the immune system during virus dissemination in the embryo as well as the ability of ZIKV to cross the blood brain barrier, and may potentially create a biased view on the infected brain cell types and timing of their infection. ZIKV intracerebral injection in WT embryos represents a better model as it more accurately mimics a congenital brain infection. However, it still faces issues due to the used inoculation route, as described above.
To overcome the issues of the above-mentioned mouse models, we conducted intraplacental (IPL) injections with ZIKV on WT dams. In this model, ZIKV is injected in the labyrinth (i.e., the fetal part) of the placenta of each embryo, thereby bypassing the placental barrier, which allows the virus to reach and subsequently disseminate in the embryo in a manner similar to that following a natural vertical transmission. Moreover, in our model, ZIKV was injected at E10.5; a time when the development of the cerebral cortex in mice corresponds to that in human embryos during the first trimester of pregnancy48. Therefore, our IPL ZIKV challenge mouse model more appropriately mimics a congenital ZIKV infection of the brain during the first trimester of human pregnancy, which is a critical period for the development of brain abnormalities in fetuses47.
The capacity of ZIKV to evade the immune system as well as to cross the placental barrier could have played important roles in the emergence of CZS epidemics. The IPL model, however, prevented us from studying the capacity of the different ZIKV strains to cross the placental barrier. Moreover, injecting the virus directly in (fetal part of) the placenta does not allow us to study ZIKV pathogenicity at the placental level, while it may lead to more miscarriages and spontaneous abortions. Furthermore, the IPL ZIKV challenge mouse model neglects the ability of ZIKV strains to evade the maternal immune system, which permits vertical transmission. Finally, our model precludes longitudinal studies, which may conceal subtle changes between individual embryos. The above limitations of the IPL ZIKV challenge mouse model, however, are also applicable to the earlier described ZIKV infection models.
In summary, we show that the different Asian ZIKV strains share neurotropic and neurovirulent properties. The sudden emergence of CZS epidemics was therefore not due to the acquisition of novel neurovirulent traits, nor to an increase in the neurovirulent potential of epZIKV. We further postulate that the pathogenic profile of ZIKV strains of the Asian lineage progressively evolved towards an attenuation, and that macrophages/microglial cells appear to be implicated in ZIKV pathogenicity, although more extensive research is needed to substantiate this hypothesis. The accelerating urbanization, particularly to the natural habitats of the Aedes mosquito vector69, and its associated expanding geographical distribution70 as well as the immune status of individuals represent additional factors contributing to the increased incidence of ZIKV outbreaks around the world. Further investigations on ZIKV-mediated nervous system damage should include the collaborative development of predictive models that integrate the multivariate impacts due to climatic, ecological, social, and cultural factors to improve the public health response.
Methods
Cells
Vero E6 cells (African green monkey kidney cells; Vero 76, clone E6; ATCC: Cat# CRL-1586) and HEK293T (human embryonic kidney cells; ATCC, Cat# CRL-3216) were cultured in DMEM supplemented with 10% fetal bovine serum (FBS; HyClone), 2 mM L glutamine (Thermo Fisher Scientific). In the endpoint titration experiments with Vero E6 cells, 2% FBS was used. BHK-21J (baby hamster kidney fibroblasts; provided by P. Bredenbeek at the Leiden University Medical Center (LUMC), the Netherlands) and SH-SY5Y (human neuroblastoma cells, ATCC, Cat# CRL-2266) were cultured in DMEM supplemented with 10% FBS (HyClone). Vero E6, BHK-21J, HEK293T and SH-SY5Y cells were incubated at 37 °C in presence of 5% CO2. All cell lines (Vero E6, BHK-21J, HEK293T and SH-SY5Y) were regularly tested for mycoplasma contamination.
Differentiation of hiPSCs to cortical hNPCs
Human induced Pluripotent Stem Cells (hiPSCs) used for the differentiation towards human cortical neural precursor cells (hNPCs) were purchased from Sigma-Aldrich (iPSC EPITHELIAL-1; Catalog Number IPSC0028). The iPSC EPITHELIAL-1 cells were produced by reprogramming of epithelial cells obtained from a Caucasian female donor 24 years of age with no known genetic disorders, as stated in the Product Information Sheet that is available on the vendor’s website (https://www.sigmaaldrich.com/NL/en/product/sigma/ipsc0028#product-documentation). Use of the hiPSCs for scientific research was approved by the Ethics Committee Research UZ/KU Leuven (project license S52426). hNPCs were generated from the hiPSCs using the protocol described by Shi and colleagues71. The hiPSCs were plated on human Matrigel-coated 6-well plates (Corning) in mTESR (StemCell) with Revitacell (Life Technologies). These hiPSCs were maintained in E8 flex medium (E8 basal medium (Gibco) complimented with E8 supplement Flex and 5 U/ml Penicillin-Streptomycin) and split twice a week with 0.5 mM EDTA (Gibco). When the hiPSC colonies reached 90% confluency, hNPC induction was done by dissociating the hiPSC colonies by accutase (Sigma) and seeding 2.5 million single cell suspension per well in a 6-well plate in neural induction media (NIM) comprising of neural maintenance medium (NMM) complemented with the dual-SMAD inhibitors SB431542 (10 µM, Tocris) and LDN193189 (1 µM, Miltenyi). The NMM comprises of a 1:1 mixture of Neurobasal medium (Gibco) and DMEM/F-12 Glutamax™ augmented with 0.5X Glutamax™ (Gibco), 50 U/ml Penicillin-Streptomycin, 0.5X B27 (Gibco), 0.5X N2 (Gibco), 0.5X MEM-NEAA (Gibco), 0.5X sodium pyruvate, 0.025 % human insulin (Sigma) and 50 µM 2-mercaptoethanol (Gibco). Media changes with NIM were performed every day for 11 days. At day 12, the neuroepithelial cells were collected using Dispase II (Sigma) and cultured for an additional 4 days with NMM spiked with 20 ng/mL bFGF. Rosette-forming neuroepithelial cells were passaged twice (after 5–7 days) using Dispase II to enrich the hNPC population. The cultured hNPCs were dissociated as single cells on DIV33 (33 days in vitro) and cryopreserved for further experimental use. For ZIKV infection, 1 million DIV33 hNPCs per well were thawed on human Matrigel-coated 6-well plates in NMM supplemented with RevitaCell and cultured in NMM for next 4 days. On day 38, these hNPCs were dissociated as single cells using accutase and replated.
ZIKV strains
Eight ZIKV strains were selected of which three carry a serine (S) at position 139 in the polyprotein protein and five contain an asparagine (N) at position 139 (Supplementary Table 1). ZIKV strains CAM_2010 (Zika virus/H.sapiens-tc/KHM/2010/FSS13025; GenBank KU955593), PR_2015 (ZIKV/Homo sapiens/PRI/PRVABC59/2015; GenBank KU501215), and MEX_2016 (ZIKV/Aedes.sp/MEX/MEX_2-81/2016; GenBank KX446950) were obtained from the World Reference Center for Emerging Viruses and Arboviruses (WRCEVA) at the University of Texas Medical Branch. ZIKV strains PHL_2012 (Zika virus/H.sapiens-tc/PHL/2012/CPC-0740; GenBank KU681082) and THA_2014 (Zika virus/H.sapiens-tc/THA/2014/SV0127-14; GenBank KU681081) were obtained from the Walter Reed Army Institute of Research (WRAIR). ZIKV PF_13 (ZIKV Pf13/251013-18; GenBank KY766069) was obtained from the Institut Louis Malardé in French Polynesia. ZIKV SUR_2016 (ZIKV SL160272; GenBank KY348640) was received from LUMC in the Netherlands. ZIKV BRA_2015 (ZIKV/H.sapiens/Brazil/PE243/201573; GenBank KX197192) was provided by the MRC-University of Glasgow Centre for Virus Research in the UK. High-titer virus stocks were grown on C6/36 mosquito cells and infectious titers (plaque forming units per ml; PFU/ml) were quantified by plaque assay on BHK cells, as described earlier74. All ZIKV stocks were sequenced prior to the in vitro and in vivo experiments. The sequencing results of ZIKV strains PF_13 and THA_201440 were deposited into the European Nucleotide Archive (ENA) database. Next-Generation Sequencing of the ZIKV strains BRA_2015, MEX_2016 and SUR_2016 was performed on a HiSeq™ 2500 platform (Illumina) following the NetoVIR (Novel enrichment technique of VIRomes) protocol75. Sequencing reads of these ZIKV strains were assembled into genomes using the ViPER pipeline v1.0 (https://github.com/Matthijnssenslab/ViPER/tree/v1.0; https://zenodo.org/records/14040438)76 on default settings, which uses Trimmomatic v0.3977 for read trimming and metaSPAdes v3.15.3 for assembly78. The sequences were deposited into the GenBank database; see Supplementary Table 1 for the ENA and GenBank accession numbers.
Viral replication experiments in cells
SH-SY5Y cells were seeded at 5 × 105 cells/ml in T25-flasks and were incubated overnight at 37 °C in the presence of 5% CO2. The following day, medium was removed, and cells were infected with one of the ZIKV strains at an MOI = 0.0001. After 3 h, the inoculum was removed, and cells were washed 3 times with PBS. Finally, 5 ml of DMEM supplemented with 2% FBS was added to the cells. In experiments using hNPCs, cells were seeded at 1.5 × 105 or 3 × 104 per well in human Matrigel-coated 24-well or 96-well plates, respectively, and incubated overnight at 37 °C in the presence of 5% CO2. Next day, medium was removed, and cells were infected with the one of the ZIKV strains at an MOI = 0.1 for 12 h after which the inoculum was removed and 2.5 ml of NMM medium was added to the cells.
Mouse experiments
Breeding couples of immunocompetent SWISS/CD1 mice were purchased from Janvier Labs, Envigo or Charles River and bred at the KU Leuven Laboratory Animal Center. The SPF status of the mice was regularly checked at the KU Leuven animal facility. Mice were housed at biosafety level 2 in individually ventilated cages (Sealsafe Plus, Tecniplast) at 21 °C, 55% humidity and 12:12 light/dark cycles. Animals were provided with food and water ad libitum as well as with cardboard play tunnels and cotton/tissues as extra bedding material. Allocation to experimental groups was performed randomly. Housing conditions and experimental procedures were approved by the ethical committee of KU Leuven (license P029/2021), following institutional guidelines approved by the Federation of European Laboratory Animal Science Associations (FELASA).
Intraplacental infection
Time-mated, SWISS mice, 8–12 weeks of age, were used for ZIKV vertical transmission experiments. All surgeries on wild-type female SWISS mice were performed at the same time, with embryonic day (E) 0.5 corresponding to the afternoon following the day of mating. At E10.5, preoperative analgesia (0.1 mg/kg of buprenorphine; Temgesic) was administered subcutaneously before induction of anesthesia. The pregnant females were anesthetized using isoflurane (Abbot Laboratories Ltd.), after which the mice were shaved. Next, a small incision (1.0–1.5 cm) was made through the lower ventral peritoneum, followed by the careful extraction of the uterine horns, which were carefully placed on warm humidified gauze pads. The developing embryos were challenged intra-placentally with 105 PFU/ml ZIKV or 2%FCS/PBS (‘mock’). After infection, incisions were sutured and disinfected. Mice were maintained on a heat pad during the whole procedure. Eight hours post-surgery, mice received a second dose of analgesia (0.1 mg/kg of buprenorphine; Temgesic).
Quantification of cytokines and chemokines
Dissected brain tissues were weighed and homogenized in protein extraction buffer, consisting of 15 mM NaCl (VWR Chemicals, Radnor), 0.05% Tween 20 (Sigma), 5% Bovine Serum Albumin (BSA, Sigma), 10 mM EDTA (Sigma) and 1% protease inhibitor cocktail (Sigma) in PBS. Homogenates were centrifuged for 20 min (1000 × g, 4 °C), supernatants were collected and concentrations of chemokines and cytokines (i.e., C-C motif chemokine ligand 3 [CCL3], CCL5, C-X-C motif chemokine ligand 1 [CXCL1], CXCL10, Interferon-γ [IFN-γ], Interleukin-1β [IL-1β], IL-4, IL-6, IL-10, Macrophage Colony-Stimulating Factor [M-CSF], and Tumor Necrosis Factor-α [TNF-α]) were quantified using the Mouse Magnetic Luminex Assay (Bio-Techne), according to the manufacturer’s protocol. The absorbance was determined using a spectrophotometer (BioTek) at 450 nm. Results are expressed as picogram (pg) per 100 mg of brain tissue.
RNA isolation and RT–qPCR
Total RNA was isolated from embryonic tissue using Trizol (Ambion, Life Technologies), according to the manufacturer’s protocol. Total RNA was isolated from cells supernatants using the NucleoSpin RNA Virus Mini kit for viral RNA isolation from cell-free fluids (Macherey-Nagel), according to the manufacturer’s protocol. Quantification of ZIKV genome copy numbers was performed by quantitative reverse transcription PCR (RT-qPCR) using the one step RT-qPCR Low ROX MasterMix (Eurogentec), according to the manufacturer’s protocol. Primer and probe sequences for ZIKV RNA quantification are provided in Supplementary Table 4. Ct values were converted into a relative number of ZIKV RNA copies/µl using the formula y = a*ln(x) + b, where a is the slope of the standard curve, b is the y-intercept of the standard curve and y is the Ct value for a specific sample.
Endpoint titrations
E18.5 brains were dissected, and half of the brain was used to perform endpoint titrations. Brain tissues were weighed and immerged in 350 µl of 2% FBS assay medium (described earlier) in a tube for homogenization. Tissues were homogenized and subsequently centrifuged for 10 min (16,200 × g, 4 °C). Vero E6 cells (pre-seeded in 96-wells plates at 1 × 104 cells/well, 1 day earlier) were infected with supernatant from either infected homogenized brains or infected cells using ten-fold dilutions series, in triplicate. After 7 days of incubation, virus-induced cytopathic effect (CPE) was microscopically quantified and the virus titer (expressed as TCID50 per ml) was determined using the method of Reed and Muench79.
Generation of ZIKV THA_139N
The THA_139N variant was generated following the Infectious Subgenomic Amplicons (ISA) method49. RNA isolation was performed on virus stock using the NucleoSpin RNA Virus Mini kit for viral RNA extraction from cell-free fluids (Macherey-Nagel), according to the manufacturer’s protocol. A first fragment (pCMV-5’- 608 bp), which contains the pCMV promotor and the S139N mutation (Supplementary Table 4), was synthesized by GenScript. Following RNA isolation, RT-PCR was performed using the SuperScript™ IV One-Step RT-PCR System (ThermoFisher, 12594025) to generate two overlapping fragments of approximately 5-kb covering the full genomic sequence (Supplementary Table 4). After RT-PCR, the proper size of the fragments was confirmed by agarose gel electrophoresis. The RT-PCR products were purified using the Monarch® PCR & DNA Cleanup Kit (New England Biolabs, T1030L), according to the manufacturer’s protocol.
On day 0, a mixture of 80% HEK293T cells and 20% BHK cells was seeded in a 96-well amine-coated plate (VWR, 734–1475). On day 1, transfection of the purified fragments was performed using Lipofectamine 3000 reagent (ThermoFisher Scientific, L3000008), according to the manufacturer’s protocol, and 12 h post-transfection the cells were washed three times with PBS and fresh medium was added to the cells. Six days post-transfection, the supernatant was passaged on pre-seeded Vero E6 cells in a 96-well plate (1 × 104 cells/well) and the transfected cells were pooled and re-seeded in a 6-well plate. Seven days post-passaging and re-seeding, the presence of the virus was confirmed by RT-qPCR and plaque assay. The newly generated virus was grown on Vero E6 cells and infectious titers (PFU/ml) were quantified by plaque assay on BHK cells. Finally, the genome integrity and the presence of the mutation was assessed by Next-Generation Sequencing.
Cryo-sectioning
E18.5 embryonic mouse heads were harvested and fixed overnight at 4 °C in 4% paraformaldehyde (PFA). Brains were dissected in 0.1 M PBS (pH 7.4) and cryoprotected by overnight immersion in 20% Sucrose in PBS before embedding in Polyfreeze (Sigma-Aldrich). Brain sections of 14 µm were obtained by cryosectioning (Leica) onto slides (Epredia™ SuperFrost Plus™ Adhesion slides, Fisher Scientific).
Immunofluorescence
For immunofluorescence of brain sections, the slides were immerged in an antigen retrieval solution (Dako Target Retrieval Solution) and heated at 95 °C for 5 min. Next, slides were incubated in blocking solution (Normal Donkey Serum 10%, Bioconnect Life Sciences) for 1 h at room temperature. The rabbit anti-cleaved caspase 3 (1:300, #9661, Cell Signaling Technologies), mouse anti-flavivirus group antigen (1:1000, MAB10216, Merck Millipore) and goat anti-Iba1 (1:300, ab5076, Abcam) were used as primary antibody, and slides were incubated overnight at 4 °C. The following day, slides were stained using the secondary antibodies (Life Technologies) donkey anti-rabbit Alexa Fluor-488 (A-21206), donkey anti-mouse Alexa Fluor-555 (A- 31570), all diluted 1:1000, or donkey anti-goat Alexa Fluor-647 (A-21447), and nuclei were counterstained with DAPI (1:1000) for 2 h at room temperature. Slides were mounted in Dako Fluorescence Mounting Medium (Agilent).
For immunofluorescence of hNPCs, supernatant was removed from the cells at the indicated time points, and cells were washed three times with PBS. Next, cells were fixed using 4% PFA for 30 min, followed by three PBS washes. Nuclei were counterstained with DAPI (1:1000) for 1 h at room temperature. Finally, cells were washed three times with PBS.
Imaging and analysis
Confocal images of sections of E18.5 mouse brains were acquired using a NIKON A1R confocal microscope at a 20X magnification, or using a Leica DMi8 confocal spinning disc microscope at a 25X magnification. Image analysis and processing were performed with ImageJ 1.42q 276 (Wayne Rasband, National Institutes of Health) or Fiji (v2.0.0-rc-54/1.51 h; https://imagej.net/Fiji) software.
Statistics
Statistical analyses were performed using GraphPad Prism v9.3.1. Results (from brain analyses, viremia and subcutaneous edema) were first tested for normality using the Shapiro-Wilk test. The Brown-Forsythe and Welch ANOVA test followed by Games-Howell’s multiple comparisons post-hoc test was performed when the assumption of equal variance was not met. Alternatively, the two-sided Kruskal-Wallis test followed by Dunn’s multiple comparisons test was used when both normality and the assumption of equal variance were not met. Differences were considered statistically significant when p < 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw sequence data of ZIKV strains PF_13 and THA_2014 are available from the European Nucleotide Archive database under accession number PRJEB39677. The coding sequences of ZIKV THA_139N, ZIKV BRA_2015, ZIKV MEX_2016 and ZIKV SUR_2016 have been deposited in GenBank (Accession number PP115590, PQ129535, PQ129534 and PQ129533, respectively). All data supporting the findings of this study are available within the article, the source data or the Supplementary Information. Supplementary Figs. are provided with this paper. Source data are provided with this paper.
Code availability
The ViPER pipeline v1.0 that was used to assemble the sequencing reads of the ZIKV strains into genomes is available at github (https://github.com/Matthijnssenslab/ViPER). This package has also been deposited at Zenodo (https://doi.org/10.5281/zenodo.5502203)76. Graphs and figures were generated using Microsoft PowerPoint, GraphPad Prism (version 9.3.1; LaJolla, USA), or Adobe Illustrator (version 25.4.1; San Jose, USA); the software is made available by KU Leuven through a group license.
References
Wikan, N. & Smith, D. R. First published report of Zika virus infection in people: Simpson, not MacNamara. Lancet Infect. Dis. 17, 15–17 (2017).
World Health Organization. Zika virus. Factsheet. WHO, Geneva, Switzerland. (2022). https://www.who.int/news-room/fact-sheets/detail/zika-virus
Morris, J. et al. Prevalence of microcephaly: the Latin American Network of Congenital Malformations 2010-2017. BMJ Paediatr. Open 5, e001235 (2021).
Freitas, D. A. et al. Congenital Zika syndrome: a systematic review. PLoS ONE 15, e0242367 (2020).
Filgueiras, I. S. et al. The clinical spectrum and immunopathological mechanisms underlying ZIKV-induced neurological manifestations. PLoS Negl. Trop. Dis. 15, e0009575 (2021).
Goodfellow, F. T. et al. Strain-dependent consequences of Zika virus infection and differential impact on neural development. Viruses 10, 550 (2018).
Anfasa, F. et al. Phenotypic Differences between Asian and African lineage Zika viruses in human neural progenitor cells. mSphere 2, e00292–17 (2017).
Cugola, F. R. et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature 534, 267–271 (2016).
Garcez, P. P. et al. Zika virus impairs growth in human neurospheres and brain organoids. Science 352, 816–818 (2016).
Li, C. et al. Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell 19, 120–126 (2016).
Manet, C. et al. Zika virus infection of mature neurons from immunocompetent mice generates a disease-associated microglia and a tauopathy-like phenotype in link with a delayed interferon beta response. J. Neuroinflamm. 19, 307 (2022).
Sher, A. A., Glover, K. K. M. & Coombs, K. M. Zika virus infection disrupts astrocytic proteins involved in synapse control and axon guidance. Front. Microbiol. 10, 596 (2019).
Stefanik, M. et al. Characterisation of Zika virus infection in primary human astrocytes. BMC Neurosci. 19, 5 (2018).
Rubio-Hernández, E. I. et al. Astrocytes derived from neural progenitor cells are susceptible to Zika virus infection. PLoS ONE 18, e0283429 (2023).
Chen, J. et al. AXL promotes Zika virus infection in astrocytes by antagonizing type I interferon signalling. Nat. Microbiol. 3, 302–309 (2018).
Limonta, D. et al. Human fetal astrocytes infected with Zika virus exhibit delayed apoptosis and resistance to interferon: implications for persistence. Viruses 10, 646 (2018).
Schultz, V. et al. Oligodendrocytes are susceptible to Zika virus infection in a mouse model of perinatal exposure: Implications for CNS complications. Glia 69, 2023–2036 (2021).
Li, C. et al. Disruption of glial cell development by Zika virus contributes to severe microcephalic newborn mice. Cell Discov. 4, 43 (2018).
Schultz, V. et al. Zika virus infection leads to demyelination and axonal injury in mature CNS Cultures. Viruses 13, 91 (2021).
Lum, F. M. et al. Zika virus infects human fetal brain microglia and induces inflammation. Clin. Infect. Dis. 64, 914–920 (2017).
Xu, P. et al. Role of microglia in the dissemination of Zika virus from mother to fetal brain. PLoS Negl. Trop. Dis. 14, e0008413 (2020).
Diop, F. et al. Zika virus infection modulates the metabolomic profile of microglial cells. PLoS ONE 13, e0206093 (2018).
Martinez Viedma, M. D. P. & Pickett, B. E. Characterizing the different effects of Zika virus infection in placenta and microglia cells. Viruses 10, 649 (2018).
Dang, J. et al. Zika virus depletes neural progenitors in human cerebral organoids through activation of the innate immune receptor TLR3. Cell Stem Cell 19, 258–265 (2016).
Li, H. et al. Zika virus infects neural progenitors in the adult mouse brain and alters proliferation. Cell Stem Cell 19, 593–598 (2016).
Gabriel, E. et al. Recent Zika virus isolates induce premature differentiation of neural progenitors in human brain organoids. Cell Stem Cell 20, 397–406.e5 (2017).
Gladwyn-Ng, I. et al. Stress-induced unfolded protein response contributes to Zika virus-associated microcephaly. Nat. Neurosci. 21, 63–71 (2017).
Saade, M. et al. Multimerization of Zika Virus-NS5 causes ciliopathy and forces premature neurogenesis. Cell Stem Cell 27, 920–936.e8 (2020).
Plociennikowska et al. TLR3 activation by Zika virus stimulates inflammatory cytokine production which dampens the antiviral response induced by RIG-I-like receptors. J. Virol. 95, e01050–20 (2021).
Jeong, G. U. et al. Zika virus infection induces Interleukin-1β-Mediated Inflammatory responses by macrophages in the brain of an adult mouse model. J. Virol. 97, e0055623 (2023).
Hu, T., Li, J., Carr, M. J., Duchêne, S. & Shi, W. The Asian lineage of Zika Virus: transmission and evolution in Asia and the Americas. Virol. Sin. 34, 1–8 (2019).
Pettersson, J. H. O. et al. How did Zika virus emerge in the Pacific Islands and Latin America? MBio 7, e01239–16 (2016).
Sheridan, M. A. et al. Vulnerability of primitive human placental trophoblast to Zika virus. Proc. Natl Acad. Sci. USA 114, E1587–E1596 (2017).
Rosinski, J. R. et al. Frequent first-trimester pregnancy loss in rhesus macaques infected with African-lineage Zika virus. PLoS Pathog. 19, e1011282 (2022).
Tripathi, S. et al. A novel Zika virus mouse model reveals strain specific differences in virus pathogenesis and host inflammatory immune responses. PLoS Pathog. 13, e1006258 (2017).
Jaeger, A. S. et al. Zika viruses of African and Asian lineages cause fetal harm in a mouse model of vertical transmission. PLoS Negl. Trop. Dis. 13, e0007343 (2019).
Shao, Q. et al. The African Zika virus MR-766 is more virulent and causes more severe brain damage than current Asian lineage and dengue virus. Development 144, 4114–4124 (2017).
Smith, D. R. et al. African and Asian Zika virus isolates display phenotypic differences both in vitro and in vivo. Am. J. Trop. Med. Hyg. 98, 432–444 (2018).
Duggal, N. K. et al. Differential neurovirulence of African and Asian genotype Zika virus isolates in outbred immunocompetent mice. Am. J. Trop. Med. Hyg. 97, 1410–1417 (2017).
Aubry, F. et al. Recent African strains of Zika virus display higher transmissibility and fetal pathogenicity than Asian strains. Nat. Commun. 12, 916 (2021).
Yuan, L. et al. A single mutation in the prM protein of Zika virus contributes to fetal microcephaly. Science 358, 933–936 (2017).
Lin, C. S. et al. A reverse mutation E143K within the PrM protein of Zika virus asian lineage natal RGN strain increases infectivity and cytopathicity. Viruses 14, 1572 (2022).
Shan, C. et al. A Zika virus envelope mutation preceding the 2015 epidemic enhances virulence and fitness for transmission. Proc. Natl Acad. Sci. USA 117, 20190–20197 (2020).
Inagaki, T. et al. Leu-to-Phe substitution at prM146 decreases the growth ability of Zika virus and partially reduces its pathogenicity in mice. Sci. Rep. 11, 19635 (2021).
Xia, H. et al. An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nat. Commun. 9, 414 (2018).
Carbaugh, D. L. et al. Two genetic differences between closely related Zika virus strains determine pathogenic outcome in mice. J. Virol. 94, e00618–e00620 (2020).
Klase, Z. A. et al. Zika fetal neuropathogenesis: etiology of a viral syndrome. PLoS Negl. Trop. Dis. 10, e0004877 (2016).
Stagni, F., Giacomini, A., Guidi, S., Ciani, E. & Bartesaghi, R. Timing of therapies for Down syndrome: the sooner, the better. Front. Behav. Neurosci. 9, 265 (2015).
Aubry, F. et al. Single-stranded positive-sense RNA viruses generated in days using infectious subgenomic amplicons. J. Gen. Virol. 95, 2462–2467 (2014).
Duong, V., Dussart, P. & Buchy, P. Zika virus in Asia. Int. J. Infect. Dis. 54, 121–128 (2017).
Biswas, A. et al. Zika outbreak in India in 2018. J. Travel Med. 27, taaa001 (2021).
Weaver, S. C. et al. Zika virus: History, emergence, biology, and prospects for control. Antiviral Res. 130, 69–80 (2016).
World Health Organization. Zika - Epidemiological Update - 25 Aug 2017. https://www.paho.org/en/documents/25-august-2017-zika-epidemiological-update-0
Brasil, P. et al. Zika virus infection in pregnant women in Rio de Janeiro. N. Engl. J. Med. 375, 2321–2334 (2016).
Melo, A. S. et al. Congenital Zika virus infection: beyond neonatal microcephaly. JAMA Neurol. 73, 1407–1416 (2016).
Grant, R. et al. In utero Zika virus exposure and neurodevelopment at 24 months in toddlers normocephalic at birth: a cohort study. BMC Med. 19, 12 (2021).
Abtibol-Bernardino, M. R. et al. Neurological findings in children without congenital microcephaly exposed to Zika virus in utero: a case series study. Viruses 12, 1335 (2020).
Sarno, M. et al. Zika virus infection and stillbirths: a case of hydrops fetalis, hydranencephaly and fetal demise. PLoS Negl. Trop. Dis. 10, e0004517 (2016).
Stanford Medicine Children’s Health. Hydrops Fetalis (2023). https://www.stanfordchildrens.org/en/topic/default?id=hydrops-fetalis-90-P02374
Cauchemez, S. et al. Association between Zika virus and microcephaly in French Polynesia, 2013-15: a retrospective study. Lancet 387, 2125–2132 (2016).
Driggers, R. W. et al. Zika virus infection with prolonged maternal viremia and fetal brain abnormalities. N. Engl. J. Med. 374, 2142–2151 (2016).
Sochocka, M., Diniz, B. S. & Leszek, J. Inflammatory response in the CNS: friend or foe? Mol. Neurobiol. 54, 8071–8089 (2017).
Kim, Y. S. & Joh, T. H. Microglia, major player in the brain inflammation: their roles in the pathogenesis of Parkinson’s disease. Exp. Mol. Med. 38, 333–347 (2006).
Pardy, R. D., Valbon, S. F., Cordeiro, B., Krawczyk, C. M. & Richer, M. J. An epidemic Zika virus isolate suppresses antiviral immunity by disrupting antigen presentation pathways. Nat. Commun. 12, 4051 (2021).
Tatara, J. M. et al. Differential proteomics of Zika virus (ZIKV) infection reveals molecular changes potentially involved in immune system evasion by a Brazilian strain of ZIKV. Arch. Virol. 168, 70 (2023).
Esser-Nobis, K. et al. Comparative analysis of African and Asian Lineage-Derived Zika virus strains reveals differences in activation of and sensitivity to antiviral innate immunity. J. Virol. 93, e00640–19 (2019).
Liu, Z. et al. A single nonsynonymous mutation on ZIKV E protein-coding sequences leads to markedly increased neurovirulence in vivo. Virol. Sin. 37, 115–126 (2022).
Gladwyn-Ng, I. et al. Stress-induced unfolded protein response contributes to Zika virus-associated microcephaly. Nat. Neurosci. 21, 63–73 (2018).
Kolimenakis, A. et al. The role of urbanisation in the spread of Aedes mosquitoes and the diseases they transmit-A systematic review. PLoS Negl. Trop. Dis. 15, e0009631 (2021).
Kraemer, M. U. G. et al. Past and future spread of the arbovirus vectors Aedes aegypti and Aedes albopictus. Nat. Microbiol. 4, 854–863 (2019).
Shi, Y., Kirwan, P. & Livesey, F. J. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc. 7, 1836–1846 (2012).
van Boheemen, S. et al. Quasispecies composition and evolution of a typical Zika virus clinical isolate from Suriname. Sci. Rep. 7, 2368 (2017).
Donald, C. L. et al. Full genome sequence and sfRNA interferon antagonist activity of Zika virus from recife, Brazil. PLoS Negl. Trop. Dis. 10, e0005048 (2016).
Kum, D. B. et al. A yellow fever-Zika chimeric virus vaccine candidate protects against Zika infection and congenital malformations in mice. npj Vaccines 3, 56 (2018).
Conceição-Neto, N. et al. Modular approach to customise sample preparation procedures for viral metagenomics: a reproducible protocol for virome analysis. Sci. Rep. 5, 16532 (2015).
De Coninck, L., Faye, L., Basler, N., Jansen, D. & Van Espen, L. ViPER. Zenodo (2024). https://doi.org/10.5281/zenodo.5502203
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Nurk, S., Meleshko, D., Korobeynikov, A. & Pevzner, P. A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834 (2017).
Reed, L. J. & Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 27, 493–497 (1938).
Acknowledgements
We thank Prof. Catherine Sadzot-Delvaux (Liège University, Uliège) for her support. We warmly thank Stijn Hendrickx for technical support. We thank Niels Cremers, Elke Maas, Johanna Bouckaert, Jill Swinnen and the staff at the Rega animal facility at KU Leuven for technical assistance. We thank Jelle Matthijnssens and Lander De Coninck (KU Leuven) for sequencing the ZIKV strains and analyzing the results. We thank Alexandre Hego and the imaging platform at Uliège for their grateful technical help. We thank Martijn van Hemert (LUMC), Alain Kohl (MRC-University of Glasgow Centre for Virus Research), Van-Mai Cao-Lormeau (Institut Louis Malardé), Richard G. Jarman (WRAIR), and Amadou A. Sall (Institut Pasteur Dakar) for kindly providing the ZIKV strains. This work was primarily funded by the European Union’s Horizon 2020 research and innovation program under ZikaPLAN grant agreement no. 734584 (to J.N.) and under ZIKAlliance grant agreement no. 734548 (to J.N.). This work was also supported by the Fonds de la Recherche Scientifique (FNRS-FRIA Ph.D. fellowship to M.D.). The funders had no role in the study design, data collection and interpretation, or the decision to submit the work for publication.
Author information
Authors and Affiliations
Contributions
M.D.: Planning, coordination and execution of the work; F.T., L.S.F., and X.d.L.: Help with the generation of the ZIKV strain containing a single mutation; E.S.: Processing of samples and performing the Luminex experiments; K.A. and C.V.: Generation of hNPCs; S.J.F.K.: Supervision of the project; S.J.F.K. and J.N.: Securing of funding. M.D. and S.J.F.K. wrote the manuscript with contributions from I.G.-N., P.E.M., and J.N., and comments from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Darmuzey, M., Touret, F., Slowikowski, E. et al. Epidemic Zika virus strains from the Asian lineage induce an attenuated fetal brain pathogenicity. Nat Commun 15, 10870 (2024). https://doi.org/10.1038/s41467-024-55155-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55155-4
