
Abstract
Accumulating evidence indicates that cellular senescence is closely associated with osteoarthritis. However, there is limited research on the mechanisms underlying fibroblast-like synoviocyte senescence and its impact on osteoarthritis progression. Here, we elucidate a positive correlation between fibroblast-like synoviocyte senescence and osteoarthritis progression and reveal that GATD3A deficiency induces fibroblast-like synoviocyte senescence. Mechanistically, GATD3A deficiency enhances the binding of Sirt3 to MDH2, leading to deacetylation and decreased activity of MDH2. Reduced MDH2 activity impairs tricarboxylic acid cycle flux, resulting in mitochondrial dysfunction and fibroblast-like synoviocyte senescence. Intra-articular injection of recombinant adeno-associated virus carrying GATD3A significantly alleviates the osteoarthritis phenotype in male mice. This study increases our current understanding of GATD3A function. In particular, we reveal a novel mechanism of fibroblast-like synoviocyte senescence, suggesting that targeting GATD3A is a potential therapeutic approach for osteoarthritis.
Similar content being viewed by others


Introduction
Osteoarthritis (OA) is a disabling disease characterised by cartilage degradation, synovial inflammation and subchondral bone sclerosis, and is the most prevalent disease among ageing populations. Presently, OA affects over 500 million people worldwide, with an annually increasing incidence1,2. The current therapeutic options for OA remain limited, and the pathological mechanism needs further investigation.
Various pathological factors, such as mechanical loading3, inflammatory mediators4, cellular senescence5 and metabolic dysregulation6, have been implicated in the progression of OA. Cellular senescence has attracted considerable attention in recent years and is characterized by cell cycle arrest and senescence-associated secretory phenotype (SASP). Senescent cells not only exhibit changes in their own functions but also affect the surrounding cells7. A strong correlation between cellular senescence and OA has been confirmed8, and cellular senescence has been shown to be a crucial driver of OA9,10,11. But further investigation is required to establish the causes of senescence and the mechanisms promoting progression of OA.
Numerous studies have been conducted to address these issues, especially studies regarding chondrocyte senescence12,13. Synovial fluid serves as the primary source of cartilage nutrition, and synoviocyte lesions inevitably result in chondrocyte dysfunction. The synovium comprises fibrous and fatty tissues, fibroblast-like synoviocytes (FLSs) and macrophages. Most studies have focused on chondrocyte senescence, with insufficient attention given to the senescence of FLSs. Recently, FLS senescence in OA has been confirmed14, and several studies have reported an OA phenotype by injecting senescent FLSs into knee joints15,16, thereby highlighting the role of senescent FLSs in promoting OA progression. Senescent FLSs impair the physiological function of the synovium, and also drive chondrocyte catabolism and accelerate cartilage degradation through the secretion of SASP factors such as IL-1β, TNF, IL-6, and IL-179,17,18. Thus, the senescence of FLSs in OA, and especially its mechanism, requires further clarification.
In addition to replicative senescence, factors such as mitochondrial dysfunction, DNA damage and oxidative stress have been identified as inducers of cellular senescence19,20,21. Mitochondria use oxygen to oxidize nutrients through the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) to produce essential ATP and metabolites. Mitochondrial dysfunction, such as decreased ATP production and increased proton leakage induced by damaged respiratory capacity and decreased membrane potential, can trigger cellular senescence22,23. Targeting mitochondrial dysfunction is therefore considered to have potential for counteracting cellular senescence24. However, there is still a need to establish whether mitochondrial dysfunction triggers senescence of OA-FLSs, as well as to identify the underlying mechanism of mitochondrial dysfunction.
RNA-sequencing (RNA-Seq) results have indicated marked downregulation of GATD3A (glutamine amidotransferase-like class 1 domain-containing 3 A) in OA-FLSs. Genome-wide association studies have provided novel insights into many diseases by identifying genetic loci. Recent studies have identified genetic variants around the GATD3A gene as risk factors for several phenotypes such as bone density25, age-related hearing impairment26, and type 1 diabetes27. However, whether GATD3A is a risk locus for OA remains unexplored. Understanding the function of GATD3A and its impact on OA is therefore of significant importance. A recent study reported that GATD3A limited the formation of advanced glycation end products (AGEs) and regulated mitochondrial dynamics28. However, the precise mechanism by which GATD3A modulates mitochondrial function has not been fully elucidated. Co-localization results indicated that GATD3A was predominantly localized to the mitochondrial matrix, which further suggested its correlation with mitochondrial function. The TCA cycle is a crucial energy and metabolic hub in the mitochondrial matrix. It generates essential NADH + H+ and FADH2 for OXPHOS, and regulates chromatin modification, redox homeostasis, and post-translational modifications of proteins through its intermediate metabolites, thereby controlling cellular function and fate29,30,31. Energy and metabolic imbalances caused by TCA cycle impairment can lead to alterations in mitochondrial function and cellular physiology. However, it is unknown whether the downregulation of GATD3A mediates senescence of OA-FLSs by disrupting mitochondrial function and metabolism through the TCA cycle.
In this study, we found that GATD3A maintains TCA cycle stability by competitively binding MDH2 to Sirt3, thereby regulating the acetylation level of MDH2. GATD3A downregulation disrupts the TCA cycle, leading to mitochondrial dysfunction and metabolic imbalance, resulting in FLS senescence and OA progression. Furthermore, overexpression of GATD3A in FLSs effectively alleviates DMM-induced FLS senescence and OA progression, highlighting GATD3A as a promising therapeutic target for OA treatment.
Results
Senescence of FLSs is associated with OA progression
To evaluate synovial senescence in OA, we collected synovial tissues from OA patients (who underwent total knee arthroplasty) and normal control individuals (who underwent arthroscopic surgery and were confirmed using arthroscopy to have no cartilage damage or synovitis). We then determined the expression of senescence marker p16INK4a. Compared with normal controls, the synovium of OA patients was thickened and accompanied by inflammatory infiltration, and expression of p16INK4a was significantly elevated (Fig. 1a and Supplementary Fig. 1a). To further assess the senescence of synovial FLSs in OA patients, immunofluorescence double staining of p16INK4a and vimentin was conducted, which showed that the percentage of p16INK4a positive FLSs in OA synovium was elevated, when compared to normal synovium, and a positive correlation was observed between FLS senescence and OA severity (Fig. 1b, c). Compared to normal synovium, the CDKN1A (encoding p21) and CDKN2A (encoding p16) mRNA expressions were also upregulated in OA synovium (Supplementary Fig. 1b). FLSs were separated from the synovium, and mRNA levels of CDKN1A and CDKN2A, as well as protein levels of p16INK4a, p21, and p53 were significantly elevated in OA-FLSs, when compared to Nor-FLSs (Fig. 1d, e). SA-β-Gal staining and immunofluorescence staining of p16INK4a further indicated the senescence of OA-FLSs (Fig. 1f, g). The secretion of SASP factors by senescent cells affects the function of normal chondrocytes, which is an important factor in promoting OA progression32. The mRNA expression and secretion of pro-inflammatory cytokines (IL-1β and TNF-α) were enhanced in OA-FLSs, when compared to Nor-FLSs (Fig. 1h). Moreover, chondrocytes co-cultured with OA-FLSs exhibited reduced matrix synthesis capability (Fig. 1i and Supplementary Fig. 1c), suggesting that OA-FLSs impaired chondrocyte function through paracrine mechanisms. A post-traumatic OA model was then constructed to confirm the senescence of FLSs in OA mice (Fig. 1j). Compared with the sham group, synovial senescence was observed in the OA group, with a significant increase in p16INK4a positive FLSs in the synovium (Fig. 1k–n). OARSI score was also highly correlated with FLS senescence (Fig. 1o). Together, the results suggest that senescence of FLSs is associated with OA progression.

a Representative images of immunohistochemistry staining of p16INK4a in normal and OA patients. n = 10 independent biological samples. Scale bar: 100 µm. b Representative images of immunofluorescence staining of p16INK4a in synovium from normal and OA patients, and quantification of the percentage of p16INK4a positive FLSs (n = 25 Nor, n = 33 OA). Scale bar: 100 µm. c Spearman’s correlation analysis was performed between Mankin score and the percentage of p16INK4a positive FLSs in OA samples (n = 33). d Quantitative PCR analysis of CDKN1A and CDKN2A in FLSs. (n = 25 Nor, n = 33 OA). e Western blotting analysis of senescent markers. n = 3 independent biological replicates. f Representative images of SA-β-Gal staining for Nor-FLSs and OA-FLSs. n = 9 independent biological replicates. Scale bar: 100 µm. g Representative images of immunofluorescence staining of p16INK4a in Nor-FLSs and OA-FLSs, and the percentage of p16INK4a positive cells. n = 15 independent biological replicates. Scale bar: 50 µm. h Quantitative PCR analysis (n = 3 independent biological replicates) and Elisa assay (n = 6 independent biological replicates) of pro-inflammatory cytokine (IL-1β and TNF-α) in Nor-FLSs and OA-FLSs. i Western blotting assay to detect the protein levels of chondrocytes co-cultured with Nor-FLSs or OA-FLSs. n = 3 independent biological replicates. j Representative images of Safranin O/Fast green staining of knee joints. n = 10. Scale bar: 200 µm. k Representative images of immunohistochemistry staining of p16INK4a. n = 10. Scale bar: 100 µm. l The severity of the OA phenotype was assessed using the OARSI score system. n = 10 independent biological animals. m Representative images of immunofluorescence in knee joints. S, synovium; C, cartilage. Scale bar: 500 µm (Left panel), 50 µm (Right panel). n Quantification of the percentage of p16INK4a positive FLSs in the synovium of mice. n = 10 independent biological animals. o Spearman’s correlation analysis was performed between OARSI score and the percentage of p16INK4a positive FLSs in OA mice. n = 10. All data were presented as the means ± SD. P values were determined by two-tailed unpaired Student’s t-test. P < 0.05 was considered statistically significant. Source data are provided as a Source Data file.
To confirm whether senescent FLSs accelerated OA progression, stress-induced senescence in mouse FLSs was induced using H2O2, as previously described in refs. 33,34. The senescence of FLSs induced by H2O2 was confirmed by western blotting and SA-β-Gal staining (Supplementary Fig. 2a, b). Then, normal mouse FLSs or H2O2-induced senescent FLSs were intra-articularly injected into mice without DMM surgery (Supplementary Fig. 2c). The results showed that, when compared to normal FLSs, the injection of senescent FLSs induced synovitis and cartilage degradation (Supplementary Fig. 2d, e). Taken together, these findings highlight the important role of FLS senescence in OA progression.
Mitochondrial function is impaired in OA-FLSs
We subsequently tried to investigate the mechanisms underlying FLS senescence in OA. FLSs from OA patients and controls were collected, and RNA-Seq analysis was conducted. A total of 349 upregulated and 386 downregulated mRNAs were detected in the OA group compared with the control group (Fig. 2a). The changes were enriched in several biological process terms, such as mitochondrial function, metabolic process, cell proliferation and aging, which were identified by Gene Ontology enrichment analysis (Fig. 2b). Gene set enrichment analysis showed impaired OXPHOS in OA-FLSs, suggesting mitochondrial dysfunction (Fig. 2c). Mitochondrial dysfunction is a major factor in cellular senescence; therefore, we further evaluated the changes in mitochondrial function of OA-FLSs. Mitochondrial network and structural changes were observed by Mitotracker staining and transmission electron microscopy (TEM), which revealed increased mitochondrial fragmentation and decreased mitochondrial cristae in OA-FLSs compared with Nor-FLSs (Fig. 2d, e). Increased mitochondrial superoxide and decreased mitochondrial membrane potential were also observed in OA-FLSs (Fig. 2f, g).

a The heatmap of RNA-Seq analysis for FLSs separated from normal and OA patients. b The biological processes of differentially expressed mRNAs were analysed by GO enrichment analysis. c GSEA of ‘OXPHOS’ gene sets in Nor-FLSs and OA-FLSs. (d) Representative images of Mitotracker staining to observe the mitochondrial network of Nor-FLSs (n = 12) and OA-FLSs (n = 11), and the statistical analysis of mean branch length. Scale bar: 40 µm. The white dashed box represents the enlarged image area in the bottom right corner. e Representative images of TEM to observe Nor-FLSs (n = 5) and OA-FLSs (n = 12) and the arrows point to the mitochondria. Scale bar: 1 µm. f Representative images of MitoSOX staining to detect mitochondrial superoxide in Nor-FLSs and OA-FLSs. n = 6 independent biological replicates. Scale bar: 100 µm. g Mitochondrial membrane potential detected by JC-1 assay and the statistical analysis of aggregate-to-monomer ratio (n = 3 Nor-FLSs, n = 7 OA-FLSs). h, i OCR of Nor-FLSs and OA-FLSs, and the analysis of mitochondrial respiration. n = 3 independent biological replicates. j, k PER of Nor-FLSs and OA-FLSs, and the analysis of basal and compensatory glycolysis. n = 4 independent biological replicates. l Quantification of ATP content in Nor-FLSs and OA-FLSs. n = 5 independent biological replicates. All data were presented as the means ± SD. P values were determined by two-tailed unpaired Student’s t-test. P < 0.05 was considered statistically significant. Source data are provided as a Source Data file.
Mitochondria are centres of energy production and metabolism, maintaining physiological activities by balancing OXPHOS and glycolysis. We conducted Seahorse assays to detect whether the balance was disrupted in OA-FLSs. Oxygen consumption rate (OCR) and proton efflux rate (PER) indicated marked impairment of mitochondrial respiration in OA-FLSs, while glycolysis, an inefficient method of energy production, was enhanced (Fig. 2h–k). ATP assay confirmed the reduced energy generation in OA-FLSs (Fig. 2l). These data indicated significant mitochondrial dysfunction in OA-FLSs, which could have contributed to OA-FLS senescence.
Decreased GATD3A contributes to mitochondrial dysfunction and promotes FLS senescence
The RNA-Seq data were used to analyse further the potential mechanisms of mitochondrial dysfunction in OA-FLSs. Expression of GATD3A was significantly decreased in OA-FLSs (Fig. 3a), which has been suggested to have a potential role in regulating mitochondrial function28. Western blotting and PCR analyses of synovium and FLSs confirmed the decrease of GATD3A in OA (Fig. 3b, c and Supplementary Fig. 5a), and a negative correlation between GATD3A and senescent marker CDKN2A was observed (Fig. 3d). Expression of GATD3A detected by immunofluorescence and immunohistochemistry confirmed these changes (Fig. 3e and Supplementary Fig. 3a and 4a). The immunofluorescent chip data of synovium from controls and OA patients further indicated the negative correlation between GATD3A and p16INK4a (Supplementary Fig. 4b).

a Volcano plot of RNA-Seq data. b Western blotting analysis of GATD3A in synovium and FLSs (whole cell lysate) from normal and OA patients. c Quantitative PCR analysis of GATD3A in FLSs (n = 25 Nor, n = 33 OA). d Pearson’s correlation between GATD3A expression and CDKN2A expression in OA-FLSs (n = 33). e Representative images of immunofluorescence of GATD3A in synovium. n = 15 independent biological samples. Scale bar: 100 µm. f Western blotting localization of GATD3A. n = 3 independent biological replicates. g Fluorescent analysis of GATD3A localization, with signal intensity profiles along a line and Pearson’s correlation analysis (n = 5 TOMM20, n = 6 ATP5A and GRP75). Scale bar: 5 µm. Mitotracker staining (n = 10 ShNC, n = 12 ShGATD3A, n = 13 Vec, n = 11 GATD3A) (h), TEM (n = 16 ShNC and ShGATD3A, n = 8 Vec, n = 9 GATD3A) (i) were conducted. Scale bar: 40 µm for Mitotracker staining, 1 µm for TEM. OCR (n = 4 ShNC and ShGATD3A, n = 5 Vec and GATD3A) (j) and PER (n = 5 ShNC and ShGATD3A, n = 6 Vec, n = 4 GATD3A) (k) assays of Nor-FLSs or OA-FLSs after different treatments. l, m Quantification of ATP content in FLSs after different treatments. n = 6 independent biological replicates. Western blotting analysis (n = 3 independent biological replicates) (n), SA-β-Gal staining (n = 6 ShNC and ShGATD3A, n = 8 Vec and GATD3A) (o) and immunofluorescence (n = 10 ShNC and ShGATD3A, n = 13 Vec and GATD3A) (p) of FLSs after different treatments. Scale bar: 100 µm for SA-β-Gal staining, 50 µm for immunofluorescence. q, r Elisa assay to detect the pro-inflammatory cytokines. n = 6 independent biological replicates. s Western blotting analysis of chondrocytes co-cultured with FLSs after different treatments. n = 3 independent biological replicates. All data were presented as the means ± SD. P values were determined by two-tailed unpaired Student’s t-test or one-way ANOVA with Tukey’s multiple-comparisons (g). P < 0.05 was considered statistically significant. Source data are provided as a Source Data file.
Subcellular fractionation confirmed that GATD3A was mainly localized to the mitochondria (Fig. 3f). Decreased GATD3A in the mitochondria of OA-FLSs was also confirmed (Supplementary Fig. 4c). Fluorescent co-localization assay showed that GATD3A was primarily co-localized with the mitochondrial matrix protein GRP75, rather than with the outer membrane marker TOMM20 or the inner membrane marker ATP5A (Fig. 3g). These results suggested that the decrease in GATD3A was correlated with mitochondrial dysfunction and senescence of OA-FLSs.
To determine whether the decrease of GATD3A was an accompanying feature of FLS senescence, or a specific characteristic of OA-FLSs, the expression of GATD3A in an in vitro replicative senescence model was determined. The results showed that GATD3A in Nor-FLSs did not decline during passage (Supplementary Fig. 4d), which suggested that the GATD3A deficiency might be a characteristic feature of OA-FLSs and could potentially drive their senescence. Then, GATD3A was depressed in Nor-FLSs and overexpressed in OA-FLSs through lentivirus infection, which was confirmed by detecting GATD3A expression in whole cell lysates and mitochondria (Supplementary Fig. 5b, d). The mitochondrial network in Nor-FLSs was disrupted by GATD3A knockdown, while overexpression of GATD3A alleviated mitochondrial fragmentation in OA-FLSs (Fig. 3h and Supplementary Fig. 5c). We also found that GATD3A suppression decreased the mitochondrial cristae and the membrane potential, and increased mitochondrial superoxide in Nor-FLSs, while GATD3A overexpression rescued these impaired functions in OA-FLSs (Fig. 3i and Supplementary Fig. 5e, f). OXPHOS and ATP production were impaired in the ShGATD3A group, accompanied by enhanced glycolysis. Conversely, overexpression of GATD3A shifted OA-FLSs from glycolysis to OXPHOS for ATP production (Fig. 3j−m). Besides alterations in mitochondrial function, the ShGATD3A group exhibited significant senescence compared with the control group, including elevated expressions of p16INK4a, p21, p53, and SA-β-Gal, while overexpression of GATD3A mitigated the senescence of OA-FLSs (Fig. 3n–p). Knockdown of GATD3A increased expressions of pro-inflammatory cytokines (IL-1β and TNF-α) in Nor-FLSs, whereas GATD3A overexpression in OA-FLSs reduced their expressions (Fig. 3q, r and Supplementary Fig. 5g). The matrix synthesis of chondrocytes co-cultured with GATD3A-knockdowned Nor-FLSs was diminished, while GATD3A overexpression attenuated the inhibitory effect of OA-FLSs on chondrocyte function (Fig. 3s and Supplementary Fig. 5h). Together, the above results demonstrated that GATD3A deficiency induced mitochondrial dysfunction and FLS senescence, while GATD3A overexpression may serve as an approach to alleviate OA-FLS senescence.
GATD3A overexpression in FLSs alleviates OA development in mice
To assess whether GATD3A overexpression in FLSs alleviated OA progression, we constructed rAAV9-GATD3A modified with HAP-1, a synovial-fibroblast-targeting peptide. First, we evaluated the targeting efficiency of rAAV9.HAP-1 in FLSs. Intra-articular injection of rAAV9.HAP-1-EGFP into mice showed significantly higher EGFP-positive expression in the synovium compared to cartilage (Supplementary Fig. 6), confirming the satisfactory targeting efficiency of rAAV9.HAP-1 in the synovium. Then, rAAV9.HAP-1-GATD3A (rAAV-GATD3A) and negative control rAAV-Vec were intra-articularly injected (Fig. 4a). The results showed that GATD3A was markedly expressed after rAAV-GATD3A injection, accompanied with reduced p16INK4a (Fig. 4b, c). Micro-CT analysis showed that GATD3A overexpression reduced osteophyte formation and subchondral bone sclerosis induced by DMM surgery (Fig. 4d–f). In the sham group, rAAV-GATD3A injection did not induce an OA phenotype, confirming the safety of physiological GATD3A overexpression. Compared to the rAAV-Vec group, the rAAV-GATD3A group showed significant alleviation of DMM-induced synovial inflammation, cartilage erosion, and loss of articular cartilage proteoglycan (Fig. 4g). The results were validated through synovial inflammation and OARSI score (Fig. 4h, i). Overall, the findings demonstrated that FLS-targeted GATD3A overexpression alleviated OA progression in mice.

a Experimental design diagram of intra-articular injection of rAAV-Vec or rAAV-GATD3A in mice (Created in BioRender. Shen, K. (2024) https://BioRender.com/w90b978). b Representative images of immunohistochemistry staining of GATD3A in the knee joint from mice injected with rAAV-Vec or rAAV-GATD3A. n = 7 independent biological samples. Scale bar: 50 µm. c Representative images of immunofluorescent staining of vimentin, GATD3A, and p16INK4a in the knee joint from sham or DMM-induced OA mice after intra-articular injection with rAAV-Vec or rAAV-GATD3A. Enlarged images were shown below. n = 7 independent biological samples. Scale bar: 50 µm. d 3D reconstructed images of micro-CT of knees and osteophytes (arrows pointed), and statistical analyses of the number of osteophytes. n = 7 independent biological animals. e Representative coronal plane images of knee joints obtained from micro-CT data analyses. f Quantification of bone volume per total volume (BV/TV), and trabecular thickness (Tb. Th) obtained from micro-CT data. n = 7 independent biological animals. g Representative images of H&E staining, Safranin O/Fast green staining and Toluidine Blue staining for knee joints from sham or DMM-induced OA mice after intra-articular injection with rAAV-Vec or rAAV-GATD3A. n = 7 independent biological animals. Scale bar: 100 µm. The severities of synovitis and OA phenotype were assessed through synovial inflammation score (h) and OARSI score system (i). n = 7 independent biological animals. All data were presented as the means ± SD. P values were determined by one-way ANOVA with Tukey’s multiple-comparisons. P < 0.05 was considered statistically significant. Source data are provided as a Source Data file.
GATD3A deficiency reduces TCA cycle flux, altering the metabolic pattern of FLSs
To explore the mechanism by which GATD3A regulated mitochondrial function leading to FLS senescence, a label-free proteomics assay was conducted. Knockdown of GATD3A caused significant changes in FLS proteins, with these proteins primarily participating in cellular and metabolic processes, among other biological processes (Fig. 5a, b). IP/MS indicated that the proteins interacting with GATD3A were mainly associated with metabolism and cellular senescence (Fig. 5c). Thus, we speculated that GATD3A deficiency induced mitochondrial dysfunction by altering metabolism, leading to FLS senescence.

a Heatmap of label-free proteomics assay for Nor-FLSs infected with ShNC or ShGATD3A lentivirus. b Number of proteins involved in biological processes was analysed through GO enrichment analysis based on proteomic assay. c KEGG pathway enrichment analysis showed the number of proteins interacting with GATD3A within these pathways based on IP/MS. OPLS-DA analysis (d) and KEGG pathway enrichment analysis (e) of the untargeted metabolomics study for Nor-FLSs infected with ShNC or ShGATD3A lentivirus. f Heatmap of significantly different metabolites in the TCA cycle, glutaminolysis and glycolysis based on the untargeted metabolomics study for Nor-FLSs infected with ShNC or ShGATD3A lentivirus. OCR (n = 5 for glucose supplementation group, and n = 4 for other two groups) (g) and PER (n = 6) (h) assays of Nor-FLSs treated with glucose or glutamine, and glucose and glutamine deprivation. i Diagram of 13C isotopomer patterns with [13C5] glutamine and [U-13C6] glucose as tracer. j Absolute isotopologue distribution of [13C5] glutamine-derived glutaminolysis and TCA cycle metabolites including glutamate, α-KG, succinate, fumarate, malate, oxaloacetate, citrate and aspartate. n = 4 independent biological replicates. The incorporation of 13C atoms was denoted as m + n (number of 13C atoms). k Absolute isotopologue distribution of [U-13C6] glucose-derived glycolysis metabolites including pyruvate and lactate. n = 4 independent biological replicates. The incorporation of 13C atoms was denoted as m + n (number of 13C atoms). l Cellular malate and oxaloacetate content were determined in Nor-FLSs infected with ShNC or ShGATD3A lentivirus. n = 5 independent biological replicates. The glucose uptake rate (n = 5 independent biological replicates) (m) and the cellular lactate content (n = 4 independent biological replicates) (n) in Nor-FLSs infected with ShNC or ShGATD3A lentivirus. All data were presented as the means ± SD. P values were determined by two-tailed unpaired Student’s t-test or one-way ANOVA with Tukey’s multiple-comparisons (g, h). P < 0.05 was considered statistically significant. Source data are provided as a Source Data file.
To investigate whether knockdown of GATD3A altered the metabolism of FLSs, an untargeted metabolomics study was conducted. Based on OPLS-DA, the metabolic profiles showed significant separation between the ShNC and ShGATD3A groups, indicating significant changes in metabolism caused by GATD3A knockdown (Fig. 5d). Among the pathways analyzed by KEGG, aspartate and glutamate metabolism and the TCA cycle were significantly changed (Fig. 5e). Further analyses revealed marked changes in metabolites related to the TCA cycle, glutaminolysis and glycolysis (Fig. 5f). As the main sources of cellular metabolism, glucose and glutamine play crucial roles in maintaining cellular physiology. Therefore, we assessed the dependency of FLSs on glucose and glutamine by measuring OCR and PER. Supplementation with glucose or glutamine alone sustained mitochondrial respiration in FLSs, while deprivation of both impaired respiratory capacity (Fig. 5g). These results suggested that both glucose and glutamine were important for maintaining the TCA cycle in FLSs. PER was also elevated under glutamine supplementation compared with glucose and glutamine deprivation (Fig. 5h). Together, these results supported the importance of glucose and glutamine metabolism in FLSs.
To determine the specific metabolic changes caused by GATD3A deficiency in FLSs, [13C5] glutamine and [U-13C6] glucose were used to monitor glutamine and glucose metabolism. Suppression of GATD3A in FLSs significantly decreased metabolism of [13C5] glutamine. Malate (m + 4) isotopologue accumulated, but other TCA cycle metabolites including α-KG (m + 5), succinate (m + 4), fumarate (m + 4), oxaloacetate (m + 4) and citrate (m + 4) were significantly reduced in the ShGATD3A group compared with the ShNC group. GATD3A knockdown also reduced the level of aspartate (m + 4), which is produced from the TCA cycle metabolite oxaloacetate (Fig. 5i, j and Supplementary Fig. 7a). Conversely, [U-13C6] glucose tracing revealed enhanced glycolysis in the ShGATD3A group, including pyruvate (m + 3) and lactate (m + 3), but not TCA-circulating metabolites such as citrate (m + 2), α-KG (m + 2), succinate (m + 2), fumarate (m + 2), malate (m + 2), and oxaloacetate (m + 2). There was no difference in the contribution of aspartate (m + 2) from [U-13C6] glucose (Fig. 5k and Supplementary Fig. 7b–d). The metabolite contents assay confirmed accumulation of malate and reduction of downstream oxaloacetate in the ShGATD3A group (Fig. 5l). We then verified that GATD3A deficiency promoted glucose uptake and lactate accumulation (Fig. 5m, n). Overall, the results suggested that GATD3A deficiency reduced TCA cycle flux, leading to mitochondrial dysfunction and FLS senescence.
GATD3A interacts with MDH2, a key enzyme of the TCA cycle
Considering the crucial role of energy and metabolite imbalances caused by TCA cycle impairment in promoting cellular senescence35,36, we tried to elucidate the mechanism by which GATD3A deficiency leads to mitochondrial TCA cycle blockade. Proteomics showed that GATD3A knockdown resulted in significant changes in TCA cycle enzymes (Fig. 6a). IP/MS suggested a potential interaction between GATD3A and the TCA cycle enzyme MDH2 (Fig. 6b). MDH2 catalyses the conversion of malate to oxaloacetate, and plays a crucial role in maintaining the TCA cycle flux and the malate–aspartate shuttle31. In the previous part of our study, we also demonstrated accumulation of malate in GATD3A-deficient FLSs. Therefore, we speculated a strong association between GATD3A and MDH2.

a The heatmap of significantly different enzymes in the TCA cycle based on proteomics assay for Nor-FLSs infected with ShNC or ShGATD3A lentivirus. b IP/MS analysis indicated that MDH2 is an interacting protein that binds to GATD3A. c Co-IP assay to verify the interaction between endogenous GATD3A and MDH2 in FLSs. n = 3 independent biological replicates. d Representative images of immunofluorescence to verify the co-localisation of endogenous GATD3A and MDH2 in FLSs. n = 5 independent biological replicates. Scale bar: 10 µm. e Co-IP assay to verify the interaction between exogenous Flag-GATD3A and His-MDH2 in HEK 293 T cells. n = 3 independent biological replicates. f Representative images of immunofluorescence to verify the co-localisation of exogenous Flag-GATD3A and His-MDH2 in HEK 293 T cells. n = 5 independent biological replicates. Scale bar: 10 µm. g Design of GATD3A and MDH2 truncations. h Co-IP assay showing the interactions between Flag-GATD3A and His-MDH2 in HEK 293 T cells. n = 3 independent biological replicates. i Molecular docking predicted the docking sites of GATD3A and MDH2. j Docking sites of GATD3A and MDH2, and the mutations of binding sites between GATD3A and MDH2. k Co-IP assay showing the interactions between Flag-tagged mutated GATD3A and His-tagged mutated MDH2 in HEK 293 T cells. n = 3 independent biological replicates. Source data are provided as a Source Data file.
Endogenous co-immunoprecipitation (Co-IP) experiments demonstrated the interaction between GATD3A and MDH2 (Fig. 6c). Immunofluorescence analysis also confirmed the co-localization of endogenous GATD3A and MDH2 (Fig. 6d). Exogenous Flag-GATD3A and His-MDH2 were transfected into HEK 293 T cells, and the Co-IP results also confirmed the interaction between GATD3A and MDH2 (Fig. 6e, f). Next, we investigated the domains of GATD3A and MDH2 contribute to the interaction with each other. Co-IP assay revealed that domain D1 (residues 1–155) of GATD3A was responsible for binding to MDH2, and the binding fragment in MDH2 was located in domain M2 (residues 169–338) (Fig. 6g, h). Molecular docking was conducted to predict the docking sites of GATD3A and MDH2 (Fig. 6i). The predicted binding sites were mutated (Fig. 6j) and the interaction between GATD3A and MDH2 was verified. The interaction between GATD3A and MDH2 was abolished by GATD3A-binding site mutations (Flag-GATD3A-MUT1) or MDH2 binding site mutations (His-MDH2-MUT2) (Fig. 6k). These results confirmed the interaction between GATD3A and MDH2, and suggested that GATD3A deficiency might reduce mitochondrial TCA cycle flux by affecting MDH2 activity.
GATD3A deficiency induces mitochondrial dysfunction by increasing Sirt3-mediated MDH2 deacetylation
After the interaction between GATD3A and MDH2 was confirmed, we evaluated whether GATD3A deficiency regulated mitochondrial function and FLS senescence by impairing MDH2 activity. The synovium of OA patients and mice with DMM-induced OA expressed a low level of MDH2, while rAAV-GATD3A injection increased MDH2 expression (Supplementary Fig. 3b and Supplementary Fig. 8). MDH2 activity of Nor-FLSs infected with ShGATD3A was impaired compared with that in the control group (Fig. 7a). Previous studies have suggested that reversible acetylation of mitochondrial enzymes plays an important role in regulating mitochondrial function and cellular senescence, and MDH2 enzyme activity is also regulated by acetylation37. Western blotting showed that GATD3A knockdown reduced acetylation of proteins in whole-cell lysates and mitochondria of FLSs (Fig. 7b). Further assays confirmed that acetylation of MDH2 was impaired by GATD3A knockdown (Fig. 7c, d). Furthermore, we also observed that loss of GATD3A dramatically reduced MDH2 acetylation, while GATD3A-FL transfection, but not GATD3A-D2, abolished deacetylation of MDH2 in HEK293T cells (Fig. 7e). These results confirmed that GATD3A deficiency resulted in deacetylation of MDH2.

a The MDH2 activity in Nor-FLSs infected with ShNC or ShGATD3A. n = 5 independent biological replicates. b Western blotting analysis of protein acetylation levels in FLSs. n = 3 independent biological replicates. c Immunoprecipitation assay assessing MDH2 acetylation in FLSs infected with ShNC or ShGATD3A. n = 3 independent biological replicates. HEK293T cells infected with ShNC or ShGATD3A were transfected with His-MDH2 alone (d), with His-MDH2 and Flag-GATD3A (e), or with His-MDH2 and treated with NAM or TSA (f). Immunoprecipitation with anti-His was conducted to assess His-MDH2 acetylation. n = 3 independent biological replicates. g GATD3A-knockdowned Nor-FLSs were treated with NAM or TSA, and the MDH2 activity was assed. n = 5 independent biological replicates. h–k OCR (n = 5 NAM + TSA, n = 6 other four groups) and PER assays (n = 3 ShGATD3A, n = 4 other groups) of Nor-FLSs after different treatment. l, m Representative images of SA-β-Gal staining (n = 7 ShNC, n = 6 DMSO, n = 4 NAM and TSA, n = 5 NAM + TSA) and immunofluorescence staining (n = 15) of Nor-FLSs after different treatment. Scale bar: 100 µm for SA-β-Gal staining, 50 µm for immunofluorescence. n Western blotting analysis of senescent markers in differently treated Nor-FLSs. n = 3 independent biological replicates. o HEK293T cells were transfected with His-MDH2 WT and 4KR acetylation-deficient mutants, and the acetylation of His-MDH2 was detected. n = 3 independent biological replicates. p Co-IP assay to verify the interaction between MDH2 and Sirt3 in GATD3A-knockdowned Nor-FLSs. n = 3 independent biological replicates. q Co-IP assay to verify the interaction between His-MDH2 and Sirt3 in GATD3A-knockdowned HEK293T cells after NAM treatment. n = 3 independent biological replicates. r GATD3A-knockdowned HEK293T cells were co-transfected with His-MDH2, Myc-Sirt3, and Flag-GATD3A to assess the effect of Flag-GATD3A on His-MDH2 binding to Myc-Sirt3 via Co-IP assay. n = 3 independent biological replicates. All data were presented as the means ± SD. P values were determined by two-tailed unpaired Student’s t-test (a) or one-way ANOVA with Tukey’s multiple-comparisons. P < 0.05 was considered statistically significant. Source data are provided as a Source Data file.
Nicotinamide (NAM; an inhibitor of sirtuins) but not trichostatin A (TSA; an inhibitor of histone deacetylases) elevated acetylation of MDH2 in the absence of GATD3A (Fig. 7f). The impaired MDH2 activity caused by GATD3A knockdown was recovered by NAM (Fig. 7g). After NAM treatment, as acetylation of MDH2 recovered, glycolysis induced by GATD3A knockdown in FLSs was decreased, prompting a transition towards OXPHOS (Fig. 7h–k). Additionally, NAM treatment reduced the accumulation of malate caused by GATD3A knockdown, and partially recovered the concentration of downstream metabolite oxaloacetate (Supplementary Fig. 9a, b). Furthermore, we observed that the acetylation of MDH2 recovered by NAM attenuated the senescence of FLSs induced by GATD3A knockdown (Fig. 7l–n and Supplementary Fig. 9c, d). To further confirm that acetylation of MDH2 was regulated by GATD3A expression, the predicted acetylation sites (K185, K301, K307 and K314) of MDH2 were mutated. Even with NAM treatment, acetylation of the 4KR mutant group could not be restored, compared with the wild-type group (Fig. 7o). These findings indicated that loss of GATD3A impaired MDH2 enzyme activity by decreasing acetylation of MDH2.
Due to the NAM treatment recovered the acetylation of MDH2, we investigated whether GATD3A deficiency regulated MDH2 deacetylation through Sirtuins. Sirt3 is one of the most prominent deacetylases that is expressed in mitochondria, and it regulates the TCA cycle, OXPHOS, and other mitochondrial functions. A recent study also confirmed that acetylation of MDH2 was regulated by Sirt338. Our results demonstrated that GATD3A and Sirt3 both bound to MDH2 in FLSs, while loss of GATD3A dramatically enhanced the interaction between Sirt3 and MDH2 (Fig. 7p). The enhanced binding between Sirt3 and MDH2 in HEK 293 T cells, which was caused by GATD3A knockdown, was attenuated by NAM (Fig. 7q). Then, endogenous GATD3A of HEK 293 T cells was inhibited and exogenous Flag-GATD3A was transfected. Co-IP demonstrated that loss of GATD3A increased Myc–Sirt3 binding to His-MDH2, while transfection of Flag-GATD3A abolished Myc–Sirt3 recruitment to His-MDH2 (Fig. 7r). These results demonstrated that GATD3A deficiency enhanced the Sirt3–MDH2 interaction, resulting in decreased MDH2 acetylation and activity. Impaired MDH2 activity disrupted the TCA cycle, leading to mitochondrial dysfunction and ultimately, FLS senescence.
Oxaloacetate attenuates GATD3A-deficiency-induced mitochondrial dysfunction and OA development
We then investigated whether supplementation of the downstream metabolite, oxaloacetate, restored mitochondrial function of GATD3A-knockdown FLSs (Fig. 8a). Our results indicated that 1 mM oxaloacetate treatment restored the decrease of oxaloacetate in FLSs caused by GATD3A knockdown (Fig. 8b) and recovered the impaired respiratory capacity and inhibited glycolysis (Fig. 8c, d). Oxaloacetate supplementation also attenuated the senescence of FLSs induced by GATD3A knockdown (Fig. 8e, f), without altering the expression of GATD3A and the acetylation of MDH2 (Fig. 8g). These results further confirmed that GATD3A deficiency inhibited the TCA cycle by reducing MDH2 activity, and supplementation of oxaloacetate, downstream of malate, restored the TCA cycle and alleviated FLS senescence.

a Role of oxaloacetate in TCA cycle and malate-aspartate shuttle (Created in BioRender. Shen, K. (2024) https://BioRender.com/w90b978). b The content of oxaloacetate in Nor-FLSs after infection with ShNC or ShGATD3A lentivirus and supplementation with oxaloacetate. n = 5 independent biological replicates. OCR (n = 4 ShNC, and n = 3 other two groups) (c) and PER (n = 4 ShGATD3A, and n = 3 other two groups) d assays of Nor-FLSs after infection with ShNC or ShGATD3A lentivirus and supplementation with 1 mM oxaloacetate. e, f Representative images of SA-β-Gal staining (n = 10) and immunofluorescence staining (n = 15) of Nor-FLSs after infection with ShNC or ShGATD3A lentivirus and supplementation with 1 mM oxaloacetate. Scale bar: 100 µm for SA-β-Gal staining and 50 µm for immunofluorescence. g Western blotting and immunoprecipitation analysis to detect the senescent markers and the acetylation level of MDH2 in Nor-FLSs after infection with ShNC or ShGATD3A lentivirus and supplementation with 1 mM oxaloacetate. n = 3 independent biological replicates. h Experimental design diagram of intraperitoneal injection of oxaloacetate in mice (Created in BioRender. Shen, K. (2024) https://BioRender.com/w90b978). i Representative images of immunofluorescent staining in the knee joint from DMM-induced OA mice with or without oxaloacetate treatment. n = 5 independent biological samples. Scale bar: 50 µm. j 3D reconstruction images of micro-CT of the knees and osteophytes (arrows pointed), and the statistical analysis of the number of osteophytes. n = 5 independent biological animals. k Representative coronal plane images of knee joints. l Quantification of BV/TV, and Tb. Th. n = 5 independent biological animals. Representative images of H&E staining (m), Safranin O/Fast green staining and Toluidine Blue staining (n) for knee joints. n = 5 independent biological samples. Scale bar: 100 µm. o The severity of synovitis and OA phenotype were assessed through synovial inflammation score and OARSI score system. n = 5 independent biological animals. MAS, malate-aspartate shuttle; OAA, oxaloacetate. All data were presented as the means ± SD. P values were determined by one-way ANOVA with Tukey’s multiple-comparisons. P < 0.05 was considered statistically significant. Source data are provided as a Source Data file.
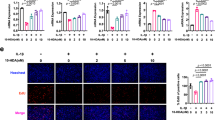
Furthermore, we evaluated the effects of oxaloacetate supplementation on synovial senescence and OA development (Fig. 8h). Immunofluorescence indicated that even though GATD3A expression decreased, oxaloacetate treatment still attenuated the expression of p16INK4a in the synovium (Fig. 8i). Micro-CT analysis showed that oxaloacetate reduced osteophyte formation and subchondral bone sclerosis induced by DMM (Fig. 8j–l). Compared with the DMM group, the oxaloacetate group showed significantly reduced synovial inflammation and cartilage erosion (Fig. 8m, n), and synovial inflammation and OARSI score confirmed these changes (Fig. 8o). The above findings indicated that oxaloacetate supplementation could alleviate the synovial senescence and OA progression caused by GATD3A deficiency.
Discussion
Cartilage degeneration is a primary characteristic of OA patients, and for a long time, synovial cell abnormalities were considered secondary changes of cartilage destruction, but were not intensely investigated. Increasing evidence confirms that OA is a joint disease closely associated with synovial tissue, and suggests that synovial abnormalities precede cartilage injury and mediate its degeneration39. However, the pathological mechanisms in the synovium in OA need further elucidation. In this study, we confirmed the senescence of FLSs in both OA patients and mice, and showed that senescent FLSs could induce the OA phenotype, highlighting their crucial role in OA pathogenesis.
Interventions targeting the mechanism of cellular senescence are expected to provide new insights into alleviating OA14,40. Mitochondria play a pivotal role in providing essential energy for cellular physiological functions and serve as metabolic hubs in regulating metabolic homeostasis, thus exerting a crucial influence on cellular fate41. Cellular senescence induced by mitochondrial dysfunction is associated with OA progression42,43. In the current study, manifestations of mitochondrial dysfunction were found in OA-FLSs, and GATD3A, a novel potential regulator of mitochondrial function28, was dramatically decreased in OA-FLSs. In vitro results showed that GATD3A did not decrease with increased passaging of Nor-FLSs, suggesting that the decline of GATD3A was not a feature of replicative senescence, but rather a specific change in FLSs under OA conditions that might drive their senescence. Our further studies confirmed the role of GATD3A in regulating the mitochondrial function and the senescence of FLSs. It is important to note that cellular senescence is traditionally considered as a static cell fate characterized by stable growth arrest44. Nonetheless, we observed that GATD3A overexpression significantly reduced the expressions of senescent markers in OA-FLSs. As senescence is increasingly understood as a dynamic, multi-step process45, we speculated that GATD3A overexpression might mitigate the mitochondrial dysfunction of quiescent or pre-senescent OA-FLSs and alleviate the senescent phenotype. Further in vivo studies confirmed that overexpression of GATD3A in FLSs significantly attenuated senescence and alleviated OA progression induced by DMM surgery, indicating that targeting GATD3A was a potential therapeutic approach for OA.
GATD3A is a Parkinson-associated DJ-1/PARK7-like protein that is considered to be associated with accumulation of advanced glycation end products and regulation of mitochondrial dynamics28. However, the regulatory mechanism of GATD3A in mitochondrial function and its impact on cellular senescence remain unclear. In this study, we demonstrated for the first time that GATD3A was primarily involved in the regulation of metabolic processes, particularly playing a beneficial role in maintaining the integrity of the TCA cycle. Proteomics and metabolomics revealed that GATD3A deficiency led to a reduction in TCA cycle flux, accompanied by increased glucose uptake and the production of glycolytic products such as lactate. Enhanced glycolysis in FLSs has also been confirmed to drive synovial macrophage infiltration and polarization in OA patients, and mediate synovitis46, which further highlights the importance of investigating the functional mechanisms of GATD3A in FLSs. The TCA cycle plays crucial roles in both energy production and metabolism, and is closely associated with mitochondrial dynamics and oxidative stress47. Numerous studies have shown that cellular senescence is closely associated with the TCA cycle44,48. Here, we confirmed that GATD3A deficiency contributed to mitochondrial dysfunction by inhibiting the TCA cycle, inducing OA-FLS senescence. However, the relationship between metabolic shift and cellular senescence remains an area that requires active exploration. Studies also find that senescent cells maintain high metabolic activity and have an increased demand for energy, relying on enhanced glycolysis to sustain their survival49,50, we cannot exclude the possibility that FLS senescence, resulting from TCA cycle blockade caused by GATD3A deficiency, further enhanced the observed metabolic shift towards glycolysis.
Despite reductions in TCA cycle metabolites such as α-KG, fumarate and oxaloacetate, it is puzzling that malate shows abnormal accumulation. Maintenance of the TCA cycle relies on enzyme activity, with MDH2 catalyzing conversion of malate to oxaloacetate, which is a crucial step in the TCA cycle51. Our study confirmed the interaction between GATD3A and MDH2, and demonstrated that GATD3A deficiency decreased acetylation of MDH2, thereby impairing MDH2 activity. Sirt3 is a mitochondrial deacetylase that primarily regulates acetylation of mitochondrial proteins, and recent studies have confirmed its close association with MDH2 activity38,52. In the present study, we demonstrated enhanced binding between MDH2 and Sirt3 after GATD3A inhibition, resulting in MDH2 deacetylation and inactivation. Studies have shown that Sirt3 deficiency in chondrocytes is associated with OA progression, and Sirt3 has been identified as a target for preserving chondrocyte function and preventing OA53,54. Our findings did not contradict the results of these studies. However, the role of Sirt3 in regulating cellular function and disease is complex and should be comprehensively considered in the context of different mechanisms of cellular dysfunction55. Our study broadened the understanding of the mechanisms by which Sirt3 regulated cellular function. Additionally, an interesting finding should also be noted: despite feedback regulation being an adaptive response following TCA cycle blockade, we observed that MDH2 expression was reduced in OA samples and subsequently recovered by GATD3A overexpression (Supplementary Fig. 8). This may be because the reduction in MDH2 can facilitated the conversion of accumulated malate via the pyruvate-malate or malate-aspartate shuttle, which then re-entered the TCA cycle, thereby partially alleviating the TCA cycle blockade caused by decreased MDH2 activity, and preventing the collapse of cellular energy56,57,58. The mechanism responsible for the decrease in MDH2 in OA needs to be further elucidated. Overall, our results elucidated the reasons for the reduction in TCA cycle flux in FLSs caused by GATD3A deficiency.
Supplementing downstream metabolites of impaired enzymes can restore the stability of the TCA cycle59. Oxaloacetate, a downstream metabolite of MDH2, is not only an important metabolite for maintaining the TCA cycle, but it also participates in the malate-aspartate shuttle to maintain its role in NAD+/NADH balance and cell proliferation60,61. Our study confirmed that 1 mM oxaloacetate recovered the oxaloacetate content in GATD3A-deficient Nor-FLSs. Since previous studies have suggested that accumulation of excessive oxaloacetate inhibits succinate dehydrogenase activity and affects cellular respiration62, we chose 1 mM instead of 10 mM for subsequent experiments. Notably, 1 mM oxaloacetate significantly enhanced the respiratory ability and attenuated the senescence of GATD3A-deficient Nor-FLSs. These findings further confirmed that GATD3A deficiency disrupted the TCA cycle through MDH2. In vivo experiments demonstrated that oxaloacetate alleviated the synovial senescence and OA progression caused by GATD3A deficiency. However, since senescent cells accumulate widely accumulated in various tissues in OA, and oxaloacetate was administered via intraperitoneal injection in this study, we could not eliminate the possibility that oxaloacetate might have a positive effect on the metabolism of cells other than FLSs (such as chondrocytes), potentially contributing to the alleviation of OA. Methods for precise supplementation of oxaloacetate in FLSs could be designed and used to further confirm the role of oxaloacetate in alleviating FLS senescence and OA progression.
In conclusion, we confirmed an association between FLS senescence and OA development. GATD3A deficiency disrupted the TCA cycle by Sirt3-mediated deacetylation of MDH2, inducing mitochondrial dysfunction, and promoting senescence of FLSs. Our study demonstrates that GATD3A is a novel potential therapeutic target for OA. However, this study had certain limitations. Beyond its important role in maintaining the TCA cycle, whether GATD3A has additional functions and whether its deficiency involves other mechanisms regulating cellular senescence (such as the accumulation of AGEs) requires further investigation. Additionally, the decrease of GATD3A was unexpectedly observed in OA chondrocytes (Supplementary Fig. 4a), and whether targeting GATD3A represents an “universal” therapeutic strategy for OA requires further investigation.
Methods
Human participants
This study has been approved by the ethics committee of the First Affiliated Hospital of Nanjing Medical University (Approval No: 2023-SR-291). All patients were informed and gave consent prior to sample and information collection, and full written informed consent was obtained. Human OA synovium tissues and cartilage were obtained from OA patients undergoing total knee arthroplasty (n = 33, age 58.79 ± 3.43, BMI 23.02 ± 2.02, Kellgren-Lawrence grade 3.73 ± 0.45). Normal synovium tissues were taken from patients undergoing arthroscopic surgery for meniscus or anterior cruciate ligament injury, and confirmed by arthroscopy to have no cartilage damage or synovitis (n = 25, age 57.36 ± 3.38, BMI 22.47 ± 1.86, Kellgren-Lawrence grade 0.60 ± 0.50). Cartilage tissues collected from OA patients were examined by histological staining to assess OA severity according to the modified Mankin scoring system (0–14)63. The synovitis score (0–9) of normal and OA synovium were also assessed as described before64. Detailed information of patients was shown in Supplementary Table 1-3.
Isolation and culture of FLSs and chondrocytes from human
After clearing the fat and other tissues, the synovium was minced and digested using 0.2% collagenase I (Gibco, USA) for 2 h at 37 °C to separate FLSs. The digestion was then filtrated through a sterile 70 μm filter (Fisherbrand, USA). After centrifugation, the cells were resuspended and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco, USA) containing 10% fetal bovine serum (FBS, Gibco, USA) and 1% penicillin-streptomycin (P/S, Sigma-Aldrich, USA). The next day, the nonadherent cells were aspirated and the adherent cells were washed with complete medium to yield FLSs. Chondrocytes were obtained from the undamaged cartilage area of OA patients, and the procedure was conducted as we described before65. After isolation, the chondrocytes were cultured in DMEM complemented with 10% FBS and 1% P/S.
All cells were passaged and cultured in new medium when they reached 80% confluence.
Recombination adeno-associated virus construction
As described before16,66, recombination adeno-associated virus 9 (rAAV9) modified with synovial fibroblast specific targeting peptide HAP-1 (rAAV9.HAP-1, peptide sequence of HAP-1: SFHQFARATLAS) was designed to overexpress the gatd3a in mice synovium. rAAV9.HAP-1-Gatd3a (rAAV-GATD3A), rAAV9.HAP-1-Vec (rAAV-Vec) and rAAV9.HAP-1-EGFP were purchased from Obio Biotechnology (Shanghai, China).
Experimental OA model and rAAV injection
C57BL/6 J mice were purchased from Nanjing Medical university and housed under pathogen-free conditions with five or fewer mice per cage. Mice were kept under constant temperature and humidity and controlled 12 h light-dark cycles. Mice had free access to standard laboratory chow and water. Due to factors such as hormone levels, model stability, and previous research practices, ten-week-old male mice were used for experimental OA model by DMM. Mice were anesthetized with sodium pentobarbital and the medial meniscotibial ligament in the right knee joints were transected. In the sham group, the joint cavity of right knee joint was also opened without menisci ligament section, and then closed the skin. The experimental protocol was approved by the Animal Research Committee of Nanjing Medical University (Approval No: IACUC-2304053). All animal experiments were performed in accordance with ARRIVE guidelines.
To assess the efficacy of rAAV9.HAP-1 in targeting synovium, 10 μL rAAV9.HAP-1-EGFP was intra-articular injected into knee joint of mice. The mice were sacrificed and the knee joints were collected to observe the EGFP expression using fluorescence microscope at 5 weeks after injection.
For local deliver of GATD3A, 10 μL rAAV (1 × 1012 vg/mL) containing GATD3A (rAAV-GATD3A) and negative control (rAAV-Vec) was administered to mice by intra-articular injection at 7-, 21- and 35-day post DMM surgery. Same procedure was also performed on sham group. All mice were euthanized at 8 weeks post-surgery.
Isolation, senescence induction, and intra-articular injection of mouse FLSs
The isolation of mouse FLSs was conducted as described before67. Briefly, the synovium (including Hoffa’s fat pad) was dissected with microscopic scissors and forceps under a microscopy. After clearing the fat and other tissues, synovial tissue was digested with 0.1% Collagenase IV (Gibco, USA) and 0.01% DNase I (Invitrogen, USA) for 30 min at 37 °C. Digestions were filtered through a sterile 70 μm filter and centrifuged to collect cells. Cells were than culture in complete DMEM medium.
For the induction of FLS senescence, mouse FLSs were treated with 200 μM hydrogen peroxide (H2O2) 2 h daily for 3 days as described before33,34. The senescence of mouse FLSs was confirmed by Western blotting assay and SA-β-Gal staining.
To assess the role of senescent FLSs in promoting OA, normal mouse FLSs and H2O2-induced senescent mouse FLSs (2 × 105 cells in 10 µL) were intra-articularly injected into mice without DMM surgery at 0-, 14- and 28-day. All mice were euthanized at 6 weeks post-injection.
Oxaloacetate treatment
Oxaloacetate (Sigma, USA) was dissolved in PBS and pH was adjusted to 7.0 with NaOH, mice in experimental group were treated daily with 0.5 g/kg oxaloacetate in 200 µL via intraperitoneal injection. The Sham and DMM groups received PBS injection. Mice were dosed for 8 weeks post-surgery.
Micro-CT, histology and immunohistochemistry
The mouse joints were collected and fixed, and the structural were scanned using SkyScan 1176 Micro-CT equipment (Bruker, USA) with an 8 µm scanning resolution. Then the data were reconstructed into 3D images, number of osteophytes, trabecular bone volume per total volume (BV/TV) and trabecular thickness (Tb. Th) were analysed using CT Analyzer software (SkyScan).
For histological staining, the deparaffinized sections of mouse knee joint and human synovium were stained with hematoxylin and eosin (H&E), Toluidine Blue, or Safranin O/Fast green (Solarbio, China) according to the manufacturer’s instructions. The severity of cartilage degeneration was scored Osteoarthritis Research Society International score (OARSI, grade 0–6) and synovial inflammation was scored on an arbitrary scale (0–3) depending on the infiltration of inflammatory cells into the synovial.
For immunohistochemistry staining, deparaffinized sections were treated with 3% H2O2 for 15 min and blocked with 5% BSA for 1 h. Then, the sections were incubated overnight at 4 °C with anti-p16INK4a, anti-GATD3A, or anti-MDH2 antibodies. After incubation, sections were washed with PBS and incubated with HRP-conjugated secondary antibodies (ZSGB-BIO, China) at room temperature for 1 h. Subsequently, the sections were stained using a Diaminobenzidine kit (ZSGB-BIO, China). All images were observed and acquired with an optical microscope (Olympus, Japan).
Immunofluorescence
Decalcified mouse knee joint and human synovium samples were fixed in 4% paraformaldehyde (PFA) and then embedded in paraffin. The embedded samples were cut into 5 μm sections and subsequently deparaffinized. The sections were subjected to antigen retrieval using Sodium citrate antigen retrieval solution (Beyotime, China), followed by treatment with 0.3% Triton X-100 for 15 min, and subsequently blocked with 5% BSA (Biosharp, China) for 1 h. Then, the sections were incubated with primary antibodies overnight at 4 °C and treated with Alexa Fluor 488 or Alexa Fluor 594-conjugated secondary antibodies (Jackson, USA) next day. Finally, the nucleus was stained with DAPI solution for 5 min. Furthermore, multiple immunofluorescence staining was also conducted through tyramide signal amplification (TSA) technology according to the manufacturer’s instructions (AiFang biological, China). The immunofluorescence staining of cells was conducted as the above steps described. All images were observed and captured by using fluorescence microscope or confocal fluorescence microscope (Leica, Germany). The intensity of p16INK4a were quantified using ImageJ software.
Lentivirus infection and cell transfection
GATD3A knockdown (ShGATD3A-1: 5’-GAAGCTCACGTGGACCAGAAA-3’, ShGATD3A-2: 5’-AGTCTACGATGGGACCGAGAT-3’), GATD3A overexpression and control lentiviruses were purchased from GenePharma (Shanghai, China). Cells were infected with lentiviruses (MOI for FLSs: 100, MOI for HEK 293 T:10) in the presence of 5 μg/mL polybrene. After infection for 48 h, stable cells were selected with 2 μg/mL puromycin for a week. FLSs infected differently were divided into ShNC, ShGATD3A, Vec and GATD3A group respectively. The human Flag-tagged GATD3A, His-tagged MDH2, Flag-tagged GATD3A-G1(1-155aa), Flag-tagged GATD3A-G2(156-268aa), His-tagged MDH2-M1(26-168aa), His-tagged MDH2-M2(169-338aa), Flag-tagged GATD3A-MUT#1, Flag-tagged GATD3A-MUT#2, His-tagged MDH2-MUT#1, His-tagged MDH2-MUT#2, His-tagged MDH24KR (K185, K301, K307 and K314) and Myc-tagged Sirt3 plasmids were all constructed by Genebay Biotech (Nanjing, China). FLSs and HEK 293 T cells were transfected using Lipofectamine 3000 reagent (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions.
Senescence associated β-galactosidase (SA-β-Gal) assay
FLSs from different groups were seeded in 24-well plates at a density of 2 × 104 cells. The SA-β-Gal Staining kit (Beyotime, China) was used to detect the senescence of FLSs according to the manufacturer’s instructions. Briefly, FLSs were fixed at room temperature for 15 min and washed with PBS, followed by treatment with SA-β-Gal staining solution overnight at 37 °C. At least four random fields per group were captured, and the percentage of positive staining was calculated using ImageJ.
Elisa assay
The contents of IL-1β and TNF-α in the supernatant of FLSs treated differently were analysed using IL-1β and TNF-α Quantikine ELISA kits (R&D, SLB50 and DTA00D, USA) according to the manufacturer’s instructions.
Western blotting and immunoprecipitation assay
Tissue samples and cells were lysed with RIPA buffer (Beyotime, China) and the BCA protein assay kit (Beyotime, China) was used to measure protein concentration. For Western blotting, protein supernatant was mixed with loading buffer and heated at 95 °C for 10 min. For immunoprecipitation (IP), an Immunoprecipitation Kit (Beyotime, China) was used based on manufacturer’s protocol. Briefly, protein A/G magnetic beads were washed and incubated with required primary antibody or the control IgG at room temperature for 1 h, followed by incubation with lysates overnight at 4 °C. The beads were separated and eluted with SDS-PAGE sample loading buffer (1X). For the elution of tag-target protein, competitive elution was performed with 3X Flag peptide, 6X His peptide or c-Myc peptide (Beyotime, China). Proteins were then separated by SDS-PAGE and transferred to polyvinylidene fluoride membranes (Millipore, USA). After being blocked with 5% BSA for 1 h, the membranes were incubated with primary antibodies overnight at 4 °C, followed by incubation with HRP-conjugated secondary antibodies at room temperature for 1 h. To avoid the interference of heavy chain or light chain, specific targeting secondary antibodies purchased from Abbkine’s IPKineTM were also used. The protein bands were visualized using ECL reagent (Thermo Fisher Scientific, USA), and detected with Tanon 5200. The primary antibodies were listed in Supplementary Table 4. Uncropped gels have been provided in Source Data file and Supplementary Materials.
IP coupled with mass spectrometry (IP/MS)
Total proteins were extracted from the FLSs and IP was performed with GATD3A antibodies and protein A/G magnetic beads as described above. The protein gel strips were proteolyzed and the peptide samples were separated with Thermo UltiMate 3000 UHPLC. Then the mass spectrometry detection was performed using Q-Exactive HF X (Thermo Fisher Scientific, USA).
Quantitative RT-PCR (qRT-PCR)
Total RNA of tissues and cells was extracted using the RNA-easy Isolation Reagent (Vazyme, China) according to the manufacturer’s instructions. 1000 ng total RNA was then reverse-transcribed into cDNA using the PrimeScript RT reagent kit (Takara, Japan). Real-time PCR was performed with SYBR Green PCR master mix (Applied Biosystems, USA) using Step One Plus PCR system (Applied Biosystems, USA). Transcript levels were normalized to the housekeeping gene (β-actin). The relative gene expression was evaluated using the 2 ˗ΔΔCT method. Each group was performed in biological triplicates. The primers used in qRT-PCR were shown in Supplementary Table 5.
RNA-sequencing study
Total RNA of Nor-FLSs and OA-FLSs were extracted with TRIzol reagent (Invitrogen, USA) and treated with RNase-free DNase I to remove contaminating genomic DNA. After cDNA synthesis and PCR amplification, the library construction and the sequence were performed on BGISEQ-500 at BGI (Shenzhen, China). The sequence data was filtered and HISAT was used to map clean reads to the genome of hg19. Then, the RSEM software was used to quantify gene expressions and differentially expressed genes (DEGs) were identified using DEGseq under the conditions of Fold change >2 or <0.5, and p-value < 0.05.
ATP content assay
To assess the content of ATP in cells, ATP assay kit (Beyotime, China) was used. Briefly, after removing the culture medium, cells were treated with lysis buffer and centrifuged at 12,000 g at 4 °C for 5 min. The supernatant was added into the ATP working solution and the luminescence of solution was tested using SynergyH1 (Biotek, USA). Then, the protein concentration of each group was detected by BCA assay, and ATP levels were normalized to nmol/mg protein. Relative ATP contents were compared between groups.
Mitotracker and MitoSOX staining
For Mitotracker and MitoSOX staining, living FLSs from different group were stained with MitoTracker Deep Red FM (Beyotime, China) or MitoSOX Red (Invitrogen, USA) to observe the mitochondrial network and the mitochondrial superoxide following the instructions. The nuclear was stained with Hoechst 33342 dye (Beyotime, China). Images were captured using confocal fluorescence microscope or fluorescence microscopy (Leica, Germany). The mitochondrial network was analysed with FIJI MiNA plug-in and the MitoSOX intensity was analysed using ImageJ.
Transmission electron microscopy
To observe the morphological and structural changes of mitochondria, FLSs were collected and fixed in glutaraldehyde/0.1 M phosphate buffer (pH 7.4) for 1 h, and then fixed in 1% osmium tetroxide for 1 h. After being dehydrated, samples were embedded in acetone/embedding resin and cut into 60 nm ultrathin sections, followed by uranyl acetate and lead citrate staining. Sections were observed under a transmission electron microscope (TEM, Hitachi, Japan).
Mitochondrial membrane potential measurement
The mitochondrial membrane potential of FLSs were measured with a JC-1 assay kit (Beyotime, China). Briefly, FLSs treated differently were digested to prepare cell suspension, followed by incubation with JC-1 working solution for 20 min at 37 °C. After incubation, cells were washed with JC-1 buffer and detected with a flow cytometer (CytoFLEX, Beckman Coulter, USA). The aggregate-to-monomer ratio was analysed using FlowJo software.
Seahorse assay
The mitochondrial respiration and glycolysis of FLSs were measured by Seahorse XF96 Mitochondrial Stress Test and Proton Efflux Rate (PER) assay (Agilent Technologies, USA). Briefly, FLSs treated differently were seeded at 15,000 cells per well in Seahorse XF96 microplates and incubated overnight at 37 °C. Next day, the medium was replaced with seahorse medium, and the cells were then incubated at 37 °C for 1 h in a CO2 free incubator. For Mitochondrial Stress Test assay, cells were measured in basal conditions, followed by 2 μM oligomycin, 3 μM FCCP and 1 μM rotenone/antimycin A (Rot/AA) treatment. For the Glycolytic Rate Assay, after measuring basal conditions, cells were treated with 0.5 μM Rot/AA and 50 mM 2-deoxyglucose (2-DG). The OCR and PER data were normalized to cell number and analysed using WAVE software (Agilent Technologies, USA).
Micromass assay
Human chondrocytes at passage 3 were trypsinized and counted for micromass culture. Briefly, 20 μL of medium containing 4 × 105 chondrocytes was spotted onto 24-well plates and incubated for 2 h. Then the medium of FLSs treated differently was added to chondrocytes, and the medium was changed every 2 days. The micromass was fixed after incubation for 7 days and stained with Alcian blue (Solarbio, China).
Mitochondria, nuclear and cytoplasmic protein extraction
To obtain the mitochondria of cells, a Mitochondrial Isolation kit (MCE, USA) was used. Briefly, cells were trypsinized and suspended in cold PBS. After centrifugation, cells were treated with mitochondria isolation reagent and placed in an ice bath for 15 min. Then, samples were homogenized and centrifuged at 600 g for 10 min. The supernatant was further centrifuged and the isolated mitochondria were treated with mitochondria lysis buffer for further research. The nuclear and cytoplasmic proteins were extracted with the Nuclear and Cytoplasmic Protein Extraction kit (Beyotime, China) according to the manufacturer’s instructions.
Fluorescence colocalization analysis
FLSs or HEK 293 T cells were seeded in glass bottom cell culture dishes (NEST, China) and fixed with 4% fresh PFA for 20 min. Then, cells were washed with PBS three times, permeabilized with 0.1% Triton X-100 for 10 min, and blocked with 10% goat serum. After incubation with primary antibodies overnight at 4 °C, fluorescence conjugated secondary antibodies were added. Images were observed and captured through a Stellaris STED confocal laser microscope (Leica, Germany). Signal intensity profiles along a line were measured and the colocalization were analysed with Pearson’s correlation using Leica LAS X software.
Label-free proteomics assay
The proteins were extracted from FLSs infected with ShNC or ShGATD3A lentiviruses (n = 3 independent biological replicates per group), and the peptides were subjected to LC-MS analysis at Allwegene Technology (Beijing, China). Label-free quantification was used to compare the abundance of proteins between the two groups, and the significant differences was identified using Student’s t-test. After database retrieval, the data was analysed bioinformatically.
Untargeted metabolomics study
The untargeted metabolomics were performed at Metabo-Profile Biotechnology (Shanghai, China). For the preparation of samples, FLSs (5 × 106/sample) collected from ShNC and ShGATD3A group (n = 6) were homogenized in 400 µL extraction solution (methanol/acetonitrile=1:1, v/v) containing 0.02 mg/mL L-2-chlorophenylalanine for 3 min and oscillated for 15 min. Samples were centrifuged at 18,000 g at 4 °C for 20 min to obtain supernatants. Then, the metabolites were analysed using UHPLC-Q Exactive HF-X system (Thermo Fisher Scientific). The original data were imported into the metabolomics software ProgenesisQI (Waters Corporation, USA) for further processing. The MS information was matched with the metabolic database (Human Metabolome Database, HMDB), and metabolites were identified based on the matching score.
Stable isotope tracing analysis
To analyse the isotope distributions in intracellular metabolites, the 13C metabolic flux was performed on FLSs. After reaching 85% confluence, the media of FLSs were replaced with glutamine-free DMEM (Gibco) supplemented with 2 mM [13C5] glutamine or glucose-free DMEM (Gibco) supplemented with 10 mM [U-13C6] glucose and incubated for 24 h. The [13C5] glutamine and [U-13C6] glucose were all purchased from Cambridge Isotope Laboratories (USA). The FLSs from ShNC and ShGATD3A group were washed with PBS and 1 ml 80% cooled methanol were added, and then the cells were scraped and stored at -80 °C. Samples were analysed at Metabo-Profile Biotechnology (Shanghai, China). Before metabolic detection, samples were broken ultrasonically and centrifuged at 18,000 g for 15 min. All supernatant was centrifugally concentrated and redissolved with 100 µL of 80% methanol solution, waiting for sample analysis. After correcting for the natural isotope, samples were analysed using ultra-performance liquid chromatography triple-quadrupole mass spectrometry (UPLC-TQ-MS, Waters Corporation, USA). Peak extraction, integration, identification and quantitative analysis of each metabolite were performed using MassLynx software (Waters Corporation, USA) for raw data.
Glucose uptake and lactate content assay
A Glucose Uptake Assay kit (Abcam) based on 2-deoxyglucose (2-DG) was used to detect the glucose consumption of FLSs according to the manufacturer’s instructions. The lactate content was detected using a CheKine Micro Lactate Assay Kit (Abbkine, China) and calculated micromoles per 1 × 106 FLSs.
Malate and Oxaloacetate content, and MDH2 activity assasy
Intracellular malate and Oxaloacetate concentration were assessed using a Malate or Oxaloacetate Assay kit (Sigma, MAK067 and MAK070, USA) according to the manufacturer’s instructions. FLSs (1 × 106/group) were collected and assayed at 450 nm in a microplate reader. The MDH2 activity was assessed using the kit purchased from Elabscience (E-BC-K561-M, China).
Statistical analysis
Results are presented as the means ± SD of at least three independent experiments and graphs were generated with GraphPad Prism 10.2.1 software. Differences between two groups were analysed using Student’s t-tests. For comparisons of more than two groups, one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test was used. For all tests, P < 0.05 was considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA sequencing data generated in this study have been deposited in the NCBI Sequence Read Archive under accession code PRJNA1149351. The proteomics data generated in this study have been deposited in the ProteomeXchange Consortium via the iProX partner repository under accession code PXD055034. All other relevant data supporting the findings of this study are available within the article and its Supplementary Information file. Source data are provided with this paper.
References
Berenbaum, F. Annals of the Rheumatic Diseases collection on osteoarthritis (2018-2023): hopes and disappointments. Ann Rheum Dis 83, 133–135 (2024).
Jin, Z. et al. Incidence trend of five common musculoskeletal disorders from 1990 to 2017 at the global, regional and national level: results from the global burden of disease study 2017. Ann Rheum Dis 79, 1014–1022 (2020).
Hodgkinson, T., Kelly, D. C., Curtin, C. M. & O’Brien, F. J. Mechanosignalling in cartilage: an emerging target for the treatment of osteoarthritis. Nat Rev Rheumatol 18, 67–84 (2022).
Knights, A. J., Redding, S. J. & Maerz, T. Inflammation in osteoarthritis: the latest progress and ongoing challenges. Curr Opin Rheumatol 35, 128–134 (2023).
Miura, Y., Endo, K., Komori, K. & Sekiya, I. Clearance of senescent cells with ABT-263 improves biological functions of synovial mesenchymal stem cells from osteoarthritis patients. Stem Cell Res Ther 13, 222 (2022).
Mobasheri, A. et al. The role of metabolism in the pathogenesis of osteoarthritis. Nat Rev Rheumatol 13, 302–311 (2017).
Childs, B. G., Durik, M., Baker, D. J. & van Deursen, J. M. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med 21, 1424–1435 (2015).
Wan, M., Gray-Gaillard, E. F. & Elisseeff, J. H. Cellular senescence in musculoskeletal homeostasis, diseases, and regeneration. Bone Res 9, 41 (2021).
Coryell, P. R., Diekman, B. O. & Loeser, R. F. Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat Rev Rheumatol 17, 47–57 (2021).
Xie, J. et al. Cellular senescence in knee osteoarthritis: molecular mechanisms and therapeutic implications. Ageing Res Rev 70, 101413 (2021).
Diekman, B. O. & Loeser, R. F. Aging and the emerging role of cellular senescence in osteoarthritis. Osteoarthritis Cartilage 32, 365–371 (2024).
Malaise, O. et al. Mesenchymal stem cell senescence alleviates their intrinsic and seno-suppressive paracrine properties contributing to osteoarthritis development. Aging (Albany NY) 11, 9128–9146 (2019).
Chen, Y. H. et al. Association of Dipeptidylpeptidase 4 (CD26) With Chondrocyte Senescence and Radiographic Progression in Knee Osteoarthritis. Arthritis Rheumatol 75, 1120–1131 (2023).
Jeon, O. H. et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med 23, 775–781 (2017).
Xu, M. et al. Transplanted Senescent Cells Induce an Osteoarthritis-Like Condition in Mice. J Gerontol A Biol Sci Med Sci 72, 780–785 (2017).
Chen, X. et al. METTL3-mediated m(6)A modification of ATG7 regulates autophagy-GATA4 axis to promote cellular senescence and osteoarthritis progression. Ann Rheum Dis 81, 87–99 (2022).
Jeon, O. H., David, N., Campisi, J. & Elisseeff, J. H. Senescent cells and osteoarthritis: a painful connection. J Clin Invest 128, 1229–1237 (2018).
Suryadevara, V. et al. SenNet recommendations for detecting senescent cells in different tissues. Nat Rev Mol Cell Biol. https://doi.org/10.1038/s41580-024-00738-8 (2024).
Wang, Y., Zhao, X., Lotz, M., Terkeltaub, R. & Liu-Bryan, R. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor gamma coactivator 1alpha. Arthritis Rheumatol 67, 2141–2153 (2015).
Copp, M. E. et al. Comet assay for quantification of the increased DNA damage burden in primary human chondrocytes with aging and osteoarthritis. Aging Cell 21, e13698 (2022).
Wiley, C. D. et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab 23, 303–314 (2016).
Nacarelli, T. et al. NAD(+) metabolism governs the proinflammatory senescence-associated secretome. Nat Cell Biol 21, 397–407 (2019).
Zhang, H. et al Reduction of elevated proton leak rejuvenates mitochondria in the aged cardiomyocyte. Elife 9, https://doi.org/10.7554/eLife.60827 (2020).
Habiballa, L., Salmonowicz, H. & Passos, J. F. Mitochondria and cellular senescence: Implications for musculoskeletal ageing. Free Radic Biol Med 132, 3–10 (2019).
He, D. et al. A longitudinal genome-wide association study of bone mineral density mean and variability in the UK Biobank. Osteoporos Int 34, 1907–1916 (2023).
Zainul, Abidin, F. N., Scelsi, M. A., Dawson, S. J. & Altmann, A. Alzheimer’s Disease Neuroimaging I. Glucose hypometabolism in the Auditory Pathway in Age Related Hearing Loss in the ADNI cohort. Neuroimage Clin 32, 102823 (2021).
Robertson, C. C. et al. Fine-mapping, trans-ancestral and genomic analyses identify causal variants, cells, genes and drug targets for type 1 diabetes. Nat Genet 53, 962–971 (2021).
Smith, A. J. et al. GATD3A, a mitochondrial deglycase with evolutionary origins from gammaproteobacteria, restricts the formation of advanced glycation end products. BMC Biol 20, 68 (2022).
Martinez-Reyes, I. & Chandel, N. S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun 11, 102 (2020).
Chakrabarty, R. P. & Chandel, N. S. Mitochondria as Signaling Organelles Control Mammalian Stem Cell Fate. Cell Stem Cell 28, 394–408 (2021).
Molinie, T. et al. MDH2 produced OAA is a metabolic switch rewiring the fuelling of respiratory chain and TCA cycle. Biochim Biophys Acta Bioenerg 1863, 148532 (2022).
Astrike-Davis, E. M., Coryell, P. & Loeser, R. F. Targeting cellular senescence as a novel treatment for osteoarthritis. Curr Opin Pharmacol 64, 102213 (2022).
Wang, X. et al. Induction of Fibroblast Senescence During Mouse Corneal Wound Healing. Invest Ophthalmol Vis Sci 60, 3669–3679 (2019).
Chen, M. et al. A p21-ATD mouse model for monitoring and eliminating senescent cells and its application in liver regeneration post injury. Mol Ther 32, 2992–3011 (2024).
Kurhaluk N. Tricarboxylic Acid Cycle Intermediates and Individual Ageing. Biomolecules 14, https://doi.org/10.3390/biom14030260 (2024).
Sharma, R. & Ramanathan, A. The Aging Metabolome-Biomarkers to Hub Metabolites. Proteomics 20, e1800407 (2020).
Zhao, S. et al. Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004 (2010).
Sang, L. et al. Mitochondrial long non-coding RNA GAS5 tunes TCA metabolism in response to nutrient stress. Nat Metab 3, 90–106 (2021).
Atukorala, I. et al. Synovitis in knee osteoarthritis: a precursor of disease? Ann Rheum Dis 75, 390–395 (2016).
Ansari, M. M., Ghosh, M., Lee, D. S. & Son, Y. O. Senolytic therapeutics: An emerging treatment modality for osteoarthritis. Ageing Res Rev 96, 102275 (2024).
Youle, R. J. & van der Bliek, A. M. Mitochondrial fission, fusion, and stress. Science 337, 1062–1065 (2012).
D’Amico, D. et al. Urolithin A improves mitochondrial health, reduces cartilage degeneration, and alleviates pain in osteoarthritis. Aging Cell 21, e13662 (2022).
Hu, S. et al. Stabilization of HIF-1alpha alleviates osteoarthritis via enhancing mitophagy. Cell Death Dis 11, 481 (2020).
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. Hallmarks of aging: An expanding universe. Cell 186, 243–278 (2023).
van Deursen, J. M. The role of senescent cells in ageing. Nature 509, 439–446 (2014).
Yang, J. et al. Targeting YAP1-regulated Glycolysis in Fibroblast-Like Synoviocytes Impairs Macrophage Infiltration to Ameliorate Diabetic Osteoarthritis Progression. Adv Sci (Weinh) 11, e2304617 (2024).
Gong, Y. et al. Newcastle disease virus degrades SIRT3 via PINK1-PRKN-dependent mitophagy to reprogram energy metabolism in infected cells. Autophagy 18, 1503–1521 (2022).
Lee, I. et al. IDH2 Deficiency Promotes Endothelial Senescence by Eliciting miR-34b/c-Mediated Suppression of Mitophagy and Increased ROS Production. Antioxidants (Basel) 12, https://doi.org/10.3390/antiox12030585 (2023).
Song, S., Lam, E. W., Tchkonia, T., Kirkland, J. L. & Sun, Y. Senescent Cells: Emerging Targets for Human Aging and Age-Related Diseases. Trends Biochem Sci 45, 578–592 (2020).
Dou, X. et al. PDK4-dependent hypercatabolism and lactate production of senescent cells promotes cancer malignancy. Nat Metab 5, 1887–1910 (2023).
Pei, X. et al. Palmitoylation of MDH2 by ZDHHC18 activates mitochondrial respiration and accelerates ovarian cancer growth. Sci China Life Sci 65, 2017–2030 (2022).
Li, Y. et al. Intermittent fasting promotes adipocyte mitochondrial fusion through Sirt3-mediated deacetylation of Mdh2. Br J Nutr 130, 1473–1486 (2023).
Chen, L. Y., Wang, Y., Terkeltaub, R. & Liu-Bryan, R. Activation of AMPK-SIRT3 signaling is chondroprotective by preserving mitochondrial DNA integrity and function. Osteoarthritis Cartilage 26, 1539–1550 (2018).
Zhang, Y. et al. Reprogramming of Mitochondrial Respiratory Chain Complex by Targeting SIRT3-COX4I2 Axis Attenuates Osteoarthritis Progression. Adv Sci (Weinh) 10, e2206144 (2023).
Zhu, S. et al. Sirt3 Promotes Chondrogenesis, Chondrocyte Mitochondrial Respiration and the Development of High-Fat Diet-Induced Osteoarthritis in Mice. J Bone Miner Res 37, 2531–2547 (2022).
Eleftheriadis, T. et al Inhibition of Malate Dehydrogenase-2 Protects Renal Tubular Epithelial Cells from Anoxia-Reoxygenation-Induced Death or Senescence. Biomolecules 12, https://doi.org/10.3390/biom12101415 (2022).
Friedman, B., Goodman, E. H. Jr, Saunders, H. L., Kostos, V. & Weinhouse, S. Estimation of pyruvate recycling during gluconeogenesis in perfused rat liver. Metabolism 20, 2–12 (1971).
Borst, P. The malate-aspartate shuttle (Borst cycle): How it started and developed into a major metabolic pathway. IUBMB Life 72, 2241–2259 (2020).
Sun, L. Y. et al. Nuclear Receptor NR1D1 Regulates Abdominal Aortic Aneurysm Development by Targeting the Mitochondrial Tricarboxylic Acid Cycle Enzyme Aconitase-2. Circulation 146, 1591–1609 (2022).
Birsoy, K. et al. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 162, 540–551 (2015).
Weisshaar, N. et al. The malate shuttle detoxifies ammonia in exhausted T cells by producing 2-ketoglutarate. Nat Immunol 24, 1921–1932 (2023).
Fink, B. D. et al. Oxaloacetic acid mediates ADP-dependent inhibition of mitochondrial complex II-driven respiration. J Biol Chem 293, 19932–19941 (2018).
Ji, M. L. et al. Sirt6 attenuates chondrocyte senescence and osteoarthritis progression. Nat Commun 13, 7658 (2022).
Krenn, V. et al. Synovitis score: discrimination between chronic low-grade and high-grade synovitis. Histopathology 49, 358–364 (2006).
Shen, K. et al. Exosomes derived from hypoxia preconditioned mesenchymal stem cells laden in a silk hydrogel promote cartilage regeneration via the miR-205-5p/PTEN/AKT pathway. Acta Biomater 143, 173–188 (2022).
Mi, Z. et al. Identification of a synovial fibroblast-specific protein transduction domain for delivery of apoptotic agents to hyperplastic synovium. Mol Ther 8, 295–305 (2003).
Liu, B. et al. Inflammatory Fibroblast-Like Synoviocyte-Derived Exosomes Aggravate Osteoarthritis via Enhancing Macrophage Glycolysis. Adv Sci (Weinh) 11, e2307338 (2024).
Acknowledgements
The authors gratefully acknowledge the support received from the National Natural Science Foundation of China (Grant No. 82202716, CZ and No. 81974332, WF). We would like to thank the Core Facility of the First Affiliated Hospital with Nanjing Medical University for its help in the experiment. We thank International Science Editing (http://www.internationalscienceediting.com) for editing this manuscript. Graphical Abstract, Fig. 4a, Fig. 8a, Fig. 8h and Supplementary Fig. 2c were created in BioRender (https://BioRender.com/w90b978) by KS.
Author information
Authors and Affiliations
Contributions
K.S., C.Z. and W.F. conceived and designed the project. K.S., H. Zhou, Q.Z. and Y.G. performed all cell and animal experiments. K.S., J.C., and K.Y. collected the specimens and data from humans and mice. H. Zhang performed immunofluorescence and confocal imaging. H.S., W.L., J.Z. and J.L. graded the histological changes. K.S. and H.Zhou analysed data and wrote the manuscript. F.L., C.Z. and W.F. supervised the project. All authors approved the final version of the manuscript. C.Z. and W.F. accepted full responsibility for the finished work, had access to the data and controlled the decision to publish.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jean-marc Brondello, Noriaki Ono and Jana Riegger for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shen, K., Zhou, H., Zuo, Q. et al. GATD3A-deficiency-induced mitochondrial dysfunction facilitates senescence of fibroblast-like synoviocytes and osteoarthritis progression. Nat Commun 15, 10923 (2024). https://doi.org/10.1038/s41467-024-55335-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-55335-2
This article is cited by
-
Targeting the mitochondrial metabolite-dynamics-MDVs-MitoEVs axis: a new frontier in osteoarthritis management
Journal of Translational Medicine (2026)
-
Hyperglycemia exacerbates osteoarthritis by impairing macrophage efferocytosis through modulation of CD11b lactylation
Nature Communications (2025)
