
Abstract
As batteries drive the transition to electrified transportation and energy systems, ensuring their quality, reliability, lifetime, and safety is crucial. While the solid electrolyte interphase (SEI) is known to govern these performance characteristics, its dynamic nature makes understanding its nucleation, growth, and composition an ambitious, yet elusive aspiration. This work employs chalcogenide fibres embedded in negative electrode materials for operando Infra-red Fibre-optic Evanescent Wave Spectroscopy (IR-FEWS), combined with Multivariate Curve Resolution by Alternating Least Squares (MCR-ALS) algorithms for spectra analysis. By establishing molecular fingerprints that can be used to identify reaction products, IR-FEWS combined with MCR-ALS enables improved understanding of SEI evolution during cell formation with notable differences stemming from electrolyte or anode material. For example, despite operating at an elevated potential, lithium titanate’s SEI has intrinsic instability, evidenced by continued carbonate formation. This approach leads the hunt for the SEI down a new path, giving empirical formulations theoretical roots.
Similar content being viewed by others

Introduction
Widespread electrification combined with low-carbon electricity production is becoming the primary strategy for mitigating climate change. Lithium-ion (Li-ion) batteries, as mature and efficient energy storage devices, play a major role in this transition as their impact on mobility is yielding unprecedented cost reductions that are further broadening their application space. Given this massive deployment, there is a pressing need for cutting-edge and cost-effective Li-ion batteries with improved energy density, autonomy, lifetime and safety1. However, battery operation is governed by a multitude of complex and constantly evolving chemical, physical, and electrochemical processes. Among them, the formation and stability of the solid electrolyte interphase (SEI) plays a key role. Resulting from electrolyte reduction over the course of the first few battery cycles, the SEI is made up of a complex mosaic of inorganic and organic species, which depends on both the electrolyte and electrode materials, as well as the thermodynamic and kinetic situation of the cell. The SEI is a double-edged sword—it is crucial for improving the initial capacity, efficiency and lifetime of batteries, but an unstable SEI can lead to parasitic reactions, consuming lithium ions, electrolyte solvents and additives, thereby increasing the thickness (and resistance) of the interface and generating deleterious gas by-products. However, with its thickness ranging from a few nanometres to almost a micrometre, understanding and characterizing the SEI continues to be a major and longstanding challenge for battery researchers.
X-ray photoelectron spectroscopy (XPS) and Fourier transform infra-red spectroscopy (FTIR) have proved to be fruitful complementary techniques for elucidating the SEI composition of recovered negative electrode materials. While XPS analysis can identify the inorganic components comprising the surface of the SEI, such as LiF or Li2O2, vibrational spectroscopy, namely Raman and mid-infra-red FTIR, provides additional insights into the organic compounds present, such as the ROCO2Li semi-carbonates3,4,5. Several sophisticated in situ methods such as XPS6 or transmission electron microscopy7, have been utilized to analyse the evolution of the interface (and overcome problems of contamination during the post-mortem treatment of samples, such as washing and glovebox handling). In this context, since the pioneering work of Goren et al. in 19918, infra-red (IR) operando monitoring of the SEI has been developed using electrochemical cells with innovative designs. For instance, reflective glassy carbon electrodes and amorphous silicon thin-films deposited on the attenuated total reflectance (ATR) crystal have been used as model systems for graphite and silicon anode surfaces, respectively9,10. In the study by Shi et al. a silicon substrate was used to observe the chemisorption of ethylene carbonate (EC), as well as formation of diethyl 2,5-dioxahexane dicarboxylate (DEDOHC) and lithium ethylene dicarbonate (LEDC) after several cycles11. Conversely, Raman spectroscopy has proven to be a valuable tool for characterizing the SEI by detecting lower-wavenumber vibrational modes of molecules. For instance, Gogoi et al. investigated in situ the reduction of EC to lithium carbonate (Li2CO3) on a gold film electrode12. However, the Raman effect is weak compared to IR absorption despite plasmonic enhancement techniques, including tip probes13, nanoparticles14 or nanostructured substrates15, and requires laser light sources operating in the visible region which can potentially damage the SEI samples. For brevity, we direct interested readers to more comprehensive reviews on progress with operando optical spectroscopy of the SEI found elsewhere, e.g. Meyer et al. 16.
Nevertheless, these highly specialized cell designs cannot be translated to commercial cells where precise design tolerances in packaging meet zero-excess-anything. Thus, cell producers and pack integrators are handling something like a fractally illuminated black box when it comes to dynamic electrochemical and chemical processes, with some understanding of ideal scenarios, but a limited ability to confirm and verify internal cell evolution post-production. This is even more true for the SEI due to its dynamic nature and complexity that depends on a wide variety of experimental conditions17 and amazing sensitivity to such conditions18.
In this context, Grey and Tarascon have highlighted a range of attempts to demonstrate the usefulness of integrating optical or other sensors into batteries19. Notably, Huang et al. monitored the heat generation associated with electrolyte reduction and SEI formation under real-world conditions20. However, this preliminary work was unable to identify the individual molecular species associated with these parasitic reactions. Using ultrafine double fibre probes and hollow-core microstructure fibres, respectively21,22,23, Yamanaka et al. and Miele et al. overcame the double challenge of sample excitation and Raman photon collection. They implemented operando fibre-based Raman spectroscopy to pouch cell configuration, evidencing electrolyte composition change and gas bubbles formation. However, their setup depends on an external microfluidic system to pump the electrolyte through the fibre and enhance the signal, limiting both continuous monitoring and the ability to track changes in the electrode materials themselves.
This limitation was overcome by our group in 2022, who succeeded in characterizing the chemistry at work in commercial 18650 batteries, under real-world conditions, by the integration of Infra-red Fibre-optic Evanescent Wave Spectroscopy (IR-FEWS) in their central cavity, within the electrolyte24. This was made possible by the use of chalcogenide glass (Te2As3Se5 or TAS) glass fibres, allowing efficient transmission of infra-red light in the wavelength range from 3 to 13 µm, where many relevant molecules show absorbance. This study affirmed the ability to monitor, in real-time, the decomposition of the electrolyte and the formation of parasitic products, such as dimethyl-2,5-dioxa-hexane carboxylate (DMDOHC), while simultaneously observing the dynamics of ion-solvation.
Therefore, by placing the IR-sensing fibre close to the interface of interest, this new technique offers exciting opportunities for SEI characterisation. However, challenges remain due to the overlapping IR signatures of electrolytes and by-products, whether soluble or solid. In addition, charge transfer events inevitably lead to local variations in the concentration of lithium ions in the electrolyte and at the interfaces, affecting the absorption and position of the IR band. These must be taken into account when analysing IR spectra25. To address these issues, mathematical tools such as correction, linear decomposition, and machine learning-based models can be employed. For instance, Alves Dalla Corte et al. performed electrolyte correction by adding a carefully scaled reference spectrum and thus were able to observe the formation of the SEI operando10. However, as the authors acknowledge, this method could not take into account the change in salt concentration and the associated dynamic solvation phenomena. Ellis et al. took the understanding of solvation phenomena further by introducing calibrated models for predicting the composition of the electrolyte from its spectrum26. More recently, Meyer et al. demonstrated that linear approaches such as Principal Component Analysis (PCA) or Multivariate Curve Resolution by Alternating Least Squares (MCR-ALS) could be used to model the effect of solvation27. This approach is based on the linear hypothesis represented by Beer-Lambert’s law, according to which the absorbance of a given species linearly depends on its concentration. Finally, excellent prediction of lithium concentration could be achieved using a convolutional neural network (CNN) to overcome small non-linear deviations27. While the latter approach is particularly effective in the context of a pristine electrolyte, it will face immediate challenges when presented with new species both in solution and forming the SEI, which are unknown and unrecognizable to the model.
Herein, we extend our preliminary work on IR-FEWS24 to real-time observations of the evolving SEI, by implementing a chalcogenide glass (TAS) optical fibre directly into the negative electrode of the battery. Using MCR-ALS analysis, we were able to distinguish the signature of the electrolyte from that of the SEI. More specifically, by first training with a linear representation of the electrolyte and then applying it to the operando data, we carry out an unsupervised approach to determining the composition of the SEI and observe its formation and evolution during cell operation thereafter.
As proof of concept, the formation of the SEI from various electrolytes is investigated first using bare copper electrodes. Then, negative electrodes were investigated operating either with a conversion mechanism (CoO) or with alloying reactions (LixSn), both known to lead to abundant SEI formation when electrochemically reduced by lithium28,29,30. In addition, by considering lithium-titanium-oxide (LTO) insertion electrode, we highlight the role of catalytic activity against electrolyte decomposition at low potential. Based on the time-resolved evolution profiles of IR spectra collected during electrochemical reduction, we demonstrate the feasibility of identifying the nature and evolution of the SEI during battery operation and suggest ways forward for others hoping to also track the complex chemical footprints in this dynamic liquid-solid landscape.
Results
Preliminary step: establishing reference curves
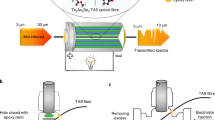
As noted, the possibility of FTIR using a chalcogenide glass (TAS) single-index multimode optical fibre was previously demonstrated to allow for indirect monitoring of the SEI formation in commercial 18650 Na-ion cells24. Recapping briefly: when the infra-red beam is guided through a single-index TAS fibre (n1 ≈ 2.931), evanescent waves develop beyond the fibre-surface interface, interacting with the surrounding media, and encoding the interaction into the infra-red signal. The spectrum resulting from the propagation of infra-red light along the fibre length integrates the absorbance of the fibre’s surroundings, such that molecules present around the fibre can be tracked and identified. Thus, an IR spectrum can simply be obtained by immersing part of the TAS fibre in a size-controlled droplet of electrolyte, as depicted in the schematic of Fig. 1a.

a Schematic representation of the liquid sample IR-FEWS testing, with a Te2As3Se5 (TAS) fibre immersed in an electrolyte droplet, illustrated with ethylene carbonate (EC) and dimethyl carbonate (DMC) molecules. b Electrolyte calibration reference spectra, with LiPF6 concentration from 0.0 M to 2.0 M (from blue to yellow), evidencing bands of free and coordinated solvent molecules. Ratios of EC:DMC are 1:2, 1:1 or 2:1 (v/v), and separately shown in Supplementary Fig. 1. c Mean square error (MSE) associated with several MCR-ALS decompositions of the electrolyte calibration dataset, up to five components. d Spectral components of the MCR-ALS decomposition using 4 components.
Using this method, 26 solutions of ethylene carbonate (EC) and dimethyl carbonate (DMC) at various solvent ratios and various hexafluorophosphate salt (LiPF6) concentrations were tested systematically to map the anticipated parameter space. The resulting IR-FEWS absorbance spectra (Fig. 1b, detailed in Supplementary Fig. 1) show that both solvents display their IR absorption fingerprint within the same spectral window. Moreover, when the LiPF6 salt is added, a separation of several bands is observed, corresponding to the free and coordinated solvent molecules to the Li+ cation and referred to as DMCfree and DMCcoor., respectively. In order to separately identify the effects of the solvent, the salt and their interactions, we have chosen to implement linear modelling, by MCR-ALS. More precisely, we assimilate our operando data to a matrix D, the rows of which are composed of the IR spectra, which we factor into two smaller matrices \({S}^{T}\) and C representing the pure spectra and their respective contributions, along with the summative remaining error E.
This algorithm, which is widely used in the chemometrics community, consists of an iterative regression of \({{{\rm{C}}}}\) from \({{{\rm{D}}}}\) and\(\,{{{{\rm{S}}}}}^{{{{\rm{T}}}}}\), and \({{{{\rm{S}}}}}^{{{{\rm{T}}}}}\) from \({{{\rm{D}}}}\) and \({{{\rm{C}}}}\), with mathematical or chemical constraints applied to the new estimates of concentration and spectral profiles between regression steps32. Lastly, a goodness of fit is assessed by calculating the mean square error (MSE).
MCR-ALS analysis was carried out on the spectra of the 26 electrolyte compositions (Fig. 1b) by applying positivity (\({{{\rm{S}}}} > 0\)) and normalisation constraints to the spectra. The spectral range was restricted to 2000 cm−1 to 900 cm−1, excluding the anion band at 840 cm−1 as this deviates from linearity at salt concentration higher than 1 molar, as shown in Supplementary Fig. 2. Several decompositions with different numbers of components nelectrolyte were performed to assess the accuracy of the fit, which improves markedly with the number of components (Fig. 1c). Within the test-space here, all the electrolyte solutions can be accurately described with four components (Fig. 1d) corresponding to DMCfree, DMCcoor.olv, ECfree and ECcoor. molecules, respectively. This result is in line with Meyer’s previous results that four principal components are necessary to explain similar electrolyte spectra27. Consequently, this data analysis method will be used to differentiate between the absorbance characteristics of the electrolyte and the SEI in a series of IR spectra.
Observing and modelling formation of the SEI
Swagelok-based electrochemical cells were designed to allow the fibre to rest directly on the current collector surface, as depicted in Fig. 2a. Thanks to this configuration, metal powders, electrode material suspensions or slurries are applied to the IR fibre by drop-casting, which improves physical adhesion of the particles to the fibre’s surface. The cell is then assembled in an argon-filled glovebox, before connecting the two ends of the fibre to a FTIR spectrometer for sequential operando spectra acquisition during the electrochemical cycling of the cell (see setup in Supplementary Fig. 3). The acquired spectra were pre-processed to correct for atmospheric water vapour contributions and baseline variations (see 'Methods' section). In addition, the initial spectrum was subtracted from the time series spectra to directly obtain the absorbance variation A(t)-A(t0), as this accounts for small spectral changes related to electrolyte concentration perturbations, as well as the formation of the SEI.

a Schematic illustration of the IR-FEWS electrochemical Swagelok cell. The inset represents the TAS fibre embedded in a copper powder electrode, covered with SEI species. b GITT voltage profile of the Cu-PVDF//LP30//Li Metal cell (OCV is highlighted in yellow) and detailed A(t)−A(t0) absorbance variations for two selected wavenumbers representative of Li2CO3 (1525 cm−1, red), and lithium semi-carbonates (1640 cm−1, purple). Purple arrows are shown as visual guide to highlight the increase during reduction events. c Absorbance reference spectrum of 1 M LiPF6 in EC:DMC (1:1 v/v) electrolyte. d A(t)−A(t0) absorbance variations spectra, highlighting the range where only semi-carbonates and Li2CO3 absorb (purple and red rectangles, respectively).
Prior to exploring SEI formation on anode surfaces themselves, we first start by assembling a cell with a mixture of copper powder/polyvinylidene fluoride (PVDF-HFP, also referred to as PVDF) on the IR fibre as positive electrode, Li metal as negative electrode (Fig. 2a) and 1M LiPF6 EC:DMC (1:1 w:w), or LP30, as electrolyte. Then, the cell was cycled using a Galvanostatic Intermittent Titration Technique (GITT) protocol with reduction pulses of 206 µA for 45 min (corresponding to 3 × 10−2 mA/cm2 of effective surface area, estimated in Supplementary Fig. 4), followed by open circuit voltage (OCV) periods of 2 h. The magnitude of the reduction current was selected with the aim of observing reduction events on a reasonable timescale. The GITT trace is shown in Fig. 2b: during reduction, the electrode material initially shows a minor capacity of around 1 mAh (Supplementary Fig. 4), which we attribute to conversion reaction of surface copper oxides, whose presence on a pristine sample was confirmed by XPS, as shown in Supplementary Fig. 5. Importantly, no mid-infra-red contribution is expected from metallic copper or copper oxides, which absorbs below 700 cm−1, beyond the transparency limit of TAS glass33.
The variation in absorbance A(t)−A(t0) over the course of the experiment is illustrated in Fig. 2d, with the original electrolyte spectrum provided for comparison in Fig. 2c. Looking at the characteristic negative and positive symmetric absorbance variations in the νC = O and νOCO regions reveals variations in Li+ concentration occurring in the electrode, with an entirely new band appearing at 1640 cm−1, attributed to the asymmetric stretching mode of the CO2 group of lithium semi-carbonate species (such as lithium methyl carbonate (CH3OCOOLi, or LMC) or lithium ethylene dicarbonate ((CH2OCOOLi)2, or LEDC)4,34,35). In the 1550 cm−1 and 1400 cm-1 regions, we also observe the emergence of the characteristic double band that is a fingerprint of Li2CO3 formation4,5,36. Thus, the formation and evolution of the SEI constituent species was evaluated by monitoring and comparing single wavenumber absorbance of semi-carbonates (i.e. LMC, LEDC) at 1640 cm−1 relative to the absorbance Li2CO3 at 1525 cm−1, where neither LMC nor LEDC shows absorbance. Figure 2b (purple line) highlights the formation of semi-carbonates that is enhanced during reduction event, in agreement with the formation of the SEI by electrochemical reduction of the electrolyte (see arrows in Fig. 2b). In contrast, the Li2CO3 band (red line in Fig. 2b) shows a more regular increase, consistent with its description as a by-product of the primarily formed semi-carbonates37. Finally, we note that both bands at 1640 cm−1 and 1525 cm−1 continue to increase at lower potential even when no current is applied (circled area in Fig. 2b), suggesting that solvent reduction is not the only mechanism at play for semi-carbonate formation and thus some degree of spontaneous chemical evolution is also observed.
As to the origins of this contribution, the spontaneous electrolyte reduction on Li metal surface of the counter electrode should be considered as a possible source. To address this a pristine half-cell was rested over a period of several hours while monitoring the IR spectra, as shown in Supplementary Fig. 8. Considering the two Whatman glass fibre separators (~500 µm) and room temperature LP30 diffusivities (roughly ~10−6 cm2/s)38, only a couple hours would be needed to observe counter electrode product migration to the sensor surface. Comparison of this background to the Cu-PVDF spectra seems to disqualify chemical oxidation of the Li-metal counter electrode as a source of any deposited products on the reference electrode.
However, outside the 1700–1500 cm−1 range, the bands of the SEI species overlap with those of the electrolyte, complicating further direct analysis. Therefore, the MCR-ALS method was used to analyse the operando IR spectra collected during the reduction of LP30 on copper using additional constraints summarized in Fig. 3a and discussed in detail in the 'Methods' section.

a Schematic description of the MCR-ALS algorithm with applied constraints. Salt concentration (blue to yellow) and time (greys) shadings are identical to Figs. 1 and 2, respectively. b GITT voltage profile of the Cu-PVDF//1 M LiPF6 in EC:DMC (LP30)//Li Metal cell (resting OCV time is highlighted in yellow). c Electrolyte spectral components (ST) from MCR-ALS. d Associated electrolyte components contributions (C). Black arrows act as a visual aide for solvation variations. e SEI spectral components from MCR-ALS; weak spectral components C7 and C8 are given in Supplementary Fig. 9. The grey box reports the MSE scores calculated on the reconstructed operando and calibration spectra. The IR bands of LEDC34,35 and LMC4 are shown below the plot for comparison. f Contributions of the SEI components contributions (C).
The resulting eight components are shown in Fig. 3c–f. Firstly, the four components comprising the electrolyte in Fig. 3c, d (C1 to C4) clearly reflect the change in electrolyte concentration during cell polarization. We observe an increase in the contributions of the components attributed to non-solvating solvents (C1, C3), while the components ascribed to solvating solvents decrease (C2, C4), indicating a decrease in salt concentration near the fibre surface, as also observed tracking the PF6− anion band using the single wavenumber approach (Supplementary Fig. 11). It should be noted that the salt gradient returns to equilibrium during the two hours of rest.
Turning to the SEI, four other components (C5 to C8) were equally assigned to describe their contribution to the operando IR spectra, based on chemical rank analysis39 and iterative increase of the parameter, as discussed in Supplementary Fig. 12. Ultimately, only two components are truly relevant to the SEI. The first component (C5, dark green) shows a strong band around 1650 cm−1, characteristic of semi-carbonates (LMC4 and LEDC34,35), as indicated with the purple marker at the bottom of Fig. 3e. Although exact identification remains difficult due to overlaps, we have attributed the band at 1355 cm−1 to LMC. Similarly, the band at 1310 cm−1 suggests the presence of LEDC, with residual uncertainty due to difficulties in perfect decoupling stemming from the overlapping DMC band at 1310 cm−1. The double band between 1550 cm−1 and 1400 cm−1, reflected by the C6 component (light green) is associated with Li2CO3 whose formation increases at low voltage. Finally, two components (C7, grey and C8, brown in Fig. 3f and Supplementary Fig. 9) only show minor contributions and likely result from imperfect electrolyte modelling and remaining baseline fluctuations.
Overall, direct wavenumber tracking and MCR-ALS prove complementary in analysing operando spectra. Given that one species or functional group only absorbs at a studied wavenumber, tracking it provides the most straightforward analysis, while multivariate regression permits comprehensiveness correlating the wavenumber of the entire spectrum inferring numerical components from statistical variations, here without prior knowledge of the SEI spectra, but for posterior comparison with established reference spectra. As a result, species can only be distinguished once formed, provided they display sufficient dynamics. Conversely, a given chemical species may spread between several MCR-ALS components, as observed here for Li2CO3 (C5 and C6) and be influenced, to some extent, by coupled concentration-intensity changes. Collectively, while the IR spectra of a single substance may indeed be considered as a unique fingerprint, the MCR-ALS components provide a footprint without the same level of definition, but now with a classification level suitable for a variety of challenging dynamic conditions and environments.
To verify that the entities identified during our operando measurements actually constitute the SEI, we used ex-situ ATR-FTIR to characterise recovered electrodes, previously washed with DMC. Similarly, we dismantled a cycled operando cell, rinsed the electrode and fibre with DMC, dried and reconnected the fibre to the spectrometer. The resulting IR-FEWS spectrum shows a decreased intensity of the EC band, indicating that the electrolyte was removed, while the bands attributed to the SEI species were not affected (Supplementary Fig. 13). This is in line with the belief that SEI components like Li2CO3 or semi-carbonates are poorly soluble in the electrolyte40 and is confirmed by the absence of new bands when comparing the IR spectra of pristine LP30 electrolyte and LP30 electrolyte saturated with either Li2CO3, LMC or LEDC (see Supplementary Figs. 14 and 15). While these observations confirm that the SEI is indeed insoluble within the accuracy of the experiment, a few literature papers have reported detecting semi-carbonate in liquid electrolytes via infra-red spectroscopy24,41. This apparent paradox can be resolved by proposing that a nucleation-precipitation process is likely occurring during SEI formation. Therefore, while precipitated species are the major contributors to the SEI’s signature, minor contributions from soluble molecules cannot be totally excluded.
Finally, while the long-term chemical stability of TAS glass in the electrolyte was previously demonstrated24, it should be further considered within the intimate contact of the electrode. Hence, the integrity of the fibre in the conditions of the article was verified on scanning-electron microscopy (SEM) images after cycling, and equally demonstrated by the stability of the optic power transmitted over the course of the experiment, as detailed in Supplementary Fig. 16.
Overall, these results demonstrate the feasibility of operando IR optical spectroscopy to monitor the formation of SEI deposits on the fibre. The complementary approach of using single wavenumber tracking and MCR-ALS marks a path for testing its applicability with respect to relevant electrolytes and anode materials, such that we might improve our prospects for understanding this critical interphase.
Tuning electrolyte composition
Several carbonated species were observed forming from the reduction of the 1 M LiPF6 in EC:DMC (LP30) electrolyte on copper (Cu) powders. To determine whether these species result from the reduction of EC, DMC, or a combination of both, the same GITT experimental protocol was carried out as implemented earlier, but this time using 1 M LiPF6 in a single DMC solvent as an electrolyte. The collected IR spectra (Fig. 4a) show the appearance of the 1640 cm−1 semi-carbonate band as well as small bands in the 1350 cm−1 to 1600 cm−1 region. As expected in the presence of DMC, the semi-carbonate observed can be identified as LMC, which is corroborated by the appearance of a band at 1355 cm−1, observed in the original spectra in Fig. 4a and confirmed via MCR-ALS (Supplementary Fig. 17). However, herein with the sole use of DMC solvent, the increase of the 1640 cm−1 semi-carbonates band is generally limited to the reduction events (highlighted with arrows in Fig. 4b), with an absorbance after 18 h of testing plateauing at 0.008 absorbance units, versus 0.016 absorbance units for the previous EC/DMC mixture electrolyte (Fig. 2d), and still increasing thereafter. Quantitative comparison between experiments should be taken cautiously, as variations in sample preparation can lead to dispersion in observed absolute intensities. However, similar trends were observed when duplicating experiments. More importantly, the low voltage steps evolution of DMC solvent contrasts with the previous reduction of EC/DMC, which not only showed an increase during reduction but also during OCV periods.

a A(t)−A(t0) absorbance variations spectra during intermittent galvanostatic electrolyte reduction on Cu electrode assembled versus Li metal with 1 M LiPF6 in DMC electrolyte. The range where only Li2CO3 and semi-carbonates absorb is highlighted with red and purple rectangles, respectively. The spectrum of the electrolyte is provided as a grey background. The IR bands of LMC4 are shown below the plot. b GITT voltage profile of the Cu-PVDF//LP30//Li Metal cell (resting OCV time is highlighted in yellow) and detailed A(t)−A(t0) absorbance variations for two selected wavenumbers representative of Li2CO3 (1525 cm−1, red), and lithium semi-carbonates (1640 cm−1, purple). Purple arrows are shown as visual guide to highlight the increase during reduction events. Subfigures (c, d) are similar to (a, b), for 1 M LiPF6 in EC:DMC (1:1 w:w) + 2%VC electrolyte. For comparison, subfigure d reproduces wavenumber tracking from Fig. 2b (shaded).
This observation further supports the speculated SEI formation mechanisms from the literature, such as those proposed by Gachot et al., regarding a reaction cascade initiated by two successive electrochemical reductions of DMC into LMC and subsequently lithium methoxide. The latter chemically reacts with EC and DMC leading to the formation of LMC as well as ethylene oxide oligomers42. Suppressing EC inhibits the cascade of spontaneous reactions and the copious formation of semi-carbonates, which is highly consistent with our experimental observations when we have removed EC from the electrolyte altogether.
In addition to changing solvents, another way of adjusting the physical-chemical properties of electrolytes is to use low-level additives, such as vinylene carbonate (VC). We explored this possibility and noted that by adding 2 wt.% of VC to LP30, the intensity of the semi-carbonate band is drastically reduced compared to the 1 M in DMC and LP30 electrolytes (Fig. 4c, d). Furthermore, the MCR-ALS decomposition, shown in Supplementary Fig. 18, confirms the absence of the LMC band at 1355 cm−1 altogether. This is in agreement with Aurbach’s original work, which demonstrated how VC polymerisation on the surface of the electrode prevents subsequent electrolyte solvent reduction, as directly deduced from ex-situ diffuse reflectance FTIR measurements43 or indirectly by measuring an improved first cycle reversibility. Finally, it should be noted how the spontaneous evolution previously observed in Fig. 2b is inhibited by VC such that LMC is no longer part of the cascade reaction scheme.
Influence of the material on SEI formation
By adjusting the electrolyte formulation, we were able to observe significant changes in the nature of the SEI and its evolution with preliminary insights into the (electro-)chemical chain of reactions and reactant-product links. Nevertheless, it is widely recognized that the formation of the SEI is complex, influenced not only by the electrolyte but also by the negative electrode material and the interface between them. To investigate how different materials influence the SEI’s nature during electrolyte reduction, electrodes made of tin (Sn), cobalt oxide (CoO), and lithium-titanium-oxide (LTO) powders were deposited at the surface of the IR fibre and assembled in half-cells against lithium using LP30 as the electrolyte, intermittently cycled as before. The reduction current was chosen with the intention of observing a sufficient number of reduction events on a reasonable timescale. For Sn, CoO and LTO, it was C/80, C/5, and C/2.5 respectively, (C/N: 1 stoichiometric lithium, \(\Delta x=1\), per N hours). It therefore corresponds to a theoretical reduction time of 10 h and 7.5 h for CoO and LTO, respectively. We note that the theoretical capacity of the tin electrode was not reached, presumably attributed to the copious SEI formation in the Sn electrode that is coupled with materials rapid and dynamic increase in surface area that is coupled with volume change. The wavenumber tracking results are reported in Fig. 5 and following the spectral analysis methods discussed previously, MCR-ALS decomposition of the operando IR spectra was conducted as shown in Fig. 6 (in detail with electrolyte components, in Supplementary Figs. 20–25).

a 1 M LiPF6 in EC:DMC (1:1 v/v) electrolyte IF-FEWS reference spectrum. b A(t)−A(t0) absorbance variations operando spectra for the Sn electrode. The range where only Li2CO3 and semi-carbonates absorb is highlighted with red and purple rectangles, respectively. c GITT voltage profile of the Cu-PVDF//LP30//Li Metal cell (resting OCV time is highlighted in yellow) and detailed A(t)−A(t0) absorbance variations for two selected wavenumbers representative of Li2CO3 (1525 cm−1, red), and lithium semi-carbonates (1640 cm−1, purple). Subfigures (d–g) are similar to (b, c), for CoO and LTO electrodes, respectively. The three shadings refer to the scale in (b). The IR bands of DMDOHC44 are shown below (d).

a 1 M LiPF6 in EC:DMC (1:1 v/v) electrolyte IR-FEWS reference spectrum; MCR-ALS SEI spectral components (C5 and C6) and associated contributions for three electrode materials with LP30 electrolyte, along with the GITT voltage profile (resting OCV time is highlighted in yellow): (b, c), tin metal powder; (d, e), CoO/SuperP; and (f, g), LTO/SuperP. For clarity, electrolyte components are not represented on the figure but are reported in Supplementary Figs. 20–25. The grey boxes report the MSE scores calculated on the reconstructed operando and calibration spectra. The range of the characteristic IR bands of Li2CO3 and semi-carbonates are highlighted with red and purple rectangles, respectively; the bands of LEDC34,35, and LMC4 and DMDOHC44 are more precisely detailed below the spectra.
In agreement with previous reports, the voltage composition curve for a Sn/Li cell shows additional capacity at 1.5 V before reaching the low-voltage plateau associated with the LixSn alloy reaction at 0.5 V28,29. The collected IR spectra (Fig. 5b, c) show a significant formation of Li2CO3 as deduced from its characteristic double band at 1525 cm−1 which reveals an increase in absorbance of +0.014 through the reduction process, while the appearance of lithium semi-carbonates at 1640 cm−1 remains limited. In addition, note that MCR-ALS reveals a second component (C6, light green) not only representative of Li2CO3 double band between 1550 cm−1 and 1400 cm−1 but also of what could be DMDOHC.
Figure 5d, e shows the IR spectra collected during the reduction of LP30 on CoO, revealing a SEI dominated by semi-carbonates. This is complementarily confirmed by MCR-ALS decomposition, showing an initial component (referred to as C5, dark green in Fig. 6d, e), which we attribute to DMDOHC (strong absorption bands at 1750 cm−1 and 1280 cm−1)44, with a contribution of the semi-carbonates band at 1640 cm−1. A second component (C6, indicated in light green in Fig. 6d, e) appears toward the end of the reduction, well separated from the electrolyte fingerprint, and assigned to LMC thanks to its band at 1355 cm−1 4,35. These results strongly suggest that the film previously reported during CoO reduction30, but whose composition was ill-defined, is indeed mainly made up of LMC.
Finally, we collected IR spectra during the reduction of LTO half-cells, a compound often considered as SEI-free due to the high cycling voltage. When deployed with the operando cell, the spectra surprisingly reveal abundant formation of carbonate species, as shown by the band at 1640 cm−1 exhibiting a sharp increase at the beginning and end of LTO lithiation, while the absorbance at 1525 cm−1 regularly increases (Fig. 5g). This assignment was confirmed via MCR-ALS which also provides additional information on the composition of the SEI. The first component dominates the main evolutionary trend (C5, dark green in Fig. 6f, g). Its broad band around 1500 cm−1 is attributed to Li2CO3, while the sharp bands at 1400 cm−1 and 1300 cm−1 are indicative of LEDC. It is worth mentioning this reference spectrum of LEDC was recently questioned by Wang et al. (Supplementary Fig. 26)45. Nevertheless, a second component (C6, light green in Fig. 6f, g) is needed to model the end of LTO lithiation which indicates the further formation of LMC (characteristic band at 1355 cm-1).
Table 1 summarizes the composition of the SEIs for the various types of electrode materials (Sn, CoO, and LTO) explored above. These values, without being calibrated species concentration, still assess the relative abundance of the SEI species within an experiment. Comparison between experiments should be taken more qualitatively than quantitatively, as observed absolute intensities may vary depending on the contact between electrode particles and the fibre (as illustrated in Supplementary Fig. 27 for LTO), but it can be noted that similar trends are observed when duplicating experiments.
LTO—longer cycling
Aware of the dynamic nature of the SEI during cycling, we attempt to explore its evolution in time by operando IR-FEWS over a few cycles. A cell using a LTO/SuperP composite as the positive electrode, Li as the negative electrode and a LP30 as the electrolyte, was assembled and cycled over the range 0.8– 2.5 V at a C/2.5 rate (2.5 h per 1 lithium, hence 7.5 h for full lithiation) for at least 5 cycles while collecting IR spectra (Fig. 7). The graphical representation of absorbance variations A(t)−A(t0) (Fig. 7c) indicates progressive changes upon cycling are better visualized considering two selected wavenumbers representative of Li2CO3 (1525 cm−1, red), and lithium semi-carbonates (1640 cm−1, purple). Remarkably, the semi-carbonate band at 1640 cm−1 (purple in Fig. 7c, d) exhibits a large increase at the beginning of the first LTO lithiation, then almost stabilizes on the following delithiation before increasing again on the next lithiation and so on. In contrast, the Li2CO3 (red in Fig. 7c, d) shows smooth rather than staircase variations upon cycling.

a 1 M LiPF6 in EC:DMC (1:1 v/v) electrolyte IR-FEWS reference spectrum. b Voltage profile during galvanostatic cycling, resting OCV time is highlighted in yellow. c A(t)−A(t0) absorbance variations spectra during cycling. The range where only Li2CO3 and semi-carbonates absorb is highlighted with red and purple rectangles, respectively. d Detailed A(t)−A(t0) absorbance variations for two selected wavenumbers representative of Li2CO3 (1525 cm−1, red), and lithium semi-carbonates (1640 cm−1, purple). e SEI spectra components resulting from MCR-ALS decomposition of the operando data, shifted along the vertical axis for readability. The IR bands of LEDC34,35 and LMC4 are shown below the plot for comparison. f Associated time-dependent contributions. This decomposition is associated to MSEoperando = 1.35e-02 and MSEcalib = 3.88e-04.
The data were exploited further by MCR-ALS analysis to determine the compositional evolution of the SEI upon cycling and it was found that three components are needed to best describe it. The first component (C5, dark green in Fig. 7e, f), in addition to representing contributions from the electrolyte (reversible solvation-desolvation changes during cycling) also includes bands that can be attributed to LEDC (1650 cm−1, 1400 cm−1) which forms rapidly during the Li+ insertion plateau, even at a potential as high as 1.55 V. The two other MCR components model long-term SEI accumulation, with a distinction between the component of the first three cycles (mainly C6, light green, enlisting both Li2CO3 and semi-carbonates, including LMC), and the next two cycles (mainly C7, grey, having a lessened Li2CO3 contribution). More precisely, we attribute C7 to contributions from LMC (bands at 1635 cm−1, 1460 cm−1, 1355 cm−1, and 1104 cm−1), predominantly formed at the end of the reduction phase.
Overall, the LEDC appears to form mainly during the reduction plateau, while the LMC emerges at a lower voltage at the end of the lithiation. Nevertheless, the SEI components appear to evolve and grow over time, suggesting ongoing electrolyte reduction. This was previously demonstrated by ex-situ XPS by Song et al., who reported repetitive formation of lithium semi-carbonates on LTO electrodes during the 1.55 V plateau, completely masking the electrode material after 50 cycles46. In addition to identifying the continued growth of the SEI, our approach has the merit of identifying the nature of the species that make it up (LEDC and LMC) and the way in which their respective ratios evolve over cycles.
Discussion
In this research, constraints of traditional ATR-FTIR spectroscopy as conventionally practised within the field of batteries are circumvented by integrating a chalcogenide IR fibre within the negative electrodes of half-cell batteries. Embedding such fibre sensors allows for localized probing of both the deposited SEI species and electrolyte components. Beyond first-order single-wavenumber absorption tracking, we show that by employing MCR-ALS as a numerical technique, it becomes possible to tackle another prevalent issue in IR spectroscopy: the overlapping of infra-red signatures from species with similar functional groups. This strategy allowed us to distinguish the IR variations stemming from the electrolytes, enabling real-time observation of SEI layer formation and growth during battery operation.
Classic curve fitting approaches cannot be applied to the obtained IR spectra because quantitative differences instantly arise between the static reference samples (synthetic/pure SEI compounds) and dynamically convoluted operando spectra. Instead, an approach without a priori knowledge on the SEI composition was adopted where the interpretation is facilitated by employing a limited number of components in the numerical analysis, although admittedly at the risk of oversimplification or exclusion of minor species. As is often the case with numerical-driven methods, a critical posture should be adopted when interpreting the decomposition results, keeping the established literature surrounding the SEI in mind. Nevertheless, we hope this opens the door for groups with stronger chemometrics expertise to pursue work in the direction of robust decomposition algorithms.
Thanks to the versatility of our setup, various electrolyte formulations, electrode materials, and electrochemical protocols were investigated. Firstly, the reduction process of electrolytes with similar formulations was tracked using the methods established. Notably from these experiments, Li2CO3 and lithium semi-carbonates (LEDC and LMC) can be identified as key SEI components. The addition of the popular additive, VC, or the exclusion of EC, shows clear changes to the formation and evolution of the SEI through electrochemically driven and spontaneous chemical reactions. Additionally, the crucial impact of the electrode material on the SEI nature and evolution was confirmed, underscoring the importance of future work with potentiostatic experiments to isolate surface-material-driven reactions from surface-potential-driven ones. Finally, the possibility to follow the SEI evolution over several cycles was demonstrated on LTO, observing a repeated formation of SEI carbonate species during prolonged cycling.
The synergy of optical fibre technology, spectroscopy, and numerical methods offers diverse opportunities in the battery domain, facilitating the resolution of intricate issues related to interactions among anodes, cathodes, and electrolytes. Specifically, fibre-based infra-red spectroscopy enables detailed exploration of the SEI nature and the impact parameters such as additives, temperature, and cycling conditions. Moreover, we believe this approach should be combined with other innovative fibre-based characterization techniques. For example, if the linearity between the techniques is strong enough, augmenting the FTIR dataset with simultaneous acquisitions of other methods, such as Raman spectroscopy, it may be possible to facilitate linear decomposition into chemically meaningful components.
As of today, several efforts remain to bring these lab-scale experiments to the level of industrial research and development teams. Notably, this work has been limited to serial, one-cell-at-a-time testing of homemade laboratory cells. The implementation of this approach to commercial pouch cells is being successfully carried out in our group, as it will be described in a forthcoming article. Additionally, designing optical and automation systems that enable sequential testing of multiple cells will elevate the technology to a higher level. Moreover, the current commercial availability of chalcogenide fibres is limited to a handful of companies, at prices that discourage their use as consumables. However, these pricing estimates stem from the modest size of the current market rather than material sourcing, which promises decreased prices upon development.
Overall, these results offer a new temporal resolution to monitor the composition of the SEI and its dynamics. With this rationalization, it is possible to guide the optimization of electrolyte composition and SEI formation protocols, to identify cost-effective material/formation schedule combinations that enhance cell longevity. While our team continues to explore SEI nucleation dynamics using the demonstrated tools, we hope to empower others to pursue intersecting research pathways to the benefit of the whole community.
Methods
Electrode material preparation
Copper powder, -100mesh, 99% (metals basis) (Alfa Aesar) was mixed with PVDF-HFP binder (Arkema) (99/1 wt.% ratio) into an N-Methyl−2-pyrrolidone (NMP, DodoChem) slurry. Tin metal powder, -325 mesh, 99.8% (metal basis) (Alfa Aesar) was used as received. Cobalt(II) oxide powder (≥99.995%, Alfa Aesar) was mixed with conductive SuperP carbon black (90/10 wt.% ratio) and ground for 5 minutes in ethanol, with a SPEX grinder. Commercial LTO powder (Nanomyte BE-10, NEI corporation, unknown purity) was mixed with conductive SuperP carbon black (90/10 wt.% ratio) and manually ground for 5 min, with a mortar and a pestle.
Electrolyte preparation
Several LiPF6 electrolytes were used in this work:
-
Commercial 1 M LiPF6 in EC:DMC (1:1 w/w), or LP30, provided by DodoChem.
-
1 M LiPF6 in EC:DMC (1:1 w/w) + 2% VC was prepared from LP30 (DodoChem) and 2 wt.% of fresh VC (TCI, stored at low temperature).
-
1 M LiPF6 in DMC was prepared from LiPF6 (Solvionic) and battery grade DMC (E-Lyte) dried with molecular sieve prior to electrolyte preparation.
Integration of the fibre in the Swagelok cell
TAS fibre was drawn in-lab (Glass&Ceramics team at Institut des Sciences Chimiques de Rennes) from synthesized TAS preform47,48. It was cut to 50 cm long fragments equipped with temporary FC-PC fibre connectors (BFT1, Thorlabs). Each end of the fibre was polished with three successive lapping sheets (3 µm, 1 µm and 0.3 µm grit, Thorlabs) and cleaned with ethanol-wetted Kimtech paper. Two 0.5 mm-deep grooves allow fibre integration in the Swagelok cell and vertically align with the top of a current collector, deposited on the bottom of the cell. Epoxycure (Buehler) resin was applied in the grooves to secure the fibre and was cured for 12 h. Additional epoxy resin fixes the current collector to the base. Posterior cell assembly involves screwing top part on the base, with top O-ring facing epoxy-filled grooves, ensuring airtightness.
Cell and electrode preparation
After integration of the fibre in the Swagelok cell, the electrode material is deposited on the fibre via drop-casting.
For the Copper-PVDF slurry, 10 drops were deposited to fully cover the section of the fibre on the current collector, with a total area of ~1 cm2. Subsequent weighing revealed a deposited mass of approximately 80 mg (i.e. 80 mg.cm−2).
Other electrode materials were previously weighed (80 mg for Sn powder, 10–15 mg for CoO/SuperP and LTO/SuperP. These powders were dispersed into anhydrous ethanol and drop-casted onto the fibre and current collector on an area of approximately 1 cm2, giving mass loadings of 80 mg.cm−2 (Sn) and 10–15 mg.cm−2 (CoO/SuperP and LTO/SuperP). Upon drying, the fibre is fully covered with particles adhering on its surface, ensuring good solid-solid contact. The electrodes were dried at 80 °C under vacuum for at least 12 h, after which the fibre was connected to the spectrometer for a background spectrum acquisition. Finally, the electrode was assembled against a lithium metal disk (diameter: 14 mm, thickness: 0.4 mm) with two Whatman borosilicate glass fibre discs (GF/D grade) as separators (diameter: 20 mm, thickness: 675 µm) and soaked with 700 µL of electrolyte, in an argon-filled glovebox.
IR-FEWS operando measurements
The operando measurements were performed with a FTIR spectrometer (Invenio S, Bruker) with an accessory connection system to focus the infra-red beam on one end of the fibre. A liquid nitrogen-cooled mercury–cadmium–tellurium (HgCdTe, or MCT) photoconductive detector with a spectral range of 12,000–600 cm−1 recorded the optical signal at the output end of the fibre. The spectrum was averaged over 128 scans recorded over approximately 30 s.
During operando measurements, spectra were recorded sequentially every 2–3 min. The experiments were performed in ambient conditions at within a 20–25 °C temperature range. A Plexiglas box covered the experiment to limit ambient humidity atmospheric changes along the IR beam path (see Supplementary Fig. 3b). Electrochemical cycling was performed using an MPG2 potentiostat (Bio-Logic), controlled with EC-Lab.
Spectra analysis framework
Data are analysed with Python language: original Opus spectra files acquired by the spectrometer were opened with opusFC49. The contribution of atmospheric water vapour is subtracted using OCTAVVS50 atmospheric correction function, and pybaselines51 was used for baselining. SpectroChemPy52 module was used to plot and interact with spectral data. MCR-ALS was performed via pyMCR53 module, in the framework of a scikit-learn pipeline54.
Pre-processing
Spectra were firstly corrected for water vapour. In accordance with the state of the art of spectroscopy chemometrics, mean-centring and standard deviation normalization were discarded, for they would overexpress the noise of non-absorbing regions. To emphasize the variations of spectra dominated by the contribution of the liquid electrolyte, the first spectrum was subtracted from absorbance spectra series, giving A(t)−A(t0). This favourably increases the weight of the SEI contributions in the least-square penalties of posterior numerical analysis, permitting more accurate fitting of these contributions.
Baselining
Baselining is a critical step in spectra processing, extracting the spectra enclosing the vibrational information from other physics-based affecting parameters. Temperature of the liquid-nitrogen-cooled-MCT detector influences the overall intensity measured, resulting in changed baseline when liquid nitrogen is refilled. This broadband intensity reduction translates into vertical shift of the spectra, which was corrected via subtraction of a reference value in a non-absorbing region of the spectrum, without affecting the intensity of the absorbance bands.
Embedding the fibre into the electrode further affects the baseline, e.g. using a conductive carbon filler (SuperP), due to its broadband absorption properties, as shown in Supplementary Fig. 32. This effect is observable when cycling electrodes of material undergoing significant volume change, such as CoO, with an increased contact of carbon particles on optic fibre surface upon electrode particle expansion. This is first corrected with a linear correction fitted in the 2200–2000 cm−1 range. To fully model this effect, a quadratic baseline correction was applied on the A(t)−A(t0) spectra, fitted in three regions of the spectra, 2000–1900 cm−1, 1580–1570 cm−1 and 940–925 cm−1; respectively weighted 1, 10 and 5. These wavenumber ranges correspond to regions of the spectra without electrolyte absorption, where no SEI peak was observed for LP30 reduction on copper experiment.
Linear decomposition via MCR-ALS
The MCR-ALS linear decomposition was performed using pymcr package53, adapted for interoperability with scikit-learn pipelines.
We first apply \({L}^{2}\)-normalisation separately on the operando spectra matrix and the matrix of concentration calibration spectra, for the MSE to be comparable throughout different datasets. To enhance data conditioning for electrolyte decoupling, the normalized operando and electrolyte calibration matrices are concatenated, resulting in a matrix \({{{\rm{D}}}}\) is of shape\(\,(N,{P})\), consisting of \(N\) rows of absorbance spectra, each composed of \({P}\) wavenumbers.
MCR-ALS decomposition is then performed by allocating nelectrolyte components to electrolyte modelling, and nSEI to SEI modelling. The allocation is reflected in the constraints applied in-between regression steps:
-
Constraints applied on concentration (\({{{\rm{C}}}}\)):
-
Non-negativity of the concentration for the SEI, while allowing negativity for electrolyte components, reflecting the initial absence of SEI and its subsequent formation (\({{{{\rm{C}}}}}_{{{{\rm{SEI}}}}} > 0\)),
-
Zero-concentration of the SEI in the calibration dataset (local rank constraint55).
-
-
Constraints applied on concentration (\({{{{\rm{S}}}}}^{{{{\rm{T}}}}}\)):
-
Non-negativity of the spectra for all components,
-
Equality of the electrolyte spectra to MCR components from initial calibration (in Fig. 1d),
-
Custom refitting of electrolyte spectra from SEI spectra, penalizing the contributions of the electrolyte spectra in the SEI components (see below),
-
Normalisation of the spectra (\({L}^{2}\)-norm).
-
To avoid overfitting with diverging intensities, a ridge regressor with a regularization parameter of 10−3 was used and weighted to account for the number of spectra in calibration or operando datasets. An additional parameter \({\alpha }_{{{{\rm{electrolyte}}}}}\) finally weights calibration data to operando data, with little effect considering the equality constraint on electrolyte component spectra.
A custom refitting constraint was employed to limit possible contribution of the electrolyte in the SEI band. We successively fit each SEI component spectrum \(y\) with the electrolyte components spectra \({X}\), minimizing the objective function \(({1-\alpha }_{{{{\rm{refit}}}}}){\|{{{\rm{y}}}}-{{{\rm{Xw}}}}\|}^{2}+{\alpha }_{{{{\rm{refit}}}}}{{||}\min ({{{\rm{y}}}}-{{{\rm{Xw}}}},0){||}}^{2}\), corresponding an ordinary least square (OLS) optimization with penalization of negative residuals. The fitted electrolyte contributions are then subtracted from the SEI components.
The goodness of the fit is assessed by calculating the coefficient of determination \({R}^{2}\) as well as the mean square error (MSE) defined as:
Considering the \({L}^{2}\)-normalisation of the data, a MSE of 1 corresponds to the null decomposition (\({{{\rm{C}}}}=0\)).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The original spectrum data generated in this study have been deposited in the Zenodo database: https://zenodo.org/records/11122131. The figure data generated in this study are provided in the Source Data file. Source data are provided with this paper.
Code availability
The computer code and associated data for MCR-ALS analysis generated in this study have been deposited in the Zenodo database: https://zenodo.org/records/11122131.
References
Larcher, D. & Tarascon, J.-M. Towards greener and more sustainable batteries for electrical energy storage. Nat. Chem. 7, 19–29 (2015).
Dedryvère, R. et al. Characterization of lithium alkyl carbonates by X-ray photoelectron spectroscopy: experimental and theoretical study. J. Phys. Chem. B 109, 15868–15875 (2005).
Aurbach, D. Electrode–solution interactions in Li-ion batteries: a short summary and new insights. J. Power Sources 119–121, 497–503 (2003).
Gireaud, L., Grugeon, S., Laruelle, S., Pilard, S. & Tarascon, J.-M. Identification of Li battery electrolyte degradation products through direct synthesis and characterization of alkyl carbonate salts. J. Electrochem. Soc. 152, A850 (2005).
Seo, D. M. et al. Reduction reactions of carbonate solvents for lithium ion batteries. ECS Electrochem. Lett. 3, A91 (2014).
Capone, F. et al. Operando observation of the dynamic SEI formation on a carbonaceous electrode by near-ambient pressure XPS. Energy Environ. Sci. 17, 1509–1519 (2024).
Zeng, Z. et al. Visualization of electrode–electrolyte interfaces in LiPF6/EC/DEC electrolyte for lithium ion batteries via in situ TEM. Nano Lett. 14, 1745–1750 (2014).
Goren, E. et al. The application of in situ FTIR spectroscopy to the study of surface films formed on lithium and noble metals at low potentials in Li battery electrolytes. J. Electrochem. Soc. 138, L6–L9 (1991).
Pérez-Villar, S., Lanz, P., Schneider, H. & Novák, P. Characterization of a model solid electrolyte interphase/carbon interface by combined in situ Raman/Fourier transform infrared microscopy. Electrochim. Acta 106, 506–515 (2013).
Alves Dalla Corte, D. et al. Spectroscopic insight into Li-ion batteries during operation: an alternative infrared approach. Adv. Energy Mater. 6, 1501768 (2016).
Shi, F., Ross, P. N., Somorjai, G. A. & Komvopoulos, K. The chemistry of electrolyte reduction on silicon electrodes revealed by in situ ATR-FTIR spectroscopy. J. Phys. Chem. C. 121, 14476–14483 (2017).
Gogoi, N., Melin, T. & Berg, E. J. Elucidating the step-wise solid electrolyte interphase formation in lithium-ion batteries with Operando Raman spectroscopy. Adv. Mater. Interfaces 9, 2200945 (2022).
Nanda, J. et al. Unraveling the nanoscale heterogeneity of solid electrolyte interphase using Tip-enhanced Raman spectroscopy. Joule 3, 2001–2019 (2019).
Gajan, A. et al. Solid electrolyte interphase instability in operating lithium-ion batteries unraveled by enhanced-Raman spectroscopy. ACS Energy Lett. 6, 1757–1763 (2021).
Ha, Y. et al. Probing the evolution of surface chemistry at the silicon–electrolyte interphase via in situ surface-enhanced Raman spectroscopy. J. Phys. Chem. Lett. 11, 286–291 (2020).
Meyer, L., Saqib, N. & Porter, J. Review—Operando optical spectroscopy studies of batteries. J. Electrochem. Soc. 168, 090561 (2021).
Verma, P., Maire, P. & Novák, P. A review of the features and analyses of the solid electrolyte interphase in Li-ion batteries. Electrochim. Acta 55, 6332–6341 (2010).
Forero-Saboya, J. et al. Influence of formation temperature on cycling stability of sodium-ion cells: a case study of Na3V2(PO4)2F3|HC cells. J. Electrochem. Soc. 170, 100529 (2023).
Grey, C. P. & Tarascon, J. M. Sustainability and in situ monitoring in battery development. Nat. Mater. 16, 45–56 (2017).
Huang, J. et al. Operando decoding of chemical and thermal events in commercial Na(Li)-ion cells via optical sensors. Nat. Energy 5, 674–683 (2020).
Yamanaka, T. et al. In situ diagnosis of the electrolyte solution in a laminate lithium ion battery by using ultrafine multi-probe Raman spectroscopy. J. Power Sources 359, 435–440 (2017).
Yamanaka, T. et al. In situ Raman spectroscopic studies on concentration of electrolyte salt in lithium‐ion batteries by using ultrafine multifiber probes. ChemSusChem 10, 855–861 (2017).
Miele, E. et al. Hollow-core optical fibre sensors for operando Raman spectroscopy investigation of Li-ion battery liquid electrolytes. Nat. Commun. 13, 1651 (2022).
Gervillié-Mouravieff, C. et al. Unlocking cell chemistry evolution with operando fibre optic infrared spectroscopy in commercial Na(Li)-ion batteries. Nat. Energy https://doi.org/10.1038/s41560-022-01141-3 (2022).
Meyer, L., Curran, D., Brow, R., Santhanagopalan, S. & Porter, J. Operando measurements of electrolyte Li-ion concentration during fast charging with FTIR/ATR. J. Electrochem. Soc. 168, 090502 (2021).
Ellis, L. D. et al. A new method for determining the concentration of electrolyte components in lithium-ion cells, using Fourier transform infrared spectroscopy and machine learning. J. Electrochem. Soc. 165, A256 (2018).
Meyer, L., Kinder, C. & Porter, J. Using machine learning and infrared spectroscopy to quantify species concentrations in battery electrolytes. J. Electrochem. Soc. 170, 100521 (2023).
Beattie, S. D., Hatchard, T., Bonakdarpour, A., Hewitt, K. C. & Dahn, J. R. Anomalous, high-voltage irreversible capacity in tin electrodes for lithium batteries. J. Electrochem. Soc. 150, A701 (2003).
Bridel, J.-S. et al. Decomposition of ethylene carbonate on electrodeposited metal thin film anode. J. Power Sources 195, 2036–2043 (2010).
Laruelle, S. et al. On the origin of the extra electrochemical capacity displayed by MO/Li cells at low potential. J. Electrochem. Soc. 8, A627–A634 (2002).
Houizot, P. et al. Infrared single mode chalcogenide glass fiber for space. Opt. Express 15, 12529 (2007).
De Juan, A. & Tauler, R. Multivariate curve resolution: 50 years addressing the mixture analysis problem—a review. Analytica Chim. Acta 1145, 59–78 (2021).
Debbichi, L., Marco de Lucas, M. C., Pierson, J. F. & Krüger, P. Vibrational properties of CuO and Cu4O3 from first-principles calculations, and Raman and infrared spectroscopy. J. Phys. Chem. C. 116, 10232–10237 (2012).
Matsuta, S., Asada, T. & Kitaura, K. Vibrational assignments of lithium alkyl carbonate and lithium alkoxide in the infrared spectra an ab initio MO study. J. Electrochem. Soc. 147, 1695 (2000).
Zhuang, G. V., Xu, K., Yang, H., Jow, T. R. & Ross, PhilipN. Lithium ethylene dicarbonate identified as the primary product of chemical and electrochemical reduction of EC in 1.2 M LiPF6/EC:EMC electrolyte. J. Phys. Chem. B 109, 17567–17573 (2005).
Buijs, K. & Schutte, C. J. H. The infra-red spectra and structures of Li2CO3 and anhydrous Na3CO3. Spectrochim. Acta 17, 927–932 (1961).
Heiskanen, S. K. et al. Generation and evolution of the solid electrolyte interphase of lithium-ion batteries. Joule 3, 2322–2333 (2019).
Ehrl, A., Landesfeind, J., Wall, W. A. & Gasteiger, H. A. Determination of transport parameters in liquid binary lithium ion battery electrolytes. J. Electrochem. Soc. 164, A826 (2017).
Meloun, M., Čapek, J., Mikšı́k, P. & Brereton, R. G. Critical comparison of methods predicting the number of components in spectroscopic data. Analytica Chim. Acta 423, 51–68 (2000).
Jones, J. et al. Solubilization of SEI lithium salts in alkylcarbonate solvents. Fluid Phase Equilibria 305, 121–126 (2011).
Hongyou, K. et al. Dynamic in situ Fourier transform infrared measurements of chemical bonds of electrolyte solvents during the initial charging process in a Li ion battery. J. Power Sources 243, 72–77 (2013).
Gachot, G. et al. Deciphering the multi-step degradation mechanisms of carbonate-based electrolyte in Li batteries. J. Power Sources 178, 409–421 (2008).
Aurbach, D. et al. On the use of vinylene carbonate (VC) as an additive to electrolyte solutions for Li-ion batteries. Electrochim. Acta 47, 1423–1439 (2002).
Yoshida, H. et al. Degradation mechanism of alkyl carbonate solvents used in lithium-ion cells during initial charging. J. Power Sources 68, 311–315 (1997).
Wang, L. et al. Identifying the components of the solid–electrolyte interphase in Li-ion batteries. Nat. Chem. 11, 789–796 (2019).
Song, M.-S. et al. Is Li4Ti5O12 a solid-electrolyte- interphase-free electrode material in Li-ion batteries? Reactivity between the Li4Ti5O12 electrode and electrolyte. J. Mater. Chem. A 2, 631–636 (2014).
Adam, J.-L. & Zhang, X. Chalcogenide Glasses: Preparation, Properties and Applications, (2014).
Hocdé, S., Boussard-Plédel, C., Fonteneau, G. & Lucas, J. Chalcogens based glasses for IR fiber chemical sensors. Solid State Sci. 3, 279–284 (2001).
opusFC: OPUS File Converter module.
Troein, C. et al. OCTAVVS: a graphical toolbox for high-throughput preprocessing and analysis of vibrational spectroscopy imaging data. Methods Protoc. 3, 34 (2020).
Erb, D. pybaselines: A Python library of algorithms for the baseline correction of experimental data https://doi.org/10.5281/zenodo.7255880 (2022).
Travert, A. & Fernandez, F. SpectroChemPy. https://doi.org/10.5281/zenodo.8103617 (2023).
Camp, C. H. pyMCR: A Python library for multivariatecurve resolution analysis with alternating regression (MCR-AR). J. Res. Natl Inst. Stand Technol. 124, 1–10 (2019).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Tauler, R., Smilde, A. & Kowalski, B. Selectivity, local rank, three-way data analysis and ambiguity in multivariate curve resolution. J. Chemom. 9, 31–58 (1995).
Acknowledgements
J.-M.T. acknowledges the International Balzan Prize Foundation and the LABEX STOREXII for funding. X.H.Z., C.B.-P. and J.-M.T. acknowledge the French Agence National de la Recherche (ANR) under reference ANR-22-CE50-0031 (project SensOLiB). We thank D. Foix (Pau, France) for her assistance with X-ray photoelectron spectroscopy. Finally, we gladly thank J. Forero-Saboya for valuable discussions and comments.
Author information
Authors and Affiliations
Contributions
C.L., Y.W., C.G.-M. and J.-M.T. conceived the idea and designed the experiments. C.L. performed the experiments. C.L., Y.W., C.G.-M. and J.-M.T. performed the data analysis. X.H.Z., S.C. and C.B.-P. provided the TAS fibre. Finally, C.L., C.G.-M., S.T.B., and J.-M.T. wrote the paper with contributions from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jason Porter, Yifei Yu, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Leau, C., Wang, Y., Gervillié-Mouravieff, C. et al. Tracking solid electrolyte interphase dynamics using operando fibre-optic infra-red spectroscopy and multivariate curve regression. Nat Commun 16, 757 (2025). https://doi.org/10.1038/s41467-024-55339-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55339-y
