
Abstract
Prime editing enables precise and efficient genome editing, but its efficacy is hindered by pegRNA’s 3’ extension, forming secondary structures due to high complementarity with the protospacer. The continuous presence of the prime editing system also leads to unintended indel formation, raising safety concerns for therapeutic applications. To address these challenges, we develop a mismatched pegRNA (mpegRNA) strategy that introduces mismatched bases into the pegRNA protospacer, reducing complementarity and secondary structure formation, and preventing sustained activity. Our findings show that mpegRNA enhances editing efficiency by up to 2.3 times and reduces indel levels by 76.5% without compromising performance. Combining mpegRNA with epegRNA further increases efficiency up to 14-fold, or 2.4-fold in PE4max/PE5max systems, underscoring its potential in research and therapy. AlphaFold 3 analysis suggests that the optimal mpegRNA structure contributes significantly to improved editing outcomes. Overall, mpegRNA advances prime editing technology, improving efficiency while reducing indels.
Similar content being viewed by others


Introduction
Gene-based disease treatment often hinges on correcting genomic variants within diseased cells1,2,3,4 or organisms5,6. Prime editing (PE) stands out as a precise genome editing method capable of various DNA conversions, including base pair substitutions, small insertions, and deletions7. Due to its comprehensive sequence-altering capabilities, prime editing theoretically addresses 90% of human genetic diseases8, and has been used in scientific research6,9,10 and gene therapy development11. However, its utility is often limited by relatively low editing efficiency and high rates of indels12.
The prime editing complex consists of a pegRNA and an editing complex, which includes an engineered reverse transcriptase (RT) fused to the nCas9-H840A nickase13,14. The pegRNA comprises a guide RNA region that targets the specific DNA sequence15, a primer binding site (PBS) region homologous to the target sequence, and an RT template region containing the desired mutation. During the editing process, the pegRNA guides nCas9 to create a nick in the target DNA strand, after which the PBS sequence binds to the nicked strand, initiating reverse transcription. The RT then extends the nicked strand using the pegRNA template, forming the edited DNA sequence, which integrates into the genome through DNA repair and replication mechanisms7. To enhance PE systems, strategies such as using different Cas9 variants16,17, optimizing the M-MLV RT18, modifying pegRNAs with 3’ RNA structural motifs19,20,21,22, employing cell repair factors23, and enhancing chromatin accessibility have been explored24. Despite the 3’ extension of the PBS providing a template for editing, its high complementarity to the protospacer sequence can form secondary RNA structures, obstructing the interaction between the pegRNA, Cas9 protein, and the target DNA sequence. Consequently, the targeting efficiency of pegRNA relative to single guide RNA (sgRNA) can be compromised25,26.
The development of PE3 and PE5 systems, which incorporate an additional sgRNA to nick the non-edited DNA strand, has aimed to resolve heteroduplex DNA27. However, these systems increase the risk of double-strand breaks (DSBs) and indel formation near the target site, and the presence of DSBs can activate DNA damage responses, posing pathogenic risks in therapeutic applications28,29.
To mitigate these issues, we propose the use of mismatched pegRNA (mpegRNA) to reduce pegRNA secondary structure formation and prevent persistent DNA nicks by nCas927. This study will evaluate the mpegRNA strategy across multiple genomic loci under various conditions to assess its potential for enhancing PE efficiency while minimizing indels.
Results
Design of the mpegRNA
Since the 3’ extension of the PBS region providing a template for editing, its high complementarity to the protospacer sequence can form secondary RNA structures, obstructing the interaction between the pegRNA, Cas9 protein, and the target DNA sequence. Consequently, the targeting efficiency of pegRNA relative to single guide RNA (sgRNA) can be compromised. In addition, after the target DNA sequence is modified by PE, the original pegRNA is no longer complementary to the edited locus, thereby terminating the editing process. However, due to the inherent design of the PE system, the protospacer sequence and the 3’ extension of the pegRNA naturally exhibit complementarity (Fig. 1a). Additionally, due to the mismatch tolerance characteristic of Cas9, even when the pegRNA is not fully complementary to the edited locus, nCas9 can still bind and continuously nick the target site30,31. Such persistent DNA damage can lead to chromosomal indels and other undesired DNA alterations. Thus, to ameliorate these two challenges, we consider introduction of mismatched bases in the pegRNA might be a solution.

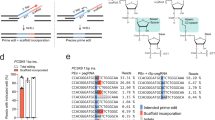
a Predicted secondary structures of pegRNA and mismatch pegRNA using AlphaFold 3. b Indel formation induced by testing pegRNA in combination with sgRNA versus sgRNA alone. Data are presented as mean ± s.d. of n = 3 independent biological replicates. Significance was determined using unpaired two-tailed Student’s t test (***p < 0.001, **p < 0.01, *p < 0.05, ns indicates no significant difference). c Schematic representation of the prime editing process facilitated by pegRNA and mpegRNA. Prior to prime editing, mpegRNA exhibits a more extended secondary structure compared to conventional pegRNA. Following prime editing, mpegRNA incorporates additional noncomplementary nucleotides relative to the target locus, which aids in concluding the prime editing process. Source data are provided as a Source Data file.
To demonstrate that traditional pegRNAs retain targeting capabilities with the edited locus sequence, we designed a pegRNA with base mismatches at positions 18-20 of the protospacer (referred to as testing pegRNA, where the proximal PAM is at position 20). This testing pegRNA, with single-base mismatches to the target site, simulates the mismatches between the edited genome and the original pegRNA after editing events. Its 3’ extension is fully complementary to the wild-type genomic locus. We co-transfected the testing pegRNA with the corresponding sgRNA for PE3 editing. Across six tested loci, the combination of testing pegRNA and sgRNA induced more indels compared to sgRNA alone (Fig. 1b). These results indicate that pegRNA can still target the genome after the locus has been edited, resulting in increased indels.
We introduce a mismatched PE gRNA (mpegRNA) strategy (Fig. 1c) that employs a pegRNA with one or more bases that are not complementary to the target locus. Before PE editing, mpegRNA exhibits a more extended secondary structure than conventional pegRNA, potentially enhancing the targeting ability of PE complexes. After PE editing, mpegRNA contains more noncomplementary nucleotides relative to the traditional pegRNA, reducing the affinity of PE complexes for the edited locus. This reduction in affinity helps terminate the PE editing process and prevents continuous DNA damage.
The mpegRNA strategy increased editing efficiency and reduced indels
Introducing mismatches into pegRNAs may affects their targeting ability. To identify the mismatch range that minimally impacts pegRNA targeting ability, we introduced mismatches across the entire protospacers at UBE3A and EMX1 loci. As illustrated in Fig. 2a, the UBE3A site exhibited greater mismatch tolerance, whereas the EMX1 site displayed a narrower range around positions N3 to N11, with the highest editing efficiency achieved at mismatch sites N6 to N10. The PE3 results were similar to those of PE2. Concurrently, emerging literature underscores the importance of the fourth to seventh bases upstream of the PAM sequence in gRNA targeting efficacy32.

a The editing efficiency of prime editing mediated by mpegRNA at various mismatch positions across the protospacer in the UBE3A and EMX1 loci. b, c The efficiency and indel outcomes of PE2 and PE3 using pegRNA and mpegRNA with mismatches at positions three to eleven within the protospacer, where ST denotes the standard pegRNA without mismatches. For a single target locus, data are presented as the mean ± s.d. from n = 3 independent biological replicates. For the combined analysis of ten loci, data are presented as the mean ± s.d. from n = 30 independent biological replicates, with three replicates per locus. Only the group with the best performance underwent unpaired two-tailed Student’s t test (***p < 0.001, **p < 0.01, *p < 0.05, ns indicates no significant difference). Source data are provided as a Source Data file.
Based on these results and previous studies13,14, we developed mpegRNAs with mismatches at positions 3 to 11 of the protospacer sequence, sequentially designated as N3 to N11 mpegRNA. We evaluated the editing efficiency and indel rates at various genomic loci using the PE2 and PE3 systems. As illustrated in Fig. 2b, the mpegRNA strategy significantly enhanced editing efficiency or reduced indel formation at 9 out of the 10 sites tested with PE2. The average editing efficiency increased by 44.97%, while the average indel rate decreased by 47.16%. For example, the mpegRNA targeting VISTA at N8 improved efficiency from 11.73% to 23.9% and reduced indels from 58.45% to 19.42%. Similarly, the mpegRNA targeting UBE3A-3 with a + 8 C to A mutation at N9 increased efficiency from 13.97% to 27.63% and decreased indels from 20.36% to 5.83%. In the PE3 system (Fig. 2c), the mpegRNA approach significantly improved editing efficiency or reduced indel formation at 8 out of the 10 sites examined, with the average editing efficiency rising by 44.97% while maintaining a stable average indel rate. At some loci, efficiency gains were accompanied by a reduction in indels. For instance, the mpegRNA at HEK4 N7 increased efficiency from 11.6% to 25.73% and reduced indels from 17.28% to 12.33%. The mpegRNA targeting VEGFA at N7 increased efficiency from 36.13% to 47.8% and decreased indels from 3.85% to 2.63%. Similar enhancement effects were replicated in HeLa cells (Fig. S1).
Overall, most sites demonstrated improved editing efficiency with mpegRNA, although the optimal mismatch location varied. Editing efficiency with mpegRNA typically increased and then decreased between N6 and N10, with the best mismatches usually between these positions. The variation trends of mpegRNA were consistent for PE2 and PE3 at certain sites. For example, mpegRNA showed a significant efficiency decline at N7 for HEK3 in both PE2 and PE3. At the VEGFA and UBE3A-3 sites, different editing positions led to different optimal mismatch results, suggesting that both the protospacer and 3’ extension influence the mismatch outcome.
Given the reduced complementarity between mpegRNA and the target site, the number of off-target sites may change. To compare off-target site numbers between mpegRNA and pegRNA, we used Cas-OFFinder33 (allowing up to 3 mismatches, no bulges). The results, shown in Supplementary Data 2, indicated that single-base mismatches could either increase or decrease off-target sites. To further validate these findings, we selected two mpegRNAs predicted by Cas-OFFinder to have lower off-target potential, targeting the HEK3 and VISTA HS267 loci, and used GUIDE-seq34 to assess off-target effects. The relative off-target rates for mpegRNA and pegRNA were consistent with those predicted by Cas-OFFinder (Fig. S2). For example, at the HEK3 locus, GUIDE-seq detected three off-target sites with pegRNA and two with mpegRNA; notably, both off-target sites detected with mpegRNA were also found with pegRNA, with no additional off-target sites identified. At the VISTA HS267 locus, pegRNA and mpegRNA identified the same off-target site, but mpegRNA showed relatively fewer off-target reads. By selecting appropriate positions for designing mpegRNA, we may be able to enhance editing efficiency and potentially reduce the number of off-target sites.
Considering the substantial variability in selecting mismatched bases, we chose eight positive sites to determine if there is an optimal mismatch base. As depicted in Fig. 3a, b, within the PE3 system, 6 out of 7 sites showed better performance when mismatched with G, 5 out of 5 with T, 5 out of 5 with A, and 6 out of 7 with C. These findings suggest that the location of the mismatched base, rather than its type, exerts the most significant influence on the effectiveness of mpegRNA.

a, b Comparison of editing efficiency across four different base types in PE2 (a) and PE3 (b). c The editing results of (m)pegRNA at various nCas9 second cut positions. The P/I ratio represents the ratio of prime editing efficiency to indels. Data are presented as mean ± s.d. of n = 3 independent biological replicates. Only the group with the best performance underwent unpaired two-tailed Student’s t test (****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05, ns indicates no significant difference). Source data are provided as a Source Data file.
The performance of PE3 editing is influenced by the position of the additional sgRNA. Researchers typically select sgRNAs at different secondary cutting positions to achieve optimal editing efficiency with PE3. To investigate whether mpegRNA can enhance editing efficiency when the spacing between the two PE3 cuts varies, we randomly selected sgRNAs at five different cutting positions at the HEK4 site and tested them with three sets of mpegRNAs to analyze their performance. As shown in Fig. 3c, the N3 mpegRNA exhibited the best editing efficiency in all tested sgRNA, increasing PE2 from 5.83% to 10.07%, and PE3 ( + 74 nick) from 6.87% to 15.4%.
The indels for PE3, but not PE2, were reduced with different sgRNAs when using N3 mpegRNA. The prime editing efficiency/indels ratio for PE3 with different sgRNAs improved significantly, from 0.40, 0.44, 0.77, and 0.26 to 1.23, 3.48, 2.11, and 2.31 at -52 nick, +74 nick, -95 nick, and -26 nick, respectively. These results demonstrate that mpegRNA strategies can universally enhance the performance of PE3 with various corresponding sgRNAs. Additionally, the optimal mismatch position of mpegRNAs does not change with different PE3 additional sgRNA cleavage positions. The influence of mpegRNA is primarily determined by the sequence of the pegRNA itself.
Enhanced editing efficiency through the combination of mpegRNA with different PE systems
The epegRNA enhances prime editing efficiency by stabilizing the pegRNA structure through the addition of a 3’ motif20. PE4max and PE5max further improve editing efficiency by incorporating MMR pathway inhibitors23. These two strategies are currently among the most effective for enhancing prime editing. We combined either epegRNA or PE4max/PE5max with mpegRNA strategy.
We tested pegRNA, mpegRNA, epegRNA, and mismatched epegRNA at three editing sites. As shown in Fig. 4a, both mpegRNA and epegRNA generally improved editing efficiency. Although epegRNA showed slightly higher efficiency than mpegRNA, the combination of epegRNA and mpegRNA strategies produced the highest editing efficiencies. Notably, at the RUNX1 site, the editing efficiency increased 14-fold with mismatched epegRNA.

a Comparison of editing results among pegRNA, epegRNA, mpegRNA, and mepegRNA. b Editing outcomes of pegRNA and mpegRNA in PE4max system. c Editing outcomes of pegRNA and mpegRNA in PE5max system. For a single target locus, data are presented as the mean ± s.d. from n = 3 independent biological replicates. For the combined analysis of twelve loci, data are presented as the mean ± s.d. from n = 36 independent biological replicates, with three replicates per locus. Significance was determined using unpaired two-tailed Student’s t test (****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05). Source data are provided as a Source Data file.
When combined with PE4max and PE5max, we initially tested the variation in editing efficiency and indel formation across the entire protospacer at the EMX1 locus, similar to tests conducted with PE2 and PE3. We found that, consistent with previous observations using PE2 and PE3, the highest editing efficiency was achieved at mismatch sites N6 to N10, while N12 and N18 had the most negative impact on efficiency. However, we also observed a modest increase in editing efficiency at mismatch sites N14 and N20 relative to other low-efficiency mismatch sites (Fig. S3). Despite this improvement, the efficiency at these sites did not surpass that of pegRNA without mismatches and remained lower than the peak efficiency observed at N6 to N10 (Fig. S3). This suggests that MLH1dn may influence changes in editing efficiency at specific mismatch sites without substantially affecting the optimal mismatch window. We randomly selected mpegRNAs between N6 and N10 across 12 loci and applied them to PE4max and PE5max. As shown in Fig. 4b, c, 10 out of the 12 loci in PE4max and 9 out of 12 in PE5max demonstrated improvements in either editing efficiency or indel reduction. For example, at VEGFA + 3 A to C, mpegRNA increased efficiency from 15.9% to 24.7% and decreased indels from 1.4% to 0.87% in PE5max.
These results indicate that the mpegRNA strategy can be effectively combined with epegRNA or PE4max/PE5max to synergistically improve prime editing results.
The optimal structure of mpegRNA might contribute to the improved editing performance
We hypothesized that the PE efficacy can be hindered by the tendency of pegRNA’s 3’ extension to form secondary structures due to high complementarity with the protospacer sequence. To analyze the structure of mpegRNAs, we conducted an extensive series of site-specific assessments by Alphafold 3. Our experimental data suggest that nucleotide positions N6 through N10 are potentially advantageous for the introduction of mismatches. To obtain a more precise 3D structure of the mpegRNAs, we utilized AlphaFold 3, a state-of-the-art artificial intelligence system for protein structure prediction35, to forecast the three-dimensional conformation of pegRNA targeted to the HEK3 locus.
For the mpegRNAs for HEK3 locus, our analysis revealed that a substantial fraction of the bases within the N20 region display complementary pairing with the 3’ extension (Fig. 5a, b). To elucidate the spatial proximity of these complementary base pairs within the predicted structure, we computed the inter-base distances for all such pairs and determined the average distance, a graphical representation of which is provided in Fig. 5a–c. The utilization of base mismatches within the pegRNA was further substantiated by AlphaFold modeling, which demonstrated a more extended secondary structure (Fig. 5d). Specifically, mismatches at nucleotide positions N6, N7, N8, and N11 significantly altered the spatial positioning of their respective complementary bases, increasing the distances from a baseline of around 5 Å to ~8.6 Å, 8.7 Å, 8.3 Å, and 8.6 Å, respectively (Fig. 5b). However, mismatches at positions N9 and N10 were less efficacious, showing minimal displacement of their corresponding complementary bases.

a Overview of pegRNA distance measurements. b Distances between complementary base pairs of various mpegRNAs, with the protospacer base position plotted on the x-axis and the corresponding distance on the y-axis. c Average distances between complementary base pairs in the protospacer region of different mpegRNAs. d Micro-structural comparison of N6-mpegRNA and standard pegRNA (ST-pegRNA). e Structural analysis of three pegRNAs targeting the HEK3 locus.
The structural dynamics of pegRNA, notably altered by mismatches at N6 and N8, directly enhanced editing efficiencies at these positions, as illustrated in Fig. 2b, c. Conversely, the lack of substantial increase in average base distance contributed by the mismatches at N9 and N10 paralleled their failure to augment editing efficiency, resulting in efficiencies that were on par with or inferior to those achieved with the standard pegRNA.
Notwithstanding the pronounced increase in base distances, the N7-pegRNA’s secondary structure undergoes significant modifications (Fig. 5e), obstructing the formation of a functional scaffold. This impedes accurate assembly with Cas9, culminating in reduced editing efficiency. Notably, the exceptionally high base pairing distance at the 20th position within the N7-pegRNA serves as an indicator of scaffold distortion. This also implies that the low PE editing efficiency at certain positions might be attributable to the loss of fundamental pegRNA structural integrity. A similar scenario is observed with the N7-mpegRNA in UBE3A-3 + 4 T to A, where substantial structural alterations result in a significant decline in efficiency (Figs. 2 & S4). Position N11 may embody a locus of enhanced mismatch tolerance at the target site. This comprehensive structural analysis, combined with functional editing outcomes, underscores the strategic importance of base mismatch location for optimizing pegRNA performance in prime editing applications.
Unexpectedly, the N5 mismatch led to the formation of a G-G base pair, which intriguingly displayed a significantly reduced inter-base distance compared to the canonical base pairs (A-U, C-G, typically within the range of 4.9–5.1 Å) evaluated in our study. Contrary to initial expectations, this anomalous G-G pairing was associated with an enhancement in editing efficiency. This counterintuitive observation could not be adequately explained by base pair spacing distance alone. We speculate that the closer spatial proximity of the G-G base pair might be attributable to forces similar to those mediating interactions within a G-quadruplex structure.
With the structure analysis, we conclude that the optimal structure of mpegRNA might be a major contributor for the improved editing performance of mpegRNA.
Discussion
Prime editing has demonstrated considerable versatility in gene editing, yet its broader application is constrained by relatively low editing efficiency and high rates of indel formation. In this study, we developed mpegRNA approach to address these challenges. By introducing mismatches at strategic positions within the protospacer sequence, we alleviated the formation of secondary structures in the pegRNA and prevented persistent DNA nicking by nCas9.
Our results showed that the mpegRNA strategy significantly enhanced editing efficiency or reduced indel formation in both PE2 and PE3 systems across more than 80% of the tested genomic sites. The optimal mismatch locations were identified around the N6 ~ N10 positions of the protospacer sequence. Utilizing AlphaFold 3 facilitated the generation of several highly suitable candidate mpegRNAs, providing a robust guideline for future applications. Moreover, the mpegRNA approach proved universally applicable, showing consistent enhancement effects regardless of the sgRNA cutting position in the PE3 system. Some studies have reported that in certain circumstances, mismatches in the protospacer can also enhance the editing efficiency of Cas936, and it is possible that part of the improvement in PE editing efficiency by mpegRNA is due to this reason. We also explored the combinatory potential of the mpegRNA strategy with other prime editing enhancement techniques, such as epegRNA and PE4max/PE5max. The combined strategies exhibited synergistic effects, further boosting editing efficiency and reducing indel formation, thereby enhancing the overall efficacy of prime editing.
Overall, the mpegRNA strategy represents a significant advancement in prime editing technology. It offers a practical and effective solution to improve editing outcomes, making it a promising tool for both research and therapeutic applications in genome editing.
Methods
Strains and culture conditions
As a cloning host, E. coli DH5α strains were cultured at 37 °C in L- broth medium (LB) with 1% (w/v) tryptone, 0.5% (w/v) yeast extract, and 1% (w/v) NaCl) or L-agar (LA), consisting of LB medium with 1.5% agar. Ampicillin (100 mg/L) was added to the media as appropriate to ensure the accuracy of the screening results.
Plasmid construction
All plasmids were assembled by the Golden Gate method. PCR primers for pegRNA and mpegRNA were designed with the desired sequences (PBS and RT template sequence) embedded in the primers7. The PBS and RT components of the template epegRNA, along with tevopreQ1, were synthesized by GenScript. The DNA templates were PCR amplified with Primerstar MAX (R045A, Takara). PCR products were gel purified with Kit (JC08KA2210, Sangon Biotech), digested with DpnI restriction enzyme (FD1704, Thermo), Utilizing DH5α (TSC-C01, Beijing Tsingke Biotech Co., Ltd.) competent cells for transformation. The plasmid construction results were verified by Sanger sequencing (BeijingTsingke Biotech Co., Ltd.).
Cell culture and transfection
HEK 293 T cells were cultured in Dulbecco’s minimal essential medium (DMEM, Gibco), supplemented with 10% (vol/vol) fetal bovine serum (FBS) and 1× penicillin streptomycin (Corning). Cells were incubated and cultured at 37 °C with 5% CO2. Before transfection, the cells were seeded in 24-well plates (Corning), incubated for ~24 h and then transfected with PEI after reaching ~40% confluence. A total of 600 ng of Cas9 plasmids and 300 ng of pegRNA or mpegRNA expressing plasmids were transfected with 50 μl of DMEM containing 3 μl of PEI, 100 ng of additional gRNA-expressing plasmids (if required) were also co-transfected. Twenty-four hours after transfection, 4 μg/ml puromycin (Merck) was added to the medium and incubated for 4 days. Then, the cells were collected for high-throughput sequencing.
High-throughput sequencing
Total genomic DNA was extracted using QuickExtract DNA extraction solution (Epicenter, USA) supplemented with proteinase K (Roche) following the manufacturer’s instructions with slight modifications. Cells were washed with PBS and lysed in 30 μl of extraction solution supplemented with 0.3 μl of proteinase K. The samples were incubated at 55 °C for 10 min and inactivated at 80 °C for 3 min. Targeted regions (180–250 bp) of interest were amplified by PCR with Es Taq MasterMix (CW0690S, Cwbio) and used for high-throughput DNA sequencing, as previously described. Libraries with different barcodes were analyzed by Illumina high-throughput sequencing (GENEWIZ, China). The data were split according to their barcodes, and the examined target sites were selected. Base substitution ratios were calculated by dividing base-substitution reads by total reads.
GUlDE-seq assay
The GUlDE-seq assay was performed as previously described34. pegRNA and Cas9 plasmids, along with double-stranded oligodeoxynucleotides (dsODN), were transfected into HEK293T cells and cultured for 72 hours prior to genomic DNA isolation. The purified genomic DNA was then subjected to fragmentation, end-repair, A-tailing, adapter ligation, and dsODN-specific amplification. Libraries were sequenced on Illumina platform, followed by analysis.
AlphaFold 3 structure analysis
Enter sequence on the website (https://golgi.sandbox.google.com/) and submit it to run the prediction. Download the computation results. Open the top-ranked file using PyMOL and observe the overall structure. Use the ‘dist’ command to calculate the distance between each complementary base pair on the protospacer
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
There is no restriction on data associated with this study. The DNA sequences of the main primers, as well as the individual components of sgRNA, pegRNA, mpegRNA, epegRNA, and mepegRNA, are provided in Supplementary Data 1. The high-throughput sequencing data generated in this study have been deposited in NCBI database under accession codes PRJNA916761 and PRJNA1145659. Source data are provided with this paper.
Code availability
The data analysis in this study utilizes publicly available tools with no restrictions on code usage.
References
Takeda, S., Clemens, P. R. & Hoffman, E. P. Exon-skipping in Duchenne muscular dystrophy. J. Neuromuscul. Dis. 8, S343–s358 (2021).
Aartsma-Rus, A. et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 30, 293–299 (2009).
Cox, D. B., Platt, R. J. & Zhang, F. Therapeutic genome editing: prospects and challenges. Nat. Med. 21, 121–131 (2015).
Chemello, F. et al. Precise correction of Duchenne muscular dystrophy exon deletion mutations by base and prime editing. Science advances 7, https://doi.org/10.1126/sciadv.abg4910 (2021).
Schene, I. F. et al. Prime editing for functional repair in patient-derived disease models. Nat. Commun. 11, https://doi.org/10.1038/s41467-020-19136-7 (2020).
Geurts, M. H. et al. Evaluating CRISPR-based prime editing for cancer modeling and CFTR repair in organoids. Life sci. alliance 4, https://doi.org/10.26508/lsa.202000940 (2021).
Anzalone, A. V. et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019).
Landrum, M. J. et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–868 (2016).
Qiao, D. X. et al. Optimized prime editing efficiently generates heritable mutations in maize. J. Integr. Plant Biol. 65, 900–906 (2023).
Lin, Q. et al. Prime genome editing in rice and wheat. Nat. Biotechnol. 38, 582–585 (2020).
Zhi, S. et al. Dual-AAV delivering split prime editor system for in vivo genome editing. Mol. Ther.: J. Am. Soc. Gene Ther. 30, 283–294 (2022).
Chen, P. J. & Liu, D. R. Prime editing for precise and highly versatile genome manipulation. Nat. Rev. Genet. 24, 161–177 (2023).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Lapinaite, A. et al. DNA capture by a CRISPR-Cas9-guided adenine base editor. Science 369, 566–571 (2020).
Walton, R. T., Christie, K. A., Whittaker, M. N. & Kleinstiver, B. P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 368, 290–296 (2020).
Kweon, J. et al. Engineered prime editors with PAM flexibility. Mol. Ther. 29, 2001–2007 (2021).
Böck, D. et al. In vivo prime editing of a metabolic liver disease in mice. Sci. Transl. Med. 14, eabl9238 (2022).
Li, X. et al. Enhancing prime editing efficiency by modified pegRNA with RNA G-quadruplexes. J. Mol. Cell Biol. 14, https://doi.org/10.1093/jmcb/mjac022 (2022).
Nelson, J. W. et al. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 40, 402–410 (2022).
Zhang, G. et al. Enhancement of prime editing via xrRNA motif-joined pegRNA. Nat. Commun. 13, 1856 (2022).
Liu, Y. et al. Enhancing prime editing by Csy4-mediated processing of pegRNA. Cell Res. 31, 1134–1136 (2021).
Chen, P. J. et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell 184, 5635–5652.e5629 (2021).
Chen, R. et al. Enhancement of a prime editing system via optimal recruitment of the pioneer transcription factor P65. Nat. Commun. 14, 257 (2023).
Feng, Y. et al. Enhancing prime editing efficiency and flexibility with tethered and split pegRNAs. Protein & Cell, pwac014, https://doi.org/10.1093/procel/pwac014 (2022).
Houseley, J. & Tollervey, D. The many pathways of RNA degradation. Cell 136, 763–776 (2009).
Shalem, O. et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014).
Wang, Y. J., Sun, H., Liu, G. T. & Chen, X. G. Advances in the study of p53 in response to DNA damage. Yao xue xue bao = Acta pharmaceutica Sin. 46, 1413–1419 (2011).
Abuetabh, Y. et al. DNA damage response revisited: the p53 family and its regulators provide endless cancer therapy opportunities. Exp. Mol. Med. 54, 1658–1669 (2022).
Xu, S. et al. CAS9 is a genome mutator by directly disrupting DNA-PK dependent DNA repair pathway. Protein Cell 11, 352–365 (2020).
Schaefer, K. A. et al. Unexpected mutations after CRISPR-Cas9 editing in vivo. Nat. Methods 14, 547–548 (2017).
Hu, W. X. et al. ExsgRNA: reduce off-target efficiency by on-target mismatched sgRNA. Briefings in Bioinformatics 23, https://doi.org/10.1093/bib/bbac183 (2022).
Bae, S., Park, J. & Kim, J.-S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475 (2014).
Tsai, S. Q. et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197 (2015).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Zheng, T. et al. Profiling single-guide RNA specificity reveals a mismatch sensitive core sequence. Sci. Rep. 7, 40638 (2017).
Acknowledgements
This research was financially supported by the National Key Research and Development Program of China (2020YFA0908000), the National Natural Science Foundation of China (32225031, 32171449, 32271483), the Tianjin Synthetic Biotechnology Innovation Capacity Improvement Project (TSBICIP-KJGG-017) and the Youth Innovation Promotion Association CAS (2022177). We gratefully acknowledge Dr. Chong Zhang and Dr. Chengfeng Ding from Westlake University for their valuable suggestions in revising the manuscript. We also extend our thanks to Zhiyi Zhao and Xiuquan Ye from Westlake University for their assistance and guidance in structural biology analysis. Additionally, we thank Dr. Jianping Zhang and Xiaobing Zhang from the Chinese Academy of Medical Sciences & Peking Union Medical College for their support with off-target analysis using GUIDE-seq. This work received computational resources support from the Intelligent computing platform at the Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences.
Author information
Authors and Affiliations
Contributions
X.Z. and C.B. designed the research, analyzed data, and wrote the manuscript. J.F., D.Z., and C.P. designed the research, performed experiments, analyzed data, and wrote the manuscript. J.L., W.Q., and J.T. designed the research. S.L. performed experiments, analyzed data.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fei, J., Zhao, D., Pang, C. et al. Mismatch prime editing gRNA increased efficiency and reduced indels. Nat Commun 16, 139 (2025). https://doi.org/10.1038/s41467-024-55578-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-55578-z
This article is cited by
-
Harnessing artificial intelligence to advance CRISPR-based genome editing technologies
Nature Reviews Genetics (2026)
-
Anti-CRISPR protein AcrIIA5 can enhance the activity and security of prime editing
Nature Communications (2025)
-
Therapeutic in vivo genome editing: innovations and challenges in rAAV vector-based CRISPR delivery
Gene Therapy (2025)
