
Abstract
Chromosomes are spatially organized and functionally folded into a specific macro-structure in the nucleus. Recently, we and others created haploid cells with chromosome fusions. However, there is still lack of an effective strategy for precisely investigating how the genome copes with fusions. Here, we developed a down-sampling method to convert the populational Hi-C dataset into single cell-like Khimaira Matrix (K-matrix). K-matrix preserves not only the most prominent functional genomic features but also cell-to-cell variations. K-matrix-originated genome 3D models display spatial approach of fused chromosomes and minor global structure alterations. Combined with a layered positional decomposition analysis, our models indicate slight re-adjustment of chromosome distributions accordingly with an increasing tendency following more fusions involved. Nevertheless, the radial distribution of the A/B compartment is not affected dramatically. By contrast, natural populations harboring Rb fusions display significant alterations of chromosome radial location. Overall, K-matrix-originated models enable visualization of chromosomal reorganization with high resolution.
Similar content being viewed by others

Introduction
In the nucleus of eutherian mammals, string-like genomic DNA macromolecule of each chromosome is folded into sub-compartments, forming chromosome territories (CT) that occupy discrete regions1,2,3. Elucidating the organizational pattern of the genome in the nucleus has been a prominent subject, as it is crucially related to its functional implications, such as DNA modification, repair, and transcriptional activity4,5,6,7,8,9. The rapid expansion of chromosome conformation capture technologies has permitted achieving deep insights into the hierarchy of genome architecture10. Based on the acquired contact information, we have known that the organization of chromosomes is largely partitioned into different compartments, including the highly active A compartment and the inactive B compartment10. Moreover, at a higher resolution, the genome is aggregated into topological-associated domains (TADs) and loops, which provide a structural framework for gene regulation11,12,13. Despite the remarkable progression of fine-mapping genome 3D organizations, the challenges of comprehending the genome architecture still remain14. One is the molecular mechanism underlying how these higher-order non-random structures are organized7,15,16,17,18,19; and the other is to decipher how the genome architecture relates to its functions20,21.
To answer these questions, large efforts have been devoted to studying the structural alteration of specific genes or chromosome segments through mutation or knock-out experiments22,23,24,25,26,27,28. However, messages obtained based on these strategies are limited around the span of target site. Dramatic chromosomal changes, such as chromosome fusions or translocations, which are accompanied by the displacement of large chromosome structures in the nucleus, provide valuable and visible genetic materials to investigate how the 3D genome is organized and the impact of these changes. Furthermore, these chromosome rearrangements are crucial during the evolution process or diseases29,30,31,32,33. For example, human chromosome 2 was formed by Rb fusion between two independent ancestral primate chromosomes at 4.0 to 6.5 Mya34. Interesting, a recent study, using male germ cells from mice with naturally occurring Robertsonian (Rb) fusions (BRbS mice), has shown that Rb fusions impact the dynamic genome topology through altering chromosomal nuclear occupancy and synapsis, and reshaping the landscapes of recombination35. To further decipher the structural and biological significances of such karyotype reorganization, we created mouse haploid ESCs (haESCs) and stable mouse strains with fused chromosomes by CRISPR-Cas9-induced cleavage at minor satellites of centromere36. We demonstrated that fused chromosomes occupy adjacent territories in the nucleus. With the bulk Hi-C and RNA-seq information, however, the overall chromosome territories, compartment distributions, and transcriptomes are largely unchanged. Moreover, inter-chromosomal interactions between fused chromosomes are elevated, and inter-chromosomal hubs are affected by the fusion events, especially in chr11 which is not involved in fusion events36.
To comprehend how the genome achieves such a robust and flexible architecture in the nucleus, it is crucial to investigate how chromosomes are reorganized after chromosome fusions. However, due to the probabilistic property of the chromosome localization, mFISH (multifluorescence in situ hybridization) could only give us partial snapshots of the genome architecture17. Meanwhile, current methods for genome modeling using bulk Hi-C dataset could return whole genome 3D models, but failed to show appropriate spatial location of fused chromosomes (Supplementary Fig. 1)37. On the other hand, single-cell Hi-C genome modeling can only detect a small fraction of interactions in a cell, and thus requires a large number of resources to describe the global characteristics of the genome38,39,40. Together, there is still an unmet need for an efficient method that can visualize chromosomal organization at single-cell level with a high global resolution. To address these limitations, we constructed a down-sampling method to convert populational Hi-C datasets into Genome Khimaira Matrix (K-matrix) mimicking single-cell Hi-C characteristics, which preserves not only the main features of the original dataset, but also displays model-to-model variations. 3D models calculated from K-matrices indicated that chromosomes occupy a relatively stable region in the nucleus, echoing the nonrandom probabilistic feature of chromosome location2,41. In the events of chromosome fusion, the spatial distribution of the A/B compartment in the nucleus is well maintained even in haESCs with multiple Rb fusions. Meanwhile, the chromosomes could readjust their locations properly to buffer the potential adverse effects of chromosome displacements. However, significant 3D organization adjustments are observed in the species carrying naturally selected Rb fusions35. These results further support the genomic organization homeostasis model and broaden our understanding of genomic architecture reorganization during evolution.
Results
Down-sampling of populational Hi-C data into genome Khimaira Matrix
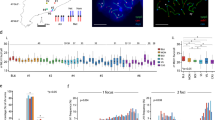
Recently, we observed that fused chromosomes display decreased spatial distance and altered inter-chromosomal interactions in the nucleus of mouse haESCs. Meanwhile, chr11 that is not involved in fusion events exhibits more changeable interactions with other chromosomes, suggesting that fused and non-fused chromosomes might adjust their locations accordingly following Rb fusions36. To investigate how each chromosome readjusts its position in the nucleus after fusion, we first attempted to build genome-wide 3D models by using populational Hi-C contacts. However, all tested methods37,42,43,44 could not return valid models for downstream analyses (See Supplement Information, Supplementary Fig. 1). On the other hand, for single-cell Hi-C data, nuc_dynamics could generate 3D genome structures with radial A/B compartment and expression patterns40, yet it’s still unknown whether nuc_dynamics could be used for modeling populational Hi-C datasets. To this end, we first tested the model calculation of O48 cells using raw populational Hi-C contacts. The results showed that nuc_dynamics returned models with clear chromosomal territories (Supplementary Fig. 2a). However, the structures showed a compacted configuration (Supplementary Fig. 2a), and the end-temp and model RMSD (ensemble root mean square deviation) were extremely high, representing highly inconsistent 3D genome structures (Raw in Fig. 1a). The potential explanation is that one genome segment digested from a single haploid cell can have only one interaction with another segment at most in a single-cell Hi-C dataset; however, each genome segment may interact with many other segments in the bulk Hi-C datasets, leading to complex interaction network that could cause excessive restraints and structural violations when using the nuc_dynamics software for modeling the 3D genome structures.

a Final Temp and RMSD of each K-matrix method returned by nuc_dynamics. Structures were calculated at 1 M resolution, and three samplings (1M reads) were used for each method using the O48 Hi-C dataset. b Diagram of K-matrix method. Three strategies have been implemented to convert bulk Hi-C data to K-matrices. Segments are abstracted as vertices with size representing contact number and contacts between segments are abstracted as edges with width representing contact frequency. Arrow and numbers, simplified selection sequences. For Max-Point, select the vertices first (orange arrow), then the edges (cyan arrow). c Correlations between original bulk data, sampled bulk data, K-matrices, single-cell data, and pooled single-cell data. d Log-scale plots of contact probability against genomic distance. 10 K-matrices each for O48, R1n13, R2n6 and R5n17 were used. e 3D genome structures calculated from K-matrices of O48, R1n13, R2n6, and R5n17. Models of two repeats for each sample were displayed here, with a resolution of 100Kb.
To address this problem, we attempted to develop a sampling method to simulate single-cell contact characteristics from the populational datasets, which produces Khimaira Matrices (referred to as K-matrices, details in Methods, Fig. 1b and Supplementary Fig. 2b). Based on the contact feature of the chromosomes in single-cell Hi-C experiments, we developed a step-wise selection procedure to simplify the populational Hi-C datasets. Generally, each populational dataset was abstracted as genome contact networks, with each chromosome segment as vertex and each interaction as edge. The weight of the vertex denotes the total number of edges sharing the vertex, and the weight of the edges denotes total number of contacts between two vertices (segments). For Random selection, the simplified genome contact network was initially started by selecting one edge randomly. Then, all other edges sharing the vertices of the selected edge were removed. Repeating the selecting procedure until all the edge are resolved. For Max-Edge selection, initially, the edge with the highest weight in the populational dataset was selected as seed, and after removing all other edges sharing the vertices of the selected edge, the network expands by selecting next edge with the highest weight in the rest of the dataset. For Max-Point selection, the vertex with the highest weight was selected first, and its edge was randomly selected and all other edges were removed, then the network was built by repeating these procedures. We tested these three sampling methods for K-matrix conversion, and found that strikingly, models generated by all three methods return better model parameters and decreased runtime than using original populational data. Especially, we observed the Max-Point method returned a more optimized model with minimal RMSD during model generation than other two methods for given read pools (Fig. 1a).
K-matrices display a high correlation with original bulk data and variations
To investigate the genome distribution and coverage of K-matrix converted by the Max-Point method, we used the dataset reported by Stevens et al’s40, which contains both bulk and single-cell datasets. We sampled 10 repeats with different numbers of reads (500Kb, 1 M, 2 M, 5 M and 10 M) from bulk Hi-C data and converted them into K-matrices with a threshold of 2 (points with contact number less than 2 were filtered out, Supplementary Fig. 2c). The results showed that the coverage at 100Kb bin increased with more initial reads in both bulk Hi-C (Supplementary Fig. 2d) and K-matrices (Supplementary Fig. 2e). Especially, with initial 1 M reads, both coverage ratio and the average contact counts in K-matrices ( ~ 94% and 4 contacts per 100Kb bin respectively) were similar to those in single cell data ( ~ 89.7% and 4.1 contacts respectively) (Supplementary Fig. 2d–f). Together, the matrix exhibits similar coverage and distribution as single-cell Hi-C data, thus being suitable for nuc-dynamics to generate 3D models.
Given that K-matrices are not real single-cell Hi-C matrices, but matrices with a combination of specific contact features from different single cells, we would like to know whether these K-matrices can reflect genome-wide features of bulk Hi-C and also display variations of single-cell Hi-C. To this, we set to investigate the similarity between K-matrices and bulk Hi-C data or single-cell data. First, we calculated correlations among K-matrices (20 repeats), single-cell data (8 cells), pooled single-cell data and original/sampled bulk data. The results showed a high correlation between K-matrices and sampled/original bulk data ( ~ 0.53/ ~ 0.7), whereas single-cell data showed a lower correlation with sampled/original bulk data ( ~ 0.2/ ~ 0.5) (Fig. 1c). This indicates that K-matrices are better representatives of the original bulk dataset. Furthermore, the correlations among our K-matrices were about 0.2, remarkably lower than the correlations among sampled bulk data ( ~ 0.77), but correlations among single-cell matrices were about -0.5, reflecting that K-matrices sustain the single-cell feature of model-to-model variation (Fig. 1c).
Next, we compared the models generated from K-matrices and single-cell Hi-C data through nuc-dynamics. The results showed that RMSD within models from single cells was higher than that within K-matrix models. Meanwhile, the RMSD between single-cell models and K-matrix models was lower than RMSD among single-cell models but higher than that of K-matrix models (Supplementary Fig. 2g, h). Together, these results further support that our K-matrix models are more stable in modeling the genome, but as expected, sustain a feature with lesser variation than that of the single-cell data.
To further demonstrate that K-matrix is capable to construct 3D genome models from bulk Hi-C dataset, we sampled 10 repeats with different numbers of reads (500Kb, 1 M, 2 M, 5 M and 10 M) for O48 and R1n13 and converted them to K-matrices. Similar to the result from Stevens’ data, the coverage at 100Kb bin of the resultant K-matrices (~ 97%) from 1 M reads was most similar to single-cell data (~ 89.7%) (Supplementary Fig. 2i, j). Importantly, in consistent with previous observations in human and Drosophila genome10,45, K-matrices displayed similar regimes that the contacts between two intra-chromosomal segments (distance at > 100 kb) decay with linear distance (Fig. 1d). 3D models built from K-matrices (10 for each K-matrices), as expected, displayed not only discrete chromosome territories but also model-to-model variations (Fig. 1e and Supplementary Fig. 2k), consistent with the observations in single-cell models38,40. Finally, to further validate the feasibility of our method, we constructed K-matrix-originated 3D models from bulk Hi-C data of haESCs carrying telomere-centromere fused chromosomes between chr1/2, chr2/1, or chr4/546. Consistently, all models displayed clear chromosome territories (Supplementary Fig. 2l).
K-matrix-originated 3D genome models preserve functional 3D structure features of the original genome
In ROB-O48 cells, two telocentric chromosomes are fused by their centromeres, which form a metacentric chromosome. The fusion event also reduced the distance between fused chromosomes36. To test the approach of two fused chromosomes in the 3D models generated from K-matrices, we colored fused chromosomes in these models and observed that two chromosomes were tethered together in all ROB-O48 models, but separated in wild-type models (Fig. 2a and Supplementary Fig 3a). Both chromosome and centromere distances between two fused chromosomes reduced dramatically (Fig. 2b, c and Supplementary Fig 3b). Interestingly, the distances between two fused chromosomes kept constant across all tested ROB-O48 models, whereas varied in individual wild-type models (Fig. 2d and Supplementary Fig. 3c), reflecting more stable chromosomal relationship between two chromosomes after fusion. In addition, all 3D models established from K-matrices indicated that while the two fused chromosomes directly linked and approached each other, they largely occupied their own chromosome territories and didn’t form a mixed territory (Fig. 2a and Supplementary Fig. 3a). These results were confirmed by the observations based on chromosome FISH (please see Fig. 1d, and 1p in paper by Zhang et al. 36), indicating that our K-matrix-originated 3D models can correctly recapitulate the relationship between fused chromosomes.

a Representative models show tethering of the fused chromosomes in R1n13 cell lines, whereas separated in O48 cells. Fused chromosomes were colored as indicated accordingly; other chromosomes were shown as gray lines; Centromeres were displayed as balls. b, c Euclidean distance between fused chromosomes (b) or centromeres (c) in R1n13 and O48 cell lines. Each data point represents the average distance in 100 models of each sample repeats (3 repeats). p-value was indicated, two-sided t-test. d Chromosome distance distribution between fused chromosomes in each model of wild-type O48 and R1n13 cell line repeats. e Representative models show tethering of the fused chromosomes in telomere-centromere fused T12 (chr1 + 2) cell line, cleavage of chr1 was clearly observed, and the proximal arm was fused with chr17. Here, telomere refers to the telomere located at the end of the chromosome opposite to the centromere end. Centromeres were displayed as balls. f, Distance between telomere of chr1 and centromere of chr2 in T12 cell line (2 repeats, 100 models each). g, h Cross-section view of expression profile (g) and A/B compartment distribution (h) in R1n13 and O48 cell lines. Blue, low expression area or B compartments; Red, high expression area or A compartments. i Surface view and cross-section view of A/B compartment distribution in Rod neurons.
To further validate our method, we colored K-matrix-originated 3D genome models from Hi-C data of haESCs carrying telomere-centromere fused chromosomes between chr1/2, chr2/1, or chr4/546. In consistent with observed karyotype and FISH data, fused chromosomes were adjacent to each other in a telomere-centromere linked fashion. The telomere-centromere and chromosome-chromosome distances between fused chromosomes were reduced dramatically compared with wild-type haESCs (Fig. 2e, f and Supplementary Fig. 3d–f). Strikingly, our models precisely recapitulated the splitting of chr1 at the 114 Mb location46, and its proximal arm was fused with chr17 in the chr1/2 cell line (T12) (Fig. 2e). Together, our method can precisely display different chromosome fusion events.
Previous studies have demonstrated that A/B compartments are organized in a spatially radial pattern in a single nucleus, with a low expression outer ring of B compartments and a high expression inner ring of A compartments, as well as an inner B compartment around the nucleoli39, 40,47. To test whether models built from K-matrices could also display such radial pattern, we then projected expression profiles and A/B compartment information on these models. In all tested cell lines, the results showed that the distribution of A/B compartments and expression regions correlated well with each other. Highly expressed A compartments are located at the nuclear interior, and lowly expressed B compartments mainly located at the peripheral regions and around the center (Fig. 2g, h and Supplementary Fig 3g–i), which are consistent with previous observations in the single-cell 3D structures of mouse ESCs38,39,40,47. In addition, these patterns were also confirmed in the K-matrix models of telomere-centromere fused cells (Supplementary Fig. 3j–l). Surprisingly, even in the T12 cells with the split of chr1, both arms of chr1 could be well posed to maintain a radial compartment distribution pattern (Supplementary Fig. 3l).
In addition to haESCs, we also investigated differentiated cells carrying centromere-centromere or centromere-telomere fusions by calculating K-matrix-originated 3D genome models. As expected, our models, consistent with DNA-FISH results, could recapitulate the displacement of two fused chromosomes in brain cells (Supplementary Fig 4a–g), similar to those in parental cells36. Meanwhile, A/B compartment distribution and expression profile remained radial in all the models (Supplementary Fig. 4h, i).
To further validate the capability of our method to recapitulate the A/B compartment distribution, we generated K-matrices 3D models from Falk et al.’s dataset, which contains inverted compartment distribution in the rod neuron nuclei48. Indeed, our K-matrix models showed that A compartment was distributed at the peripheral of the nuclei, while B compartment occupied the central region (Fig. 2i and Supplementary Fig 4j). Together, all these results indicate that K-matrix-originated models preserve the most prominent functional 3D structure features of the original genome in cells and thus could be used as valid models for further analyses.
Fusion-induced chromosome location changes support the probabilistic model of proximity patterns for 3D genome organization
Previous studies have suggested a probabilistic model of proximity patterns for 3D genome organization, rather than absolute random in some species41,49,50. However, due to the limitation of FISH technology, insufficient snap-shots of the nucleus do not allow conclusions on the functional implications of the probabilistic model51,52,53,54,55,56. Since fusions can not only directly alter the chromosome location but also induce the genome-wide readjustment of inter-chromosomal interaction correspondingly36,46, we proposed that fusion-mediated chromosome location changes might be an appropriate system to validate the probabilistic model of proximity patterns. To this, we attempted to investigate the location adjustment of chromosomes by analyzing spatial relationships between all chromosomes in the nucleus. Thus, we calculated the Euclidean distances between all chromosome pairs using the spherical coordinates of all chromosome segments in the nucleus (Fig. 3a). The results showed that the average chromosomal distance tended to be stable across sample repeats (Supplementary Fig. 5a), reflecting that K-matrix-originated 3D models are stable and consistent. Meanwhile, models of a same cell type also displayed distance variations (Supplementary Fig. 5a).

a Averaged chromosome distance between all chromosomes in O48 and ROB-O48 cell lines. Red, chromosomes are close to each other; blue, chromosomes are distanced. 100 models for each sample repeat. b Percentage changes of chromosome distance between each chromosome pair in R1n13, R2n6 and R5n17 cell lines. Except for the fused chromosomes, chromosome distances were either decreased (blue) or increased (red) slightly. c Distribution of chromosome distance disturbances in ROB-O48 cell lines, 190 chromosome pairs. d Volcano plot of chromosome distance percentage change and p-value in R1n13, R2n6 and R5n17 cell lines. Cyan (*), p < 0.05, |PC | > 0.2; yellow (**), p < 0.01, |PC | > 0.2; red, p < 0.05 only; gray, p > 0.05; two-sided t-test; round dots, distances involved fused chromosomes; triangle dots, the distance between non-fused chromosomes; numbers of chromosome pair in each section are indicated. e, f Chromosome distance between chr17 and chr18/chr19 in O48 and R5n17 cell lines (3 samples, 100 models each). p-values were indicated, two-sided t-test.
Except for distance changes between fused chromosomes, we only noticed minor changes of the inter-chromosomal distance in ROB-O48 cell lines when compared with wild-type cells (Fig. 3b–d). Most of the alterations occurred between fused chromosomes and non-fused chromosomes, while the distances between non-fused chromosomes remain similar to wild-type (Fig. 3d). Interestingly, consistent with previous observation17,57, we found that the smallest chromosomes, including chr17, chr18, and chr19, tended to be closely distributed in the nucleus (Fig. 3a). However, fusion between chr5 and 17 in R5n17 cells led to separate chr17 from chr18 and chr19 (Fig. 3e, f). Meanwhile, we found that fusion of chr5 and 17 had more notable effect on chromosome distance with chr17 than chr5 (Fig. 3b–d), probably due to that chr17 had a short average distance with other chromosomes (Fig. 3a, and Supplementary Fig. 5b, c). Moreover, we observed that fusion between chr1 and chr13 caused the increased distances between chr13 and chr19 in the R1n13 cells (Supplementary Fig. 5d). Similar changes were also observed in T12, T21, and T45 cells (Supplementary Fig. 5f–i). Distances between chr17 and several chromosomes were altered after chr17 fused with the proximal arm of chr1 in the T12 cell line (Supplementary Fig. 5i). These results indicate that fusion events can only induce minor chromosome location changes between fused chromosomes and non-fused chromosomes, thus supporting the probabilistic model of proximity patterns.
Layered Positional Decomposition analysis indicates stable radial position of fused chromosomes in the nucleus
Given that A/B compartments and gene expression are organized in a spatially radial pattern in the nucleus7,39,40,47, we next attempted to investigate radial location changes of all chromosomes induced by fusions in the nucleus. Since the nucleus is not a typical spherical structure51, we developed a layered positional decomposition method (LPD, details in methods, Fig. 4a and Supplementary Fig. 6a, b) to inspect the radial location of each chromosome relative to the whole nucleus, represented by InterDepth (0-1, from the periphery to the center of the nucleus). The results showed that the InterDepth of each chromosome was stable across sample repeats (Fig. 4b). We then calculated the InterDepth of A/B compartments. As expected, we observed an increasing A compartment ratio and a decreasing B compartment ratio from outside to inside of the nucleus (Supplementary Fig. 6c, d). In consistent with previous observations40, we noticed a drop in A compartment ratio and an increase in B compartment ratio at the InterDepth range of 0.6 ~ 0.8 (Supplementary Fig. 6c, d), a region around the nucleoli. The internal B compartment region consisted of the B compartment of chromosomes that are located in the center of the nucleus, including chr4, 5, 9, 11, 17 in wildtype O48 cells58 (Supplementary Fig. 6e). In addition, we observed internal A compartment instead of a hollow nucleolus in the nucleus center, which could reflect that the K-matrix is a combination of segment contacts from different single cells.

a Diagram of layered positional decomposition and calculation of InterDepth. Different color of dots indicates different chromosomes. b Pearson correlation between sample repeats in ROB-O48 cell lines (3 samples). c Centromere InterDepth of fused chromosomes in ROB-O48 cell lines. p-values were indicated, two-sided t-test, 3 samples each. d Averaged chromosome InterDepth in ROB-O48 haESCs and brain cells. Black boxes, fused chromosomes; white, nucleus peripheral; red, nucleus center. e InterDepth shifts of each chromosome segment between R1n13/T45 cell lines (InterDepth > 0, upper points) and control O48 cell lines (InterDepth <0, lower points). Resolution, 100Kb; bars, InterDepth shifts ( > 0, shifts toward inside, <0, shifts toward outside); points and bars were colored by chromosomes. f, g Chromosome InterDepth (f) and telomere_centromere InterDepth (g) between chr4 and chr5 in T45 cell lines, 2 samples each. h Averaged chromosome InterDepth in telomere-centromere fused haESCs and brain cells. Black boxes, fused chromosomes; white, nucleus peripheral; red, nucleus center. i Telomere distance change between each chromosome in T45 haESCs and T45 brain cells. Here, telomere refers to the telomere located at the end of the chromosome opposite to the centromere end. Telomere distances were either decreased (blue) or increased (red). j, k, Chromosome InterDepth (j) and telomere_centromere InterDepth (k) between chr4 and chr5 in T45 brain cells, 2 samples each. l InterDepth shifts of each chromosome segment between T45 brain cells (InterDepth > 0, upper points) and control brain cells (InterDepth <0, lower points). Resolution, 100Kb; bars, InterDepth shifts ( > 0, shifts toward inside, <0, shifts toward outside); points and bars were colored by chromosomes.
We next investigate the InterDepth changes induced by Rb fusions. Expectedly, the InterDepth of neo-centromeres shifted in comparison with the centromeres before fusion (Fig. 4c and Supplementary Fig. 6f). However, although inducing some minor shifts, Rb fusions overall had a small effect on the InterDepth of fused or non-fused chromosomes in all ROB-O48 haESCs and brain cells, resulting in relatively stable chromosomal radial position (Fig. 4d, e and Supplementary Fig. 6g, h). In contrast, in T12 and T45 cells generated by telomere-centromere fusions, we observed more radial shifts of chromosome segments (Fig. 4e–h and Supplementary Fig. 7a–c). Especially in the fused chr4 and chr5, large chromosome regions shifted to the outside of the nucleus after fusion (Fig. 4e, f). The differences between ROB-O48 cells and telomere-centromere ligated cells could be explained by Rabl configuration in the ES cells, which describes that centromeres and telomeres are clustered on opposite sides of the nucleus39,40. In the ROB-O48 cells, centromere fusions occur on the same side, which causes minor disturbances in the nucleus (Supplementary Fig. 7d, e); however, telomere-centromere fusions involve segments located on opposite sides, which may lead to more violent alterations among telomeres (Fig. 4i and Supplementary Fig. 7f, g). These changes also echo the results that more differential expressed genes (DEGs) and changed TADs are detected in telomere-centromere fused cell lines compared to minor alterations in ROB-O48 cells36,46.
Surprisingly, in contrast with significant radial shifts in T45 haploid ESCs, T45 brain cells (T45B) only displayed minor chromosome shifts (Fig. 4h, j) and altered relationship among telomeres when compared with wild-type brain cells (Fig. 4i–l), suggesting that brain cells could tolerate the ligation of chr4 and chr5. This could also be reflected by the normal phenotype of T45 pups in anxiety tests46. Together, these observations suggest that unlike the chromosome distribution in haESCs, brain cells may exhibit different chromosome configurations in the nucleus. To this, we compared the centromere and telomere distance in the 3D models between brain cells and their parental cells. The results showed decreased distances in brain cells (Supplementary Fig. 7h). To further confirm this, we performed FISH analysis and found that centromeres and telomeres trend to cluster together more in brain cells compared with haESCs (Supplementary Fig. 7i, j).
In summary, Rb fusions do not cause dramatic chromosomal InterDepth shifts, implying that the reorganization of chromosomes in the nucleus might be parallel but not perpendicular to the circumference, which in turn could preserve radial A/B compartment distribution. In addition, cell type-specific chromosome configurations may affect radial position shifts caused by chromosome fusions.
Stable radial distribution of A/B compartment in ROB-O48 cells with multiple-fusion events
With the accumulation of chromosome fusion events in cells (multiple-fusion ROB-O48, mROB-O48), we previously observed an increased tendency of the number of DEGs compared with wild-type cells, which suggests accumulated readjustment of 3D genome organizations within the nucleus36. To further address this, we performed sequential CRISPR-Cas9-based Rb fusions in R1n13.R2n9.R7n19 cells (M3) that were derived from R1n13.R2n9 (M2) cells and generated ROB-O48 cell lines with four or five fused chromosomes, including R1n13.R2n9.R7n19.R3n6 (M4) and R1n13.R2n9.R7n19.R3n6.R17n18 (M5). Similar to transcriptome changes36, we detected a tendency of more changes in TADs following with more chromosomes fused (Supplementary Fig. 8a, b). However, the strength changed TADs distributed in all chromosomes (Supplementary Fig. 8c), without especial enrichment in fused chromosomes. Nevertheless, more TAD changes occurred on the additional fused chromosomes (Supplementary Fig. 8c). Moreover, we observed that increased inter-chromosomal interaction mainly located at the fused chromosomes (Supplementary Fig. 8d), which, however, couldn’t be related to significant expression changes (Supplementary Fig. 8e).
As expected, K-matrix-originated 3D genome models could correctly predict the approaching of each fused chromosome pair (Fig. 5a); chromosome and centromere distances between fused chromosomes were diminished (Supplementary Fig. 8f). The genome structure difference between mROB-O48 and O48 models increased as the number of fusion events increased (Supplementary Fig. 8g, h). Consistently, similar to ROB-O48 cells with a single-fusion event, most of the distance changes in mROB-O48 cells occurred on the fused chromosomes, and the distances between non-fused chromosomes only slightly readjusted (Fig. 5b and Supplementary Fig. 8i, j). For example, distance between chr17/chr18 and chr19 changed after chr19 fused with chr7 in M3 (Fig. 5b and Supplementary Fig. 8k, l). Strikingly, although the number of DEGs increased with more chromosomes fused36, the overall A/B compartment distribution and expression pattern in the nucleus maintained the radial distribution profile across all the mROB-O48 cells (Fig. 5c and Supplementary Fig. 8m).

a Representative models show tethering of all fused chromosome pairs in each engineered mROB-O48 fusion cell line. Balls, centromeres of each indicated chromosome. b Percentage changes of chromosome distance between each chromosome pair in mROB-O48 cells, using O48 as control. Heatmaps show decreased (blue) or increased (red) chromosome distances. c Distribution of A/B compartment (Left, surface view and cross-section view) and expression pattern (Right) in M5.R17n18 3D genome models. d Averaged chromosome InterDepth of mROB-O48 cells and O48 cells. Colored boxes, fused chromosome pairs are displayed with the same color; white, nucleus peripheral; red, nucleus center. e Left, representative FISH images of chr11 and chrX in O48 cell lines, Right, centroid distance of chr11 and chrX. p-value was indicated, two-sided t-test. f Representative models show tethering of each fused chromosome pair in naturally occurring Rb mouse fibroblast cells. Fused chromosomes were colored as indicated accordingly; other chromosomes were shown as gray lines; Centromeres were displayed as balls. g Percentage changes of chromosome distance between each chromosome pair in naturally occurring Rb mouse fibroblast cells. Heatmaps show decreased (blue) or increased (red) chromosome distances. h Distribution of A/B compartment (surface view and cross-section view) in naturally occurring Rb mouse fibroblast cells. i InterDepth shifts of each chromosome segment in M5.R17n18 cell line and naturally occurring Rb mouse fibroblast cells. Resolution, 100Kb; bars, InterDepth shifts ( > 0, shifts toward inside, <0, shifts toward outside); points and bars were colored by chromosomes.
Interestingly, we found that chr11 tended to locate at nuclear center, whereas chrX mainly occupied the periphery space26,59 in all models of ROB-O48 cells with single or multiple fusions and differentiated cells (Fig. 4d, i and Fig. 5d). To validate these observations, we performed DNA-FISH experiments with the chr11- and chrX-specific probes in O48 cells. Indeed, the results showed that chr11 was mainly located at the nuclear interior, while chrX maintained its location at the periphery of the nucleus (Fig. 5e and Supplementary Fig. 8n). These data are consistent with a recent observation that chr11 has strong interactions with other chromosomes based on single-cell Hi-C data60, which may echo previous results that chr11 is part of nucleolar organizer regions58 and has highest gene density among all chromosomes (Supplementary Fig. 8o)7,61,62,63. Meanwhile, our previous study has shown that, although chr11 was not involved in fused chromosomes, it exhibited significant inter-chromosomal interaction changes and harbored a large number of DEGs in haploid cells with two or three metacentric chromosomes36. Together, our results suggest that chr11 may play an important role during this process through readjusting its location in the nucleus accordingly.
Long-period effects of Rb fusions cause 3D genome organization alterations during evolution
Having shown that Rb fusions can’t dramatically change 3D genome organization in the resultant ROB-O48 cells with single or multiple metacentric chromosomes in a short period of time, we next attempted to investigate the long-term effects of Rb fusions on the genome organization during the process of evolution. To this, we reanalyzed Hi-C data collected from somatic and germ cells of mice with naturally occurring Rb fusions (BRbS mice)35. The BRbS mice analyzed here have six Rb fusions (chr3/chr8, chr4/chr14, chr5/chr15, chr6/chr10, chr9/chr11 and chr12/chr13). Consistently, K-matrix-originated 3D genome models recapitulated all the chromosome fusion pairs in the nucleus of BRbS fibroblasts and germ cells precisely (Fig. 5f and Supplementary Fig. 9a, b). Meanwhile, distances between fused chromosomes were decreased in all cells (Fig. 5g and Supplementary Fig. 9c, d). Interestingly, while fibroblast chromosomes were folded into a compact configuration same as observations in ESCs (Fig. 5f), chromosomes in pachynema/diplonema (PD) cells displayed an elongated structure and were intertwined together, reflecting the features of meiosis (Supplementary Fig. 9b). In round spermatids (RS), while chromosomes were folded into territories, their structure appeared to be loosened (Supplementary Fig. 9a, e), probably providing a structural basis for the displacement of histone by protamine. Together, these results indicate that our K-matrix-originated 3D genome models can recapitulate not only chromosome fusions but also chromatin structure features of a specific cell type.
Consistently, projection of A/B compartments and expression profiles showed radial distribution patterns in fibroblast cells (Fig. 5h). However, they were scattered in the nucleus of PD cells and RS, probably due to the reorganization of TADs and A/B compartments during cell divisions (Supplementary Fig. 9f)64. Finally, we investigated radial shifts in naturally occurring Rb fusions cells by comparing with engineered mROB-O48 cells. In mROB-O48 cells, with the increase of fusion events, the InterDepth only had minor alterations (Fig. 5i and Supplementary Fig. 9g, h). Surprisingly, in the 3D models of BRbS fibroblasts, we observed dramatic radial shifts of several chromosomes, especially chr6 and chr18 in fibroblast cells, in which, chr6 was pulled into the center of the nucleus, and chr18 that is not involved in fusion was squeezed out of nuclear center (Fig. 5i and Supplementary Fig. 9i, j). In contrast, PD cells and RS didn’t show whole chromosome InterDepth changes, probably due to that they are in the cell division cycle or dramatic genome remodeling stage (Supplementary Fig. 9k). Taken together, in contrast with lab-made cells with Rb fusions, these results suggest that 3D genome organization changes induced by Rb fusions might be preserved and aggregated in BRbS mice during long-term natural evolution.
Discussion
CRISPR-Cas9-induced chromosome fusions in haESCs provide a unique system to study the relationship between the structure and spatial position of chromosomes and their functionalities. However, there is still a technical challenge to recapitulate precise chromosome reorganization with a high global confidence. Here, we developed a sampling strategy to convert the bulk Hi-C dataset into K-matrix, from which, 3D genome models could be built through nuc-dynamics. These models can not only precisely recapitulate the tethering of fused chromosomes in different cell types with different fusion events, but also accurately predict radial distribution of A/B compartment. Instead of nuc_dynamics, Single-Cell Chromosome Conformation Calculator (Si-C) developed by Meng et al. 65 can also be used to reconstruct intact genome 3D structures from single-cell Hi-C data. We thus used K-matrices as input for Si-C method and found that the resulting models exhibited similar genome structures as those from nuc_dynamics, such as the tethering of fused chromosomes (Supplementary Fig. 10a, b), radial distribution of A/B compartment (Supplementary Fig. 10c), and central localization of chr11 in the nucleus (Supplementary Fig. 10d–e). Together, these results have shown that K-matrices are feasible for different tools to model the genome structure.
With K-matrix, we have built 3D genome models from different cell types, including haESCs, brain cells, fibroblast, PD and RS cells. The generated models display different genome configurations and different compaction (Supplementary Fig. 9e). Meanwhile, K-matrix-originated 3D genome models can recapitulate chromatin structure features of a specific cell type, such as intertwined chromosomes in PD cells and loosened chromosomes in RS. Moreover, subtle genome structure changes can also be recapitulated in the resultant 3D models (Fig. 2e). Together, these results indicate that our method has a broad applicability in regard to cell types with various chromatin states. Nevertheless, we found that the RMSD among models of the same single-cell sample is much lower than that of K-matrix (Supplementary Fig. 2h). Therefore, a future task is to optimize our sampling method through modifying selecting procedure to reduce the RMSD of models from the same sample.
Our previous study showed that only ~0.1% of total genes were detected as DEGs in haESCs with single Rb fusion. With the accumulation of fused chromosomes in engineered ROB-O48 cells, while more spatial adjustments were as observed, the number of DEGs is still only 4% of total genes in cells with three Rb fusions36. Interestingly, DEGs are distributed throughout the genome with no tendency to enrich at fusion sites or fused chromosomes36. Meanwhile, in balancers (fly with highly rearranged chromosomes), a small-scale gene expression change (9.6% of total testable genes) was also witnessed, in which, only a relatively small proportion (~ 18%) can be explained by genetic variation directly impacting the genes themselves66. Both studies indicate the robustness of 3D genome organization. However, the underlying reason is still unclear. Here, by calculating the genome 3D models of ROB-O48 cells, we observed that although translocations lead to significant displacements between fused chromosomes, the distance between non-fused chromosomes are overall stable. Meanwhile, the location of chromosomes in the nucleus is also well-maintained. Moreover, the global chromosomal radial position and A/B compartment distribution are also well kept. These observations together suggest that the preferred readjustment of the fused chromosomes might be parallel but not perpendicular to the nuclear circumference. We hypothesize that the parallel movement of chromosomes may minimize the uncertainty induced by fusion events, leading to the maintenance of compartment distribution and gene expression profile, which probably underlies the robustness of 3D genome organization.
Previous studies indicate the Rabl configuration in the ES cells39,40. Interestingly, our 3D genome models show that telomere-centromere fusions can cause more structural changes and adjustments than centromere-centromere fusions in haploid ES cells, which could be explained by the Rabl configuration of chromosomes in ESCs. However, the telomere-centromere fusion-caused changes were attenuated in T45 mouse brain cells, which suggests the difference in chromosome configuration between ES and brain cells. Consistently, our further immunostaining analysis support this difference. Meanwhile, previous studies indicate that a preferred chromosome configuration in different tissues is crucial for compartment distribution and tissue-specific gene expression67,68,69. Together, cell type-specific chromosome configuration may alleviate or enhance the side effects caused by chromosome fusions. Thus, one intriguing application of our methods is to describe genome configuration atlas of all cell types, which may help to establish the correlation between 3D genome and functionality.
In addition, our models show that chromosomes have their preferences of spatial location in the nucleus. For example, chr11 as a part of the nucleolar organization center prefers to locate at the center58. Even after fusion events, the centralized location of chr11 shows consistency across all the engineered ES cells and tested differentiated cells, suggesting that chr11 may play a critical role in 3D genome organization in ES cells and some differentiated cells. However, in fibroblast cells, chr10 was found at the center of the nucleus (Supplementary Fig. 9i). Together, these results suggest that with distinct genome structure configuration, chromosomes may have preferred locations in different cell types to establish unique expression profile70,71. Further investigation on how central chromosomes stably sustain its spatial location and function in genome expression hub formation will aid our understanding of the mechanisms underlying whole genome 3D structure organization in the nucleus.
Surprisingly, significant radial location shift of several chromosomes has been observed in mouse cells carrying naturally occurring Rb fusions35. These results suggest that the spatial location adjustments of chromosomes bring small changes at the beginning, which enables the survival of the individuals carrying fused chromosomes, and provides the chance to gradually preserve and accumulate spatial adjustments during the long-period time-course of natural selection. Studies of the genome structure of muntjac deer has been also shown that significant interactions, especially long-range interactions between same compartment type, has been established after chromosome fusion during evolutional process72. The abundance of significant interactions across fusion sites is positively associated with the ages of fusion sites72. Together, K-matrix strategy enables visualization of 3D genome organization dynamics induced by chromosomal changes, providing an efficient method to comprehensively understand functional genome architecture that may expand our perspectives on chromosomal evolution.
Methods
Animal use and care
The specific pathogen-free grade mice (SPF) grade mice, including C57BL/6j, DBA/2 and B6D2F1 mice were used in this study. All mice were housed with a temperature of 22 °C, humidity at 30% ~ 40%, and light cycle of 12 h (light) / 12 h (dark). All animal procedures in this study were approved by the Animal Care and Use Committee of Center for Excellence in Molecular Cell Science, Shanghai Institute of Biochemistry and Cell Biology (SIBCB), Chinese Academy of Sciences under the ethical guidelines for the Care and Use of Laboratory Animals of SIBCB.
Brain sample preparation
Whole brain of C57 and chromosome fused strains was dissected and chopped with blade on dish cooled on ice. After cutting up, the brain pastes were collected and suspended with 20 ml 1 × HBSS (with 1%BSA). For crosslinking, 16 ml suspension was separated into two groups, and each 8 mL suspension was crosslinked with 222ul formaldehyde (37%) for 20 min on rotator at room temperature. Then, cross-linking reaction was terminated with 714ul glycine (2.5 M) for 10 min on rotator at room temperature. After termination, suspension was centrifuged under 300 G for 5 min at 4 °C and the cell pellet was washed with cold PBS (pH = 7.5). Brain pastes were suspended with 6 ml tissue lysis buffer (10 mM Tris-HCl pH8.0, 10 mM NaCl, 1% Triton-X100 (Thermo Scientific, 85111), protease inhibitors (Sigma, P8340)) and dissociated with glass tissue grinder homogenizer on ice. The brain homogenate was filtered with 70 um and 40 um filter sequentially to obtained single cell suspension and pelleted by centrifuge. The cell pellet could be stored at -80 °C after quick-freezing by liquid nitrogen, and the following process was followed standard in situ Hi-C process mentioned above.
Hi-C library preparation and data analysis
In situ Hi-C was performed identically to the previous report36. Raw reads were processed by HiC-Pro (v2.11.4)73 to generate valid pairs and contact matrices. Detection of the A/B compartment at various bins was executed by HOMER software (v4.11)74 with runHiCpca.pl command at 100Kb resolution.
Calculation of the PASTIS model
We used 100Kb resolution contact count matrices and a Poisson model (PM2) in PASTIS to simulate the 3D structure of the chromosomes. Default parameters were used. Models were displayed by using either the plotly package in R or pyMOL (v2.5.0a0).
Calculation of the superRec model
We also used 100Kb resolution contact count matrices from HiC-Pro as the input for SuperRec to simulate the 3D structure of the chromosomes. Default parameters were used. Models were displayed by using plotly in R.
Calculation of the PGS model
For calculating PGS models, we used the 100Kb resolution matrix in cool format75 and followed the procedure to produce 1000 models37. Input domain information was calculated by Topdom package76. Default parameters were used. Models were displayed and colored by using plotly in R.
Calculation of the IGM model
For calculating IGM models, we used the 1 Mb resolution matrix in cool format75 and followed the procedure to produce 10 testing models44. Default parameters were used. Models were displayed and colored by using plotly in R.
Convert populational Hi-C data to K-matrices
In a haploid single-cell Hi-C dataset, after enzyme digestion, one digested genome segment can have only one interaction with another segment at most. Based on this principle, we implemented a step-wise selection procedure to convert the populational Hi-C data to K-matrices. The benefit of this is that the populational contact feature or the interaction structure is preserved in the K-matrices, and also some extent of heterogeneity of single cells is also observed through the randomness of the selection. Currently, three methods were coded (deposited at GitHub: https://github.com/DiabloRex/Kmatrixize). The first one is Random selection. In this procedure, selection starts from a randomly selected valid contact pair, then removes all the pairs from the rest of the data which share either segment in the selected contact pair. Repeat this step until reaches the required thresholds. The second one is Max-Edge random selection. Considering each chromosome segment as a vertex, and each valid contact pair (their relationship) as an edge between the two vertices. Count all edges for every point pair. Then, sort all the edge counts decreasingly, and start selection by choosing the edge with the most count. Remove all the edges that share either one of the segments. Repeat this step until reaches the required thresholds. The third one is Max-Point random selection. Similar to the Max-Edge selection, we count how many edges share a vertex (segments). Selection starts by selecting the point with the most edges, and randomly selecting an edge that shares the vertex based on contact frequency. Remove all edges sharing either point of the edge. Repeat this step until reaches the required thresholds. To build the 3D structures, we sampled 1 million reads from total raw reads. Obtained valid pair using nuc_process in populational mode. Then, we tested all three methods to simulate single-cell Hi-C data. 3D models were built by using nuc_dynamics at 100Kb resolution. Max-Point method shows the best results among the tested. So, we used Max-Point for the following analysis.
Calculation of the Si-C model
For calculating Si-C models, we used the K-matrices of O48 and R1n13 with 10 repeats. K-matrices converted to input format of Si-C, then for each repeat, we built 10 models with default parameters at 100Kb resolution, however, some of models cannot be returned. Plot of the Si-C models were done in R with plotly package.
Correlation of Hi-C matrices
Raw and sampled populational data were processed in nuc_process using populational mode (-p). Single-cell data were processed in nuc_process using default single-cell mode. The results of sampled populational data were converted to K-matrices using Max-Point method. All.ncc files from the preceding calculation were converted to cool file using cooler (v0.9.2)75. The correlation of these samples was calculated by hicCorrelate in HiCExplorer (v3.7.2)77.
Building and visualizing genome 3D structure
For each populational Hi-C data, we sampled 1 million reads for 10 rounds, and built 10 models for each round of sampling. A total of 100 models per sample repeat were built. Models are ported to pyMOL (v2.5.0a0) to view their structures. Colorization of models (A/B compartment, gene expression, and chromosome locations) was projected using customized python scripts.
RMSD between different models
For calculating the difference between models, we used the align function embedded in pyMOL to acquire the RMSD values. To calculate RMSD between models of same sample and between models of different samples, customized bash scripts were used to parallelize the calculation.
Centromere location and centromere distance
Due to a lack of sequence knowledge around the centromeres, we cannot know the exact location of the centromeres in the 3D models, so we evaluated the centromere location by using the first segment position of the chromosome string (about +3 M position in each chromosome in mm10 genome). The Euclidean distance between the centromeres is calculated by using their coordinates.
Telomere location and telomere distance
In mice, all chromosomes are acrocentric and telomere located at both end of the chromosome. Here, the telomere refers to the one that located at the end of the chromosome opposite to the centromere end. In the 3D models, telomere location was evaluated by the position of the last segment of the chromosome (about the length of the chromosome) and the Euclidean distance between the telomeres was calculated using these coordinates.
Chromosome distance
Distance between two chromosomes is calculated by averaging the Euclidean distance from each segment in chromosome A to each segment in chromosome B. Percentage changes were calculated by: PCAB = (Distance_RbAB – Distance_WTAB)/ Distance_WTAB.
Layered positional decomposition
To identify which chromosome segments are located at the periphery or center of the nucleus, we employed the boundary function in MATLAB (vR2018b). For each model, we started by identifying the boundary points (segments) of the model (using the default parameter), then recorded those points as layer 1 and removed them. In the next iteration, we assigned the position as layer 2. Such iteration repeated until only a small number of segments remains, and signed those point layer N. Radial position of each segment (Depth) is calculated by: N_Depthi = (Layeri – 1) / (Total Layer Number (N) - 1). Depth ranges from 0 to 1, segments with a depth of near 0 indicate that it locates at the periphery of the nucleus, and a depth of near 1 means it sits at the core of the nucleus.
Segments per cube
A cube with side length of 2 and the segment as center were drew for each segment in the model, and segments per cube for this segment is calculate by counting the total number of segments within the cube. Average segments per cube is the average of all segments per cube within each model. If the model is more compacted, average segments per cube is higher, and vice versa.
RNA-seq data analysis
Raw RNA-seq reads were cleaned using fastp (v0.20.0)78 program, and aligned to mm10 using STAR (v2.7.3a)79. Only unique mapped reads were kept and counted by featureCounts (v2.0.0)80, and FPKM and TPM were calculated by stringtie (v2.0)81,82,83. Genome coverage was counted by bamCoverage (v3.3.1)84. Differential gene expression was carried out using DESeq2 (v1.30.0)85.
DNA fluorescent in situ hybridization (DNA-FISH) and image analysis
Chromosome territories (CTs) location of chrX and chr11 in the nucleus were measured based on DNA-FISH images with ImageJ. DNA-FISH method was followed in the previous research36. Briefly, for haploid ES cells, the cell suspension was fixed with 4% PFA and attached to slides. After penetration with 1% Triton X-100 (in PBS pH=7.5), and digestion with RNase to remove RNA, the sides were co-incubated with chrX (D-1420-050-FI, Metasystem) and chr11 (D-1411-050-OR, Metasystem) paint probe and followed the incubation program, 80 °C for 5 min and hybridization under 37 °C for 16 h. After hybridization, the slides were washed with 0.4 × SSC at 72 °C for 1 min and 2 × SSC at room temperature for 1 min. For brain cells, whole brain was dissected and chopped up. Then the tissue pastes were collected and resuspended in 15 ml corning-tube with 3 ml digestion buffer I (DMEM medium, SLM-220-B, Millipore) supplemented with 1 mg/ml collagenase IV (17104019, Gibco) and 0.1 mg/ml DNase I (11284932001, Sigma). The brain was digested at 37 °C on shaker for 15 min. After digestion, the suspension was centrifuged at 120 g for 5 min and supernatant was discarded. The cell pellet was resuspended with 3 ml 0.25% Trypsin-EDTA (25200072, Gibco) and digested at 37 °C on shaker for 10 min. Cell clumps were pipetted up, and the cell suspension was filtered with 40μm nylon mesh. The single cell suspension was centrifuged and washed with 1 × PBS (A1286301, Gibco) to remove debris and fat. Finally, the cell pellet was resuspended with PBS and dropped and fixed on slides with 4% PFA. Fixed cells were penetrated with 0.03% TritonX-100 (85111, Thermo Scientific) in PBS at room temperature for 15 min and followed by washing with PBS twice. Then, samples were treated with Pepsin Antigen Retrieval Kit (E673007, Sangon Biotech) at 37 °C for 15 min and washed with PBS twice. After that, slides were dehydrated with grade ethanol and dried. Chromosome paint probes (chr1, D-1401-050-FI, Metasystem, chr5, D-1405-050-FI, Metasystem, chr13, D-1413-050-OR, Metasystem, chr17, D-1417-050-OR, Metasystem) were mixed and applied to slides. Samples and probes were denatured together at 80 °C for 5 min and hybridized at 37 °C for 16 hours. The washing steps were similar as mentioned above.
The images were captured with FV3000 and analyzed with FV3000 (Olympus) and ImageJ. The edge of the nucleus and chromosome territories were captured manually, and coordinates of the centroid of chromosome territories and nucleus were calculated with ImageJ. The distance between the centroid of CTs and the centroid of the nucleus was calculated using the Euclidean distance. A similar method was used to measure the distance between mass centers. The perimeter of CTs and the edge of CTs merged with the edge of the nucleus were measured manually with ImageJ, the ratio between which was used as the edge approaching score.
Statistics & reproducibility
All boxplot in the paper were generated in R using ggplot function geom_boxplot with default settings. The center line represents the median value, the lower and upper hinges correspond to the first and third quartiles (the 25th and 75th percentiles), the whisker extends from the hinge to the largest or smallest value at most 1.5 * IQR (interquartile range) of the hinge. The two-sided Student’s t test was used to analyze the significant difference between different groups in all the figures. Number of samples were obtained from exist data sources listed in data availability section.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The datasets generated during the current study are available in the GSA repository: accession number CRA009787. HiC data were obtained from GSA: CRA004756 (single-event Rb fusion cell lines)36, GSA: CRA006835 (engineered telomere-centromere fused cell lines)46, GEO: GSE145978 (naturally occurring Rb mice)35 and GEO: GSE132054 (wild type mice)86, GEO: GSE80280 (single cell dataset and bulk dataset), GEO: GSE111032 (inverted nuclei of rods). All source data are provided with this paper [https://doi.org/10.5281/zenodo.14129270].
Code availability
Program for converting populational data to K-matrix was deposited at GitHub: https://github.com/DiabloRex/Kmatrixize [https://doi.org/10.5281/zenodo.14263925]87.
References
Cremer, T. & Cremer, C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet 2, 292–301 (2001).
Rowley, M. J. & Corces, V. G. Organizational principles of 3D genome architecture. Nat. Rev. Genet 19, 789–800 (2018).
Misteli, T. Beyond the sequence: cellular organization of genome function. Cell 128, 787–800 (2007).
Bickmore, W. A. The spatial organization of the human genome. Annu Rev. Genom. Hum. G 14, 67–84 (2013).
Osborne, C. S. et al. Active genes dynamically colocalize to shared sites of ongoing transcription. Nat. Genet 36, 1065–1071 (2004).
Spilianakis, C. G., Lalioti, M. D., Town, T., Lee, G. R. & Flavell, R. A. Interchromosomal associations between alternatively expressed loci. Nature 435, 637–645 (2005).
Cremer, M. et al. Non-random radial higher-order chromatin arrangements in nuclei of diploid human cells. Chromosome Res. 9, 541–567 (2001).
Kupper, K. et al. Radial chromatin positioning is shaped by local gene density, not by gene expression. Chromosoma 116, 285–306 (2007).
Spector, D. L. The dynamics of chromosome organization and gene regulation. Annu Rev. Biochem 72, 573–608 (2003).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009).
Nora, E. P. et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385 (2012).
Dixon, J. R. et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012).
Rao, S. S. P. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Misteli, T. The self-organizing genome: principles of genome architecture and function. Cell 183, 28–45 (2020).
Sexton, T., Schober, H., Fraser, P. & Gasser, S. M. Gene regulation through nuclear organization. Nat. Struct. Mol. Biol. 14, 1049–1055 (2007).
Lanctot, C., Cheutin, T., Cremer, M., Cavalli, G. & Cremer, T. Dynamic genome architecture in the nuclear space: regulation of gene expression in three dimensions. Nat. Rev. Genet 8, 104–115 (2007).
Bolzer, A. et al. Three-dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes. Plos Biol. 3, 826–842 (2005).
Quinodoz, S. A. et al. Higher-order inter-chromosomal hubs shape 3d genome organization in the nucleus. Cell 174, 744–757 (2018).
Arrastia, M. V. et al. Single-cell measurement of higher-order 3D genome organization with scSPRITE. Nat. Biotechnol. 40, 64–73 (2022).
Fraser, P. & Bickmore, W. Nuclear organization of the genome and the potential for gene regulation. Nature 447, 413–417 (2007).
Finn, E. H. & Misteli, T. Molecular basis and biological function of variability in spatial genome organization. Science 365, eaaw9498 (2019).
Zuin, J. et al. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc. Natl Acad. Sci. USA 111, 996–1001 (2014).
Foti, R. et al. Nuclear architecture organized by rif1 underpins the replication-timing program. Mol. Cell 61, 260–273 (2016).
Guo, Y. et al. CRISPR inversion of ctcf sites alters genome topology and enhancer/promoter function. Cell 162, 900–910 (2015).
Minajigi, A. et al. Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science 349, aab2276 (2015).
Galupa, R. & Heard, E. X-chromosome inactivation: A crossroads between chromosome architecture and gene regulation. Annu. Rev. Genet. 52, 535–566 (2018).
Lupiáñez, DaríoG. et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012–1025 (2015).
Franke, M. et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538, 265–269 (2016).
Zhang, Y. et al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell 148, 908–921 (2012).
Dubois, F., Sidiropoulos, N., Weischenfeldt, J. & Beroukhim, R. Structural variations in cancer and the 3D genome. Nat. Rev. Cancer 22, 533–546 (2022).
Ferguson-Smith, M. A. & Trifonov, V. Mammalian karyotype evolution. Nat. Rev. Genet 8, 950–962 (2007).
Graphodatsky, A. S., Trifonov, V. A. & Stanyon, R. The genome diversity and karyotype evolution of mammals. Mol. Cytogenet 4, 22 (2011).
Klaasen, S. J. et al. Nuclear chromosome locations dictate segregation error frequencies. Nature 607, 604–609 (2022).
Ijdo, J. W., Baldini, A., Ward, D. C., Reeders, S. T. & Wells, R. A. Origin of human chromosome-2 - an ancestral telomere telomere fusion. P Natl Acad. Sci. USA 88, 9051–9055 (1991).
Vara, C. et al. The impact of chromosomal fusions on 3D genome folding and recombination in the germ line. Nat. Commun. 12, 2981 (2021).
Zhang, X. M. et al. Creation of artificial karyotypes in mice reveals robustness of genome organization. Cell Res 32, 1026–1029 (2022).
Hua, N. et al. Producing genome structure populations with the dynamic and automated PGS software. Nat. Protoc. 13, 915–926 (2018).
Nagano, T. et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59 (2013).
Tan, L. Z., Xing, D., Chang, C. H., Li, H. & Xie, S. Three-dimensional genome structures of single diploid human cells. Science 361, 924–928 (2018).
Stevens, T. J. et al. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 544, 59 (2017).
Cremer, T. & Cremer, M. Chromosome territories. Cold Spring Harb. Perspect. Biol. 2, a003889 (2010).
Varoquaux, N., Ay, F., Noble, W. S. & Vert, J. P. A statistical approach for inferring the 3D structure of the genome. Bioinformatics 30, 26–33 (2014).
Zhang, Y. L., Liu, W. W., Lin, Y., Ng, Y. K. & Li, S. C. Large-scale 3D chromatin reconstruction from chromosomal contacts. Bmc Genomics 20, 186 (2019).
Boninsegna, L. et al. Integrative genome modeling platform reveals essentiality of rare contact events in 3D genome organizations. Nat. Methods 19, 938–949 (2022).
Sexton, T. et al. Three-Dimensional Folding and Functional Organization Principles of the Drosophila Genome. Cell 148, 458–472 (2012).
Wang, L. B. et al. A sustainable mouse karyotype created by programmed chromosome fusion. Science 377, 967–975 (2022).
Wang, S. Y. et al. Spatial organization of chromatin domains and compartments in single chromosomes. Science 353, 598–602 (2016).
Falk, M. et al. Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature 570, 395–399 (2019).
Habermann, F. A. et al. Arrangements of macro- and microchromosomes in chicken cells. Chromosome Res 9, 569–584 (2001).
Parada, L. A., Roix, J. J. & Misteli, T. An uncertainty principle in chromosome positioning. Trends Cell Biol. 13, 393–396 (2003).
Khalil, A. et al. Chromosome territories have a highly nonspherical morphology and nonrandom positioning. Chromosome Res. 15, 899–916 (2007).
Nagele, R. G. et al. Chromosomes exhibit preferential positioning in nuclei of quiescent human cells. J. Cell Sci. 112, 525–535 (1999).
Marella, N. V., Bhattacharya, S., Mukherjee, L., Xu, J. & Berezney, R. Cell type specific chromosome territory organization in the interphase nucleus of normal and cancer cells. J. Cell Physiol. 221, 130–138 (2009).
Mayer, R. et al. Common themes and cell type specific variations of higher order chromatin arrangements in the mouse. BMC Cell Biol. 6, 44 (2005).
Parada, L. A., McQueen, P. G. & Misteli, T. Tissue-specific spatial organization of genomes. Genome Biol. 5, R44 (2004).
Kuroda, M. et al. Alteration of chromosome positioning during adipocyte differentiation. J. Cell Sci. 117, 5897–5903 (2004).
Tanabe, H., Habermann, F. A., Solovei, I., Cremer, M. & Cremer, T. Non-random radial arrangements of interphase chromosome territories: evolutionary considerations and functional implications. Mutat. Res 504, 37–45 (2002).
Britton-Davidian, J., Cazaux, B. & Catalan, J. Chromosomal dynamics of nucleolar organizer regions (NORs) in the house mouse: micro-evolutionary insights. Heredity (Edinb.) 108, 68–74 (2012).
Jegu, T., Aeby, E. & Lee, J. T. The X chromosome in space. Nat. Rev. Genet 18, 377–389 (2017).
Nagano, T. et al. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 547, 61–67 (2017).
Boyle, S. et al. The spatial organization of human chromosomes within the nuclei of normal and emerin-mutant cells. Hum. Mol. Genet 10, 211–219 (2001).
Croft, J. A. et al. Differences in the localization and morphology of chromosomes in the human nucleus. J. Cell Biol. 145, 1119–1131 (1999).
Cremer, M. et al. Inheritance of gene density-related higher order chromatin arrangements in normal and tumor cell nuclei. J. Cell Biol. 162, 809–820 (2003).
Naumova, N. et al. Organization of the mitotic chromosome. Science 342, 948–953 (2013).
Meng, L., Wang, C., Shi, Y. & Luo, Q. Si-C is a method for inferring super-resolution intact genome structure from single-cell Hi-C data. Nat. Commun. 12, 4369 (2021).
Ghavi-Helm, Y. et al. Highly rearranged chromosomes reveal uncoupling between genome topology and gene expression. Nat. Genet 51, 1272–1282 (2019).
Tian, W. et al. Single-cell DNA methylation and 3D genome architecture in the human brain. Science 382, eadf5357 (2023).
Liu, H. et al. Single-cell DNA methylome and 3D multi-omic atlas of the adult mouse brain. Nature 624, 366–377 (2023).
Schmitt, A. D. et al. A Compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep. 17, 2042–2059 (2016).
Girelli, G. et al. GPSeq reveals the radial organization of chromatin in the cell nucleus. Nat. Biotechnol. 38, 1184–1193 (2020).
Tjong, H. et al. Population-based 3D genome structure analysis reveals driving forces in spatial genome organization. Proc. Natl Acad. Sci. USA 113, E1663–E1672 (2016).
Yin, Y. et al. Molecular mechanisms and topological consequences of drastic chromosomal rearrangements of muntjac deer. Nat. Commun. 12, 6858 (2021).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Heinz, S. et al. Simple Combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and b cell identities. Mol. Cell 38, 576–589 (2010).
Abdennur, N. & Mirny, L. A. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics 36, 311–316 (2020).
Shin, H. et al. TopDom: an efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res 44, e70 (2016).
Ramirez, F. et al. High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat. Commun. 9, 189 (2018).
Chen, S. F., Zhou, Y. Q., Chen, Y. R. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890 (2018).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, stringtie and ballgown. Nat. Protoc. 11, 1650–1667 (2016).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with stringtie2. Genome Biol. 20, 278 (2019).
Ramirez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–W165 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Vara, C. et al. Three-dimensional genomic structure and cohesin occupancy correlate with transcriptional activity during spermatogenesis. Cell Rep. 28, 352–367 (2019).
Meng, Y. et al. Visualization of chromosomal reorganization induced by heterologous fusions in the mammalian nucleus. Kmatrixize, https://doi.org/10.5281/zenodo.14263925,2024.
Acknowledgements
This work was supported by Genome Tagging Project (GTP) and grants from the National Key R&D Program of China and the National Natural Science Foundation of China (32488101, 2024YFA1800010, 31821004, 32030029, 32293231, and 2020YFA0509000 to J.L.), and partly supported by Shanghai Municipal Science and Technology Major Project (22YS1400900 and 23HC1401000 to J.L.), the Major Special Projects of Basic Research of Shanghai Science and Technology Commission, and China Postdoctoral Science Foundation (2021 M703343 to M.Y., 2021TQ0329 to X.M.Z.).
Author information
Authors and Affiliations
Contributions
J.L., M.Y. and X.M.Z. conceived of the project. X.M.Z., Z.Y. and M.J. performed the experiments. M.Y., X.M.Z. and R.L. analyzed the high-throughput data and interpreted the data with the help from all other authors. J.L. and M.Y. wrote the manuscript. J.L. supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Peng Hua and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yan, M., Zhang, X.M., Yang, Z. et al. Visualization of chromosomal reorganization induced by heterologous fusions in the mammalian nucleus. Nat Commun 16, 1485 (2025). https://doi.org/10.1038/s41467-024-55582-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55582-3
