
Abstract
Accurate gametogenesis requires the establishment of the telomere bouquet, an evolutionarily conserved, 3D chromosomal arrangement. In this spatial configuration, telomeres temporarily aggregate at the nuclear envelope during meiotic prophase, which facilitates chromosome pairing and recombination. The mechanisms governing the assembly of the telomere bouquet remain largely unexplored, primarily due to the challenges in visualizing and manipulating the bouquet. Here, using Schizosaccharomyces pombe as a model system to elucidate telomere bouquet function, we reveal that centromeres, traditionally perceived as playing a passive role in the chromosomal reorganization necessary for bouquet assembly, play a key role in the initiation of telomere bouquet formation. We demonstrate that centromeres are capable to induce telomere mobilization, which is sufficient to trigger the first stages of bouquet assembly and the meiotic transcription program in mitotic cells. This discovery highlights the finely tuned control exerted over long-distance heterochromatic regions and underscores a pivotal step in the mechanism of eukaryotic telomere bouquet formation and meiotic transcriptional rewiring.
Similar content being viewed by others


Introduction
Recent advances in genome mapping have strongly supported the idea that chromosome architecture is actively regulated both spatially and temporally. Chromosomes exhibit distinct spatial organizations, ranging from well-defined chromosome territories at genome-wide scale to finely resolved interaction loops at the nucleosomal level1. Chromosome-wide imaging has revealed a crucial yet enigmatic role for centromeres in chromosome architecture, as they cluster in specific nuclear regions, frequently near the nuclear envelope (NE). However, the functional relevance of this clustering is not fully understood. Notably, certain plant and fungi species limit centromere clustering at a single chromosomal focus, culminating in the Rabl or Rabl-like chromosome conformation. In other species, such as Drosophila melanogaster, centromeres are clustered at multiple foci next to the NE2.
The intricate re-arrangement of chromosome architecture takes center stage during cell differentiation, especially in gametogenesis. Fundamental to this phenomenon are changes in chromosome positioning within the nucleus, which is a prerequisite for the initiation and progression of the meiotic program3. A key event in the 3D chromosomal reorganization during meiosis onset is the formation of the telomere bouquet, which is characterized by the clustering of the telomeres at the NE. The bouquet configuration serves as a crucial facilitator for homologous chromosome pairing and recombination by effectively transmitting to the chromosomes the nuclear forces that support the meiotic processes4. Nevertheless, understanding the transition in chromosome architecture from interphase to meiotic prophase poses a challenge, given its swift nature and difficulties in observing and manipulating it. To unravel these processes, researchers have focused on fission yeast, which has only three chromosomes. This model system enables dynamic tracking of specific loci in chromosomes, centromeres and telomeres throughout mitosis and meiosis, alongside the manipulation of the Rabl conformation and the telomere bouquet.
In fission yeast, the interphase Rabl chromosome configuration largely depends on the kinetochores, which organize the clustering of all three centromeres at a specific focus behind the spindle pole body (SPB)5,6,7,8. The linkage between kinetochores and the SPB is mediated by the LINC (LInker of Nucleoskeleton and Cytoskeleton) complex, a partnership between the SUN- and the KASH-domain proteins9. This kinetochore-LINC interaction is well established in fission yeast as relying on Sad1, the SUN-domain protein10. Additionally, the mitotic centromere-SPB clustering protein Csi1 and the inner nuclear membrane (INM) protein Lem2 support the connection between Sad1 and outer kinetochore proteins (Fig. 1a)11,12. Simultaneously, telomeric regions are positioned opposite the SPB microenvironment at the NE. Telomeres are associated with the INM, which is mediated by the interaction between two telomeric proteins, Taz1 and Rap1, and two INM proteins, Bqt3 and Bqt413; the telomere-INM interactions are supported by Lem2 and other INM proteins like Man114.

a Schematic representation of the centromere-SPB interaction in fission yeast. b State of the interphase centromere (Mis6-GFP)-SPB (Ppc89-CFP) interaction in Rabl and Rabl-deficient (sad1-2 csi1Δ) cells; the nuclear envelope (NE) was visualized by Ish1-mRFP, scale bars, 2 μm). c Flow diagram of the synthetic genetic analysis used to screen for genetic interactors of sad1-2 csi1Δ. d Selected gene interaction data after filtering non-differential phenotypes, between wt (control) and sad1-2 csi1Δ settings (27 out of 2,988). The colony growth rate was analyzed using hierarchical clustering with Euclidean distance and the complete linkage method to identify gene clusters (two-sided p value < 0.001, abs (log2(fold change)) > 2). e Representation of the role of each analyzed chromatin anti-silencing factor in the regulation of centromeric, euchromatic, subtelomeric and telomeric silencing.
Upon meiosis initiation in fission yeast, a chromosomal reorganization occurs, resulting in the formation of the bouquet configuration, where telomeres and centromeres undergo a strategic positional switch15,16. This rearrangement is coordinated by the recruitment of the meiosis-specific proteins Bqt1 and Bqt2 to the telomeres17. These two proteins begin locating at the interphase position of the telomeres, opposite the SPB, facilitated by the interaction with Rap117,18 during meiotic entry. Bqt1/2-Rap1 interaction occurs in coordination with the formation of a meiosis-specific microtubule organization in the cytoplasm next to the telomeres, referred to as the telocentrosome19. The telocentrosome, along with nuclear movements driven by dynein and the meiosis-specific protein Hrs1/Mcp620,21,22, brings telomeres closer to the SPB through the interaction of Bqt1/Bqt2 complex with Sad1, which is enriched beneath the SPB17. This process culminates in a stable telomere-SPB interaction during meiotic prophase. At the end of prophase, Bqt1 and Bqt2 are eliminated, disrupting the interaction between Rap1 and Sad117,23 and, consequently, dismantling the telomere-SPB association and initiating the first meiotic division.
During telomere bouquet assembly, it is known that centromeres disengage from the SPB as the outer kinetochore structure disassembles, aided by vigorous nuclear movements that weaken their interaction5,24. The detachment of centromeres from the SPB is crucial to facilitate the reassembly of the outer kinetochore, which is essential for preparing the kinetochores for the specialized meiotic divisions. Indeed, defects in the dissociation of centromeres from the SPB during meiotic prophase can lead to abnormalities in chromosome segregation5. However, little attention has been given to the role of centromeres in initiating the formation of the telomere bouquet, as it is believed that they do not participate in the initial signals that mobilize the telomeres to form the bouquet.
Here, we show the role played by the Rabl chromosome configuration in regulating the initiation of the meiotic program. By manipulating centromere positioning through a combination of sad1-2 allele and the deletion of csi1, which disrupts the centromere cluster at the SPB8,12,25, we have uncovered the presence of a long-distance signal originating from centromeres to telomeres. We demonstrate that sad1-2 csi1∆ cells initiate bouquet formation and activate the meiotic program in mitotic cells. This can be attributed to the loss of centromere-SPB interaction, as these phenotypes can be rescued by restoring this interaction. Consequently, a signal exists between two specialized chromosome regions, spatially separated in distinct microenvironments inside the nucleus, to coordinate the initiation of bouquet formation. Considering the widespread conservation of centromere clustering during mitosis and telomere bouquet formation during meiosis in eukaryotes, our results provide insights into the fundamental orchestration of bouquet assembly and, more broadly, the control of the initiation of meiotic differentiation.
Results
Anti-silencing factors mitigate growth impairment in cells lacking Rabl chromosome configuration
To identify genes whose absence exacerbates growth impairments upon the loss of the Rabl chromosome configuration, we conducted a genome-wide screening using synthetic genetic array (SGA) technology26, which involved mating haploid Schizosaccharomyces pombe strains containing single-deletion mutations of non-essential genes with strains possessing or lacking the Rabl chromosome configuration, i.e., wild-type (control, Rabl) and sad1-2 csi1∆ (Rabl-deficient) (Fig. 1b, c). By leveraging the thermosensitive allele of sad1, known as sad1-28 (Supplementary Fig. 1a), at a semi-permissive temperature of 32 °C, and depleting Csi112 (Fig. 1a), we were able to induce declustering of all three centromeres from the SPB during mitotic interphase (Fig. 1b and Supplementary Fig. 1b), allowing comprehensive exploration of this scenario without severely compromising cellular growth25. The subsequent examination of mutants derived from SGAs involved the precise measurement of colony size as a proxy for the organism’s fitness (Fig. 1c). This allowed us to assess the growth impairment in individual mutants and the corresponding triple mutants, facilitating a quantitative evaluation of genetic interactions. Our analysis examined 2,988 genetic interactions based on the colony growth assessment. By applying a specific threshold (abs (log2(fold-change))> 2) to isolate robust genetic interactions, we pinpointed 27 deletion mutants with a clearly discernible synthetic growth defect phenotype when the normal Rabl chromosome configuration was absent (p-value < 0.001) (Fig. 1d). Notably, key factors such as the microtubule plus end polymerase Alp14 and the DASH complex proteins Spc19 and Dad2 were found among mutants showing strong genetic interactions in the screens (Fig. 1d). This suggests a critical role in countering the high rate of chromosome loss due to disrupted centromere clustering to the SPB27, which likely affects proper chromosome segregation.
Unexpectedly, our analysis identified several genes whose functions are mainly linked to anti-silencing processes. For example, Swd2, Swd3, and Spf1 are subunits of the Set1/COMPASS (Set1/C) complex, which mediates H3K4 methylation in euchromatin28,29,30 and counteracts heterochromatin spreading in gene-rich areas31,32 (Fig. 1d, e). We also found interaction with the histone acetyltransferase Mst2, which acetylates histone H3 at K14 and the non-histone substrate Brl1, a component of the histone H2B ubiquitin ligase complex. These modifications are also associated with active chromatin and contribute to maintaining euchromatin-heterochromatin boundaries33,34,35,36,37 (Fig. 1d, e).
To further support the idea that the anti-silencing factors Set1/C complex and Mst2 play a crucial role when the Rabl chromosome configuration is compromised, we assessed in more detail the impact resulting from their loss in the sad1-2 csi1∆ background. We carried out spore dissection analysis (Supplementary Fig. 1c) that included the single sad1-2 allele and the sole csi1 deletion. We examined the roles of the Set1/C complex subunits, Swd3 and Spf1, and also included Set1, which is the catalytic member of this complex38. We observed a robust growth defect when sad1-2 csi1∆ cells were combined with deletions of swd3 or set1 (Supplementary Fig. 1d and 1e). By contrast, sad1-2 csi1∆ spf1∆ spores did not exhibit noticeable growth impairments (Supplementary Fig. 1f). Similarly, and given that the role of Mst2 in maintaining heterochromatin boundaries at centromeres and euchromatin regions34 synergizes with the putative histone demethylase Epe134,39,40,41,42 (Fig. 1e), we also incorporated strains in which epe1 is deleted into our analysis. We observed that the absence of mst2 or epe1 resulted in significant and severe growth impairments in sad1-2 csi1∆ cells (Supplementary Figs. 1g and 1h).
The above analyses suggest that cells without proper Rabl chromosome configuration display chromatin silencing defects which must be alleviated by anti-silencing factors such as the Set1/C complex, Mst2 or Epe1.
The meiotic transcriptional program is initiated in Rabl-deficient mitotic cells
Considering our observations regarding the potential influence of altered centromere positioning at the SPB on the transcription regulation, we embarked on a comprehensive exploration of gene expression levels by comparing cells with and without proper Rabl chromosome conformation by transcriptome analysis using RNA sequencing (RNA-seq). Our results showed that the majority of genes that exhibited differential transcriptional levels are upregulated in the sad1-2 csi1∆ mutant (326 upregulated genes versus 12 downregulated genes, from a total of 6642 transcripts; Fig. 2a and b and Supplementary Figs. 2a and b). Intriguingly, our RNA-seq analysis revealed a profound increase in transcripts enriched for Gene Ontology (GO) terms associated with meiotic genes (Fig. 2c–e, and Supplementary Fig. 2c)43. Real time-quantitative PCR (RT-qPCR) analysis further validated the upregulation of genes with crucial roles in the initiation of the meiotic program and early meiosis. These include genes encoding the DNA-binding transcription factor Ste11, critical regulator of sexual development44, the RNA-binding protein Mei245, the meiosis-specific lncRNA sme2 locus46, the meiotic recombination protein Rec1047,48 and the telomere bouquet protein Bqt217,49 (Fig. 2f).

a Volcano plot depicting RNA-seq data from sad1-2 csi1Δ versus control cells. Genes significantly up- (red) or downregulated (blue) are highlighted (log2(fold change) > 0.5 or < −0.5 with two-sided p value < 0.05 by the Wald test, as implemented within the DESeq2 framework). b M (log ratio) and A (mean average) (MA) plot depicting RNA-seq data. c Venn diagram depicting the comparison between RNA-seq data from sad1-2 csi1Δ versus control cells (log2(fold change) > 0.3 or < −0.3 with two-sided p value < 0.05 by the Wald test, as implemented within the DESeq2 framework) and RNA-seq data from meiotic cells versus vegetative cells (from43). The p value from one-sided binomial test analysis is shown. d Table of the most upregulated meiotic-specific genes from previous RNA-seq data. Two-sided p values by Wald test are shown. e Plot of reads-per-million (RPM)-normalized read density at the indicated loci. Individual signals for each biological replicate are shown in a lighter color, and the average of these is shown in a darker color. f RT-qPCR analysis of relevant meiotic-specific genes showing RNA levels relative to wt after normalization to act1+. Data are represented as mean ± SEM from 3 independent experiments; the numbers above the columns represent p values from two-tailed Student’s t-test analysis. Source data are provided as a Source Data file.
We speculated that the activation of the meiotic program during vegetative cell growth might have implications for meiotic entry, as pre-activation of the meiotic signal could potentially disrupt the transition into meiosis50. To address this possibility, we assessed meiotic entry and efficiency by monitoring the number of meiocytes produced over a period of 48 hours after meiosis induction. This revealed a significant reduction in the efficiency of meiocyte formation in Rabl-deficient cells (Supplementary Fig. 2d).
All these data revealed that the disconnection of centromeres from the SPB serves as an unexpected trigger for the activation of the meiotic transcriptional program.
Centromere positioning influences transcription at the end of the chromosome
To gain a deeper understanding of the repercussions of disrupting the Rabl chromosome configuration, we investigated whether the elimination of centromere-SPB interaction leads to significant changes in the transcriptional landscape, particularly affecting specific regions that lose their connection with the LINC, such as centromeric and pericentromeric regions. A topological enrichment analysis from our RNA-seq data considering the genomic context of genes with altered transcriptional levels and their relationship to the overall chromosomal transcription activity showed that these dysregulated genes were randomly distributed across the chromosomes (Supplementary Figs. 3a, b). Indeed, we did not observe a notable enrichment of genes in proximity to the centromeres—the chromosome regions theoretically most affected in sad1-2 csi1∆ cells (Supplementary Figs. 3a, b). To further assess the status of centromere silencing, we performed RT-qPCR analysis targeting various loci within the conserved pericentromeric heterochromatic regions and the centromere core51. While the centromere core (cnt1/3) exhibited significantly increased transcription compared with control cells, several locations in the pericentromeric regions that are highly regulated by heterochromatin factors52 displayed no major changes (Supplementary Figs. 3c, d). These findings indicate that centromere disconnection from the SPB does not drastically affect centromere transcriptional regulation, which is congruent to previous observations in lem2∆ settings11.
Next, we aimed to investigate the transcriptional state of another crucial specialized chromosome region, in addition to centromeres, which is also finely regulated by silencing factors and tightly connected to the NE—the telomeres. To study possible telomeric regulation defects in Rabl-deficient cells, we analyzed by RT-qPCR the transcription of the telomere-associated sequences (TAS) and the endogenous subtelomeric non-coding RNAs (TERRA, telomeric repeat-containing non-coding RNA)53. These experiments revealed no significant changes in the transcription levels of TAS1, TAS2, and TERRA loci, with only a slight increase in TAS3 transcription levels observed in Rabl-deficient cells compared to wt settings (Supplementary 3e and 3f). This suggests that the telomeric regions, where silencing is preserved by shelterin complex and heterochromatin mechanisms28,54,55,56, remain relatively unaffected when centromeres are disconnected from the SPB.
Conversely, we uncovered substantial defects in transcriptional levels within subtelomeric regions, at both homologous (SH) and unique (ST) chromatin domains53. Specifically, we identified noteworthy differences in the transcriptional levels of the proximal subtelomeric regions of chromosomes I and II, which were silenced by heterochromatic pathways28,55 (Fig. 3a, b). Particularly notable was the higher disparity in expression levels between cells with and without proper Rabl configuration at the distal subtelomeric regions of both chromosomes I and II compared with those in the absence of the sole histone lysine H3K36 methyltransferase Set2, which regulates distal subtelomeric transcription57, with the most significant changes observed in chromosome II subtelomeric regions (Figs. 3a, c). These observations align with our RNA-seq data, where these genes exhibited clear upregulation (Fig. 3d). Also, we found that sad1-2 or csi1Δ single mutants display only partial subtelomeric silencing defects at both proximal and distal regions (Supplementary Fig. 3g). This unexpected finding highlights that the absence of proper centromere positioning has a pronounced effect on the silencing of subtelomeric regions, especially at the knob regions (Fig. 3a, c), as occurs during meiosis57, which are characterized by highly condensed chromatin regulated by Set2 and the centromeric adapter, shugoshin (Sgo2)57,58. Accordingly, Sgo2 tagged with GFP exhibits a slight tendency to lose its connection with some telomeres from chromosomes I and II in Rabl-deficient cells57 (Supplementary Fig. 4a and b). By contrast, chromosome III, which lacks specific heterochromatin and knob features, exhibited no significant transcriptional differences in telomeric and subtelomeric regions when centromere positioning at the SPB was altered (Supplementary Fig. 4c and d). These results indicate an unexpected regulatory role for the centromere-SPB connection in subtelomeric transcription and highlight transcriptional similarities with the meiotic program, during which centromeres are naturally disengaged from the SPB upon meiotic entry.

a S. pombe subtelomeric and telomeric regions of chromosomes I and II, showing the shelterin domain (pink triangle), heterochromatic domain (purple semicircle), and knob domain (light green semicircle). The locations of the primers used are shown with red lines. b RT-qPCR analysis of proximal subtelomeric regions of chromosomes I and II showing RNA levels relative to wt after normalization to act1+. tlh1+ and tlh2+ are located on the left and right arms of chromosomes I and II, respectively, but share 100% identity. c RT-qPCR analysis in distal subtelomeric regions of chromosomes I and II, as in (b). For all quantitative experiments, data are represented as mean ± SEM from three independent experiments in all cases, except for SPAC869.04, pfl9, ght7, subtel2R-51.8 kb, and gal1, which were conducted with four independent experiments. The numbers above the brackets represent p values from two-tailed Student’s t-test analysis. d Plot of reads-per-million (RPM)-normalized read density at the indicated loci. Individual signals for each biological replicate are shown in a lighter color, and the average of these is shown in a darker color. Source data are provided as a Source Data file.
Telomeres are declustered but still associated with the nuclear envelope upon alteration of centromere positioning
The prominent silencing defect at chromosomal ends in Rabl-deficient cells prompted us to explore the telomeric dynamics by pointing to a potential link between centromere repositioning and changes in telomere location within the nucleus, akin to what occurs upon meiosis induction. We used endogenous GFP-tagging of the telomeric protein Taz1, along with the nuclear membrane marker Ish1-mRFP. In wt cells, telomeres typically appeared as 1-3 foci, with 2 foci being the most common phenotype, as previously described13,15 (Fig. 4a, b). Remarkably, sad1-2 csi1∆ cells exhibited a higher number of telomere foci, ranging from 1 to more than 4 (Fig. 4a, b). This increase in the number of foci was attributed to a significant declustering of telomeres, ruling out any changes in Taz1 protein levels as a cause (Supplementary Fig. 5a–c). To confirm that telomere declustering is a direct consequence of the loss of centromere-SPB interaction, we examined the positions of centromeres (using Mis6-GFP) and telomeres (Taz1-mCherry) within the same cell. We observed a tight correlation between the extent of centromere declustering and the number of telomere foci, confirming that only cells with complete centromere declustering from the SPB (in sad1-2 csi1∆ conditions) displayed 3- > 4 telomere foci (Supplementary Figs. 5d and e). These findings support a robust repositioning of telomeres that arises from the manipulation of centromere-SPB interactions.

a Representative pictures of two-color live-cell imaging of control, sad1-2, csi1Δ and sad1-2 csi1Δ strains during interphase. The telomeric protein Taz1 is visualized in green (Taz1-GFP), while the NE is shown in magenta (Ish1-mRFP). b Quantification of telomere foci in the indicated strains. c Representative pictures of two-color live-cell imaging as in (a). d Quantification of cells with number of telomere foci in the indicated strains. e Zone designation (I-III) and distribution of telomeres within 15 confocal planes with illustrative pictures (scale bars, 2 μm). f One z plane of live-cell imaging of control, sad1-2 csi1Δ and lem2Δ strains during interphase. g Quantification of telomeres (Taz1-GFP) distribution relative to the NE, showing the percentage of telomeres for each nuclear zone. Data in (b, d and g) were collected from three independent experiments, and the one-sided p values from the χ² test analysis are shown above the brackets. Scale bars, 5 μm; 2 μm for magnified nucleus.
We also examined other mutants where an increase in the number of Taz1 foci was previously described, including mutants of heterochromatin factors such as Dcr1 and Ago159, and the INM protein Lem211. Notably, at a comparable stage in the cell cycle, telomere declustering was more pronounced in sad1-2 csi1∆ cells than in the other mutants analyzed (Fig. 4c, d; Supplementary Figs. 5f and 5g).
The telomere declustering shown by Rabl-deficient cells could indicate a detachment of telomeres from the NE, analogous to what happens in mutants like lem2∆11. To investigate this hypothesis, we analyzed the positioning of telomeres in the nucleus with respect to the NE by measuring the distance between the Taz1-GFP signal and the Ish1-mRFP envelope marker (Fig. 4e). Unlike in lem2∆ mutant, we discovered that the increase in the number of telomere foci did not consistently correlate with telomere detachment from the NE in sad1-2 csi1Δ cells (Fig. 4f and g). Consequently, the crucial distinction in telomere mobilization along the NE observed when manipulating centromere positioning unveiled a qualitatively different phenotype from previous observations that is characterized by the highest penetrance of telomere declustering yet steadfast telomere-NE associations, as demonstrated during the first stages of the meiotic cycle.
Forcing centromere clustering at the nuclear envelope can reinstate proper telomere clustering and silence subtelomeric and meiotic genes
A potential explanation for the results described above might be topological constraints. Both, Sad1 and Csi1 are present in wt cells at the NE8,10,12. Thus, their mutation or loss might directly affect telomere clustering. To test this possibility, we investigated the consequences of artificially tethering centromeres to the SPB employing fusion proteins with GFP and the GFP-binding protein (GBP) to recruit GFP-tagged proteins60 for centromere clustering at the NE (Fig. 5a). Previously, this approach was used successfully to tether the outer kinetochore component Ndc80 to NE-bound Sad1-225. We verified the functionality of this construct by assessing cellular growth under microtubule depolymerizing drug conditions, where sad1-2 csi1∆ cells exhibited a hypersensitive response25(Supplementary Fig. 1b). The sad1-2-GBP csi1∆ cells harboring ndc80-GFP subsequently demonstrated the restoration of cellular viability (Fig. 5b), consistent with the use of this strategy in previous genetic backgrounds25. Using this strategy, we observed that the number of clustered telomeres was comparable as in wt cells, with the majority of cells displaying 2-3 telomere foci (Fig. 5c and 5d; Supplementary Fig. 6a). In line with these results, RT-qPCR experiments comparing Rabl-deficient cells with and without forced centromere clustering at the SPB revealed that the transcriptional levels of proximal and distal subtelomeric regions in chromosomes I and II restored to wt settings in most of analyzed loci (Fig. 5e, f and g). More importantly, the intervention of this GBP-GFP system also restored the expression of meiotic genes such as ste11, mei2, sme2, rec10, and bqt2 to control levels in sad1-2 csi1∆ settings (Fig. 5h). In summary, our findings provide strong evidence that the relocation of telomeres along the NE, the subtelomeric silencing defects, and the initiation of the meiotic transcriptional program are indeed linked to the loss of normal centromere-SPB associations.

a GFP-GBP system used to force the interaction between Sad1-2 and Ndc80. b Serial dilutions (5-fold) of log-phase cultures of the indicated strains spotted onto YE4S plates containing MBC or TBZ and incubated at 32 °C for 48 h. c Outer kinetochore (Ndc80-GFP), telomeres (Taz1-mCherry), and the SPB (Cut12-CFP). Scale bars, 5 μm; 2 μm for magnified nucleus. d Data were collected from three independent experiments, and the one-sided p values from the χ² test analysis are shown above the brackets. e–h RT-qPCR analysis. Data are represented as mean ± SEM from three independent experiments; p value from two-tailed Student’s t-test analysis is shown above the brackets. Source data are provided as a Source Data file.
Telomeres acquire the capability to host the bouquet proteins Bqt1 and Bqt2 and induce the telocentrosome formation when centromere positioning is modified
Dissociation of centromeres from the SPB, as we observed, particularly affects the chromosome ends, leading to transcriptional changes and the mobilization of telomeres along the NE while maintaining their strong envelope association (Figs. 2, 3 and 4). Typically, telomeres are intricately tethered to the NE through the interaction between the INM Bqt3-Bqt4 complex and the telomeric Rap1-Taz1 complex13,61. The best-studied event involving telomere mobilization is the formation of the telomere bouquet, a phenomenon driven by the meiotic prophase-specific proteins Bqt1 and Bqt217.
To investigate whether centromere declustering alone is sufficient to prepare telomeres for bouquet formation in haploid cells, we ectopically expressed bqt1-GFP and bqt2 under the control of the nmt1 promoter in cells with and without normal centromere positioning. In Rabl settings, ectopic expression of bqt1 and bqt2 causes co-localization of Bqt1-GFP with the SPB in most of the cases and only around 20% of cells display additional Bqt1-GFP foci at the telomeric regions (Fig. 6a–c). This is because in mitotic cells the affinity of Bqt1 is higher by the SPB (via the direct interaction with the population of Sad1 associated to the SPB) than with the telomeres (via interaction with Rap1)13,17,18. By contrast, we observed a notable increase in the presence of Bqt1-GFP at telomeres in Rabl-deficient cells, with roughly 85% of cells exhibiting the recruitment of Bqt1-GFP protein (Fig. 6b, c). This event suggests that telomeres display an enhanced affinity for recruiting Bqt proteins in the absence of centromere-SPB association.

a-b Bqt1 and Bqt2 were ectopically expressed in haploid mitotic cells and localization of Taz1-mCherry, Bqt1-GFP, and Ppc89-CFP was examined along with key telocentrosome components. Scale bars: 2 μm. c Quantification of phenotypes observed in (a-b), 150 cells per genotype from three independent experiments were analyzed. P value derived from two-sided Fisher’s exact test is shown. d sad1-2 csi1Δ cell where the telomere is stably associated with the SPB is shown. e Quantification of the phenotype observed in (d); 150 cells per genotype from three independent experiments were scored and mean value +/- SEM shown, p value from two-tailed Student’s t-test analysis. f-g SGA experiment as in Fig. 1d. Colony growth rate was analyzed using hierarchical clustering with Euclidean distance and the complete linkage method to identify gene clusters (two-sided p value < 0.001).
The foregoing results suggest that centromere declustering facilitates the recruitment of telomere bouquet proteins to the telomeres. However, a key question is whether these cells can form the telocentrosome, the protein complex required for telomere migration to the SPB upon meiosis induction19. The telocentrosome comprises an enrichment of the LINC complex at the telomere environment and the recruitment of several protein complexes involved in microtubule formation and organization, including the microtubule-organizing center formed by Mto1, the gamma-tubulin complex, and dynein19. To explore the telocentrosome formation in the absence of normal centromere-SPB interaction, we examined the behavior of key telocentrosome components: the LINC complex (Sad1 and Kms1), the gamma-tubulin component Alp4, the dynein heavy chain Dhc1, and the gamma-tubulin complex linker Mto119. We found that both the LINC complex, Alp4 and Mto1 were recruited to the telomere environment specifically in Rabl-deficient cells—a phenotype not observed in control strains (Fig. 6a–c). This indicates that manipulating centromere positioning alone can enable telomeres to recruit bouquet proteins and initiate at least partial telocentrosome formation, even during mitosis in haploid cells.
Additionally, a more detailed analysis revealed that some telomeres harboring Bqt1 were even able to reach the SPB and remain stably associated there (Fig. 6d, e; Supplementary movie 1). Nonetheless, we did not observe complete formation of the telomere bouquet, where all three telomeres associate with the SPB in mitosis. The most likely explanation is the absence of the characteristic nuclear movements driven by dynein that occur during meiotic prophase and are essential for bouquet formation in meiosis. Indeed, we did not detect Dhc1 in mutant conditions, suggesting the absence of these motor proteins under our experimental conditions.
Our previous observations indicate that upon the absence of centromere-SPB interaction, mitotic haploid cells can induce the telomere bouquet formation with even some telomeres reaching the SPB. These observations together with the fact that telomeres present a higher declustering but still being associated to the NE might indicate that the telomere environment displays a greater propensity to migrate along the NE (Fig. 4). Therefore, we investigated whether changes occur in the telomeric protein composition when centromeres dissociate from the SPB, focusing on Rap1. Rap1 plays a crucial role in the transition from telomere-NE attachment, facilitated by Bqt3 and Bqt4 proteins, to the orchestrated recruitment of Bqt1 and Bqt2 during meiotic entry13,18,61. In comparison to wt settings, we observed that although Rap1 is normally positioned at the telomeres displaying similar numbers of foci, intensity, and localization to the NE, there was a slight reduction in total Rap1 protein levels in Rabl-deficient cells (Supplementary Figs. 6b–f). This reduction may affect NE proteins, leading to a more relaxed interaction between telomeres and the NE when centromeres are not interacting with the SPB. Ongoing studies in the lab are aimed at exploring this hypothesis. Consistent with this result, subtelomeric regions approximately 50–100 kb from chromosome ends exhibited significant changes in proximity to the NE during interphase, subtly increasing their distance (Supplementary Fig. 6g and h).
Collectively, these findings suggest that alterations in centromere positioning act as a prelude, preparing chromosome ends for the moving from their mitotic location to the SPB along the NE and thereby facilitating the formation of the telomere bouquet.
The loss of kms1 improves mitotic cell growth in Rabl-deficient mutants
Our previous results show that total declustering of the centromeres from the SPB triggers chromosomal mobilization to form the telomere bouquet and activates a crucial group of meiotic genes. Presumably, cells in which the telomere bouquet is induced experience growth arrest, which may be one of the primary reasons for the growth defects observed in sad1-2 csi1∆ cells. If this hypothesis is correct, some growth defects could be alleviated by preventing the telomeres from reaching the SPB. To investigate this, we conducted a second SGA screening using a similar strategy to that in Fig. 1 but focusing on identifying suppressors of growth defects. The goal was to find mutants that improve growth of sad1-2 csi1∆ cells. To enhance the penetrance of the growth defects in Rabl-deficient cells and facilitate identification of relevant mutants, we performed the SGA on rich media containing TBZ (20 μM)(see Supplementary Fig. 1b). We identified only six mutants that allowed the sad1-2 csi1∆ cells to improve growth (Fig. 6f and g). Four of these six genes showed particularly interesting suppression effects. Ash2, a regulatory subunit of the Set1/C complex62, confirmed the link between gene expression regulation mediated by the Set1/C complex and the growth defects observed in Rabl-deficient cells. Klp6, a member of the Kinesin-8 family, whose deletion hyperstabilizes microtubules63, may improve SPB-centromere interactions. Interestingly, two of the six identified genes encode nuclear membrane proteins: the nucleoporin Nup40 and the KASH-domain protein Kms1. Given these findings, we decided to further explore the effect of deleting kms1, as it is a crucial component of the LINC complex in meiosis and telocentrosome19,64. We confirmed that the sad1-2 csi1∆ kms1∆ mutant restored control-like behavior in terms of telomere migration to the SPB (Fig. 6e), suggesting that the growth defects in sad1-2 csi1∆ cells are, at least partially, due to telomere bouquet induction. Taken together, these findings support the hypothesis that loss of the centromere-SPB interaction triggers the initiation of differentiation.
The removal of the outer kinetochore occurs prior to telomere mobilization and is necessary for bouquet formation during meiotic entry
Our previous findings imply that the initial signal for bouquet formation and the onset of the meiotic program may result from changes in centromere positioning even during mitotic cell division. While it is understood that centromere declustering is necessary for proper bouquet formation, centromere reassembly, and meiotic progression5,65,66, the intricate choreography of these events and the precise timing of bouquet assembly remain unexplored. Previously, it has been observed that the convergence of centromeres and telomeres into the same microenvironment is essential for transmitting heterochromatin factors between them24. In this context, using inner centromere markers such as Mis6, we confirmed the convergence between centromeres and telomeres near the SPB. However, we observed that the arrival of telomeres at the SPB is preceded by the declustering of some centromeres in meiotic cells (Supplementary Fig. 7a–c), which require the loss of the outer kinetochore5. To gain further insight into the natural process of bouquet formation, we monitored this process in diploid strains expressing Taz1-mCherry and Ndc80-GFP, a highly conserved component of the outer kinetochore. Removal of Ndc80 and Nuf2 from the kinetochores is essential for disengaging centromeres from their association with the SPB during mitosis and meiosis5,25. We observed that a decrease in the Ndc80-GFP signal preceded the mobilization of telomeres toward centromere clustering (Fig. 7a, b, Supplementary movie 2). Crucially, the complete disappearance of the Ndc80-GFP signal was a prerequisite for all telomeres to complete the bouquet formation, marking the inception of the vigorous nuclear movements, characteristic of meiotic prophase (Fig. 7a, c, Supplementary movie 2). To confirm the necessity of outer kinetochore loss for inducing bouquet formation, we artificially maintained the Ndc80 protein at the SPB upon meiosis induction in diploid strains using the GBP system, as shown in Fig. 5. We observed that these cells were unable to form the bouquet at a similar time as seen in Fig. 7a, b. We compared the behavior of a population of diploid cells with and without Sad1-GBP and found that forcing the interaction of Ndc80 with the SPB prevented diploid cells from progressing in meiosis, even after 12 hours of induction (Fig. 7d and f). In summary, our findings suggest that during the natural induction of the meiotic program, the initial signal to form the telomere bouquet originates from the centromeres, consistent with our observations that the disconnection of centromeres from the SPB may serve as a necessary and sufficient trigger for bouquet formation, even during vegetative growth.

a-b Frames from time-lapse experiment of meiotic diploid cells expressing endogenously tagged Ndc80-GFP and Taz1-mCherry. c Quantification of Ndc80-GFP intensity throughout the experiment as depicted in panels (a) and (b), with each color representing the behavior of 9 independent cells. d Meiotic diploid cells were analyzed as in (a-b), but with one copy of Sad1 endogenously tagged with GBP. e-f Exponentially growing diploid cells were induced to undergo meiosis. The percentage of cells with the indicated number of nuclei per cell is shown for 200 cells at each time point across three independent experiments. g Frames from a time-lapse experiment of meiotic diploid cells with endogenously tagged Cdc2-GFP and Taz1-mCherry. Yellow arrows indicate early and late co-localization between telomeres and Cdc2. h Quantification of Cdc2-GFP intensity throughout the experiment as depicted in panel (g), with each color representing the behavior of 7 independent cells. Scale bars, 5 μm.
CDK1 mediates long-distance communication between centromeres and telomeres to initiate meiotic differentiation
Two major, non-mutually exclusive hypotheses may explain the long-distance communication between centromere and telomeres: i) a biophysical mechanism, as the loss of tension from the centromere-SPB connection might impact the connection of telomeres with the NE; or ii) a chemical signal traveling from centromeres to telomeres, potentially triggered by centromere dissociation from the SPB. To distinguish between these hypotheses, we first investigated whether the detachment of a specific centromere from the SPB affects the expression of subtelomeric regions on the same chromosome, or alternatively, impacts telomeric regions on both chromosomes. To explore this, we forced the interaction of centromere 2 (cen2) with the SPB via GFP-tagging and the GBP nanobody system, as shown in Fig. 5. Using this approach, we found that Cen2-GFP recruitment restored not only gene expression in the subtelomeric regions of the same chromosome but also in the distal subtelomeric region of chromosome I and in meiotic genes (Supplementary Fig. 8). These results suggest that communication between centromeres and telomeres might involve a chemical signal released from the centromere to the telomeres.
A few molecules are shared between telomeres and centromeres in fission yeast, two of the most specialized regions of chromosomes. One of these molecules is Cdc2/Cdk1, which has been observed at the telomeres during bouquet formation in meiotic prophase as well as the centromeres during late meiotic prophase67,68. However, the behavior of Cdc2 during the transition from mitosis to meiosis, specifically upon the induction of telomere bouquet formation, has not been analyzed so far. To investigate this, we tracked the behavior of Cdc2-GFP and confirmed its localization at the telomeres during the horsetail stage (yellow arrows in Fig. 7g) and at the centromeres approximately one hour after bouquet formation began (Fig. 7g)68. Interestingly, we observed a previously uncharacterized phenotype: an increase in the nucleoplasmic signal of Cdc2 during the initial stages of bouquet formation, as telomeres begin migrating toward the SPB (Fig. 7g, h). Additionally, some signals appeared to localize with the telomeres just before bouquet formation (yellow arrow, -3 min in Fig. 7g). These observations led us to explore Cdc2 in mitotic cells upon the disruption of the proper Rabl chromosome configuration.
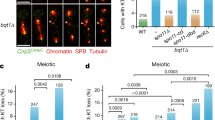
In late G2, Cdc2 displays a nucleoplasmic signal and localizes to the SPB68. However, in Rabl-deficient cells, we found that some telomeres showed a clear co-localizing signal with Cdc2 (Fig. 8a, b). These data suggest that the loss of centromere-SPB interaction may promote the recycling of Cdc2 to the telomeres. To investigate whether this correlation might be involved in mediating communication between centromeres and telomeres, we used the Cdc2.RL (cdc2.F84GY15F) allele, a Cdc2-GBP fusion protein that remains unaffected by Mik1 or Wee1 inhibition and is responsive to ATP analogs69. Activation of Cdc2.RL is achieved by removing the analog in strains where the chimeric kinase was directed to telomeres via Taz1-GFP (Fig. 8c). Using this approach, we observed that activation of Cdc2 at the telomeres triggered a substantial declustering of telomeres, which remained associated with the NE (Fig. 8d–f). These findings indicate that the accumulation of Cdc2 induces telomere dissociation, recapitulating the telomere declustering seen in Rabl-deficient cells (Fig. 4).

a Images of haploid cells during vegetative growth, showing endogenous tagging of Taz1 (telomeres), Cdc2/Cdk1, and Ppc89 (SPB) with mCherry, GFP, and CFP, respectively. The yellow arrow indicates colocalization between telomeres and Cdc2/Cdk1. b Quantification of the phenotype shown in (a), based on analysis of 201 control cells and 294 mutant cells in three independent repetitions. Data are presented as mean values +/- SEM. p value from two-tailed Student’s t-test analysis is shown above the column. c Verification of efficient recruitment of Cdc2.F84GY15F-GBP to the telomeres. Telomeres were visualized via GFP-tagged Taz1. d Vegetative mitotic haploid cells carrying the cdc2.F84GY15F-GBP allele under the control of the nmt81 promoter were grown without thiamine for 15 hours. The ATP analog (40 µM 3MB-PP1) was either removed (ON) or maintained (OFF) one hour before imaging. e–f Quantification of telomere foci and their localization relative to the NE, as in Fig. 4b and g. g–h A similar strategy to that in (d) was used to direct the Cdc2 chimera to the SPB via Sad1-2-GFP in csi1∆ cells, with endogenous tagging of mis6-mCherry or taz1-mCherry to visualize and quantify centromere and telomere behavior, respectively. Controls in (g) and (h) represent wild-type cells carrying sad1-GFP mis6-mCherry and sad1-GFP taz1-mCherry, respectively. Scale bars: 5 μm (full images) and 2 μm (single-nucleus windows). One-sided p value from χ² test analysis is indicated above the brackets.
To further explore the potential role of Cdc2 in mediating centromere-telomere connections, we attempted to sequester Cdc2 at the SPB in Rabl-deficient cells using a similar approach. We engineered an interaction between Cdc2 and the SPB by endogenously GFP-tagging Sad1-2. Under these conditions, we observed that both centromere and telomere clustering were restored to control (Rabl) conditions (Fig. 8g, h). This strongly suggests that long-distance communication from centromeres to telomeres, necessary to initiate telomere bouquet formation, might be mediated by the trafficking of Cdc2 molecules from centromeres to telomeres within the nucleoplasmic environment.
Altogether, these findings reveal an unexpected communication between two distinct specialized regions of the chromosomes, enabling a precise transition of the 3D chromosome conformation. This communication allows centromeres and telomeres to interchange positions, initiating the differentiation program.
Discussion
Tethering telomeres to the nuclear envelope is crucial for preserving genome integrity during vegetative growth. During meiosis, they undergo a significant change in localization, assembling into the telomere bouquet. The formation of the meiotic bouquet is a prerequisite for numerous meiotic processes, making it essential for the faithful progression of gametogenesis4. Centromeres were traditionally perceived as playing a passive role in telomere bouquet formation, and the modification of telomeres to form a bouquet was thought to be independent of the centromeric regions. Our study challenges these traditional views by revealing that centromeres play an active and pivotal role, likely serving as the initial trigger for bouquet formation and the initiation of the meiotic program. We have unveiled significant consequences of centromere dissociation from the NE during interphase in S. pombe. Upon removal of the proper Rabl chromosome conformation, cells exhibited an attempt to enter meiosis, as evidenced by the pre-activation of meiotic program genes such as ste11 and mei2, the long non-coding RNA sme2, rec10, and the telomere bouquet gene bqt2 (Fig. 2), increased transcription of subtelomeric genes (Fig. 3), and telomere declustering along the NE (Fig. 4). Additionally, we observed a proper initiation of the meiotic program upon disconnection of the centromeres from the SPB in interphase, as indicated by a specific meiotic feature—a heightened affinity of telomeres for bouquet proteins (Fig. 6), the formation of part of the telocentrosome (Fig. 6a–c) and the attachment of some telomeres to the SPB (Fig. 6d, e).
The recruitment of Bqt1 and Bqt2 proteins at the telomeres upon meiotic entry is crucial for the migration of the telomeres from their localization in interphase, opposite to the SPB, to beneath this structure17. Ectopic expression of bqt1/bqt2 in Rabl-deficient mitotic cells showed that most telomeres displayed a Bqt1-GFP signal. This suggests that, under these conditions, Bqt1 has a higher affinity for Rap1 than when centromeres are normally located at the SPB (Fig. 6a–c). Rap1 appears to be a ‘scaffolding’ protein suffering a hyper-phosphorylation during meiosis and uses its negatively charged amino acid residues to bind the Bqt1-2 complex18. An altered modification on the phosphorylation state of Rap1 might be behind the slight reduction of the Rap1 levels detected by western blot in the absence of normal centromere positioning (Supplementary Figs. 6e and f). One possibility is that this reduction of the Rap1 levels weaken its interaction with Bqt4 leading to the telomere declustering along the NE and increasing the separation between subtelomeric regions and the NE (Supplementary Figs. 6g and h).
The consequences of these premature activation of several meiotic genes and the chromosome architecture likely result in Rabl-deficient cells having severe problems in normal meiotic entry upon nitrogen starvation (Supplementary Fig. 2d).
However, we did not observe a complete formation of the telomere bouquet and the completion of the differentiation to spores. The full formation of the bouquet requires nuclear movements that bring the telomeres together at the SPB19. These movements are driven by cytoskeletal forces, mainly dynein in S. pombe22, and the meiosis-specific protein Hrs1/Mcp620,21. The absence of this vigorous movement in mitosis likely hinders the arrival of the telomeres at the SPB. Consistently, although we observed partial formation of the telocentrosome, dynein was not detected (Fig. 6). Therefore, it could be interesting to combine the loss of the Rabl chromosome configuration with the ectopic recreation of strong nuclear movement in mitosis. Current studies in our lab aim to develop this system.
We think that our results highlight the normal meiotic differentiation in terms of the role of the centromere positioning in the formation of the telomere bouquet. It is known that, during the formation of the bouquet, telomeres and centromeres share the same environment (Supplementary Fig. 7 and stage iii in Fig. 9). This is important to transmit the heterochromatin properties between centromeres and telomeres; the absence of this event leads to problems in outer kinetochore reassembly24. However, little is known about the first stages of the mobilization of the telomeres to the SPB, likely due to the difficulty in exploring this process in vivo. The formation of the telocentrosome has been shown to support migration to the SPB, functioning as a microtubule-organizing center19, which may be an evolutionarily equivalent structure to the cilium recently observed in zebrafish and mouse meiosis70. In this context, the prior positioning of centromeres may be essential as we observed in Supplementary Fig. 7.

Working model for telomere bouquet formation upon meiotic entry: i) During mitotic interphase, fission yeast cells display a conserved Rabl chromosome configuration. In this conformation, centromeres are positioned beneath the SPB thanks to the interaction between the LINC complex (Kms1/2-Sad1) and the kinetochore complex (inner and outer kinetochore), with the aid of Csi1 and Lem2 proteins. Also, telomeres are located in the NE, at the opposite site of the nucleus, based on the interaction between the Bqt3-Bqt4 complex and the Rap1-Taz1 complex. ii) At the onset of meiotic prophase, centromeres decluster from the SPB as the outer kinetochore disassembles, a process that may require a decrement of Cdk1 activity in the outer kinetochore region to facilitate centromere detachment. Following this, Cdc2/Cdk1 may be recycled and mobilized to the telomeres to promote their declustering. At this stage, telomere declustering facilitates their movement towards the SPB and the recruitment of bouquet proteins. iii) During prophase progression, centromere dissociation from the SPB is a prerequisite for telomere-SPB interaction, thanks to the recruitment of bouquet proteins (Bqt1 and Bqt2) to the LINC complex with the aid of the Bqt3-Bqt4 complex. iv) Bouquet formation occurs when centromeres are completely dissociated from the SPB. Then, telomeres are positioned beneath the SPB, based on the interaction between the LINC complex and the Rap1-Taz1 complex, which depends on the bouquet proteins (Bqt1 and Bqt2) and is aided by the Bqt3-Bqt4 complex.
We think that the loss of the outer kinetochore precedes the declustering of the telomeres and their clustering to the SPB. Two pieces of evidence reinforce this hypothesis: first, the compromise of the Rabl chromosome configuration leads to the loss of Ndc80 and Nuf2 signals from the centromeres during interphase in sad1-2 csi1∆ cells25. In this genetic background, telomere declustering is observed (Fig. 4), along with an increased affinity for bouquet proteins and the formation of the telocentrosome (Fig. 6a–c). Secondly, a more detailed in vivo analysis of the meiotic entry in diploid cells shows that the reduction of the Ndc80-GFP signal precedes the formation of the telomere bouquet (Fig. 7a–c). Preventing this reduction halts bouquet formation (Fig. 7d–f). We think that upon meiotic signal, the outer kinetochore is disassembled, leading to centromeres starting to separate from the SPB, and then telomeres become competent to recruit the bouquet proteins Bqt1 and Bqt2 (stage ii in Fig. 9). Then, the initiation of the vigorous meiotic movement facilitates the arriving of the telomeres at the SPB and the complete declustering of the centromeres (stage iii in Fig. 9), forming the telomere bouquet throughout meiotic prophase (stage iv in Fig. 9).
It is important to highlight that centromere disconnection from the SPB appears to act as a trigger for meiotic differentiation, as issues such as cell viability problems, telomere dispersion, and transcriptional silencing defects in subtelomeric and meiotic genes are rescued in Rabl-deficient cells when centromeres are forced to position beneath the SPB (Fig. 5). However, a crucial question is how this long-distance communication from centromeres to telomeres could be mediated. We propose that it is facilitated by the recycling of Cdk1 molecules within the nucleoplasm between these specialized regions (Figs. 8 and 9). Our hypothesis is that, upon meiotic induction, a population of Cdk1 molecules is released from the nucleoplasmic face of the SPB and this decrease in Cdk1 activity leads to the disassembly of the outer kinetochore and centromere declustering (Fig. 8d–h and stage i and ii in Fig. 9). This population of Cdk1 is likely recruited by telomeric proteins, which may be sufficient to induce telomere declustering (Fig. 8d–f). The presence of Cdk1 at the telomeres during the horsetail stage might be necessary to maintain proper bouquet formation (stage iv in Fig. 9).
In summary, our data strongly suggest that centromere positioning plays a crucial role in initiating the bouquet formation process and likely serves as the primary factor triggering the onset of the meiotic program. Due to the high conservation of the telomere bouquet in plants, fungi, and animals, the identification of a long-distance signal from centromeres to telomeres as a controller of meiosis reveals an additional layer of regulation orchestrated by the centromere positioning for the meiotic program in eukaryotes.
Methods
Yeast strains, genetic procedures, and growth conditions
Strains used are listed in Supplementary data 1. Growth conditions and molecular biology approaches were used as described previously71. Gene deletion and C-terminal tagging were performed as described72,73. Insertions of mCherry-Atb2 at the aur1 locus74 utilized pYC19-mCherryAtb275 provided by T. Toda (Hiroshima University). GBP C-terminal tagging60 was performed as was described previously8. Unless otherwise stated, experiments were performed with haploid cells, that were usually grown in rich media (YE4S) at 32 °C or EMM2 media at 32 °C until exponential mid-log phase. Final concentrations of aureobasidin A (0.5 µg/mL) (ClonTech), nourseothricin (100 µg/mL clonNAT) (Werner Bioreagents), G418 (100 µg/mL geneticin) (Invitrogen) and hygromycin B (300 µg/mL) (Invitrogen) were added for selection purpose.
Strain crosses and sporulation were performed in sporulation agar (SPA) plates at 28 °C, and genetic dissection was performed using a dissection microscope (MSM 400; Singer Instruments). Meiosis progression and efficiency assays were carried out in sporulation agar (SPA) plates at 28 °C during 48 h incubation. In each type of experiment, at least 300 meiotic cells were quantified.
Diploid strain in Fig. 7 were initially grown in liquid YE during at least 4 generation times. Afterwards, cells were transferred to EMM with supplements at OD600 = 0.02 and incubated overnight at 28 °C. The next day, cells were collected by centrifugation, washed twice with MM-N low glucose (0.5% Glucose), inoculated in the same medium and incubated at 28 °C for 4 h before time-lapse acquisition.
SGA analysis
Large-scale crosses by SGA assay were performed as described previously26,76 using as query strains control and sad1-2 csi1Δ mutant. Using the Singer RoToR HAD (stands for High-density arrayer), control and sad1-2 csi1Δ cells were crossed with 2,988 gene deletion mutants (Bioneer haploid deletion mutant library, v. 3.0 (Fig. 1) and v. 5.0 (Fig. 6)) and haploid cells were selected by incubation at 42 °C for 4 days (temperature method77). After haploid selection, additional steps of triple mutant selection were performed, spotting onto YE4S supplemented with 300 µg/mL hygromicin B (Invitrogen) and 100 µg/mL ClonNat (Werner Bioreagents) for selection of sad1-2 csi1Δ mutant, and YE4S supplemented with 100 µg/mL G418 (Invitrogen) for selection of deletion mutants. Finally, cells were spotted on YE4S plates and incubated at 32 °C. The growth of single and triple mutants was quantified in YE4S medium, and the ratio with the median colony size inside each plate was compared11. Genetic interactions of triple mutants were analyzed based on colony size (area) as a readout of cellular fitness.
GO and gene expression enrichment analysis was performed using the AnGeLi tool78 with a two-tailed Fisher’s Exact test type and a False-Discovery Rate of 0.01, searching for biological process or gene expression pattern.
MBC and TBZ sensitivity drop assay
Strains were grown in YE4S to exponential midlog phase (1 × 107 − 1.4 × 107 cells/mL; OD600 = 0.5–0.7) at 32 °C, were normalized to 107 cells/mL, and 5-fold serial dilutions were spotted onto YE4S plates containing DMSO or YE4S plates containing different concentrations of MBC (carbendazim, Sigma-Aldrich) or TBZ (tiabendazole, Sigma-Aldrich). The plates were incubated at 32 °C for 48 h.
Pictures were taken as a 48-bit color, reflective document type, at 300 dpi resolution in and Epson scan (Epson scan v. 3.04E) and tiff images were cropped using Adobe Photoshop CS5 Extended for representation purpose.
Fluorescence microscopy, live image analysis, and quantification
Fluorescence microscopy images for telomere foci formation experiments, telomere-NE and subtelomeric loci-NE distance experiments, and centromere declustering assays were generated from cells in exponential midlog phase in all cases, using the DeltaVision microscope system (Applied Precision, Seattle, WA). In brief, cells collected by centrifugation and immobilized to 35 mm glass culture dishes (Ibidi) using 0.2 mg/mL soybean lectin (Sigma-Aldrich) and immersed in EMM2 (with required supplements). Live-cell imaging was carried out at 32 °C in an environmental chamber for temperature control under the microscope, with a DeltaVision Spectris (Applied Precision) comprising an Olympus IX70 widefield inverted epifluorescence microscope, an Olympus UPlanSapo 100x NA 1.4 oil immersion objective, and a Photometrics CCD CoolSnap HQ camera. Images were acquired over 15 focal planes at a 0.4 μm step size. Unless otherwise stated, image processing for foci quantification, distance quantification and representation were performed by deconvolving and combining each color channel into a 2D image using the maximum intensity projection setting in softWoRx (Applied Precision) from raw microscopy data. Combined maximum Z-projection images were treated using the open ImageJ software. Quantification of Taz1-GFP in Figs. 4g, 8f was performed in single-central z sections. Quantification of telomere declustering was conducted in mononucleated cells, identified by the presence of a single SPB when possible or confirmed by FACS analysis.
Signal quantification of Taz1 and Rap1 was carried out using Fiji software on images captured across 15 focal planes, with a step size of 0.4 µm. The intensity of each area containing a given signal was measured, and the intensity of a signal-free region within the same cell was subtracted. The resulting signal intensities were normalized to the average intensity per pixel in the background outside the cell.
For experiments to rescue telomere foci formation using the GBP-GFP system in exponentially growing cells (Fig. 5), a Zeiss Axio Observer 7 inverted microscope was used, with Zeiss Plan-Apochromat 63x/1,40 Oil DIC and Alpha Plan-Apochromat 100x/1,46 Oil DIC lenses, coupled to Spinning Disk Confocal Yokogawa CSUW1 head with excitation lasers and filters from 3i (Intelligent Imaging Innovations). SlideBook 6 software was used for device control and image capturing. In brief, cells collected by centrifugation and immobilized to 35 mm glass culture dishes (Ibidi) using 0.2 mg/mL soybean lectin (1 mg/ml, Sigma-Aldrich) and immersed in EMM2 (with required supplements). Live-cell imaging was carried out at 32 °C in an environmental chamber for temperature control under the microscope. Images were acquired over 15 focal planes at a 0.4 μm step size. All images were processed using the open Image J software. Presented images correspond to combined maximum Z-projections.
To observe bouquet formation in diploid cells (Fig. 7), cells were immobilized in 8 multi-well Ibidi slides coated with soybean lectin (1 mg/mL, Sigma-Aldrich) as described in ref. 79 and imaged at 3 minutes interval during 2 hours. 13 Z-planes 0.4 µm apart were acquired for each fluorescence channel (GFP and Cherry) and time-point. Maximum projections were used for Fig. 7a, b while sum projections were made in order to quantify Ndc80-GFP intensity (Fig. 7c) along the time-lapse.
RNA-seq library preparation and data analysis
An asynchronic culture of 30 mL of exponentially growing cells in liquid YE4S medium at 32 °C until an OD600 = 0.5 is spined down and washed twice with equal volume of milliQ water and the pellet is deep-frozen in liquid nitrogen. The frozen pellet is covered by liquid nitrogen in a pestle and gently fragmented and crushed with a mortar down to dust. The crashed pellet is deep-frozen again and submitted to RNA extraction protocol from the Rneasy Kit (QIAGEN, Cat. No. / ID: 74104), obtaining a sample of total RNA in 50 μL of Rnase-free water. This total RNA sample was submitted to ribosomal RNA depletion and the rest of RNA was used for library preparation and sequencing by the BGI platform. Generated sequencing reads where quality control-tested and -trimmed and adapters sequence were removed by the BGI platform. Quality control-filtered sequences reads were mapped to the Schizosaccharomyces pombe (strain 972 h-) reference genome (assembly ASM294v2.47 from Pombase) using HISAT280. Feature reads counts were obtained with htseq-count81 using the genome annotation file for the corresponding assembly from Pombase. Generated SAM/BAM files were used to perform the differential gene expression analysis with DESeq282.
RNA-seq reads density plots
Raw reads were subjected to quality control using ‘fastqc’ (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and trimmed for removal of near-end low-quality positions and adapter sequences with ‘cutadapt’. Read quality improvement after this processing was checked again with ‘fastqc’. Index of the reference genome, Schizosaccharomyces pombe (assembly ASM294v2.47), was built with ‘STAR’83 with the command: ‘STAR --runThreadN 16 --runMode genomeGenerate --genomeDir./index --genomeFastaFiles /S_pombe_genome.fa --sjdbGTFfile /S_pombe_genome.gtf --sjdbOverhang 100 --genomeSAindexNbases 12’, using the genome sequence (.fa file) and the genome features annotations (.gtf file) for the indicated assembly. Quality reads were mapped to the reference genome using the built index with ‘STAR’ aligner with the command: ‘STAR --genomeDir./index --runThreadN 16 --readFilesIn./read_1./read_2 --outFileNamePrefix sampleName’. From the output files generated by this latter command, ‘samtools’84 was used to convert SAM files to BAM format with the command: ‘samtools view -S -b sampleNameAligned.out.sam > sampleNameAligned.out.bam’ and sorted by genome coordinate with the command: ‘samtools sort -o sampleNameAligned.out.sorted.bam sampleNameAligned.out.bam’. ‘STAR’ was used to generate the BedGraphs normalized per million of reads from the sorted BAM files with the command: ‘STAR --runMode inputAlignmentsFromBAM --inputBAMfile sampleNameAligned.out.sorted.bam --outWigType bedGraph --outWigStrand Unstranded --outWigNorm RPM’. Plots of reads density as reads-per-million were generated from individual and global average BedGraphs of all biological replicates for each sample and condition using R software (https://www.R-project.org/) and Rstudio.
Positional enrichment analysis
We assessed the overrepresentation of differentially expressed features at specific genome locations via positional enrichment analysis with the PGE tool (REF: PMID18346969). This tool tests the enrichment of a set of query genes at chromosomal regions of any width. The number of genes found at a specific location is modeled as a hypergeometric distribution, which is approximated by sampling. The gene annotation and coordinates of the S. pombe genome were restricted to the genes detected by RNA-seq and fed into PGE as the reference dataset. Then, the list of genes passing the filtering criteria for the RNA-seq analysis (abs(log2FC) > 0.5 and p-value < 0.05) were fed into PGE. The threshold for significance in the positional analysis was set to p-value < 0.05, adjusted for multiple testing. Two lists of genome ranges were obtained, for upregulated and downregulated genes from the RNA-seq analysis, respectively. shinyCircos 2.0 (REF: 10.1002/imt2.109) was used to generate the visualizations of RNA-seq and positional enrichment statistics.
RT-qPCR
Cells were grown in liquid YE4S medium at 32 °C to exponential midlog phase, and total RNA was isolated from cells lysed by bead beating (FastPrep 24, MP BioMedical) using TRIzol reagent (Life Technologies) and zirconia/silica beads (BioSpec), followed by centrifugation at 13,000 r.p.m. for 10 min at 4 °C. Recovered supernatant was extracted with chloroform and re-precipitated with isopropyl alcohol. After removing DNA contamination from 20 µg of resuspended RNA with TURBO DNA-free (Applied Biosystems), 2 µg of RNA samples were subjected to cDNA synthesis using iScript cDNA Synthesis Kit (Bio-Rad). RT-qPCR reactions were carried out in 10 µL of volume, with 5 µL TB Green Premix Ex Taq II (Tli RNase H Plus) (Takara), 1.7 µL of a mixture of forward and reverse primers (1.5 µM), and 3.3 µL cDNA, previously diluted (1:25 for each analyzed loci and 1:250 for act1+ locus), and using CFX96 Touch Real-Time PCR Detection System (Bio-Rad). The primers used for qPCR experiments are listed in Supplementary data 2.
Mean expression values for control and mutants, and standard error from independent experiments were calculated by normalizing to act1+ and then dividing by the mean of a sample pool of mutants from each experiment (group normalization)35. The mean of data from 3-4 independent biological replicates are shown as relative to the mean value of the control (which was set to 1). Multiple two-tailed Student’s t-test analysis was performed using ANOVA followed by a Tukey’s post hoc test at a 0.05 significance level using GraphPad Prism software (Dotmatics).
Western blot analysis
Samples of interphase cells were obtained from 10 mL of exponential growing cultures at OD600 = 0.5 at 32 °C. Cells were collected by centrifugation at 4 °C and cellular pellets were flash frozen in liquid nitrogen and stored at -80 °C until use.
Protein extracts were prepared from trichloroacetic acid-treated cells as described85. Briefly, cell pellets corresponding to 1 × 108 cells per condition and strain were removed from storage at -80 °C and mixed with cold 20% TCA and cold acid washed glass beads (Sigma-Aldrich). Cell integrity was disrupted by FastPrep-24 (MP Biomedicals) for 4 cycles of 20 s at 4 m/s. Then, ice-cold 5% TCA was added, and cell lysate was recovered. Next, samples were centrifuged, and supernatant was discarded. Protein pellet was washed with ice-cold 100% acetone and centrifuged. All previous manipulations were performed at 4 °C. Supernatant was discarded and protein pellet was resuspended at room temperature in SDS loading buffer and 1 M Tris-HCl pH 8 to raise to neutral the pH of the sample. Samples were denatured by heating at 100 °C, centrifuged and loaded in a 10% SDS-PAGE gel (Biorad) to separate proteins. HA-tagged proteins were detected with an anti-HA antibody (Biolegend, 9011501) (1:1000) and anti-tubulin antibody (Sigma-Aldrich T6199) (1:20000) were used in Supplementary Fig. 6e; living Colors a.v. Monoclonal Antibody (JL-8, Clontech 632380)(1:3000) and anti-tubulin monoclonal antibody, clone DM1A, (Sigma-Aldrich T9026)(1:5000) were used for Supplementary 5b. The secondary antibody was horseradish peroxidase (HRP)-coupled anti-mouse IgG (Sigma-Aldrich). Visualization was performed using the SuperSignal WestFemto Maximun Sensitivity Substrate (ThermoFisher) in a Chemidoc MP Imaging System (Biorad). Images processing for representation was performed using Adobe Photoshop CS5 Extended, signal quantifications were performed in ImageJ and a two-tailed Student’s t-test analysis was performed at a 0.05 significance level using GraphPad Prism software (Dotmatics).
FACS
107 cells from exponentially growing cultures (OD ~ 0.3–0.4) were centrifuge at 1000 × g for 5 min. The supernatant was discarded, 1 mL cold 70% EtOH was added per sample, and the tubes were vortexed (cells could be stored indefinitely at 4 °C). For sample processing, 300 µL (~ 2–3 ×106 cells) were transferred to 1 mL of 50 mM Na citrate in an eppendorf tube, mixed, and spinned down at 1000 × g for 5 min. After discarding the supernatant, the pellet was resuspended in 0.5 mL 50 mM Na citrate containing 0.1 mg/mL RNase A. Samples were then incubated at 37 °C for at least 2 h (or overnight if needed). For DNA staining, samples were centrifuged at 1000 × g for 5 min, the supernatant was discarded and, each pellet was resuspended in 1 mL 50 mM Na citrate containing 4 µg/mL propidium iodide. Samples were either processed immediately by sonicating at 50% amplitude for 10 s to separate cells or stored overnight at 4 °C in the dark for processing the next day. The settings used in the FACS Calibur cytometer were as follows: detector FSC E00 Gain:3; Detector FL2-A Voltage: 890 Gain: 2.
Statistics and reproducibility
Micrographs of representative images, as shown in Figs. 1b, 4e, and 8c, d, were obtained from three independent experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq data generated in this study have been deposited in NCBI’s Gene Expression Omnibus under GEO Series accession number GSE252561. Source data are provided with this paper.
References
Dekker, J. et al. Spatial and temporal organization of the genome: current state and future aims of the 4D nucleome project. Mol. Cell 83, 2624–2640 (2023).
Muller, H., Gil, J. Jr & Drinnenberg, I. A. The impact of centromeres on spatial genome architecture. Trends Genet 35, 565–578 (2019).
Zheng, H. & Xie, W. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 20, 535–550 (2019).
Zickler, D. & Kleckner, N. Meiosis: Dances between homologs. Annu Rev Genet, https://doi.org/10.1146/annurev-genet-061323-044915 (2023).
Asakawa, H., Hayashi, A., Haraguchi, T. & Hiraoka, Y. Dissociation of the Nuf2-Ndc80 complex releases centromeres from the spindle-pole body during meiotic prophase in fission yeast. Mol. Biol. Cell 16, 2325–2338 (2005).
Nabetani, A., Koujin, T., Tsutsumi, C., Haraguchi, T. & Hiraoka, Y. A conserved protein, Nuf2, is implicated in connecting the centromere to the spindle during chromosome segregation: a link between the kinetochore function and the spindle checkpoint. Chromosoma 110, 322–334 (2001).
Takahashi, K., Chen, E. S. & Yanagida, M. Requirement of Mis6 centromere connector for localizing a CENP-A-like protein in fission yeast. Science 288, 2215–2219 (2000).
Fernández-Álvarez, A., Bez, C., O’Toole, E. T., Morphew, M. & Cooper, J. P. Mitotic nuclear envelope breakdown and spindle nucleation are controlled by interphase contacts between centromeres and the nuclear envelope. Developmental Cell 39, 544–559 (2016).
Rothballer, A., Schwartz, T. U. & Kutay, U. LINCing complex functions at the nuclear envelope what the molecular architecture of the LINC complex can reveal about its function. Nucl. (U. S.) 4, 1–8 (2013).
Hagan, I. & Yanagida, M. The product of the spindle formation gene sad1+ associates with the fission yeast spindle pole body and is essential for viability. J. Cell Biol. 129, 1033–1047 (1995).
Barrales, R. R., Forn, M., Georgescu, P. R., Sarkadi, Z. & Braun, S. Control of heterochromatin localization and silencing by the nuclear membrane protein Lem2. Genes Dev. 30, 133–148 (2016).
Hou, H. et al. Csi1 links centromeres to the nuclear envelope for centromere clustering. J. Cell Biol. 199, 735–744 (2012).
Chikashige, Y. et al. Membrane proteins Bqt3 and -4 anchor telomeres to the nuclear envelope to ensure chromosomal bouquet formation. J. Cell Biol. 187, 413–427 (2009).
Ebrahimi, H., Masuda, H., Jain, D. & Cooper, J. P. Distinct ‘safe zones’ at the nuclear envelope ensure robust replication of heterochromatic chromosome regions. Elife 7, https://doi.org/10.7554/eLife.32911 (2018).
Funabiki, H., Hagan, I., Uzawa, S. & Yanagida, M. Cell cycle-dependent specific positioning and clustering of centromeres and telomeres in fission yeast. J. Cell Biol. 121, 961–976 (1993).
Chikashige, Y. et al. Meiotic nuclear reorganization: switching the position of centromeres and telomeres in the fission yeast Schizosaccharomyces pombe. EMBO J. 16, 193–202 (1997).
Chikashige, Y. et al. Meiotic proteins Bqt1 and Bqt2 tether telomeres to form the bouquet arrangement of chromosomes. Cell 125, 59–69 (2006).
Amelina, H. et al. Telomere protein Rap1 is a charge resistant scaffolding protein in chromosomal bouquet formation. BMC Biol. 13, 37 (2015).
Yoshida, M. et al. Microtubule-organizing center formation at telomeres induces meiotic telomere clustering. J. Cell Biol. 200, 385–395 (2013).
Saito, T. T., Tougan, T., Okuzaki, D., Kasama, T. & Nojima, H. Mcp6, a meiosis-specific coiled-coil protein of Schizosaccharomyces pombe, localizes to the spindle pole body and is required for horsetail movement and recombination. J. Cell Sci. 118, 447–459 (2005).
Tanaka, K., Kohda, T., Yamashita, A., Nonaka, N. & Yamamoto, M. Hrs1p/Mcp6p on the meiotic SPB organizes astral microtubule arrays for oscillatory nuclear movement. Curr. Biol. 15, 1479–1486 (2005).
Miki, F. et al. The 14-kDa dynein light chain-family protein Dlc1 is required for regular oscillatory nuclear movement and efficient recombination during meiotic prophase in fission yeast. Mol. Biol. Cell 13, 930–946 (2002).
Tomita, K. & Cooper, J. P. The telomere bouquet controls the meiotic spindle. Cell 130, 113–126 (2007).
Klutstein, M., Fennell, A., Fernandez-Alvarez, A. & Cooper, J. P. The telomere bouquet regulates meiotic centromere assembly. Nat. Cell Biol. 17, 458–469 (2015).
Jimenez-Martin, A. et al. The Rabl chromosome configuration masks a kinetochore reassembly mechanism in yeast mitosis. Mol. Biol. Cell 33, br8 (2022).
Verrier, L. et al. Global regulation of heterochromatin spreading by Leo1. Open Biol 5, https://doi.org/10.1098/rsob.150045 (2015).
Hou, H., Kallgren, S. P. & Jia, S. Csi1 illuminates the mechanism and function of Rabl configuration. Nucleus 4, 176–181 (2013).
Cam, H. P. et al. Comprehensive analysis of heterochromatin- and RNAi-mediated epigenetic control of the fission yeast genome. Nat. Genet 37, 809–819 (2005).
Noma, K. & Grewal, S. I. Histone H3 lysine 4 methylation is mediated by Set1 and promotes maintenance of active chromatin states in fission yeast. Proc. Natl Acad. Sci. USA 99, 16438–16445 (2002).
Shilatifard, A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev. Biochem 81, 65–95 (2012).
Noma, K., Allis, C. D. & Grewal, S. I. Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science 293, 1150–1155 (2001).
Greenstein, R. A. et al. Set1/COMPASS repels heterochromatin invasion at euchromatic sites by disrupting Suv39/Clr4 activity and nucleosome stability. Genes Dev. 34, 99–117 (2020).
Reddy, B. D. et al. Elimination of a specific histone H3K14 acetyltransferase complex bypasses the RNAi pathway to regulate pericentric heterochromatin functions. Genes Dev. 25, 214–219 (2011).
Wang, J., Reddy, B. D. & Jia, S. Rapid epigenetic adaptation to uncontrolled heterochromatin spreading. Elife 4, https://doi.org/10.7554/eLife.06179 (2015).
Georgescu, P. R., Capella, M., Fischer-Burkart, S. & Braun, S. The euchromatic histone mark H3K36me3 preserves heterochromatin through sequestration of an acetyltransferase complex in fission yeast. Micro. Cell 7, 80–92 (2020).
Flury, V. et al. The histone acetyltransferase Mst2 protects active chromatin from epigenetic silencing by acetylating the ubiquitin ligase Brl1. Mol. Cell 67, 294–307 e299 (2017).
Gomez, E. B., Espinosa, J. M. & Forsburg, S. L. Schizosaccharomyces pombe mst2+ encodes a MYST family histone acetyltransferase that negatively regulates telomere silencing. Mol. Cell Biol. 25, 8887–8903 (2005).
Roguev, A. et al. High conservation of the Set1/Rad6 axis of histone 3 lysine 4 methylation in budding and fission yeasts. J. Biol. Chem. 278, 8487–8493 (2003).
Braun, S. et al. The Cul4-Ddb1(Cdt)(2) ubiquitin ligase inhibits invasion of a boundary-associated antisilencing factor into heterochromatin. Cell 144, 41–54 (2011).
Zofall, M. & Grewal, S. I. Swi6/HP1 recruits a JmjC domain protein to facilitate transcription of heterochromatic repeats. Mol. Cell 22, 681–692 (2006).
Ayoub, N. et al. A novel jmjC domain protein modulates heterochromatization in fission yeast. Mol. Cell Biol. 23, 4356–4370 (2003).
Wang, J. et al. Epe1 recruits BET family bromodomain protein Bdf2 to establish heterochromatin boundaries. Genes Dev. 27, 1886–1902 (2013).
Mata, J., Lyne, R., Burns, G. & Bahler, J. The transcriptional program of meiosis and sporulation in fission yeast. Nat. Genet 32, 143–147 (2002).
Sugimoto, A., Iino, Y., Maeda, T., Watanabe, Y. & Yamamoto, M. Schizosaccharomyces pombe ste11+ encodes a transcription factor with an HMG motif that is a critical regulator of sexual development. Genes Dev. 5, 1990–1999 (1991).
Watanabe, Y., Shinozaki-Yabana, S., Chikashige, Y., Hiraoka, Y. & Yamamoto, M. Phosphorylation of RNA-binding protein controls cell cycle switch from mitotic to meiotic in fission yeast. Nature 386, 187–190 (1997).
Ding, D. Q. et al. Meiosis-specific noncoding RNA mediates robust pairing of homologous chromosomes in meiosis. Science 336, 732–736 (2012).
Lin, Y. & Smith, G. R. Molecular cloning of the meiosis-induced rec10 gene of Schizosaccharomyces pombe. Curr. Genet 27, 440–446 (1995).
De Veaux, L. C., Hoagland, N. A. & Smith, G. R. Seventeen complementation groups of mutations decreasing meiotic recombination in Schizosaccharomyces pombe. Genetics 130, 251–262 (1992).
Tang, X., Jin, Y. & Cande, W. Z. Bqt2p is essential for initiating telomere clustering upon pheromone sensing in fission yeast. J. Cell Biol. 173, 845–851 (2006).
Harigaya, Y. & Yamamoto, M. Molecular mechanisms underlying the mitosis-meiosis decision. Chromosome Res 15, 523–537 (2007).
Tong, P. et al. Interspecies conservation of organisation and function between nonhomologous regional centromeres. Nat. Commun. 10, 2343 (2019).
Allshire, R. C., Nimmo, E. R., Ekwall, K., Javerzat, J. P. & Cranston, G. Mutations derepressing silent centromeric domains in fission yeast disrupt chromosome segregation. Genes Dev. 9, 218–233 (1995).
Yadav, R. K., Matsuda, A., Lowe, B. R., Hiraoka, Y. & Partridge, J. F. Subtelomeric Chromatin in the Fission Yeast S. pombe. Microorganisms 9, https://doi.org/10.3390/microorganisms9091977 (2021).
Wang, J. et al. The proper connection between shelterin components is required for telomeric heterochromatin assembly. Genes Dev. 30, 827–839 (2016).
Kanoh, J., Sadaie, M., Urano, T. & Ishikawa, F. Telomere binding protein Taz1 establishes Swi6 heterochromatin independently of RNAi at telomeres. Curr. Biol. 15, 1808–1819 (2005).
van Emden, T. S. et al. Shelterin and subtelomeric DNA sequences control nucleosome maintenance and genome stability. EMBO Rep 20, https://doi.org/10.15252/embr.201847181 (2019).
Matsuda, A. et al. Highly condensed chromatins are formed adjacent to subtelomeric and decondensed silent chromatin in fission yeast. Nat. Commun. 6, 7753 (2015).
Tashiro, S. et al. Shugoshin forms a specialized chromatin domain at subtelomeres that regulates transcription and replication timing. Nat. Commun. 7, 10393 (2016).
Hall, I. M., Noma, K. & Grewal, S. I. RNA interference machinery regulates chromosome dynamics during mitosis and meiosis in fission yeast. Proc. Natl Acad. Sci. USA 100, 193–198 (2003).
Rothbauer, U. et al. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat. Methods 3, 887–889 (2006).
Fujita, I. et al. Telomere-nuclear envelope dissociation promoted by Rap1 phosphorylation ensures faithful chromosome segregation. Curr. Biol. 22, 1932–1937 (2012).
Mikheyeva, I. V., Grady, P. J., Tamburini, F. B., Lorenz, D. R. & Cam, H. P. Multifaceted genome control by Set1 Dependent and Independent of H3K4 methylation and the Set1C/COMPASS complex. PLoS Genet 10, e1004740 (2014).
Pinder, C., Matsuo, Y., Maurer, S. P. & Toda, T. Kinesin-8 and Dis1/TOG collaborate to limit spindle elongation from prophase to anaphase A for proper chromosome segregation in fission yeast. Journal of Cell Science 132, https://doi.org/10.1242/jcs.232306 (2019).
Niwa, O., Shimanuki, M. & Miki, F. Telomere-led bouquet formation facilitates homologous chromosome pairing and restricts ectopic interaction in fission yeast meiosis. EMBO J. 19, 3831–3840 (2000).
Hayashi, A., Asakawa, H., Haraguchi, T. & Hiraoka, Y. Reconstruction of the kinetochore during meiosis in fission yeast Schizosaccharomyces pombe. Mol. Biol. Cell 17, 5173–5184 (2006).
Hou, H. et al. Centromeres are dismantled by foundational meiotic proteins Spo11 and Rec8. Nature 591, 671–676 (2021).
Moiseeva, V. et al. The telomere bouquet facilitates meiotic prophase progression and exit in fission yeast. Cell Discov. 3, 17041 (2017).
Decottignies, A., Zarzov, P. & Nurse, P. In vivo localisation of fission yeast cyclin-dependent kinase cdc2p and cyclin B cdc13p during mitosis and meiosis. J. Cell Sci. 114, 2627–2640 (2001).
Grallert, A. et al. Centrosomal MPF triggers the mitotic and morphogenetic switches of fission yeast. Nat. Cell Biol. 15, 88–95 (2013).
Mytlis, A. et al. Control of meiotic chromosomal bouquet and germ cell morphogenesis by the zygotene cilium. Science 376, eabh3104 (2022).
Moreno, S., Klar, A. & Nurse, P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 194, 795–823 (1991).
Bahler, J. et al. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14, 943–951 (1998).
Fennell, A., Fernández-álvarez, A., Tomita, K. & Cooper, J. P. Telomeres and centromeres have interchangeable roles in promoting meiotic spindle formation. J. Cell Biol. 208, 415–428 (2015).
Hashida-Okado, T., Yasumoto, R., Endo, M., Takesako, K. & Kato, I. Isolation and characterization of the aureobasidin A-resistant gene, aur1R, on Schizosaccharomyces pombe: roles of Aur1p+ in cell morphogenesis. Curr. Genet 33, 38–45 (1998).
Nakamura, Y., Arai, A., Takebe, Y. & Masuda, M. A chemical compound for controlled expression of nmt1-driven gene in the fission yeast Schizosaccharomyces pombe. Anal. Biochem 412, 159–164 (2011).
Barrales, R. R. & Braun, S. Chromatin binding and silencing: two roles of the same protein Lem2. Micro. Cell 3, 185–188 (2016).
Dixon, S. J. et al. Significant conservation of synthetic lethal genetic interaction networks between distantly related eukaryotes. Proc. Natl Acad. Sci. USA 105, 16653–16658 (2008).
Bitton, D. A. et al. AnGeLi: a tool for the analysis of gene lists from fission yeast. Front Genet 6, 330 (2015).
Pineda-Santaella, A., Martin-Garcia, R. & Fernandez-Alvarez, A. Analyzing self-assembled spindle dynamics in fission yeast meiosis using in vivo fluorescence imaging. STAR Protoc. 4, 102655 (2023).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Anders, S., Pyl, P. T. & Huber, W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Grallert, A. & Hagan, I. M. Preparation of Protein Extracts from Schizosaccharomyces pombe using trichloroacetic acid precipitation. Cold Spring Harb Protoc 2017, https://doi.org/10.1101/pdb.prot091579 (2017).
Acknowledgements
We thank all lab members for their critical comments on the manuscript and express our gratitude to all participants of the IBFG and CABD seminars for their valuable input. Special thanks to Matías Capella for his support with the SGA experiment, Iain Hagan lab for cdc2.RL strains, Víctor Álvarez Tallada for his support in managing the logistics of the CABD's transition to the IBFG, and to the Shiv Grewal lab for providing the script for (RPM)-normalized read density plots. Additionally, we express our gratitude to INLEXIO for English grammar corrections and to the NBRP resource in Japan for providing strains. This work was supported by PID2021-127232NB-I00 funded by the MCIN/ AEI /10.13039/501100011033 and by the “FEDER, Una manera de hacer Europa”, awarded to A.F.-A. AJ-M was funded by Juan de la Cierva Formación 2020 Grant FJC2020-046386-I; AT was supported by the EUR G.E.N.E. (reference #ANR-17-EURE-0013) and is part of the Université Paris Cité IdEx #ANR-18-IDEX-0001 funded by the French Government through its “Investments for the Future” program. R.R.D. was supported by PID2021-1284080B-I00 funded by MCIN/ AEI. The IBFG is an institution funded by the Salamanca University and Consejo Superior de Investigaciones Científicas (CSIC).
Author information
Authors and Affiliations
Contributions
A.F-A. and A.J-M. conceived the study. A.J-M. (Figs. 1–5, 8a, 9; Supplementary Fig. 1c-h, 2, 3c-g, 4, 5f-g, 6a, e, f-h, 8) and A.F-A. (Figs. 6 and 8; Supplementary Fig. 1a-b, 5d-e, 6b-e; Supplementary movie 1) conducted most of the experiments and data analysis. The initial SGA screening was performed by A.F-A. and S.B., while the second SGA was carried out by R.E-V. and J.P-C. RNA-seq experiments and data analysis were performed by A.P-S. (Fig. 2a, b, e; Supplementary Figs. 2a–b, 3a) and A.J-M. D.L-P. handled the analysis of SGA and the positional enrichment of transcription from RNAseq data (Supplementary Fig. 3b). R.M-G. performed the experiments in Fig. 7 and Supplementary Figs. 5a–c and 7, with support from S.C-C. A.T. and A.M. contributed to the validation of SGA genetic interactions. A.F-A. and A.J-M. wrote the manuscript with significant input from R.R.D. and S.B.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Kayoko Tanaka and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jiménez-Martín, A., Pineda-Santaella, A., Martín-García, R. et al. Centromere positioning orchestrates telomere bouquet formation and the initiation of meiotic differentiation. Nat Commun 16, 837 (2025). https://doi.org/10.1038/s41467-025-56049-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56049-9
