
Abstract
The generation of radicals through photo-Fenton-like reactions demonstrates significant potential for remediating emerging organic contaminants (EOCs) in complex aqueous environments. However, the excitonic effect, induced by Coulomb interactions between photoexcited electrons and holes, reduces carrier utilization efficiency in these systems. In this study, we develop Cu single-atom-loaded covalent organic frameworks (CuSA/COFs) as models to modulate excitonic effects. Temperature-dependent photoluminescence and ultrafast transient absorption spectra reveal that incorporating acenaphthene units into the linker (CuSA/Ace-COF) significantly reduces exciton binding energy (Eb). This modification not only enhances peroxymonosulfate adsorption at Cu active sites but also facilitates rapid electron transfer and promotes selective hydroxyl radical generation. Compared to CuSA/Obq-COF (Eb = 25.6 meV), CuSA/Ace-COF (Eb = 12.2 meV) shows a 39.5-fold increase in the pseudo-first-order rate constant for sulfamethoxazole degradation (0.434 min−1). This work provides insights into modulating excitonic behavior in single-atom catalysts via linker engineering for EOCs degradation.
Similar content being viewed by others


Introduction
In recent years, the proliferation of emerging organic contaminants (EOCs) in aquatic environments has become a significant concern, primarily due to the extensive use of pharmaceuticals and personal care products1,2. These contaminants exhibit high chemical stability and resistance to biodegradation, rendering conventional municipal wastewater treatment plants (WWTPs) ineffective3,4. Consequently, EOCs pose substantial risks to ecosystems and public health5. Advanced oxidation processes (AOPs) have shown considerable promise in mitigating these persistent pollutants by generating highly reactive oxygen species (ROS), such as hydroxyl radicals (•OH) and sulfate radicals (•SO4−)6,7. Among these AOPs, the photo-Fenton-like reaction system, which employs photocatalysts to activate peroxymonosulfate (PMS) for the production of potent ROS, is regarded as a promising technology for sustainable water treatment8,9. However, the current photoactivated PMS catalysts are hindered by suboptimal light absorption efficiency, electron-hole recombination, and a paucity of catalytically active sites10. These deficiencies impede the effective utilization of photogenerated carriers and limit the overall reaction efficiency. Thus, the development of highly efficient catalysts for photo-Fenton-like reactions remains a pivotal obstacle in advancing sustainable water treatment technologies.
Covalent organic frameworks (COFs), as a novel class of crystalline porous materials, exhibit excellent porosity, large surface area, broad-spectrum light absorption, and exceptional stability, rendering them an ideal platform for the photo-Fenton reaction11,12,13. The incorporation of single-atom metal into COFs (MSA/COF) not only broadens the photoresponse range but also homogenizes metal atoms, thereby shortening electron transfer distances, promoting electron accumulation, and accelerating carrier transfer kinetics14,15. Furthermore, atomically dispersed active metals maximize the number of reactive sites, while electron-rich metal centers serve as high-energy active sites that stabilize intermediates during the photo-Fenton-like reaction, facilitating efficient PMS activation16. However, the intrinsically low dielectric constant of COFs categorizes them as strong exciton systems17. Upon photoexcitation, MSA/COFs generate strong excitonic effects due to substantial electrostatic Coulomb interactions, which hinder exciton dissociation and free carrier generation, consequently reducing the catalytic activation efficiency for PMS18. Therefore, it remains a formidable challenge to rationally design and construct effective MSA/COFs at the molecular level to attenuate excitonic effects and promote exciton dissociation within catalytic systems.
To mitigate the excitonic effect in MSA/COFs, creating energetically disordered domains within the catalyst is a reliable strategy19,20. Techniques such as heterojunction formation, heteroatom doping, and defect engineering can destabilize excitons and expedite their dissociation into free carriers21,22,23. However, the implementation of heterojunctions is constrained by the necessity for precise energy level alignment between electron donors and acceptors, while heteroatom doping and defect engineering often require severe calcination or reduction conditions, compromising material stability. Constructing donor-acceptor (D-A) type COFs by integrating diverse donor and acceptor units can enhance unit polarity and facilitate rational molecular design24,25. Despite efforts to reinforce the conjugated structure within COFs to augment electron-hole separation, there is a paucity of research elucidating the impact of these modifications on catalytic activity from an excitonic perspective. Thus, developing a straightforward and effective strategy to modulate the intrinsic excitonic behavior of MSA/COFs through precise structural tuning is critical for optimizing their performance in photo-Fenton-activated PMS applications, a domain that remains underexplored.
Herein, we employ a linker engineering strategy to synthesize two imine-linked COFs incorporating distinct electron acceptor units: ace-naphthenequinone (Ace-COF) and o-benzoquinone (Obq-COF). Atomically dispersed Cu species were successfully anchored within the COF substrates, forming CuSA/COFs via post-synthetic modifications. As an environmentally benign material, the acenaphthene units effectively mitigate the intrinsic excitonic effect in CuSA/Ace-COF and modulates the electronic structure of the Cu single-atoms. This modification not only enhances PMS adsorption at the Cu sites but also facilitates rapid and efficient electron transfer to the adsorbed PMS, thereby promoting the selective generation of •OH radicals. The optimized CuSA/Ace-COF exhibited exceptional potential for the degradation of EOCs via photoactivated PMS, demonstrating rapid pseudo-first-order degradation kinetics (0.434 min-¹ for sulfamethoxazole). The system exhibited resilience against interference from co-existing ions and dissolved organic matter (DOM) and proved to be remarkably cost-effective (EE/O = 0.35 kWh·m−3). This study elucidates the critical role of excitonic behavior in pollutant degradation by photoactivated PMS and offers insights into the design of catalysts for photo-Fenton-like reactions from an excitonic perspective.
Results
Synthesis and characterization of COFs and CuSA/COFs
Based on a linker engineering strategy, Cu single-atoms were immobilized onto two distinct COF substrates, resulting in the development of single-atom catalytic materials with superior photoactive PMS performance, as depicted in Fig. 1 and Supplementary Fig. 1. Initially, 4,4′4″-(1,3,5-triazine-2,4,6-triyl)trianiline (TTA) was employed as the core unit, with either ace-naphthenequinone (Ace) or o-benzoquinone (Obq) serving as the linker, to synthesize reticular COFs (Ace-COF or Obq-COF) with analogous topologies via Schiff base and Knoevenagel condensations26.

Illustrations for the structure and synthesis of COFs and CuSA/COFs.
Fourier transform infrared spectroscopy (FTIR) spectra (Fig. 2a) comparison of the COFs and linker monomers revealed characteristic peaks at ~1510 cm−1 and ~1366 cm−1 in TTA, Obq-COF, and Ace-COF, which are attributed to the triazine units27. After polymerization, a new peak at ~1608 cm−1, corresponding to the imine bond, was observed in Obq-COF and Ace-COF but was absent in the individual linker monomers26. This confirms the successful polymerization of the precursor monomers. To provide more detailed characterization of the chemical structure of the polymerized COFs, 13C solid-state nuclear magnetic resonance (ss-NMR) tests were further conducted (Fig. 2b). The peaks observed at 111-140 ppm (maked a, b, and c) and ~170 ppm (maked e) correspond to the benzene ring and triazine ring structures, respectively24. Notably, Ace-COF exhibited distinct signal peaks at ~185 ppm and ~155 ppm, corresponding to the tertiary carbons in α and β positions of acenaphthene moiety28. Additionally, characteristic peaks typical of imine carbons were detected at 146–152 ppm for both COFs, confirming the formation of covalent structure via amidine-formaldehyde polycondensation reactions29. Thermogravimetric analysis (TGA) (Supplementary Fig. 2) indicates that significant weight loss occurs only above ~500 °C, demonstrating excellent thermal stability of the material. To further confirm the stability of the imine bonds, extended thermal treatment at 180 °C for 5.0 h was conducted. The FTIR spectra (Supplementary Fig. 3) continued to show the characteristic imine peak at ~1608 cm−1, confirming the retention of imine bonds after prolonged thermal exposure.

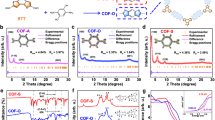
a FTIR spectra of linker monomers and COFs. b 13C ss-NMR spectra of Obq-COF and Ace-COF. SEM images of c CuSA/Obq-COF and d CuSA/Ace-COF; the insets show the corresponding TEM and EDS elemental mapping images. AC-HAADF-STEM images of e CuSA/Obq-COF and f CuSA/Ace-COF, with the yellow circle highlighting the single-atom Cu feature.
Subsequently, energetic Cu single atoms were successfully integrated into the 2D framework of COFs via a mild post-synthetic impregnation method to form CuSA/Ace-COF and CuSA/Obq-COF. Inductively coupled plasma mass spectrometry (ICP-MS) and X-ray photoelectron spectroscopy (XPS) spectra (Supplementary Table 1 and Supplementary Fig. 4) verified the successful incorporation of elemental Cu into COFs, resulting in Cu concentrations of 3.35 wt% for CuSA/Ace-COF and 3.09 wt% for CuSA/Obq-COF. X-ray diffraction (XRD) results (Supplementary Fig. 5) revealed characteristic COF peaks in both CuSA/COFs, with diffraction peaks at ~12.4° and ~15.0° corresponding to the (130) and (400) facets of COFs, respectively. Additionally, a high and broad peak at ~25.0° arose from π-π stacking between COF layers, corresponding to the (001) plane16. All diffraction peaks follow the P-6 space group, indicative of the hexagonal two-dimensional laminar network structure. Importantly, the absence of Cu-related diffraction peaks suggests that Cu elements in the CuSA/COFs are likely atomically dispersed. Rietveld refinement and structural simulations were further conducted based on the experimental XRD patterns of CuSA/Ace-COFs and CuSA/Obq-COFs (Supplementary Fig. 6). The results indicate that the unit cell parameters for CuSA/Ace-COFs are a = b = 29.57 Å and c = 3.66 Å, with α = β = 90° and γ = 120°. The simulated PXRD pattern using the AA stacking model aligns closely with the experimental data, and the difference curve reveals a weighted-profile R factor (Rwp = 2.64%) and an unweighted-profile R factor (Rp = 2.03%). Additionally, the XRD patterns of both Obq-COF and Ace-COF demonstrate that the incorporation of Cu single atoms does not significantly alter the overall crystal structure or crystallinity of the substrates. This indicates that Cu loading does not adversely affect the structural integrity of the COFs frameworks. Moreover, scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HR-TEM) were employed to observe the morphological and structural alterations in the COFs before and after Cu incorporation (Fig. 2c, d and Supplementary Figs. 7 and 8). SEM micrographs displayed a distinctive striped irregular porous structure for both Obq-COF and Ace-COF, with increased surface pore density following Cu incorporation. TEM images elucidated the interlayer texture characteristic of the 2D network architecture in both materials, which remained intact post-Cu loading. Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) images (Fig. 2e, f) displayed atomic dispersion of metallic elements in CuSA/Ace-COF and CuSA/Obq-COF, with individual Cu single-atoms appearing as isolated bright spots. Energy-dispersive spectroscopy (EDS) mapping further corroborated the homogeneous distribution of C, N, and Cu elements within CuSA/COFs.
To better elucidate the dispersion and coordination environment of elemental Cu within the COF structure, X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) analyses were conducted. The Cu K-edge XANES spectra (Fig. 3a) revealed that the absorption edge of CuSA/Ace-COF is positioned between that of Cu-foil and CuO, resembling the characteristic absorption edge of CuPc, suggesting a Cu valence state within the range of 0 to +230. The Fourier-transformed k3-weighted EXAFS (FT-EXAFS) analysis (Fig. 3b) in the R space of CuSA/Ace-COF and CuPc revealed a similar peak at 1.54 Å, corresponding to the first-shell coordination of Cu−N. The absence of metallic bonding, analogous to Cu-Cu interactions (2.24 Å) in the standard Cu-foil, suggests potential monoatomic dispersion of Cu species within the COF structure. Subsequently, the monodispersity of Cu atoms in CuSA/Ace-COF was further confirmed by the wavelet transform (WT) signal displayed at 4.15 Å−1 contrasting with the WT-EXAFS profiles (Supplementary Fig. 9) of other reference systems. Quantitative EXAFS fitting analysis (Fig. 3c and Supplementary Table 2) revealed a first-shell coordination number of 2.0 for the central Cu atom, anchored to the COF nanosheets by two N atoms, with an average Cu−N bond length of 1.94 Å. Re-fitting of the EXAFS data ruled out Cu−Cl coordination, confirming that the Cu atoms in CuSA/Ace-COF are primarily coordinated as Cu−N2 due to the controlled synthesis conditions and thermal treatment.

a Cu K-edge XANES spectra and b FT k2-weighted χ(k)-function of EXAFS of CuSA/Ace-COF, Cu-foil, CuO, and CuPc. c EXAFS fitting curves of CuSA/Ace-COF in R space; the insets show the corresponding structure. d N 1 s XPS spectrum and e Cu 2p XPS spectrum of CuSA/COFs. f Calculated charge density difference for CuSA/Obq-COF and CuSA/Ace-COF with the isosurfaces value of 0.001 e/Å, where yellow represents electron-rich regions and green represents electron-deficient regions.
Furthermore, XPS analysis provided comprehensive insights into the chemical states and detailed bonding configurations of the CuSA/COFs. The C 1 s spectrum (Supplementary Fig. 10) exhibited peaks corresponding to π-π*, C = N, and C − N moieties31. The N 1 s spectrum (Fig. 3d) showed imine-N at ~400.3 eV and triazine-N at ~398.6 eV32. Importantly, the imine-N species in CuSA/Ace-COF exhibited a propensity to shift towards higher binding energies compared to CuSA/Obq-COF, indicating lower electron density33. In the high-resolution Cu 2p XPS spectra (Fig. 3e), the binding energies of Cu 2p3/2 and Cu 2p1/2 are observed at 933.1 eV and 952.7 eV, respectively, with satellite peaks indicating ionic state Cu species34. Interestingly, compared to CuSA/Obq-COF, the peaks corresponding to Cu species in CuSA/Ace-COF exhibit a shift trend towards lower binding energy, as opposed to the observed trend in imine-N species. This suggests that the imine-N atoms experience heightened interatomic interactions, likely attributable to the presence of acenaphthene units during coordination with Cu single-atoms. Moreover, differential charge density analysis from density functional theory (DFT) provides deeper insights into the discrete charge distribution within CuSA/COFs (Fig. 3f). In comparison to CuSA/Obq-COF, CuSA/Ace-COF reveal significant electron aggregation regions (marked as yellow color) surrounding the Cu metal centers, alongside accentuated electron depletion regions (marked as green color) within the acenaphthene and imine units. This result underscores the enhancement effect of the π-conjugation system of acenaphthene units on the electron-withdrawing characteristics of Cu atoms, thereby facilitating dynamic electron transfer from the delocalized electrons within the COF frameworks to the Cu single-atoms via Cu−N channel.
Overall, a novel Ace-COF material incorporating acenaphthene units was synthesized using a mild strategy, achieving atomically dispersed anchoring of Cu species within the COF frameworks to form CuSA/Ace-COF. Various physicochemical characterizations confirmed the presence of single-atom Cu, predominantly coordinated as Cu−N2 within the pore structure of CuSA/Ace-COF. Compared to the CuSA/Obq-COF, the introduction of acenaphthene units in CuSA/Ace-COF disrupts the electronic equilibrium between the COF frameworks and the Cu metal centers, facilitating the transfer of delocalized π-electrons to the Cu sites via the Cu−N channels, thereby potentially enhancing the catalytic performance of CuSA/Ace-COF.
Photo-driven PMS activation for pollutants degradation
A proof-of-concept investigation was conducted utilizing CuSA/Ace-COF and CuSA/Obq-COF as model catalysts for the photo-Fenton-like treatment of wastewater containing typical EOCs. Specifically, sulfamethoxazole (SMX), ibuprofen (IBU), bisphenol A (BPA), and benzophenone (BP) were selected as model EOCs to evaluate the degradation performance of CuSA/COFs through photo-driven PMS activation under various experimental conditions (Fig. 4a and Supplementary Fig. 11). In the PMS/Vis system, the removal percentages of all four pollutants within 20 min remained below 2.8%, indicating negligible activation of PMS by visible light alone. Additionally, both bare Ace-COF and Obq-COF demonstrated no photocatalytic degradation of EOCs (Supplementary Fig. 12), even when the reaction time was extended to 4.0 h, underscoring the essential role of Cu single-atom in enhancing photocatalytic activity. In the CuSA/COFs/Vis system, CuSA/Ace-COF demonstrated significantly higher photodegradation rates for EOCs compared to CuSA/Obq-COF, suggesting the superior photocatalytic performance of CuSA/Ace-COF. In addition, in the CuSA/COFs/PMS system, CuSA/Ace-COF also exhibited effective catalytic activity towards EOCs degradation. Specifically, the degradation rates of SMX, IBU, BPA, and BP in CuSA/Ace-COF/PMS system were 0.035 min−1, 0.032 min−1, 0.029 min−1, and 0.032 min−1, respectively (Fig. 4b and Supplementary Fig. 13). Interestingly, CuSA/Ace-COF exhibited exceptionally rapid degradation rates for all four EOCs in the CuSA/COFs/PMS/Vis integrated system. Within a mere 12 min, CuSA/Ace-COF achieved removal percentages of 99.5%, 94.7%, 95.5%, and 98.7% for SMX, IBU, BPA, and BP, respectively. Notably, focusing on SMX removal, the pseudo-first-order rate in the CuSA/Ace-COF/PMS/Vis system (0.434 min−1) surpassed those in both the CuSA/Ace-COF/Vis (0.024 min−1) system and CuSA/Obq-COF/PMS/Vis (0.011 min−1) system by 18.1-fold and 39.5-fold, respectively. This result underscores the synergistic coupling of CuSA/Ace-COF-mediated photocatalysis and non-homogeneous PMS activation, which exerts an accelerated degradation effect on the pollutants. In both the Cu2+/PMS and Cu2+/PMS/Vis systems (Supplementary Fig. 14), the efficiency of SMX removal was notably constrained, coupled with sluggish degradation kinetics. This indicates that the superiority of non-homogeneous (0.434 min−1) over homogeneous (0.004 min−1) reaction in the photo-Fenton-like process facilitated by CuSA/Ace-COF. The mineralization efficiencies of the two catalysts for SMX in CuSA/COFs/PMS/Vis system were assessed through total organic carbon (TOC) analysis. As depicted in Fig. 4c, CuSA/Ace-COF achieved a notable reduction in solution TOC to 27.3% within 180 min, whereas CuSA/Obq-COF exhibited antioxidative properties, maintaining the TOC above 90.4% even after 300 min. This result suggests the exceptional capacity of CuSA/Ace-COF for deep oxidation, facilitating the decomposition of pollutants into CO2 and H2O. CuSA/Ace-COF exhibits notable performance superiority surpassing state-of-the-art catalysts, as evidenced by comparative analysis against previously reported references (Fig. 4d and Supplementary Table 3). Additionally, an exhaustive assessment of stability for CuSA/Ace-COF under diverse environmental conditions was conducted. As depicted in Supplementary Fig. 15, CuSA/Ace-COF demonstrates robust pollutant removal efficiency across a broad pH range of 3–11, with SMX removal consistently exceeding 97.6%. Notably, the leaching of Cu species from the catalyst remained below 34.5 μg L−1 across varying pH levels after 10 h, representing only 1.03% of the total Cu content in the material (Supplementary Fig. 16). Under neutral conditions, continuous monitoring of CuSA/Ace-COF for 80.0 h showed no significant increase in Cu species (Supplementary Fig. 17), indicating that leaching saturation was reached. This concentration is well below the threshold associated with aquatic biotoxicity, underscoring the eco-environmental safety of the material. Moreover, over 98.9% of SMX was effectively removed by CuSA/Ace-COF even after undergoing 10 consecutive catalytic degradation cycles (Supplementary Fig. 18). Subsequent characterization of the catalyst using XRD, FTIR, and AC-HAADF-TEM following these cycles revealed no discernible structural or chemical alterations, suggesting the exceptional stability and reusability of the CuSA/Ace-COF catalyst (Supplementary Fig. 19).

a SMX degradation efficiency and b the corresponding kinetic rate constants in the various systems. c Time-dependent TOC removal in CuSA/COFs/PMS/Vis system. d Comparison of the Kobs and degradation percentage of state-of-the-art catalysts in photoactivated PMS systems. e Quenching experiments and f corresponding removal percentage for the degradation of SMX in CuSA/Ace-COF/PMS/Vis system under various quenching conditions. g DMPO spin-trapping EPR spectra for •OH and •SO4−. h In situ Raman spectroscopy for the various oxidation systems. i Toxicity indices of SMX and its intermediates are expressed on numerical scales: acute toxicity (0 ~ 10,000), bioaccumulation factor (0 ~ 18), mutagenicity (−0.5 ~ 1.2), and developmental toxicity (0 ~ 1), with higher values indicating greater risk. Error bars are standard error values of three tests (n = 3).
Furthermore, to elucidate the primary ROS involved in pollutant degradation within CuSA/COFs/Vis and CuSA/COFs/PMS/Vis systems, ROS quenching experiments were conducted. Methanol (MeOH) and tert-butanol (TBA) were selected as •OH quenchers, with MeOH also serving as a scavenger for •SO4−. Nitroblue tetrazolium (NBT) and furfuryl alcohol (FFA) were employed as scavengers for •O2− and 1O2, respectively35,36. In the photocatalytic systems (Supplementary Figs. 20 and 21), the introduction of NBT resulted in a decline in SMX removal from 39.1% and 9.2% to 13.4% and 3.5% within 20 min in the CuSA/Ace-COFs/Vis and CuSA/Obq-COFs/Vis systems, respectively. This indicates that the degradation process in these systems is primarily mediated by •O2− radicals. In the CuSA/Obq-COF/PMS/Vis system (Supplementary Fig. 22), the inhibition of SMX degradation by excess ROS scavengers remained relatively stable, with removal percentages maintained at 14.2–23.7%. Conversely, the efficacy of pollutant degradation in CuSA/Ace-COF/PMS/Vis system (Fig. 4e, f) was significantly hindered upon the addition of MeOH and TBA. Specifically, the removal of SMX within 12.0 min decreased from 99.5% to 8.7% and 16.7% by these two scavengers, respectively, indicating that •OH were the predominant ROS in the CuSA/Ace-COF/PMS/Vis system. This shift is attributed to the efficient decomposition of PMS by CuSA/Ace-COF, which reallocates the majority of photogenerated electrons from activating molecular oxygen to generating substantial •OH radicals, thereby dominating the degradation process of EOCs. To eliminate the contribution of non-radical oxidation pathways to SMX degradation, the role of electron transfer and surface-activated ROS in CuSA/Ace-COF/PMS/Vis system was investigated. As shown in Supplementary Fig. 23, the contribution of these two non-radical pathways to SMX degradation was deemed negligible. The aforementioned results suggest that multiple radicals (•OH, •SO4−, •O2−, and 1O2) involved pollutants degradation in the CuSA/Obq-COF/PMS/Vis system, while •OH emerge as the predominant radical driving the degradation process in the CuSA/Ace-COF/PMS/Vis system. The active species generated in different systems were further assessed using electron paramagnetic resonance (EPR) spectroscopy. DMPO-•OH, DMPO-•SO4−, DMPO-•O2−, and TEMP-1O2 signals were identified using DMPO and TEMP as spin trapping agents, respectively37. As depicted in Fig. 4g and Supplementary Fig. 24, no signals were detected in CuSA/Obq-COF/PMS system, indicating that CuSA/Obq-COF alone is inert in activating PMS. Weaker DMPO-•O2− and TEMP-1O2 signals were observed in CuSA/Obq-COF/PMS/Vis and CuSA/Ace-COF/PMS systems. Interestingly, the DMPO-•O2− and TEMP-1O2 radical signals in CuSA/Ace-COF/PMS/Vis system nearly disappeared, while a significantly amplified DMPO-•OH signal was observed compared to CuSA/Obq-COF/PMS/Vis system. In contrast to CuSA/Obq-COF/PMS/Vis system, the concentration of quantified •OH increased by a factor of 2.21 in CuSA/Ace-COF/PMS/Vis system. The in-situ Raman spectra were further were employed to track the surface chemical evolution of the catalyst during PMS activation under visible light conditions (Fig. 4h and Supplementary Fig. 25). In the PMS solution, peaks at ~887 cm−1 and ~1066 cm−1 were attributed to the presence of HSO5−, while the peak at ~984 cm−1 was indicative of SO42−38. Following the addition of CuSA/Ace-COF, the Raman peaks associated with adsorbed PMS (*PMS) underwent a discernible shift, specifically from 774 cm−1 to 780 cm−1, underscoring the robust interaction between PMS and active sites on the CuSA/Ace-COF surface. Upon the introduction of visible light into the system, the HSO5− peak sharply declined, while the SO42− peak increased, suggesting rapid PMS decomposition under light conditions in the CuSA/Ace-COF/PMS/Vis system, thus generating reactive radicals. The addition of PMS significantly enhanced the open circuit potential signals (Supplementary Fig. 26), demonstrating improved electron transfer from Cu single-atom sites to PMS, particularly in the CuSA/Ace-COF/PMS/Vis system. These findings confirm that the introduction of acenaphthene units effectively modifies the electronic structure of both COF substrates and Cu species, leading to enhanced selectivity and yield of •OH generation by PMS under visible light, as demonstrated by the ROS quenching experiments. To investigate the CuSA/Ace-COF-mediated photo-Fenton-like degradation of SMX, DFT calculations were performed to analyze its electronic structure (Supplementary Figs. 27 and 28, and Supplementary Table 4), revealing electron-rich regions around specific atoms and identifying reactive sites. Theoretical predictions and liquid chromatograph mass spectrometer (LC-MS) analysis elucidated four distinct degradation pathways (Supplementary Figs. 29 and 30) involving electrophilic attacks by •OH radicals, ultimately transforming SMX into low-toxicity intermediates. Toxicity assessments (Fig. 4i) indicated that while SMX and its intermediates exhibited low mutagenicity, the acute toxicity and bioaccumulation potential of SMX were significantly higher, highlighting the efficacy of the CuSA/Ace-COF system in reducing the ecological impact of SMX.
Exciton dissociation and free charge carrier dynamics
Degradation of pollutants through photoactivated PMS involves processes such as light absorption, electron transfer, and interfacial reactions. Evaluating the optical properties of catalysts, particularly focusing on exciton dissociation and carrier dynamics, is crucial for comprehending their catalytic performance. UV-Vis diffuse reflectance spectra (UV-Vis DRS) (Fig. 5a) of the synthesized CuSA/COFs revealed a notable extension of the light absorption range of CuSA/Ace-COF into the near-infrared region upon the integration of acenaphthene units, thereby enhancing its utilization of visible light. Employing the Kubelka-Munk-transformed equation (Supplementary Fig. 31), optical bandgap (Eg) values of CuSA/Ace-COF and CuSA/Obq-COF were calculated to be ~1.88 eV and ~2.34 eV, respectively. The reduced Eg suggests a stronger D-A interaction in CuSA/Ace-COF39. Analysis of the energy band structure of CuSA/COFs was conducted based on Eg values and estimated conduction band values derived from Mott−Schottky (M − S) measurements (Supplementary Fig. 32). Consequently, all CuSA/COFs fit the minimum reduction potential required for PMS activation, with the reduction in the bandgap primarily attributed to the elevation of the valence band in CuSA/Ace-COF.

a UV-Vis diffuse reflectance spectra of CuSA/COFs. Steady-state PL spectra with a function of the reciprocal temperature of b CuSA/Obq-COF and c CuSA/Ace-COF; the insets display the PL spectra at varying temperatures. 2D pseudo-color maps of d CuSA/Obq-COF and e CuSA/Ace-COF, and their corresponding fs-TA spectra under several representative probe delays: f CuSA/Obq-COF and g CuSA/Ace-COF. Kinetics decay profile probed at 520 nm of h CuSA/Obq-COF and i CuSA/Ace-COF.
To further investigate the influence of energy band structure variations on the excitonic effect in CuSA/COFs, the exciton binding energy (Eb) was measured using temperature-dependent photoluminescence (PL) spectroscopy, a key parameter for characterizing the exciton Coulomb interaction. As illustrated in Fig. 5b, c, the integrated PL intensity of both materials exhibited a monotonically decreasing trend with increasing temperature (from 80 K to 280 K), corresponding to the thermally activated nonradiative recombination proces. The Eb of CuSA/COFs was computed using the formula I(T) = I0/(1 + A exp(-Eb/kBT))18,40, where I0 represents the PL intensity at 0 K, T denotes the thermodynamic temperature (ranging from 80 to 280 K), and kB is the Boltzmann constant. Consequently, the Eb of CuSA/Ace-COF and CuSA/Obq-COF were determined to be 12.2 and 25.6 meV, respectively. A smaller Eb promotes the dissociation of excitons into long-lived free charge carriers, suggesting that excitons in CuSA/Ace-COF can readily surmount thermodynamic constraints and dissociate into free electrons and holes at this temperature. Room-temperature photoluminescence (PL) emission spectra (Supplementary Fig. 33) unveiled a notable reduction in the fluorescence emission intensity of CuSA/Ace-COF. A more detailed spectrum reveals a new emission peak attributed to the acenaphthene unit near 627 nm in CuSA/Ace-COF. Furthermore, time-dependent PL decay curves (Supplementary Fig. 34) demonstrated that the carrier lifetime of CuSA/Ace-COF extended from 0.56 ns to 16.57 ns compared to that of CuSA/Obq-COF, confirming that the incorporation of the acenaphthene moiety enhances the effective charge transfer in CuSA/Ace-COF. Subsequent photoelectrochemical tests (Supplementary Fig. 35) revealed that CuSA/Ace-COF, with a smaller Eb, exhibited lower electrochemical impedances, higher carrier densities, and stronger photocurrent responses. These results suggest that the incorporation of the acenaphthene unit effectively promotes exciton dissociation in CuSA/Ace-COF, facilitating the migration of photogenerated electrons from the interior to the surface of materials for interfacial catalytic reactions.
To elucidate the photogenerated free charge carrier dynamics of CuSA/Ace-COF on a precise time scale, femtosecond time-resolved transient absorption spectra (fs-TAS) were conducted. Figure 5d,e and Supplementary Fig. 36 present two-dimensional and three-dimensional pseudo-contour images, while representative decay curves at different delay times (0.05 ps ~ 5000 ns) are depicted in Fig. 5f, g. The decay kinetics analysis revealed broad absorption bands in the visible region for both CuSA/COFs, comprising broader negative bands corresponding to band-edge ground state bleach (GSB) and positive bands indicative of photoinduced excited-state absorption (PIA)39. Within the initial 3.5 ps, CuSA/Ace-COF exhibits a prominent negative signal at 470–530 nm, indicating ultrafast carrier separation, followed by a rapid decay and transition to a positive signal, possibly due to exciton dissociation and any charge transition41. The decay of photoinduced transient absorption or recovery to equilibrium reflects the reduction of photoexcited carriers at the conduction band (CB). Notably, the fs-TAS spectra of CuSA/Ace-COF markedly differ from that of CuSA/Obq-COF at 470-530 nm, attributed to the presence of acenaphthene units. Decay kinetics were analyzed using tri-exponential fitting (Fig. 5h, i) at 520 nm, yielding radiative/nonradiative lifetimes (τ1), exciton annihilation (τ2), and shallow trapping state (τ3)18. In CuSA/Ace-COF (14.6 ps), there was a notable reduction in the exciton annihilation lifetime compared to CuSA/Obq-COF (61.5 ps), suggesting a significant effect on charge carrier kinetics. The decreased τ2 of CuSA/Ace-COF is attributed to reduced exciton binding energy, leading to a shorter lifetime. Additionally, τ3 of CuSA/Ace-COF extends to 578.5 ps, indicating that the presence of acenaphthene units triggers singlet exciton dissociation and the trapping of long-lived shallow electrons, accelerating the transfer of active electrons, in contrast to CuSA/Obq-COF (146.0 ps). The enhanced charge separation in CuSA/Ace-COF promotes increased electron participation in activated PMS reactions, facilitating the efficient and selective generation of target radicals for pollutant degradation.
Enhanced mechanisms for pollutant removal
To unravel the intricate mechanism of intramolecular excited electron transfer promoted by the acenaphthene moiety within the CuSA/Ace-COF framework, DFT simulations on characteristic fragments of both COFs using the B3LYP/6-311 G(d,p) methodology were conducted. As depicted in Fig. 6a, while the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of Obq-COF exhibited uniform distribution across benzene ring structures in both the main and side chains. Conversely, Ace-COF displayed a distinct pattern, with the HOMO was predominantly localized within the benzene ring of main chain, while the LUMO primarily resided in the acenaphthene units of the side chain. Computational analyses revealed a notable decrease in the band gap (Eg) upon integration of acenaphthene units, consistent with experimental results. Ace-COF exhibited a reduction of 0.5 eV in HOMO compared to Obq-COF, indicating an increased electron affinity. The polarized distribution of the frontier orbitals predicted a discernible D-A characteristic in Ace-COF, facilitating efficient charge spatial separation25. Electrostatic potential analyses provided insights into the spatial distribution of electron density within the COFs (Fig. 6b). The incorporation of the acenaphthene unit resulted in a reorganization of the electron density profile across the COF substrate. Specifically, Ace-COF exhibited a more pronounced charge depletion region, particularly within the acenaphthene moiety, compared to Obq-COF. Moreover, electrons displayed significant localization around the imine nitrogen in Ace-COF, potentially facilitating the delivery of the N-ligand to the Cu active site. The dipole moment of Ace-COF is calculated to be 3.58 Debye, which is 1.43 Debye higher than that of Obq-COF. This elevated molecular dipole moment facilitates efficient charge transfer within the molecule. Theoretical analyses of frontier orbital distributions and molecular dipole moments indicate that the incorporation of the acenaphthene unit induces localized polarization within the Ace-COF, enhancing exciton dissociation and generation of photogenerated carriers under visible light.

a HOMO/LUMO surfaces for two COFs, with yellow indicating positive phase and green indicating negative phase. b Electrostatic potential distribution of two COFs, red and blue colors represented electron accumulation and depletion. c COHP analysis of the Cu−Oβ bond in CuSA/Obq-COF and CuSA/Ace-COF with PMS adsorption; the inset shows the corresponding adsorption configuration. d Optimized charge density difference configurations and Bader charge analyses of adsorption of PMS on CuSA/Obq-COF and CuSA/Ace-COF, where yellow represents electron-rich regions and green represents electron-deficient regions. e The density of states of 4 s and dz2 orbitals in CuSA/COFs. f Free-energy diagram for PMS activation to •OH radicals on CuSA/Obq-COF and CuSA/Ace-COF. g Mechanism of photocatalytic PMS activation over CuSA/COFs. h Schematic illustration of the continuous-flow photo-Fenton-like reaction device and its application for wastewater treatment.
After analyzing the enhancement of electron transfer, the interfacial reaction process involved in the activation of PMS at the active site were further investigated through DFT calculations. Initially, the adsorption configuration of PMS on CuSA/COFs was optimized (Supplementary Fig. 37), revealing single-atom Cu as the primary adsorption site for PMS, where Oβ in PMS bonded to the individual Cu atoms. The adsorption energies reveal that the introduction of acenaphthene moieties notably enhances the adsorption strength of single-atom Cu sites for PMS. The chemical bonding alterations upon PMS adsorption were scrutinized via crystal orbital Hamiltonian population (COHP) analysis. The value of integrated COHP (ICOHP) up to Fermi surface facilitated the assessment of bonding contributions between neighboring atoms, thereby quantifying the bonding strength12. As depicted in Fig. 6c and Supplementary Fig. 38, for CuSA/Obq-COF, the ICOHP values of the Cu−Oβ bond and Oβ − Oγ bond were −1.80 and −3.77 eV, respectively. Conversely, in CuSA/Ace-COF, the ICOHP value of the Cu−Oβ bond exhibited an increase of −2.24 eV, indicating strengthened adsorption stability between PMS and the material interface, facilitating PMS adsorption and activation reactions. Furthermore, the ICOHP value of Oβ − Oγ in CuSA/Ace-COF exhibited a decrease of −3.15 eV, implying an increased propensity for Oβ − Oγ bond cleavage and consequent activation of PMS into *SO4 and *OH intermediates. Differential charge density analysis revealed that the formation of more evident electron depletion regions (metal sites on the catalyst) and electron accumulation regions (O atoms on PMS) in CuSA/Ace-COF. Bader charge calculations (Fig. 6d) further yielded values of −0.63 |e| for CuSA/Ace-COF and −0.37 |e| for CuSA/Obq-COF, respectively. The higher value for CuSA/Ace-COF indicates an increased electron migration from the Cu metal center to PMS.
To elucidate the intrinsic mechanisms underlying the heightened adsorption and activation of PMS on CuSA/Ace-COF, a comprehensive analysis of the partial density of states (PDOS) were conducted to discern the d-orbital differentiation of CuSA/COFs. As illustrated in Fig. 6e, the PDOS peaks within the eg* band of the d orbitals exhibit a discernible broadening trend with diminishing intensity from CuSA/Obq-COF to CuSA/Ace-COF. This trend indicates the efficacy of acenaphthene unit in augmenting the broadening of the eg* band, enlarging the non-overlapping region between dz2 and a1g*. Consequently, more antibonding state of the adsorbed intermediates is elevated further above the Fermi energy level, thereby enhancing the binding affinity between the single-atom Cu and the adsorbed PMS molecules42,43. However, despite the enhance of adsorption and electron transfer mechanisms, a complete understanding of the preferential formation of •OH by activated PMS at the CuSA/Ace-COF interface remains elusive. Gibbs free energy (∆G) calculations (Fig. 6f) for the PMS activation step on the CuSA/COFs surface unveiled the spontaneous exothermic process of the generation of *OH and *SO4 from the self-decomposition of PMS on CuSA/Ace-COF (∆G = −0.70 eV), in contrast to the endothermic process on CuSA/Obq-COF (∆G = 0.35 eV). Consequently, during the activation of PMS, the Cu site of CuSA/Ace-COF selectively binds to the Oβ site in PMS. Subsequent electron transfer to the *OH intermediate via the Cu-Oβ bond pathways assumes a pivotal role in the preferential generation of •OH (HSO5− + e− → SO42− + •OH).
Overall, in CuSA/Ace-COF, the single-atom Cu functions as the catalytic site for PMS activation, facilitating electron transfer within the photo-Fenton-like process (Fig. 6g). The incorporation of the acenaphthene units effectively mitigates the excitonic effects inherent in COFs, regulating the broadening of the eg-band associated with the Cu metal centers. There units not only serve as an “electron booster”, augmenting the electron density in Cu single-atom sites and enhancing PMS adsorption. They also reduce the exciton binding energy of Ace-COF substrate, acting as a “pump” to drive the rapid and efficient electron migration. Consequently, this promoting the selective generation of •OH radicals through PMS activation in CuSA/Ace-COF/PMS/Vis system.
Environmental applications in natural water matrices
To evaluate the stability of CuSA/Ace-COF in practical water treatment scenarios employing photo-Fenton-like method, replication experiments were conducted under various environmental conditions. Firstly, the influence of various ions on the performance of the CuSA/Ace-COFs/PMS/Vis system in degrading SMX was investigated (Supplementary Fig. 39), revealing that 3.0 mM cations and NO3− had negligible effects, while Cl− and CO32− slightly inhibited the process due to interactions generating low-redox-potential radicals (•Cl and •CO3−)44. Additionally, the presence of humic acid (HA) and fulvic acid (FA) as models for dissolved organic matter (DOM) demonstrated competitive adsorption with SMX for ROS (Supplementary Fig. 40), yet even at concentrations up to 6.0 mgC L−1, the degradation efficiency remained high, achieving 99.5% and 98.1% SMX removal within 20 min. Secondly, to evaluate the real-world applicability of CuSA/Ace-COFs, SMX was tested in various natural water matrices (Supplementary Fig. 41 and Supplementary Table 5), including river water, lake water, groundwater, and treated wastewater. The results indicated that river water, lake water, and groundwater minimally affected SMX removal, with reductions of only 1.7–4.3%. Although there was a slight decrease in degradation efficiency in WWTPs effluent, 91.2% removal was still achieved within 15 min, with complete removal possible by extending treatment time or increasing PMS dosage.
Furthermore, to explore the potential application scenarios of the CuSA/Ace-COFs/PMS/Vis system in treating real wastewater, a continuous-flow photo-Fenton-like reaction device mediated by CuSA/Ace-COFs was constructed (Fig. 6g and Supplementary Fig. 42). The prepared solutions, with one containing PMS and the other comprising both catalyst and pollutant (Methylene blue), were simultaneously pumped into a mixer and then flowed in a bottom-to-top direction through a spiral reactor. Under the illumination of LED lamp positioned at the center of spiral up-flow reactor, the activation of PMS by the CuSA/Ace-COFs was promptly triggered, leading to the generation of abundant •OH radicals conducive to pollutant degradation. Obviously, the progressive colorless of solution from the bottom to the top of the reactor denoted effective degradation, followed by the segregation of the catalyst and treated effluent facilitated by a membrane filtration process. This highlights the availability of the pollutants flow system in water treatment, which can be tailored for pollutants with varying degradation rates by adjusting the wastewater pumping rate. After continuous operation for 240 min (Supplementary Fig. 43), CuSA/Ace-COFs maintained a high removal percentage of pollutants, reaching 100%, while the leaching of metal Cu remained consistently below 0.9 μg L−1. To further evaluate the suitability of this system in practical engineering applications, we conducted an assessment of the energy consumption (electrical energy per order (EE/O) values) involved in treating micropollutants (500 μg L−1 of SMX) using the CuSA/Ace-COFs/PMS/Vis photo-Fenton-like system. Detailed methodology and information are present in the supplementary information. Generally, EE/O values < 0.5 − 10 kWh m−3 are deemed competitive within the realm of AOPs for water pollution control15,45. Lower EE/O values (0.35 kWh·m−3) observed for the removal of emerging pollutants in CuSA/Ace-COFs/PMS/Vis system, compared to CuSA/Obq-COFs/PMS/Vis system (Supplementary Table 6), indicate that the photo-Fenton-like reaction achieved superior micropollutant removal with reduced energy consumption.
Discussion
Overall, we synthesized two types of CuSA/COFs, incorporating and lacking acenaphthene units, via linker engineering to elucidate the critical role of excitonic behavior in the photo-Fenton degradation of EOCs. Temperature-dependent PL spectroscopy revealed that the incorporation of acenaphthene units significantly reduced the intrinsic exciton binding energy in CuSA/Ace-COF. The ultrafast carrier dynamic of carriers in CuSA/Ace-COF was corroborated by fs-TAS. As a result, the low-excitonic effect CuSA/Ace-COF (12.2 meV) demonstrated a 39.5-fold enhancement in kinetic performance and a 22.5-fold reduction in economic cost for SMX degradation compared to CuSA/Opa-COF (25.6 meV). This work establishes a paradigm for modulating excitonic behavior in CuSA/COFs to optimize the photo-Fenton reaction through linker engineering strategy.
Methods
Synthesis of Ace-COF and Obq-COF
The synthesis of pristine COF involved a conventional Schiff base reaction. Initially, 4,4′4″-(1,3,5-triazine-2,4,6-triyl)trianiline (TTA, 0.2 mmol, ≥99.5%) and 1,2-acenaphthenequinone (Ace, 0.3 mmol, ≥99.5%) were mixed in a 10 mL Pyrex tube, followed by the addition of a solvent mixture consisting of dioxane/acetonitrile/6 M acetic acid (1.0 mL/1.0 mL/0.2 mL). The Pyrex tube was subsequently treated with ultrasonication for 15 min and degassed using three freeze-pump-thaw cycles, then sealed and heated at 120 °C for 72 h. The resulting precipitate was isolated by filtration, washed thoroughly with acetone and THF, and then stirred in DMF solution for 3.0 h to eliminate any unreacted monomers. The obtained powder was subsequently collected and dried under vacuum at 80 °C overnight, yielding a dark-red Ace-COF material (104 mg, 82.8% yield). A similar procedure was employed, replacing 1,2-Acenaphthenequinone (Ace, 0.3 mmol, ≥99.5%) with o-Benzoquinone (Obq, 0.3 mmol, ≥99.5%), to obtain brownish-yellow Obq-COF material (87 mg, 84.2% yield).
Synthesis of CuSA/Ace-COF and CuSA/Obq-COF
The aforementioned COF (200 mg) were dispersed in deionized water (70 mL) with stirring under ultrasonication for 30 min. Then, CuCl2·2H2O (40 mM; 10 mL, ≥99.0%) was gradually added dropwise to the solution while continuously stirring for 2.0 h to ensure the uniform dispersion of COF in the solution. The mixture was then transferred to a Teflon-lined stainless-steel autoclave and heated at 100 °C for 24.0 h. After cooling to room temperature, the solid precipitate was separated by centrifugation and washed with H2O and CH3OH. The precipitate was vacuum dried at 60 °C overnight. Afterward, the powder was transferred to a tube furnace and heated at 180 °C for 2.0 h to obtain CuSA/Ace-COF or CuSA/Obq-COF catalysts.
Characterization of COFs and CuSA/COFs
The physical structure and surface properties of the samples were investigated using XRD and FTIR. SEM, HR-TEM, and AC-HAADF-STEM were employed to analyze the microstructure. ss-NMR and XPS were utilized for in-depth analysis of the chemical structure and electronic environment of the samples. The distribution and coordination structure of Cu elements in the samples were investigated by synchrotron-based XAFS spectroscopy. The recombination and kinetics of carriers in the photoexcited state of the samples were studied using photoluminescence spectroscopy and Ultra-fast fs-TA spectroscopy. The UV-Vis DRS, Mott-Schottky plots, periodic on/off photocurrent responses, and electrochemical impedance spectroscopy were evaluated by photoelectrochemical tests. The radical species in the reaction process were detected by EPR spectroscopy. Detailed methods are provided in the Supporting Information.
Catalytic degradation of pollutants
SMX, IBU, BPA, and BP were selected as representative EOCs to elucidate the degradation behavior within a photoactive PMS system. The reaction solution (50 mL) containing 10.0 mg L−1 pollutant and 0.1 g L−1 catalyst was subjected to stirring in darkness for 30 min to attain adsorption/desorption equilibrium. Subsequent to pH adjustment with 0.1 M HCl or 0.1 M NaOH, 0.5 mM of PMS was introduced, followed by light irradiation using a 300 W xenon lamp fitted with a cutoff filter (λ ≥ 420 nm, 50 Mw cm−2). At required intervals, the reaction solution (500 μL) was sampled and promptly quenched with MeOH (100 μL). Subsequently, the solution was filtered through a 0.22 µm membrane to separate the solid catalyst. The residual concentration of pollutant was determined using an Agilent high-performance liquid chromatography (HPLC) system equipped with a C18 chromatographic column and a UV-vis detector. TOC concentration was quantified utilizing a SHIMADZU TOC-L analyzer with an ASI-L autosampler. Intermediate products were analyzed by an Agilent HPLC time-of-flight mass spectrometer (LC-MS Triple TOF 5600, AB Sciex, Foster City, CA) equipped with an electrospray ionization (ESI) source operating in negative ion mode. Detailed methods are provided in the Supporting Information.
Computational methods and data analysis
The periodic electronic structures of COFs and CuSA/COFs were performed at the DFT + U level using the Vienna ab initio simulation package (VASP 5.4.1)46,47. The Perdew-Burke-Ernzerhof (PBE) functional with generalized gradient approximation (GGA) was employed to handle the exchange-correlation interactions48,49. The calculations were performed with a cutoff energy of 450 eV and convergence thresholds of 10-4 eV and 0.02 eV Å−1. Structural optimization was carried out using a 3 × 3 × 1 Monkhorst-Pack (MP) k-point mesh sampling of the Brillouin zone, followed by electronic property calculations with a denser 6 × 6 × 1 MP k-point grid. For the analysis of non-covalent interactions (NCI) of the molecular models, Gaussian 16 software was employed to optimize the structures at the B3LYP/6-311 G (d, p) level with the DFT-D3 empirical dispersion correction50,51. Detailed methods are provided in the Supporting Information.
Data availability
All data supporting the findings of the study are available within the main text, the Supplementary Information and the Source Data files that accompany this article. Source Data file has been deposited in Figshare under accession code DOI link: https://doi.org/10.6084/m9.figshare.27186216 Source data are provided with this paper.
Code availability
We have provided a full code availability statement in the manuscript.
References
Yang, Y. et al. Which micropollutants in water environments deserve more attention globally? Environ. Sci. Technol. 56, 13–29 (2022).
Xu, J. et al. Elucidating the charge redistribution in bimetallic metal-organic frameworks: High-electron-density reactive center for removal of organic micropollutants in water. Chem. Eng. J. 468, 143723 (2023).
Zhu, C. et al. Optimized pore configuration in solar-driven regenerable adsorbent for organic micro-pollutants removal. Chem. Eng. J. 426, 131244 (2021).
Yao, W. et al. Seasonal variation and dissolved organic matter influence on the distribution, transformation, and environmental risk of pharmaceuticals and personal care products in coastal zone: A case study of Tianjin, China. Water Res. 249, 120881 (2024).
Zhong, M. et al. Emerging organic contaminants in Chinese surface water: Identification of priority pollutants. Engineering 11, 111–125 (2022).
Xu, J. et al. Curved strain-induced modulation of potential difference optimizes electron-mediated persulfate activation for pollutant removal. Appl. Catal. B Environ. Energy 357, 124285 (2024).
Xu, J. et al. Unveiling enhanced electron-mediated peroxymonosulfate activation for degradation of emerging organic pollutants. Appl. Catal. B Environ. 341, 123356 (2024).
Qian, M. et al. Modulation of charge trapping by island-like single-atom cobalt catalyst for enhanced photo-Fenton-like reaction. Adv. Funct. Mater. 33, 202208688 (2023).
Wu, X.-L. et al. Directional and ultrafast charge transfer in oxygen-vacancy-rich ZnO@single-atom cobalt core-shell junction for photo-Fenton-like reaction. Angew. Chem. Int. Ed. 135, 202305639 (2023).
Yang, J., Zhu, M. & Dionysiou, D. D. What is the role of light in persulfate-based advanced oxidation for water treatment? Water Res. 189, 116627 (2021).
Shen, Y. et al. Pyridine-linked covalent triazine frameworks with bidirectional electron donor-acceptor for efficient organic pollution removal. J. Hazard. Mater. 444, 130428 (2023).
Shen, Y. et al. Mechanistic insights into the atomic distance effect on adsorption and degradation of aromatic compounds. ACS Catal. 13, 8943–8954 (2023).
Shen, Y. et al. Defect-abundant covalent triazine frameworks as sunlight-driven self-cleaning adsorbents for volatile aromatic pollutants in water. Environ. Sci. Technol. 53, 9091–9101 (2019).
Zhu, C. et al. Unveiling the dynamic evolution of single-atom Co sites in covalent triazine frameworks for enhanced H2O2 photosynthesis. ACS Catal. 14, 2847–2858 (2024).
Zhu, C. et al. Unveiling spin state-dependent micropollutant removal using single-atom covalent triazine framework. Adv. Funct. Mater. 33, 202210905 (2023).
Ran, L. et al. Engineering single-atom active sites on covalent organic frameworks for boosting CO2 photoreduction. J. Am. Chem. Soc. 144, 17097–17109 (2022).
Liu, F. et al. Regulating excitonic effects in covalent organic frameworks to promote free charge carrier generation. ACS Catal. 12, 9494–9502 (2022).
Shen, R., Li, X., Qin, C., Zhang, P. & Li, X. Efficient photocatalytic hydrogen evolution by modulating excitonic effects in Ni-intercalated covalent organic frameworks. Adv. Energy Mater. 13, 202203695 (2023).
Shi, Y. et al. Surface boronizing can weaken the excitonic effects of BiOBr nanosheets for efficient O2 activation and selective NO oxidation under visible light irradiation. Environ. Sci. Technol. 56, 14478–14486 (2022).
Zhu, C. et al. Bidirectional progressive optimization of carbon and nitrogen defects in solar-driven regenerable adsorbent to remove UV-filters from water. ACS ES&T Engg. 1, 456–466 (2021).
Xu, J. et al. A nanocubicle-like 3D adsorbent fabricated by in situ growth of 2D heterostructures for removal of aromatic contaminants in water. J. Hazard. Mater. 423, 127004 (2022).
Shen, Y. et al. Unveiling excitonic effect-regulated singlet oxygen generation towards efficient photocatalytic H2O2 production. Chem. Eng. J. 475, 146383 (2023).
Wang, H. et al. Oxygen-vacancy-mediated exciton dissociation in BiOBr for boosting charge-carrier-involved molecular oxygen activation. J. Am. Chem. Soc. 140, 1760–1766 (2018).
Wu, C. et al. Polarization engineering of covalent triazine frameworks for highly efficient photosynthesis of hydrogen peroxide from molecular oxygen and water. Adv. Mater. 34, 202110266 (2022).
Yang, C., Wan, S., Zhu, B., Yu, J. & Cao, S. Calcination-regulated microstructures of donor-acceptor polymers towards enhanced and stable photocatalytic H2O2 production in pure water. Angew. Chem. Int. Ed. 61, 202208438 (2022).
Chen, H. et al. A visible-light-harvesting covalent organic framework bearing single nickel sites as a highly efficient sulfur-carbon cross-coupling dual catalyst. Angew. Chem. Int. Ed. 60, 10820–10827 (2021).
Shen, Y. et al. Enhanced photocatalytic performance of S-doped covalent triazine framework for organic pollutant degradation. J. Zhejiang Univ. Sci. A 23, 988–997 (2022).
Jena, H. S. et al. Acetylacetone covalent triazine framework: An efficient carbon capture and storage material and a highly stable heterogeneous catalyst. Chem. Mater. 30, 4102–4111 (2018).
Yang, J. et al. Protonated imine-linked covalent organic frameworks for photocatalytic hydrogen evolution. Angew. Chem. Int. Ed. 60, 19797–19803 (2021).
Chen, X. et al. Multivalent Cu sites synergistically adjust carbonaceous intermediates adsorption for electrocatalytic ethanol production. Nat. Commun. 15, 7691 (2024).
Shen, Y. et al. Bifunctional covalent triazine frameworks based on Ti-ON bonds for micropollutants removal: Effects of 3D extended structure and electron transport bridges. Chem. Eng. J. 465, 143026 (2023).
Zhu, Q., Shi, L., Li, Z., Li, G. & Xu, X. Protonation of an imine-linked covalent organic framework for efficient H2O2 photosynthesis under visible light up to 700 nm. Angew. Chem. Int. ed. 136, e202408041 (2024).
Shen, Y. et al. A hybrid block consisting of covalent triazine frameworks and GO aerogel with switchable selectivity between adsorption of UV filters and regeneration under sunlight. Chem. Eng. J. 395, 125074 (2020).
Zang, Y. et al. Ionic covalent organic nanosheet anchoring discrete copper for efficient quasi-homogeneous photocatalytic proton reduction. Appl. Catal. B Environ. 302, 120817 (2022).
Miao, J. et al. Single-atom MnN5 catalytic sites enable efficient peroxymonosulfate activation by forming highly reactive Mn(IV)-oxo species. Environ. Sci. Technol. 57, 4266–4275 (2023).
Zhu, C. et al. Metal monovacancy-induced spin polarization for simultaneous energy recovery and wastewater purification. Chem. Eng. J. 451, 138537 (2023).
Xu, X. et al. Identifying the role of surface hydroxyl on FeOCl in bridging electron transfer toward efficient persulfate activation. Environ. Sci. Technol. 57, 12922–12930 (2023).
Li, X. et al. CoN1O2 single-atom catalyst for efficient peroxymonosulfate activation and selective cobalt(IV)=O generation. Angew. Chem. Int. Ed. 62, e202303267 (2023).
Hou, Y. et al. Efficient photosynthesis of hydrogen peroxide by cyano-containing covalent organic frameworks from water, air and sunlight. Angew. Chem. Int. Ed. 63, e202318562 (2024).
Chen, Y. et al. Achieving long-lived shallow trapping states in carbon nitride through the n-π*electronic transition for enhanced photocatalytic hydrogen generation. Appl. Catal. B Environ. Energy 342, 123453 (2024).
Wang, W. et al. Fullerene-graphene acceptor drives ultrafast carrier dynamics for sustainable CdS photocatalytic hydrogen evolution. Adv. Funct. Mater. 32, 202201357 (2022).
Zhong, H. et al. Key role of eg* band broadening in nickel-based oxyhydroxides on coupled oxygen evolution mechanism. Nat. Commun. 14, 7488 (2023).
Zhong, H. et al. Optimization of oxygen evolution activity by tuning eg* band broadening in nickel oxyhydroxide. Energy Environ. Sci. 16, 641–652 (2023).
Wang, J. & Wang, S. Effect of inorganic anions on the performance of advanced oxidation processes for degradation of organic contaminants. Chem. Eng. J. 411, 128392 (2021).
Loeb, S. K. et al. The technology horizon for photocatalytic water treatment: Sunrise or Sunset? Environ. Sci. Technol. 53, 2937–2947 (2019).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter Mater. Phys. 54, 11169–11186 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 59, 1758–1775 (1999).
Blochl, P. E. Projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Kresse, G. & Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kadam, A. et al. Comparative performance evaluation and systematic screening of solvents in a range of Grignard reactions. Green Chem. 15, 1880–1888 (2013).
Acknowledgements
This research was supported by the Zhejiang Provincial Natural Science Foundation of China (LR24B070001 to Y.S.), National Natural Science Foundation of China (22276171 to Y.S., 22406170 to C.Z., 22006131 to Y.S., and 22106136 to L.L.), Fundamental Research Funds for the Provincial Universities of Zhejiang (RF-C2023009 to Y.S.), Zhuhai Science and Technology Bureau (ZH22017003210025PWC to Q.L.F.), Natural Science Foundation of Hubei Province (2022CFB820 to Y.Z.).
Author information
Authors and Affiliations
Contributions
Y.S. and C.Z. conceived and designed the study, supervised the project, and wrote the original manuscript. C.Z., M.Z.Y. and B.J. conducted the synthesis, characterization, and catalytic performance tests. L.L. and Q.L.F. performed the theoretical studies. Y.Z. contributed to experimental discussions and data analysis. S.S., B.L.C. and Y.S. provided input for the discussion and revised the manuscript. All authors contributed to discussion of the results and the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Kai Xi and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, C., Yang, M., Jiang, B. et al. Insights into excitonic behavior in single-atom covalent organic frameworks for efficient photo-Fenton-like pollutant degradation. Nat Commun 16, 790 (2025). https://doi.org/10.1038/s41467-025-56103-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56103-6
This article is cited by
-
Dynamic oxygen vacancy engineering on CuO via refreshable catalytic surface for high-efficient water decontamination
Nature Communications (2026)
-
Enhanced photocatalytic degradation of polyethylene microplastics under simulated sunlight using Fe3O4@TiO2/Ag nanocomposite and optimization via response surface methodology
Journal of Materials Science: Materials in Electronics (2025)
-
Fabrication of flexibility-rigidity alternately assembled solid-state luminescence covalent organic frameworks: application to rifampicin antibiotics detection and latent fingerprint recognition
Microchimica Acta (2025)
-
Facile fabrication of CoFe2O4/CNPs activation of peroxymonosulfate toward efficient p-nitrophenol destruction: the role of light irradiation
Journal of Materials Science: Materials in Electronics (2025)
