
Abstract
Immune checkpoint inhibitors (ICIs) have limited efficacy in microsatellite stable (MSS) metastatic colorectal cancer (mCRC), and combination therapy needs to be further explored. In this single-arm, open-label, phase II trial (NCT04745130), we evaluate the efficacy and safety of the combination therapy of antiangiogenesis (regorafenib) and ICI (sintilimab) in patients with MSS mCRC. The primary endpoint is overall survival (OS). Secondary endpoints include progression free survival (PFS), objective response rate (ORR), disease control rate (DCR), duration of response (DoR) and safety. The median OS and PFS are 14.1 months (95% CI: 10.5–17.7) and 4.1 months (95% CI: 3.4–4.8), respectively. The ORR is 21.4%, DCR is 63.1%, and DoR is 13.0 months (95% CI: 2.5–23.5). Patients with RAS/RAF wild-type exhibit significantly longer median OS (23.3 months, 95% CI: 10.0–36.6) compared to those with mutations (12.1 months, 95% CI: 8.4–15.8). The combination therapy is well tolerated and has limited toxicity. Biomarker analysis, including transcriptome sequencing and multiplex immunohistochemistry staining are performed. The efficacy of this combination treatment is tied to specific gene expressions governing tumor metabolism. Moreover, the effectiveness of immunotherapy depends on the abundance of immune cells, as well as the distance between immune cells and tumor cells.
Similar content being viewed by others

Introduction
Colorectal carcinoma (CRC) is the third most prevalent malignancy and represents the second leading cause of cancer-associated deaths globally, accounting for an estimated over 1.9 million new cases (including anal cancer) and 935,000 deaths in 20201. Approximately 23% of patients with CRC present with metastasis at the time of diagnosis, contributing to the overall unfavorable prognosis of metastatic CRC (mCRC), with a modest 5-year survival rate of only 15.6%2. Systemic treatment for mCRC is based on palliative fluorouracil-based chemotherapy regimens associated with agents targeting vascular endothelial growth factor (VEGF) or epidermal growth factor receptor (EGFR)3. However, there are still only a handful of targeted therapeutic agents for mCRC patients who have progressed upon second-line treatment. A few agents (e.g., regorafenib, trifluridine/tipiracil, and fruquintinib) have recently been tested for their therapeutic effect on mCRC, each of which showed inferior efficacy in mCRC, with an objective response rate (ORR) ranging from 1% to 4%4,5,6,7.
Immune checkpoint inhibitors (ICIs) targeting the programmed cell death receptor-1/ligand-1 (PD-1/PD-L1) pathway have revolutionized the landscape of cancer treatment and are now recognized as evolving regimens for various cancers. In the management of mCRC, ICIs have been specifically demonstrated to improve the clinical outcomes of patients with microsatellite instable (MSI). However, this subgroup represents a minimal portion of the broader mCRC patient population8,9,10,11. Consequently, there persist unmet clinical needs in the third-line treatment for mCRC patients with a microsatellite stable (MSS)/mismatch repair proficient (pMMR) status, who do not exhibit sensitivity to ICIs.
Sintilimab, a fully human IgG4 monoclonal antibody, selectively binds to PD-1 on the surface of T cells to block the interaction of PD-1 with its ligands (PD-L1 and PD-L2) and subsequently restores the endogenous antitumor response of T cells12. On the other hand, regorafenib functions as a multi-targeting tyrosine kinase inhibitor (TKI) to target a diverse range of tyrosine kinases, including VEGFRs, TIE2, PDGFR, FGFR, RET, and CSF1R. Through the inhibition of these kinases, regorafenib exerts antitumor effects by downregulating angiogenesis, tumor cell proliferation, invasion, and metastasis. It has been approved for the treatment of mCRC13. Accumulating evidence indicates that the combination of antiangiogenetic agents and PD-1 checkpoint inhibitors can collaboratively impact the tumor microenvironment by modulating angiogenesis and antitumor immunity, leading to synergistic antitumor effects14,15,16,17,18.
Variable clinical responses have been documented with the use of TKIs targeting VEGFRs in combination with ICIs in the third-line setting for patients with MSS mCRC. The REGONIVO study reported an ORR of 36% (95% CI: 18.0–57.5%) with the combination of regorafenib and nivolumab (a PD-1 inhibitor) in Japanese patients19. And in American patients, the combination of regorafenib and nivolumab showed an ORR of 7% (95% CI: 2.4–15.9%)20. In contrast, the REGOMUNE study showed no objective response among the 43 patients who were evaluated for assessing the treatment efficacy (an ORR of 0%) of regorafenib combined with avelumab (a PD-L1 inhibitor)21. Additionally, two other phase Ib/II studies involving a limited cohort of Chinese patients explored the combination of fruquintinib plus sintilimab and regorafenib plus toripalimab and exhibited ORRs of 23.8% (95% CI: 8.2–47.2%) and 15.2% (95% CI: 5.7–32.7%), respectively22,23.
In terms of the third-line treatment of mCRC with targeted combination immunotherapy, clear predictors of efficacy are lacking. Subgroup analyses in the REGONIVO and REGOTORI studies revealed varied ORRs based on clinicopathological characteristics. In the REGONIVO study, patients with lung metastases had a higher ORR (50%), while those with liver metastases had a lower ORR (15.4%)19. In the REGOTORI study, patients with liver metastases had a lower ORR (8.7%) than those without (30.0%). Gut microbiome analysis identified a higher abundance of Clostridium difficile in non-responders22,24. Molecular markers also showed differences, whereby CPS > 1 and a high TMB were correlated with a higher ORR, while the impact of RAS mutations varied between studies. Immune microenvironment analysis in the REGOMUNE study suggested there were associations between macrophage infiltration and a poor progression free survival (PFS)/overall survival (OS), and CD8+ T-cell infiltration and improved outcomes21. However, the results may have been biased due to the limited sample size, requiring confirmation from larger populations.
In this work, we evaluate the efficacy and safety of sintilimab plus regorafenib for the treatment of MSS mCRC. The combination regimen is well tolerated and offers a longer survival benefit, particularly in patients with RAS/RAF wild-type. Sequential biomarker analysis, including transcriptome sequencing and multiplex immunohistochemistry staining between the treatment-sensitive and treatment-resistant patients reveal several putative biomarkers and immune microenvironment characteristics.
Results
Baseline patient characteristics
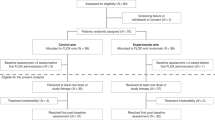
A total of 103 patients were enrolled in the study between November 2020 and February 2023 (Fig. 1). The median patient age was 57 years old, ranging from 28 to 75 years old. Among the enrolled patients, 61 (59.2%) were male, and 87 (84.5%) were patients with left-side CRC. Additionally, 53 (51.5%) patients expressed RAS/RAF wild-type, while 49 (47.6%) had multiple organ metastasis. Notably, 83.5% of the patients had undergone prior anti-VEGF, and 45.6% had received anti-EGFR therapy. The patients’ detailed baseline characteristics are presented in Table 1.

Patients meeting the enrollment criteria received regorafenib and sintilimab until disease progression or the occurrence of an intolerable adverse event.
Efficacy
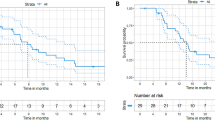
The trial enrollment process is visually depicted in Fig. 1. As of June 7, 2023, the median follow-up length was 19.9 months (95% CI: 12.8–27.0). All the trial participants had at least one oncological imaging assessment. Scheduled visit has all been completed in the trial and the survival follow up is still in progress. Among the 103 patients, 55 patients experienced a fatal event, leading to a median OS (mOS) of 14.1 months (95% CI: 10.5–17.7) (Fig. 2a), and 6-month OS rate of 85.4% (95% CI: 78.5–92.4%). Furthermore, univariate analysis of OS revealed significant differences in gender, Eastern Cooperative Oncology Group performance scale (ECOG PS) score, liver metastasis, and RAS/RAF gene status (Fig. 2b). RAS/RAF gene subgroups, as depicted through Kaplan-Meier survival curves and the Log-rank test, showed mOS of 23.3 months (95% CI: 10.0–36.6) in patients with RAS/RAF wild-type, significantly outperforming the mutant patients with mOS of 12.1 months (95% CI: 8.4–15.8) (Fig. 3a). It was found that patients without liver metastasis had mOS of 19.2 months (95% CI: 15.2–23.2) and median PFS (mPFS) of 5.4 months (95% CI: 1.1–9.7), both of which were significantly better than those with liver metastasis (Fig. 3b, c).

a Kaplan-Meier curves of OS. The mOS was 14.1 months (95% CI: 10.5–17.7) and 6-month OS rate was 85.4% (95% CI: 78.5–92.4%). The 95% CIs for point estimates are shown in gray shading. Data are presented as median (95% CI). b Forest plot for the subgroup analysis of OS. Univariate analysis of OS revealed significant differences in gender, ECOG, liver metastases, and RAS/RAF gene mutations. Data are presented as Hazard Ratio (95% CI). Source data are provided as a Source Data file. OS overall survival, mOS median OS, ECOG Eastern Cooperative Oncology Group, VEGF vascular endothelial growth factor, EGFR epidermal growth factor receptor.

a Kaplan-Meier curves of OS by RAS/RAF mutation status. The mOS was 23.3 months (95% CI: 10.0–36.6) in patients with wild-type RAS/RAF genes, the mOS was 12.1 months (95% CI: 8.4–15.8) in patients with mutant-type RAS/RAF genes. b Kaplan-Meier curves of OS by live metastasis status. The mOS was 19.2 months (95% CI: 15.2–23.2) in patients with liver metastasis, the mOS was 12.4 months (95% CI: 11.0–13.9) in patients without liver metastasis. c Kaplan-Meier curves of PFS by live metastasis status. The mPFS was 5.4 months (95% CI: 1.1–9.7) in patients with liver metastasis, the mPFS was 3.1 months (95% CI: 1.7–4.5) in patients without liver metastasis. Two-sided Log-rank test was used for survival comparison in (a–c). Source data are provided as a Source Data file. OS overall survival, mOS median OS, PFS progression free survival, mPFS median PFS.
Seventy-eight patients experienced disease progression or death, leading to a mPFS of 4.1 months (95% CI: 3.4–4.8) (Fig. 4a), and the outcomes of the univariate analysis of mPFS are detailed in Fig. 4b. One patient (1.0%) achieved complete response (CR), 21 patients (20.4%) achieved partial remission (PR), and 43 patients (41.7%) achieved stable disease (SD) according to the RECIST 1.1 criteria, with an ORR of 21.4% (95% CI: 13.3–29.4%) and a disease control rate (DCR) of 63.1% (95% CI: 53.6–72.6%) (Fig. 5a). The patients who achieved an objective response had a duration of response (DoR) of 13.0 months (95% CI: 2.5–23.5) (Fig. 5b). In this study, 41 patients underwent follow-up therapy, including 36 patients (35.0%) on targeted therapy, 31 patients (30.1%) on chemotherapy, 6 patients (5.8%) on immunotherapy, 3 patients (2.9%) on radiation therapy, and 4 patients (3.9%) on other therapy.

a Kaplan-Meier curves of PFS. The mPFS was 4.1 months (95% CI: 3.4–4.8). The 95% CIs for point estimates are shown in gray shading. Data are presented as median (95% CI). b Forest plot for the subgroup analysis of PFS. Univariate analysis of PFS revealed significant differences in gender. Data are presented as Hazard Ratio (95% CI). Source data are provided as a Source Data file. PFS progression free survival, mPFS median PFS.

a Best percentage change in target lesion size from baseline. According to the RECIST 1.1 criteria, ORR was 21.4% (95% CI: 13.3–29.4%) and DCR was 63.1% (95% CI: 53.6–72.6%). b Kaplan–Meier curves for the DoR. The patients who achieved an objective response had a DoR of 13.0 months (95% CI: 2.5–23.5). The 95% CIs for point estimates are shown in blue shading. Data are presented as median (95% CI). Source data are provided as a Source Data file. ORR objective response rate, DCR disease control rate, DoR duration of response.
Safety
The median duration of exposure was 4.1 (2.1–7.2) months for regorafenib and 4.1 (2.2–7.2) months for sintilimab. Nine patients discontinued the treatment or had their dose adjusted due to adverse events. A comprehensive safety analysis of all the treated patients showed that 96.1% experienced at least one treatment-related adverse event (TRAE), with 8.7% experiencing a grade 3 TRAE. There was no grade 4–5 TRAE or serious adverse event (SAE), and no death attributed to TRAE. Common TRAE (i.e., those with an incidence of >10%) were asthenia (29.1%), hand-foot syndrome (27.2%), gastrointestinal symptoms (25.2%), mucositis (17.5%), skin rash (16.5%), hypertension (16.5%), and anorexia (14.6%). The most common grade 3 TRAE included hand-foot syndrome (2.9%) and skin rash (2.9%) (Table 2).
Biomarker analysis
We analyzed 20 samples (ten treatment-sensitive patients achieved disease remission RECIST responders, while ten treatment-resistant patients experienced rapid disease progression RECIST non-responders), and tissues were selected from the diagnostic biopsy or initial resection of CRC lesion, using the DNBSEQ platform, yielding an average of 6.19G of data per sample, with a genome match rate of 23.95% and a gene set match rate of 5.05%. A total of 17,196 genes were detected, and differential expression analysis identified 4135 genes as differentially expressed genes (DEGs). Furthermore, a gene heatmap (Fig. 6a) was used to illustrate the relative expression levels of genes in the treatment-sensitive and resistant groups, whereas a volcano plot (Fig. 6b) showed that there were 2497 upregulated and 1638 downregulated genes in the treatment-sensitive group compared to the treatment-resistant group. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis highlighted the differential expression genes were involved in metabolic pathways, acute myeloid leukemia, platinum drug resistance, P53 signaling pathway, ErbE signaling pathway, etc., with the metabolic pathway being the most significant (Fig. 6c). The metabolic pathways included lipid, carbohydrate, and amino acid (Fig. 6d). Gene Set Enrichment Analysis (GSEA) analysis showed the differential genes were enriched in multiple metabolism-related and immune-related pathways (Fig. 6e).

a A total of 4135 differentially expressed genes were identified in this study, and the gene heatmap shows the relative expression levels of the genes in the treatment-sensitive and treatment-resistant groups. b Volcano plot showing that a total of 2497 genes were upregulated and 1638 genes were downregulated in the treatment-sensitive group compared with the treatment-resistant group. c, d KEGG analysis showing the gene enrichment related to multiple metabolic pathways, including lipid metabolism, carbohydrate metabolism, amino acid metabolism. e GSEA analysis showing the gene enrichment related to multiple metabolism-related and immune-related pathways. Source data are provided as a Source Data file.
The top 20 DEGs underwent immune microenvironment correlation analysis (Fig. 7a), validated with The Cancer Genome Atlas (TCGA) data. The immunity scores were higher for the downregulated genes (ULBP1, HOXB9) in the treatment-sensitive group with low expression (Fig. 7b). A comparison of tumorous and para-tumorous tissues revealed that the expression patterns of the upregulated genes (DYRK2, ID4) in the treatment-sensitive group were low expressed in the tumor tissues, and the downregulated genes (TRMT112, ULBP1) in the treatment-sensitive group were high expressed in the tumor tissues (Fig. 7c). GSEA immune cell analysis demonstrated higher immune cell expression levels, for the upregulated genes (DYRK2, ID4) in the treatment-sensitive group with high expression (Fig. 7d). Metabolic pathway analysis indicated the glycine, serine, and threonine metabolic pathways were negatively correlated with the upregulated genes in the treatment-sensitive group (DYRK2), and positively correlated with the downregulated genes in the treatment-sensitive group (ULBP1) (Fig. 7e). Validation with the TCGA database suggested a role of these genes in regulating the immune microenvironment and influencing treatment outcome.

a The 20 most statistically significant molecules were selected based on the gene heatmap. b Immunity scores derived from TCGA showing that the downregulated genes (ULBP1, HOXB9) in the treatment-sensitive group with low expression had higher immunity scores (n = 454). c Comparison of the tumorous (n = 454) and para-tumorous (n = 41) tissues derived from TCGA showing that the upregulated genes (DYRK2, ID4) and downregulated genes (TRMT112, ULBP1) in the treatment-sensitive group were low and high expressed in tumor tissues, respectively. d GSEA immune cell analysis derived from TCGA showing that the upregulated genes (DYRK2, ID4) in the treatment-sensitive group with high expression had higher immune cell expression (n = 454). Centers, boxes, and whiskers indicate medians, quantiles, and minima/maxima, respectively, in (b–d), and two-sided Wilcoxon rank-sum test was used for comparison in (b–d). e Metabolic pathway analysis derived from TCGA showing that the glycine, serine and threonine metabolic pathways were negatively correlated with the genes upregulated in the treatment-sensitive group (DYRK2) and positively correlated with the genes downregulated in the treatment-sensitive group (ULBP1), with Spearman’s rank correlation test applied.
In order to delve deeper into the correlation between immune cell infiltration and therapeutic effect, we selected tissue samples from 15 patients each in both treatment-sensitive and treatment-resistant groups for comprehensive immunohistochemical analysis. Using DAPI, panCK, FoxP3, CD103, CD68, PD-L1, CD20, and CD8 staining, we elucidated the tumor immune microenvironment (Fig. 8a, b). Subsequent analysis of various marker-positive cell density showed a significant higher density of CD8+ T cells in the treatment-sensitive group compared to the treatment-resistant group, without significant difference found in other marker-positive cells between the two groups (Fig. 8c). Furthermore, we conducted an advance analysis on cell distance within the tumor immune microenvironment. For example, the distance from CD8+ to CK+ signifies the proximity between the nearest CD8+ cell and CK+ cell (Fig. 8d). Our findings indicated that the mean distance of CD8+ cells to CK+ cells was significantly shorter in the treatment-sensitive group than in the treatment-resistant group. Conversely, the mean distance of other immune cell marker-positive cells (FoxP3, CD103, CD68, PD-L1, CD20) to CK+ cells did not exhibit significant difference between the two groups (Fig. 8e).

a, b Among all stains, DAPI (dark blue) for nucleus, panCK (light blue) for cancer cells, FoxP3 (red) for FoxP3 (+) Treg cells, CD103 (orange) for CD103 (+) T cells, CD68 (green) for CD68 (+) macrophages, PD‐L1 (purple) for PD-L1 (+) cells, CD20 (yellow) for CD20 (+) B cells, and CD8 (pink) for CD8 (+) T cells. Staining was performed only once in (a, b). c Comparison of cell density between treatment-sensitive (n = 15) and treatment-resistant groups (n = 15). d Cell distance within the tumor immune microenvironment. e Comparison of cell distance to CK+ cells between treatment-sensitive (n = 15) and treatment-resistant groups (n = 15). Centers, boxes, and whiskers indicate medians, quantiles, and minima/maxima, respectively, in (c, e), and two-sided Wilcoxon rank-sum test was used for comparison. Source data are provided as a Source Data file.
Discussion
Combining antiangiogenic therapy targeting with immunotherapy presents a promising approach for treating “cold tumor” MSS CRC patients. However, the efficacy of antiangiogenic agents combined with immunotherapy in MSS CRC needs further clarification, as previous studies have shown mixed results. The present study focused on sintilimab in combination with regorafenib for treating mCRC, involving the largest Chinese sample size (103 participants) yet in this area. The present study suggests that combining regorafenib with sintilimab in the treatment of mCRC surpasses the efficacy of regorafenib alone (ORR: 1–4%; OS: 6.4–8.8 months)4, achieving a higher ORR and prolonged OS (ORR: 21.4%; OS: 14.1 months). In addition, regorafenib has many other sites besides antiangiogenesis, and whether inhibition of these sites can increase the efficacy of immunotherapy needs to be further explored.
In the American study, the ORR of regorafenib combined with nivolumab was 7%, the mPFS was 1.8 months, and the mOS was 11.9 months, and no more than 3 systemic treatment regimens were required before enrollment20. In the LEAP-017 study, lenvatinib combined with pembrolizumab had an ORR of 10.4%, mPFS of 3.8 months, and mOS of 9.8 months25. Although the result of the LEAP-017 study was negative, the subgroup analysis suggested that the Asian population was more likely to benefit from this treatment mode. In our study, there were at least two previous systemic treatment regimens, but the effective rate and survival data were further improved, suggesting that Chinese patients could benefit more from regorafenib with sintilimab. We found patients without liver metastasis had an mOS of 19.2 months (95% CI: 15.2–23.2) and mPFS of 5.4 months (95% CI: 1.1–9.7), better than those with liver metastasis. The same conclusion was addressed in the American study, and none of the patients with ORR had liver metastasis at baseline20. The subgroup analysis in LEAP-017 study also suggested that patients without liver metastasis were more likely to benefit from lenvatinib combined with pembrolizumab25. Translational research has shown that the expression of positive immune cells (such as CD8+ T cells) was significantly reduced in patients with liver metastasis compared with patients without liver metastasis, leading to an inhibitory immune microenvironment26, which may contribute to the poor outcomes in patients with liver metastasis.
This combination regimen demonstrated effectiveness, especially in patients with RAS/RAF wild-type CRC, exhibiting a mOS of 23.3 months (95% CI: 10.0–36.6) comparing with 12.1 months (95% CI: 8.4–15.8) in RAS/RAF mutant-type patients. However, there was no significant difference in mPFS between the two groups, but as time went on, the survival benefits of RAS/RAF wild-type patients became more and more obvious, and the PFS rates at 12 months and 18 months were higher than those of RAS/RAF mutant-type patients. Since the majority (60.2%) of patients did not receive follow-up therapy, we believe that the survival benefit was due to the combination of regorafenib and sintilimab. Some patients have long-lasting therapeutic benefits and imaging data of two patients with PFS over 24 months were included in the Supplementary Note 1 in the Supplementary Information file. However the subgroup analysis in LEAP-017 study suggested that patients with RAS mutant-type were more likely to benefit from lenvatinib combined with pembrolizumab25. Regorafenib had relatively clear combined immune targets, which improved the survival benefits of RAS/RAF wild-type patients enrolled in this study. However the targets of regorafenib and lenvatinib are different, and it is reasonable to assume that the combined effect of lenvatinib with immunotherapy was limited due to its target.
Analyses suggested that CRC samples with NRAS mutations had an increased infiltration of M1/M2 macrophages and CD56 bright NK cells, while SMAD4 mutations were correlated with higher CD8+ T cells and M1 macrophages. Conversely, PIK3CA mutation led to more M2 macrophages and fewer CD8+ T cells. The presence of other immune checkpoints in PD-L1 negative CRC tumor immune macrophages suggested diverse tumor immune microenvironment types were influenced by different mutations27. Investigating the molecular underpinning of this divergence, the existing literature highlights the negative correlation between KRAS G12D point mutation in non-small cell lung cancer and PD-L1 levels, as well as reduced CD8+ T cells due to the secretion of chemokine CXCL10/CXCL11. CRC specimens have exhibited diminished cytotoxic CD8+ T-cell infiltration in KRAS mutant tumors, leading to poor responses to PD-1 monoclonal antibody and T-cell therapy. This may be related to the sensitization of cytotoxic T cells by lactate produced by KRAS mutant tumor cells through the NF-κB pathway, leading to tumor cell death28. Another study exploring the efficacy of cetuximab in combination with immunotherapy in KRAS wild-type patients showed a 47% augmentation in the number of cytotoxic T cells in the tumor after such treatment. The tumor microenvironment was characterized by a large number of TIM3+ and CTLA4+ cells and fewer activated OX40+ cells29. Side by side, it was elucidated that KRAS wild-type patients exhibit a more robust immune microenvironment.
The effectiveness of antiangiogenic-targeted drugs combined with immunotherapy in MSS CRC relies on the immune microenvironment, which exhibits distinct phenotypes, broadly categorized as immune inflamed, immune exclusion, and immune desert types30. Most MSS CRC falls into the latter two categories, limiting the efficacy of immunotherapy. To further explore the interplay between immune cell infiltration and therapeutic response, we conducted extensive immunohistochemical staining, which showed a significantly higher density of CD8+ T cells and shorter distance of CD8+ cells to CK+ cells in the treatment-sensitive group compared to the treatment-resistant group. The REGOMUNE trial further corroborated the pivotal role of CD8+ T cells in combination immunotherapy, demonstrating that increased tumor infiltration of CD8+ T cells during cycle 2, day 1, was associated with superior outcomes compared to baseline21. In the first-line ATEZOTRIBE study of mCRC, the Immunoscore Immune-Checkpoint (IC) were evaluated with patient outcomes, which including densities, proximity, and clustering of PD-L1 cells and CD8 T cells in the tumor core. And patients with Immunoscore IC-high tumors derived higher benefit from the addition of atezolizumab31. This suggests that the effectiveness of immunotherapy is influenced not only by the quantity of immune cells but also by their spatial position proximity to tumor cells.
GSEA analysis was performed and showed there were genetic differences in the activation and depletion pathways of CD8+ T cells between the treatment-sensitive and treatment-resistant groups in the trial, emphasizing the crucial role of CD8+ T cells in immunotherapy. Previous literature supports the idea that regorafenib enhances immunotherapy efficacy by increasing the CD8+ T-cell proportion and decreasing the Treg cell ratio in the tumor immune microenvironment. This study’s findings, which are consistent with existing conclusions from previous studies, affirm the significance of CD8+ T cells in the explored targeted therapy combination immunotherapy, highlighting their pivotal role in the treatment approach for patients with MSS CRC. KEGG analysis was performed and revealed metabolic pathway-specific expressions, especially in glycine, serine, and threonine metabolism, the metabolism of linoleic acid, the metabolic differences of retinol, and so on. Malignant cells reprogram metabolic processes for unrestricted survival, with glycine and serine potentially exerting immunosuppressive effects32,33. Abnormalities in tumor metabolic pathways may contribute to resistance in MSS CRC. This study proposes that regorafenib, by interacting with multiple targets in malignant cells and inhibiting metabolic pathways, can enhance targeted combined immunity against cancer cells, addressing potential resistance mechanisms, which has been further explored in the ongoing research.
In the initial design of this study, we assumed an event rate of 70% based on data from the literature. However, the actual event rate observed in the study was 53%, a difference that may be attributed to factors such as patient characteristics, treatment efficacy, or follow-up duration. The lower-than-expected event rate has the potential to impact the statistical power of the study. To address this challenge, we reassessed the sample size and adjusted it according to the actual event rate. Despite the lower event rate, we implemented appropriate measures, including increasing the sample size, to ensure the scientific rigor and statistical reliability of the study’s conclusions. Future research should consider the potential variability in event rates and incorporate sensitivity analyses during the design phase to manage similar uncertainties.
A major limitation of this study is its nonrandomized design. Randomized controlled clinical trials could provide the most reliable evidence about the effects of a new drug. However, with a mPFS and a mOS of 4.1 and 14.1 months, respectively, our results compared favorably with those of regorafenib in the CORRECT study (mPFS: 1.9 months, mOS: 6.4 months) and CONCUR study (mPFS: 3.2 months, mOS: 8.8 months)4,5. However, only a randomized controlled trial could provide the most credible efficacy of regorafenib combined with sintilimab versus regorafenib alone. On the other hand, there may be bias in the enrollment, with a small number of patients with poor physical status excluded, and these patients may be at higher risk for adverse events that could negatively affect outcomes. In addition, during our enrollment period, there was no unified standard for PD-L1 expression detection scheme in China, so the expression of PD-L1 was not required in our research scheme. And the predictive effect of PD-L1 expression on immunotherapy efficacy in mCRC patients remains unclear, so we did not report the results regarding PD-L1 expression.
In conclusion, the combination of regorafenib and sintilimab was well tolerated in advanced MSS CRC patients, showing improved antitumor effects compared to the monotherapy of either agent. Moreover, the combination regimen offered a longer survival benefit, particularly in patients with the RAS/RAF wild-type gene. The observed outcomes suggest a potentially favorable tumor microenvironment for immunotherapy, warranting further trial through a larger cohort of this regimen in patients with this subtype of CRC.
Methods
Study design
This prospective phase II clinical trial was designated as a single-arm, single-center study, and was conducted at the Department of Gastrointestinal Oncology, Tianjin Medical University Cancer Institute and Hospital. Ethical approval was obtained from the Ethics Committee of Tianjin Medical University Cancer Institute and Hospital (Number: E2020608), aligned with the Declaration of Helsinki. This study was registered at https://www.clinicaltrials.gov before patient enrollment (clinical trial identifier NCT04745130). The trial primarily enrolled patients aged ≥18 years old with confirmed inoperable recurrent or mCRC and progression on at least two lines of standard systemic therapy. The inclusion criteria comprised status of MSS, ECOG PS score of 0–2, and presence of at least one measurable lesion according to RECIST 1.1 criteria, along with an adequate bone marrow and organ function; whereas the exclusion criteria were prior treatment with regorafenib or anti-PD-L1/PD-L2/CTLA4 antibodies, with details available in the Supplementary Note 2 in the Supplementary Information file. The first patient was enrolled on March 1st, 2021, and the last was recruited on February 25, 2023. Data are recorded by clinical research associates, based on medical files in Tianjin Medical University Cancer Institute and Hospital from March 1st, 2021 to June 7, 2023.
Sample size estimation for this exploratory single-arm trial was conducted utilizing the one-sample Log-rank test. Based on historical data, the mOS for CRC patients treated with regorafenib monotherapy is 8.8 months. Notably, a clinical phase II study, REGOMUNE, reported a mOS of 10.8 months for patients with advanced CRC receiving regorafenib combined with avelumab. In the initial design of this study, based on previous literature and related research, it was assumed that the addition of sintilimab to regorafenib could elevate the median OS from 8.8 months to 13.5 months, with an anticipated hazard ratio of 0.65 and event rate of 70%. This assumption was used to calculate the sample size to ensure that, with a two-sided significance level (α) of 0.05 and a power of 0.80, the number of events was 48, the initially calculated sample size was 68 participants. Considering a dropout rate of 20%, the initially sample size was determined to be 82 patients. However, during the actual study, we observed that the event rate was ~60%, which was significantly lower than the initially assumed 70%. To ensure the statistical power and reliability of the study results, we recalculated the sample size based on the actual event rate. The new calculation indicated that, with an anticipated hazard ratio of 0.65, event rate of 60%, a two-sided significance level (α) of 0.05, and a power of 0.80, the minimum required sample size was calculated to be 80 patients. Considering a dropout rate of 20%, the final sample size was determined to be 96 patients. A total of 103 patients were included in this study, which met the sample size required for the study.
In the initial stage of this study, we intended to enroll two cohorts of patients, one with wild-type RAS/ RAF and no prior treatment with cetuximab, receiving treatment with regorafenib in combination with cetuximab and sintilimab. However, with the recommendation of guideline and release of results from relevant phase III trials, the value and survival benefits of EGFR antibodies in frontline treatment for patients with wild-type RAS/ RAF mCRC have been approved. As a result, almost all of these patients received frontline treatment with cetuximab in clinical practice, making it very difficult to enroll patients into this cohort, ultimately leading to a suspension of enrollment for this cohort.
Study procedure
Patients meeting the enrollment criteria were administered regorafenib orally at 80 mg daily for 3 weeks followed by a 1-week break, and intravenous sintilimab at 200 mg on day 1 and then once every 3 weeks. Treatment persisted until disease progression, the occurrence of an intolerable adverse event, the investigator’s decision to discontinue, or patient withdrawal from the study (see Fig. 1 for the study flow chart). Sintilimab was used for no more than 24 months. Fixed doses were used for both drugs, with dose adjustments of regorafenib allowed during its use based on patient tolerance, while no dose adjustment was allowed for sintilimab. Depending on the patient expiring an adverse event, the use of either or both drugs could be suspended at the discretion of the patient. Permanent discontinuation was mandated if the suspension of regorafenib exceeded 4 weeks, and if sintilimab had two or more suspensions. Tumor assessments were performed using contrast-enhanced CT/MRI according to RECIST 1.1 criteria every two cycles at baseline, and every 3 months after completion of the study treatment. The patients would be followed up for survival status every 3 months up to 24 months (or until death). Safety profiles were assessed based on National Cancer Institute Common Terminology Criteria for Adverse Events Version 5.0 (NCI CTCAE5.0) at baseline and at the end of each treatment cycle until 90 days post-study treatment. Selected patients’ tumor tissue samples underwent baseline RNA sequencing and multiplex immunohistochemistry analysis to characterize the combination’s effects on the tumor and immune microenvironment and identify potential clinical benefit predictive biomarkers.
Outcomes
This study chose OS as its primary endpoint, which was measured from the initiation of study treatment to death from any cause, whereas the secondary study endpoints were designated as the ORR, DCR, DoR, PFS, and safety. ORR was determined by the proportion of patients achieving a confirmed CR and PR based on RECIST1.1 criteria; DCR was defined as the proportion of patients who showed a CR, PR, or SD state per the RECIST1.1 criteria; DoR was calculated from the first assessment of CR or PR to the first occurrence of disease progression or death, whichever came first; and PFS was defined as the duration from the initiation of study treatment to the earliest instance of progression or death from any cause, whichever occurred first. Lastly, safety evaluations were graded using the common toxicity criteria outlined in the NCI CTCAEV5.0. Since PD-1 checkpoint inhibitors have residual efficacy for a longer duration, in addition, recommendations issued by the NHS in 2020 regarding clinical cancer research during the COVID-19 period34, we changed primary endpoint to OS before enrollment, which was also been amended in our protocol.
Statistical analysis
The statistical analysis was performed utilizing SPSS 22.0 software, with measurement information presented through the median and quartiles [M(P25–P75)] based on the data distribution, while categorical information was delineated using constitutive ratios. The Kaplan-Meier method was used to estimate the OS, PFS, and DoR. Survival comparison between groups utilized the Log-rank test, and the Cox proportional hazard risk model facilitated between-group analyses for the OS and PFS, along with estimating the corresponding 95% confidence intervals (95% CI). The median follow-up time for survival was calculated with the reverse Kaplan-Meier method. The Clopper-Pearson method was also used to assess the 95% CI for the ORR, with differences evaluated using Chi-squared or Fisher’s exact tests. The significance level was set at α = 0. 05 (two-sided).
Sequencing
Rolling cycle amplification was employed for the replication of single-stranded circular DNA molecules, resulting in the generation of DNA nanoballs (DNB) harboring multiple DNA copies. These high-quality DNBs were then loaded into patterned nanoarrays using a high-intensity DNA nanochip technique. The sequencing process was facilitated through combinatorial probe-anchor synthesis.
Gene quantification differential expression analysis
The gene expression levels were computed using RSEM (v1.3.1). Heatmaps visualizing the gene expression differences across various samples were generated with pheatmap (v1.0.8). Differential expression analysis utilized DESeq2(v1.4.5), employing a significance threshold Q ≤ 0.05.
Gene annotation
To comprehend the phenotypic changes, the annotated differentially expressed genes underwent KEGG and GSEA enrichment analysis conducted with Phyper (https://en.wikipedia.org/wiki/Hypergeometric_distribution) based on a hypergeometric test. The significant levels for the terms and pathways were corrected by Q value with a rigorous threshold of Q ≤ 0.05.
Multiplex immunohistochemistry staining
The investigate of tumor cells and immune cells in the immune microenvironment involved conducting multiplex immunohistochemistry following the Opal 7-Color Manual IHC Kit protocol. Primary antibodies used included panCK, FoxP3, CD103, CD68, PD-L1, CD20, and CD8. Imaging and scanning of Slides were performed using a PerkinElmer VectraX® platform. The results were meticulously chosen and batch analyzed using PerkinElmer Inform software, with the threshold value of each marker identified and illustrated by the Inform Score, which allowed for potential adjustment of cut-offs based on the score map and original staining images. Quantification of positively stained cells was accomplished using an R script.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Transcriptome sequencing in our study are available in the Genome Sequence Archive under the accession code HRA009402. Due to data privacy laws related to patient consent to data sharing, sequencing data is available under controlled access and data should only be used for research purposes. Due to patient privacy concerns, CT scans and pathology images are not shared publicly. Within 3 years of the publication of this paper, personal data and statistical analysis plans may be made available upon reasonable request. Qualified researchers with Physician Qualification Certificate may make requests for data by contacting corresponding author at liurui9003@163.com. The study protocol is included as Supplementary Note 2 in the Supplementary Information file. All remaining data are available within the article, Supplementary Information or Source Data file. Source data are provided with this paper.
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 71, 209–249 (2021).
National Cancer Institute Surveillance, Epidemiology, and End Results Program. Colon and Rectum SEER relative survival rates by time since diagnosis by stage at diagnosis. Accessed November 2023. https://seer.cancer.gov/statfacts/html/colorect.html.
National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: Rectal Cancer (version 4. 2023). Available at: https://www.nccn.org/professionals/physician_gls/pdf/rectal.pdf.
Grothey, A. et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381, 303–312 (2013).
Li, J. et al. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 16, 619–629 (2015).
Mayer, R. J. et al. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N. Engl. J. Med. 372, 1909–1919 (2015).
Li, J. et al. Effect of fruquintinib vs placebo on overall survival in patients with previously treated metastatic colorectal cancer: the FRESCO randomized clinical trial. JAMA 319, 2486–2496 (2018).
Marcus, L., Lemery, S. J., Keegan, P. & Pazdur, R. FDA approval summary: pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin. Cancer Res. 25, 3753–3758 (2019).
Fan, A. et al. Immunotherapy in colorectal cancer: current achievements and future perspective. Int. J. Biol. Sci. 17, 3837–3849 (2021).
Jin, Z. & Sinicrope, F. A. Mismatch repair-deficient colorectal cancer: building on checkpoint blockade. J. Clin. Oncol. 40, 2735–2750 (2022).
Pang, K. et al. Research progress of therapeutic effects and drug resistance of immunotherapy based on PD-1/PD-L1 blockade. Drug Resist. Updat. 66, 100907 (2023).
Lou, B. et al. Preclinical characterization of GLS-010 (Zimberelimab), a novel fully human anti-PD-1 therapeutic monoclonal antibody for cancer. Front. Oncol. 11, 736955 (2021).
Arai, H. et al. Molecular insight of regorafenib treatment for colorectal cancer. Cancer Treat. Rev. 81, 101912 (2019).
Fukumura, D., Kloepper, J., Amoozgar, Z., Duda, D. G. & Jain, R. K. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat. Rev. Clin. Oncol. 15, 325–340 (2018).
Khan, K. A. & Kerbel, R. S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 15, 310–324 (2018).
Li, S. J., Chen, J. X. & Sun, Z. J. Improving antitumor immunity using antiangiogenic agents: Mechanistic insights, current progress, and clinical challenges. Cancer Commun. 41, 830–850 (2021).
Lee, W. S., Yang, H., Chon, H. J. & Kim, C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp. Mol. Med. 52, 1475–1485 (2020).
Hu, H. et al. The research progress of antiangiogenic therapy, immune therapy and tumor microenvironment. Front. Immunol. 13, 802846 (2022).
Fukuoka, S. et al. Regorafenib plus nivolumab in patients with advanced gastric or colorectal cancer: an open-label, dose-escalation, and dose-expansion phase ib trial (REGONIVO, EPOC1603). J. Clin. Oncol. 38, 2053–2061 (2020).
Fakih, M. et al. Regorafenib plus nivolumab in patients with mismatch repair-proficient/microsatellite stable metastatic colorectal cancer: a single-arm, open-label, multicentre phase 2 study. EClinicalMedicine 58, 101917 (2023).
Cousin, S. et al. Regorafenib-avelumab combination in patients with microsatellite stable colorectal cancer (REGOMUNE): a single-arm, open-label, phase II trial. Clin. Cancer Res. 27, 2139–2147 (2021).
Wang, F. et al. Regorafenib plus toripalimab in patients with metastatic colorectal cancer: a phase Ib/II clinical trial and gut microbiome analysis. Cell Rep. Med. 2, 100383 (2021).
Guo, Y. et al. Phase 1b/2 trial of fruquintinib plus sintilimab in treating advanced solid tumours: the dose-escalation and metastatic colorectal cancer cohort in the dose-expansion phases. Eur. J. Cancer 181, 26–37 (2023).
Yao, Y., Jin, Y. & Wang, M. P-170 Exploratory biomarker findings from regorafenib plus toripalimab in patients with refractory metastatic colorectal cancer (REGOTORI study). Ann. Oncol. 33, S310 (2022).
Kawazoe, A. et al. Lenvatinib plus pembrolizumab versus standard of care for previously treated metastatic colorectal cancer: final analysis of the randomized, open-label, phase III LEAP-017 study. J. Clin. Oncol. 42, 2918–2927 (2024).
Yu, J. et al. Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat. Med. 27, 152–164 (2021).
Shi, C. et al. 481P Different gene mutation of colorectal cancer was tightly associated with various types of tumor immune microenvironment. Ann. Oncol. 31, S445 (2020).
Liu, H. et al. Mutant KRAS drives immune evasion by sensitizing cytotoxic T-cells to activation-induced cell death in colorectal cancer. Adv. Sci. 10, e2203757 (2023).
Fountzilas, C. et al. Phase Ib/II study of cetuximab plus pembrolizumab in patients with advanced RAS wild-type colorectal cancer. Clin. Cancer Res. 27, 6726–6736 (2021).
Chen, D. S. & Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 541, 321–330 (2017).
Antoniotti, C. et al. Upfront fluorouracil, leucovorin, oxaliplatin, and irinotecan plus bevacizumab with or without atezolizumab for patients with metastatic colorectal cancer: updated and overall survival results of the ATEZOTRIBE study. J. Clin. Oncol. 42, 2637–2644 (2024).
Amelio, I., Cutruzzolá, F., Antonov, A., Agostini, M. & Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 39, 191–198 (2014).
Ma, E. H. et al. Serine Is an essential metabolite for effector T cell expansion. Cell Metab. 25, 482 (2017).
de Paula, B. H. R., Araújo, I., Bandeira, L., Barreto, N. & Doherty, G. J. Recommendations from national regulatory agencies for ongoing cancer trials during the COVID-19 pandemic. Lancet Oncol. 21, 624–627 (2020).
Acknowledgements
We thank all patients and their families for participating in this trial. We thank Dr. Jing Han (Tianjin Haihe Hospital) for her guidance on statistics. There were no sponsors for this study. This work was supported by Tianjin Science and Technology Commission Key Project (21JCZDJC00980, R.L.), Special Research Project of Post-Marketing Clinical Research of Innovative Drugs (WKZX2024CX102217, R.L.), and Tianjin Key Medical Discipline (Specialty) Construction Project (TJYXZDXK-009A, J.H.).
Author information
Authors and Affiliations
Contributions
R.L., Y.B., and J.H. conceived the study. R.L., Z.J., and X.W. wrote the protocol. Z.J., X.W., S.G., L. Zhang, Y.Y., T.N., M.B., and J.D. were responsible for patient visits and treatment with help from H.L., T.D., and R.L. H.L., T.D., R.L., Y.B. and J.H. were responsible for patient recruitment. L. Zhu, J.X., and L.M. were responsible for collecting data and samples and coordinating procedures. R.L. and Z.J. were responsible for analyzing and interpreting clinical data with help from Y.B. and J.H. L. Zhu, J.X., L.M., and J.Z. were responsible for performing bioanalysis and statistical analysis with help from F.W. and Y.S. R.L., Z.J., and X.W. wrote the manuscript. All authors reviewed the manuscript, interpreted data, and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, R., Ji, Z., Wang, X. et al. Regorafenib plus sintilimab as a salvage treatment for microsatellite stable metastatic colorectal cancer: a single-arm, open-label, phase II clinical trial. Nat Commun 16, 1481 (2025). https://doi.org/10.1038/s41467-025-56748-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56748-3
This article is cited by
-
Immune checkpoint inhibitor in third-line treatment of microsatellite stable metastatic colorectal cancer: an observational and retrospective study
BMC Cancer (2026)
-
Exploring research frontiers and emerging trends of tyrosine kinase inhibitors in the treatment of colorectal cancer
BMC Cancer (2025)
