
Abstract
Bacillus subtilis uses cytoplasmic complexes called stressosomes to initiate the σB-mediated general stress response to environmental stress. Each stressosome comprises two types of proteins — RsbS and four paralogous RsbR proteins — that are thought to sequester the RsbT protein until stress causes RsbT release and subsequent σB activation. RsbR proteins have been assumed to sense stress, but evidence for their sensing function has been elusive, and the identity of the true sensor has remained unknown. Here, we conduct an alanine-scanning analysis of the putative sensing domain of one of the RsbR paralogs, RsbRA. We find that single substitutions impact but do not abolish the σB response, suggesting that RsbRA has a key role in σB response dynamics and is “tunable” and robust to substitution, but not directly supporting a sensing function. Surprisingly, deletion of the stressosome does not abolish environmental stress-inducible σB activity and instead leads to a stronger and longer-lived response than in strains with stressosomes. Finally, we show that RsbT is necessary for the stressosome-independent response and that its kinase activity is also important. RsbT thus has a previously unappreciated role in initiating stress responses and may itself be a stress sensor in the general stress response.
Similar content being viewed by others

Introduction
Bacteria constantly experience various fluctuating conditions in nature, including many conditions that cause cellular stress. Appropriate sensing and adaptation to stressors is therefore important for cell survival and growth. The soil bacterium Bacillus subtilis and related gram-positive bacteria have evolved a wide variety of ways to sense, respond to, and survive environmental stresses. One prominent way that these bacteria protect against non-lethal stress is termed the general stress response (GSR), which is regulated by the alternative sigma factor σB 1,2,3,4,5. In B. subtilis, σB activates the general stress regulon, comprising some 200 genes6,7,8, in response to energy or environmental stress. Energy stress includes starvation for nutrients and factors such as glucose, phosphate, and oxygen as well as treatments that limit biological processes, such as rifampicin, azide, and mycophenolic acid9,10,11,12,13,14. Environmental and physical stressors include salt, ethanol, heat, acid, sodium nitroprusside, and blue light9,10,14,15,16,17,18. σB controls the GSR in several other gram-positive bacteria, such as Listeria monocytogenes19,20,21,22,23, B. licheniformis24,25, B. cereus26, and Staphylococcus aureus27,28,29,30.
In B. subtilis, σB activity is ultimately controlled by the RsbVW partner-switching mechanism. In unstressed cells, σB is held inactive by the anti-sigma factor RsbW, which prevents its interaction with core RNA polymerase31,32,33,34. RsbW is also the kinase of RsbV, the anti-anti-sigma factor of σB 35. Upon stress exposure, RsbV becomes dephosphorylated, making it a preferred partner for RsbW; RsbW releases σB in favor of binding RsbV, allowing σB to activate its regulon. RsbV is dephosphorylated by two known serine/threonine phosphatases, RsbP and RsbU (Fig. 1A)36,37,38. Energy stress activates RsbP, whereas environmental stress activates RsbU9,37,39. RsbU phosphatase activity is activated by interaction with RsbT (Fig. 1A)40,41,42. The availability of RsbT to activate RsbU is controlled by sequestration of RsbT in large cytoplasmic complexes termed stressosomes43,44, which are only known to control σB in a subset of species, including B. subtilis, B. licheniformis, and L. monocytogenes, but are also present in a number of other bacteria45,46.

A Schematic of the B. subtilis general stress response. Environmental stress is thought to be sensed by stressosome complexes composed of a mixed population of 40 RsbR paralogs (different shades of red) and 20 RsbS proteins (gray) that sequester RsbT proteins (blue). Stress sensation leads to release of RsbT, which then activates downstream steps leading to release of σB. Energy stress is sensed separately, by the proteins RsbQ and RsbP, but the final steps leading to σB release are the same. B Schematic of the RsbRA protein showing the region of residues from 11 to 58 (red) that was subjected to individual Ala substitutions. Mutant rsbRA genes were added back at amyE together with rsbU to a strain background lacking all rsbR genes, ytvA, rsbP (to deactivate the energy stress pathway), and rsbU. The strain carried a σB-responsive PrsbV-lacZ reporter to assess the general stress response. C Cartoon of predicted results, with a nonfunctional or unstable RsbRA predicted to yield a constitutive σB response even without stress (green line), and a sensing-dead version of RsbRA unable to activate in the presence of stress (red line). The black line is the same wild-type data shown in other figure panels, showing the mean of biological triplicate experiments, with the shading indicating standard deviation. D–F Graphs showing β-galactosidase activity from PrsbV as a proxy for σB activity (measured at 15-min intervals for 3 h) upon stimulation of the general stress response with 3% ethanol. WT indicates complementation of the reporter strain with rsbU and wild-type rsbRA at amyE as the only source of RsbR in the cell (CSS744; black trace). No RsbRA or U indicates the uncomplemented recipient strain (CSS716; gray trace). Substitutions (e.g., “T38A”) indicate complementation of the reporter strain with rsbU and Ala-substituted rsbRA as indicated at amyE as the only source of RsbR in the cell. All traces are means of biological triplicate experiments, with the shading indicating standard deviation. D Substitutions resulting in increased (>10% increased from wild-type) peak σB activity. E Substitutions resulting in decreased (>10% decreased from wild-type) peak σB activity. F Substitutions resulting in unchanged (within 10% of wild-type) peak σB activity. G Color-coded sequence logo showing the impact of Ala substitutions. Red, decreased; blue, unchanged; green and bold, increased; black, untested; gray, unsubstituted (already Ala). Source data are provided as a Source Data file.
Each stressosome is a 1.8-MDa protein complex, comprising 20 RsbR dimers and 20 copies of RsbS, that can bind and sequester up to 20 copies of RsbT that are released upon onset of stress43,47. RsbR and RsbS proteins both include a STAS (sulfate transporter and anti-sigma factor antagonist) domain48; these domains form the core structure of the stressosome complex, with the RsbR N-terminal domains oriented outward into the cytoplasm47. In contrast to RsbS, which contains only the STAS domain, RsbR proteins bear conserved (45–50% sequence identity) STAS domains in their C-terminal half, and less-conserved (17–22% sequence identity) N-terminal non-heme globin dimers (Fig. 1B)47,49,50. B. subtilis encodes five intact RsbR paralogs, RsbRA, RsbRB, RsbRC, and RsbRD, and a blue light sensor called YtvA; these are presumably mixed within stressosomes at an unknown stoichiometry51. The stressosome is thought to somehow sense environmental stress and to activate the serine/threonine kinase activity of RsbT to phosphorylate RsbS at S59 coincidently with the release of RsbT from the stressosome (Fig. 1A) to activate RsbU and, consequently, σB 40,41,42,43,52. In this model, the release of RsbT is the central event in the environmental stress sensing pathway. The elevated σB levels resulting from environmental stress are reversed by the activity of a phosphatase, RsbX, which dephosphorylates RsbS and presumably allows RsbT recapture42,53.
Despite the availability of detailed structural information on the stressosome, it is not clear how stress is sensed by the stressosome or how RsbT kinase activity is activated. One attractive model is that RsbR proteins serve as sensors and as activators of RsbT kinase activity towards S59 of RsbS54. The outward orientation of the RsbR globin dimers in the cryo-EM structure of the wild-type B. subtilis stressosome47 is consistent with a sensing function. Additionally, YtvA senses blue light using a PAS/LOV (light-oxygen-voltage) domain in the N-terminal half of the protein51,55,56, and heme-binding RsbR orthologs in Vibrio species sense oxygen57,58. The RsbR paralogs are distinct with respect to σB response profiles (i.e., timing and magnitude) when subjected to different environmental stressors59,60, showing that RsbR proteins impact σB responses; this is at least consistent with a sensory function. However, substitutions in the linker that would presumably be important for communicating sensory information between the N-terminal globin and C-terminal STAS domains of RsbRA impacted steady-state output but not stress sensitivity61, and a similar result was obtained for specific point mutations in the N-terminal domain50, including mutations that interfered with RsbT-stressosome interactions in vitro49. Moreover, the putative outcome of RsbR stress sensing, namely stimulation of RsbT kinase activity, is not necessary for σB activation, as reduced signaling still occurs even in mutant strains with unphosphorylatable RsbS62. Hence, experimental evidence for stress sensing by RsbRs has remained elusive, despite several well-designed inquiries.
Here, we sought experimental evidence for RsbR-mediated stress sensing using RsbRA as a model. We reasoned that the N-terminal regions of RsbR proteins might include amino acid residues that are important for environmental stress sensing, and we systematically engineered a larger number of substitutions in the putative N-terminal sensing domain than in previous studies. We found that these substitutions influenced the magnitude and duration of the σB response to ethanol, a classic stressor, but that none was critical for stress sensing. Our results prompted us to reexamine the requirement for the stressosome (i.e., RsbRs and RsbS) in environmental stress sensing. Surprisingly, we found that cells deleted for stressosomes retained an environmental stress-inducible σB response that strictly depended on RsbT. Collectively, our data refine the model for stressosome function, showing that RsbT can mediate stressosome-independent environmental sensing and consistent with a sensing role for RsbT, with the stressosome primarily serving to modulate the strength and timing of the σB response.
Results
A complementation strategy to identify RsbRA residues with critical roles in stress sensing
We reasoned that if RsbR proteins have a sensory function in their N-terminal globin domain, we might identify specific amino acids in the N-terminus of an RsbR with critical roles in stress sensing. We used RsbRA as a model, as its gene resides in an operon with the downstream signaling components and because the σB responses of cells bearing RsbRA as the only RsbR paralog closely resemble those of wild-type cells59,60. We began by systematically substituting residues in the N-terminal half of the N-terminal domain of RsbRA with alanine (Fig. 1B), thus probing a more N-terminal region than in previous studies50,61. To focus exclusively on the σB response mediated by RsbRA, we used a strain deleted for the other RsbR paralogs (RsbRB, RsbRC, RsbRD, and YtvA). To avoid constitutive σB activity due to RsbT no longer being sequestered in this background44, we first deleted rsbU to disable environmental stress sensing. We also deleted rsbP to block any σB activation due to energy stress. To monitor σB activity using blue-white screening or kinetic β-galactosidase assays, we used a chromosomally encoded PrsbV -lacZ reporter. A major advantage of this complementation strategy is that sensing-dead mutants, which would show no σB response upon stressor treatment, are easily distinguished from mutants with unstable or misfolded RsbRA, which would be incompetent for stressosome formation and hence would show a constitutive response irrespective of the presence of stressor (Fig. 1C). The uncomplemented base strain, as expected, showed no response to 3% ethanol stress (Fig. 1D, gray line). We then reconstituted RsbRA-mediated environmental stress signaling by integrating a vector (pDG1730) containing both wild-type rsbRA and rsbU, each under the control of the native PrsbRA promoter (Fig. 1B). This strain showed low σB activity in the absence of stress and a characteristic σB response that peaked about 1 h after stressor addition and then subsided thereafter (Fig. 1C–F).
Complemented RsbRA point mutants impact the timing and magnitude of the σB response
We next systematically tested mutants of RsbRA with Ala substitutions at each position between I11 and K58 in the putative stress-sensing domain (Fig. 1B). In accord with the idea that this domain is important for the environmental σB response, many of the substitutions altered the timing or magnitude of the response to 3% ethanol. One mutation, T38A, increased the response magnitude (Fig. 1D, Fig. S1). The most common effect of Ala substitution was to decrease the magnitude of the response (e.g., I11A, E13A, N14A, N16A, L18A, T23A L26A, Q32A, E33A, D39A, S46A, I52A, L54A, V57A, K58A, and many more) (Fig. 1E, Figs. S1–S3). Some substitutions also altered response timing. For instance, N16A showed a faster response than the wild type, whereas the E33A and L18A substitutions showed a markedly delayed response (Fig. 1E, Fig. S1). Several other substitutions had minimal effects on the σB response, such as E17A and L37A (Fig. 1F, Fig. S1). Overall, our initial testing revealed that most Ala substitutions in this region indeed impact the σB response, consistent with a role in the overall stress response. However, we recovered neither truly sensing-dead mutants nor constitutively active mutants. To further verify the phenotypes of selected Ala substitutions, we examined them using markerless allelic complementation as a more stringent and less perturbative way to examine the function of those residues and learn whether any was sensing-dead or constitutively active.
Natively replaced RsbRA point mutants tune rather than break the σB response
For our markerless allelic replacements, we began with a strain deleted for all the RsbR paralogs except RsbRA, deleted for rsbP and with PrsbV-lacZ as a σB reporter (Fig. 2A). We then replaced native rsbRA with Ala-substituted point mutants, such that the only change from the parental strain was the Ala-encoding codon in rsbRA. We again observed impacts on the σB response timing and magnitude (Fig. 2B–D, Fig. S4). Substitutions at some positions reduced (e.g., I11, N14, V57, and K58) or increased (T38) the magnitude of the σB response in both experimental systems (Figs. 1D, E, 2B, C), increasing our confidence that these residues are truly important for influencing the magnitude of the response (Fig. 2E). Other substitutions (e.g. at E17 and L37) had a minimal effect in the complemented system (Fig. 1F) but either increased (E17) or decreased (L37) the response in the native system (Fig. 2B, C, E). Still others (N16 and L26) showed opposite effects in the two systems. In such cases of disagreement, we consider the markerless allelic replacements the more reliable system owing to the minimal perturbation to the native sigB operon (Fig. 2A). Ala substitution at the native locus also changed the timing of the σB response. For instance, the I11A variant had a delayed and decreased but longer-lived response than the wild type (Fig. 2C, Fig. S4), whereas the E17A variant had a stronger and much longer-lived response (Fig. 2B, Fig. S4).

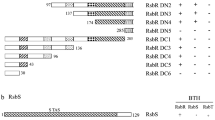
A Schematic of the allelic replacement strategy, wherein Ala-substituted RsbR was encoded at the native rsbR locus in cells deleted for all other RsbR paralogs and carrying a σB-responsive PrsbV-lacZ reporter. B–D Graphs showing β-galactosidase activity from PrsbV as a proxy for σB activity (measured at 15-min intervals for 3 h) upon stimulation of the general stress response with 3% ethanol. “True WT” (CSS1113) cells carry all four RsbR paralogs, whereas “WT” denotes a strain (CSS1480) with wild-type RsbRA as the only RsbR paralog in the cell. Substitutions (e.g., “E17”) indicate allelic replacement of the native rsbRA gene with Ala-substituted rsbRA as indicated as the only source of RsbR in the cell. All traces are means of biological triplicate experiments, with the shading indicating standard deviation. B Substitutions resulting in increased (>10% increased from wild-type) peak σB activity. C Substitutions resulting in decreased (>10% decreased from wild-type) peak σB activity. D Substitutions resulting in unchanged (within 10% of wild-type) peak σB activity. E Color-coded sequence logo showing the impact of Ala substitutions. Red, decreased; blue, unchanged; green and bold, increased; black, untested; gray, unsubstituted (already Ala). Asterisks appear below a position when the impact of that substitution was different in the complementation than in the allelic replacement, with the color of the asterisk denoting the result from the complementation strategy. F Crystal structure of the dimeric N-terminal region of RsbRA (PDB 2BNL)49 with positions impacting the σB response highlighted using the same color scheme as in panel E. The left panel shows a ribbon model, and the right panels show space-filling models. For simplicity, we highlighted only those positions whose results agreed in the complementation and allelic replacement experiments. Source data are provided as a Source Data file.
Because some substitutions displayed a minimal response to 3% ethanol stress and others showed enhanced responses, we asked whether such weakened or strengthened responses were truly stress-responsive relative to unstressed cells. Thus, we subjected a subset of substitutions (three each for complemented and native-locus rsbRA) to non-stress conditions or to stronger stress (4% ethanol). In all cases, stress increased the response over non-stress conditions, and stronger stress (4% vs 3% ethanol) elicited a stronger response, even for substitutions that showed an elevated baseline response without stress (e.g., native I11A and E17A) (Fig. S5). These results imply that the observed σB responses reflect true stress responses and not simply altered basal responses. When we mapped only those substitutions whose effects in the two experimental systems agreed (i.e., no asterisk in Fig. 2E) onto the crystal structure of the N-terminal domain of RsbRA49, we observed that they tended to cluster near unstructured regions at the junctions of alpha helices on the exposed surface of the protein (Fig. 2F, Supplementary Movie 1). Collectively, these data show that the N-terminal region of RsbRA impacts both the magnitude and the timing (onset and duration) of the σB response (a summary of the phenotypic changes is given in Table S1), but none of the tested substitutions fully abolished the response or elicited constitutive activity. Such a system, which is “easy to tune” but “hard to break”, suggests a remarkable evolutionary flexibility and robustness of the stressosome in the general stress response, where mutations can alter the response without eliminating it. In support of this notion, an alignment of 50 RsbRA protein sequences from B. subtilis genomes in NCBI showed more-frequent amino acid substitutions in the N-terminal region of RsbRA than in its C-terminal STAS domain or in the N-terminal region of its paralog RsbRB (Fig. S6).
Cells deleted for all RsbR paralogs retain an ethanol stress-inducible σB response
Even with the remarkable functional robustness of RsbRA to Ala substitution, we found that none of the 42 substitutions we had made thus far resulted in total abrogation of sensing or constitutive signaling. This finding prompted us to stop making additional substitutions and to critically re-examine the presumed model (Fig. 1A), which posits that the stressosome (i.e. RsbRs and RsbS) is required for stress sensing, leading to phosphorlyation of RsbS by RsbT and subsequent release of RsbT from its sequestered state36,41,42,63,64. A key prediction of this model is that in the absence of a functional stressosome, RsbT would never be sequestered, leading to a constitutive σB response. Indeed, a strong basal response was previously observed in a fully RsbR-deleted strain44. Nonetheless, we reasoned that one possible explanation for not finding sensing-dead or constitutively active mutants among the Ala-substituted variants is that RsbR proteins in the stressosome modulate σB response dynamics but are not strictly required for stress sensing or for preventing constitutive σB activation.
To test the idea that the stressosome is required to sense stress and to sequester RsbT, we constructed a mutant strain with all 5 known active RsbR paralogs (rsbRA, rsbRB, rsbRC, rsbRD, and ytvA)51 deleted using markerless, in-frame allelic replacement and with our standard PrsbV-lacZ σB reporter. Strikingly, when we subjected this rsbR-deleted strain to 3% ethanol stress, we observed a clear σB response to the onset of stress, rather than the expected constitutive response (Fig. 3A). The response was delayed relative to the wild-type or RsbRA-only response and was stronger and longer-lived, eventually subsiding after approximately 2 h (Fig. 3A). Might there be another source of RsbR protein in the cell? A sixth RsbR paralog, termed YetI/YezB, was evidently split by a frameshift mutation (Fig. 3B)51. While we considered it unlikely that YetI and YezB could substitute for the other RsbR paralogs, we deleted the yetI-yezB coding sequences (Fig. 3B). Adding this deletion to the rsbR-deleted quintuple mutant resulted in a response to 3% ethanol that was indistinguishable from that of the rsbR-deleted mutant (Fig. 3C). We, therefore, concluded that RsbR proteins are not necessary for mounting a σB response to 3% ethanol stress. Because the RsbR proteins are important components of the stressosome, these results also suggested that the stressosome itself may be dispensable for a σB response to environmental stress.

A Graph showing β-galactosidase activity from PrsbV as a proxy for σB activity (measured at 15-min intervals for 3 h) upon stimulation of the general stress response with 3% ethanol. WT, strain (CSS1113) carrying all four RsbR paralogs. RsbRA only, strain (CSS1480) producing RsbRA as the only RsbR paralog in the cell. No RsbR paralogs, strain (CSS1531) deleted for all four RsbR paralogs. B Schematic showing the yetI and yezB genes, which encode a putative split RsbR paralog, to scale with the rsbRA gene. The deleted region in the ΔyetI-yezB strain is shown with a dashed box. C Graph as in A but also including the trace from a strain (CSS1760) lacking all four rsbR paralogs and deleted for yetI-yezB. In panels A and C, traces are means of biological triplicate experiments, with the shading indicating standard deviation. Source data are provided as a Source Data file.
Cells deleted for RsbS display a similar σB response to RsbR-deleted cells
We next aimed to test more thoroughly the requirement for the stressosome by eliminating RsbS. RsbS is a scaffold protein in the stressosome43,47,51,65; the 20 RsbS proteins in each stressosome each bind an RsbR dimer43,47. Upon exposure to environmental stress, RsbS is specifically phosphorylated by RsbT, which is then released to bind and activate the RsbU phosphatase by direct protein-protein interaction42,63,64. We considered the formal possibility that, even in the absence of all RsbR proteins, RsbS might be able to sequester RsbT on its own to preserve the stress inducibility we observed. Conversely, deletion of rsbS, even in the presence of RsbR proteins, ought to abolish stressosome formation due to loss of the scaffolding function of RsbS36,42,43,44. We, therefore, initially deleted rsbS from wild-type (ΔytvA ΔrsbP) cells (Fig. 4A) and challenged the ΔrsbS cells with 3% ethanol. We observed a strong, longer-lived response (Fig. 4B) that was remarkably similar in magnitude and timing to the RsbR-deficient strains (Fig. 3A, C). This similarity suggested that we had uncovered a characteristic σB response of B. subtilis lacking stressosomes.

A Schematic of the 5’ portion of the rsbRA operon showing the regions deleted in the individual and combined rsbRA and rsbS deletions. B Graph showing β-galactosidase activity from PrsbV as a proxy for σB activity (measured at 15-min intervals for 3 h) upon stimulation of the general stress response with 3% ethanol. WT, strain (CSS1113) carrying all four RsbR paralogs. ΔrsbS, strain (CSS1168) deleted for rsbS as an alternative way to block stressosome formation. C Graph as in B (note different Y-axis scale) but also comparing the ΔrsbRAΔrsbS strain (CSS1392) to a strain harboring the same deletion, but in a background lacking the other RsbR paralogs and RsbP (CSS1441). All traces are means of biological triplicate experiments, with the shading indicating standard deviation. Source data are provided as a Source Data file.
Cells with a clean deletion spanning both rsbRA and rsbS no longer respond to ethanol stress
Because we had separately tested cells lacking RsbRs and RsbS, it remained a formal, albeit unlikely, possibility that RsbR and RsbS were functionally redundant with respect to σB activation. We thus built strains deleted for both the RsbR proteins and RsbS. The components of the environmental stress pathway are encoded by the genes of the eight-gene sigB operon, which contains a second, σB-driven promoter ahead of rsbV that allows positive feedback (i.e., PA rsbR rsbS rsbT rsbU, PB rsbV rsbW sigB rsbX)32,66,67. To simultaneously delete rsbRA and rsbS, we engineered a clean deletion spanning both genes’ entire coding sequences (Fig. 4A), placing this deletion in a strain background deleted for the other RsbR paralogs and RsbP. We were initially surprised that this strain showed no stress response to ethanol (Fig. 4C). We further tested the ΔrsbRAΔrsbS deletion in an otherwise wild-type background, and again observed no response except for a late response as cells approached stationary phase, which we attribute to RsbP-mediated energy stress due to its late appearance and its absence in the ΔrsbP background (Fig. 4C). Given the operonic position of rsbT just downstream of rsbRA and rsbS, we reasoned that the ΔrsbRAΔrsbS deletion might be interfering with RsbT expression or production. Hence, we examined rsbT expression by qRT-PCR. We observed that, while the ΔrsbRAΔrsbS strain showed less basal rsbT expression than the wild-type, it showed more expression than the strain deleted for all RsbRs (Fig. S7), which had shown a strong σB response (Fig. 3A). This result argues that reduced rsbT transcription is not responsible for the absent σB response in ΔrsbRAΔrsbS. Instead, subsequent proteomic analysis suggested reduced RsbT protein levels in this strain (Table 1; see below). However, this strain, unlike wild-type or RsbRs-deleted cells, showed no increase in rsbT expression after 15 min of treatment with ethanol (Fig. S7), implying the existence of σB-mediated positive feedback on rsbT expression.
Cells blocked for RsbRA and RsbS production with early nonsense mutations display a long-lived, stress-inducible σB response
To block RsbRA and RsbS production without interfering with rsbT expression or production, we introduced early stop codons in rsbRA (CAG → TAG at codon 4) and rsbS (AAA → TAA at codon 5) in different strain backgrounds (Fig. 5A). We initially tested this approach by translationally blocking only RsbS in a strain deleted for all RsbRs except RsbRA. When challenged with 3% ethanol, this strain showed a strong, delayed, long-lived response (Fig. 5B) that qualitatively resembled our deletion mutants lacking all RsbRs (Fig. 5B) or RsbS (Fig. 4B). We observed the same response when both RsbRA and RsbS were translationally blocked (Fig. 5C), offering strong support to the ideas that the stressosome is dispensable for the environmental σB response to ethanol and that the result with the ΔrsbRAΔrsbS deletion was due to a deficiency of RsbT. While the phenotypic distinction between the strain blocked for RsbS production but producing RsbRA and the strain producing both RsbRA and RsbS (Fig. 5C, blue vs. black trace) implied that RsbS was translationally blocked, it remained formally possible that a TTG codon at position 9 of rsbS could be used as an alternative translation initiation codon. To rule out this possibility, we built a strain with both codon 5 and codon 9 (TTG → TAA) of rsbS mutated and with RsbRA translationally blocked. This strain qualitatively resembled the strain blocked only at codon 5 of rsbS (Fig. S8), arguing against an effect of alternative initiation.

A Schematic of the 5’ portion of the rsbRA operon showing the points at which stop codons were introduced to the rsbRA and rsbS genes at their native chromosomal loci. B Graph showing β-galactosidase activity from PrsbV as a proxy for σB activity (measured at 15-min intervals for 3 h) upon stimulation of the general stress response with 3% ethanol. WT, strain (CSS1113) carrying all four RsbR paralogs. RsbRA only, strain (CSS1480) producing RsbRA as the only RsbR paralog in the cell. No RsbR paralogs, strain (CSS1531) deleted for all four rsbR paralogs. RsbRA only RsbSSTOP, strain (CSS1681) encoding RsbRA as the only RsbR in the cell and bearing an early stop codon in rsbS. C Graph as in B but also showing the response (orange trace) of a strain (CSS1686) with early stop codons in rsbRA and rsbS. D Graph as in C but comparing the listed strains (WT, CSS1113; RsbRA only, CSS1480; No RsbRs, CSS1531; ΔrsbRAΔrsbS, CSS1441; RsbRASTOP RsbSSTOP, CSS1686) with respect to their σB responses to 0.6 M NaCl. All traces are means of biological triplicate experiments, with the shading indicating standard deviation. Source data are provided as a Source Data file.
Stressosome-deleted cells respond to other environmental stressors and activate other σB-driven genes
As a further test of the environmental stress response in the absence of a stressosome, we asked whether stressosome-deleted cells (i.e., cells deleted for all RsbRs, RsbP, and with RsbRASTOP RsbSSTOP) could respond to stressors other than ethanol. We thus subjected this strain along with a strain lacking all RsbRs, the ΔrsbRAΔrsbS strain, and control strains (wild type and RsbRA-only) to another commonly used stressor, 0.6 M NaCl. All the tested strains showed salt-inducible σB responses, with the wild-type showing the strongest response and RsbRA-only cells showing the weakest response (Fig. 5D). The stressosome-deleted strains showed clear, stress-inducible responses were distinct in timing, with a later response being more prominent, corresponding to their longer-lived response in ethanol (Fig. 5D). In contrast, ΔrsbRAΔrsbS again showed a minimal response (Fig. 5D). We also subjected the same set of strains to acid stress in LB medium (downshift to pH 5.0). The overall response across all the strains was much weaker, with only the WT showing a delayed response after 60 min that was distinct from a no-stress control (Fig. S9A, B). Other strains, including a RsbRA-only strain and stressosome-deleted strains, showed a minimal difference from unstressed controls (S9A-B), while the ΔrsbRAΔrsbS strain showed no response (Fig. S9A). The weakness of this acid response, and the lack of a response in RsbRA-only (stressosome-replete) cells, makes it difficult to draw conclusions about the stressosome independence of the σB acid response at present. As a different test of the breadth of the stressosome-independent response, we examined the response of another previously used σB reporter, Pctc-lacZ68, to ethanol stress in the same set of strains. We again observed responses in all the strains except for ΔrsbRAΔrsbS, and the stressosome-deleted cells showed their characteristically slower but longer-lived response (Fig. S9C), in general agreement with the PrsbV results. Stressosome-deleted cells mock-induced with LB medium instead of a stressor showed little to no difference from the wild type, irrespective of which reporter was used (Fig. S9D, E). These data strengthen the case that RsbT is sufficient to sense and/or respond to multiple environmental stressors on its own, even in the absence of a functional stressosome, and that the subsequent σB response activates multiple genes in its regulon.
RsbT, and its kinase activity, are required for the stressosome-independent σB response
To examine the idea that RsbT is necessary for the stressosome-independent σB response, we first performed proteomic analyses comparing a wild-type strain to ΔrsbRAΔrsbS and RsbRASTOP RsbSSTOP strains (both of which were deleted for all other RsbR proteins and RsbP) both before and 1 h after 3% ethanol treatment. Satisfyingly, proteins encoded by deleted genes were never detected, whereas most of the other Rsb proteins were readily detected, including RsbU and RsbV, which are encoded downstream of rsbT (Table 1). RsbS was never detected, even in the wild type, suggesting that it is below the threshold of detection. RsbT was detected in both unstressed and stressed wild-type cells and in stressed RsbRASTOP RsbSSTOP cells but never in the ΔrsbRAΔrsbS deletion, lending further support to the idea that the σB response defect in ΔrsbRAΔrsbS (Fig. 4C) is specifically due to a deficiency in RsbT production.
To test the requirement of RsbT for the σB response in the absence of the stressosome, we engineered strains either deleted for or containing an early stop codon (CAA→ TAA at codon 4; Fig. 6A) in rsbT in addition to the early stop codons in rsbRA and rsbS (and deletions of all other RsbR proteins and RsbP). We again used the early-stop strategy to minimally perturb the sigB operon. Whether rsbT was deleted (Fig. S10A) or translationally blocked (Fig. 6B), σB activation upon ethanol stress was completely abrogated, consistent with RsbT being strictly required for the stressosome-independent response. This requirement for RsbT was also clearly visible for the response to NaCl (Fig. S10B), to acid (Fig. S10C), and for the Pctc response (Fig. S10D). Next, we asked whether RsbT serine kinase activity was important for the stressosome-independent response using a kinase-dead D78N substitution that was previously shown to interact with RsbS and RsbU in two-hybrid assays and to activate RsbU in vitro but was unable to transduce environmental stress in wild-type cells41. As expected, replacement of native rsbT with the D78N mutant abolished the σB response to ethanol in wild-type (ΔytvA ΔrsbP) cells (Fig. S10E). However, the D78N substitution also blocked σB activity in stressosome-deleted cells (Fig. 6C), suggesting that kinase activity is important for stressosome-independent signaling by RsbT. Consistent with earlier work41, RsbTD78N appeared to be produced at similar levels to the wild-type protein (Fig. S11). We then used plasmid-based IPTG-inducible constructs69 to test whether ectopic production of RsbT could rescue the σB response in RsbT-blocked cells. IPTG induction restored a σB response that scaled with induction level, though it was not reliably stress-inducible (Fig. 6D). Induction of rsbTD78N at the minimal level showing a response with wild-type rsbT (20 µM IPTG) did not elicit a σB response, consistent with the D78N substitution reducing the ability of RsbT to respond to stress41. We obtained similar results with plasmid-borne rsbTU, with σB activity scaling with induction level (Fig. S12A, C–E). Importantly, a plasmid bearing only rsbU yielded no response, even at 1 mM IPTG (Fig. S12B), confirming that the absence of a response in RsbT-deleted or translationally blocked cells was due to the absence of RsbT and not a polar effect on rsbU. Collectively, these results suggest that RsbT can by itself mediate stress sensing in the absence of a functional stressosome, thereby ascribing to RsbT a stressosome-independent stress sensing or transduction function. Further, RsbT kinase activity appears to be an important aspect of this function.

A Schematic of the 5’ portion of the rsbRA operon showing the point at which stop codons were introduced to the rsbRA, rsbS, and rsbT genes at their native chromosomal loci and showing the region deleted in ΔrsbT. B Graph showing β-galactosidase activity from PrsbV as a proxy for σB activity (measured at 15-min intervals for 3 h) upon stimulation of the general stress response with 3% ethanol. WT, strain (CSS1113) carrying all four RsbR paralogs. RsbRASTOP RsbSSTOP, strain (CSS1686) blocked for all stressosome components. RsbRASTOP RsbSSTOP RsbTSTOP, a strain (CSS1974) blocked for all stressosome components and with an early stop codon in rsbT. C Graph as in B but showing the response of a strain (CSS2285) blocked for all stressosome components and with an Asp-to-Asn substitution at position 78 of RsbT (RsbTD78N) to inactivate its kinase activity. D Graph as in B but showing the response of CSS1974 complemented with IPTG-inducible rsbT or rsbTD78N from pKH001 (CSS2179 or CSS2300, respectively) at the IPTG concentrations shown. All traces are means of biological triplicate experiments, with the shading indicating standard deviation. Source data are provided as a Source Data file.
Stressosome-deleted cells display repeated and amplitude-modulated σB responses at the single-cell level
As a final analysis of the environmental responses of stressosome-deleted B. subtilis cells, we deployed microfluidics-coupled fluorescence microscopy to investigate how stressosome-deleted strains responded to stress at the single-cell level under constant exponential-phase growth conditions. What do the long-lived responses we saw in bulk (Fig. 5B–D) look like in individual stressosome-deleted cells? Do stressosome-deleted cells adjust their response magnitude according to stressor concentration, as wild-type cells do59? To monitor σB activity, all the mutant strains carried the PrsbV-driven mNeonGreen (mNG) fluorescent reporter we used in previous work59,60. We challenged cells with sublethal concentrations (2–4%) of ethanol that activate the σB response without broadly killing the cells59,60.
We previously reported that wild-type cells display a transient and synchronous σB response irrespective of the type of stressor59,60. When we challenged stressosome-deleted cells with 4% ethanol stress, we observed a pulsatile, repeated σB response that began upon exposure to the stressor (Fig. 7A and Supplementary Movies 2–4); such single-cell pulsatile responses have become a hallmark of sustained responses at the average level, such as those we observed in Fig. 5C or in cells with stressosomes containing only RsbRC or RsbRD59,60. Pulsatile responses were sustained for the duration of stress exposure and at least qualitatively appear to have a relatively regular period (Fig. 7A). Importantly, exposure to different ethanol concentrations (2, 3, or 4%) provoked concordant increases in the average magnitude of the σB response (Fig. 7A), indicating that RsbT is sufficient to sense different stressor concentrations and accordingly increase the amplitude of the response. An increased average response could be due to stronger pulses in individual cells (amplitude modulation) or to more frequent pulses (frequency modulation). To distinguish these effects, we examined the magnitudes of and intervals between individual response peaks in single cells. As the ethanol concentration rose, we observed a significant increase in peak magnitudes but no decrease in inter-peak times (Fig. S13), implying that the increased mean response magnitude is mostly, if not entirely, attributable to amplitude modulation. Cells deleted for all RsbRs but not for RsbS displayed a qualitatively similar, pulsatile, sustained response to 4% ethanol (Fig. 7B and Supplementary Movie 5), in accord with the data from Fig. 3 and consistent with these cells lacking functional stressosomes due to the absence of any RsbR proteins. Meanwhile, the ∆rsbRA∆rsbS cells that showed minimal responses in our bulk assays (Fig. 4C) similarly exhibited a very small shift in average reporter fluorescence levels that was unaccompanied by detectable pulsing at the single-cell level (Fig. 7C and Supplementary Movie 6), consistent with a lack of RsbT to sense or transduce stress and provoke the σB response. These results are in full agreement with our bulk data and strongly show that stressosome-deleted cells respond to stress in ways that are qualitatively quite similar to stressosome-replete cells59,60. Collectively, these results support the idea that RsbT is capable of mediating environmental stress sensing and the consequent σB response independently of the stressosome.

A Data from a strain (CSS1719) lacking all stressosome components and analyzed using microfluidics-coupled fluorescence microscopy. Left panel, ensemble of 25 individual-lineage traces. Center panel, two representative single-lineage traces. Right panel, average traces (each assembled from 25 single-cell responses) of cells exposed to the listed concentrations of ethanol. B Data as in A but for a strain (CSS1778) lacking all RsbR paralogs but still encoding RsbS. C Data as in A and B but for a strain (CSS1779) with full deletion of rsbRA and rsbS. Ensemble traces (left) and the average trace (right) are shown. Source data are provided as a Source Data file.
Discussion
Extensive work in the past 3 decades has led to the present model in which each stressosome, composed of 20 RsbS proteins and 20 RsbR dimers, sequesters up to 20 RsbT molecules, preventing any σB activity until stress is sensed. Exposure to environmental stress coincides with activation of RsbT kinase activity, RsbT-mediated phosphorylation of RsbS (and in some cases, RsbR) proteins, and release of RsbT. However, fundamental questions have endured. Which protein(s) are primarily responsible for stress sensing and activation of RsbT kinase activity? It has been presumed, based on excellent structural models and by analogy with the blue-light sensor YtvA, that RsbR proteins are sensors that encourage RsbS phosphorlyation by RsbT in the presence of stress. However, studies aimed at identifying how B. subtilis RsbR proteins sense stress have not uncovered direct evidence for a sensing function. Our present results strengthen the case that stressosomes, including RsbR proteins, may not be the primary environmental stress sensors, as they are not necessary for a σB response to environmental stress. Furthermore, our data showing that RsbT can mediate environmental stress sensing in the absence of RsbR and RsbS proteins imply that RsbT may be a stress sensor. In a revised model accounting for our new results, the onset of stress might directly activate stressosome-bound RsbT, thereby activating its kinase activity and leading to its release from the stressosome. RsbT kinase activity would be modulated, or “advised”, by association with the stressosome, such that the stressosome ensures a quicker σB response upon stressor exposure and earlier and sharper deactivation following exposure. Such a role is also concordant with our RsbRA mutagenesis results (Figs. 1, 2); perhaps changes to the RsbRA N-terminal sequence alter not sensing activity but the dynamics of RsbT release and recapture. We envision that a faster initial response may be attributed to cooperativity in the stressosome complex, as previously postulated70,71, and that recapture of RsbT by the stressosome yields a faster shut-off. Curiously, our data also suggest that the kinase activity of RsbT is important not only for phosphorylation of RsbS and RsbR proteins in conjunction with its release but also for its innate signaling function, even without stressosome proteins to phosphorylate (Fig. 6C, D). By analogy with Vibrio brasiliensis RsbT, which autophosphorylates57, it is at least possible that RsbT may autophosphorylate under stressosome-absent conditions, a possibility warranting future study. Alternatively, perhaps stressosome-independent signaling involves other RsbT kinase targets.
Notably, while the data argue that RsbT does not require the stressosome to initiate an environmental stress response, none of the data conclusively rules out a sensing role for RsbR proteins, whose gram-negative Vibrio counterparts sense oxygen via heme57,58 and whose B. subtilis paralog YtvA senses blue light51,55,56. Additionally, phosphorylation of the RsbR and RsbS stressosome constituents impacts stress sensing differently across species and stressors, with non-phosphorylatable RsbS blocking the L. monocytogenes σB response to acid72 but only decreasing the B. subtilis response to ethanol62. One potential benefit of a regime in which stress sensing is distributed across multiple proteins (e.g., RsbRs and RsbT) is that the system can then be evolutionarily “tuned” for different responses via mutation. The fact that alanine-scanning mutagenesis of RsbRA as the only source of RsbR in the cell resulted in altered, but not absent or constitutive, σB responses constitutes evidence for such a robust but tunable system. Our results also suggest that RsbR proteins are generally accommodating of mutations, at least when one residue at a time is mutated. In the structure of RsbRA, the substitutions with the most consistent phenotypes tended to cluster in unstructured regions at or near the ends of alpha helices (Fig. 2F). We speculate that such substitutions may impact response dynamics (possibly via RsbT capture and release) by altering the helical geometry at the N terminus of RsbRA, an idea that will require further inquiry.
We were initially surprised to find that stressosome-deleted cells did not display the anticipated constitutive σB response. However, the consistency of the results for RsbR-deleted, RsbS-deleted, and early stop codon-blocked strains increases our confidence that there is a bona fide RsbT-mediated response in stressosome-deleted cells. In previous work on cells lacking RsbRA, RB, RC, and RD, despite much higher overall basal σB activity than in the wild type, a response to the addition of stressor was nonetheless visible44, in accord with our results. We suspect that our observation of a lower baseline and more prominent stress-induced σB response in stressosome-deleted strains is related to a reduced production level of RsbT relative to the wild type, as suggested by our proteomic (Table 1) and qRT-PCR (Fig. S7) data. We propose a speculative model in which a low initial concentration of RsbT prevents excessive σB activity in the absence of stress. However, the absence of a stressosome to sequester RsbT makes the existing RsbT more available to activate RsbU. When stress is encountered, RsbT is somehow activated so that it now activates RsbU and the downstream steps of σB release. We do not yet understand the mechanism of RsbT activation, but our data suggest that it may involve RsbT kinase activity (Fig. 6C). Once σB is initially activated, there appears to be at least some σB-mediated positive feedback on rsbT transcription (Fig. S7) and translation (Table 1), potentially explaining how a long-lived, strong response can be achieved with initially scarce RsbT. An impact of such positive feedback is supported by our observation of constitutive σB responses in stressosome-deleted strains with plasmid-borne, IPTG-inducible rsbT (Figs. 6D, S12), where RsbT is initially produced at higher levels but is not subject to σB-mediated feedback. We presume that, in wild-type cells, sequestration of RsbT by stressosomes suppresses the σB response before stress is encountered and prevents a sustained response despite much higher initial and post-stress levels of RsbT (Table 1), highlighting the importance of stressosomes in controlling σB response dynamics.
At the single-cell level, we observed in stress-induced, stressosome-deleted cells repeated, stochastic pulsing that has become a hallmark of strains that exhibit a sustained response at the bulk level59,60. The similarity between the pulsing seen here in stressosome-deleted cells and the pulsing seen in certain strains containing only one of the RsbR paralogs (e.g., RsbRC)59,60 tempts us to speculate that RsbT is not recaptured under stress conditions in those single-RsbR strains, thus mimicking the stressosome-deleted condition. Such pulsatility is most probably reflective of the internal dynamics of σB activation and inactivation, which displays mixed (positive and negative) feedback loops73,74. σB controls its own four-gene operon (rsbV-rsbW-sigB-rsbX), located downstream of rsbT and rsbU67, and pulsing is caused by feedback of rsbV-rsbW-sigB with a input signal from the RsbT-activated RsbU phosphatase75. Once σB is initiated, its activity increases due to increased expression both of the anti-anti-sigma factor RsbV and of σB itself. Next, the increased expression of RsbW—the anti-sigma factor and RsbV kinase—results in RsbV phosphorylation and σB recapture by RsbW, marking attenuation of σB activity and the end of the pulse75. Previous work examining σB pulse dynamics with differing levels of inducible RsbTU input signal concluded that the single-cell response was primarily frequency-modulated75. Importantly, our results show that amplitude modulation at the single-cell level is also possible, and in fact appears to be the main driver of increased mean σB responses by cells facing higher stressor concentrations (Fig. S13).
Overall, a revised picture of σB activation in B. subtilis and L. monocytogenes is emerging that goes beyond the canonical models for environmental and energy stress sensing. For instance, previous work implies the existence of other pathways for σB activation that totally bypass all putative stress sensors by operating independently of the anti-anti-sigma factor RsbV; these pathways are active at low and high temperatures76,77,78. Moreover, there are connections between the ribosome and σB such that protein L11 in B. subtilis is required for the environmental stress response79, and a G50C mutant of S21 in L. monocytogenes can activate σB independently of RsbV80. Meanwhile, our study sheds new light on the environmental, stressosome-mediated branch of the σB response, positioning RsbT as an independent stress sensor rather than merely a messenger. New questions also arise: what is the molecular basis for stress sensing by RsbT? Does RsbT directly sense stress, or does it interact with one or more proteins in the absence of a stressosome, perhaps Obg77? How is RsbT kinase activity related to stress sensing? What parameters with respect to cellular RsbT concentration and the presence of other stressosome components allow a stress-inducible response? Does RsbT have the same stressosome-independent function in other stressosome-encoding species? We look forward to tackling these puzzles.
Methods
Strains and growth conditions
All B. subtilis strains and their genotypes used are listed in Table S2. All B. subtilis strains were derived from strain PY79 by transformation or phage transduction. Transformed strains were selected on LB-Lennox agar plates (10 g/l tryptone, 5 g/l yeast extract, 5 g/l NaCl, and 15 g/l agar). When appropriate, antibiotics were added at the following concentrations: carbenicillin (100 µg/ml for E. coli), chloramphenicol (5 µg/ml), spectinomycin (100 µg/ml), streptomycin (5 µg/ml), zeocin (20 µg/ml), and 1X MLS (100X MLS: 0.5 µg/ml erythromycin and 2.5 µg/ml lincomycin). Plasmids were cloned and propagated in NEB5α E. coli cells.
Strains with markerless deletions of rsb genes were constructed using the pMiniMAD vector for allelic replacement. A pMiniMAD plasmid harboring ~600 bp of homology on either flank of the desired site of deletion was introduced into the PY79 strains via competence and selected on LB-MLS. The MLS-resistant transformants were grown overnight in LB at 25 °C for 1-2 days and then grown in LB at 37 °C to allow second crossover recombination for excision of plasmid. The transformants were plated on LB and colonies were screened for the presence of the correct clean deletion of gene by colony PCR and verified by sequencing. The verified colonies were patched on LB-MLS plates to verify MLS sensitivity indicating the loss of plasmid, restreaked for single colonies, grown in liquid LB and stored at -80 °C. Reporter strains contained a transcriptional fusion of the σB -dependent PrsbV -promoter to lacZ to measure σB activity. For environmental stress-conditions, ethanol or NaCl was added to LB/LBK to the desired final concentration. To prevent motile cells from swimming out of the device, all strains used for microfluidics analysis contained a hagA233V flagellin point mutation, which impairs the ability of the flagellum to generate force without interfering its structure81. All constructed strains for microfluidics analysis also contained a transcriptional fusion of the σB-dependent PrsbV promoter to an mNeonGreen fluorescent reporter gene.
Complementation of rsbRA and rsbU
PrsbRA, rsbRA and rsbU gene were amplified by PCR using chromosomal DNA of B. subtilis 3610 as a template with the appropriate set of forward and reverse primers containing the 5’ flanking region upstream of PrsbRA and 3’ flanking region downstream of rsbU. The PCR fragments had an overlapping region with the adjacent fragment to stitch the fragments. The fragments were stitched together, gel purified and assembled into integration vector pDG1730 (digested with EcoRI and HindIII) by using isothermal assembly82. To construct the reporter plasmid, PrsbV promoter was amplified by PCR using 3610 gDNA as template with the appropriate forward and reverse primers and assembled into pDG268 (digested with EcoRI and HindIII) by using isothermal assembly so that PrsbV-lacZ fusion whose expression was fully dependent on σB. The plasmids were cloned and propagated in the E. coli NEB5α and confirmed by colony PCR and sequencing. The pDG1730-rsbRA-rsbU plasmid was then transformed into B. subtilis CSS 716 strains via competence with selection for spectinomycin and streptomycin resistance. The transformants were screened for spectinomycin-streptomycin resistance and screened for the presence of rsbRA-rsbU insert by PCR. The transformants were also screened for starch hydrolysis, indicating the integration of plasmid at the amyE locus. This strain was transduced with pDG268-PrsbV-lacZ reporter via transduction with selection for chloramphenicol resistance. Since both the vectors used were amyE locus integration vectors, the disruption of the amyE gene at an alternative genomic locus (marked with kanamycin resistance) by single-site homologous recombination was verified by retention of endogenous α-amylase activity but loss of kanamycin resistance. We then tested the ability to form stressosome and reconstitution of environmental stress sensing by blue-white screening and β-galactosidase assays.
Site Directed Mutagenesis and Ectopic integration of rsbRA
The pDG1730-PrsbRA-rsbRA-rsbU plasmid was used as a template for alanine scanning by using a forward primer containing the desired mutation and a reverse primer. Primers for site-directed mutagenesis were designed by using the NEBaseChanger tool. The amino acids at selected positions (I11 to K58) were systematically substituted for alanine at the sensor coding sequence of RsbRA (N-terminal region) using the Q5 ® Site-Directed Mutagenesis kit (NEB). After PCR, the amplified materials were mixed and incubated for 5 min with Kinase-Ligase-DpnI (KLD) enzyme mix for rapid circularization and template removal. The KLD-treated mixture was transformed into E. coli NEB5α cells as per standard procedure (NEB, Q5 site-directed mutagenesis kit protocol) and selected on carbenicillin plates. pDG1730-rsbRA plasmid was verified by colony PCR and point mutation at selected position on N-term RsbRA was confirmed by sequencing. Plasmids were then transformed into CSS1384. All constructed strains were transduced with lysate from PY79 ywrK::pDG268-PrsbV-lacZ to add the σB β-galactosidase reporter and selected on LB-chloramphenicol plates. The effect of each amino acid substitution was assessed for its influence on stress signaling by β-Galactosidase activity.
Allelic replacement of point mutant rsbRA
Strains with allelic replacement of WT rsbRA and point mutant versions of rsbRA were constructed using pMiniMAD; the rsbRA gene was amplified by PCR using the pDG1730-rsbRA point mutation plasmid as a template with the appropriate set of forward and reverse primers. The upstream fragment contains the 5’ flanking region and gene sequence encoding the variable N-terminal domain of rsbRA harboring point mutation at desired position. The downstream fragment contains the gene sequence encoding the conserved C-terminal domain of rsbRA and 3’ flanking region. The PCR fragments had an overlapping region with the adjacent fragment to stitch the fragments. The fragments were stitched together, gel purified and assembled into pMiniMAD plasmid (digested with EcoRI and HindIII) by using isothermal assembly82. The plasmid was cloned and propagated in E. coli NEB5α and confirmed by colony PCR and sequencing. The plasmid was then transformed into B. subtilis CSS1384 strains via competence with selection for MLS. The MLS-resistant transformants were grown overnight in LB at 25 °C for 1–2 days and then grown in LB at 37 °C to allow second crossover recombination for excision of plasmid. The transformants were plated on LB and colonies were screened for the allelic replacement of point mutant rsbRA by colony PCR and verified by sequencing. The verified colonies were patched on LB-MLS plates to verify MLS sensitivity indicating the loss of plasmid, restreaked for single colonies, grown in liquid LB and stored at −80 °C. All constructed strains were transduced with lysate from PY79 ywrK::pDG268-PrsbV-lacZ to add the σB β-galactosidase reporter and selected on LB-chloramphenicol plates.
Kinetic β-Galactosidase assay
Strains carrying lacZ reporter constructs were grown to exponential phase in LB or LBK at 37 °C in a shaking incubator. When the cultures reached an OD600 of 0.1 (T0) sampling was started. Stress was induced by adding either ethanol (final concentration 3% v/v) or 1 M NaCl in LBK (final concentration 600 mM) to exponentially growing cultures at T0. An equal volume of water was added to controls without stress addition to correct for any dilution effects. Samples were collected at various time points (every 15 mins) for 3 h. 1 ml of culture was centrifuged at 14,000 rpm for 4 mins and stored at −80 °C for later processing. Cell pellets were resuspended in 500 µl of Z-buffer (60 mM Na2HPO4.7H2O, 40 mM NaH2PO4.H20, 10 mM KCl. 1 mM MgSO4.7H2O, 1.35 µl β-mercaptoethanol at pH 7.0). 50 µl of 0.4 mg/ml lysozyme dissolved in Z-buffer with βME was added to the individual wells of a 96-well plate containing 50 µl of the resuspended cells. Plate was incubated at 37 °C for 20 mins to allow cell lysis. 20 µl of 4 mg/ml 2-Nitrophenyl β-D-galactopyranoside (ONPG; RPI Research Products International) dissolved in Z-buffer was then added to each well and mixed thoroughly. For each reaction, absorbance at 420 nm was read once per 50–60 s for 45 mins at 37 °C with continuous shaking in a Synergy H1 microplate reader (BioTek) and recorded in Gen5 software v. 3.09. β-Galactosidase activity (in arbitrary units) is reported as the rate of ONPG hydrolysis (i.e. Vmax with units of OD420 per minute) divided by the OD600 of the culture at the time of collection. The Vmax/OD600 calculation occasionally produced small negative numbers, which were set to zero for graphing. Graphs were constructed in GraphPad Prism 10.
qRT-PCR
Cultures (MTC 52, CSS 1384, and CSS 1432) were grown in LB at 37 °C up to an OD600 of ~0.1 (T0). Stress was induced by adding ethanol (final concentration 3% v/v) to exponentially growing cultures at T0. Samples were collected every 15 mins before (T0) and after (T15 & T30) adding stress for 30 min and centrifuged at 16,000 g. The cell pellet was then resuspended in 250 µl of TE buffer [10 mM Tris (pH 8.0), 1 mM EDTA], treated with 10 mg/ml lysozyme, and incubated at room temperature for 10-15 mins to allow cell lysis. Total RNA was extracted using the Monarch Total RNA extraction kit (New England Biolabs). The purified RNA was diluted to 100 ng/µl and reverse-transcribed with the RevertAid RT Reverse transcription kit (Thermo Fisher). The resulting cDNA was further diluted 1:10 in nuclease-free water, and 1 µl of diluted cDNA was used in a 10 µl qPCR reaction mix. Appropriate concentrations (0.8 µM) of gene-specific primers for rsbT (1692 & 1693) and for the housekeeping gene gyrB (1660 & 1661) were used. Quantitative reverse-transcription-PCR (qRT-PCR) assays were performed with the Powertrack SYBR Green dye-based Master mix (Thermo Fisher) on a CFX96 Touch real-time PCR thermocycler (Bio-Rad). The PCR was performed by using the following cycling conditions: Initial denaturation was performed at 95°C for 2 mins, and 40 cycles were performed by using a 15-second denaturation step at 95°C followed by annealing and extension at 60°C for 1 min. Expression levels of rsbT were normalized to the B. subtilis gyrB gene, and the copies of the rsbT reverse-transcribed RNA present in the cDNA sample were calculated using the standard curve method. All samples were analyzed in three technical replicates. The standard error was calculated by using the error in the mean of three technical replicates, both for the rsbT and gyrB genes.
Mass spectrometry
Strains MTC52 (WT control), CSS1432 (ΔrsbRAΔrsbS), and CSS1646 (RsbRASTOP RsbSSTOP) were treated with 3% ethanol as for β-galactosidase assays, and cells were collected before or 60 min after ethanol treatment. Cell pellets were lysed in 6 M guanidine hydrochloride, 0.1 M Tris-HCl, 10 mM tris(2-carboxyethyl)phosphine pH 8.2, boiled for 5 min, and sheared by sonication in a Diogenode Bioruptor. The resultant cell lysates were quantified by tryptophan fluorescence and digested with trypsin/LysC (Promega V5072) using the filter-aided sample preparation technique83. For this, 30-kDa centrifugal filter units (Micron/Sigma MRCF0R030) were used to perform three centrifugation exchanges into 8 M urea, 0.1 M Tris-HCl pH 8.5, followed by three centrifugation exchanges into 0.1 M Tris-HC, pH 8.5. Proteins retained in the top unit of the spin filter device were diluted with 50 µl of 0.1 M Tris-HCl pH 8.5, containing one microgram of trypsin/LysC. After digestion overnight at 37 °C, the digestion volume was harvested by centrifugation, followed by two additional elutions of the top chamber with 0.1 M Tris-HCl pH 8.5. Peptides were further desalted using C18 spin filter devices (HMM S18R, The Nest Group), following the manufacturer’s recommendations.
The desalted peptides were analyzed by LC-MS/MS using an Orbitrap Fusion mass spectrometer equipped with an Easy-nLC 1200 nano UPLC system (Thermo). For this, peptides were dissolved in mobile phase A (0.1 % aqueous formic acid) and injected onto a 2-cm C18 trap column (Thermo PN 164946) plumbed in a vented column configuration to a 50-cm analytical column (Thermo PN 164942). Peptides were separated using 80:20:0.1 acetonitrile/water/formic acid as mobile phase B, delivered as a linear 4 to 32 percent gradient over 120 min at a net solvent flow of 250 nL/min. Peptides were eluted into a Nanospray Flex ion source (Thermo) equipped with a stainless steel emitter. Peptide ions were analyzed by “high/low” MS/MS using data-dependent acquisitions and dynamic exclusion. For this, parent ions were analyzed in the Orbitrap sector, followed by collisional dissociation in the ion routing multipole using 32% HCD energy, and analysis of ion fragments in the ion trap sector.
Peptides were identified and quantified by using MaxQuant84 v2.2.0.0 to search the raw instrument files against a database of 4,260 B. subtilis reference protein sequences downloaded from UniProt on 06/23/23. Searches utilized the default MaxQuant settings, supplemented with the modifications deamidation of N and Q, and cyclization of N-terminal Q to pyroglutamate. Peptide identifications were propagated among chromatograms using the MaxQuant Match Between Runs algorithm.
Proteins were quantified on the basis of normalized LFQ protein intensities85. Rsb proteins of interest were manually selected, and normalized protein intensities were compared, using GapA (glyceraldehyde 3-phosphate dehydrogenase) as a housekeeping control protein.
rsbT and rsbT D78N complementation
An IPTG-inducible expression vector carrying rsbT, rsbU, or both under the control of the LacI-repressible IPTG-inducible promoter Pspank (pKH001-Pspank-rsbTU69 or pKH001-Pspank-rsbT) was used for complementation of rsbT. Plasmid pKH001-Pspank-rsbU69 was used as a control vector to verify the necessity of rsbT for complementation. An IPTG-inducible expression vector carrying the mutant rsbTD78N under the control of the LacI-repressible IPTG inducible promoter Pspank (pKH001-Pspank-rsbTD78N) was also used for the complementation with an RsbT kinase activity mutant. The mutation at Asp 78 to Asn (D78N) disrupts the kinase activity of RsbT; the variant can still activate RsbU but is unable to phosphorylate RsbS41,86. Plasmids were introduced into the CSS1974 parental strain (PY79 ΔrsbRB ΔrsbRC ΔrsbRD ΔytvA ΔrsbP rsbS (STOP) rsbRA (STOP) rsbT (STOP) ywrk::DG268-PrsbV-lacZ) using standard B. subtilis transformation techniques. Cultures were grown in a selective medium (LB-MLS) containing IPTG at 37°C in a shaking incubator. Different concentrations of IPTG from 0.05 mM-1 mM were added to test the induction. When the cultures reached an OD600 of 0.05 (T−30 min), sampling was started. Stress was induced by adding ethanol (final concentration 3% v/v) to exponentially growing cultures at T0. An equal amount of growth medium (LB) was added to controls without stress addition to correct for any dilution effects. Samples were collected every 15 mins for 3 h, and σB reporter activity was measured by β-galactosidase assay.
Anti-RsbT immunoblotting
B. subtilis strains were inoculated into 100 mL of liquid culture. When the culture optical density was approximately 1.0, cell pellets were produced by centrifugation at 4100 g for 45 min at 4 °C. The cell pellet was washed 2–3 times in 50 mM Tris (pH 8.4)-1M KCl solution. To make crude cell extracts, the washed cell pellet was resuspended in a buffer mix (10 mM Tris [pH 8], 100 mM EDTA, 10 mM MgCl2, 100 mM dithiothreitol, 5% [vol/vol] of phenylmethylsulfonylfluoride solution). The resuspended cell pellets were lysed using a bead beater homogenizer at 3000 rpm for 4 min (15 sec on and 1 min off cycle) to lyse the cell. The lysed cell pellets were separated from the supernatant by centrifugation at 18,400 g for 15 min at 4 °C. The protein concentration of each crude cell extract was determined by Bradford Assay. At least 50 µg protein of crude cell extracts were loaded into 15% SDS-polyacrylamide gels and electrophoretically separated. Proteins were transferred electrophoretically to a mini-size PVDF membrane using a Bio-Rad Trans-blot Turbo. The membrane was blocked into a blocking solution (5% nonfat dry milk in TBST) for one hour at room temperature before being placed into a plastic bag with diluted (1:1000) polyclonal rabbit anti-RsbT antibody and incubated for one hour at room temperature. After incubation with the primary antibody, the membrane was washed 3–5 times with TBST solution. The blot was then incubated with diluted (1:3000) horseradish peroxidase-conjugated goat immunoglobulin G (Cell Signaling Technology 7074S) in TBST for one hour at room temperature. Finally, the membrane was washed once again 4-5 times with TBST solution and enhanced chemiluminescence reagents were added to the membrane to image on a Bio-Rad ChemiDoc. The primary antibody was validated by showing its ability to detect purified recombinant RsbT proteins and the disappearance of a band at the approximate size of RsbT in rsbT-deleted strains.
Microfluidics apparatus setup and media
Microfluidics experiment and apparatus setup methods were conducted as described previously59,60,81. Cured PDMS (polydimethylsiloxane) casted on silicon wafer master lacking the shallow surrounding channels were punched with a 0.75 mm biopsy punch to create holes to connect the channels using the 21-guage blunt needles. The chips were bonded to coverslip cleaned with isopropyl alcohol using oxygen plasma treatment at 200 mTorr O2 for 15 S at 30 W and baked at 60–65 °C for an hour before use. The microfluidics device was passivated (to prevent cell sticking) with LB containing 1 mg/ml bovine serum albumin (BSA) before cell loading. The cells were grown to a stationary phase in LB and then filtered through a 5 µm filter to remove cell chains. The cells were concentrated by centrifugation at 5000 g for 10 min and injected into the channel. The chip loaded with cells was centrifuged by using a custom-designed microcentrifuge adaptor at 6000 g for 10 min to force the cells into the side channels. The fluidics were connected to the device and run at 35 µl/min for approximately 20 mins to flush out excess cells before being run at 2.5 µl/min for imaging.
The LB media used for fluidics contained 0.1 mg/ml BSA as a passivation agent to prevent cells from adhering to the channel during flow. The fluidics were fed by 20-mL syringes in six-channel syringe pumps (New Era Pump Systems, Farmingdale, NY) that were connected by 21-gauge blunt needles to Tygon flexible tubing with an inner diameter (ID) of 0.02 in. To permit medium switches, two banks of syringes were used: one for the one for the prestress phase containing plain medium and the other for the stress phase containing the stressor. Each pair of syringes (minus and plus stressor) was joined with a polypropylene 1.6-mm-ID Y connector with 200-series barbs; 2-cm lengths of flexible silicone tubing (0.04-in. ID, 0.085-in. outer diameter) were used to connect the Tygon lines to the two input branches of the Y connector. The lengths of silicone tubing facilitated the placement of small binder clips to one or the other branch to make pinch valves. A 1-cm length of silicone tubing was used to connect a 10-cm length of Tygon tubing to the output of the Y connector. The output Tygon tubing was directly connected to a bent 21-gauge blunt needle that was then connected to the PDMS device, and a similarly constructed needle-tube combination was used to carry the outflow of the device to a waste beaker.
Ethanol (2%, 3%, and 4%) was used as an environmental stressor. The prestress medium was LB with 0.1 mg/mL BSA. For stress media, 4% ethanol was added to the LB containing 0.1 mg/mL BSA.
Medium switching
The initial growth phase experiments were conducted in stressor-free medium (LB + 0.1 mg/mL BSA) which lasted for 10-12 hr before the switch. In the initial phase, pinch valves were closed by using binder clips on the stressor-containing branch of the fluidics, and the corresponding syringe pump was paused. During switch, the syringe pump with stressor-free medium was paused, the valves were closed by binder clips on the stressor-containing branch of the fluidics, and the syringe pump was activated at 2.5 µl/min. The switch apparatus was housed within a temperature-controlled microscope enclosure during imaging59,60.
Automated imaging
Imaging was performed with a Nikon Eclipse Ti inverted microscope equipped with a Photometrics Prime 95B sCMOS camera, a 100 Plan Apo oil objective (NA 1.45, Nikon), an automated stage (Nikon), a Lumencor SOLA SE II 365 Light Engine fluorescent illumination system, and an OKO temperature-controlled enclosure in which the temperature was maintained at approximately 37 °C during imaging. Image acquisition was performed using NIS-Elements AR 5.11.03 64-bit. A filter cube for GFP was used to image mNeonGreen (used for σB reporters) at 33% illumination power with 200-ms exposures. The images were captured at 10-min intervals. Phase-contrast images were also captured59,60.
Lineage tracking and curation
The average mNeonGreen average intensity in mother cells was used to generate σB reporter traces. Lineages were manually filtered to retain only lineages that were tracked for >150 continuous frames. Images of cells were then stabilized to eliminate any movement or shifts during the time lapse by using ImageJ 1.52p with the plugin “Template Matching and Slice Alignment.” The mean mNeonGreen fluorescence value was then determined for each lineage by drawing a region of interest (ROI) box slightly smaller than the size of a cell in ImageJ and using the ImageJ plugin “Time Series Analyzer.” The position of the ROI, which otherwise remained static at each time point, was manually reviewed and corrected when necessary to ensure that the ROI contained the mother cell of each lineage for the duration of the experiment. The mean pixel intensity in the mNeonGreen channel in the ROI of the cell at each 10-min interval was collected and then corrected by subtraction of the average background adjacent to the cell to eliminate any fluctuations during the time lapse59,60. The average fluorescence value of each lineage prior to stressor onset (50 frames) and after stressor onset (150 frames) was obtained. This process was repeated for each lineage. The curated lineages were used to plot average traces and overlaid single-cell trace59,60.
Frequency and amplitude assessment
Single-cell traces from lineages curated as described above were manually analyzed to identify peaks in the PrsbV-mNG signal, and the intensity of each peak (i.e., the maximal mean pixel intensity in the ROI) was manually recorded. The intervals between identified peaks (to the nearest 10 min, as images were acquired at 10-min intervals) were also manually recorded.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided with this paper for all the graphs (Figs. 1–7, S1–S5, S7–S10, S12–13) and images (Fig. S11) shown in this study. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE87 partner repository with the dataset identifier PXD059924. Source data are provided with this paper.
References
Binnie, C., Lampe, M. & Losick, R. Gene encoding the sigma 37 species of RNA polymerase sigma factor from Bacillus subtilis. Proc. Natl Acad. Sci. USA 83, 5943–5947 (1986).
Duncan, M. L., Kalman, S. S., Thomas, S. M. & Price, C. W. Gene encoding the 37,000-dalton minor sigma factor of Bacillus subtilis RNA polymerase: isolation, nucleotide sequence, chromosomal locus, and cryptic function. J. Bacteriol. 169, 771–778 (1987).
Hecker, M. & Völker, U. General stress response of Bacillus subtilis and other bacteria. Adv. Micro. Physiol. 44, 35–91 (2001).
Haldenwang, W. G. The sigma factors of Bacillus subtilis. Microbiol Rev. 59, 1–30 (1995).
Hecker, M., Pané-Farré, J. & Uwe, V. SigB-dependent general stress response in Bacillus subtilis and related gram-positive bacteria. Annu Rev. Microbiol 61, 215–236 (2007).
Price, C. W. et al. Genome-wide analysis of the general stress response in Bacillus subtilis. Mol. Microbiol 41, 757–774 (2001).
Petersohn, A. et al. Global analysis of the general stress response of Bacillus subtilis. J. Bacteriol. 183, 5617–5631 (2001).
Nannapaneni, P. et al. Defining the structure of the general stress regulon of Bacillus subtilis using targeted microarray analysis and random forest classification. Microbiol Read. Engl. 158, 696–707 (2012).
Voelker, U. et al. Separate mechanisms activate sigma B of Bacillus subtilis in response to environmental and metabolic stresses. J. Bacteriol. 177, 3771–3780 (1995).
Volker, U. et al. Analysis of the induction of general stress proteins of Bacillus subtilis. Microbiology 140, 741–752 (1994).
Bernhardt, J., Weibezahn, J., Scharf, C. & Hecker, M. Bacillus subtilis during feast and famine: visualization of the overall regulation of protein synthesis during glucose starvation by proteome analysis. Genome Res. 13, 224–237 (2003).
Zhang, S. & Haldenwang, W. G. Contributions of ATP, GTP, and redox state to nutritional stress activation of the Bacillus subtilis sigmaB transcription factor. J. Bacteriol. 187, 7554–7560 (2005).
Bandow, J. E., Brötz, H. & Hecker, M. Bacillus subtilis tolerance of moderate concentrations of rifampin involves the sigma(B)-dependent general and multiple stress response. J. Bacteriol. 184, 459–467 (2002).
Maul, B., Völker, U., Riethdorf, S., Engelmann, S. & Hecker, M. sigma B-dependent regulation of gsiB in response to multiple stimuli in Bacillus subtilis. Mol. Gen. Genet MGG 248, 114–120 (1995).
Avila-Pérez, M., Hellingwerf, K. J. & Kort, R. Blue light activates the sigmaB-dependent stress response of Bacillus subtilis via YtvA. J. Bacteriol. 188, 6411–6414 (2006).
Benson, A. K. & Haldenwang, W. G. The sigma B-dependent promoter of the Bacillus subtilis sigB operon is induced by heat shock. J. Bacteriol. 175, 1929–1935 (1993).
Boylan, S. A., Redfield, A. R., Brody, M. S. & Price, C. W. Stress-induced activation of the sigma B transcription factor of Bacillus subtilis. J. Bacteriol. 175, 7931–7937 (1993).
Suzuki, N., Takaya, N., Hoshino, T. & Nakamura, A. Enhancement of a sigma(B)-dependent stress response in Bacillus subtilis by light via YtvA photoreceptor. J. Gen. Appl Microbiol 53, 81–88 (2007).
Becker, L. A., Cetin, M. S., Hutkins, R. W. & Benson, A. K. Identification of the gene encoding the alternative sigma factor sigmaB from Listeria monocytogenes and its role in osmotolerance. J. Bacteriol. 180, 4547–4554 (1998).
Chaturongakul, S., Raengpradub, S., Wiedmann, M. & Boor, K. J. Modulation of stress and virulence in Listeria monocytogenes. Trends Microbiol 16, 388–396 (2008).
Chaturongakul, S. & Boor, K. J. RsbT and RsbV contribute to sigmaB-dependent survival under environmental, energy, and intracellular stress conditions in Listeria monocytogenes. Appl Environ. Microbiol 70, 5349–5356 (2004).
Chaturongakul, S. & Boor, K. J. SigmaB activation under environmental and energy stress conditions in Listeria monocytogenes. Appl Environ. Microbiol 72, 5197–5203 (2006).
Shin, J. H., Brody, M. S. & Price, C. W. Physical and antibiotic stresses require activation of the RsbU phosphatase to induce the general stress response in Listeria monocytogenes. Microbiol Read. Engl. 156, 2660–2669 (2010).
Brody, M. S. & Price, C. W. Bacillus licheniformis sigB operon encoding the general stress transcription factor sigma B. Gene 212, 111–118 (1998).
Voigt, B. et al. The response of Bacillus licheniformis to heat and ethanol stress and the role of the SigB regulon. Proteomics 13, 2140–2161 (2013).
van Schaik, W., Tempelaars, M. H., Wouters, J. A., de Vos, W. M. & Abee, T. The alternative sigma factor sigmaB of Bacillus cereus: response to stress and role in heat adaptation. J. Bacteriol. 186, 316–325 (2004).
Gertz, S. et al. Regulation of sigmaB-dependent transcription of sigB and asp23 in two different Staphylococcus aureus strains. Mol. Gen. Genet MGG 261, 558–566 (1999).
Giachino, P., Engelmann, S. & Bischoff, M. Sigma(B) activity depends on RsbU in Staphylococcus aureus. J. Bacteriol. 183, 1843–1852 (2001).
Pané-Farré, J., Jonas, B., Förstner, K., Engelmann, S. & Hecker, M. The sigmaB regulon in Staphylococcus aureus and its regulation. Int J. Med Microbiol IJMM 296, 237–258 (2006).
Senn, M. M. et al. Molecular analysis and organization of the sigmaB operon in Staphylococcus aureus. J. Bacteriol. 187, 8006–8019 (2005).
Alper, S., Dufour, A., Garsin, D. A., Duncan, L. & Losick, R. Role of adenosine nucleotides in the regulation of a stress-response transcription factor in Bacillus subtilis. J. Mol. Biol. 260, 165–177 (1996).
Boylan, S. A., Rutherford, A., Thomas, S. M. & Price, C. W. Activation of Bacillus subtilis transcription factor sigma B by a regulatory pathway responsive to stationary-phase signals. J. Bacteriol. 174, 3695–3706 (1992).
Benson, A. K. & Haldenwang, W. G. Characterization of a regulatory network that controls sigma B expression in Bacillus subtilis. J. Bacteriol. 174, 749–757 (1992).
Benson, A. K. & Haldenwang, W. G. Bacillus subtilis sigma B is regulated by a binding protein (RsbW) that blocks its association with core RNA polymerase. Proc. Natl Acad. Sci. USA 90, 2330–2334 (1993).
Dufour, A. & Haldenwang, W. G. Interactions between a Bacillus subtilis anti-sigma factor (RsbW) and its antagonist (RsbV). J. Bacteriol. 176, 1813–1820 (1994).
Kang, C. M., Brody, M. S., Akbar, S., Yang, X. & Price, C. W. Homologous pairs of regulatory proteins control activity of Bacillus subtilis transcription factor sigma(b) in response to environmental stress. J. Bacteriol. 178, 3846–3853 (1996).
Vijay, K., Brody, M. S., Fredlund, E. & Price, C. W. A PP2C phosphatase containing a PAS domain is required to convey signals of energy stress to the sigmaB transcription factor of Bacillus subtilis. Mol. Microbiol 35, 180–188 (2000).
Voelker, U., Voelker, A. & Haldenwang, W. G. Reactivation of the Bacillus subtilis anti-sigma B antagonist, RsbV, by stress- or starvation-induced phosphatase activities. J. Bacteriol. 178, 5456–5463 (1996).
Brody, M. S., Vijay, K. & Price, C. W. Catalytic function of an alpha/beta hydrolase is required for energy stress activation of the sigma(B) transcription factor in Bacillus subtilis. J. Bacteriol. 183, 6422–6428 (2001).
Delumeau, O. et al. Functional and structural characterization of RsbU, a stress signaling protein phosphatase 2C. J. Biol. Chem. 279, 40927–40937 (2004).
Kang, C. M., Vijay, K. & Price, C. W. Serine kinase activity of a Bacillus subtilis switch protein is required to transduce environmental stress signals but not to activate its target PP2C phosphatase. Mol. Microbiol 30, 189–196 (1998).
Yang, X., Kang, C. M., Brody, M. S. & Price, C. W. Opposing pairs of serine protein kinases and phosphatases transmit signals of environmental stress to activate a bacterial transcription factor. Genes Dev. 10, 2265–2275 (1996).
Chen, C. C., Lewis, R. J., Harris, R., Yudkin, M. D. & Delumeau, O. A supramolecular complex in the environmental stress signalling pathway of Bacillus subtilis: Large protein complex regulates stress response. Mol. Microbiol 49, 1657–1669 (2003).
Kim, T. J., Gaidenko, T. A. & Price, C. W. A multicomponent protein complex mediates environmental stress signaling in Bacillus subtilis. J. Mol. Biol. 341, 135–150 (2004).
Pané-Farré, J., Lewis, R. J. & Stülke, J. The RsbRST stress module in bacteria: a signalling system that may interact with different output modules. J. Mol. Microbiol Biotechnol. 9, 65–76 (2005).
Pané-Farré, J., Quin, M. B., Lewis, R. J. & Marles-Wright, J. Structure and function of the stressosome signalling hub. Subcell. Biochem 83, 1–41 (2017).
Marles-Wright, J. et al. Molecular architecture of the “stressosome,” a signal integration and transduction hub. Science 322, 92–96 (2008).
Aravind, L. & Koonin, E. V. The STAS domain — a link between anion transporters and antisigma-factor antagonists. Curr. Biol. 10, R53–R55 (2000).
Murray, J. W., Delumeau, O. & Lewis, R. J. Structure of a nonheme globin in environmental stress signaling. Proc. Natl Acad. Sci. USA 102, 17320–17325 (2005).
Gaidenko, T. A., Bie, X., Baldwin, E. P. & Price, C. W. Substitutions in the presumed sensing domain of the Bacillus subtilis stressosome affect its basal output but not response to environmental signals. J. Bacteriol. 193, 3588–3597 (2011).
Akbar, S. et al. New family of regulators in the environmental signaling pathway which activates the general stress transcription factor sigma(B) of Bacillus subtilis. J. Bacteriol. 183, 1329–1338 (2001).
Kim, T. J., Gaidenko, T. A. & Price, C. W. In vivo phosphorylation of partner switching regulators correlates with stress transmission in the environmental signaling pathway of Bacillus subtilis. J. Bacteriol. 186, 6124–6132 (2004).
Chen, C. C., Yudkin, M. D. & Delumeau, O. Phosphorylation and RsbX-dependent dephosphorylation of RsbR in the RsbR-RsbS complex of Bacillus subtilis. J. Bacteriol. 186, 6830–6836 (2004).
Eymann, C. et al. In vivo phosphorylation patterns of key stressosome proteins define a second feedback loop that limits activation of Bacillus subtilis σB. Mol. Microbiol 80, 798–810 (2011).
Losi, A., Polverini, E., Quest, B. & Gärtner, W. First evidence for phototropin-related blue-light receptors in prokaryotes. Biophys. J. 82, 2627–2634 (2002).
Van Der Steen, J. B. et al. Differentiation of function among the rsbr paralogs in the general stress response of bacillus subtilis with regard to light perception. J. Bacteriol. 194, 1708–1716 (2012).
Jia, X., Wang, J. B., Rivera, S., Duong, D. & Weinert, E. E. An O2-sensing stressosome from a Gram-negative bacterium. Nat. Commun. 7, 12381 (2016).
Heinz, V. et al. The Vibrio vulnificus stressosome is an oxygen-sensor involved in regulating iron metabolism. Commun. Biol. 5, 622 (2022).
Cabeen, M. T., Russell, J. R., Paulsson, J. & Losick, R. Use of a microfluidic platform to uncover basic features of energy and environmental stress responses in individual cells of Bacillus subtilis. PLOS Genet 13, e1006901 (2017).
Hamm, C. W., Butler, D. R. & Cabeen, M. T. Bacillus subtilis stressosome sensor protein sequences govern the ability to distinguish among environmental stressors and elicit different σ b response profiles. mBio 13, e02001–e02022 (2022).
Gaidenko, T. A., Bie, X., Baldwin, E. P. & Price, C. W. Two surfaces of a conserved interdomain linker differentially affect output from the rst sensing module of the bacillus subtilis stressosome. J. Bacteriol. 194, 3913–3921 (2012).
Gaidenko, T. A. & Price, C. W. Genetic evidence for a phosphorylation-independent signal transduction mechanism within the Bacillus subtilis stressosome. PloS One 9, e90741 (2014).
Akbar, S., Kang, C. M., Gaidenko, T. A. & Price, C. W. Modulator protein RsbR regulates environmental signalling in the general stress pathway of Bacillus subtilis. Mol. Microbiol 24, 567–578 (1997).
Gaidenko, T. A., Yang, X., Lee, Y. M. & Price, C. W. Threonine phosphorylation of modulator protein RsbR governs its ability to regulate a serine kinase in the environmental stress signaling pathway of Bacillus subtilis. J. Mol. Biol. 288, 29–39 (1999).
Delumeau, O., Chen, C. C., Murray, J. W., Yudkin, M. D. & Lewis, R. J. High-molecular-weight complexes of RsbR and paralogues in the environmental signaling pathway of Bacillus subtilis. J. Bacteriol. 188, 7885–7892 (2006).
Wise, A. A. & Price, C. W. Four additional genes in the sigB operon of Bacillus subtilis that control activity of the general stress factor sigma B in response to environmental signals. J. Bacteriol. 177, 123–133 (1995).
Kalman, S., Duncan, M. L., Thomas, S. M. & Price, C. W. Similar organization of the sigB and spoIIA operons encoding alternate sigma factors of Bacillus subtilis RNA polymerase. J. Bacteriol. 172, 5575–5585 (1990).
Carniol, K., Kim, T. J., Price, C. W. & Losick, R. Insulation of the sigmaF regulatory system in Bacillus subtilis. J. Bacteriol. 186, 4390–4394 (2004).
Ho, K. & Bradshaw, N. A conserved allosteric element controls specificity and activity of functionally divergent PP2C phosphatases from Bacillus subtilis. J. Biol. Chem. 296, 100518 (2021).
Miksys, A. et al. Molecular insights into intra-complex signal transmission during stressosome activation. Commun. Biol. 5, 621 (2022).
Liebal, U. W., Millat, T., Marles-Wright, J., Lewis, R. J. & Wolkenhauer, O. Simulations of stressosome activation emphasize allosteric interactions between RsbR and RsbT. BMC Syst. Biol. 7, 3 (2013).
Guerreiro, D. N. et al. Acid stress signals are integrated into the σB-dependent general stress response pathway via the stressosome in the food-borne pathogen Listeria monocytogenes. PLOS Pathog. 18, e1010213 (2022).
Igoshin, O. A., Brody, M. S., Price, C. W. & Savageau, M. A. Distinctive topologies of partner-switching signaling networks correlate with their physiological roles. J. Mol. Biol. 369, 1333–1352 (2007).
Narula, J., Tiwari, A. & Igoshin, O. A. Role of autoregulation and relative synthesis of operon partners in alternative sigma factor networks. PLoS Comput Biol. 12, e1005267 (2016).
Locke, J. C. W., Young, J. W., Fontes, M., Jiménez, M. J. H. & Elowitz, M. B. Stochastic pulse regulation in bacterial stress response. Science 334, 366–369 (2011).
Holtmann, G. et al. RsbV-independent induction of the SigB-dependent general stress regulon of Bacillus subtilis during growth at high temperature. J. Bacteriol. 186, 6150–6158 (2004).
Utratna, M., Cosgrave, E., Baustian, C., Ceredig, R. H. & O’Byrne, C. P. Effects of growth phase and temperature on σB activity within a Listeria monocytogenes population: evidence for RsbV-independent activation of σB at refrigeration temperatures. BioMed. Res Int 2014, 641647 (2014).
Brigulla, M. et al. Chill induction of the sigb-dependent general stress response in bacillus subtilis and its contribution to low-temperature adaptation. J. Bacteriol. 185, 4305–4314 (2003).
Zhang, S., Scott, J. M. & Haldenwang, W. G. Loss of ribosomal protein L11 blocks stress activation of the Bacillus subtilis transcription factor sigma(B). J. Bacteriol. 183, 2316–2321 (2001).
Ma, X. et al. A single point mutation in the Listeria monocytogenes ribosomal gene rpsU enables SigB activation independently of the stressosome and the anti-sigma factor antagonist RsbV. Front Microbiol 15, 1304325 (2024).
Norman, T. M., Lord, N. D., Paulsson, J. & Losick, R. Memory and modularity in cell-fate decision making. Nature 503, 481–486 (2013).
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009).
Wiśniewski, J. R., Zougman, A., Nagaraj, N. & Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 (2009).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Cox, J. et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell Proteom. MCP 13, 2513–2526 (2014).
Woodbury, R. L., Luo, T., Grant, L. & Haldenwang, W. G. Mutational analysis of RsbT, an activator of the Bacillus subtilis stress response transcription factor, sigmaB. J. Bacteriol. 186, 2789–2797 (2004).
Perez-Riverol, Y. et al. The PRIDE database at 20 years: 2025 update. Nucleic Acids Res 53, D543–D553 (2025).
Acknowledgements
We gratefully acknowledge funding from the National Institute of General Medical Sciences (GM138018 to M.T.C.). Special thanks are due to Chet Price, who gave us invaluable comments on a previous version of the manuscript. We also benefitted from discussions with our colleagues in the department. We are grateful to Steve Hartson and colleagues, who prepared and analyzed proteomic samples in the Oklahoma State University Mass Spectrometry/Proteomics Core Facility. We thank Niels Bradshaw and Kristin Ho for the generous gift of pKH001 plasmids.
Author information
Authors and Affiliations
Contributions
M.T.C. conceived and designed the project. R.K. built strains. R.K., B.M., N.D., M.H., A.T., and N.K.D. performed experiments and analyzed data. The manuscript was written by M.T.C. and R.K. with input from N.K.D.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Khadka, R., Maravich, B., Demarest, N. et al. Stressosome-independent but RsbT-dependent environmental stress sensing in Bacillus subtilis. Nat Commun 16, 1591 (2025). https://doi.org/10.1038/s41467-025-56871-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56871-1
