
Abstract
Crop domestication has revolutionized food production but increased agriculture’s reliance on fertilizers and pesticides. We investigate differences in the rhizosphere microbiome functions of wild and domesticated rice, focusing on nitrogen (N) cycling genes. Shotgun metagenomics and real-time PCR reveal a higher abundance of N-fixing genes in the wild rice rhizosphere microbiomes. Validation through transplanting rhizosphere microbiome suspensions shows the highest nitrogenase activity in soils with wild rice suspensions, regardless of planted rice type. Domesticated rice, however, exhibits an increased number of genes associated with nitrous oxide (N2O) production. Measurements of N2O emissions in soils with wild and domesticated rice are significantly higher in soil with domesticated rice compared to wild rice. Comparative root metabolomics between wild and domesticated rice further show that wild rice root exudates positively correlate with the frequency and abundance of microbial N-fixing genes, as indicated by metagenomic and qPCR, respectively. To confirm, we add wild and domesticated rice root metabolites to black soil, and qPCR shows that wild rice exudates maximize microbial N-fixing gene abundances and nitrogenase activity. Collectively, these findings suggest that rice domestication negatively impacts N-fixing bacteria and enriches bacteria that produce the greenhouse gas N2O, highlighting the environmental trade-offs associated with crop domestication.
Similar content being viewed by others

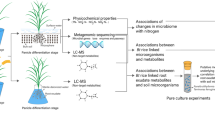

Introduction
Nitrogen (N), an essential constituent of key biomolecules such as DNA, RNA, ATP, protein, chlorophyll, auxin, and cytokinins, plays a pivotal role in crop production1,2. The nitrogen cycle is a critical factor in agroecosystems, affecting both the nitrogen source utilization rate and soil nitrogen retention, thus impacting the soil N sink3. While the intensive application of inorganic nitrogen fertilizer has enhanced crop yields, it negatively impacts the environment, leading to soil acidification4, loss of biodiversity5, and greenhouse gas emissions. This underscores an urgent need to improve crop nitrogen-use efficiency for sustainable, pollution-free agriculture.
A promising approach to enhance crop performance while reducing the use of fertilizers and pesticides, is to maximize the beneficial functions of the plant microbiome. Microbial communities associated with plant roots metabolize various forms of carbon and nitrogen, influencing plant nutrient acquisition6,7,8,9. The most well-known are the symbiotic rhizobia that fix nitrogen for leguminous plant species10, but also various other microbial taxa play a significant role in the nitrogen cycle11,12. Over the past decade, various studies have shown that plant domestication led to a reduction in the genetic diversity of the plant cultivars, substantially changed land use and concomitantly impacted the microbiome composition13,14. For example, long-term nitrogen fertilization resulted in the evolution of less-mutualistic rhizobia, providing fewer benefits to the host14,15. Nitrogen amendments also suppress soil respiration and microbial biomass, promoting copiotrophs such as Firmicutes and Actinobacteria while reducing the abundance of oligotrophs such as Acidobacteria and Verrucomicrobia16.
For rice, domestication has made the modern varieties highly dependent on nitrogen fertilizers, reducing the need for microbes that normally provide nitrogen to the plants. These microbes may still be present on the roots of wild rice varieties, yet this remains uninvestigated17,18,19. Recent studies indicated that Oryza sativa ssp. indica has a higher nitrogen use efficiency than O. sativa L. ssp. japonica due to the recruitment of a larger proportion of bacteria (FDR adjusted p < 0.05, Wilcoxon rank-sum test) that are regulated by a rice nitrate transporter and sensor (NRT1.1B)20. In Taoyuan, Hunan Province, China, the famous worldwide special ecosystem site for ultrahigh rice yield, soil microbiomes exhibit a greater abundance of genes related to the nitrification process, which induces high expression of nitrate transporters in domesticated rice (O. sativa L.) roots, ultimately leading to ultrahigh productivity21. The rhizosphere microbiomes (rhizomicrobiomes) of wild and domesticated rice can vary in many aspects, including carbon and methane metabolisms22 and bacterial chemotaxis systems23. With carbon and nitrogen metabolism being interdependent, the microbial N-metabolic functions likely differ between the two rice types. Although previous research has highlighted differences in the relative abundance of Nitrobacteriaceae, a family of bacteria that plays a significant role in the nitrogen cycle, between domesticated and wild rice24, a more comprehensive understanding of the changes in nitrogen metabolism functions of rice rhizosphere microbiomes under rice domestication is needed.
We hypothesize that the excessive use of fertilizers in rice cultivation makes domesticated rice varieties less reliant on their rhizosphere microbiomes to acquire nitrogen. More specifically, rice domestication presumably diminishes the interdependence between plants and rhizosphere microbiomes, especially concerning functional genes associated with nitrogen metabolism. To address our hypothesis, we conduct a series of experiments with wild and domesticated rice to (i) assess whether differences in rhizosphere microbiomes are reflected in the nitrogen content of five wild and seven domesticated rice accessions grown in the same soil conditions; (ii) compare the diversity and abundance of genes related to nitrogen metabolism in the rhizosphere microbiomes of these rice accessions by shotgun metagenomics sequencing; (iii) quantify the abundance of key genes in the N cycle identified through metagenomic analyses using real-time qPCR; (iv) perform transplant of rhizosphere microbiome suspensions inoculation experiments to validate the nitrogenase activity associated with the higher abundance of N-fixing genes in the rhizosphere microbiome of wild rice; (v) validate the higher abundance of specific genes associated with N2O production, given by qPCR, in domesticated rice compared to wild rice by measuring the N2O emissions in two different soils; and (vi) gain insights into how differences in root exudate composition between wild and domesticated rice affect the abundance of specific N-cycle genes; (vii) validate the effects of root exudates from wild and domesticated rice on the abundance of N-fixing genes, and nitrogenase enzyme activity.
By integrating bioassays with multiple wild and domesticated rice accessions, greenhouse gas emissions, qualitative and quantitative microbiome analyses, and root exudate metabolomics, we are able to pinpoint disparities in the nitrogen cycle between the rhizosphere microbiomes of wild and domesticated rice. Collectively, our results provide insights into the impact of rice domestication on nitrogen cycling, paving the way for future breeding strategies to maximize the beneficial effects of the root microbiome and minimize greenhouse gas emissions.
Results
Nitrogen contents of the leaves and stems of wild and domesticated rice
To determine the effectiveness of plant nitrogen uptake, we measured the nitrogen contents of the leaves, stems, roots, and entire plants of both wild and domesticated rice cultivated in identical soil conditions. The average nitrogen content of leaves, stems, roots, and total plants of the five wild rice accessions were significantly higher than the average of the seven domesticated rice accessions (Fig. 1, p = 2.75E-09 for leaves, p = 1.59E-08 for steams, p = 4.28E-08 for roots, and p = 1.49E-07 for total plant).

Asterisks indicate the leaves’ nitrogen content and stems’ nitrogen content significant differences between wild and domesticated rice based on a two-sided Welch’s t-test, p = 4.28E-08 for roots’ nitrogen content, p = 2.75E-09 for leaves’ nitrogen content, p = 1.59E-08 for steams’ nitrogen content, p = 1.49E-07 for total nitrogen content (n = 3 biological replicates). Data were presented as mean values ± SD. Source data are provided as a Source Data file.
Rhizobacterial nitrogen metabolism functions of wild and domesticated rice
Rhizosphere samples of five wild and seven domesticated rice accessions with five replicates (n = 59; one replicate discarded due to low read numbers) were subjected to shotgun metagenomic sequencing. Following post-quality control filtering, we retained a total of 9,554,978,912 reads for subsequent analyses, averaging 136,499,699 reads per sample (n = 59). From the predicted 198,319,043 open reading frames (ORFs), about 60% were classified using the KEGG database (for more detailed sequencing statistics, we refer to Table S1). The analyses identified 101,290 reads related to N-cycle functions, averaging 8441 reads and 1784 contigs per sample. From these contigs, we found an average of 2033 ORFs per sample related to N-cycle functions (Table S2), falling into 27 functional orthologs involved in N metabolism. A significant difference was found in N metabolism functional profiles in the rhizosphere microbiomes of wild and domesticated rice, with dissimilar clustering patterns evident in the Principal Coordinates Analysis (PCoA) plots (Fig. S1) (PERMANOVA, R2 = 0.42, p < 0.001). This separation accounts for 21.28% of the total variation in functional profiles associated with N metabolism as revealed by between-class analysis (BCA) (Table 1, p = 0.001).
Profiles of nitrogen-cycle genes in the rhizosphere of wild and domesticated rice
The cumulative transcripts per million (TPM) of narH (p = 4.75E-08), narG (p = 4.89E-08), napA (p = 0.0001), napB (p = 0.001), and nasA (p = 1.75E-06), key bacterial genes involved in nitrate reduction to nitrite, were significantly more enriched in the rhizosphere microbiome of domesticated rice than in those of wild rice (Fig. 2 and Table S3). For the oxidation from nitrite to nitrate, a higher cumulative TPM of the bacterial genes was observed for the rhizosphere microbiome of domesticated rice. Also, the cumulative TPM for nirD (p = 6.21E-06), nrfA (p = 0.325), nrfH (p = 0.003), and nirA (p = 0.0004), essential genes for nitrite reduction to ammonium, were enriched in the rhizosphere microbiome of domesticated rice. In contrast, the TPM of amoA (p = 0.201), amoB (p = 0.126), and amoC (p = 0.349) genes involved in the conversion of ammonium to hydroxylamine, was slightly higher in the rhizosphere microbiome of wild rice, albeit statistically insignificant. The gene hao (p = 0.122), involved in reducing hydroxylamine to nitrite, showed a similar trend. Additionally, we found a significantly higher TPM of nirK (p = 2.88E-09) and nirS (p = 0.003), genes essential for nitrite reduction to nitric oxide, in the microbiome of domesticated rice. The genes norB (p = 0.082) and norC (p = 0.568) involved in nitric oxide reduction to nitrous oxide, also occurred more frequently in the rhizomicrobiome of domesticated rice. The gene nosZ (p = 2.90E-06), responsible for converting nitrous oxide to nitrogen, showed significantly higher TPM in domesticated rice. Conversely, the genes nifD (p = 7.00E-12), nifK (p = 2.28E-17), and nifH (p = 3.37E-13), involved in nitrogen fixation, were more enriched in the microbiome of wild rice. Last but not least, the between-class analysis (BCA) showed that nitrate reduction, nitrification, denitrification, and nitrogen fixation contributed 18.03, 18.60, 25.12, and 61.59%, respectively, to the total variance in functional profiles between the wild and domesticated rice accessions (Table 1).

Asterisks indicate significant differences between the two groups (two-sided Welch’s t-test, narG p = 4.89E-08, narH p = 4.75E-08, narI p = 0.429, napA p = 0.0001, napB p = 0.001, narB p = 0.056, NR p = 0.398, nasA p = 1.75E-06, nasB p = 0.629, nirB p = 0.265, nirD p = 6.21E-06, nrfH p = 0.003, nrfA p = 0.325, NIT-6 p = 0.285, nirA p = 0.0004, amoA p = 0201, amoB p = 0.126, amoC p = 0.349, hao p = 0.122, nirK p = 2.88E-09, nirS p = 0.003, norB p = 0.082, norC p = 0.568, nosZ p = 2.90E-06, nifD p = 7.00E-12, nifH p = 3.37E-13, nifK p = 2.28E-17), group wild rice (n = 24) and group domesticated rice (n = 35), data were presented as mean values ± SD; TPM (transcripts per million); Blue arrows indicate that the TPM of genes involved in the pathway is higher in the domesticated rice rhizosphere, and red arrows indicate that the TPM of genes involved in that pathway is higher in the wild rice rhizosphere. Source data are provided as a Source Data file.
Taxonomic assignment of nitrogen-cycling genes
The taxonomic assignment of the functional genes involved in N-cycling identified by KEGG for wild and domesticated rice was obtained by searching for the last common ancestor of the hits for each query gene using the results of the Diamond search against the GenBank nr database. To ensure the robustness of the assignments, we included hits having at least 80% of the bit-score of the best hit and less than 10% identity difference. PCoA showed a significant difference between the rhizobacterial communities of wild and domesticated rice accessions (Fig. 3a, PERMANOVA, F = 65.05, R2 = 0.53, p < 0.001). The variance was primarily explained by axis1 (57.3%) and axis2 (24.9%). A t-test applied to the Shannon index highlighted significantly greater evenness in the rhizobacterial community of domesticated rice than in wild rice (Fig. 3b, p < 0.05).

a Compositional changes of rhizomicrobial taxonomic assignment of genes involved in N-cycling of wild and domesticated rice. PCoA was performed based on the Bray-Curtis dissimilarity matrix at the family level across all the samples. Ellipses demonstrate the mean ± SD, wild rice in pink and domesticated rice in blue (F = 65.05, R2 = 0.53, p < 0.001). b Alpha diversity of rhizomicrobial communities with specific roles in N-cycling of wild and domesticated rice. Wild rice rhizosphere samples, n = 24; Domesticated rice rhizosphere samples, n = 35. Asterisks indicate significant differences between wild and domesticated rice based on two-sided Welch’s t-test, p = 8.95E-20. The data were displayed using boxplots, which show the median, interquartile range, and whiskers indicating the minimum and maximum values. c Relative abundances of microbial taxonomic assignment of genes involved in N-cycling of wild and domesticated rice; d These taxonomies contained different N-cycling genes. Source data are provided as a Source Data file.
Taxonomic analysis of all N-cycling genes revealed 19 bacterial families in both wild and domesticated rice rhizospheres (Fig. 3c). Wild rice had dominance of Geobacteraceae, Syntrophaceae, Bradyrhizobiaceae, and Nitrospiraceae, while domesticated rice was characterized by Bradyrhizobiaceae, Syntrophaceae, Nitrospiraceae, and Anaeromyxobacteraceae. Several N-cycling genes were identified across various families, such as Pseudomonadaceae, Methylococcaceae, Methylocystaceae, Oxalobacteraceae, Sphingomonadaceae, Burkholderiaceae, Geobacteraceae, Anaeromyxobacteraceae, Nitrospiraceae, Syntrophaceae, and Bradyrhizobiaceae. In contrast, the Enterobacteriaceae, Thiobacillaceae, Polyangiaceae, Xanthomonadaceae, Rhodanobacteraceae, Gallionellaceae, Methanosarcinaceae, and Comamonadaceae families each contained a single nitrogen-cycling gene. Specifically, key N-cycling genes like nifK, narG, nifD, and narH were detected within Geobacteraceae, nasA, narI, and hao within Syntrophaceae, nrfA, nrfH, napA, napB, norB, norC, and narG within Nitrospiraceae, nrfA, nrfH, norB, narG, and narH within Anaeromyxobacteraceae, and nifH, nifK, nosZ, napA, napB, nasA, narG, nirS within Bradyrhizobiaceae (Fig. 3d), emphasizing the marked contrast in rhizobacterial communities between wild and domesticated rice and the indicate nature of their respective N-cycling pathways.
Quantification of N-cycle metagenome data by qPCR
Metagenome data allowed assessment of the N-cycle gene counts in the rice rhizosphere but did not reflect quantitative gene abundances25. Therefore, we validated the findings of the metagenome data by quantitative PCR (qPCR). The results revealed differential gene abundances related to N2O production and nitrogen fixation (Fig. 4). Notably, while the abundance of nifH (Fig. 4a, p = 3.98E-08) was higher in wild rice rhizomicrobiome, nosZ (Fig. 4b, p = 0.00001), nirK (Fig. 4c, p = 2.55E-13), nirS (Fig. 4d, p = 1.16E-22) and AOA-amoA (Fig. 4e, p = 1.64E-25) were significantly higher in domesticated rice. It should be noted that for the AOA-amoA gene the qPCR results did.

a nifH, b nosZ, c nirK, d nirS, e AOA-amoA, and f AOB-amoA. Asterisks indicate significant differences between the two groups (two-sided Welch’s t-test, nifH p = 3.98E-08, nosZ p = 0.00001, nirK p = 2.55E-13, nirS p = 1.16E-22, AOA-amoA p = 1.64E-25, AOB-amoA p = 0.194). The figure depicts data for wild rice, consisting of five accessions, and domesticated rice, comprising seven accessions. The gene abundances are based on measurements from three biological replicates per accession, with each replicate including three technical replicates. In addition to the scatter plot, the data are displayed using boxplots, which show the median, interquartile range, and whiskers indicating the minimum and maximum values. Source data are provided as a Source Data file.
The rhizomicrobiome of wild rice enhances nitrogenase activity and plant growth
To evaluate the positive effects of the wild rice rhizomicrobiome on nitrogen fixation, we performed an experiment in which wild and domesticated rice were grown in soil inoculated with rhizosphere soil suspensions from either wild or domesticated rice. Regardless of whether wild rice or domesticated rice was planted, soil nitrogenase activity was significantly higher when the soil was inoculated with the wild rice rhizosphere soil suspension compared to the domesticated rice rhizosphere soil suspension (Fig. 5a, b, p < 0.05). Furthermore, the plant growth of both domesticated (Fig. 5c) and wild (Fig. 5d) rice was enhanced when grown in soil inoculated with the wild rice rhizosphere suspension.

Nitrogenase activity of rhizosphere soil of a domesticated rice seedlings and b wild rice seedlings after 30 days inoculated with PBS buffer (Germ-free), rhizosphere soil suspension of domesticated rice (Dome-RS) and rhizosphere suspension of wild rice (Wild-RS). Growth status of c domesticated and d wild rice seedlings after 30 days inoculation of rhizosphere soil suspensions from domesticated (Dome-RS) and wild rice (Wild-RS). The error bars represent the standard deviation of the mean (n = 4 biological replicates), and different letters indicate significant differences between different treatments (one-way ANOVA, a domesticated rice seedings: Germ-free vs Dome-RS p = 1.24E-12, Germ-free vs Wild-RS p = 3.68E-12, Dome-RS vs Wild-RS p = 0.039; b wild rice seedings: Germ-free vs Dome-RS p = 3.55E-12, Germ-free vs Wild-RS p = 1.18E-12, Dome-RS vs Wild-RS p = 0.010). Data were presented as mean values ± SD. The unit of nitrogenase activity IU/L: indicates how many micromoles of nitrogen gas are converted into ammonia per minute per liter of the enzyme solution. Source data are provided as a Source Data file.
Validation of qPCR N-cycle data with nitrous oxide emissions in wild and domesticated rice
To validate the higher abundance of genes associated with N2O production, given by qPCR, in domesticated rice compared to wild rice, we measured N2O emissions. To mitigate the potential soil effect, we planted both wild and domesticated rice accessions in two distinctly different soils, i.e. black and saline-alkaline soils. For the wild rice accession, the N2O emission fluxes were similar in the two soils with approximately 2.5 mg m−2 day−1 (3.05 mg m−2 day−1 and 2.01 mg m−2 day−1, respectively). These emission fluxes were significantly (p < 0.05) lower than those measured for the domesticated rice (Fig. 6). The N2O emission fluxes from domesticated rice O. sativa L. ssp. japonica and O. sativa L. ssp. indica, when planted in black soil, were approximately 4.5 mg m−2 day−1 (4.46 mg m−2 day−1 and 4.49 mg m−2 day−1, respectively), while in the saline-alkali soil, N2O emissions fluxes were around 3.5 mg m−2 day−1 (3.73 mg m−2 day−1 and 3.39 mg m−2 day−1, respectively).

Asterisks indicate significant differences between wild and domesticated rice (one-way ANOVA, black soil: wild rice vs indica rice p = 0.011, wild rice vs japonica rice p = 0.012, indica rice vs japonica rice p = 0.965, Saline-alkali soil: wild rice vs indica rice p = 0.008, wild rice vs japonica rice p = 0.002, indica rice vs japonica rice p = 0.47, n = 4 biological replicates). Data were presented as mean values ± SD. Source data are provided as a Source Data file.
Correlation between abundances of nitrogen fixation gene and root metabolites of wild and domesticated rice
Root metabolomics of wild and domesticated rice accessions were determined to obtain insight into the root exudate constituents associated with changes in the abundance of specific N-cycle genes. For that, we adopted Spearman correlation analysis to investigate the associations between N-cycle genes abundances and root metabolite constituents, aiming to elucidate the relationship between wild rice higher abundance of nitrogen fixation genes and the root metabolites. The correlation between nifH and exudates in wild rice encompassed 22 exudate constituents, including fumaric acid, quinic acid, succinic acid, shikimic acid, and others, classified as organic acids, amino acids, phosphates, alcohols, fatty acids, and sugars. Contrastingly, only seven exudates (i.e., amino acids and sugars), correlated with nifH in the rhizosphere microbiome of domesticated rice (Fig. 7, r > 0.4, p < 0.05). Notably, wild rice showed 21 positive and only one negative relationship, whereas domesticated rice exhibited more negative (5) than positive (2) correlations. Succinic acid, shikimic acid, and maleic acid were common in both wild and domesticated rice accessions.

a Pie charts illustrating the number of metabolites and the number of positive and negative relationships correlated with nifH in wild and domesticated rice within the different compositional categories, wild rice in pink and domesticated rice in blue. b A hierarchically structured network depicting the correlation between nifH and metabolites of wild and domesticated rice by Spearman (|r| > 0.4, p < 0.05), the metabolites nodes of wild rice in pink and domesticated rice in blue, the shared metabolites of wild and domesticated rice in gray. The size of the nodes is the abundance of metabolites. The edges in red lines mean the significantly positive correlations and the edges in blue lines mean the significantly negative correlations. wild rice rhizosphere samples, n = 24; domesticated rice rhizosphere samples, n = 35. Source data are provided as a Source Data file.
Root metabolites of wild rice promote the abundance of the N-fixation gene
To further demonstrate the influence of wild rice root metabolites on nitrogen fixation genes in the microbiome, we conducted a pot experiment where root exudates collected from wild and domesticated rice accessions were amended to the soil, followed by monitoring the dynamics of nifH gene abundances at intervals of 0, 4, 8, 12, 24, and 36 h, as well as 2, 4, 6, and 8 days. A total of 231 metabolites were collectively identified in the root metabolite solutions of both wild and domesticated rice (Fig. 8). Following the incubation with the root metabolite solutions in soil, we observed differences in the dynamics of nifH abundances, with the most significant distinction occurring at 36 h after addition of the metabolites where nifH gene abundance in the wild rice root exudate treatment was significantly higher than in the domesticated rice root exudate treatment (Fig. 8a). In addition, nitrogenase activity was significantly higher in the wild rice root exudate treatment compared to the domesticated rice root exudate treatment (Fig. 8b).

a Pie charts illustrating the number of differentially abundant metabolites in wild and domesticated rice within the different compositional categories. The differentially abundant root metabolites were identified by a comparison of wild and domesticated rice (Welch’s t-test, p < 0.05). b A line chart shows the dynamics of the abundance of nifH gene after root metabolites inoculation in soil. The treatments are as follows: CK: sterilized distilled water; Wild: wild rice root metabolites solution; Domesticated: domesticated rice root metabolites solution. Vertical bars indicate the standard error of the mean (n = 5 biological replicates). c Nitrogenase activity at 36 h. The error bars represent the standard deviation of the mean (n = 4 biological replicates), and different letters indicate significant differences between wild and domesticated rice (one-way ANOVA, p = 2.03E-09). Data were presented as mean values ± SD. Source data are provided as a Source Data file.
Discussion
Our study utilized shotgun metagenomics sequencing to contrast the N-cycling capacities and corresponding bacterial families in the rhizosphere of a collection of five wild and seven domesticated rice accessions. This comparison revealed significant disparities between wild and domesticated rice in plant nitrogen content, microbial nitrogen pathways, and gene abundances as measured by qPCR, nitrous oxide emissions, and a link with differences in root exudates composition. Thousands of years ago, early human societies relied on diets composed of wild, native plants. With the growing global population, the demand for food increased, resulting in the domestication of wild plant species and subsequent breeding together with modern agriculture. Plant domestication and breeding typically involve genetic modifications to create new genotypes better suited to human needs. In rice, traits like shattering and lodging have been modified to improve grain yield26. However, this has also made the plant largely dependent on external inputs for growth and health. One of the most significant is the reliance of domesticated crops on excessive nitrogen fertilizer to maintain or boost yields. The results of our study confirm and extend earlier results that wild and domesticated rice have different microbiome compositions in the rhizosphere, including bacteria and fungi17,18,19,27,28. To our knowledge, our study is the first to systemically compare the microbial N-cycling functions of the rhizosphere microbiomes of wild and domesticated rice accessions, focusing on both Asian and African domesticated rice and their wild relatives. Here, we demonstrated that the N metabolic profiles of the rhizomicrobiomes of wild rice and their domesticated derivatives were significantly different. One possible explanation for these findings is that rice domestication, which encompasses a long history of directional selection and breeding aimed at developing genotypes with increased yields that are directly linked to nutrient uptake from fertilizers, has led to the elimination of the co-dependency between microbes and plants. As a consequence, some N-cycling rhizosphere microbial groups present in wild rice accessions have been gradually diminished24. This study highlights the specific effect of rice domestication on rhizobacterial nitrogen functions and demonstrates that domestication led to significant changes in the frequency of nitrogen functional traits of the rhizosphere microbial community.
Under low-oxygen conditions, such as the condition of the rhizosphere soil in our experiments, denitrification is the main process of nitrogen loss by which NO, N2O, and N2 are emitted into the atmosphere29. The nirK and nirS genes of the nitrite-to-NO pathway were more abundant in the microbiome of domesticated rice than in that of wild rice, suggesting that the rhizobacteria in domesticated rice have a greater potential to transform nitrite into NO gas form than those of wild rice. Besides, the abundance of nirK was higher than that of nirS in the rhizosphere of domesticated rice. Although both nirK and nirS are nitrite reductase genes, studies have shown that nirK-harboring denitrifiers are more responsive to denitrification induced in rice paddy soil than nirS-harboring bacteria30. Interestingly, ref. 31 found that nitrite can be bypassed during the denitrification process, leading to the conversion of two nitric oxide molecules to dinitrogen and oxygen, which is used to oxidize methane. In fact, in our previous study, we found distinct differences in methane metabolism between the rhizosphere microbiomes of wild and cultivated rice accessions, with domesticated rice showing an overrepresentation of key enzymes associated with methane production22. Combining these findings with our results predicts that the more NO production, the more methane could be oxidized, playing an important role in linking the global carbon and nitrogen cycles in low-oxygen environments.
Similarly, norB and norC belonging to the N2O production pathway were also more enriched in the rhizosphere of domesticated rice than that of wild rice, indicating greater nitrogen loss in domesticated rice. Nitric oxide reductase, encoded by norB and norC, catalyzes the reduction of NO to N2O32. However, N2O emission is also related to nosZ, which encodes for nitrous oxide reductase participating in the N2O reduction pathway. Our study showed that the nosZ gene was enriched in the rhizosphere of domesticated rice, indicating a different ecological balance in the rhizosphere microbiomes of domesticated rice compared to that of wild rice. Furthermore, our results showed that the abundance of AOA-amoA was significantly higher in the rhizosphere of domesticated rice than in that of wild rice, suggesting nitrification by archaea in the rhizosphere of domesticated rice.
In conclusion, we provided evidence that the proportion of functional genes related to nitrogen metabolism, especially nitrogen fixation gene nifH, is lower in domesticated rice, as confirmed by BCA analyses and quantified by qPCR (Fig. 9). The BCA confirmed this difference, showing that 61.59% of the total variation between the N-fixation pathways is related to different rice genotypes, while for the total variation between other pathways involved in nitrogen cycling, only about 20% is related to different accessions. In addition, the nitrogen contents in leaves, stems and roots of wild rice were significantly higher than those of related domesticated rice. These results suggest that wild rice has an efficient nutrient acquisition strategy, which may rely, at least in part, on N-fixing members in the rhizosphere microbiome. These findings complement the results of our previous study, where 16S rRNA amplicon sequencing predicted nitrogen fixation as a key ecological function of the core rhizosphere bacteriome of wild rice33. We found that Geobacteraceae, Methanosarcinaceae, and Bradyrhizobiaceae have specific roles in N-fixation. Geobacteraceae strains have been reported, including mRNA transcript abundance of the nitrogen fixation gene, nifD, and the ammonium transporter gene, amtB34. Bradyrhizobium, a member of the Bradyrhizobiaceae family, comprises diverse bacteria capable of forming symbiotic and endophytic relationships with both leguminous and nonleguminous plants, including rice35. Validation through inoculation experiments with rhizosphere microbiome suspensions revealed that soils inoculated with wild rice suspensions exhibited the highest nitrogenase activity, regardless of the type of rice planted. The tryptic soy broth (TSB) enrichment method was employed to rapidly obtain rhizosphere microbial communities, though it favors fast-growing and aerobic microorganisms, potentially excluding slow-growing or anaerobic species. These inoculation experiments highlighted the ecological role of the wild rice rhizomicrobial community in enhancing nitrogen uptake. Future research will focus on optimizing cultivation conditions, such as extending incubation times or incorporating anaerobic environments, to achieve a more comprehensive representation of rhizomicrobial diversity.

During rice domestication, a higher abundance of nitrogen-fixing genes in the rhizosphere of wild rice, while domesticated rice exhibited an increased number of genes associated with nitrous oxide (N2O) production and reduction in the rhizosphere of domesticated rice. Created in BioRender. Chang, J. (2025) https://BioRender.com/r38f406.
Finally, we conducted root metabolic analyses to begin to understand the differential assembly and functions of the rhizosphere microbiomes of wild and domesticated rice accessions. During the process of domestication, plants have been selectively bred to enhance yield, which has led to changes in their exudate profiles. These changes may subsequently influence the function and activity of nitrogen-cycling microbes on the roots. For example, in wild rice, we observed a significant correlation between nifH and 22 specific exudates, including fumaric acid, 4-hydroxycinnamic acid, citric acid, and aspartic acid. However, in domesticated rice, these relationships with the aforementioned compounds were not detected. Certain bacteria, such as Bradyrhizobium, can utilize fumaric acid, maleic acid, and 4-hydroxycinnamic acid as carbon and energy sources for nitrogen fixation36. Azospirillum is a N-fixing bacterium that can utilize citric acid and aspartic acid37. While almost all correlations between metabolites and nifH were positive in wild rice, domesticated rice exhibited only seven metabolites related to nifH, and many of these connections were negative. These results suggest that wild rice releases metabolites to attract N-fixing microbiome members, whereas during domestication, the composition of the root exudates changed with a concomitant change in microbiome assembly and abundance of N-fixing microbiome members.
Considering the potential use of beneficial microbes from wild populations as bioinoculants for domesticated crops, a process called rewilding38, our findings caution against a straightforward application of microorganisms from wild crops to domesticated ones. These effects may be visible in short-term experiments, as we showed in the transplanting rhizosphere microbiome suspensions inoculation experiments, but for longer-term effects, the introduced microbial inoculants should be able to establish and survive over longer time periods. Hence, the host plant needs to provide the necessary metabolites to regulate the survival and activities of the introduced microbial inoculants. The importance of this host support was exemplified in our experiments, where the addition of root exudates from wild and domesticated rice applied to soil exhibited differential effects on N fixation. These results support and extend the results of a previous study in which the addition of root exudates, including carbohydrates, significantly increased soil N fixation39. In our study, the observed differences in nitrogenase activity between the transplanting rhizosphere microbiome suspension inoculation experiment and the experiment inoculating root metabolites extracted from wild and domesticated rice into soil arise from their distinct experimental designs. The transplanting experiment simulates long-term plant-microbe interactions, where the increase in nitrogenase activity reflects the combined effects of these interactions. The microbes from the enriching, fast-growing, and aerobic microorganisms required time to adapt to the new environment and form functional communities. At the same time, rice roots gradually influenced the colonization and activity of rhizosphere microbial communities by secreting metabolites into the soil. As a result, nitrogenase activity reached 18 IU/L after 30 days, representing a dynamic equilibrium. Conversely, the root metabolite experiments emphasize the immediate short-term stimulatory effects of water-based collected root exudate directly on soil native microbes. Without the competition or interference of plants, the metabolites rapidly activated native nitrogen-fixing microbes, causing nitrogenase activity to rise sharply to 60 IU/L. Although direct comparisons between the two experiments are challenging due to differences in experimental conditions and designs, both consistently demonstrate that root exudate significantly enhances the activity of nitrogen-fixing microbes. Moreover, the two approaches offer complementary perspectives on the complex interactions between root exudates and microbial communities, highlighting both the short-term dynamic effects of metabolite-driven activation of soil native microbes and the long-term stimulation through plant-mediated re-colonization of fast-growing microbial communities. Our study marks the initial discovery that wild rice and domesticated rice exhibit distinct differences in the composition of root exudates, which in turn affects the abundances of nifH and nitrogenase activity present in the soil. This finding extends our knowledge of how root exudates change with plant nutrient status and subsequently influence microbial-mediated nitrogen fixation. To mechanistically disentangle how these root exudates change, leading to differences in nitrogen fixation in rice, future studies are necessary.
Rice fields are considered a significant non-point nitrogen source for both atmospheric and aquatic environments, posing a high risk of environmental pollution due to reactive nitrogen losses through ammonia volatilization, denitrification, surface runoff, and leaching40,41. The importance of plant genomes shaping the rhizosphere microbiome is increasingly apparent while breeding for yield-related traits has ignored the impact on the taxonomic and functional composition in the rhizosphere of modern crop microbiomes42. Research on food production is beginning to clarify the mechanisms by which wild crops harbor compositionally different microbiomes. In conclusion, incorporating the depleted microbiome members and their functions present in wild rice linked to specific root exudates into breeding of modern domesticated crops should be considered to maximize the success of sustainable rice production and reducing environmental pollution and greenhouse gas emissions.
Methods
Plant materials, site description, sample collection, and plant nitrogen content determination
The 12 accessions of wild and domesticated rice including five wild rice accessions: African wild rice IRGC 106238 (O. barthii), common wild rice IRGC 106452 and IRGC 106286 (O. rufipogon), and Indian wild rice IRGC 86655 and IRGC 88949 (O. nivara); and seven domesticated rice accessions: African domesticated rice LM8, No. 3 African domesticated rice and WH20 (O. glaberrima), Asian domesticated rice japonica Jiangxi (O. sativa L. ssp. japonica), Asian domesticated rice japonica Daohuaxiang (O. sativa L. ssp. japonica), and Asian domesticated rice cultivars indica 106 and Meitezhen (O. sativa L. ssp. indica) were used for this study22.
In order to avoid the influence of environment and soil, wild and domesticated rice were planted in the same soil and climate conditions, located in the rice experimental station of the Chinese Academy of Sciences (18°19’57 N, 109°27’ E) in Sanya, Hainan Province, a typical Tropical Ocean monsoon climate. The values for soil properties and the experimental design have been described in a comparative study of the rhizosphere fungal communities of wild and domesticated rice accessions17.
At the flowering stage of rice, rhizosphere soil was sampled for each rice accession. The loose soil adhering to the roots was shaken off, and approximately 1 mm of soil attached to the roots was left. Then, we separated the 1 mm of soil from the roots by immersing the root with soil in 5 mL of sterile water and vortexed to collect the rhizosphere soil that was tightly attached to root43. After a short centrifugation and removal of supernatant, each rhizosphere soil sample was stored at −80 °C. Part of the roots were separated and stored at −20 °C for root metabolites extraction. Leaves and stems were collected, air-dried at 60 °C to constant weight, grinded, and passed through a 0.15-mm sieve. A total of 50.0 mg leaves and stem samples were weighted and tightly wrapped in tin foil cones. An automatic injection tray of element analyzer (Vario Macro CN, German) was used to determine nitrogen contents.
DNA extraction, shotgun metagenomics sequencing, bioinformatics analysis, and quantitative real-time PCR
A 0.5 g rhizosphere soil per sample was ground into powder in liquid nitrogen, and the total DNA was extracted using Fast DNA SPIN Kit (MPBio, Santa Ana, USA). DNA concentrations were measured using a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Waltham, USA). The quality of the DNA was checked by 1.2% agarose gel electrophoresis, and only the DNA with a clear band was chosen for further metagenome shotgun sequencing. The sequences of 12 wild and domesticated rice accessions with five replicates, in a total of 59 samples, were deposited in the National Center for Biotechnology Information (NCBI /) under the bioproject number PRJNA632564 described by ref. 22.
The shotgun metagenomics sequences were processed using the SqueezeMeta pipeline in seqmerge mode44. Briefly, Trimmomatic was used for adapter removal, trimming, and quality filtering45. Megahit and prinseq were used to assemble each sample separately and filter contigs46,47. Prodigal and barrnap were used for ORF prediction and rRNA gene sequence retrieval, respectively48,49. Diamond software was used for taxonomic classification of the ORFs against the latest publicly available version of KEGG database for the kyoto encyclopedia of genes and genomes (KEGG) orthology (KO) numbers50. The N-cycling genes were identified and annotated using the automatic annotation server of the KEGG. The average coverage and normalized TPM values for information on gene and function abundances were computed by SqueezeMeta script SQM2tables.py. Therefore, we evaluated the differences between treatments as differences in the proportion of the functional genes. Normalized TPM SqueezeMeta ORF related to nitrogen metabolism dataset and taxonomic assignment of the N-cycling genes were performed at ORF level for the assembled contigs, and results recovered from MGNify analysis were used for statistical analyses, performed in RStudio version 1.1.423 running R version 4.1.251. The quality controlled contigs larger than 1000 kb were used for binning using Concoct, Maxbin, and Metabat52,53,54; DAS tool was used to refined resulting bins55, and genome dereplication was performed with dRep56. The ORFs of the genomes were predicted using Prodigal, and DIAMOND software57 was used for functional annotation with eggNOG (KO numbers)58. The overall effect of domestication in the variability of the functional genes related to the nitrogen-cycle pathways were evaluated using the between-class analysis (BCA) of the clr-transformed data in the ade4 package59.
The quantitative real-time PCR (qPCR) was applied to quantify the rhizomicrobial genes involved in the nitrogen cycle, including the ammonia-oxidizing genes (AOA-amoA, AOB-amoA), denitrifying genes (nirK, nirS, nosZ), and nitrogen fixation gene (nifH)60,61,62. qPCR was performed in a 96-well plate using the MX3000P qPCR system (Agilent, La Jolla, CA, USA). The primer combinations, reaction descriptions, and thermal cycler conditions for each gene amplification are listed in Table S4. Plasmid DNA from microbiomes containing the target genes from wild rice rhizosphere samples was used to construct standard curves and then cloned into vectors. Standard curves for each gene were performed ten-fold serial dilutions from 101 to 107 with three technical replicates. The reaction efficiencies from 90 to 105% were obtained for all these genes' quantification with R2 values above 0.99. qPCR was performed in biological triplicates with three technical replicates. A minus-template qPCR that used distilled water as a template instead of rhizosphere DNA was conducted simultaneously.
Rhizosphere microbiome harvesting and inoculation
The rhizosphere microbial community was obtained from 10 g of rhizosphere soil either from wild rice (O. rufipogon) or domesticated rice (O. sativa L. ssp. japonica) by culturing them in 120 mL of tryptic soy broth (TSB) with shaking at 60 rpm for 2 days. The supernatant of the rhizosphere soil suspension was then collected and incubated for another 3 days at 28 °C63. These rhizosphere soil suspensions, from wild and domesticated rice, were washed with PBS buffer, and 1.0 ml of rhizosphere soil suspension (OD600 of 0.5) was added to 100 ml of solution and 100 g of sterilized black soil mixed with vermiculite (1:1). As a control, germ-free PBS buffer was used in place of the suspension20. Wild and domesticated rice seedlings (8 days from seeds) were grown in the inoculated soil at 35 °C during the 15 h light period and 30 °C during the 9 h dark period; the relative humidity was maintained between 70 and 80%. Each treatment had four biological replicates.
Rhizosphere soil samples were collected 30 days post-inoculation (dpi) to measure nitrogenase activity. For each sample, 0.5 g of rhizosphere soil was added to PBS at a ratio of 1:10, vortexed for 40 s, and centrifuged for 15 min. The supernatant was used to measure nitrogenase activity using the Soil Nitrogenase (NITS) ELISA Kit (ADANTI, Wuhan, China).
Nitrous oxide (N2O) measurement
To validate the highest abundance of genes associated with N2O production, given by qPCR, in domesticated rice compared to wild rice, we measured the N2O emissions in two different soils under field experiment conditions. Three accessions were used: common wild rice (O. rufipogon), Asian domesticated rice japonica (O. sativa L. ssp. japonica), and Asian domesticated rice indica (O. sativa L. ssp. indica) in two fields, at Northeast Institute of Geography and Agroecology, Chinese Academy of Sciences, which is located at the National Agricultural Station in Liaoheyuan, Jilin Province, China, and in Daan, Jilin Province, China. The site is characterized by a typical temperate and monsoonal climate, and the soil are classified as black soil and saline-alkali soil, respectively, according to the United States Department of Agriculture (USDA) soil classification system.
The flux of N2O was quantified using PVC static chambers with a height of 80 cm and a diameter of 25 cm. At the flowering stage of rice, gas samples were collected in the morning from 9 am to 12 am. These chambers were inserted into the soil to a depth of 5 cm. Each chamber cap featured two openings, one connected to a valve for gas sampling and the other for pressure equilibrium. The chambers remained open until gas sampling. Gas was sampled with plastic syringes (60 ml of gas) at five intervals (0, 10, 20, 30, and 40 min) after the chambers were closed. The samples were analyzed in a gas chromatograph (model GC-2014, Shimadzu Co.) with an electron capture detector for N2O determination. The overall N2O flux was calculated by linear interpolation of the five sampling times.
Root metabolites extraction and gas chromatography time-of-flight mass spectrometry (GC-MS) analysis
The root metabolomics of five wild and seven domesticated rice accessions were determined. Root metabolites were extracted from 10 mg of each root sample placed in 2 mL tubes, which were treated with 1 mL of pre-cold extraction solution (v/v, methanol/ddH2O = 3/1) containing 10 μL of the internal standard L-2-Chlorophenylalanine (1 mg/mL stock), vortexed for 30 s, homogenized for 4 min at 35 Hz, and subjected to ultrasonication for 5 min in an ice-water bath, repeated three times. After centrifuged for 15 min at 4 °C, the supernatant was dried by methoxyamine hydrochloride and incubated at 80 °C for 30 min. Then, derivatized by 40 μL of BSTFA-TMCS regent (bis(trimethylsilyl)trifluoroacetamide-trimethylchlorosilane, v/v, 99/1) at 70 °C for 1.5 h. The samples were transferred into vials for GC-TOF-MS analysis using an Agilent 7890 gas chromatograph coupled with a time-of-flight mass spectrometer and a DB-5MS capillary column (J&W Scientific, Folsom, USA)23. Raw data, including peak extraction, baseline adjustment, deconvolution, alignment, and integration, were processed using Chroma TOF (V 4.3x, LECO) software, and metabolites were identified by matching the mass spectrum and retention index using the LECO-Fiehn Rtx5 database. These metabolites were then correlated (Pearson correlation) with N-cycle gene abundance obtained by qPCR, with the aim of gaining insight into the root exudate constituents associated with changes in the abundance of specific N-cycle genes.
Effect of wild and domesticated rice root metabolites on nifH and nitrogenase activity in soil
Because we observed a correlation between the root exudates of wild rice and the high abundances of nifH, we conducted a pot experiment using root exudates from both wild and domesticated rice in non-sterile soil without planting rice seedlings to further support our findings.
We planted common wild rice IRGC 106452 (O. rufipogon) and Asian domesticated rice japonica Daohuaxiang (O. sativa L. ssp. japonica) in soil that is exactly the same soil where the 12 accessions (5 wild- and 7 domesticated rice) were cultivated previously for rhizosphere metagenome analysis. At the stage of rice with three leaves and one heart development, the root system of common wild rice IRGC 106452 (O. rufipogon) and Asian domesticated rice japonica Daohuaxiang (O. sativa L. ssp. japonica) was carefully washed with sterilized de-ionized water and soil was completely removed. About 250 ml of sterilized water was used for root exudate collection per three plants, each accession at once. Plants were cultivated in sterilized water for 48 h (35 °C during the 15 h light period and 30 °C during the 9 h dark period; the relative humidity was maintained between 70 and 80 %) to ensure adequate accumulation of root metabolites. The root exudate solution was filtered through a 0.22 µm sterile membrane to remove particulate matter. The filtrate was then freeze-dried using a freeze-drier (FD-1A-50, temperature at −60 °C) to obtain metabolite powder64. The dried metabolite powder was accurately weighed and dissolved in sterile water to achieve a final concentration of 5 mg ml−1, using the formula C = m/V, where “m” represents the quantity of root exudate, and “V” denotes the volume. The solution was homogenized with a vortex mixer. Subsequently, the above solution containing root metabolites at a concentration of 5 mg ml−1 was introduced into 2 kg of non-sterile soil of the same type. This was done to achieve a moisture level of approximately 20% in the inoculated soil, which is roughly similar to the average level of the prevailing climatic conditions at the site from where the soil was taken. In total, ten pots of soil (five replicates for wild and five for domesticated rice) were used to assess the variance. Soil samples were collected at time points: 0 h, 4 h, 8 h, 12 h, 24 h, 36 h, 2 days, 4 days, 6 days, and 8 days post-incubation, and total DNA extracted to monitor the dynamics of nifH gene by qPCR, following previously described methods in the methods section above. Nitrogenase activity was measured at the time points at which significant differences in nifH abundances were observed. To measure nitrogenase activity, 0.5 g of rhizosphere soil was added to PBS at a 1:10 ratio, vortexed for 40 s, and centrifuged for 15 min. The supernatant was used to measure nitrogenase activity using the Soil Nitrogenase (NITS) ELISA Kit (ADANTI, Wuhan, China).
Statistical analysis
Principal coordinates analysis (PCoA) was used to visualize the difference in the composition of rhizobacterial functional genes involved in nitrogen metabolism identified by KEGG and the taxonomic assignment of all these genes involved in N-cycling of wild and domesticated rice based on the Bray-Curtis dissimilarity matrix with the vegan package in R (v4.1.2)65. Then the statistical significances of the clustering patterns in ordination plots were evaluated by using Permutational ANOVA (PERMANOVA)66. Shannon index of taxonomic assignment of all these genes involved in N-cycling of wild and domesticated rice was calculated by vegan package64. Significant differences in the estimators between wild and domesticated rice were evaluated through two-sided Welch’s t-test (p < 0.05) in SPSS v20.0 (IBM, Chicago, IL, USA). The correlation analysis between gene abundances and root exudates of wild and domesticated rice were performed using the pairwise Spearman’s rank correlation based on the results of qPCR in R (v4.1.2).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The shotgun metagenomics sequencing data have been deposited in the National Center for Biotechnology Information (NCBI) under accession number PRJNA632564. Source data are provided with this paper.
References
Andrews, M., Raven, J. & Lea, P. Do plants need nitrate? The mechanisms by which nitrogen form affects plants. Ann. Appl. Biol. 163, 174–199 (2013).
Marschner, H. Marschner’s Mineral Nutrition of Higher Plants (Academic Press, 2011).
Lu, J. et al. Nitrogen fertilizer management effects on soil nitrate leaching, grain yield and economic benefit of summer maize in northwest China. Agric. Water Manage. 247, 106739 (2021).
Liu, Y. et al. Genomic basis of geographical adaptation to soil nitrogen in rice. Nature 590, 600–605 (2021).
Beltran-Garcia, M. J. et al. Nitrogen fertilization and stress factors drive shifts in microbial diversity in soils and plants. Symbiosis 84, 379–390 (2021).
Qin, H. et al. A few key nirK-and nosZ-denitrifier taxa play a dominant role in moisture-enhanced N2O emissions in acidic paddy soil. Geoderma 385, 114917 (2021).
Henneron, L. et al. Rhizosphere control of soil nitrogen cycling: a key component of plant economic strategies. New Phytol. 228, 1269–1282 (2020).
Kennedy, I. R., Choudhury, A. & Kecskés, M. L. Non-symbiotic bacterial diazotrophs in crop-farming systems: can their potential for plant growth promotion be better exploited? Soil Biol. Biochem. 36, 1229–1244 (2004).
Zhu, S., Vivanco, J. M. & Manter, D. K. Nitrogen fertilizer rate affects root exudation, the rhizosphere microbiome and nitrogen-use-efficiency of maize. Appl. Soil Ecol. 107, 324–333 (2016).
Remigi, P., Zhu, J., Young, J. P. W. & Masson-Boivin, C. Symbiosis within symbiosis: evolving nitrogen-fixing legume symbionts. Trends Microbiol. 24, 63–75 (2016).
Steenhoudt, O. & Vanderleyden, J. Azospirillum, a free-living nitrogen-fixing bacterium closely associated with grasses: genetic, biochemical and ecological aspects. FEMS Microbiol. Rev. 24, 487–506 (2000).
Orr, C. H. et al. Diversity and activity of free-living nitrogen-fixing bacteria and total bacteria in organic and conventionally managed soils. Appl. Environ. Microb. 77, 911–919 (2011).
Pérez-Jaramillo, J. E., Mendes, R. & Raaijmakers, J. M. Impact of plant domestication on rhizosphere microbiome assembly and functions. Plant Mol. Biol. 90, 635–644 (2016).
Porter, S. S. & Sachs, J. L. Agriculture and the disruption of plant-microbial symbiosis. Trends Ecol. Evol. 35, 426–439 (2020).
Weese, D. J., Heath, K. D., Dentinger, B. T. & Lau, J. A. Long–term nitrogen addition causes the evolution of less-cooperative mutualists. Evolution 69, 631–642 (2015).
Ramirez, K. S., Craine, J. M. & Fierer, N. Consistent effects of nitrogen amendments on soil microbial communities and processes across biomes. Global Change Biol. 18, 1918–1927 (2012).
Chang, J. et al. The structure of rhizosphere fungal communities of wild and domesticated rice: changes in diversity and co-occurrence patterns. Front. Microbiol. 12, 610823 (2021).
Tian, L. et al. Root-associated bacterial diversities of Oryza rufipogon and Oryza sativa and their influencing environmental factors. Arch. Microbiol. 199, 1–9 (2017).
Tian, L. et al. Co-evolutionary associations between root-associated microbiomes and root transcriptomes in wild and cultivated rice varieties. Plant Physiol. Biochem. 128, 134–141 (2018).
Zhang, J. et al. NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat. Biotechnol. 37, 676–684 (2019).
Zhong, Y. et al. Soil microbial mechanisms promoting ultrahigh rice yield. Soil Biol. Biochem. 143, 107741 (2020).
Tian, L. et al. Comparison of methane metabolism in the rhizomicrobiomes of wild and related cultivated rice accessions reveals a strong impact of crop domestication. Sci. Total Environ. 803, 150131 (2022).
Sun, Y. et al. Rice domestication influences the composition and function of the rhizosphere bacterial chemotaxis systems. Plant Soil 466, 81–99 (2021).
Chang, J. et al. Self-crossing leads to weak co-variation of the bacterial and fungal communities in the rice rhizosphere. Microorganisms 9, 175 (2021).
Leite, M. F. A. & Kuramae, E. E. You must choose, but choose wisely: Model-based approaches for microbial community analysis. Soil Biol. Biochem. 151, 108042 (2020).
Liu, H. & Yan, J. Rice domestication: an imperfect African solution. Nat. Plants 3, 1–2 (2017).
Shenton, M., Iwamoto, C., Kurata, N. & Ikeo, K. Effect of wild and cultivated rice genotypes on rhizosphere bacterial community composition. Rice 9, 42 (2016).
Shi, S. et al. Comparative analysis of the rhizomicrobiome of the wild versus cultivated crop: insights from rice and soybean. Arch. Microbiol. 201, 879–888 (2019).
Xing, G. X. et al. Denitrification in underground saturated soil in a rice paddy region. Soil Biol. Biochem. 34, 1593–1598 (2002).
Yoshida, M., Ishii, S., Otsuka, S. & Senoo, K. nirK-harboring denitrifiers are more responsive to denitrification-inducing conditions in rice paddy soil than nirS-harboring bacteria. Microbes Environ. 25, 45–48 (2009).
Ettwig, K. F. et al. Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 464, 543–548 (2010).
Lourenço, K. S. et al. Nitrosospira sp. govern nitrous oxide emissions in a tropical soil amended with residues of bioenergy crop. Front. Microbiol. 9, 674 (2018).
Chang, J. et al. Nitrogen, manganese, iron, and carbon resource acquisition are potential functions of the wild rice Oryza rufipogon core rhizomicrobiome. Microbiome 10, 1–13 (2022).
Mouser, P. J. et al. Influence of heterogeneous ammonium availability on bacterial community structure and the expression of nitrogen fixation and ammonium transporter genes during in situ bioremediation of uranium-contaminated groundwater. Environ. Sci. Technol. 43, 4386–4392 (2009).
Piromyou, P. et al. Potential of rice stubble as a reservoir of bradyrhizobial inoculum in rice-legume crop rotation. Appl. Environ. Microbiol. 83, e01488–17 (2017).
Ramdani, N., Belhadi, D., Kaci, Y. & Benallaoua, S. Symbiotic, phenotypic and genotypic characterization of Bradyrhizobium sp. nodulating Spartium junceum L. from Bejaia, northeastern Algeria. Symbiosis 81, 25–37 (2020).
Xu, C. H. et al. Azospirillum Aestuarii sp. nov., a novel nitrogen-fixting and aerobic denitrifying bacteria isolated from an estuary of a freshwater river. Curr. Microbiol. 80, 113–122 (2023).
Raaijmakers, J. M. & Kiers, E. T. Rewilding plant microbiomes. Science 378, 599–600 (2022).
Liu, Y., Evans, S. E., Friesen, M. L. & Tiemann, L. K. Root exudates shift how N mineralization and N fixation contribute to the plant-available N supply in low fertility soils. Soil Biol. Biochem. 165, 108541 (2022).
Liu, L. et al. Ammonia volatilization as the major nitrogen loss pathway in dryland agro-ecosystems. Environ. Pollut. 265, 114862 (2020).
Xia, Y., Ti, C., She, D. & Yan, X. Linking river nutrient concentrations to land use and rainfall in a paddy agriculture–urban area gradient watershed in southeast China. Sci. Total Environ. 566, 1094–1105 (2016).
Favela, A., O Bohn, M. & Kent, D. A. maize germplasm chronosequence shows crop breeding history impacts recruitment of the rhizosphere microbiome. ISME J. 15, 2454–2464 (2021).
Edwards, J. et al. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 112, E911–E920 (2015).
Tamames, J. & Puente-Sánchez, F. SqueezeMeta, a highly portable, fully automatic metagenomic analysis pipeline. Front. Microbiol. 9, 3349 (2019).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Schmieder, R. & Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864 (2011).
Li, D. et al. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 1–11 (2010).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Team, R. C. R: a language and environment for statistical computing. (2013).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146 (2014).
Wu, Y. W. et al. MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome 2, 1–18 (2014).
Kang, D. D., Froula, J., Egan, R. & Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3, e1165 (2015).
Sieber, C. M. et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 3, 836–843 (2018).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868 (2017).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Huerta-Cepas, J. et al. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293 (2016).
Thioulouse, J. et al. Multivariate Analysis of Ecological Data with Ade4 (Springer, 2018).
Lourenço, K. S. et al. Dominance of bacterial ammonium oxidizers and fungal denitrifiers in the complex nitrogen cycle pathways related to nitrous oxide emission. GCB Bioenergy 10, 645–660 (2018).
Suleiman, A. K. A. et al. From toilet to agriculture: fertilization with microalgal biomass from wastewater impacts the soil and rhizosphere active microbiomes, greenhouse gas emissions and plant growth. Resour. Conserv. Recycl. 161, 104924 (2020).
Lourenço, K. S., de Assis Costa, O. Y., Cantarella, H. & Kuramae, E. E. Ammonia-oxidizing bacterial and fungal denitrifier diversity are associated with N2O production in tropical soils. Soil Biol. Biochem. 166, 108563 (2022).
Yue, H. et al. Host genotype-specific rhizosphere fungus enhances drought resistance in wheat. Microbiome 12, 44–63 (2024).
Williams, A. et al. Comparing root exudate collection techniques: an improved hybrid method. Soil Biol. Biochem. 161, 108391 (2021).
Oksanen, J. et al. Package ‘vegan’. community ecology package. version 2, 1–295 (2013).
Schlemper, T. R., van Veen, J. A. & Kuramae, E. E. Co-variation of bacterial and fungal communities in different sorghum cultivars and growth stages is soil dependent. Microb. Ecol. 76, 205–214 (2018).
Acknowledgements
This research was supported by the National Key R&D Program of China (2024YFD1501003 to C.T., J.C., and Y.S.), the National Natural Science Foundation of China (U22A20593, 41920104008 to C.T., 32401436 to J.C., 32371734 to Y.S.), and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA28020400 to C.T.), and the Major Science and Technology R&D project of Jiangxi Province, China (20213AAF01001 to J.W.). We would like to express our sincere gratitude to Wen Zhou, who provided invaluable assistance with the African domesticated rice materials, at the Wuhan Botanical Garden, Chinese Academy of Sciences. We thank Marcio F.A. Leite from the Netherlands Institute of Ecology (NIOO-KNAW) for valuable suggestions during the analysis process. We are also thankful to Johannes A. van Veen from the Netherlands Institute of Ecology (NIOO-KNAW) for his valuable discussions and insightful suggestions, which greatly enhanced the quality of our research.
Author information
Authors and Affiliations
Contributions
J.C., J.M.R., C.T., and E.E.K. planned and designed the research; S.S., J.W., L.T., E.W., Y.S., H.C., L.J., Z.Y., C.W., J.Z., Y.P., L.Y., H.C., Y.C., D.C., Z.S., and J.R. contributed to the experiments; J.C. and O.Y.A.C. analyzed the data; J.C., J.M.R., C.T., and E.E.K. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Mika Tarkka and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chang, J., Costa, O.Y.A., Sun, Y. et al. Domesticated rice alters the rhizosphere microbiome, reducing nitrogen fixation and increasing nitrous oxide emissions. Nat Commun 16, 2038 (2025). https://doi.org/10.1038/s41467-025-57213-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57213-x
