
Abstract
Known fetal haemoglobin (HbF)-modulating loci explain 10–24% variation of HbF level in Africans with Sickle Cell Disease (SCD), compared to 50% among Europeans. Here, we report fourteen candidate loci from a genome-wide association study (GWAS) of HbF level in patients with SCD from Cameroon, Tanzania, and the United States of America. We present results of cell-based experiments for FLT1 candidate, demonstrating expression in early haematopoiesis and a possible involvement in hypoxia associated HbF induction. Our study employed genotyping arrays that capture a broad range of African and non-African genetic variation and replicated known loci (BCL11A and HBS1L-MYB). We estimated the heritability of HbF level in SCD at 94%, higher than estimated in unselected Europeans, and suggesting a robust capture of HbF-associated loci by these arrays. Our approach, which involved genotype imputation against six reference haplotype panels and association analysis with each of the panels, proved superior over selecting a best-performing panel, evidenced by a substantial proportion of panel-specific (up to 18%) and a low proportion of shared (28%) imputed variants across the panels.
Similar content being viewed by others


Introduction
Sickle-cell disease (SCD) is caused by a biallelic single nucleotide substitution in the beta-globin gene resulting in an amino acid substitution, HBB (Glu7Val, formerly known as Glu6Val)1. As a result of the partial protection conferred by the heterozygosity of the sickle variant against severe malaria, SCD has become prevalent in areas of the world where malaria is endemic2. It is estimated that ~300,000 babies are born worldwide each year with SCD, with nearly 75% of these births being in sub-Saharan Africa3. In Africa, at least 30–50% of children with untreated SCD die before the age of 5 years4,5. Therefore, accelerating the path for novel therapies for SCD through genomics research on fetal haemoglobin (HbF; α2γ2) is critical.
During fetal life, HbF is the most predominant haemoglobin subtype. After birth, the level of HbF decreases progressively to ~1% in ~8–12 weeks, and it is replaced by adult haemoglobin (HbA; α2β2)6. The regulation of Hb production is controlled by repressive transcription factors (TFs) including BCL11A and ZBTB7A that bind to the HBG1 and HBG2 gene promoters7. Genetic variations in HbF‐modulating genes allow some individuals the capacity to continue producing HbF in adult life. SCD patients that produce higher levels of HbF (>8%) after birth have longer life expectancy8, because the presence of HbF in sickle RBCs delays deoxy‐HbS polymerisation and thus reduces clinical complications. A successful gene‐editing strategy for treating individuals with the most common and severe subtype of SCD is the induction of HbF expression through downregulation of the TF BCL11A9,10.
Variants in the currently known HbF‐modulating genes/loci, i.e., BCL11A, HBS1L-MYB, and XmnI-HBG2, explain only 10–20% of the variation of HbF levels in African individuals with SCD11,12, compared with nearly 50% of the variation in HbF levels among Europeans13. Expanding genomic research in populations of African ancestry could uncover the missing heritability of HbF-promoting loci14.
In this study, we used the Human Heredity and Health (H3Africa) consortium SNP genotyping array developed from whole genome enriched for common variants in sub-Saharan Africans with 3280 individuals from 17 African countries to identify genomic variations associated with HbF levels in a discovery cohort of 827 patients living with Sickle Cell Anaemia from Cameroon. This was followed by a meta-analysis with previously published data from 884 SCD samples from Tanzania15 and summary statistics from four African American SCD cohorts (2040 samples)16, reaching a combined sample size of 3751. We used a multi-panel approach for genotype imputation and association testing, employing six reference haplotype panels. Our strategy led to improved detection of associations, identifying fourteen novel candidate loci for investigating therapeutic interventions for SCD. We present additional experiments for the FLT1 locus, one of the 14 significant signals.
Results
The dataset
Our study included 3751 individuals with sickle-cell anaemia (SCA) of African ancestry from Cameroon, Tanzania, and the United States of America (USA) (see Methods for a description of the cohorts). The basic demographic and clinical characteristics of Cameroonian and Tanzanian participants, as well as haematological features, alpha-thalassaemia genotypes, and the HBB gene cluster haplotypes of Cameroonian participants are presented in the Supplementary Tables 1 & 2 and Supplementary Fig. 1. We restricted our analyses to participants aged five years and older and we normalised HbF level in both cohorts by cubic root transformation to match the age distributions and transformations in the USA-based studies (see Code Availability section for more information). In-depth quality control for the Cameroonian and Tanzanian genotype datasets and the results are provided in Supplementary Figs. 2–4. A total of 827 samples were analysed from Cameroon after quality control, 50.8% were females, and median age was 15 years (ranging from 5 to 66 years). From Tanzania, 884 samples were analysed, 52.8% were females, and median age was 13 (ranging from 5 to 44 years). Only samples for which there was concordance between reported and genotyped sex were considered. The USA-based cohorts involved summary statistics from previously published studies16 (see Methods).
Comparative performance across different imputation panels
We separately imputed genotypes in each cohort using six reference panels (Supplementary Table 3) and filtered out variants with imputation accuracy (R2) <0.3 before assessing imputation performance. Genotypes from the TOPMed panel were imputed in GRCh38 coordinates, while the others remained in GRCh37 coordinates to ensure comparability and prevent loss of variants due to reference build migration. A positive correlation between panel size and the number of imputed variants was observed (Fig. 1a), except when comparing the H3A panel with the smaller CAAPA and KGP panels, suggesting low accuracy for many H3A variants (R2 < 0.3). Both SNPs and INDELs were imputed from the CUSTOM, KGP, and TOPMed panels while only SNPs were imputed from the H3A, AGR (Sanger), and CAAPA panels. H3A and CAAPA panels supported only autosomes (Supplementary Table 3). Panel size correlated positively with imputation accuracy, with TOPMed performing best (Fig. 1b, c). The CUSTOM and KGP panels outperformed H3A and AGR, possibly due to the genetic and phenotypic proximity of CUSTOM to the study population, and trio information utilisation in KGP17. Exclusion of related individuals likely impacted the performance of AGR18.

a Number of variants imputed per panel with imputation accuracy (R2) ≥ 0.3 for the Cameroonian (blue) and Tanzanian (green) cohorts. b Dot and line plots of R2 per chromosome for the Cameroonian cohort. Only autosomes (chromosome 1–22) were used since some of the panels (H3A and CAAPA) did not support the sex chromosomes. c Dashed-line plots of R2 within different minor allele frequency (MAF) bins per autosome in the Cameroonian cohort. A bin size of 0.01 was used to bin the variants into fifty (50) bins from 0 to 0.5. d Five overlapping Venn diagrams showing proportions of shared and panel-specific variants in the Cameroonian cohort. Only panels in GRCh37 coordinates are shown. b–d are statistics for the Cameroonian cohort only. Supplementary Fig. 5 presents similar statistics for the Tanzanian cohort.
Comparing the mean R2 per chromosome, our custom panel outperformed H3A, CAAPA, and AGR in the Cameroonian cohort (Fig. 1b). AGR ranked second in the Tanzanian cohort due to its enrichment with haplotypes from eastern and southern African populations similar to those from Tanzania (Supplementary Fig. 5a). Zooming into each chromosome by minor allele frequency (MAF) bins, H3A generally performed better overall, especially at lower MAFs (<0.1; Fig. 1c; Supplementary Fig. 5b). Imputation accuracy was slightly higher for the Cameroonian cohort, likely due to differences in genotyping chips used, with H3A having tags that more accurately match African haplotypes. Panel-specific variants were observed across GRCh37 panels, with <30% overlap (Fig. 1d; Supplementary Fig. 5c). This suggests varied accuracies in imputing the same variant across panels due to differences in haplotype structures from different tagging schemes17, highlighting the panels’ complementary use. Moreover, it implies different association patterns when utilising different panels, therefore the absence of a signal in one panel should not dismiss its significance if observed in another.
Association testing supports complementary use of multiple imputation panels
Following the above observations, we utilised datasets from all six panels for downstream association analyses in three stages (Fig. 2a; see Methods). The total number of variants analysed per panel for each dataset is presented in Supplementary Table 4. Genome-wide significance was defined by P < 5e-08. Variants for which the Benjamini-Hochberg false discovery rate (FDR) was less than 0.05 and P > 5e-08 were considered of marginal significance. Suggestive associations were considered at FDR [0.05–0.10) or P < 5e-06. Evidence of association was also inferred when a locus had marginally significant signals in association testing and meta-analysis. Figure 2b shows loci with significant associations, and varying performance among the various imputation panels. Different association patterns were observed, with AGR and CUSTOM panels exhibiting best overall performance. KGP and CAAPA panels showed suboptimal performance cumulatively, while H3A showed the least significance without meta-analysis. More loci were identified in the Tanzanian cohort, indicating improved capture of genetic variations in African populations by recent imputation panels. The well-characterised BCL11A and HBS1L-MYB loci were replicated. A third significant locus, FLT1, was identified, along with thirteen additional marginally significant loci (Table 1). Figure 2c displays Q-Q plots and genomic control inflation factors indicating no residual population structure, while Fig. 2d shows Manhattan plots highlighting significant signals. Supplementary Data 1 and Supplementary Figs. 6 and 7 include the full list of significant and suggestive signals.

a Association testing was performed in three stages: in stage 1 (red circle), we performed single variant association tests for Cameroonian discovery (n = 827) and Tanzanian re-analysis cohort (n = 884) imputed datasets filtered to include only biallelic SNVs (SNPs and INDELs) with R2 ≥ 0.6. A generalised linear mixed model was run using SAIGE93, with multiple testing correction by the Benjamini–Hochberg false discovery rate (FDR) method; stage 2 (green circle) involved a meta-analysis of two Africa-based cohorts (n = 1711), while in stage 3 (purple circle), we included summary statistics from HbF GWAS involving sickle cell anaemia cohorts of African ancestry based in the United States of America (n = 2040) to perform an overall or a global meta-analysis (n = 3751). Meta-analysis was performed by the inverse variance method of METAL95. Summary statistics from all three stages were then used to perform functional GWAS and fine-mapping. b Overview of significant loci detected per analysis unit. Green circles indicate presence while red diamond indicates absence of significant signal in the corresponding locus. Blue coloured loci represent the major known HbF-influencing loci, while the rest in black are novel loci. c The quantile-quantile (Q-Q) plots of the expected against the observed p values, as well as the genomic control inflation factors (λ) demonstrate the robustness of our association results and show that our test statistics were not inflated. d Manhattan plots showing the significant signals in the Cameroon, Cameroon-Tanzania meta-analysis, and global meta-analysis.
Replication of the major HbF-influencing loci: BCL11A and HBS1L-MYB
Across all analyses, BCL11A and HBS1L-MYB were the most significant loci, and they remain the largest contributors to HbF variability in these cohorts. rs1427407 and rs9399137 are the most widely and frequently reported sentinel variants in BCL11A and HBS1L-MYB respectively. Multi-ancestry fine-mapping has suggested rs1427407 as the likely functionally relevant variant within the BCL11A locus19. Yet, the sentinel variants in these loci usually differ amongst cohorts, including in our study (see Supplementary Data 1): in the Cameroonian cohort, rs7606173 emerged as the BCL11A sentinel variant (P = 8.25e-20). This is likely because it was almost twice as frequent as rs1427407 (MAFrs7606173 = 0.45; MAFrs1427407 = 0.26), since the two variants had similar effects (βrs7606173 = −0.22; βrs1427407 = −0.23). rs7606173 therefore contributed the largest proportion (9.1%) in HbF variability, and together, the two variants accounted for 8.85% in HbF variance; in the Tanzanian cohort, rs1896294 and rs1427407 were the BCL11A sentinel variants with similar significance (P = 4.26e-36). rs1896294 was more frequent (MAFrs1896294 = 0.28; MAFrs1427407 = 0.22) while rs1427407 had a larger effect (βrs1896294 = 0.28; βrs1427407 = 0.30). Individually and together, the variants contributed ~15.2% in HbF variability in the cohort similar to previous reports13, and substantially higher than the variance explained by BCL11A sentinel variants in Cameroonians. In a meta-analysis of the two cohorts, rs1427407 was the sentinel BCL11A variant with the largest effect on HbF level (P = 2.48e-50, β = 0.27), and it contributed ~11.6% in HbF variability in the joint cohort. In the global meta-analysis, rs766432 emerged as the BCL11A sentinel variant (P = 2.42e-100), contributing 10.7% in HbF variability in the combined cohorts. Yet, rs1427407 still had the largest effect (βrs766432 = −0.24; βrs1427407 = 0.26), supporting the attribution of functional relevance to it within the BCL11A locus19, although it was slightly less frequent (MAFrs766432 = 0.28 versus MAFrs1427407 = 0.24). rs1427407 therefore contributed a smaller proportion (7.6%) in HbF variability.
In the HBS1L-MYB intergenic region, rs9399137 and rs35786788 were the sentinel variants in the Cameroonian cohort (cm) with identical significance (P = 1.76e-08, β = 0.38). In the Tanzanian cohort (tz), as well as in the Cameroon-Tanzania meta-analysis, the rs55634702 INDEL was the sentinel variant (Ptz = 1.13e-09, βtz = 0.35; Pcm_tz_metal = 2.32e-16, βcm_tz_metal = 0.36). Generally, these HBS1L-MYB sentinel variants explained ~4% of HbF variance, consistent with previous findings. rs9399137 was the sentinel variant in the global meta-analysis and contributed 3.2% in HbF variability. The relatively low proportion of HbF variance explained by the HBS1L-MYB variants notwithstanding their relatively large effects is due to the low frequencies of these sites in African ancestries (MAF ≤ 0.03) compared with other ancestries where their MAF is greater than 0.10. Replication of signals within other genomic regions that have been associated with HbF level, including HBG219,20,21,22,23, is presented in the Supplementary Information and Supplementary Data 2.
Identification of novel candidate HbF-associated loci
In the Cameroonian cohort, a third signal that reached genome-wide significance was mapped to a novel locus upstream of the FMS related receptor tyrosine kinase 1 gene (FLT1, also known as vascular endothelial growth factor receptor 1–VEGFR1) on chromosome 13 (13q12.3). The sentinel variant rs115695442 (P = 4.18e-08, β = 0.21; Supplementary Fig. 8a) was relatively common in the cohort (MAF = 0.10), and it contributed 3.5% in HbF variance. FLT1 significant variants occurred at higher frequencies than HBS1L-MYB associations (FLT1; MAF = 0.076–0.105 Vs HBS1L-MYB; MAF < 0.04) and had similar effects as BCL11A associations (β = 0.20–0.23). No significant FLT1 associations were observed in the Tanzanian cohort (Supplementary Data 2 & 3). However, multiple variants were observed at p value < 5e-03 and with appreciable effects (β = 0.14–0.16) within 100 kb of the FLT1 signal (Supplementary Data 2). Similarly, variants within the genomic region of FLT1 were observed in the Cooperative Study of Sickle Cell Disease (CSSCD) cohort at P = 6.9e-03 (rs61763174, intronic variant, β = −0.24, MAF = 0.06)24, and in the Silent Cerebral Infarct Transfusion Trial (SITT) cohort at P = 2e-04 (rs9578046, 94 kb upstream, β = 0.14, MAF = 0.12)25 (Supplementary Data 2).
Meta-analysis of the Cameroonian and Tanzanian cohorts revealed five novel candidate loci that included FLT1 and OPCML, of which FLT1 had the strongest associations (Table 1). rs74617914 emerged as the FLT1 meta-analysis sentinel variant (P = 4.38e-08, β = 0.20) although it was not significant in the independent association tests of the two cohorts. Global meta-analysis identified two additional loci, namely GFRA1 and LINC01898. Seven novel candidate loci were observed in the Tanzanian re-analysis (see Supplementary Fig. 7), of which OPCML was previously reported in the cohort albeit it was not significant15, hence classified here as novel candidate. Each of the loci contributed ~3% in HbF variance. ZNF804A variants demonstrated the largest effect across our entire analysis (P = 8.97e-08, β = 0.46). The relatively small proportion in HbF variance (3.1%) that they contributed could be attributed to their low MAF < 0.02. The variants are indeed rare in Africans generally, whilst absent in other ancestries based on the dbSNP and EMSEMBL resources. In the Tanzanian cohort, the derived alleles were only observed in heterozygotes and were associated with higher HbF levels (Supplementary Fig. 8).
Of the likely new loci, FLT1 was particularly interesting because it was identified in a population that has not been previously studied genome-wide, was replicated in meta-analysis with consistent signals across all the imputation panels and was the third strongest signal after BCL11A and HBS1L-MYB. We therefore focused on the FLT1 signal for further functional characterisation.
Functional mapping of the FLT1 signal
Fine mapping of the functionally relevant FLT1 variants in the Cameroonian cohort revealed a single 95% credible set that included nine variants of which rs115695442 had the highest posterior inclusion (causal) probability (PIP = 0.36) (Fig. 3a). In the meta-analysis, a single 95% credible set that included only rs74617914 with causal probability of 0.99 was detected. Six of the variants with identical significance and in perfect linkage disequilibrium (LD = 1) had identical causal probabilities to one another (PIP = 0.089) which summed to >50% (Fig. 3b). These variants additionally had larger effect sizes (β = 0.23) than the FLT1 sentinel variants rs115695442 and rs74617914, suggesting that the most probable causal variant(s) might be among the six. All FLT1 fine-mapped variants were in a 40 kb interval (chr13:29069272– 29110372; GRCh37) spanning the FLT1 promoter and a candidate enhancer region of ~30 kb upstream of the FLT1 transcription start site (TSS) (Fig. 3c). Most of the variants occurred within TF binding sites (TFBSs), including five of the six aforementioned variants in perfect LD (Fig. 3b, c). Neither of the variants nor their tags have been reported in Genotype Tissue Expression (GTEx) as either expression or splicing quantitative trait loci (eQTL/sQTL), in line with the observation that they are virtually absent in non-African ancestries that make up the bulk of data of the GTEx project (Fig. 3d). The binding motifs of six TFs were disrupted by the minor allele variants. Three of the TFs implicated (STAT5A, GFI1, and MXI1) play crucial roles in haematopoiesis/erythropoiesis26,27,28,29 and their binding motifs were disrupted by three of the six perfect-LD variants (rs11840478, rs75294023, and rs11843606 respectively) (Fig. 3e–g), thus supporting the attribution of functional relevance to these.

a Regional association plot of FLT1 signal in the Cameroon-Tanzania meta-analysis. Linkage disequilibrium (LD or r2) of the lead variant (rs74617914) and the rest of the variants is represented as a coloured key. The middle window presents the relative position of the fine-mapped variants; pink represents the meta-analysis fine-mapped variant, blue represents the fine-mapped variants in the Cameroonian cohort (Supplementary Fig. 8a presents the regional association for the Cameroonian cohort FLT1 signal). b Statistics of the fine-mapped variants (BETA, effect size; SE, standard error of effect size estimate; P, unadjusted p value) from the association test described in Fig. 1 and Table 1. PIP (posterior inclusion probability) of each fine-mapped variant being causal. Functional annotations from the ENSEMBL resource and the JASPAR algorithm (TFBS transcription factor binding site) are shown. The distance of each variant from the FLT1 transcription start site is indicated as dTSS. NA, information not available. Transcription factors in bold have known roles in erythropoiesis (see Supplementary Information). c Genomic map of the FLT1 regulatory region showing chromatin state predictions in different cell lines, the promoter (HS1) and candidate enhancer (HS2), the relative position of the fine-mapped variants (light blue vertical lines), and relevant TFBSs (visualised in the UCSC Genome Browser using the hg19 reference sequence). Hypoxia response elements (HREs) bound by the hypoxia-inducible factors (HIFs; HIF1A/2 A), are highlighted in yellow. d Minor allele frequency (MAF) distribution of the fine-mapped variants and other variants looked up in the Tanzanian, CSSCD, and SITT cohorts. The MAFs are displayed for Cameroonian and Tanzanian sickle cell anaemia populations, as well as unascertained global ancestries from the 1000 Genomes dataset. One of the six variants in perfect LD is used to represent the rest. e–g Sequence logo of the TFB motifs disrupted (retrieved from https://jaspar.genereg.net/ and reverse complemented to the forward strand to reflect the base change presented throughout our text). rs75294023 disrupts the absolutely required GFI1 binding core AATC (reverse complement: GATT)115. The box plots show additive effects of rs11840478, rs75294023, and rs11843606 on fetal haemoglobin (HbF) level and mean corpuscular volume (MCV). Centre line in box plots denotes the median, the lower and upper ends of the boxes denote the lower and upper quartile. Whiskers extend from the ends of the boxes to the minimum (lower whisker) and maximum (upper whisker) values. Violin plots describe the density of the distribution. h ATAC-seq data from 3 datasets of erythropoietic cell lines provided visual overlap, showing the FLT1 signal to be enhancer-associated. BCL11A signal is used here as control for both Cameroon and Tanzania GWAS. Source data are provided as Source Data Fig. 3.
Chromatin accessibility data revealed two DNase I hypersensitive sites (HS) within the 40 kb region (Fig. 3c; see Methods): HS1 corresponded to the FLT1 promoter which showed strong activity in human umbilical vein endothelial cells (HUVECs) and weak activity in human embryonic stem cells (hESCs); HS2 had methylation and acetylation patterns marking an active promoter or strong enhancer in the human erythroleukemic K562 cells, HUVECs, and lymphoid-specific GM12878 cells. Strong TF ChIP-seq peaks for GATA2, MYC, IKZF1, and CTCF were present at HS2 in K562 cells, suggesting a restricted activity of this region in the erythro-lymphopoietic system, potentially involving loop formation30. There were strong HDAC2 peaks at HS1 and HS2 in hESCs, as well as polycomb-repressive complex (PRC) marks in most cell lines including K562, consistent with predicted polycomb repression of the promoter (HS1) in K562 cells. The HS1 chromatin marks are indicative of bivalent promoters associated with developmentally regulated genes31. The general chromatin accessibility pattern in the 40 kb region suggests a tight cell-type and stage-specific regulation of FLT1 along the developmental axis32. There are not much experimental data on HS2 as an enhancer evidenced by its absence in the ENCODE project, the VISTA and FANTOM5 enhancer browsers, hence it has no predicted interaction by GeneHancer. However, the ENSEMBL resource suggest some experimental evidence, while ENCODE classifies the region as “distal enhancer-like”, hence our classification as “candidate enhancer”. These support a hypothesis that HS2 is only transiently active, leading to a brief upregulation of FLT1. The fine-mapped variants occurred between HS1 and HS2, flanking the promoter and the candidate enhancer (Fig. 3c). ATAC-seq peaks from three datasets of erythropoietic lineages indicate the variants are enhancer-associated (Fig. 3h). The variants exhibited additive effects on HbF level with the genotypes carrying the minor alleles associated with higher HbF levels (Fig. 3e–g; Supplementary Fig. 9).
Association of FLT1 fine-mapped variants with other blood traits
We further tested association of the fine-mapped FLT1 variants with other blood traits listed in Supplementary Table 1 in the Cameroonian cohort. We observed significant associations of rs74617914 and the six perfect-LD variants with mean corpuscular volume (MCV; P < 0.05) (Supplementary Data 4). The derived alleles of the variants were associated with a slight increase in red blood cell (RBCs) size (Fig. 3e–g). In addition, rs74617914 alone was significantly associated with RBC count (P < 0.05), although no marked difference was observed for the distribution of RBC count amongst the different genotypes.
Haplotype structure provides a reason for absence of FLT1 association in the Tanzanian cohort
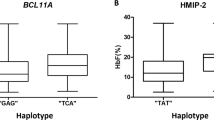
The difference in FLT1 sentinel variants observed in the Cameroonian association and Cameroon-Tanzania meta-analysis (Fig. 4a), as well as significant heterogeneity in effect sizes observed at all the fine-mapped variants (heterogeneity p value < 0.01) with the exception of rs74617914 (heterogeneity p value > 0.29) (Fig. 4b), suggest different haplotype structures within the FLT1 40 kb region between Cameroonians and Tanzanians. We thus analysed haplotype blocks (haploblocks) within 25 kb upstream and downstream of the region (see Methods). At similar SNP densities, we observed higher and longer-range LD with lower haplotype diversity in Cameroonians than in Tanzanians. The fine-mapped variants were distributed across three haploblocks (blocks 5, 6, and 7) in Cameroonians (Fig. 4c); rs181503970 and rs76296165 flanking the FLT1 promoter occupied block 5, rs115695442 had no haploblock participation, occurring between blocks 6 and 7, and the rest (perfect-LD variants) occupied block 7, flanking the candidate enhancer. Remarkably, all three haploblocks were in strong LD, evidenced by high D’ values (D’ > 0.96) indicative of little historical recombination, making rs115695442 an excellent tag for this locus in Cameroonians. The haplotype structure was different in Tanzanians: (i) the fine-mapped variants were distributed within five haploblocks (blocks 8-12), and (ii) low D’ values were observed among the haploblocks (D’ < 0.90) indicating high historical recombination, which suggests the variants are evolving independently in this cohort. Indeed, LD between rs115695442 and all the haploblocks was less than 0.2 in Tanzanians, even though the variant occurred between haploblocks 11 and 12 which are in strong LD (D’ = 0.95). There was high intra-block (short range) LD between rs181503970 and rs76296165 (LD = 1) and between rs11840478 and rs114243330 (LD = 0.99), all pairs of variants that are remarkably close to each other. Similar substantial heterogeneity in effect sizes was observed within the major HbF-influencing loci, and some of the novel loci (Fig. 4d), thus potentially explaining the difference in sentinel variants observed in different populations.

a Forest plot of FLT1 lead associations in the Cameroonian cohort (rs115695442; N = 827) and Cameroon-Tanzania meta-analysis (rs74617914; N = 1711). Black points represent effect size estimates. Error bars represent confidence intervals of effect size estimates. The values are shown in red below each point. The unadjusted p value of each estimate is shown in blue. Rs115695442 was only significant in Cameroonians and suggestive in the meta-analysis. rs74617914 was only significant in the meta-analysis and insignificant in the respective cohorts. b Heterogeneity plot of FLT1 fine-mapped variants shows significant heterogeneity (greater than 80%) at all the variants except rs74617914, indicating complex haplotype structure within the region, that was confirmed by haplotype analysis: three haploblocks (black triangles) in Cameroonians (c) harbouring the fine-mapped variants were in strong linkage disequilibrium (LD) evidenced by high D’ values (>0.6) between rs115695442 and the blocks 5 and 7 variants (red and orange bars); Low LD among haploblocks harbouring the fine-mapped variants in Tanzanians (d) explained low LD (<0.2) between rs115695442 and all other fine-mapped variants. The probability of historical recombination between blocks is shown as D’ (the higher the value, the lower the probability). A recombination map of the region, generated using the hapmap recombination map116 in GRCh37 coordinate, is shown as red line-plot above the haplotype map. A recombination hotspot is evidenced by the tallest peak. e Heterogeneity plot of sentinel variants of all the significant loci. Variants are coloured by locus. f Estimate of heritability of fetal haemoglobin level in a merged Cameroon and Tanzania genotype dataset (n = 1682) after additional quality control as described in the Supplementary Information. Additive and dominance genetic variance components were jointly estimated with eight categories of covariates. For each category, heritability was estimated thirty times to demonstrate non-randomness in the estimation given the modest sample size. The insert boxplots show the distribution of the estimates. The centre line denotes the median, the lower and upper ends of the boxes denote the lower and upper quartiles. The whiskers are shown extending from the ends of the boxes to the minimum (lower whisker) and maximum (upper whisker) values. Violin plots describe the density of the distribution. Source data is provided as Source Data Fig. 4.
Haploblock analysis for 25 non-SCD Cameroonian individuals (Supplementary Fig. 10) and HbS-negative genomes from populations in the 1000 Genomes Project (Supplementary Fig. 11) revealed lower LD with smaller haploblocks in African populations. Against an MAF of 5%, the fine-mapped variants were present in African ancestries only and had no consistent pattern in their haploblock participation; many had no haploblock participation. In addition, there was high historical recombination among the haploblocks as expected under neutral evolution. These suggest that an evolutionary force, such as natural selection, might be preserving haplotypes in the FLT1 40 kb region in Cameroonian SCA populations. Indeed, haplotype association revealed a strongly suggestive haplotype carrying the derived alleles of rs7989474-A and rs1967786-T (Padjusted = 0.053) in Cameroonians that also flank the FLT1 candidate enhancer and occurred within GATA1 peaks.
Gene-based, gene set, and heritability analyses further support the association results
Gene-based analysis revealed multiple genomic regions with significant (P < 2e-06) or strongly suggestive (P < 2e-04) evidence of association across all the datasets (p value threshold 2.5e-06; Supplementary Fig. 12). BCL11A and HBS1L-MYB were the most significant loci. The HBB gene cluster signal was evident, particularly in the global meta-analysis. It spanned the HBG1, HBG2, HBE1 genes, and the locus control region and involved >3000 variants, indicating extensive evolutionary activity related to the sickle cell allele. An additional significant locus, MMP26, that mapped immediately downstream of the HBB-3’HS1 was observed in Tanzanians (P = 8.32e-07). In Cameroonians, FLT1 demonstrated signs of replication (P = 0.005). Below the suggestive threshold (P < 2e-04), there were few overlaps in the results of the different association and meta-analysis datasets. At a less stringent threshold (P < 0.002), many common signals were detected across the datasets, leading to a highly similar pattern of gene set enrichment. The most enriched pathway, haematopoietic stem cell differentiation (Supplementary Fig. 13a), overwhelmingly featured known erythropoietic factors including GATA1, KLF1, cMYB, RUNX1, STAT5A, HIF1A, and HDAC (Padjusted <0.05). The hypoxia pathway was also significantly enriched in all the datasets. Myeloid cell differentiation and gas (oxygen) transport were the most significantly enriched biologically processes (Padjusted < 2e-3), while the haemoglobin complex was the most significantly enriched cellular component (Padjusted < 9.60e-6; Supplementary Fig. 13b, c). Unsurprisingly, RBC traits, including MCV and RBC count, were among the most significantly enriched phenotypes (Padjusted < 6e-08) (Supplementary Fig. 13d). Erectile dysfunction (priapism), an important sub-phenotype of SCD, was the most significantly enriched trait in the Tanzanian cohort and the CAM-TZN meta-analysis. In line with these observations, the blood and spleen were the sites with the most significant differentially upregulated genes, mostly erythropoiesis-related genes including GATA1, KLF1, and HBG1 (Supplementary Fig. 14).
Our results hint at a robust capture of haematopoietic factors, potentially involving HbF-modifying loci with many small-effect-size variants that did not reach genome-wide significance. In line with this observation, we estimated HbF SNP heritability in a combined cohort of Cameroonian and Tanzanian SCD populations at 0.94 (SE 0.01; 95% confidence interval [CI] 0.92–0.96; Fig. 4f), slightly higher than the 0.89 for unselected Europeans33, and substantially higher than 0.30–0.50 previously estimated for SCA populations of African ancestry living in Europe and North America34. Only a moderate reduction in the estimate was observed by increasing the number of principal components (PCs) from 20 to 100, thus capping the estimate at ~96%. Notably, our approach jointly estimated the additive and dominance genetic variance components, as opposed to only the additive component (narrow-sense heritability) estimated in previous studies. Attempts at estimating narrow-sense heritability produced highly variable outcomes (mean = 0.70; 95% CI = 0.37–1.04; SE = 0.24) (Supplementary Fig. 15), reflecting the low power associated with our small sample dataset. Besides, association analysis of the merged genotype data used to estimate heritability mirrored the results of the meta-analysis of the two populations, indicating that our heritability estimates were unlikely to be due to spurious associations (Supplementary Fig. 16).
Assessment of editing and gene expression in erythroid and erythroleukemia cells
To assess the impact of genomic FLT1 variation on HbF expression under hypoxia and normoxia, we edited the genome of the immortalised human erythroid progenitor cell line HUDEP-2 that, in the default state, expresses primarily the adult haemoglobin35. We used Cas9 nuclease to disrupt FLT1 and, as a positive control for HbF induction, the +58 kb erythroid BCL11A enhancer36. We used base editors to introduce rs76296165 and rs74993145 which we identified as FLT1-proximal SNPs associated with increased HbF and isolated clonal cultures with homozygous edits (Supplementary Fig. 17a). BCL11A disruption, but not FLT1 disruption, led to an increase in F-cells (Supplementary Fig. 17b). While BCL11A disruption led to the expected increase in HbF transcripts, neither FLT1 disruption nor the installed SNPs impacted HbF transcript levels in normoxia or hypoxia (Supplementary Fig. 17c–f). Digital polymerase chain reaction (PCR) did not detect a change in HbF induction following FLT1 knockout (Supplementary Fig. 17g). Notably, the three SNPs predicted to be functionally relevant could not be efficiently base edited, and although rs74993145 is in perfect LD with the three functionally relevant SNPs, it does not appear to have any functional consequence.
We then assessed the pattern of FLT1 expression, as well as other hypoxia- and erythropoiesis-related genes (Supplementary Fig. 18) in HUDEP-2 and the human erythroleukemia cell line, K562, which displays embryonic erythropoiesis following erythroid differentiation37, primarily expressing embryonic haemoglobin (HBE1) by default, and fetal γ-globin (HBG1/2) upon induction, but not adult β-globin (HBB)38,39, and has previously been reported to express FLT140. FLT1 transcript levels in HUDEP-2 cells were not detected under hypoxia or normoxia using qPCR and digital PCR35 (Supplementary Fig. 17c–f), thus preventing any robust quantitative comparisons with K562 cells, and possibly explaining the reason for the lack of impact of FLT1 disruption in HUDEP-2 cells. However, we estimated at least 30-fold greater FLT1 expression in K562 cells relative to HUDEP-2 cells. FLT1 and KDR transcript levels in K562 cells were >8-fold induced under hypoxic conditions (Fig. 5a). Expression of FLT1 reached a maximum between 6 and 24 hours under hypoxia and remained stable over 10 days. Known HIF1A target genes ALAS2 and CA9 were also induced under hypoxic conditions as expected (Fig. 5b). HBG1/2 were induced under long-term hypoxic conditions as previously reported41, as was the TF KLF1 that is involved in HbF regulation42 (Fig. 5c).

a K562 cells were exposed to 1% oxygen over 10 days in culture. RT-qPCR was used to evaluate gene expression at predetermined time points. FLT1 (VEGFR1) and KDR (VEGFR2) gene expression are depicted, normalized to expression during culture in normoxic conditions. b RT-qPCR measurements of ALAS2 and CA9 expression, known markers induced by hypoxia. c RT-qPCR measurements of adult beta globin HBB, fetal haemoglobin genes HBG1/2, and a transcription factor involved in fetal haemoglobin regulation, KLF. RT-qPCR samples n = 3. Mean values of technical triplicates are presented. Error bars denote the standard error as calculated by Applied Biosystems qPCR software RQmax and RQmin. RQ relative quantitation of transcript levels.
Assessment of editing and gene expression in human CD34 + HSPCs
To further assess the involvement of FLT1 in HbF expression, we used G-CSF mobilised peripheral blood purified human CD34+ HSPCs obtained from four healthy donors (see Methods). We used Cas9 nuclease to disrupt FLT1 and the +58 BCL11A enhancer as a positive control and induced in vitro erythroid differentiation under normoxic and hypoxic (2% O2) conditions in the presence or absence of 50 ng/mL VEGF or 100 nM SU5416 VEGF inhibitor (Supplementary Fig. 19a). Erythroid maturation progression was measured at days 8, 13, and 18 (D8, D13, and D18), with marked differences between D13 and D18, and between normoxic and hypoxic conditions, as well a high inter-individual variability (the donors notably spanned three ancestral backgrounds: European, American, and African) (Supplementary Fig. 19b–d). Hypoxia delayed maturation which is consistent with the role of HIFs in HSPCs proliferation43,44. Editing efficiency throughout differentiation, as well as cell viability and recovery, are presented in Supplementary Fig. 20a–d. There was no significant difference in the frequency of cells expressing HbF (F-cells) and bulk HbF between D13 and D18 (Supplementary Fig. 21a, b). Additionally, we observed that one of the donors, an African American male, was heterozygous for two of the FLT1 proximal SNPs (rs115695442 and rs76296165) albeit not functionally relevant (Supplementary Fig. 21c). Stranded mRNA sequencing revealed expression of FLT1 at D0 and the expected downregulation at D13, with a modest restoration under hypoxia in untreated cells (Supplementary Fig. 22a). Hypoxia was confirmed by upregulation of ALAS1 (Supplementary Fig. 22b). Hypoxic regulation of erythroid-specific ALAS2 was inconsistent among the donors; upregulation occurred in only one donor, with the average mRNA level across the donors showing an insignificant difference between normoxic and hypoxic conditions (Supplementary Fig. 23). Notably, FLT1 knockout (FLT1-KO) resulted in only about 30% reduction of FLT1 mRNA levels measured at D0, which was two days after electroporation. There was significant downregulation of FLT1 mRNA at D13 (day 15 after electroporation), and no significant expression of KDR throughout differentiation. Fetal-type γ-globin (HBG1 and HBG2) and adult-type β-globin (HBB) mRNAs were observed at D13 in all media conditions (Supplementary Fig. 22c, d), as well as the α-globin genes, HBA1 and HBA2. BCL11A enhancer disruption had the strongest impact on HBG1/2 and HBB mRNA levels as expected. (Supplementary Fig. 22c, d). HBG1 was more strongly induced, and its mRNA levels were highly variable within and between groups in contrast to HBG2. This suggests the differentiation conditions favoured HBG1 promoter accessibility. There was no significant difference in F cell and HbF levels between the untreated and FLT1-KO groups which could be explained by the small reduction of FLT1 mRNA levels after Cas9 targeting. VEGF inhibition markedly reduced HBG1 mRNA levels and F cells, as well as HbF to a lesser extent. This impact of VEGF signalling on γ-globin was consistent across hypoxia and normoxia, with and without BCL11A knockout. Thus, our results indicate that basal VEGF signalling plays a role in γ-globin regulation even in cultured hematopoietic cells, which could serve as a model system to further probe the mechanism of this interaction.
Discussion
Summary of key points
Our study investigated genetic variations influencing HbF level in an African sickle cell disease cohort using a multi-panel imputation and association strategy against two distinct genotyping arrays tailored to capture a broad spectrum of Africa-specific and non-African genetic variations. This is expected to contribute to the much-needed new data from populations with African ancestry45,46,47. Specific advantages of our study include: (a) a discovery cohort made of hydroxyurea-naive patients living with SCD from Cameroon which provides a proxy for the natural disease history; (b) the identification of fourteen novel candidate loci enhanced by the reanalysis of previously reported data from Tanzania, and global meta-analysis including data from individuals of African ancestry living with SCD in the United States of America; (c) in particular, the description of variants in FLT1 which are largely specific to African populations with apparent functional impact, as well as our elucidation of the complex haplotype architecture of FLT1 which provides support for substantial genomic variability that can be extended to other loci to explain the difference in sentinel variants observed in different populations; and (d) a detailed in silico and in vitro cell-based functional exploration of the potential mechanism for FLT1 involvement in erythropoiesis and HbF induction.
Heterogeneity in imputation panels
To the best of our knowledge, only one study has attempted the use of multiple imputation panels for association analysis48. Researchers typically select a best-performing panel for association testing. Although the dissection of the comparative performance of imputation panels for sub-Saharan African populations has been performed in previous studies48,49, our study presents comparisons for a larger variety of panels with variants that are relevant to populations of African ancestry. Our observations were largely similar to previous reports; differential imputation performance, substantial panel-specific variants, and relatively low proportion of shared variants (less than 30%). The TOPMed panel showed the best performance as expected. However, the freeze 8 release used here has a known limitation for African ancestry populations as revealed in our malaria GWAS study48; it fails to impute critical Africa-specific functional variants (including the sickle cell mutation, rs334) that are imputed with high accuracies (>90%) using other panels. Importantly, our findings underscore the complementarity of the panels, particular in highly diverse populations (reflected in panel-specific signals), and support the utilisation of all the panels for association analysis as an optimal approach. The recently developed meta-imputation procedure for combining multi-panel imputed datasets50 (which was unavailable at the time of our analyses) would be a more computationally tractable way of handling such datasets given the enormous challenges accompanying separate analyses. Alternatively, future association studies involving highly diverse populations should consider whole-genome sequencing (WGS), as much as possible, to alleviate the large inconsistencies and complexities that come with utilising multiple imputation panels.
Heterogeneity in association signals
Disparities in haplotype structure were the major reason for differences in imputation performance and association signals, but also differences in sentinel variants of significant loci amongst the cohorts in our study, as demonstrated in the FLT1 −40 kb region. While genetic admixture can account for haplotype differences, it could also mean that genetic loci influencing HbF level, and other modifiers of sickle cell disease, have been through different evolutionary trajectories, especially in sub-Saharan African populations. These populations have been exposed to vastly different ecologies which have shaped their genetic material differently over the roughly 300,000 years of modern human existence on the continent45. The continued revelation of extensive uncaptured genomic variations within African populations51,52, some of which are population-specific, such as the FLT1 variants reported here, reflect the enormous selective pressures that the populations have had to contend. The lack of these, sometimes functionally relevant, genetic variants in notable and European-ancestry-enriched databases such as the GTEx study reflects a current limitation for global genetic medicine. For instance, we recently reported malaria protective associations in the enhancer region of CHST15 which tag strong eQTLs in tissues relevant to the disease biology but are absent in the GTEx portal48. In cases where none of the tag variants is present in such databases, such as in the current study, a critical piece of functional information would be lost. The importance of increasing the representation of understudied populations in global omics databases could, therefore, not be overstated.
Heritability supported by association results, pathway enrichment, and potentially, selection pressure (at least in Cameroonians) evidenced by high LD and haplotype conservation in the FLT1 40 kb regulatory region
HbF heritability has previously been estimated in a twin population unselected for any disease or trait in the United Kingdom at 89%33, in sickle cell anaemia patients of African ancestry based in the USA at ~50%34, and ~32% in SCD patients of African ancestry older than 15 years of age and living in France53. Even lower estimates have recently been suggested for SCD patients of African ancestry19. In the European unselected population, half of the total HbF heritability is explained by just the three major loci i.e., BCL11A, HBS1L-MYB, and HBG213. Our estimate of 94% HbF heritability is unsurprisingly higher than previous estimates for several reasons: (i) our approach jointly estimated additive and dominance genetic variance components, whereas previous approaches estimated only the additive variance component (narrow-sense heritability), suggesting that a substantial portion of HbF heritability in selected patients from Africa could be explained by a dominance genetic variance component54, (ii) our cohorts are fundamentally different from the other cohorts in that our samples represent individuals with the most severe form of sickle cell disease who have escaped childhood mortality largely without healthcare strategies such as newborn screening and comprehensive care with penicillin prophylaxis and hydroxyurea treatment. Considering the historically high excess of under-five mortality (50–90%) of sickle cell anaemia in Africa5, therefore this group of patients likely represent a naturally selected population enriched with genetic variants that favour “long survival” such as has been previously shown in patients from Cameroon55. It is therefore reasonable to imagine that HbF-induction is among the most enriched pathways given that it is the most potent modifier of SCD severity known to date. However, larger sample sizes of patients living in Africa with SCD, and standardised measurements of HbF, would be needed to confirm the true heritability of HbF in SCD in Africa.
Functional relevance of FLT1 associations
FLT1 (VEGFR1) and the kinase insert domain receptor (KDR or VEGFR2) transduce mitogenic signals from VEGF necessary for regulating angiogenesis and vascular permeability43. There is growing evidence for the involvement of FLT1 in haematopoiesis such as in the proliferation of HSPCs43,44 and the differentiation of megakaryocytes (Mk; which share a common progenitor with erythroid cells)56 in a hypoxia-induced manner. A study that investigated the mechanism of HbF induction under hypoxia-induced stress erythropoiesis implicated HIF1A as a direct mediator that targets chromatin accessibility to favour transcription of the γ-globin genes41. FLT1 is a known target of hypoxia inducible factors (HIFs: HIF1A/2 A), demonstrated by hypoxia response elements (HREs) in the FLT1 regulatory region57 (Fig. 3c). Interestingly, a hypoxia-driven autocrine loop between VEGF, FLT1, and phosphorylated extracellular-signal regulated kinase 1/2 (ERK1/2; two mitogen-activated protein kinases–MAPKs) in a neuroblastoma model has been shown to activate HIF1A, favouring its nuclear localisation, accumulation, and transcriptional activity58. This suggests FLT1 might be implicated in the HIF1A-HbF induction nexus in erythroid cells (see Supplementary Fig. 18b). Our results indicate that the association of FLT1 with HbF level in the Cameroonian cohort might be driven by at least one of three variants that interfere with the binding motifs of three TFs active in the haematopoietic system (see Supplementary Information). GFI1 in particularly is a major repressor that regulates chromatin state and is necessary for human endothelial-to-haematopoietic transition (EHT)59.
Although HUDEP-2 cells are a common model for adult haemoglobin regulation and its perturbation, we were unable to detect substantial FLT1 expression in these cells in normoxia or hypoxia, as opposed to readily detected expression in K562 cells in which it is strongly induced by hypoxia. This supports our hypothesis of a tightly controlled cell-type and stage-specific expression of FLT1 and suggests that it might play a role during primitive erythropoiesis. Notably, GFI1 represses gene transcription in myeloid progenitors through recruitment of other major co-repressors including the Corepressor of RE1 silencing transcription factor (CoREST) and the nucleosome remodelling and deacetylating (NuRD: a key repressor of the γ-globin gene) complex60. Our experiments in CD34+ cells did not generate definitive proof of FLT1 involvement in HbF production. However, they confirmed the expression of FLT1 in primary human hematopoietic stem cells consistent with our model of an involvement in early erythropoiesis, and an induction under hypoxia during erythroid maturation, while another VEGFR gene KDR was not induced by hypoxia. FLT1 expression in bone marrow-derived mesenchymal cells dependent on HIF1A has been previously demonstrated61. Failure to detect significant FLT1 mRNA levels during erythroid maturation might thus be associated to its predicted tight regulation and transient expression similar to the HIFs. However, the apparent negative regulation of HbF and F cells by VEGF inhibition implicates FLT1 in the haemoglobin synthesis pathway through a VEGF-FLT1-HIF1A axis (Supplementary Information Fig. 18). The basal HbF levels observed across our differentiation conditions and in the general human population could therefore be associated to this axis, which seems plausible considering that the bone marrow microenvironment is relative hypoxic62.
Given the data presented, we propose a model for the regulation of FLT1 in erythroid cells in Supplementary Fig. 18. The combination of hypoxia and the disruption of the GFI1 binding motif therefore provides a reasonable model for FLT1 reactivation, and possible recapitulation of embryonic/fetal erythropoiesis, which is further supported by the association of FLT1 variants with slightly larger erythrocytes. Previous studies involving SCD patients in the USA63 and beta-thalassaemia patients from Greece64 showed FLT1 to be associated with improved hydroxyurea-induced HbF level65. Also, data from Flt1 and Flk1 (Kdr) knock-out mice show disruption of erythropoiesis66,67. Therefore, additional experiments involving primary haematopoietic progenitors from the bone marrow of SCD patients and/or healthy donors, as well as detailed phenotyping of surviving Flt1−/−, Flt1 +/−, Flk1 +/− and other knock-in mice model, will be needed to fully characterise the impact of FLT1 and the functionally relevant variants described herein in erythropoiesis, F-cells, and HbF production.
Methodological considerations
Several points lend support to the robustness of our strategy: (i) the enrichment of variants in our study in the haematopoietic pathway, particular in genes involved in haemoglobin synthesis; (ii) the suggestive variants observed in loci that were recently detected through specialised techniques, e.g., in ZNF410 and JAZF1 through CRISPR screening and RNA interference respectively21,22; (iii) the recent detection of a putative novel erythropoietin QTL on chromosome 15 with evidence of association at P = 1.05e-0768; (iv) the OPCML gene which was detected in the Tanzanian cohort in 2014 at P < 1e-0615 was replicated in this reanalysis at P < 3e-07 in both Tanzania and CAM-TZN meta-analysis, suggesting some functional relevance in HbF production although the evidence of association falls short of the conventional significance threshold; (v) the SLC4A4 gene observed in the Tanzanian cohort at P = 5.75e-07 is a bicarbonate cotransporter that is involved in regulating intracellular pH, a major factor that determines HbS polymerisation and red blood cell sickling and may therefore be involved in HbF regulation. The absence of large-effect novel associations in the meta-analysis suggests that we are approaching saturation in the discovery of major HbF level-associated loci with variants of MAF > 1%. It could also mean the saturation of loci that contribute to HbF variability additively in these cohorts.
There are limitations to our study potentially impacting the strength of evidence of the putative associations, e.g., small sample size of the study cohorts. Replication is further restricted by high genetic diversity in populations of African descent, and this was demonstrated with lipid traits in African cohorts69. Hence, additional functional characterisation is needed to support our findings. SNP ascertainment bias imposed by the availability of only about 1.1 million variants in the USA-based cohorts (see Methods and Supplementary Table 4) likely restricted the observation of additional associations. Increasing sample size and population coverage could enhance the signals and uncover additional loci as supported by the recent report of the novel BACH2 locus19 for which we observed suggestive variants. Nevertheless, the high genetic heterogeneity observed with cohort-specific sentinel variants highlights the importance of investigating larger African populations from multiple countries.
Methods
Ethical approvals
The research was performed in accordance with the Declaration of Helsinki. Approval was obtained from the University of Cape Town, Faculty of Health Sciences Human Research Ethics Committee, Cape Town, South Africa (HREC/REF: R015/2018), and National Ethical Committee of the Ministry of Public Health of Cameroon (No 193/CNE/SE/15). All patients older than 18 years signed consent forms, while informed consent was given by the parents or guardians of participants younger than 18 years old. Written and signed informed consent forms were obtained from adult participants and parents/guardians of minor patients. An assent was also obtained from the participants of more than 7 years old. The present study involved a secondary analysis of existing data and was reviewed and approved by the University of Cape Town, Faculty of Health Sciences Human Research Ethics Committee, Cape Town, South Africa (HREC REF: 606/2021).
Patient participants
The data were collected from nine hospitals in five cities in Cameroon, including Yaoundé, Douala, Bafoussam, Bertoua, and Maroua, from May 2016 to July 2018. Socio-demographic and clinical events were collected by means of a structured questionnaire administered to parents/guardians and adult SCD patients. Patients’ medical records were reviewed, to delineate their clinical features over the past 3 years. Only patients older than 5 years of age (to avoid age-related changes in the complete blood count and HbF level), who had not received a blood transfusion or hospitalisation in the past 6 weeks were included. None was currently treated with hydroxycarbamide or opioids. The sampling strategy was not restricted to hospital-based patients to avoid the bias that might result from including only the sickest patients. To accomplish this goal, two SCA patients’ associations in Cameroon were engaged in collaboration, and additional patients were recruited during their monthly meetings. No incentive was provided for participation in the study.
Measurements of haematological indices
Haemoglobin electrophoresis and complete routine blood count of the SCA patients were conducted upon arrival at the hospital. High performance liquid chromatography (HPLC) was used for the measurement of HbF levels at the haematological laboratory of the Centre Pasteur in Yaoundé, as previously described70,71. No patients had HbA measurements with HPLC.
Molecular methods
Genotyping of the sickle cell anaemia mutation, HBB cluster haplotypes, and 3.7 kb HBA1/HBA2 deletion
DNA was extracted from peripheral blood following the manufacturer’s instructions (Puregene Blood Kit; Qiagen, Hilden, Germany). Molecular analysis to determine the presence of the sickle mutation was carried out on 200 ng DNA by PCR to amplify a 770 bp segment of the HBB, followed by DdeI restriction analysis of the PCR product72. The present analysis was restricted to sickle cell anaemia (homozygous HbS) due to the well-known differences in laboratory parameters73,74, and to allow single sickle genotype (HbSS) for genetic associations. Using published primers and methods, five restriction fragment length polymorphism sites in the HBB cluster were amplified to analyse the XmnI (5’Gγ), HindIII (Gγ), HindIII (Aγ), HincII (3ψβ’) and HinfI (5’β) for the HBB haplotype background75. The 3.7 kb HBA1/HBA2 deletion was successfully screened, using the expand-long template PCR (Roche Diagnostics, Basel, Switzerland), as previously published76.
Cameroonian cohort
Two batches of samples of sickle cell anaemia patients from Cameroon (batch 1: n = 1199, batch 2: n = 403) were genotyped on the 2.3 M H3Africa SNP array at Illumina® FastTrackTM Microarray services (Illumina, San Diego, USA) between 2018 and 2019. Genotype calling was performed for each batch using the Illumina gencall algorithm from the Illumina Array Analysis Platform Genotyping Command Line Interface (IAAP-CLI) version 1.1 (IAAP Genotyping command line interface: https://emea.support.illumina.com/downloads/iaap-genotyping-cli.html). Briefly, gencall was used to process intensity data in IDAT format to GTC formats, utilising manifest and cluster files specific to the H3Africa chip retrieved from https://chipinfo.h3abionet.org/downloads (Accessed: December 5, 2021). Thereafter, the per-sample GTC files were converted to a single VCF file for the separate batches of samples using the gtc2vcf plugin of bcftools version 1.15.1, while aligning to the human reference sequence in build 37 (hg19) coordinates.
Tanzanian cohort
The dataset consisted of genotypes for 1213 Tanzanian SCA patients generated using the Illumina Human Omnichip 2.3 platform (Illumina Inc., San Diego, CA, USA), and available at the European Genome Phenome Archive (EGA) under the accession number EGAD0001000065015. The genotype data mapped to the human reference in build 37 coordinates, and in PLINK binary format, as well as clinical data were obtained from the Tanzanian investigators. The data contributed to the first GWAS of HbF in Africa published in 201415.
USA-based cohorts
We obtained meta-analysed summary statistics of HbF GWAS involving seven cohorts of sickle cell anaemia (HbSS) patients based in the United States of America (USA) from the study by Harold T. Bae et al., totalling 2040 samples16. The cohorts included: Cooperative Study of Sickle Cell Disease (CSSCD: n = 841), Multicenter Study of Hydroxyurea (MSH: n = 178), Pulmonary Hypertension and the Hypoxic Response in Sickle Cell Disease (PUSH) study (n = 73), Comprehensive Sickle Cell Centers Collaborative Data (C-data) project (n = 127), Treatment of Pulmonary Hypertension and Sickle Cell Disease with Sildenafil Treatment (Walk-PHaSST) trial (n = 181), Duke University Outcome Modifying Genes study (n = 152), and Silent Infarct Transfusion (SIT) trial (SITT: n = 488). The meta-analysis was performed using the inverse variance method of the METAL software. Apart from SNP coordinates (chromosome and position), the summary statistics included all information necessary to perform meta-analysis, including dbSNP and Illumina SNP identifiers (1,198,700 SNPs in total). We also obtained complete GWAS summary statistics from the SITT cohort in which HbF was cubic root normalised. The summary statistics included SNP coordinates in the human reference build 36 (hg18), as well as dbSNP and Illumina SNP identifiers (1,138,137 SNPs in total).
Quality control (QC)
Genotype quality control was performed for batch 1 and 2 of our stage 1 GWAS data set separately. First, each batch of samples with gencall call rate ≥90% (batch1 n = 1137, batch2 n = 367) was converted to plink binary file sets using PLINK277 while excluding duplicate SNPs. Samples that failed missingness criteria (outlying heterozygosity and missing genotype rate >10%; see Supplementary Fig. 2) were excluded. Duplicate and related individuals (up to 2nd degree relationships) were identified using the Kinship-based INference for Genome-wide association studies (KING v2.2.4) software78, and one individual from each pair of duplicate or related individuals was excluded. Apparently mislabeled samples were also identified using the KING software, and all samples that failed QC were excluded. SNP QC was performed by excluding SNPs with missing genotype rate >5%, MAF < 1%, and SNPs that failed the Hardy-Weinger equilibrium (HWE) test at a p value threshold of 1e-6, as well as palindromic [A/T] and [C/G] SNPs were also excluded. The two batches of genotype data were then merged using PLINK v1.9 –bmerge77. Additional quality control on the merged data set was performed to exclude samples that failed missingness criteria, duplicate and/or related samples, SNPs with MAF < 1%, SNPs with missing genotype rate >5%, and SNPs that failed the HWE test at P = 1e-6. To control for potential batch effects, SNPs with significant (p value < 0.001) allele frequency difference (differential missingness) between the batch 1 and batch 2 data sets were excluded. In addition, PC analysis (PCA) was performed on a set of high-quality independent SNPs using smartpca of the EIGENSOFT package (version 7.2.1)79 to investigate batch effects and to remove population and ancestry outliers. The independent set of SNPs was obtained by linkage disequilibrium (LD) pruning using the following parameters: linkage disequilibrium <0.2, window size of 50 bp, and step size of 10 bp. Population outliers were pruned with smartpca using the following pruning parameters: 10 PCs along which to remove outliers with 5 outlier removal iterations and specifying 6.0 standard deviations which an individual must exceed along one of the top 10 PCs to be excluded as an outlier. Only the merged genotype data was considered for subsequent analyses. We applied the same quality control procedure to the Tanzanian cohort genotype data.
Haplotype estimation (phasing) and genotype imputation
Haplotypes were estimated for the stage 1 and 2 genotype data sets separately using the 1000 Genomes reference panel80,81 for all autosomes and the X chromosome. First, the genotype data were aligned to the 1000 Genomes haplotype reference panel (phase 3, version 5) to ensure allele overlap with the reference panel using the conform-gt programme from the BEAGLE utils (https://faculty.washington.edu/browning/conform-gt.html). SNPs that were absent in African populations in the reference panel, as well as SNPs with inconsistent strand and allele mismatch as compared to the reference panel were excluded. We then used the EAGLE v2.4.2 software82 to phase the data sets with the combined hapmap recombination map used to provide genetic distance, and set 20,000 conditioning haplotypes (-Kpbwt, default 10,000) to improve phasing accuracy. Genotypes were imputed from six different panels: a custom panel created from whole genome sequence data of 50 individuals of Cameroonian origin (see Creation of custom imputation panel), the H3Africa panel consisting of ~3280 individuals from 17 African countries, the TOPMed panel consisting of ~180,000 individuals pooled from the NHLBI’s studies of which 29% are of African ancestry74, the 1000 Genomes reference panel (KGP, phase 3 version 5) consisting of 661 individuals from West and East Africa81, the Consortium on Asthma among African-ancestry populations in America (CAAPA) panel consisting of 883 individuals of African ancestry75, and the African Genome Resource (AGR) consisting of 4,956 individuals, 62% (~3061) of whom are of African ancestry mostly from eastern and southern Africa (~2501, 82% of all the African samples)18 (Supplementary Table 3). The TOPMed panel was accessed via the TOPMed imputation web service, the KGP and CAAPA panels were accessed via the Michigan imputation web service83, the AGR was accessed via the Sanger imputation web service84, while access to the H3Africa panel via the H3Africa imputation web service was granted upon request. In our in-house procedure, we used BEAGLE v585 for the imputation of each chromosome separately, leaving all default parameters and using the single chromosome hapmap recombination map. For the TOPMed and Michigan imputation web services86, we selected the MINIMAC4 software87 for imputation and retrieved only variants with imputation accuracy, R2 ≥ 0.3. For the Sanger imputation web service, we selected the Positional Burrows-Wheeler Transform (PBWT) package for imputation88. Imputed data from each panel and for each analysis stage were processed separately. Quality control of the imputed data included the removal of variants with imputation accuracy (R2) < 0.60 and genotype call rate <95%. Only biallelic SNPs and INDELs were retained for subsequent analysis.
Creation of custom imputation panel
We used whole-genome sequencing (WGS) data from 24 Cameroonian SCD patients, as well as WGS data of 26 individuals of Cameroonian origin who contributed to the H3Africa Trypanogen project for the custom panel creation. First, the quality of the FASTQ reads were checked using FastQC, and then mapped to the human reference genome in build 37 coordinate (http://hgdownload.cse.ucsc.edu/goldenpath/hg19/bigZips/latest/hg19.fa.gz) using BWA-MEM89. The resulting SAM files were converted to BAM and sorted by coordinate (chromosome and base pair position) using samtools90. Duplicate reads were marked, and base quality scores recalibrated using GATK version 4.2.5.091. For variant calling, we used a recently optimised pipeline involving DeepVariant version 1.3.0 for single-sample variant calling and GLNexus version 1.4.3 for joint variant calling (DV-GLN-OPT)92. In their optimisation and benchmarking study, Yun et al. developed a variant filtration scheme based on four tunable parameters, which gave the DV-GLN-OPT an edge over the popular GATK-VQSR Best Practices pipeline for all data types analysed (including whole-exome). These parameters have now been coded into the GLNexus package as the default settings, and they were therefore utilised in our study. In addition, Yun et al. showed that the reference imputation panel created using variant calls from the optimized pipeline outperformed that created using call sets from GATK best practices pipeline. To create our custom panel, we applied additional filters on our DV-GLN-OPT joint call set: we excluded variants with read depth (DP) < 10, genotype quality (GQ) < 20, as well as monoallelic and singleton sites. We then phased each chromosome separately without reference using EAGLE v2.4.2 as previously described.
Association analysis
Association testing was performed using the Scalable and Accurate Implementation of generalised mixed model (SAIGE) software, version 0.3893. First, we extracted independent SNPs for each non-imputed dataset of the Cameroonian and Tanzanian cohort through linkage disequilibrium pruning in PLINK2 according to the following parameters: window size of 500,000 base pairs (bp), step-size of 50 markers, and pairwise LD (r2) < 0.2. Next, 20 PCs were computed for each of the datasets using the high-quality independent SNPs. Thereafter, a null generalised linear mixed model (GLMM) was fitted for each of the full non-imputed datasets including only SNPs with minor allele count (MAC) ≥ 20 as recommended93. A full genetic relationship matrix (GRM) calculated on the fly from the plink binary file sets was used to fit the null GLMM on the cubic root transformed HbF quantitative trait while including the top 10 PCs, as well as age and sex as covariates. Using the fitted null GLMMs for each cohort, single variant association tests were next performed for each imputed dataset filtered to include only biallelic SNPs and INDELs with MAF ≥ 0.01, imputation accuracy ≥0.6, genotype call rate ≥95%, as well as SNPs that passed the HWE test at P = 1e-06. Association analysis in the Cameroonian cohort involved 827 samples, 52% of whom were females, and the average age of the participants was 17.61. In the Tanzanian cohort, 884 samples were analysed, 53% of whom were females, and the average age of the participants was 13.19. The Benjamin-Hockberg FDR method implemented in the p.adjust function of the R statistical package94 was then used to correct for multiple testing.
Meta-analysis
We performed fixed effects meta-analysis using the METAL software95 on the basis that all the populations were of the same ethnic background. We used a two-step approach which constituted Stage two and Stage three of our GWAS analysis; (i) GWAS Stage two: involved a meta-analysis of Cameroonian and Tanzanian cohorts using summary statistics from the Stage one GWAS, (ii) GWAS Stage three: involved meta-analysis of Cameroonian, Tanzanian, and the USA-based cohorts. For accurately matching of markers across the studies, we standardised the variant IDs (“MarkerName”) using “chromosome:position:SNV” (e.g., 2:60718043:SNV). Considering that summary statistics for the USA-based cohorts lacked coordinates (that is chromosome and position), and that the SITT cohort with coordinate information was mapped to build 36, we first updated the coordinates in the SITT cohort to build 37 (as well as to build 38 for meta-analysis with the TOPMed panel) using the UCSC liftOver tool (https://genome.ucsc.edu/cgi-bin/hgLiftOver). Briefly, we created a bed file from the build 36 coordinates and used it as input for liftOver. A total of 1,137,886 and 1,137,522 SNPs were successfully updated to build 37 and build 38, respectively. We then used the updated SITT cohort coordinates to update the USA-based cohorts summary statistics using the variant ID column as the primary key. Therefore, only the 1,137,886 and 1,137,522 SNPs that were successfully updated in the SITT cohort were retained in the updated USA-based cohorts for meta-analysis. We noted similar effect size estimates and standard errors across the studies indicating similar phenotyping and normalisation, and we therefore used the inverse variance method of METAL for meta-analysis. We used genomic control to account for population stratification, as well as allele frequency tracking to help identify allele flips. We also enabled heterogeneity analysis in which METAL computes the I2 statistic (and corresponding p values) which measures the amount of effect size variation across the studies that is due to heterogeneity rather than chance.
Statistical and functional fine mapping
We used the ‘sum of single effects’ (SuSiE) model implemented in the SusieR package96 to fine-map functionally relevant variants in each region that showed significant association(s) in our analyses. SusieR employs an iterative Bayesian stepwise selection (IBSS) procedure that affords it the advantage of capturing uncertainty in which variable to select in its variable selection scheme and is thus well suited for highly correlated data. That is, the estimate of uncertainty provides a framework for determining which variant is most probably ‘causal’ in a scenario of completely (highly) correlated variants. For significant associations in the stage 1 GWAS results, we computed 95% credible sets using in-sample correlation (LD) matrices for Cameroon (discovery) and Tanzania (replication) respectively. We used out-sample correlation matrices—computed from African samples in the 1000 Genomes reference panel—to compute 95% credible sets for significant associations from the meta-analysis results. All correlation matrices (specifically r as recommended) were calculated using Plink1.9. Regional association plots were then generated for each signal, highlighting the fine-mapped variants using LocusZoom97. For loci that were significant in the independent cohort association tests and meta-analysis, fine mapping was performed for both results, and the credible sets were merged for functional mapping. Functional fine-mapping involved: (i) searching in the GTEx (https://www.gtexportal.org/home/) portal whether the fine-mapped variants were expression quantitative or splicing quantitative trait loci (eQTLs and sQTLs respectively), (ii) searching in the ENSEMBL database for functional classifications, (iii) mapping their locations relative to the nearest gene, taking into account any evidence of recombination hotspots within the genomic area as represented in the regional association plots, (iv) investigating their occurrence in functionally relevant regions using the University of California Santa Cruz (UCSC) genome browser tracts (https://genome.ucsc.edu/index.html), including chromatin state segmentation by the ChromHMM algorithm, TFBS by the JASPAR algorithm, gene-enhancer interaction by the GeneHancer algorithm, TF chromatin immunoprecipitation sequencing (ChIP-seq) peaks from the ENCODE project, etc, and (v) investigation of enhancer classifications in the ENCODE and VISTA Enhancer (https://enhancer.lbl.gov/) databases. Sequence logos of binding motifs for TFs whose motifs were affected by the fine-mapped variants were obtained from the JASPAR website (https://jaspar.genereg.net/).
Association of FLT1 fine-mapped variants with other blood traits in Cameroonians
Association test of the FLT1 fine-mapped variants was performed for each of the imputed datasets separately using PLINK v1.90b6.26 64-bit. We specified 1,000,000 maximum permutations to account for population structure and adjust for multiple testing.
Gene set analysis and functional mapping
The FUMA v1.5.498 online platform available at https://fuma.ctglab.nl/ was used for functional annotation of the GWAS results in a two-step approach: (i) all summary statistics in GRCh37 coordinate were uploaded to the SNP2GENE algorithm using the following parameters: p value threshold for lead SNP = 5e-7 and minimum LD for defining lea SNP = 0.4. We selected the African populations (AFR) of the 100 Genomes projects (KGP) as reference, while all other default parameters were used (see Web resources). SNP2GENE uses the ANNOVAR tool to functionally annotate independent lead SNPs and their LD tags and map them to their corresponding genes. Prioritised genes based on positional, eQTL, and chromatin interaction mapping, are then processed with the GENE2FUNCTION algorithm to obtain insight into putative biological mechanisms and pathways. In addition to functional mappings, FUMA also performs gene-based tests and gene set analysis as implemented in the MAGMA v1.08 tool99. Specifically, for gene-based tests, MAGA uses the SNP p values from the summary statistics to compute Chi-Square statistics for a gene with LD generated from a reference panel, and the mean or the top Chi-Square statistic is taken as the gene test statistic. FUMA implements the mean model in which a gene p value is obtained by using a known approximation of the sampling distribution, and the significance threshold is calculated by 0.05/number of mapped genes (Bonferroni correction). In our study, the threshold was 2.517e-6 (P = 0.05/19867). For gene-set analysis, the gene p value for each gene from the gene-based analysis is converted to a Z value that reflects the strength of the association of each gene with the phenotype.
FLT1 haplotype structure and association analysis
Haplotype analysis was performed for the Cameroonian and Tanzanian cohorts using haploview v4.2100. First, the FLT1 40 kb region and 25 kb upstream and downstream were extracted for each of the imputed datasets using PLINK1.9 according to the following coordinates: GRCh38 28476218–28549906, GRCh37 29050355–29124043. At the same time, the HbF phenotype was transformed into case-control whereby cases were defined as HbF level ≥8.6% and controls as HbF level ≤3.1% as previously described101. A total of 408 variants and 520 samples were retained in the Cameroonian cohort, of which 413 were cases, and 107 were controls (240 males and 280 females). A total of 464 variants and 448 samples were retained in the Tanzanian cohort, of which 152 were cases, and 296 were controls (228 males and 220 females). PLINK1.9 was used to recode the data into haploview format. The default quality filters of haploview we used; no sample or SNP failed any of the filters. Haplotype blocks were computed using the Gabriel block definition102, i.e., 95% confidence bounds on D prime (D’) are generated and each comparison is called “strong LD”, “inconclusive” or “strong recombination”. A block is created if 95% of informative (i.e. non-inconclusive) comparisons are “strong LD”. Variants with MAF < 0.05 were not included in block calculations, and blocks were non-overlapping. LD plots showing haplotype blocks and the FLT1 fine-mapped variants were generated by haploview. The chromosome 13 hapmap recombination map was used as a track file to generate a recombination plot alongside the LD plot, as well as to highlight the FLT1 fine-mapped variants. Finally, single variant and haplotype association tests were performed using 100,000 permutations. Haplotype structure analysis was also performed for the non-SCD Cameroonians (n = 25) that contributed to the custom panel creation, as well as for genomes of individuals that were negative for the sickle mutation from the 1000 Genomes Project.
Assessment of transferability/replication of signals
Genomic regions associated with HbF level were identified from the respective summary statistics. Recent genetically nominated HbF-influencing loci were also identified through a literature search. From each cohort in which the significant signal was absent, we extracted the loci (genes), including 100 kb downstream and 100 kb upstream, given that some cis-regulatory elements could be tens of thousands of bases away. We next looked up variants within the extracted region, and replication or transferability of the signal was defined as the occurrence of variants at P < 0.05.
Estimation of HbF heritability
We first combined the VCF files from both cohorts that were aligned to the 1000 Genomes panel during preparation for imputation. There were 1711 samples all together. Next, we extracted only biallelic SNPs across autosomes that passed the following filtering criteria: MAF > 1%, missing genotype rate <5%, individual missingness <10%, HWE p value of 1e-06, and individuals with average heterozygosity within three standard deviations of the mean heterozygosity. Twenty-nine individuals with outlying heterozygosity were excluded, while no SNPs were excluded based on missingness criteria. To assess potential batch effects, we calculated differential missingness among the two cohorts. No SNPs were excluded due to differential missingness test at p value < 0.01. We then estimated HbF heritability in the resulting cohort of 1682 high-quality samples of Cameroonian and Tanzanian SCA patients using the Randomised Haseman–Elston regression for Multi-variance Components (RHe-mc) software103, which jointly estimates additive and dominant genetic variance components. We set the number of random vectors (K) to 10, and the number of block Jackknifes (B) for standard error estimation to 1000 as recommended. In addition, we included four sets of PCs (20PCs, 30PCs, 50PCs, and 100PCs), as well as age, sex, and country as covariates. To accurately capture the effect of age, we performed another set of analyses in which we used the square of age as an additional covariate. Box plots were generated using ggplot2 in R104.
HUDEP-2 cell culture, differentiation, and hypoxia treatment
HUDEP-2 cells were expanded in SFEM media (Stem Cell Technologies, 09650) supplemented with 50 ng/mL recombinant human SCF (Peprotech, 300-07) 3 units/mL EPO, 1 μg/mL doxycycline (Sigma Aldrich, D9891), 0.4 μg/mL dexamethasone (Sigma Aldrich, D4902), and 1% Penicillin-Streptomycin solution. HUDEP2 cells were differentiated using a 2-phase protocol. During phase 1 (days 0-3), cells were cultured at 0.5E6 cells/mL-1.5E6 cells/mL in IMDM with 2% fetal bovine serum, 2% human blood type AB plasma (Seracare, 1810-0001), 1% penicillin/streptomycin, 3 units/mL heparin, 10 μg/mL insulin (Sigma, I9278), 3 units/mL EPO, 1 mg/mL holo-transferrin (Millipore Sigma, T0665), 50 ng/mL SCF and 1 µg/mL doxycycline. After 3 days of culture in phase 1, the media was replaced with fresh media containing the same ingredients but without SCF, and cultured at 1E6 cells/mL-2E6 cells/mL for 7 additional days (10 days total). For hypoxic treatment, cells were differentiated, and sample collections and media changes were performed within a Whitley H35 HEPA Hypoxystation incubator at 2% O2. RNA was extracted using the RNeasy Plus Mini Kit (Qiagen) following the manufacturer’s protocol and eluted into 50 µL 10 mM Tris-HCl.
Isolation and culture of CD34+ human HSPCs
Circulating G-CSF-mobilised human mononuclear cells were obtained from de-identified healthy adult donors (Charles River, StemExpress). We complied with all relevant ethical regulations and all participants provided informed consent. CD34+ cells were enriched by immunomagnetic bead selection using a CliniMACS Plus or AutoMACS instrument (Miltenyi Biotec). CD34+ cells were maintained in stem cell culture medium: X-VIVO-10 (Lonza, BEBP02-055Q) medium supplemented with 100 ng/μl human SCF (Peprotech, 300-07), 100 ng/μl human TPO (Peprotech, 300-18) and 100 ng/μl human FLT-3 ligand (Peprotech, 300-19). Cells were seeded and maintained at a density of 1–2 × 106 cells per ml.
Erythroid differentiation of CD34+ cells was performed using a three-phase protocol105,106. Phase 1 (days 1–8): Iscove’s modified Dulbecco’s medium (IMDM; Thermo Fisher Scientific, 12440061) with 2% human blood type AB plasma (SeraCare, 1810-0001), 3% human AB serum (Atlanta Biologicals, S40110) 1% penicillin/streptomycin (Thermo Fisher Scientific, 15070063), 3 units/ml heparin (Sagent Pharmaceuticals, NDC 25021-401-02), 3 units/ml EPO (Amgen, EPOGEN NDC 55513-144-01), 200 μg/ml holo-transferrin (Millipore Sigma, T0665, 10 ng/ml human SCF (R&D systems, 255-SC/CF), and 1 ng/ml human interleukin IL-3 (R&D systems, 203-IL/CF). Phase 2 (days 8–13): phase 1 medium without IL-3. Phase 3 (days 13–18): phase 2 medium without SCF and with holo-transferrin concentration increased to 1 mg/ml. Cells were maintained daily at a density of 0.1 × 106 per ml (phase 1), 0.2 × 106 per ml (phase 2), and 1.0 × 106 per ml (phase 3).
Erythroblast maturation was monitored by immuno-flow cytometry for the cell surface markers CD235a (BD Pharmingen Cat. No. 559943, 1:100 dilution), CD49d (BioLegend Cat. No. 304304, 1:20 dilution), and BAND3 (gift from X. An, 1:100 dilution). For hypoxic treatment, cells were differentiated, and sample collections and media changes were performed within a Whitley H35 HEPA Hypoxystation incubator at 2% O2.
Cas9 nuclease purification
We transformed 3xNLS-SpCas9 plasmid36 plasmid28 into BL21 (DE3) competent cells (MilliporeSigma, 702353) and grew the cells in TB medium at 37 °C until the density reached OD600 = 2.4–2.8. Cells were induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside per litre for 20 h at 20 °C. Cell pellets were lysed in 25 mM Tris, pH 7.6, 500 mM NaCl, 5% glycerol by homogenisation and centrifuged at 45,000 × g for 1 h at 4 °C. Cas9 was purified with Nickel-NTA resin and treated with TEV protease (1 mg TEV per 40 mg of protein) and benzonase (100 units/ml, Novagen 70664-3) overnight at 4 °C. Subsequently, Cas9 was purified using a size-exclusion column (Amersham Bio-sciences HiLoad 26/60 Superdex 200 17-1071-01) followed by a 5-mlSP–HP ion exchange column (GE 17-1151-01) according to the manufacturer’s instructions. Proteins were dialysed in 20 mM Hepes buffer pH 7.5 containing 400 mM KCl, 10% glycerol, and 1 mM TCEP buffer. Contaminants were removed using a Toxin Sensor Chromogenic LAL Endotoxin Assay Kit (GenScript, L00350). Purified proteins were concentrated and filtered using Amicon ultrafiltration units with a 30-kDa MWCO (MilliporeSigma, UFC903008) and an Ultrafree-MC centrifugal filter (MilliporeSigma, UFC30GV0S). Protein fractions were further assessed using TGX stain-free 4–20% SDS–PAGE (Biorad, 5678093) and quantified by BCA assay.
Base editor mRNA transcription
Base editor plasmids were PCR-amplified with NEB Next polymerase (NEB) using primers that add an active T7 promoter upstream of the editor gene and a 120nt poly(A) tail to the 3’ end. PCR products were purified with the QIAquick PCR Purification Kit (QIAgen) and were used as a template for in vitro transcription. The HiScribe T7 High-Yield RNA Synthesis Kit (NEB) was used with co-transcriptional capping by CleanCap AG (Trilink Biotechnologies) and full substitution of uracil for N1-methylpseudouridine-5’-triphosphate (Trilink Biotechnologies). mRNA was purified by precipitation in 2.5 M LiCl and incubation at −20 °C for 30 minutes. Precipitated mRNA was washed twice in 70% ethanol and reconstituted in nuclease-free water. mRNA concentration was quantified using a NanoDrop One UV-Vis spectrophotometer, normalised to a concentration of 2 micrograms per microlitre, and stored at −80 °C.
Cas9 nuclease and base editor electroporation in HUDEP-2 and CD34+ cells
Electroporation was performed using the Lonza 4D Nucleofector and P3 Primary Cell 4D-Nucleofector Kit (Lonza, V4SP-3096) according to the manufacturer’s instructions. Ribonucleoprotein (RNP) complexes were prepared by mixing Cas9-3xNLS protein and gRNA at a final reaction concentration of 2.5 µM and 7.5 µM, respectively, and incubating at room temperature for 20 min. For base editor electroporation, evoAPOBEC or evoCDA mRNA and gRNA were combined at 4 µg and 2.5 µg, respectively. gRNA sequences are listed in Supplementary Data 5. 5 million HUDEP2 cells per reaction were washed with Phosphate Buffered Saline (PBS) (Corning, 21-031-CV), resuspended in Lonza P3 solution, mixed with the RNPs or the mRNA/gRNA mixture, transferred to a 20-μl Nucleocuvette Strip, and electroporated in the Lonza 4D Nucleofector using programme DS-130. Electroporated cells were recovered in supplemented SFEM media as described in HUDEP2 cell culture, differentiation, and hypoxia treatment). Genomic DNA was extracted on culture days 3 and 5 using QIAquick Gel extraction Solution (Qiagen, 28704) and then analyzed by next-generation sequencing for editing efficiency.
High-throughput sequencing and analysis of edited HUDEP-2 and CD34+ cells
Targeted amplicons were generated using gene-specific primers with partial Illumina adaptor overhangs (overhangs not shown) and sequenced as previously described107. Specific primer sequences are listed in Supplementary Data 5. Cell pellets were lysed, and the extracted genomic DNA was used as a template to amplify the target site and add Illumina adaptors. Amplicons were indexed in a second PCR reaction and pooled for sequencing. 10% PhiX Sequencing Control V3 (Illumina) was added to the pooled amplicon library prior to running the sample on an Miseq Sequencer System (Illumina) to generate paired 2 × 250 bp reads. Samples were demultiplexed using the index sequences, fastq files were generated, and NGS analysis was performed using CRIS.py108.
Illumina Stranded mRNA-seq
RNA was harvested from CD34+ cell-derived erythroid cells using an RNEasy RNA Isolation Kit (Qiagen, 74134) at Day 0 and Day 13 of differentiation. RNA was quantified using the Quant-iT RiboGreen RNA assay (ThermoFisher) and quality checked by the 2100 Bioanalyzer RNA 6000 Nano assay (Agilent) or 4200 TapeStation High Sensitivity RNA ScreenTape assay (Agilent) prior to library generation. Libraries were prepared from total RNA with the Illumina Stranded mRNA Library Prep Kit according to the manufacturer’s instructions (Illumina PN20040534). Libraries were analysed for insert size distribution using the 2100 BioAnalyzer High Sensitivity kit (Agilent), 4200 TapeStation D1000 ScreenTape assay (Agilent), or 5300 Fragment Analyzer NGS fragment kit (Agilent). Libraries were quantified using the Quant-iT PicoGreen ds DNA assay (ThermoFisher) or by low-pass sequencing with a MiSeq nano kit (Illumina). Paired-end 100 cycle sequencing was performed on a NovaSeq X+ (Illumina).
Total stranded RNA sequencing data were processed by the internal AutoMapper pipeline. Briefly the raw reads were first trimmed (Trim-Galore version 0.60), mapped to human (GRCh38) (STAR v2.7)109 and then the gene level values were quantified (RSEM v1.31)110 based on GENCODE annotation (v31). Genes with low counts (CPM < 0.1) were removed from the analysis, and only protein-coding genes were used for differential expression analysis. Normalisation factors were generated using the TMM method111, counts were then transformed using voom112 and then analysed using the lmFit and eBayes functions (R limma package version 3.42.2)113. The FDR was estimated using the Benjamini–Hochberg method.
Fraction of CD235a + HUDEP2-derived erythroid cells expressing fetal haemoglobin (F-cell) measurement by flow cytometry
1.0−3.0E5 CD34+ cell-derived erythroid cells were incubated with Hoechst 33342 for 20 min at 37 °C, fixed with 0.05% glutaraldehyde (Millipore Sigma, G5882), and permeabilized with 0.1% Triton X-100 (Millipore Sigma, 93443). Subsequently, cells were stained with CD235a and anti-human HbF, then analysed by flow cytometry. 1.0−3.0E5 HUDEP2-derived erythroid cells were incubated with Hoechst 33342 for 20 min at 37 °C, fixed with 0.05% glutaraldehyde (Millipore Sigma, G5882), and permeabilized with 0.1% Triton X-100 (Millipore Sigma, 93443). Subsequently, cells were stained with CD235a and anti-human HbF, then analysed by flow cytometry. Flow cytometry gating strategy is presented in Supplementary Information Fig. 23.
Globin HPLC measurements in edited HUDEP-2 cells
Analytical high-performance liquid chromatography (HPLC) quantification of haemoglobin tetramers and individual globin chains was performed using ion-exchange and reverse-phase columns on a Prominence HPLC System (Shimadzu Corporation). Proteins eluted from the column were identified at 220 and 415 nm with a diode array detector. The relative amounts of haemoglobins or individual globin chains were calculated from the area under the 415-nm peak and normalised based on the dimethyl sulfoxide control. The percentage of HbF was calculated as follows from ion-exchange HPLC: %HbF = [HbF/(HbA + HbF)] × 100. The percentage of g-globin haemoglobin subunits was calculated as follows from reverse-phase HPLC: % g-globin = [(Gg-chain + Ag-chain)/b -like chains (b + Gg + Ag)] × 100.
K-562 cell culture and hypoxia treatment
K-562 cells were expanded in IMDM media (Gibco, 12440061) supplemented with 10% fetal bovine serum. For hypoxic treatment cells were maintained within a Plas-Labs hypoxia chamber at 1% O2.
Edited HUDEP-2 cell culture, differentiation, and hypoxia treatment
Edited HUDEP-2 cells were expanded in SFEM media (Stem Cell Technologies, 09650) supplemented with 50 ng/mL recombinant human SCF (Peprotech, 300-07), 3 units/mL recombinant EPO (Peprotech, 100-64), 1 μg/mL doxycycline (R&D Systems, 4090-50), 0.4 μg/mL dexamethasone (R&D Systems, 1126/100). HUDEP-2 cells were differentiated for ten days using a 2-phase protocol. During phase 1 (days 0-3), cells were cultured at 1.0e6 cells/mL in IMDM with 5% human blood type AB plasma (GemCell, 100-512-100), 1% penicillin/streptomycin, 3 units/mL heparin (Sigma-Aldrich, H3393-10KU), 10 μg/mL insulin (Sigma, I9278), 3 units/mL recombinant EPO (Peprotech, 100-64), 100 μg/mL holo-transferrin (Bio-Techne, 2914-HT-001G), 50 ng/mL recombinant human SCF (Peprotech, 300-07) and 1 µg/mL doxycycline (R&D Systems, 4090-50). At the onset of phase 2 (days 4-10), cells were counted and adjusted to 1.5e6 cells/mL. The media was replaced with fresh media containing the same supplements minus the recombinant SCF. For hypoxic treatment, cells were differentiated within a Plas-Labs hypoxia chamber at 1% O2.
Real-Time qPCR analysis
RNA was extracted using the RNeasy Plus Mini Kit (Qiagen) following the manufacturer’s protocol and eluted into 50 µL 10 mM Tris-HCl. RNA was quantified with the Qubit® RNA BR Assay (Life Technologies). 25.0 ng of total RNA was used for reverse transcription followed by quantitative real-time PCR using IDT’s PrimeTime One-Step RT-qPCR master mix (Coralville, IA) following the manufacturer’s recommended protocol. Gene expression was evaluated using IDT PrimeTime qPCR Assays following both the protocol and suggested cycling conditions for 10 µL reactions. qPCR was performed on the QuantStudio 12 K Flex Real-Time PCR System (Applied Biosystems) and analysed with the QuantStudio 12 K Flex Software V1.5 (Applied Biosystems). RT-qPCR Ct values for graphed transcripts were all below 36; Ct values above the cutoff of 36 (such as when amplifying FLT1 transcripts in HUDEP-2 cells and BCL11A transcripts in K562 cells) were considered background variation with unreliable sensitivity.
Digital PCR analysis
RNA extracted from HUDEP-2 cells using the RNeasy Plus Mini Kit (Qiagen) was reverse transcribed using the QuantiTect Reverse Transcription kit (Qiagen) according to manufacturer’s instructions. 10 microliters of extracted RNA per sample, less than 1 microgram per sample, was used as the reverse transcription template. One microliter of cDNA was used as a template per digital PCR reaction to detect FLT1, and one microliter of 10x diluted cDNA in nuclease-free water was used as a template per digital PCR reaction to detect ACTB and HBG2. Digital PCR mixes were assembled in a 15-microliter volume using the QIAcuity EvaGreen PCR Kit (QIAgen) according to manufacturer’s instructions. Twelve microliters of each PCR reaction were added to one well of a 96-well QIAcuity digital PCR plate, 8500 partitions per sample (Qiagen). Cycling conditions were 95 degrees for 2 minutes, followed by 40 cycles of [30 seconds at 95 degrees followed by one minute at 60 degrees] before imaging. QIAcuity software was used to analyse each outcome and calculate the concentration of transcripts per microliter of PCR mix. FLT1 and HBG2 transcripts were each normalised to the concentration of ACTB transcripts for the same sample. LNA primers to detect each of the three transcripts were ordered from the Qiagen GeneGlobe catalogue. ACTB GeneGlobe ID: SBH1220543. FLT1 GeneGlobe ID: SBH0131380. HBG2 GeneGlobe ID: SBH0481164.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw data from Cameroon used in this study have been deposited in the dbGaP database under the accession code phs003748.v1.p1. The data is available under Controlled Access through the National Heart, Lung, and Blood Institute (NHLBI) Data Access Committee (DAC), and limited to not-for-profit organisations through the General Research Use consent group. The timeframe for response will be determined by the NHLBI DAC. Raw data from Tanzania are available from the EGA database under the accession code EGAD00010000650. Source data are provided with this paper.
Code availability
All codes used in this study have been deposited in Zenodo and can be accessed via114. A detailed description of the HbF transformation procedure can be found at https://genemap-research.github.io/docs/projects/hbfgwas/. Our FUMA Job parameters are available at https://github.com/GeneMAP-Research/hbf-gwas-scripts/blob/main/functionalmapping/params.config.
References
Antonarakis, S. E. et al. Origin of the beta S-globin gene in blacks: the contribution of recurrent mutation or gene conversion or both. Proc. Natl. Acad. Sci. 81, 853–856 (1984).
Allison, A. C. Protection afforded by sickle-cell trait against subtertian malarial infection. Br. Med. J. 4857, 290–294 (1954).
Piel, F. B. et al. Global epidemiology of Sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet 381, 142–151 (2013).
Ranque, B. et al. Estimating the risk of child mortality attributable to sickle cell anaemia in sub-Saharan Africa: a retrospective, multicentre, case-control study. Lancet Haematol. 9, e208–e216 (2022).
Grosse, S. D. et al. Sickle cell disease in africa: a neglected cause of early childhood mortality. Am. J. Prev. Med. 41, S398–S405 (2011).
Steinberg, M. H. & Nagel, R. L. Hemoglobins of the embryo, fetus, and adult. In: Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management (eds. Forget, B. G., Weatherall, D. J., Higgs, D. R. & Steinberg, M. H.) https://doi.org/10.1017/CBO9780511596582.011. 119–136 (Cambridge University Press, Cambridge, 2009).
Shen, Y. et al. A unified model of human hemoglobin switching through single-cell genome editing. Nat. Commun. 12, 4991 (2021).
Platt, O. S. et al. Mortality in sickle cell disease - life expectancy and risk factors for early death. N. Engl. J. Med. 330, 1639–1644 (1994).
Esrick, E. B. et al. Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. N. Engl. J. Med. 384, 205–215 (2021).
Frangoul, H. et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med. 384, 252–260 (2021).
Makani, J. et al. Genetics of fetal hemoglobin in Tanzanian and British patients with sickle cell anemia. Blood 117, 1390–1392 (2011).
Wonkam, A. et al. Association of variants at BCL11A and HBS1L-MYB with hemoglobin F and hospitalization rates among sickle cell patients in Cameroon. PloS One 9, e92506 (2014).
Menzel, S. et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat. Genet. 39, 1197–1199 (2007).
Wonkam, A. The future of sickle cell disease therapeutics rests in genomics. Dis. Model. Mech. 16, dmm049765 (2023).
Mtatiro, S. N. et al. Genome wide association study of fetal hemoglobin in sickle cell anemia in Tanzania. PloS One 9, e111464 (2014).
Bae, H. T. et al. Meta-analysis of 2040 sickle cell anemia patients: BCL11A and HBS1L-MYB are the major modifiers of HbF in African Americans. Blood 120, 1961–1962 (2012).
Marchini, J. & Howie, B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 11, 499–511 (2010).
Gurdasani, D. et al. Uganda genome resource enables insights into population history and genomic discovery in Africa. Cell 179, 984–1002.e36 (2019).
Cato, L. D. et al. Genetic regulation of fetal hemoglobin across global populations. Preprint at https://doi.org/10.1101/2023.03.24.23287659 (2023).
Vinjamur, D. S. et al. ZNF410 represses fetal globin by singular control of CHD4. Nat. Genet. 53, 719–728 (2021).
Wongborisuth, C. et al. Down-regulation of the transcriptional repressor ZNF802 (JAZF1) reactivates fetal hemoglobin in β0-thalassemia/HbE. Sci. Rep. 12, 4952 (2022).
Lan, X. et al. ZNF410 uniquely activates the NuRD component CHD4 to silence fetal hemoglobin expression. Mol. Cell 81, 239–254.e8 (2021).
Ojewunmi, O. O. et al. The genetic dissection of fetal haemoglobin persistence in sickle cell disease in Nigeria. Hum. Mol. Genet. 33, 919–929 (2024).
Solovieff, N. et al. Fetal hemoglobin in sickle cell anemia: genome-wide association studies suggest a regulatory region in the 5′ olfactory receptor gene cluster. Blood 115, 1815–1822 (2010).
Bhatnagar, P. et al. Genome-wide association study identifies genetic variants influencing F-cell levels in sickle-cell patients. J. Hum. Genet. 56, 316–323 (2011).
Grebien, F. et al. Stat5 activation enables erythropoiesis in the absence of EpoR and Jak2. Blood 111, 4511–4522 (2008).
Thambyrajah, R. et al. GFI1 proteins orchestrate the emergence of haematopoietic stem cells through recruitment of LSD1. Nat. Cell Biol. 18, 21–32 (2016).
Zhang, L., Flygare, J., Wong, P., Lim, B. & Lodish, H. F. miR-191 regulates mouse erythroblast enucleation by down-regulating Riok3 and Mxi1. Genes Dev. 25, 119–124 (2011).
Corn, P. G. et al. Mxi1 is induced by hypoxia in a HIF-1–dependent manner and protects cells from c-Myc-induced apoptosis. Cancer Biol. Ther. 4, 1285–1294 (2005).
Xi, W. & Beer, M. A. Loop competition and extrusion model predicts CTCF interaction specificity. Nat. Commun. 12, 1046 (2021).
Blanco, E., González-Ramírez, M., Alcaine-Colet, A., Aranda, S. & Croce, L. D. The bivalent genome: characterization, structure, and regulation. Trends Genet. 36, 118–131 (2020).
Yu, Y. et al. H3K27me3-H3K4me1 transition at bivalent promoters instructs lineage specification in development. Cell Biosci. 13, 66 (2023).
Garner, C. et al. Genetic influences on F cells and other hematologic variables: a twin heritability study. Blood 95, 342–346 (2000).
Galarneau, G. et al. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat. Genet. 42, 1049–1051 (2010).
Kurita, R. et al. Establishment of immortalized human erythroid progenitor cell lines able to produce enucleated red blood cells. PLoS One 8, e59890 (2013).
Wu, Y. et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 25, 776–783 (2019).
Uchida, N. et al. High-level embryonic globin production with efficient erythroid differentiation from a K562 erythroleukemia cell line. Exp. Hematol. 62, 7–16.e1 (2018).
Testa, U. et al. Hemoglobin expression in clones of K562 cell line. Eur. J. Biochem. 121, 649–655 (1982).
Jawaid, K., Wahlberg, K., Thein, S. L. & Best, S. Binding patterns of BCL11A in the globin and GATA1 loci and characterization of the BCL11A fetal hemoglobin locus. Blood Cells Mol. Dis. 45, 140–146 (2010).
Amini, R. et al. Soluble Flt-1 Gene delivery in acute myeloid leukemic cells mediating a nonviral gene carrier. BioMed. Res. Int. 2013, e752603 (2013).
Feng, R. et al. Activation of γ-globin expression by hypoxia-inducible factor 1α. Nature 610, 783–790 (2022).
Siatecka, M. & Bieker, J. J. The multifunctional role of EKLF/KLF1 during erythropoiesis. Blood 118, 2044–2054 (2011).
Ferrara, N., Gerber, H.-P. & LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 9, 669–676 (2003).
Florentin, J. et al. VEGF receptor 1 promotes hypoxia-induced hematopoietic progenitor proliferation and differentiation. Front. Immunol. 13, 882484 (2022).
Wonkam, A. et al. Five priorities of African genomics research: the next frontier. Annu. Rev. Genomics Hum. Genet. 23, 499–521 (2022).
Ju, D., Hui, D., Hammond, D. A., Wonkam, A. & Tishkoff, S. A. Importance of including non-European populations in large human genetic studies to enhance precision medicine. Annu. Rev. Biomed. Data Sci. 5, 321–339 (2022).
Sirugo, G., Williams, S. M. & Tishkoff, S. A. The missing diversity in human genetic studies. Cell 177, 26–31 (2019).
Esoh, K. K. et al. Genome-wide association study identifies novel candidate malaria resistance genes in Cameroon. Hum. Mol. Genet. 32, 1946–1958 (2023).
Sengupta, D. et al. Performance and accuracy evaluation of reference panels for genotype imputation in sub-Saharan African populations. Cell Genomics 3, 100332 (2023).
Yu, K. et al. Meta-imputation: an efficient method to combine genotype data after imputation with multiple reference panels. Am. J. Hum. Genet. 109, 1007–1015 (2022).
Choudhury, A. et al. High-depth African genomes inform human migration and health. Nature 586, 741–748 (2020).
Fan, S. et al. Whole-genome sequencing reveals a complex African population demographic history and signatures of local adaptation. Cell 186, 923–939.e14 (2023).
Bao, E. L. et al. Heritability of fetal hemoglobin, white cell count, and other clinical traits from a sickle cell disease family cohort. Am. J. Hematol. 94, 522–527 (2019).
Milner, P. F. et al. Increased HbF in sickle cell anemia is determined by a factor linked to the βS gene from one parent. Blood 63, 64–72 (1984).
Wonkam, A. et al. Genetic modifiers of long-term survival in sickle cell anemia. Clin. Transl. Med. 10, e152 (2020).
Casella, I. et al. Autocrine-paracrine VEGF loops potentiate the maturation of megakaryocytic precursors through Flt1 receptor. Blood 101, 1316–1323 (2003).
Semenza, G. L. The genomics and genetics of oxygen homeostasis. Annu. Rev. Genomics Hum. Genet. 21, 183–204 (2020).
Das, B. et al. A hypoxia-driven vascular endothelial growth factor/Flt1 autocrine loop interacts with hypoxia-inducible factor-1α through mitogen-activated protein kinase/extracellular signal-regulated kinase 1/2 pathway in neuroblastoma. Cancer Res. 65, 7267–7275 (2005).
Kang, B. et al. GFI1 regulates chromatin state essential in human endothelial-to-haematopoietic transition. Cell Prolif. 55, e13244 (2022).
Helness, A. et al. GFI1 tethers the NuRD complex to open and transcriptionally active chromatin in myeloid progenitors. Commun. Biol. 4, 1–16 (2021).
Okuyama, H. et al. Expression of vascular endothelial growth factor receptor 1 in bone marrow-derived mesenchymal cells is dependent on hypoxia-inducible factor 1*. J. Biol. Chem. 281, 15554–15563 (2006).
Johnson, R. W., Sowder, M. E. & Giaccia, A. J. Hypoxia and bone metastatic disease. Curr. Osteoporos. Rep. 15, 231–238 (2017).
Ma, Q. et al. Fetal hemoglobin in sickle cell anemia: genetic determinants of response to hydroxyurea. Pharmacogenomics J. 7, 386–394 (2007).
Kolliopoulou, A. et al. Role of genomic biomarkers in increasing fetal hemoglobin levels upon hydroxyurea therapy and in β-thalassemia intermedia: a validation cohort study. Hemoglobin 43, 27–33 (2019).
Ataga, K. I. et al. Association of soluble fms-like tyrosine kinase-1 with pulmonary hypertension and haemolysis in sickle cell disease. Br. J. Haematol. 152, 485–491 (2011).
Fong, G.-H., Rossant, J., Gertsenstein, M. & Breitman, M. L. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 376, 66–70 (1995).
Fong, G.-H., Zhang, L., Bryce, D.-M. & Peng, J. Increased hemangioblast commitment, not vascular disorganization, is the primary defect in flt-1 knock-out mice. Development 126, 3015–3025 (1999).
Corre, T. et al. Heritability and association with distinct genetic loci of erythropoietin levels in the general population. Haematologica 106, 2499–2501 (2021).
Choudhury, A. et al. Meta-analysis of sub-Saharan African studies provides insights into genetic architecture of lipid traits. Nat. Commun. 13, 2578 (2022).
Wonkam, A. et al. Clinical and genetic factors are associated with pain and hospitalisation rates in sickle cell anaemia in Cameroon. Br. J. Haematol. 180, 134–146 (2018).
Nguweneza, A. et al. Clinical characteristics and risk factors of relative systemic hypertension and hypertension among sickle cell patients in Cameroon. Front. Med. 9, 924722 (2022).
Saiki, R. K. et al. Enzymatic amplification of β-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230, 1350–1354 (1985).
Platt, O. S. et al. Pain in sickle cell disease. N. Engl. J. Med. 325, 11–16 (1991).
Darbari, D. S. et al. Severe painful vaso-occlusive crises and mortality in a contemporary adult sickle cell anemia cohort study. PloS One 8, e79923 (2013).
Bitoungui, V. J. N. et al. Beta-globin gene haplotypes among cameroonians and review of the global distribution: is there a case for a single sickle mutation origin in Africa? Omics J. Integr. Biol. 19, 171–179 (2015).
Rumaney, M. B. et al. The co-inheritance of alpha-thalassemia and sickle cell anemia is associated with better hematological indices and lower consultations rate in Cameroonian patients and could improve their survival. PLoS One 9, e100516 (2014).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7 (2015).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. PLoS Genet. 2, 2074–2093 (2006).
Gibbs, R. A. et al. A Global Reference for Human Genetic Variation. Nature 526 (Nature Publishing Group, 2015).
Sudmant, P. H. et al. An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81 (2015).
Loh, P. R., Palamara, P. F. & Price, A. L. Fast and accurate long-range phasing in a UK Biobank cohort. Nat. Genet. 48, 811–816 (2016).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016).
Browning, B. L. & Browning, S. R. Genotype imputation with millions of reference samples. Am. J. Hum. Genet. 98, 116–126 (2016).
Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. bioRxiv 2, 563866 (2019).
Fuchsberger, C., Abecasis, G. R. & Hinds, D. A. Minimac2: faster genotype imputation. Bioinformatics 31, 782–784 (2015).
Durbin, R. Efficient haplotype matching and storage using the positional Burrows–Wheeler transform (PBWT). Bioinformatics 30, 1266–1272 (2014).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
McKenna, A. et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. https://doi.org/10.1101/gr.107524.110 (2010).
Yun, T. et al. Accurate, scalable cohort variant calls using DeepVariant and GLnexus. Bioinformatics 36, 5582–5589 (2020).
Zhou, W. et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 50, 1335–1341 (2018).
R Core Team. R: a language and environment for statistical computing. R Found. Stat. Comput. Vienna Austria https://www.r-project.org (2023).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Zou, Y., Carbonetto, P., Wang, G. & Stephens, M. Fine-mapping from summary data with the “Sum of Single Effects” model. PLOS Genet. 18, e1010299 (2022).
Pruim, R. J. et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337 (2010).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLOS Comput. Biol. 11, e1004219 (2015).
Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics https://doi.org/10.1093/bioinformatics/bth457 (2005).
Liu, L. et al. Original research: a case-control genome-wide association study identifies genetic modifiers of fetal hemoglobin in sickle cell disease. Exp. Biol. Med. 241, 706–718 (2016).
Gabriel, S. B. et al. The structure of haplotype blocks in the human genome. Science 296, 2225–2229 (2002).
Pazokitoroudi, A. et al. Efficient variance components analysis across millions of genomes. Nat. Commun. 11, 4020 (2020).
Wickham, H. Ggplot2: elegant graphics for data analysis. https://doi.org/10.1007/978-0-387-98141-3. (Springer, New York, NY, 2009).
Traxler, E. A. et al. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat. Med. 22, 987–990 (2016).
Hu, J. et al. Isolation and functional characterization of human erythroblasts at distinct stages: implications for understanding of normal and disordered erythropoiesis in vivo. Blood 121, 3246–3253 (2013).
Sentmanat, M. F., Peters, S. T., Florian, C. P., Connelly, J. P. & Pruett-Miller, S. M. A survey of validation strategies for CRISPR-Cas9 editing. Sci. Rep. 8, 888 (2018).
Connelly, J. P. & Pruett-Miller, S. M. CRIS.py: a versatile and high-throughput analysis program for CRISPR-based genome editing. Sci. Rep. 9, 4194 (2019).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010).
Law, C. W., Chen, Y., Shi, W. & Smyth, G. K. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Esoh, K. GeneMAP-Research/hbf-gwas-scripts: v1. Zenodo https://doi.org/10.5281/zenodo.14607341 (2025).
Zweidler-Mckay, P. A., Grimes, H. L., Flubacher, M. M. & Tsichlis, P. N. Gfi-1 encodes a nuclear zinc finger protein that binds DNA and functions as a transcriptional repressor. Mol. Cell. Biol. 16, 4024–4034 (1996).
Frazer, K. A. et al. A second generation human haplotype map of over 3.1 million SNPs. Nature 449, 851–861 (2007).
Acknowledgements
We thank the study participants from Cameroon and Tanzania who provided their samples that contributed to this study. We thank the St. Jude Children’s Research Hospital Centre for Advanced Genome Engineering for designing and validating nuclease guide RNAs as well as measuring editing outcomes. The study was funded by the National Institutes of Health, USA grants 1U01HG007459‐01 and U24‐HL‐135600 to AW. SEA was partially supported by a grant from the Childcare Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The research was supported in part by ALSAC and the National Cancer Institute grant P30 CA021765.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: A.W., K.E., E.R.C., S.E.A. Performed the experiments: A.W., K.E., R.L. K.N., N.N., E.A.D.D., V.J.N.B., S.N., R.S., V.N., J.M., F.O., M.A.B., J.M., N.M., G.L., M.H.S., J.Y., G.N., R.L., J.F.C., D.E.A., E.R.C., S.E.A. Patient recruitment, samples, and clinical data collection and processing: A.W., V.J.N.B., K.M., S.N., R.S., J.M. Analyzed the data: K.E., A.W., G.A.N., R.M.L., D.E.A., M.A.B., E.R.C., M.H.S., G.L., S.E.A. Contributed reagents/materials/analysis tools: A.W., K.E., E.R.C., S.N., N.M. Produced the first draft of the manuscript: K.E., A.W. Revised and approved the manuscript: A.W., K.E., K.N., V.J.N.B., R.L., S.N., N.N., E.A.D.D., R.S., V.N., J.M., F.O., M.A.B., J.M., N.M., G.L., M.H.S., J.Y., G.N., R.L., J.F.C., D.A., E.R.C., S.E.A. The corresponding author confirms that he has full access to all the data in the study and has final responsibility for the decision to submit for publication.
Corresponding author
Ethics declarations
Competing interests
J.S.Y. is an equity owner of Beam Therapeutics, and consultant for Orna, Merck, and Portal Bio. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Vivien Sheehan, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41467_2025_57413_MOESM5_ESM.xlsx
Supplementary data 3. FLT1 significant associations and fine-mapped variants in the Cameroon, Tanzania, and Cameroon-Tanzania meta analysis datasets
41467_2025_57413_MOESM6_ESM.xlsx
Supplementary data 4. Association test of fetal haemoglobin level and mean corpuscula volume (MCV), and red blood cell (RBC) in Cameroonian sickle cell anaemia patients with 1,000,000 permutations
41467_2025_57413_MOESM8_ESM.xlsx
Supplementary data 6. Associations for the meta-analysis and the merged of Cameroon and Tanzania non-imputed datasets with FDR < 0.20
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wonkam, A., Esoh, K., Levine, R.M. et al. FLT1 and other candidate fetal haemoglobin modifying loci in sickle cell disease in African ancestries. Nat Commun 16, 2092 (2025). https://doi.org/10.1038/s41467-025-57413-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57413-5
