
Abstract
The next-generation gene editing tool, prime editing (PE), is adept at correcting point mutations precisely with high editing efficiency and rare off-target events and shows promising therapeutic value in treating hereditary diseases. Retinitis pigmentosa (RP) is the most common type of inherited retinal dystrophy and is characterized by progressive degeneration of retinal photoreceptors and, consequently, visual decline. To date, effective treatments for RP are lacking. Herein, a PE system is designed to target the PDE6B Y347X mutation in the rd1 mouse strain, a preclinical RP model. We screen and develop the PE system with epegRNA and RTΔRnH, which is delivered via dual-AAV in vivo with an editing efficiency of 26.47 ± 13.35%, with negligible off-target effects confirmed by AID-Seq and PE-tag. Treatment with the PE system in vivo greatly restores PDE6B protein expression and protects rod cells from degeneration. Mouse behavioural experiments also show that compared with no treatment, prime editing inhibits vision deterioration in littermate rd1 mice. This study provides a therapeutic opportunity for the use of PE to correct mutated RPs at the genomic level.
Similar content being viewed by others

Introduction
Point mutations account for half of known pathogenic human genetic variants, inducing a variety of hereditary diseases1. Despite extensive efforts, most of these mutations remain challenging to correct, leaving their associated inherited diseases without effective treatments. Prime editing (PE), a next-generation precise gene editing technology, offers a solution by modifying gene mutations with high efficiency and minimal off-target effects1. A distinctive feature of PE is its ability to operate without requiring double-strand breaks (DSBs) or homology sequence templates, making it versatile for various applications, including the correction of point mutations, as well as insertions or deletions1,2. Compared to traditional CRISPR/Cas9, PE excels in precise editing with low rates of indels and off-target effects1,3. As a result, PE has been widely applied in correcting pathogenic mutations of human diseases, including Leber congenital amaurosis3, Type I tyrosinemia4, and Duchenne muscular dystrophy5. This breakthrough technology enables the development of new applications in other disorders.
Retinitis pigmentosa (RP), the most prevalent form of inherited retinal disease (IRD) involving retinal degeneration, stands as the leading cause of visual impairment globally, affecting approximately 1 in 4000 individuals6. This condition is primarily marked by the loss of rod photoreceptor cells, which enables achromatic vision in low-light conditions such as starlight or moonlight. This is followed by the second death of cone cells, which mediates color vision and fine acuity in daylight6. Consequently, patients typically experience bilateral night vision loss in adolescence, side vision loss in young adulthood, and central vision loss later in life. Unfortunately, there is currently no effective therapy for RP, with only a few symptomatic and exploratory therapies available, including neuroprotective agents, gene therapy, stem cell therapy, optogenetics, retinal implants and prosthetics, or chemical photoswitches7.
The mode of inheritance of RP can be categorized into three main types: autosomal recessive (approximately 40% of cases), autosomal dominant (25%), and X-linked (15%)6,8,9,10,11,12 (Fig. 1a). While the majority of RP cases are monogenic, the disorder exhibits remarkable genetic complexity. Current research has catalogued more than 3000 distinct genetic variations across 70 identified genes, which are implicated in crucial visual processes including phototransduction pathways, retinoid recycling mechanisms, photoreceptor cellular architecture, protein conformation processes, and gene regulatory functions13 (RetNet, https://sph.uth.edu/RetNet/). The causative genes of autosomal-recessive RP (arRP) are illustrated in Fig. 1a. Notably, mutations of PDE6B (OMIM 180072) is one of the major causes of arRP7.

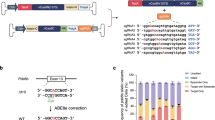
a Genetic background of RP. b Schematic representation of the Pde6b nonsense mutation in rd1 mice. The C-to-A mutation in exon 7 of the Pde6b locus induces a stop codon and inhibits normal expression of the PDE6B protein. Prime editing was designed to correct the A base to the C base. c Flowchart of pegRNA and nsgRNA design. The sequence of exon 7 of the mouse Pde6b gene was input into three websites (PE-Designer, pegIT and PrimeDesign). Among the output results, four pegRNA and nsgRNA pairs were randomly selected from each website. The corresponding pairs were named pn1-12. The sequence of each pair was illustrated below.
Pde6b encodes the β subunit of rod cGMP-phosphodiesterase (PDE6), an enzyme located in the photoreceptor outer segment that hydrolyzes cGMP. The PDE6 complex is essential for phototransduction activation by modulating cGMP-gated cation channels in rods and cones. Hence, mutations in Pde6b cause arRP by eliciting calcium influx into rods, ultimately resulting in cell apoptosis. The rodless (rd1) mouse carrying a nonsense mutation (c.1041 C > A, p.Y347X) in exon 7 of the Pde6b gene, serves as a classic preclinical mouse model for RP. This mutation triggers severe degeneration of rods starting at postnatal Day 8 (P8) and progresses to complete loss of the rods by P2014. This degenerative process induces visual loss and ultimately leads to total blindness in rd1 mice.
Gene augmentation therapy has been utilized to treat rd1 mice via the delivery of PDE6B15. Furthermore, a phase I/II trial (NCT03328130) is currently underway, involving subretinal administration of AAV2/5-hPDE6B to explore rod-directed gene augmentation16. CRISPR/Cas9, a traditional gene editing tool, has been applied to target Pde6b mutants (c.1041 C > A, p.Y347X) in rd1 mice, achieving moderate editing efficiency17,18. Another precise genome editing tool, base editing (BE), has been applied to target the Pde6b mutants (c.1678 C > T, p.R560C) in rd10 mice. However, this base editing strategy leads to BE-specific bystander editing effects19. To date, there is a lack of research on the application of prime editing to correct the PDE6B Y347X mutation in rd1 mice.
In this study, we utilize a dual-AAV prime editing system to correct point mutations in Pde6b in rd1 mice. The editing efficiency is high (26.47 ± 13.35%), and genome-wide off-target sequencing analysis demonstrates minimal off-target editing with our selected prime editing strategy. The results also demonstrate a significant restoration of PDE6B protein expression and vision in vivo. This research shows great promise as a precise genome editing tool for treating hereditary diseases, especially those for which effective therapeutic strategies are currently lacking.
Results
Design of the PE system to target the PDE6B Y347X mutation
The rd1 mouse model is recessively inherited and carries a nonsense mutation in exon 7 of the Pde6b locus (c.1041 C > A, p.Y347X), which induces a premature stop codon and inhibits normal translation of the PDE6B protein. The genomic organization of the murine Pde6b gene, particularly in terms of its intron-exon architecture, shows significant conservation with its human counterpart, PDE6B17. We aimed to apply the PE system to correct the premature stop codon mutation (TAA) to tyrosine (TAC) in the Pde6b locus (Fig. 1b). Three websites that were specifically established for the PE system were used to design pegRNA and nsgRNA, namely, PE-Designer20 (http://www.rgenome.net/pe-designer/), pegIT21 (https://pegit.giehmlab.dk), and PrimeDesign22 (https://primedesign.pinellolab.partners.org/). The exon 7 sequences in the Pde6b gene (67 bp) of wild-type (WT) and rd1 mice were used as the desired and template sequences, respectively. Among the output results from the three websites, we randomly selected four pairs of pegRNAs and nsgRNAs from each website according to the guidelines23, hereafter referred to as pn1-12. A flowchart of the design strategy and the corresponding sequences of the pegRNAs and nsgRNAs are shown in Fig. 1c and Supplementary Data 1.
In vitro screening of efficient pegRNAs and nsgRNAs targeting PDE6B Y347X
To select the most effective pegRNA and sgRNA pair for prime editing, a reporter system was established to mimic the Pde6b DNA template. The reporter plasmid contained the coding sequences of the Pde6b gene (including exon 7) in the WT or mutant type (Mut), followed by a FLAG-tag and the coding sequence of green fluorescent protein (GFP) (Fig. 2a). In human embryonic kidney (HEK) 293 T cells, the reported plasmid containing the mScarlet element for indicating transfection efficiency, together with combinations of pegRNA and nsgRNA (pn1-12) with prime editors (Cas9 nickase fused to the RT domain), were co-transfected (Fig. 2b). Due to the premature stop codon caused by the point mutation of Pde6b, the detection of the PDE6B-FLAG protein and GFP signal indicated that one-base-pair editing (A to C) was successfully achieved by the PE system in vitro. Notably, cells transfected with pn4 exhibited highest expression levels of PDE6B-FLAG protein and GFP compared to other pairs of pegRNA and nsgRNA (Fig. 2c, d and Supplementary Figs. 1a, 2a). Deep sequencing further confirmed that the on-target editing efficiency of pn4 was 27.96 ± 6.27% (Fig. 2e and Supplementary Fig. 2b). Therefore, pn4 was chosen for subsequent studies. In addition, state-of-the-art PE5 and PE5max system featured by supplement of MLH1dn (2264 bp) to inhibit DNA mismatch repair (MMR) were further compared with PE3 system, which showed comparable results in HEK293T cells (Supplementary Fig. 3a–c). Since HEK293T cells are partially MMR deficient due to hypermethylation of the MLH1 promoter24, we used MMR-proficient (MMR + ) cell types, HeLa to further evaluate the effects of PE5 and PE5max system. PE6a and b (improvements in editor size)25, PE6c and d (improvements in RT activity)25, as well as PE7 system (the prime editor protein fused to the RNA-binding domain of La)26 were also included for pan-PE validation in HeLa cells. The advanced PE systems exhibited great performances in vitro, which can be considered in further PE studies (Supplementary Fig. 3d, e). Considering the larger sizes of PE5 and PE5 max system than PE3, and the lack of evidence of PE6 and PE7 in treating ocular diseases, we decided to use the PE3 system in vivo.

a Schematic representation of the reporter system. The template carrying exon 7 of the Pde6b gene with (mut template) or without the PDE6B Y347X mutation (WT template) was followed by sequences encoding a 3xFlag and EGFP. After removal of the stop codon via prime editing, the Flag protein tag and EGFP signal were detected. b Flowchart of the in vitro screening for efficient guide RNAs. 293 T cells were co-transfected with plasmids expressing the WT/mut template (containing mScarlet for indicating transfection efficiency) and PE system (prime editors, pegRNA and nsgRNA). The pn1-12 plasmids were individually added to independent groups. After transfection for 48 h, the cells were harvested for Western blotting, immunostaining, flow cytometry and DNA amplicon sequencing. Created in BioRender60. c Western blot analysis of PDE6B-FLAG expression in 293 T cell lysates after 48 h of transfection. Uncropped blots in Source Data. d Relative GFP ratio was quantified from Immunofluorescence images of in vitro screening. Representative data are presented as the mean ± SD, n = 6 biologically independent replicates. One-way ANOVA with Tukey’s multiple comparisons test was used. ***p < 0.001. e Editing efficiency of the prime editing system for pn4. Representative data are presented as the mean ± SD, n = 6 biologically independent replicates. One-way ANOVA with Tukey’s multiple comparisons test was used. ***p < 0.001.
In vivo PE precisely corrects the PDE6B nonsense mutation and prolongs rod survival in rd1 mice
The adeno-associated virus 2 (AAV2) serotype, which has high retinal tropism, was chosen for the efficient delivery of the PE system in vivo27. Considering the limited loading capacity (approximately 4.8 kb) of AAV for delivering the PE system (approximately 6.3 kb), we engineered a dual-AAV2 system by splitting nCas9 into two AAV vectors, which were then reconstituted to full-length PE by a trans-splicing intein in vivo3,28,29,30. To maximize the in vivo editing efficiency, an engineered pegRNA (epegRNA) with an additional 3’ RNA structural motif that prevents degradation of the 3’ extension31 was constructed in this AAV vector. Moreover, because the function of the RNaseH domain (approximately 0.6 kb) was verified to be negligible in the context of prime editing32,33, a size-reduced PE2ΔRnH variant was also employed in this vector for comparable performance with full-length PE and to enable more flexible vector designs for in vivo delivery. Therefore, AAV2-PE-N contained epegRNA, sgRNA and the amino-terminus (encoding amino acids 1-713) of nCas9, whereas AAV2-PE-C contained the carboxy-terminal half (encoding amino acids 714-1368) of nCas9 and RTΔRnH (Fig. 3a).

a Schematic diagram of the PE system delivered by dual-AAV. Cas9(H840A) was split at site 713, at which the N-terminus followed by the epegRNA and nsgRNA was packaged in one AAV, whereas the C-terminus linked to the RTΔRnH domain was packaged in the other AAV. A trans-splicing intein system was employed to reconstitute the two AAVs into the full-length PE. b Timeline of the in vivo AAV-PE treatment. One-week-old rd1 mice were subjected to subretinal injection of the dual-AAV system. Three weeks after injection, PE-treated rd1 mice were subjected to a series of functional assays for vision evaluation. Then, the PE-treated rd1 mice were sacrificed at five weeks of age, and their eyeballs were collected for molecular biology and morphological analyses. Untreated littermates and WT mice of the same age were used as negative and positive controls, respectively. Created in BioRender61. c The editing efficiency of the PE system in vivo. Representative data are presented as the mean ± SD, n = 6. One-way ANOVA with Tukey’s multiple comparisons test. *p < 0.05; ***p < 0.001. d Western blot analysis of PDE6B expression in mouse eyeball lysates after treatment at P35. Uncropped blots in Source Data. e Representative immunofluorescence image of the rod photoreceptor layer marked with rhodopsin (red) at P35. ONL, outer nuclear layer; INL, inner nuclear layer; OS, outer segment. Scale bar = 25 μm. f Quantification of OS thickness in WT, rd1, and PE-edited mice at P35. The means ± SDs are shown, n = 6. One-way ANOVA with Tukey’s multiple comparisons test was used for comparisons. **P < 0.01; ***p < 0.001. g Representative retinal section of WT, rd1, and PE-edited mice with H&E staining at P35. ONL, outer nuclear layer; INL, inner nuclear layer; and GCL, ganglion cell layer. Above scale bar = 100 μm. Below scale bar = 50 μm. h Quantification of ONL thickness in WT, rd1, and PE-edited mice at P35. One-way ANOVA with Tukey’s multiple comparisons test was used for comparisons. The means ± SDs are shown, n = 6. ***p < 0.001.
The rd1 mice present a nonsense mutation in exon 7 of the Pde6b locus, which leads to absence of PDE6B protein expression and consequent accelerated degeneration of rod cells and vision loss at three weeks of age (Supplementary Fig. 4a-e). Therefore, subretinal injection of a mixture of the two AAV2 vectors (AAV2-PE-N term and AAV2-PE-C term, in a total volume of 1 μl) was performed in rd1 mice at one week of age, a time point prior to the onset of the disease, and no obvious intravitreal hemorrhage or bullous retinal detachment were observed (Supplementary Fig. 5a). Then, a series of functional assays were performed on mice at 4-5 weeks of age to evaluate visual function. Mice were sacrificed for further assessment of the therapeutic effect 4 weeks post injection, when photoreceptor death was nearly complete in untreated rd1 mice (Fig. 3b). The expression of the dual-AAV was confirmed at RNA level both in vitro and in vivo, especially at 7 days and 4 weeks post injection (Supplementary Fig. 5b–f). The Cas9 protein was also detected positive at 4 weeks post injection (Supplementary Fig. 5g). Meanwhile, the systemic leakage of dual-AAV system was not found (Supplementary Fig. 6a). Genomic DNA was extracted from mouse eyeball lysates to determine the editing efficiency at 5 weeks of age (4 weeks after injection). The deep sequencing results revealed up to 26.47 ± 13.35% (mean ± SD; n = 6) of only A-to-C conversion at the target site (Fig. 3c). Western blot analysis was performed to evaluate restoration of PDE6B protein expression in PE-edited rd1 mice. Surprisingly, PDE6B expression in PE-edited rd1 mice was restored to 39% of WT mice, in striking contrast to what was observed in the rd1 control mice, in which PDE6B protein levels were undetectable (Fig. 3d).
To further evaluate the effect of prime editing on rod survival, retinal sections were immunolabelled with an anti-rhodopsin antibody, a rod-specific marker. Significant rods survival was observed in retinal tissue following PE administration, contrasting sharply with the extensive rod cell loss characteristic of untreated rd1 mice (Fig. 3e, f). To quantify the extent to which the rods and cones were rescued, we measured the thickness of the outer nuclear layer (ONL), which consists of photoreceptor nuclei, via haematoxylin and eosin (H&E) staining. In PE-treated mice, the ONL was rescued and maintained at four layers of nuclei, in contrast to the single layer of incomplete nuclei left in untreated rd1 mice (Fig. 3g). The ONL of PE-treated rd1 mice was significantly thicker than that of untreated mice (20.48 ± 2.69 µm vs. 5.00 ± 0.99 µm, n = 6) (Fig. 3h), achieving approximately 50% of the ONL thickness in WT mice. Representative images of the whole retina from three groups were shown in Supplementary Fig. 6b. The quantitative measurement on the different sites of retinal sections provided a comprehensive evaluation of PE effect (Supplementary Fig. 6c). These results demonstrated that PE treatment was able to correct the PDE6B Y347X mutation successfully, restore the expression of the wild-type PDE6B protein and promote photoreceptor survival in vivo.
Additionally, we also observed long-term effect of PE in vivo, as ONL was significantly preserved and PDE6B protein could still be detected 8 weeks post injection (Supplementary Fig. 7a–c). These findings further revealed the therapeutic potential of prime editing delivered by AAV.
Apart from AAV, the virus-like particles (VLPs) have also been used for the transient delivery of prime editor ribonucleoprotein complexes in vivo34. VLPs assemble viral proteins which infect cells but lack viral genetic material, circumventing the limited load capacity of AAV and omitting the need for a dual-AAV-mediated gene reconstruction via an intein-split system in vivo. Herein, we applied engineered virus-like particles (eVLPs) delivering PE system in rd1 mice. In view of the successful package and expression of eVLPs (Supplementary Fig. 7d–f), the ONL of retina were preserved after eVLPs-PE treatment. However, according to the quantification of ONL thickness, eVLPs-PE group did not represent better performance than AAV-PE group (13.18 ± 1.91 µm vs. 18.28 ± 1.86 µm, n = 6) (Supplementary Fig. 7g, h). Therefore, we further explore the therapeutic efficiency and safety of AAV-PE in vivo.
In vivo PE induces negligible off-target events in the genome
Off-target editing poses safety challenges in the applications of genome editing agents. To analyze the off-target effects of PE strategy for PDE6B Y347X, we employed AID-seq (adaptor-mediated off-target identification by sequencing), an in vitro off-target detection method35, to nominate potential off-target editing sites. This off-target method uses two different adaptors, a hairpin i7 adaptor and a biotin i5 adaptor, to reduce false-positives and improve sensitivity (Fig. 4a). AID-seq used untreated rd1 mice DNA extracted from eyeballs and nominated a list of potential off-target sites for peg4 and nsg4 (Fig. 4b–d) and we further performed amplicon sequencing analysis to validate genomic modifications in these sites. Samples from treated animals did not show significantly greater indel rates than the background in untreated rd1 mice, suggesting that the PE system specifically targets the intended sites (Fig. 4e). Meanwhile, PE-tag as a latest and sensitive approach for the genome-wide identification of prime editor activity was used to find the potential off-target sites caused by pegRNA36 (Supplementary Fig. 8a). The in vitro PE-tag on gDNA of rd1 mice showed the leading unique molecular identifier (UMI) counts of on-target site but few off-target sites, indicating that they experienced low prime editing activity (Supplementary Fig. 8b). The Venn diagram exhibited that one sequence was overlapped from AID-seq and PE-tag analysis (Supplementary Fig. 8c). Further deep sequencing from treated samples also revealed no off-target events on potential sites from PE-tag, strengthening the safety of prime editing in vivo (Supplementary Fig. 8d). Taken together, two orthogonal off-target sequencing analyses suggested that PE system corrected the pathogenic Pde6b mutation efficiently in vivo and generated only negligible off-target effects.

a Schematic representation of AID-seq for assessing potential off-target sites. Created in BioRender62. b Potential off-target sites detected by AID-seq. c Schematic representation of potential off-target sites for peg4. d Schematic representation of potential off-target sites for nsg4. e Amplicon sequencing assessment of potential off-target sites induced by the PE system. Representative data are presented as the mean ± SD. Generally, n = 6 replicates (otherwise n = 5). Two-tailed unpaired t tests were used. ns, no significance.
In vivo PE upregulates the expression of genes relevant to phototransduction
To evaluate the effect of in vivo PE on the phototransduction transcriptome, high-throughput RNA sequencing was performed using the retinas from the WT, rd1 and PE-edited groups. KEGG enrichment analysis of the rd1 group versus the WT group (Fig. 5a) and the PE-edited group versus the rd1 group (Fig. 5b) revealed that compared with no treatment, PE treatment strongly upregulated the phototransduction pathway. In addition, the differentially expressed gene (DEGs) between the rd1 and WT mice and between the PE-edited and rd1 mice were analysed. The expression levels of most phototransduction-related genes were significantly greater in the PE-edited group than in the untreated rd1 group, due to the death of photoreceptor cells (Fig. 5c, d). To verify the expression levels of these genes, we performed RT‒qPCR and confirmed significant increases in the expression levels of rod-specific genes (Pde6b, Cnga1 and Rho), cone-specific genes (Arr3, Opn1mw and Pdc), other phototransduction-related genes (Rbp3, Grk1 and Gnat137,38) and RGC regeneration genes (Cryga, Cryge and Crygf39) (Fig. 5e). These data indicated that prime editing can rescue the expression of phototransduction-related genes to support photoreceptor cell survival.

a KEGG enrichment analysis: downregulated phototransduction pathway (rd1 versus the WT group). Benjamini Hochberg were used. FDR < 0.05. b KEGG enrichment analysis: upregulated phototransduction pathway (the PE-edited group versus the rd1 group). Benjamini Hochberg were used. FDR < 0.05. c Volcano plot of genes differentially expressed between rd1 and WT mice. The red dots are upregulated genes, whereas the blue dots are downregulated genes. (t tests were used; |log2(fold change)|> 1 and p < 0.05). Genes relevant to the phototransduction pathway are highlighted. d Volcano plot of differentially expressed genes between PE-edited and rd1 mice. (t tests were used; |log2(fold change)|> 1 and p < 0.05). e The mRNA expression levels of genes relevant to the phototransduction pathway were detected in RNA-seq samples by RT‒qPCR. Representative data are presented as the mean ± SEM. Generally, n = 6 replicates (otherwise n = 4). Two-tailed unpaired t tests were used. ns, not significant; *p < 0.05; **p < 0.01.
In vivo PE rescues the visual function of rd1 mice
To explore whether the rescue of rod degeneration and retinal structure via prime editing can indeed improve mice vision, we first performed scotopic ERG on the WT, rd1 and PE-edited groups at 4 weeks post injection. Mice received dark adaption overnight to evaluate rod-dominated retinal function. The representative scotopic ERG traces were demonstrated in Supplementary Fig. 9a. While the a- and b- wave amplitudes of age-matched rd1 mice were found at a negligible level, PE-edited mice restored evident a-wave at 78.14 ± 38.62 μV and b-wave at 155.80 ± 47.76 μV (Supplementary Fig. 9b-c). Next, behavioural experiments were performed on three groups. The pupil light reflex (PLR) was evaluated to further assess whether PE-treated rd1 mice exhibited a preserved light response. Along with the loss of photoreceptors, the PLR was visibly attenuated in the rd1 mice, whereas the wild-type mice exhibited obvious pupil constriction under light stimulation. Quantification of the pupil area indicated that the PE-treated rd1 mice had a significantly greater PLR than the age-matched untreated mice (Fig. 6a, b), indicating that PE promoted photoreceptor survival. To further investigate that the visual function was improved in the PE-treated rd1 mice, passive avoidance was evaluated via the light–dark transition test, an assessment that relies on the intrinsic propensity of the animals to stay in a dark environment (Fig. 6c). The PE-treated rd1 mice showed an apparent preference for staying in the dark chamber (averaging 194.00 ± 23.60 s), which was slightly inferior to that of the wild-type mice (averaging 224.00 ± 17.75 s) but much better than that of the untreated rd1 mice (averaging 149.80 ± 16.88 s) (Fig. 6d). The visual cliff test utilized the natural avoidance response exhibited by rodents when encountering a depth-perceiving apparatus with contrasting visual cues. Experimental groups included wild-type controls alongside rd1 mice receiving either therapeutic intervention or serving as untreated controls. The preferred movements on the bench side were intuitively presented in terms of the traveling trajectories and heatmaps of the recorded times spent on the whole apparatus (Fig. 6e). The results revealed that, unlike the rd1 mice, the PE-treated mice preferred to stay on the bench side (Fig. 6f). The cumulative time that each mouse spent on the cliff was also measured. The untreated rd1 mice had no preference for the bench or cliff side (cliff side: 159.00 ± 18.74 s), while the WT mice spent less time on the cliff (95.83 ± 23.65 s). Compared with those of the untreated mice, the visual function of the PE-treated mice significantly improved, as the PE-treated mice spent less time on the cliff (129.80 ± 9.15 s vs. 159.00 ± 18.74 s, P = 0.036) (Fig. 6g). Additionally, PE-treated mice still exhibited similar behavior as WT mice in light-dark box test after editing for 8w. Mice in PE-treated group spent more time in the dark chamber than the untreated rd1 mice (191.80 ± 27.47 s vs. 146.70 ± 13.29 s, P = 0.008) (Supplementary Fig. 9d). Collectively, these data suggest that in vivo prime editing can efficiently improve vision function in rd1 mice.

a Representative images of eye constriction in WT, rd1 and PE-treated mice. b Quantification of pupil constriction proportions in WT, rd1 and PE-treated mice. Representative data are presented as the mean ± SD, n = 6. One-way ANOVA with Tukey’s multiple comparisons test was used for comparisons. WT group vs rd1 group, p < 0.001; rd1 group vs PE-treated group, p < 0.001; PE-treated group vs WT group, p = 0.002. **p < 0.01; ***p < 0.001. c Schematic representation of the light–dark transition test. Mice could shuttle between the bright and dark boxes through the aperture. Created in BioRender63. d Quantification of the amount of time spent in the dark zone in the WT, rd1 and PE-treated groups. Representative data are presented as the mean ± SD, n = 6. One-way ANOVA with Tukey’s multiple comparisons test was used for comparisons. WT group vs rd1 group, p < 0.001; rd1 group vs PE-treated group, p = 0.004; PE-treated group vs WT group, p = 0.046. *p < 0.05; **p < 0.01; ***p < 0.001. e Schematic representation of the visual cliff test. Created in BioRender64. f Representative traveling trajectories (above) and heatmap recordings for time spent in distinct regions of the whole platform (below) in the WT, rd1 and PE-treated groups. g Quantification of the amount of time spent on the cliff side by WT, rd1 and PE-treated mice. Representative data are presented as the mean ± SD, n = 6. One-way ANOVA with Tukey’s multiple comparisons test was used for comparisons. WT group vs rd1 group, p < 0.001; rd1 group vs PE-treated group, p = 0.036; PE-treated group vs WT group, p = 0.015. *p < 0.05; ***p < 0.001.
Discussion
In this study, prime editing was applied to treat rd1 mice, a preclinical model of retinitis pigmentosa, demonstrating its promising therapeutic effect on the hereditary disease. With the aid of three websites that tailored design of PE strategies, 12 pairs of pegRNA and nsgRNA were selected for in vitro screening. The goal of in vitro prime editing is to select the optimal pair of pegRNA and nsgRNA. Therefore, we focused on the basic component of prime editing system, including pegRNA and nsgRNA and prime editors. The editing efficiency of each pair of pegRNA and nsgRNA was compared under the same circumstances. Next, for higher editing efficiency and better delivery in vivo, we updated the editing system with epegRNA and truncated RTΔRnH in a dual-AAV system. It was injected into rd1 mice to correct the pathogenic mutation. Surprisingly, PE treatment inhibited the progressive death of photoreceptors and retinal degeneration and rescued mouse vision (Fig. 7).

Schematic representation of the flowchart for prime editing application in treating retinitis pigmentosa. The pegRNA and nsgRNA were first designed and cloned for the target site. Then in vitro transfection was performed to screen out the most efficient pairs of pegRNA and nsgRNA. Deep sequencing confirmed the editing efficiency in vitro. Next, AAV delivering PE system was applied in vivo through subretinal injection. In vivo editing was confirmed by deep sequencing and rescued retinal degeneration. Created in BioRender65.
PE has demonstrated an excellent capacity for precise genome editing, resulting in high editing efficiency and few off-target effects. It possesses versatility to modify all 12 point mutations, including transition and transversion mutations, and can also enable insertions and deletions whereas base editing is only capable of modifying point mutations and can induce bystander editing within the editing window40. RP features with photoreceptor death represent the most prevalent cause of visual handicap among working populations in developed countries41. In this study, we first designed and constructed a PE system to target PDE6B Y347X in RP, which produces a premature stop codon, preventing PDE6B expression and resulting in detrimental retinal degeneration. Moreover, we treated mice with efficient PE at one week of age, a timepoint immediately before disease onset and demonstrated a promising effect for early disease intervention. PE has shown substantial potential in inhibiting such seemingly irreversible disease progression via permanent correction of the disease-causing mutation at DNA level.
In addition, a dual-AAV system was constructed to carry the prime editing system featuring an engineered pegRNA and truncated reverse transcriptase domain. An epegRNA with an additional 3’ RNA structural motif for the prevention of degradation has been proven to enhance editing efficiency31. The size-reduced RTΔRnH domain has also been used to construct a more compact prime editing system32,33. We combined epegRNA with a PE2 structure containing RTΔRnH to target PDE6B Y347X, which achieved effective genome editing in vivo, and verified the feasibility of the design.
Off-target effects are highly important considerations in genome editing. Herein, we applied AID-seq and PE-tag to experimentally predict potential off-target sites. The PE system induced rare off-target events, as evaluated via AID-seq and subsequent deep sequencing analysis. The results from in vitro PE-tag on gDNA of rd1 mice also demonstrated that off-target sites underwent low prime editing activity. It is noteworthy that the potential off-target events in human genome could not be deduced from those in mouse genome. The off-target rates across species needs to be re-evaluated in the future.
Our study has several limitations. First, we provided pan-PE validation in vitro and the updated PE systems exhibited great performances. Considering the larger sizes of PE5 and PE5max than PE3, lack of in vivo evidence of PE6 and PE7 in treating ocular disease none but only mouse brain editing25, while numerous safe and efficient PE3-based in vivo ocular studies3,34, therefore, we selected PE3 system in vivo. Nevertheless, future studies could further investigate the efficiency of state-of-art PE systems in vivo. In addition, although we used RTΔRnH to reduce the size of the PE system, it still exceeds optimal loading capacity of AAV. Therefore, a minimized PE in a single AAV is expected to develop in the future to reduce viral load in vivo. Second, the AAV serotype could be engineered to have greater tropism for the retina, and even with photoreceptors, to further promote delivery efficiency. Additionally, through the analysis of scRNA-seq data of both human and mouse, PDE6B/Pde6b gene was found mainly expressed in photoreceptor cells, rods in particular (Supplementary Fig. 10a–b; g–h). The distribution of PDE6B/Pde6b gene was consistent with the classical markers of photoreceptors (Supplementary Fig. 10c–f; i–l). To specifically correct mutant genome sites in photoreceptors, it would be better to utilize photoreceptor-specific promoters like rhodopsin kinase (RK) promoter42 to facilitate the expression of the transgene of interest into photoreceptors. Moreover, programmable protein delivery with a bacterial contractile injection system has been used to deliver Cas9 and base editors and may also have potential in prime editing applications43. Lately, the phage-assisted continuous evolution enhances prime-editing-assisted site-specific integrase gene editing (PASSIGE) system and yields eePASSIGE, allowing precise large gene integration exceeding 10 kilobases44,45. Given the diverse spectrum of functional mutations underlying retinitis pigmentosa pathology, targeted genomic integration of functional gene sequences or their cDNA counterparts at native chromosomal locations may represent a universal therapeutic approach capable of addressing multiple disease-causing genetic variants. To date, PASSAGE and eePASSIGE have not been reported in vivo. Further investigation is required for in vivo PASSIGE delivery by viral and non-viral vectors due to its large size and the need to mitigate potential cellular toxicity and preventing activation of innate immune responses triggered by exogenous genetic material in primary cell cultures.
The many advantages of the eye bring it to the forefront of gene therapy, such as its immune-privileged environment without systemic leakage, the low doses needed for delivery vectors and the various administration methods available for reaching targeted ocular tissues. Extensive preclinical investigations in ocular gene-based interventions have demonstrated significant therapeutic potential, with multiple successful transitions from experimental models to clinical-stage human studies46,47. Therefore, prime editing-mediated treatments are anticipated to be applied to the prosperous gene therapy field, in which one injection can achieve lifelong benefits.
Methods
Ethics statement
The study did not involve human data. All animal procedures were approved by the Institutional Review Board of Shanghai Jiao Tong University and conducted in accordance with the animal policies of Shanghai Jiao Tong University and the guidelines established by the National Health and Family Planning Commission of China.
Evaluation and estimation of the genetic background of RP
Retinitis pigmentosa can be categorized into autosomal-recessive RP (arRP) (approximately 40% of cases), autosomal-dominant RP (adRP) (25%), or X-linked RP (15%)6,8,9,10,11,12, wherein arRP encompasses syndromic and systemic RP, such as Usher syndrome and Bardet–Biedl syndrome6. Our estimation of the proportions of causative genes for arRP was based on data from the following articles: RP25 (EYS)12,48,49, USH2A6,7,48,49,50, PDE6B48,49, PDE6A49, ABCA47,49, RPE6548,51, and CNGA17,48. The frequency may vary depending on the geographic region.
pegRNA and nsgRNA design
Three online primer editing tools, namely, PE-Designer20 (http://www.rgenome.net/pe-designer/), pegIT21 (https://pegit.giehmlab.dk), and PrimeDesign22 (https://primedesign.pinellolab.partners.org/), were used to design pegRNAs and nsRNAs. We randomly selected pegRNAs among the provided candidates according to the manufacturer’s guidelines that appropriate GC content (40–60%), recommended length of the PBS (8–15 nt) and of the RT sequence (10–74 nt)23. Moreover, the websites provided probable mismatches in the whole genome of each pegRNA to assist our decision-making. The nsgRNAs were selected upstream or downstream of the pegRNA-guided nick23. The corresponding sequences of the pegRNAs and nsgRNAs are shown in Fig. 1c and Supplementary Data 1.
Plasmid construction
The cloning of the pegRNA and nsgRNA plasmids was performed according to previously published protocols23. Briefly, the pU6-pegRNA-GG-Vector (Addgene #132777) was digested overnight with BsaI-HFv2 (New England BioLabs, NEB Cat# R3733S) to isolate the 2.2 kb fragment. Oligonucleotide duplexes containing the desired spacer, scaffold and 3’ extension sequences of the pegRNA were ordered and annealed. Then, the duplexes were phosphorylated with T4 PNK and ligated to the linearized vector using Golden Gate assembly with BsaI-HFv2 (NEB) and T4 DNA ligase (NEB Cat# M0202S) in a protocol of 12 cycles of 5 min at 16 °C and 5 min at 37 °C. To construct nsgRNA-expressing plasmids, complementary oligos representing the target sequences were annealed, cloned, and inserted into the BsmBI-digested vector BPK1520 (Addgene #65777).
The in vitro PDE6B-GFP reporter plasmid contained coding sequences of the Pde6b gene (including exon 7) in the wild type or mutant type, followed by sequences encoding FLAG-tag and green fluorescent protein (GFP). The mScarlet element was also constructed in this plasmid with an independent promoter. Details of the in vitro PDE6B-GFP reporter system sequences are provided in Supplementary Data 4.
Cell culture and transfection
Human embryonic kidney 293 T (HEK293T) cells (American Type Culture Collection, ATCC, Cat# CRL-3216) were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Cat# 11965092) supplemented with 10% fetal bovine serum (FBS; Gibco, Cat# 37520). The seeding density of HEK293 cells was about 4 × 105 cells/ well of 6-wells plate. Cells were transfected at approximately 60% confluency. The cells were co-transfected via Lipofectamine 2000 (Invitrogen, Cat#11668500) using the manufacturer’s suggested protocol with 2000 ng of prime editor plasmid, 600 ng of pegRNA plasmid, 200 ng of nsgRNA plasmid and 1000 ng of plasmid from the reporter system containing the WT/mut template and the mScarlet element simultaneously. Six hours later, the transfection medium was replaced with fresh regular culture medium. Two days later, the cells were collected for immunocytochemistry (ICC), Western blotting, RT‒qPCR, flow cytometry and genomic DNA extraction.
Flow cytometry
Flow cytometry was performed to quantify cells with EGFP-positive signals to estimate the in vitro primer editing efficiency of the reporter system. In brief, after two days of transfection with the PE plasmid, HEK293T cells were washed with PBS (pH 7.4), trypsinized and harvested in a microcentrifuge tube. Then, the cells were centrifuged and resuspended in PBS containing 0.5% BSA and 2 mM EDTA (pH 8.0). A BD LSR II flow cytometer (BD Biosciences) was used to conduct flow cytometry using mock-transduced cells as controls for establishing background fluorescence thresholds. FlowJo 10.5.0 software (Tree Star) was used for the data analysis.
Quantitative real-time PCR
Total RNA was extracted from cells with an EZpress RNA purification kit (EZBioscience, Cat#B0004) according to the manufacturer’s protocol. The RNA was eluted from the columns with RNase-free water and quantified by Nanodrop. Next, cDNA was generated with PrimeScript™ RT Master Mix (Takara, Cat# RR036A) according to the manufacturer’s instructions. Quantitative real-time PCR was performed using SYBR Green PCR master mix (Thermo Fisher Scientific Cat# 4309155) and an RT‒qPCR system (Applied Biosystems, Irvine, CA, USA). Gene expression levels were normalized to GAPDH levels. The primers used for real-time PCR are listed in Supplementary Data 2.
High-throughput DNA sequencing
Genomic DNA was extracted from cells or mouse tissues with a TIANamp Genomic DNA Kit (Tiangen Cat# 4992254) according to the manufacturer’s protocol. The DNA concentration was measured using a Nanodrop (Thermo Fisher Scientific). Q5 high-fidelity polymerase (NEB, Cat# M0491S) was used to amplify the genomic region of interest (98 °C, 10 s; 60 °C 20 s; 72 °C 30 s) for 30 cycles. The resulting amplicons were purified with AMPure XP beads (Beckman Coulter Cat# B23318) and subsequently subjected to PCR barcoding using Illumina tag-specific primer pairs with unique sequence combinations for demultiplexing and sample identification (The barcoding reactions were carried out as follows: 98 °C, 10 s; 60 °C 20 s; 72 °C 30 s for 10 cycles). Libraries were quantified and subjected to paired-end read sequencing using the Illumina MiniSeq platform. For sequence analysis, alignment of the raw reads to reference sequences was performed using CRISPResso2 (http://crispresso.pinellolab.org/). The prime editing efficiency was calculated as the percentage (number of reads with the desired allele editing/number of total aligned reads). The results were loaded into GraphPad Prism 8.4 for data visualization. The primers used for amplicon sequencing are shown in Supplementary Data 3.
AID-seq analysis
Potential off-target sites were experimentally identified using AID-seq35. Purified unedited genomic DNA of rd1 mice (2 µg) was sheared with a Bioruptor Pico (Diagenode) to an average length of 500 bp, end-repaired, A-tailed and ligated to a self-annealed hairpin i7 adaptor. Adaptor-ligated DNA was treated with a cocktail of 40 U of Lambda Exonuclease (NEB), 40 U of E. coli Exonuclease I (NEB) and 100 U of E. coli Exonuclease III (NEB) at 37 °C for 2 hours to eliminate linear and single-tailed DNA. Then, in vitro cleavage reactions were performed in a 50 μL volume. Notably, the gRNA was incubated for 2 min at 95 °C and allowed to equilibrate to room temperature before being incubated with SpCas9. It was recommended that the samples be incubated at 37 °C for 2 hours to ensure low-frequency off-target detection. After incubation, the reaction was terminated by incubation with Proteinase K (NEB) at 37 °C for 15 min. Digested genomic DNA was purified with 1x DNA Clean Beads (Vazyme) and then A-tailed and ligated to a preannealed biotin i5 adaptor. The dual adaptor-ligated DNA was then enriched with 25 μL Invitrogen Dynabeads MyOne Streptavidin T1 beads, followed by USER enzyme digestion to linearize the hairpin adaptor. Finally, two rounds of nested PCR amplification were performed using KAPA HiFi HotStart ReadyMix to recover DNA from the streptavidin beads and to add indices for sequencing. The completed libraries were quantified and sequenced on the HiSeq 2500 platform. The predicted off-target sites were then PCR amplified and detected via high-throughput sequencing using genomic DNA extracted from mice treated with primer-editing components. CRISPResso2 was used to analyse the indel frequencies at off-target sites. Top sites captured by AID-seq are shown in Fig. 4b–d. The primers used for off-target sequencing are shown in Supplementary Data 3.
PE-tag
PE-tag was conducted according to the published procedures36. The pegRNA was incubated with 200 pmol prime editor protein in 15 μl PBS for 20 min at room temperature to form PE2 RNP. Then the complex was mixed with the reaction buffer (10 mM dNTP, 5% glycerol, 100 mM KCl, 10 mM HEPES, pH 7.5, 0.2 mM EDTA, 3 mM MgCl2 and 5 mM DTT final concentration) and 2 μg purified rd1 gDNA in a total volume of 30 μl for 8 h at 37 °C. Then the mixture was treated with 10 μl RNase A (50 μg ml–1) to remove the pegRNA, and the PE2 RNP-treated gDNA was purified using a DNeasy Blood & Tissue Kit (Qiagen). The Tn5 tagmentation and library preparation were then performed on purified gDNA. Completed libraries were quantified and sequenced with 150-bp paired-end reads on an Illumina MiniSeq instrument. The tag sequence was TAGCAGAAAGTGG. Off-target sites captured by PE-tag are shown in Supplementary Fig. 8b. The primers used for off-target sequencing are shown in Supplementary Data 3.
Western blotting
Tissues were crushed using a liquid nitrogen–frozen crushing apparatus and then centrifuged for 10 min at 4 °C. The resulting supernatants were collected and stored at −80 °C for further assays. Tissues or cells were lysed in Laemmli buffer (8.0% glycerol, 3% SDS and 200 mM Tris–HCl [pH 6.8]). Then, BCA assays were performed to measure the protein concentrations. Equal amounts of proteins were separated by SDS‒polyacrylamide gel electrophoresis (SDS‒PAGE), and the separated proteins were transferred to 45-m polyvinylidene difluoride (PVDF) membranes (Merck Millipore; IPVH00010) using a Trans-Blot® Turbo machine (Bio-Rad) according to the manufacturer’s protocol. The membranes were subsequently blocked with 5% milk dissolved in Tris-buffered saline with Tween 20, followed by incubation with primary antibody at 4 °C overnight (anti-PDE6B, Invitrogen, Car# PA1-722; anti-FLAG, Sigma-Aldrich, Car# F1804; anti-β-Actin, Cell Signaling Technology, Car# 3700S). Then, the membranes were washed with TBST, and the antigens were probed at RT for 2 h with fluorophore-conjugated secondary antibodies raised against mouse IgG (Cell Signaling Technology; Car#5470S) or rabbit IgG (Cell Signaling Technology; Car#5151S). The band signals were visualized using an Odyssey Infrared Imaging System (LI-COR, USA).
Immunostaining
For immunocytochemistry, cells were fixed with 4% PFA for 15 min at RT and then washed with PBS three times before being permeabilized in 0.5% Triton X-100 (Sigma Aldrich, Cat# T8787) in TBS (pH 7.6) for 15 min at RT. Next, the cells were blocked with 10% bovine serum albumin (Gibco) in PBS for 30 min at RT. Nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI; Sigma‒Aldrich) or hoechst (Sigma‒Aldrich). Digital images were obtained with a ZEISS Axio Scope A1 upright microscope. Relative GFP ratio was calculated as GFP/mScarlet signals via ImageJ software, which was then standardized by WT group.
For immunohistochemistry, the rd1 mice were euthanized 4 weeks after subretinal injection. Enucleated eyes were fixed in 4% paraformaldehyde overnight at 4 °C, dehydrated through an ethanol series and xylene, and then embedded in paraffin. The retinal sections were incubated in blocking buffer for 1 h at room temperature and then incubated with primary anti-rhodopsin antibody (Abcam, Car# ab221664,1:500) or anti-Cas9 antibody (Cell Signaling Technology, Car#65832) at 4 °C overnight. After washing with PBS three times, the sections were incubated with Alexa Fluor 594-conjugated goat anti-rabbit secondary antibody (Invitrogen, Car# A-11012,1:500) for 2 h at room temperature. Nuclei were stained using DAPI (Sigma‒Aldrich, Cat# D9542,1:10000), and sections were mounted with Canada balsam (Sigma‒Aldrich). Images were viewed under an Olympus IX83 microscope.
Animals
The rd1 mice (GemPharmatech, Strain ID T049868) were housed and maintained under a 12-hour:12-hour light: dark cycle. AAV subretinal injection was performed on one-week-old rd1 mice. C57BL/6 mice, littermates of the rd1 mice, were also maintained under a 12 h:12 h light: dark cycle. The temperature ranged from 23–25 °C, and the humidity from 40% to 60%. The animals were assigned to experimental groups according to genotype. The n number of each group was at least 6. The sex of mice was not taken into account in the study design. All animal experiments were authorized by the Shanghai Jiao Tong University Animal Care and Use Committee and conducted in accordance with the animal policies of Shanghai Jiao Tong University and the guidelines established by the National Health and Family Planning Commission of China.
AAV vector production
AAV2-PE-N contained epegRNA, sgRNA and the amino terminus (encoding amino acids 1-713) of nCas9, whereas AAV2-PE-C contained the carboxy-terminal half (encoding amino acids 714-1368) packed with RTΔRnH. The detailed dual-AAV PE sequences are listed in Supplementary Data 5. The dual-AAV virus used was packaged and produced by PackGene Biotech. In brief, HEK293T cells were co-transfected with plasmids for Rep/Cap (2/2) and the desired genes along with the Helper plasmid. After transfection for 48 h, the cells were harvested and lysed with lysis buffer. Then, the benzonase (Sigma‒Aldrich)-treated supernatants were ultracentrifuged in an iodixanol gradient for purification. Purified AAV vectors were subsequently concentrated with a Millipore Amicon filter unit (UFC910008, 100 kDa) to a final titre of 1 × 1013 vg/ml.
eVLPs production
The eVLPs were produced according to the published procedures34. In detail, for production of eVLPs, plasmids expressing VSV-G (800 ng), wild-type MMLV Gag-Pol (5626 ng), Gag-MCP-Pol (2250 ng), Gag-PE (1126 ng) and MS2-guide RNA (7040 ng MS2-epegRNA and 1760 ng MS2-ngRNA for v3 PE3-eVLPs) were co-transfected to each 10 cm dish. The eVLPs were concentrated at approximately 4.2 × 1013 eVLPs ml−1.The epegRNA and nsgRNA contents of eVLPs-PE were quantified by RT-qPCR. The detailed eVLPs-PE sequences are listed in Supplementary Data 6.
H&E staining
Enucleated eyes were fixed in 4% paraformaldehyde overnight at 4 °C, dehydrated through an ethanol series and xylene, and then embedded in paraffin. The paraffin-embedded retinas were sectioned at a thickness of 5 μm with a Leica microtome. After deparaffinization and rehydration, the retinal sections were stained with H&E and mounted with Canada balsam (Sigma‒Aldrich). Images were captured with an Olympus IX83 microscope.
Subretinal injection
Before injection, deep anesthesia was induced by intraperitoneal injection of a Zoletil and xylazine anesthetic cocktail (Zoletil 50, 1 ml; xylazine, 0.05 ml; and saline, 18.95 ml). For AAV injection, AAV2-PE-N term (1 × 1013 virus genomes (vg ml-1)) and AAV2-PE-C term (1 × 1013 vg ml-1) (Fig. 3a) were 1:1 mixed in a total volume of 1 μl and injected into the subretinal space of the mouse eye. For eVLPs injection, each eye was injected with 1 μl of eVLPs-PE (containing approximately 4.2 × 1010 eVLPs). In detail, a sterile and bevelled 30-gauge needle was used to make an incision. Next, a microsyringe equipped with a 32-gauge blunt-end needle (Hamilton) was passed through the retina to achieve subretinal injection.
RNA sequencing
The eyes of wild-type, rd1 and PE-treated rd1 mice were collected for RNA sequencing. In brief, total RNA was extracted from cells with an EZpress RNA purification kit (B0004) according to the manufacturer’s protocol. Libraries were then constructed from 100 ng of total RNA using an Illumina TruSeq RNA sample prep kit (San Diego, CA, USA) and sequenced using an Illumina HiSeq 2500 platform (San Diego, CA, USA). We identified the mRNA levels of the unigenes with TopHat v2.0.9 and Cufflinks and normalized them to the fragments per kilobase of exon model per million mapped reads (FPKM) values. The criteria of |log2(fold change)|>1 and p < 0.05 were used to identify DEGs.
scRNA-seq data analysis
The scRNA-seq data (10x Genomics) were collected from the Gene Expression Omnibus (GEO) database, under accession number GSE13222952 and GSE11610653. The data processing and analysis were performed using the Seurat v5 R package54. All functions were executed with default parameters unless otherwise stated. The scRNA-seq data for all retina samples were first integrated using the merge function, and low-quality cells ( < 200 genes/cell or >5000 genes/cell and >10% mitochondrial genes) were removed. The potential batch effect between samples was corrected using the Harmony method55. Principal component analysis was used to reduce dimensionality, and the top 20 major components were utilized to identify distinct groups of cells using the graph-based clustering method with the FindClusters function (resolution = 0.5). Major cell clusters were annotated using known cell-type marker genes and visualized using a Uniform Manifold Approximation and Projection (UMAP) scatter plot. Mouse rod cells were annotated by Cnga1, Rho and Nrl56. Human photoreceptor cells were annotated by NRL, CNGA1 and GNGT257. Differentially expressed genes (DEGs) between each cell type and all other cell types were identified using the FindAllMarker function in Seurat, and significance was determined using the Wilcoxon rank sum test with Bonferroni correction. Genes were selected as DEGs based on the threshold of |logFC| > 0.5 and an adjusted P value < 0.05.
Electroretinography
ERG was performed on WT, rd1 and PE-treated mice at 4 weeks post suggestion. Before conducting electroretinography, mice were kept in the dark overnight. Then mice were deeply anesthetized by intraperitoneal injection of the Zoletil and xylazine anesthetic cocktail. Pupils of mice were topically dilated with tropicamide eyedrops and anesthetized with lidocaine under dim red illumination. ERGs (RetiMINER-C, IRC) were recorded from the corneal surface using a pair of gold ring electrodes, a ground electrode placed in the tail and a reference electrode in the anterior scalp between the eyes. A drop of Genteal Gel (Alcon) was applied on the cornea to keep it moistened. For scotopic ERG, the eyes were stimulated with a white flash of increasing light intensity. All data were analyzed via RetiMINER4.0. The a-wave amplitudes were measured from the baseline to the lowest negative-going voltage and the b-wave amplitudes were measured from the a-wave trough to the highest peak of the positive b-wave.
Optical coherence tomography
Optical coherence tomography (OCT) images were acquired with a Spectralis HRA + OCT (Heidelberg engineering, Heidelberg, Germany), and a 30-angle lens was used to determine the in vivo retinal thickness. Retinal and ONL thicknesses were observed, and images were captured using OCT system software.
Pupillary light reflex
The rd1 mice were maintained on 12:12-hour light/dark cycles. After 2 hours of dark adaptation, the pupils of unanaesthetized mice were imaged under an infrared digital camera (Sony) as the initial state. Then, the mice were subjected to light exposure for 10 s at an intensity of 200 lx and imaged again to record the consensual pupil response. The pupil area for each eye was measured before and after light exposure with ImageJ software, which was then calibrated with the square of the distance between the inner and outer canthus of the same eye. The constriction rate of the pupil was represented by the difference between the pupil area measured in the dark and in the light.
Visual cliff test
The visual cliff test was performed to evaluate visual function58, with slight modifications. An open-topped box (60 × 60 cm square, 15 cm high; Fig. 6e) was placed on the edge of a laboratory bench, with half of the box suspended over the bench edge 90 cm above the floor (‘cliff side’), whereas the other half was on the bench (‘bench side’). The position created a visual and artificial cliff rather than an actual drop-off. A checkerboard pattern with black and white squares was set on the bottom of the bench side as well as on the floor underneath the box to emphasize the cliff. The bench and cliff sides of the box were homogenously illuminated. During the test, a mouse was placed in the centre of the box and allowed to move freely through the entire open field for 5 minutes. Its behavior and movement trajectory were recorded by a video camera and subsequently analysed by ANY maze software (Global Biotech Inc., Shanghai, China). The cumulative time spent on the cliff by each mouse was also recorded.
Light-dark box test
A 26 × 26 × 26 cm box consisting of equally sized light and dark chambers was used in this test. The two chambers were connected by a 4 × 5 cm opening through which the mice could move freely. The bright half of the box was illuminated from above by white fluorescent light with an intensity of 200 lx measured at floor level. When placed in the middle of the box, the rd1 mice were allowed to adapt to the environment for 2 min and then tested for another 5 min. The total time spent in the dark half was recorded. After each trial, the testing apparatus was cleaned with 70% ethanol to remove olfactory cues.
Statistics and reproducibility
Statistical analysis was performed using GraphPad Prism (8.4) and Microsoft Excel (Professional Plus 2013) software. The statistical tests and sample numbers used for each experiment are stated in the corresponding figure legends. Unless otherwise stated, quantitative data are presented as the means ± standard deviations (SDs). Statistical differences between different experimental groups were typically analysed by Student’s t test, one-way ANOVA, or two-way ANOVA with Tukey’s multiple comparisons. P values < 0.05 were considered to indicate statistical significance. Otherwise stated, western blotting was performed at least three times and similar results were observed.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data needed to support the conclusions are presented in the paper or the Supplementary Information. RNA-seq data have been deposited into the database have been deposited in the Genome Sequence Archive (GSA)59 (https://ngdc.cncb.ac.cn/gsa.) (accession number: CRA016583). Deep sequencing data have also been deposited into the GSA database (Accession number: In vitro prime editing: HRA007463, HRA008939; In vivo prime editing: CRA016585, CRA019625; Off-target verification from AID-seq: CRA016586, CRA019627; AID-seq: peg4: CRA016582, nsg4: CRA016595; Off-target verification from PE-tag: CRA021786; PE-tag: CRA021006). Source data are provided with this paper.
References
Anzalone, A. V. et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019).
Fu, Y. et al. Prime editing: current advances and therapeutic opportunities in human diseases. Sci. Bull. 68, 3278–3291 (2023).
Jang, H. et al. Application of prime editing to the correction of mutations and phenotypes in adult mice with liver and eye diseases. Nat Biomed Eng, https://doi.org/10.1038/s41551-021-00788-9 (2021).
Jiang, T., Zhang, X. O., Weng, Z. & Xue, W. Deletion and replacement of long genomic sequences using prime editing. Nat. Biotechnol. 40, 227–234 (2022).
Happi Mbakam, C. et al. Prime editing optimized RTT permits the correction of the c.8713C>T mutation in DMD gene. Mol. Ther. Nucleic Acids 30, 272–285 (2022).
Hartong, D. T., Berson, E. L. & Dryja, T. P. Retinitis pigmentosa. Lancet 368, 1795–1809 (2006).
Dias, M. F. et al. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Prog. Retinal Eye Res. 63, 107–131 (2018).
Bird, A. C. Retinal photoreceptor dystrophies LI. Edward Jackson Memorial Lecture. Am. J. Ophthalmol. 119, 543–562 (1995).
Ferrari, S. et al. Retinitis pigmentosa: genes and disease mechanisms. Curr. Genomics 12, 238–249 (2011).
Martin-Merida, I. et al. Genomic Landscape of Sporadic Retinitis Pigmentosa: Findings from 877 Spanish Cases. Ophthalmology 126, 1181–1188 (2019).
Bravo-Gil, N. et al. Unravelling the genetic basis of simplex Retinitis Pigmentosa cases. Sci. Rep. 7, 41937 (2017).
Barragan, I., Marcos, I., Borrego, S. & Antiñolo, G. Mutation screening of three candidate genes, ELOVL5, SMAP1 and GLULD1 in autosomal recessive retinitis pigmentosa. Int J. Mol. Med 16, 1163–1167 (2005).
Appell, M. B. et al. Next generation therapeutics for retinal neurodegenerative diseases. J. Controlled Release 367, 708–736 (2024).
Pennesi, M. E. et al. Long-Term Characterization of Retinal Degeneration inrd1andrd10Mice Using Spectral Domain Optical Coherence Tomography. Investigative Opthalmology & Visual Science 53, https://doi.org/10.1167/iovs.12-9611 (2012).
Nishiguchi, K. M. et al. Gene therapy restores vision in rd1 mice after removal of a confounding mutation in Gpr179. Nat. Commun. 6, https://doi.org/10.1038/ncomms7006 (2015).
Georgiou, M. et al. Phenotyping and genotyping inherited retinal diseases: Molecular genetics, clinical and imaging features, and therapeutics of macular dystrophies, cone and cone-rod dystrophies, rod-cone dystrophies, Leber congenital amaurosis, and cone dysfunction syndromes. Progress in Retinal and Eye Res. 100, https://doi.org/10.1016/j.preteyeres.2024.101244 (2024).
Wu, W.-H. et al. CRISPR Repair Reveals Causative Mutation in a Preclinical Model of Retinitis Pigmentosa. Mol. Ther. 24, 1388–1394 (2016).
Cai, Y. et al. In vivo genome editing rescues photoreceptor degeneration via a Cas9/RecA-mediated homology-directed repair pathway. Sci. Adv. 5, eaav3335 (2019).
Wu, Y. et al. AAV-mediated base-editing therapy ameliorates the disease phenotypes in a mouse model of retinitis pigmentosa. Nat. Commun. 14 https://doi.org/10.1038/s41467-023-40655-6 (2023).
Hwang, G. H. et al. PE-Designer and PE-Analyzer: web-based design and analysis tools for CRISPR prime editing. Nucleic Acids Res 49, W499–W504 (2021).
Anderson, M. V., Haldrup, J., Thomsen, E. A., Wolff, J. H. & Mikkelsen, J. G. pegIT - a web-based design tool for prime editing. Nucleic Acids Res 49, W505–W509 (2021).
Hsu, J. Y. et al. PrimeDesign software for rapid and simplified design of prime editing guide RNAs. Nat. Commun. 12, 1034 (2021).
Doman, J. L., Sousa, A. A., Randolph, P. B., Chen, P. J. & Liu, D. R. Designing and executing prime editing experiments in mammalian cells. Nat. Protoc. 17, 2431–2468 (2022).
Trojan, J. et al. Functional analysis of hMLH1 variants and HNPCC-related mutations using a human expression system. Gastroenterology 122, 211–219 (2002).
Doman, J. L. et al. Phage-assisted evolution and protein engineering yield compact, efficient prime editors. Cell 186, 3983–4002 e3926 (2023).
Yan, J. et al. Improving prime editing with an endogenous small RNA-binding protein. Nature 628, 639–647 (2024).
He, X. et al. AAV for Gene Therapy in Ocular Diseases: Progress and Prospects. Res. (Wash. D. C.) 6, 0291 (2023).
Zhi, S. et al. Dual-AAV delivering split prime editor system for in vivo genome editing. Mol. Ther. 30, 283–294 (2022).
Liu, P. et al. Improved prime editors enable pathogenic allele correction and cancer modelling in adult mice. Nat. Commun. 12, 2121 (2021).
Qin, H. et al. Vision rescue via unconstrained in vivo prime editing in degenerating neural retinas. J. Exp. Med. 220 https://doi.org/10.1084/jem.20220776 (2023).
Nelson, J. W. et al. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 40, 402–410 (2022).
Bock, D. et al. In vivo prime editing of a metabolic liver disease in mice. Sci. Transl. Med 14, eabl9238 (2022).
Gao, Z. et al. A truncated reverse transcriptase enhances prime editing by split AAV vectors. Mol. Ther. 30, 2942–2951 (2022).
An, M. et al. Engineered virus-like particles for transient delivery of prime editor ribonucleoprotein complexes in vivo. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-02078-y (2024).
Tian, R. et al. Massively parallel CRISPR off-target detection enables rapid off-target prediction model building. Med 4, 478–492 e476 (2023).
Liang, S. Q. et al. Genome-wide profiling of prime editor off-target sites in vitro and in vivo using PE-tag. Nat. Methods 20, 898–907 (2023).
den Hollander, A. I. et al. A homozygous missense mutation in the IRBP gene (RBP3) associated with autosomal recessive retinitis pigmentosa. Invest Ophthalmol. Vis. Sci. 50, 1864–1872 (2009).
Pearson, R. A. et al. Restoration of vision after transplantation of photoreceptors. Nature 485, 99–103 (2012).
Piri, N., Kwong, J. M. & Caprioli, J. Crystallins in retinal ganglion cell survival and regeneration. Mol. Neurobiol. 48, 819–828 (2013).
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016).
Farrar, G. J., Kenna, P. F. & Humphries, P. On the genetics of retinitis pigmentosa and on mutation-independent approaches to therapeutic intervention. EMBO J. 21, 857–864 (2002).
Fischer, M. D. et al. Codon-Optimized RPGR Improves Stability and Efficacy of AAV8 Gene Therapy in Two Mouse Models of X-Linked Retinitis Pigmentosa. Mol. Ther. 25, 1854–1865 (2017).
Kreitz, J. et al. Programmable protein delivery with a bacterial contractile injection system. Nature https://doi.org/10.1038/s41586-023-05870-7 (2023).
Pandey, S. et al. Efficient site-specific integration of large genes in mammalian cells via continuously evolved recombinases and prime editing. Nat. Biomed. Eng. https://doi.org/10.1038/s41551-024-01227-1 (2024).
Anzalone, A. V. et al. Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing. Nat. Biotechnol. 40, 731–740 (2022).
Wang, J. H. et al. Gene therapy for diabetic retinopathy: Are we ready to make the leap from bench to bedside? Pharm. Ther. 173, 1–18 (2017).
Meng, X. et al. In vivo genome editing via CRISPR/Cas9-mediated homology-independent targeted integration for Bietti crystalline corneoretinal dystrophy treatment. Nat. Commun. 15, 3773 (2024).
Oishi, M. et al. Comprehensive molecular diagnosis of a large cohort of Japanese retinitis pigmentosa and Usher syndrome patients by next-generation sequencing. Invest Ophthalmol. Vis. Sci. 55, 7369–7375 (2014).
Kim, M. S. et al. Genetic Mutation Profiles in Korean Patients with Inherited Retinal Diseases. J. Korean Med Sci. 34, e161 (2019).
Reurink, J. et al. Molecular Inversion Probe-Based Sequencing of USH2A Exons and Splice Sites as a Cost-Effective Screening Tool in USH2 and arRP Cases. Int. J. Mol. Sci. 22, https://doi.org/10.3390/ijms22126419 (2021).
Morimura, H. et al. Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or leber congenital amaurosis. Proc. Natl Acad. Sci. USA 95, 3088–3093 (1998).
Heng, J. S. et al. Comprehensive analysis of a mouse model of spontaneous uveoretinitis using single-cell RNA sequencing. Proc. Natl Acad. Sci. USA 116, 26734–26744 (2019).
Lu, Y. et al. Single-Cell Analysis of Human Retina Identifies Evolutionarily Conserved and Species-Specific Mechanisms Controlling Development. Dev. Cell 53, 473–491.e479 (2020).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e3529 (2021).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Choi, J. et al. Spatial organization of the mouse retina at single cell resolution by MERFISH. Nat. Commun. 14, 4929 (2023).
Cowan, C. S. et al. Cell Types of the Human Retina and Its Organoids at Single-Cell Resolution. Cell 182, 1623–1640.e1634 (2020).
Glynn, D., Bortnick, R. A. & Morton, A. J. Complexin II is essential for normal neurological function in mice. Hum. Mol. Genet 12, 2431–2448 (2003).
Chen, T. et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genom. Proteomics Bioinform., https://doi.org/10.1016/j.gpb.2021.08.001 (2021).
Xu, Y. https://BioRender.com/b56h536 (2025).
Xu, Y. https://BioRender.com/u25j054 (2025).
Xu, Y. https://BioRender.com/m81c419 (2025).
Xu, Y. https://BioRender.com/v75y612 (2025).
Xu, Y. https://BioRender.com/z32s281 (2025).
Xu, Y. https://BioRender.com/r72e953 (2025).
Acknowledgements
The current study was supported by the National Natural Science Foundation of China (82200961 to Z.Y.), the National Basic Science Center Program of China (82388101 to X.Q.F.), the Science and Technology Commission of Shanghai (20DZ2270800 to X.Q.F.), Shanghai Key Laboratory of Orbital Diseases and Ocular Oncology (2022SKLE-KFKT004 to Z.Y.), and the China Postdoctoral Science Foundation (2022M720091 to Z.Y., 2024T170566 to Z.Y.). We thank Professor Jihong Wu, Qian Li, and Xuejin Zhang from Eye & ENT Hospital of Fudan University for equipment and technical guidance of ERG, OCT, and fundus camera.
Author information
Authors and Affiliations
Contributions
X.Q.F. and Z.Y. designed and supervised this study. Y.D.F. and Z.Y. performed most involved experiments and drafted the original manuscript. X.Y.H. was responsible for rd1 mice behavior test. L.M. contributed to cell culture and scRNA analysis, and H.H.S. were involved in AAV intraocular injection. X.D.G. assisted the design of the experiments. P.P.L. performed PE-tag assay. X.D.G., S.F.G., R.B.J., D.R.L., P.W.C., X.Q.F and Z.Y. revised the draft. All authors discussed and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
D.R.L. is a co-founder and consultant for Prime Medicine, Beam Therapeutics, Pairwise Plants, and nChroma Bio, and owns equity in these companies. The other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Stephen Tsang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fu, Y., He, X., Ma, L. et al. In vivo prime editing rescues photoreceptor degeneration in nonsense mutant retinitis pigmentosa. Nat Commun 16, 2394 (2025). https://doi.org/10.1038/s41467-025-57628-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57628-6
This article is cited by
-
Spatial profiling of gene editing by in situ sequencing in mice and macaques
Nature Biomedical Engineering (2025)
