
Abstract
Accurate and robust prediction of drug-target interactions (DTIs) plays a vital role in drug discovery but remains challenging due to limited labeled data, cold start problems, and insufficient understanding of mechanisms of action (MoA). Distinguishing activation and inhibition mechanisms is particularly critical in clinical applications. Here, we propose DTIAM, a unified framework for predicting interactions, binding affinities, and activation/inhibition mechanisms between drugs and targets. DTIAM learns drug and target representations from large amounts of label-free data through self-supervised pre-training, which accurately extracts their substructure and contextual information, and thus benefits the downstream prediction based on these representations. DTIAM achieves substantial performance improvement over other state-of-the-art methods in all tasks, particularly in the cold start scenario. Moreover, independent validation demonstrates the strong generalization ability of DTIAM. All these results suggest that DTIAM can provide a practically useful tool for predicting novel DTIs and further distinguishing the MoA of candidate drugs.
Similar content being viewed by others

Introduction
Accurately predicting drug-target interactions (DTIs) is an essential step in drug discovery and development1,2. The biochemical experimental method for identifying new DTIs on a large scale is still expensive and time-consuming3,4,5, despite the wide application of various experimental assays in drug discovery. Various computational methods have been applied to drug discovery and successfully predict novel DTIs, and they can substantially reduce development time and costs6,7,8,9. Current computational methods mainly focus on the binary prediction of DTI or the regression prediction of drug-target binding affinity (DTA).
In binary classification-based DTI prediction studies, the goal is to predict whether there is an interaction between the drug and the target or not. Generally, the approaches for in silico DTI prediction can be divided into four major categories: structure-based approaches, ligand-based approaches, network-based approaches, and machine learning-based approaches. Structure determination of compound-protein complexes can provide insights into the mode of action and thus significantly facilitate lead compound selection and optimization in the target-based drug discovery10,11. There are many structure-based approaches, such as molecular docking12, molecular dynamics simulations13, pharmacophore modeling14 and GOLD15, which are widely applied in virtual screening of drugs binding with proteins. However, these methods generally fail to predict binding affinities when the three-dimensional (3D) structure of the target protein is unknown, and require tremendous computational resources. And ligand-based approaches, such as quantitative structure activity relationship (QSAR), compares a candidate ligand with the known ligands of a specific target protein to predict the potential interaction. However, the predictive power of such methods is limited by the number of known ligands for a specific target protein. To overcome the current limitations of the structure-based and ligand-based approaches, various computational models have been developed for DTI prediction16,17,18,19. An example is the network-based inference (NBI) methods that construct reliable networks from several data resources (e.g., chemical, genomics, proteomics, and pharmacology) and exploit the topological and structural information in the networks for potential association prediction20,21,22,23,24. For instance, Luo et al.25 develop a computational pipeline, called DTINet, to predict novel DTIs from a heterogeneous network constructed by integrating diverse drug-related information. Another promising approach for predicting DTIs is the machine learning-based methods that mainly consist of two steps: feature extraction and DTI prediction26,27,28,29. This type of approach fully exploits the latent features from input data of known drug compounds and target proteins to predict their interactions30,31. The binary classification-based DTI prediction models can successfully predict whether there is an interaction between a drug and a target and have strong performance. The drug-target interaction is not a simple binary on-off relationship, but a continuous value named binding affinity, which reflects how tightly the drug compound binds to a particular target protein32.
In order to further predict the putative strengths of the interactions, various regression-based models have been proposed to infer the binding affinities between drugs and targets32,33,34,35. Binding affinity reflects how tightly the drug binds to a particular target, which is quantified by measures such as inhibition constant (Ki), dissociation constant (Kd), and the half-maximal inhibitory concentration (IC50). The DTA prediction approaches focus on affinity scoring, which is frequently used after virtual screening and docking campaigns. Recently, deep learning methods have emerged as a successful alternative to scoring functions, employing various deep neural network architectures such as convolutional neural network (CNN) and recurrent neural network (RNN). These methods fully extract contextual features and learn the representations of drugs and targets from the input raw data for DTA prediction. For example, DeepDTA36 proposed by Öztürk et al. used CNN to learn representations from the simplified molecular-input line-entry system (SMILES) strings of compounds and amino acid sequences of proteins, and fed into fully connected layers to predict their affinities. Karimi et al.37 presented a semi-supervised deep learning model, named DeepAffinity, which unifies RNN and CNN to jointly encode molecular and protein representations and predict affinities. Although these methods can successfully predict the binding affinity between drugs and targets, their interpretability remains limited. The attention mechanism has therefore been applied to increase the interpretability of the model by assigning greater weights to the “important” features38,39,40. As an example, Li et al.41 developed a multi-objective neural network called MONN, which uses non-covalent interactions as additional supervision information to guide the model to capture the key binding sites.
While much effort has been devoted to predicting DTI and DTA, there are still several limitations in the previous studies. First, most existing methods heavily depend on the scale of the high-quality labeled data. Only large-scale labeled data can help models achieve great performance. Unfortunately, existing labeled data is insufficient, and data labeling is expensive and time-consuming. In addition, these methods often exhibit limited generalization when new drugs or targets are identified for a complicated disease, which is similar to the cold start problem in recommendation systems. More importantly, recent approaches fail to elucidate the mechanism of action (MoA) of the compound. The MoA refers to how a drug works on its target to produce the desired effects, which involve two major roles: activation and inhibition mechanisms. Distinguishing the activation and inhibition MoA between drugs and targets is critical and challenging in the drug discovery and development process, as well as their clinical applications42. It helps pharmaceutical scientists identify potential drug interactions and adverse effects, and develop safe and effective treatments for diseases43,44. For example, drugs that activate dopamine receptors can treat Parkinson’s disease, while drugs that inhibit dopamine receptors can treat psychosis45. Recently, some proteochemometrics modeling and gene expression profiles-based approaches were proposed to predict the activation and inhibition relationships between drugs and targets45,46,47,48. However, there is a manifest need for developing a more powerful deep learning model to predict and decipher MoA, which is crucial and challenging scientific research in drug development.
In this work, we develop DTIAM, a unified framework for predicting DTI, DTA, and MoA. DTIAM learns drug and target representations from a large amount of unlabeled data via multi-task self-supervised pre-training, which requires only the molecular graph of drug compounds and primary sequences of target proteins as input. It accurately extracts the substructure and contextual information from massive compound and protein data during pre-training, which improves generalization performance and provides benefits for downstream tasks. In comprehensive comparison tests across different types of tasks and under three common and realistic experiment settings (i.e., warm start, drug cold start, and target cold start), DTIAM outperforms other baseline methods in all tasks, especially in the cold start scenario. Furthermore, we successfully identify effective inhibitors of TMEM16A from a high-throughput molecular library (10 million compounds), which are verified by the whole-cell patch clamp experiment. Besides, independent validation on EFGR, CDK 4/6, and 10 specific targets indicates that DTIAM can provide a practically useful tool for predicting novel DTIs and further distinguishing the action mechanisms of potential drugs. In addition, the robustness experiments demonstrate that the representations learned by the pre-training models transfer well to downstream tasks, even with limited labeled data for training. All these results suggest that DTIAM can provide accurate representations for effectively predicting potential drug molecules or target proteins, and thus greatly facilitate the drug discovery process.
Results
Overview of DTIAM
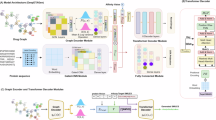
Our proposed DTIAM is a general framework used for predicting DTI, DTA, and MoA based on self-supervised learning. The overall architecture of DTIAM is illustrated in Fig. 1. DTIAM is not an end-to-end neural network model, which consists of three modules: (1) a drug molecular pre-training module based on multi-task self-supervised learning for extracting the features of both individual substructures and the whole compound from massive amounts of the molecular graph (Fig. 1A), (2) a target protein pre-training module based on Transformer attention maps for extracting the features of individual residues directly from protein sequences (Fig. 1B), and (3) a unified drug-target prediction module for predicting DTI, DTA, and MoA between the given pair of drug and target, using the previously learned features of drug molecular and target protein (Fig. 1C).

The framework mainly consists of three modules. A The drug molecular pre-training module. The module segments the molecular graph into several substructures and learns its representation through three self-supervised models from massive amounts of label-free data. B The target protein pre-training module. The module uses Transformer attention maps to learn the representations and contacts of proteins based on unsupervised language modeling from large amounts of protein sequence data. C The downstream drug-target prediction module. The module incorporates drug and target representation and predicts DTI, DTA, and MoA via an automated machine learning model.
The drug molecule pre-training module takes the molecular graph as input, which is then segmented into several substructures. The module then learns the representation of the drug molecule based on multiple self-supervised models. Specifically, for a drug molecule with n substructures, their representations are defined as a n × d embedding matrix, in which each substructure is embedded into a d-dimensional vector. These embeddings are fed into a Transformer encoder for feature extraction and learned through three self-supervised tasks: Masked Language Modeling, Molecular Descriptor Prediction, and Molecular Functional Group Prediction. The drug molecule pre-training module leverages the power of attention mechanism and self-supervised learning from vast amounts of unlabeled data to effectively extract contextual information and implicit features between molecular substructures. This process enables the module to learn meaningful representations of drug molecules without relying on explicit labels. By using the attention mechanism, the module can prioritize relevant substructures and relationships between them during training, leading to more effective feature extraction. Similarly, the target protein pre-training module uses Transformer attention maps to learn the representations and contacts of proteins based on unsupervised language modeling from large amounts of protein sequence data. The basic idea of the drug-target prediction module is to integrate information from both drugs and targets to improve the prediction of DTI, DTA, and MoA. The module combines representations of compounds and proteins to capture their complex interactions and uses various machine learning models, such as neural networks, to learn their relationship and properties for accurate and reliable predictions. These models are integrated within an automated machine learning framework that utilizes multi-layer stacking and bagging techniques. Details about each module of DTIAM and the training process can be found in the “Methods” section.
Performance of DTIAM on the DTI prediction task
In the DTI prediction task, the goal is to predict whether a given drug-target pair interacts with each other, which is a binary classification problem. We compared DTIAM with four baseline methods, including CPI_GNN49, TransformerCPI50, MPNN_CNN51, and KGE_NFM9, on the Yamanishi_08’s and Hetionet benchmark datasets under three commonly used and more realistic cross-validation settings: the warm start setting, the drug cold start setting, and the target cold start setting (Fig. 2, Supplementary Materials Table 2). The training data and test data are split via 10-fold cross-validation, and the ratio between the positive and negative samples is 1:10.

The performances of DTIAM and baseline models were evaluated in terms of AUPR and AUROC on the Yamanishi_08's and Hetionet datasets under three experiment settings. A, B AUPR of different prediction models on the Yamanishi_08's and Hetionet datasets under three experiment settings. C, D AUROC of different prediction models on the Yamanishi_08's and Hetionet datasets under three experiment settings. All results were obtained by 10-fold cross-validation. The ratio between the positive and negative samples is 1:10. Box plots show the median as the center lines, upper and lower quartiles as box limits, whiskers as maximum and minimum values, and dots represent outliers.
First, on the smaller Yamanishi_08’s dataset, we observed that DTIAM achieved higher and more robust predictive performance under three different experiment settings, especially in the cold start settings. Specifically, in the scenario of the warm start, DTIAM (AUPR = 0.931) significantly outperformed all the other baselines with a significant leading margin of 50% in terms of AUPR when compared to CPI_GNN (AUPR = 0.431). While for the end-to-end methods, TransformerCPI (AUPR = 0.816) and MPNN_CNN (AUPR = 0.802), and the network-based method KGE_NFM (AUPR = 0.817) achieved comparable predictive performance. These results indicate that the end-to-end methods and network-based methods require more labeled data, while DTIAM can partly overcome this limitation thanks to the knowledge learned in the pre-training stage. In the scenario of the cold start, we observed that the AUPR and AUROC values of all methods get reduced by different degrees, while DTIAM still achieves relatively high predictive performance, especially in the target cold start. This result highlights DTIAM’s potential capability to capture the latent features of compound substructures and protein subsequences from the large-scale unlabeled data, thus enables higher accuracy and more robust prediction even for unknown drugs or targets.
On the other hand, in the larger Hetionet dataset, we observed that DTIAM achieved the better, the best, and the second best predictive performance in the warm start, the drug cold start, and the target cold start, respectively. Specifically, in the scenario of the warm start, the average AUPR score achieved by DTIAM (AUPR = 0.808) was higher than other baseline methods. While for the network-based method, KGE_NFM (AUPR = 0.789) achieved comparable predictive performance due to the increased volume of available data. In the scenario of the drug cold start, DTIAM (AUPR = 0.529) significantly outperformed CPI_GNN (AUPR = 0.219), MPNN_CNN (AUPR = 0.453), and KGE_NFM (AUPR = 0.391). This phenomenon demonstrates the powerful expressive and feature learning ability of the proposed drug pre-training model, which provides a huge advantage for DTIAM in the situation of unknown drug prediction. In the scenario of the target cold start, KGE_NFM (AUPR = 0.651) performed better than CPI_GNN (AUPR = 0.433), MPNN_CNN (AUPR = 0.470), and DTIAM (AUPR = 0.614). This is mainly attributed to the sufficient target-related association information for the network-based method KGE_NFM. While DTIAM and the end-to-end methods only take the compound SMILES and the protein sequences as input without extra association information. Additionally, we found a similar phenomenon on the Yamanishi_08’s and Hetionet datasets that all methods achieved better predictive performance in the target cold start than the drug cold start. It seems possible that this finding is attributed to the volume of available data for targets, where both datasets have more targets than drugs. For example, there are 5763 targets while only 1384 drugs are in the Hetionet dataset (Supplementary Materials Table 1).
Performance of DTIAM on the DTA prediction task
The goal of the DTA prediction task is to predict the binding affinity between a given pair of drug targets, which is a regression task. And four baseline models were used in the performance comparison, including DeepDTA36, MONN41, BACPI40, and GraphDTA34. We evaluated our model and all the baseline methods on two benchmark datasets, the Kinase dataset Davis and KIBA dataset, under three experiment settings (Fig. 3, Supplementary Materials Table 3). For each experiment setting, we use 5-fold cross-validation to evaluate the DTA prediction performance of DTIAM and baseline methods.

The performances of DTIAM and baseline models were evaluated in terms of MSE and Pearson correlation on the Davis and KIBA datasets under three experiment settings. A, B MSE of different prediction models on the Davis and KIBA datasets under three experiment settings. C, D Pearson correlation of different prediction models on the Davis and KIBA datasets under three experiment settings. All results were obtained by 5-fold cross-validation. Box plots show the median as the center lines, upper and lower quartiles as box limits, whiskers as maximum and minimum values, and dots represent outliers.
As can be seen from Fig. 3, DTIAM achieved better predictive performance under all experimental settings on both datasets, especially in the cold start settings. For the scenario of the warm start, DTIAM and three graph-based methods, MONN, BACPI, and GraphDTA, achieved relatively high predictive performance on both datasets. While for the sequence-based method, DeepDTA did not perform as well due to the limitation of the model structure, which fails to extract accurate features from sequence information. In the scenario of the cold start, we observed a similar situation with the DTI prediction task in that the predictive performance gets reduced by different degrees for all methods. GraphDTA achieved relatively high predictive performance in the drug cold start setting, but do not perform as well in the target cold start setting. In contrast, MONN performed better in the target cold start setting than in the drug cold start setting. These results suggested that GraphDTA is more suitable for the binding affinity prediction of new drugs, while MONN is better for the situation of the cold start for targets. DeepDTA and BACPI behaved more stably in two cold start scenarios, which shows the robustness of the predictions. For the pre-training model, DTIAM performed the best in both the warm start setting and two cold start settings. All these comparative results supported the strong predictive power of DTIAM, which can successfully predict the binding affinities between drugs and targets, and has a strong generalization ability even for predictions on novel drugs or targets.
Performance of DTIAM on the MoA prediction task
To evaluate the prediction performance of DTIAM in the MoA prediction task, we approach the MoA prediction as two distinct binary classification tasks. Specifically, we aim to predict whether a given drug-target pair exhibits activatory or inhibitory effects. Thus, our two classification problems are predicting the presence of activation and inhibition for a given drug-target pair. The evaluations were conducted using two distinct types of MoA (i.e., activation and inhibition), and each type of MoA has a corresponding dataset collected from the Therapeutic Target Database. We compared DTIAM with AI-DTI48 using a 5-fold cross-validation on these two datasets under three experiment settings (Fig. 4A–D, Supplementary Materials Table 4).

A–D Performance comparison of DTIAM with AI-DTI on the Activation and Inhibition datasets under three experiment settings in terms of AUPR and AUROC. All results were obtained by 5-fold cross-validation. The ratio between the positive and negative samples is 1:10. Box plots show the median as the center lines, upper and lower quartiles as box limits, whiskers as maximum and minimum values, and dots represent outliers. E–H Validating DTIAM on 10 specific targets. E Prediction accuracy for each target, with the score at the top of the bar and the ratio inside indicating the number of correctly predicted drugs out of the total number. F–G The recall of activation and inhibition, respectively, with the top value indicating recall and the ratio inside indicating the number of correctly predicted activators or inhibitors out of the total number. H AUROC score for each target.
As shown in Fig. 4A–D, DTIAM significantly outperformed AI-DTI in all three experiment settings, on both Activation and Inhibition datasets. Specifically, on the smaller Activation dataset, DTIAM outperformed AI-DTI with 16.1%, 17.9%, and 26.8% improvement in terms of AUPR in the scenario of the warm start, the drug cold start, and the target cold start, respectively. These results indicate that the representations learned by self-supervised pre-training transfer well to downstream tasks with a small amount of labeled training data. In particular, with a larger size of labeled drug-target pairs on the Inhibition dataset, the evaluation performance of DTIAM and AI-DTI increase greatly compared to that on the Activation dataset. And DTIAM performed slightly better than AI-DTI in the scenario of the warm start and the drug cold start. While DTIAM outperformed AI-DTI with a significant leading margin of 34% in terms of AUPR when the experiment setting is the target cold start. This result demonstrated that the target protein pre-training model has the potential to learn patterns from large-scale protein amino acid sequences, and thus benefits the downstream prediction for DTIAM in the situation of the target cold start. In addition, we also observed an interesting phenomenon in the Activation and Inhibition datasets that both methods achieved better predictive performance in the drug cold start than the target cold start. This result may be explained by the fact that there are more drugs than targets in the two MoA datasets. This finding manifests the influence of the size of the predicted object in the scenario of the cold start, and a larger number of the predicted object enable better prediction performance.
Distinguishing the activation/inhibition mechanism between a drug and its target is of great biological significance because it can determine the type of biological response produced by the drug. Take the alpha-1A adrenergic receptor (ADRA1A) for example, drugs such as metaraminol activate ADRA1A for the treatment of hypotension52, whereas drugs inhibit ADRA1A used for benign prostatic hyperplasia, hypertension, schizophrenia, and bipolar disorder53,54. To demonstrate the reliability of DTIAM in distinguishing activation and inhibition interactions, we combined all activating and inhibiting DTIs to train DTIAM, which is applied to predict the activation and inhibition relationships for 10 specific targets (including ADRA1A, ADRA1B, ADRA1D, CYP3A43, CYP3A5, CYP3A7, DRD1, HTR1B, HTR1D, and HTR2A). These targets can be categorized into four distinct subfamilies: alpha-1 adrenergic receptors (ADRA1), cytochrome P450 3A enzymes (CYP3A), dopamine receptors (DR), and 5-hydroxytryptamine receptors (HTR). Each of these subfamilies possesses a considerable repertoire of known agonists and antagonists, along with distinct mechanisms of action that align with diverse therapeutic indications. The exploration of the mechanisms of interaction between these subfamilies and drugs holds paramount importance in the realms of nervous system regulation, catalytic reactions, and beyond. We collect the activation/inhibition relationships for these 10 targets from DrugBank, and all of these relationships are independent of all the training data used for DTIAM.
We list all prediction results in Supplementary Data 1 and show the results in terms of accuracy, recall of activation, recall of inhibition, and AUROC in Fig. 4E–H. We found that the prediction accuracy exceeded 93% for all targets, including 100% for HTR1D, and 9 of the targets had AUROC values above 0.96. These results suggest that DTIAM can accurately distinguish the activation and inhibition relationships between drugs and targets. In addition, we observed that the recall of activation was significantly lower than that of inhibition. This is because the samples of the dataset used in DTIAM are out of balance (far fewer samples for activation than for inhibition), which leads to a more biased prediction result of the model with inhibition.
DTIAM identifies potential inhibitors for TMEM16A
To better demonstrate the effectiveness of DTIAM, we predicted the potential inhibitors for TMEM16A from the high-throughput screening molecular library using the DTI, DTA, and MoA prediction model and outputted the list of top predictions. TMEM16A is widely expressed in various cells, including epithelial, smooth muscle, endothelial, and neuronal cells, and regulates key functions such as fluid secretion, smooth muscle contraction, gut motility, cell volume regulation, apoptosis, and pain55,56. Its dysfunction can lead to many diseases, including various cancers, gastrointestinal disorders, hypertension, and cystic fibrosis57. TMEM16A inhibitors have potential therapeutic uses in hypertension, stroke, and overactive bladder, while activators may treat epithelial dysfunction disorders like cystic fibrosis, and dry eye syndrome58. Consequently, TMEM16A represents a significant pharmacological target. Identifying potential drugs targeting TMEM16A through computational models is a challenging task with important implications for the treatment of various diseases. First, DTI prediction model of DTIAM was applied to score the compounds from the commercial off-the-shelf high-throughput screening molecular library, which contains approximately 10 million compounds, and the top 50,000 molecules (~top 0.5%, ensuring compound diversity) were selected based on predicted interaction probabilities. Second, we filtered these compounds by the Lipinski rules and filtered pan-assay interference compounds, and clustered these molecules based on their fingerprint similarities, obtaining approximately 350 clusters. Finally, 75 representative compounds were selected from the top-ranked clusters based on the predicted interaction, affinity, and inhibition scores.
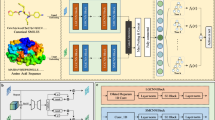
The predicted potential compounds inhibiting the TMEM16A channel are shown in Supplementary Data 2. We found that daidzein (ranked second in the predicted potential compounds) has been verified by Wang et al.59 to be a high-efficiency TMEM16A inhibitor, with an IC50 of 1.39 ± 0.59 μM, while DTIAM inferred that its interaction probability with TMEM16A is 0.994, its inhibition probability is 0.999, and its affinity is 0.121 μM. As we can see, there is a certain discrepancy between 0.121 μM and 1.39 μM. The lower value of affinity indicates higher strength binding. The predicted affinity value between daidezein and TMEM16A is lower than the actual value suggests that DTIAM has higher confidence in daidezein. This is also illustrated by the fact that the predicted interaction and inhibition probabilities both exceed 0.99. Furthermore, we use Auto-Docking Vina software to validate the predicted potential compounds to select candidate drugs for further experimental evaluation, and dehydrocostus lactone with better Vina scores and relatively easier synthetic accessibility was selected to carry out wet lab validation. We found that dehydrocostus lactone (ranked 12th in the predicted potential compounds) was verified its inhibitory effect with TMEM16A by wet-lab experiments, and DTIAM inferred that its interaction probability with TMEM16A is 0.977, its inhibition probability is 0.989, and its affinity is 205.84 nM. Specifically, to investigate the inhibitory effect of dehydrocostus lactone on TMEM16A channels, we performed whole-cell patch clamp recordings in HEK293T cells transfected with TMEM16A plasmids. The pipette solution containing 600 nM Ca2+ was adopted to elicit TMEM16A currents in the transfected HEK293T cells. As illustrated in Fig. 5B, C, the characteristic outward rectification and slow activation kinetics of the TMEM16A current were obvious. The dose–response relationship was established by plotting normalized current values at a holding potential of +80 mV following exposure to various concentrations of dehydrocostus lactone. Our findings revealed an IC50 value of 111.97 ± 22.96 nM for dehydrocostus lactone in inhibiting in TMEM16A transfected HEK293T cells (Fig. 5D). We further verified the inhibition effect of dehydrocostus lactone on TMEM16A with an outside-out patch clamp. It was demonstrated that 100 μM of dehydrocostus lactone can completely inhibit the current of TMEM16A (Fig. 5EF), indicating that the inhibition of dehydrocostus lactone on TMEM16A was directly exerted. In addition, we performed experiments on LA795 cell with high endogenous expression of TMEM16A, which also proved the inhibitory effect of dehydrocostus lactone on TMEM16A (Supplementary Materials Fig. 1). The IC50 of dehydrocostus lactone inhibiting TMEM16A in LA795 cell was 161.64 ± 61.96 nM. All these experimental results demonstrate that dehydrocostus lactone predicted by DTIAM is a highly effective inhibitor of TMEM16A.

A Voltage stimulation waveform scheme. B Representative TMEM16A current in whole-cell patch clamp in HEK293T recombination system is activated by 600 nM Ca2+ and inhibited by different concentrations of Dehydrocostus Lactone (n = 3), with 20 μM CaCCinh-A01 as the positive control. C I–V curve of TMEM16A inhibited by various concentrations of dehydrocostus lactone (n = 3), with three independent biological replicates performed for each group. Data are presented as mean values ± SD. D Dose–response curve of TMEM16A currents at +80 mV incubated with indicated concentrations of dehydrocostus lactone (n = 3), with three independent biological replicates performed for each group. The IC50 of dehydrocostus lactone on TMEM16A channels was 111.97 ± 22.96 nM. Data are presented as mean values ± SD. E Representative TMEM16A current in outside-out patch-clamp in HEK293T recombination system, which is activated by 600 nM Ca2+ and inhibited by 100 μM dehydrocostus lactone (n = 3), with 20 μM CaCCinh-A01 as the positive control. F I–V curve of TMEM16A inhibited by 100 μM dehydrocostus lactone (n = 3), with 20 μM CaCCinh-A01. n = 3 for each group, with three independent biological replicates performed for each group. Data are presented as mean values ± SD.
To evaluate the performance of our framework on known inhibitors of TMEM16A, we also applied DTIAM to infer the interactions, affinities, and inhibition scores between TMEM16A and five known inhibitors, including 1PBC60, niclosamide61, benzbromarone62, crofelemer63, and CaCCinh-A0164. The prediction results are shown in Supplementary Data 2. We found that all five inhibitors were predicted to have a high likelihood of inhibiting TMEM16A, and the interaction probability of four inhibitors exceeded 0.95. Moreover, the predicted binding affinity scores for the five inhibitors were closely aligned with experimental measurements, demonstrating the robust predictive capability of DTIAM in screening TMEM16A inhibitors.
Prediction of potential drugs for EGFR and CDK 4/6 virtual screening
To further test the applicability for drug virtual screening, we tested whether DTIAM could identify the DTIs of potential drugs for epidermal growth factor receptor (EGFR) and cyclin-dependent kinase 4/6 (CDK 4/6). EGFR is a transmembrane protein that is found at abnormally high levels in cancer cells, and its inhibitors are known for the treatment of cancers caused by EGFR up-regulation, such as non-small-cell lung cancer and pancreatic cancer. CDK is a type of enzyme that regulates the progression of cells through the cell cycle. CDK 4/6 inhibitors work by binding to and blocking the activity of CDK4 and CDK6 enzymes and are commonly used to treat breast cancer and other types of cancer that are driven by overactive CDK 4/6 activity.
We used Yamanishi_08’s dataset (removing DTIs containing EGFR) to train DTIAM and predict potential interactions between EGFR and all drugs in the dataset. We also predicted the interactions between EGFR and 13 approved drugs (Afatinib, Osimertinib, Gefitinib, Erlotinib, Lapatinib, Neratinib, Brigatinib, Dacomitinib, Mobocertinib, Vandetanib, Fostamatinib, Zanubrutinib, and Lidocaine) from DrugBank which are used as EGFR inhibitors. The predicted results of the top-15 potential drugs are listed in Table 1. We found that 9 of the 13 EGFR inhibitors were successfully rediscovered in the top-15 potential drugs by our method, and 12 of 13 EGFR inhibitors ranked in the top 50 of 802 results (more details in Supplementary Data 3). In addition, the other 6 drugs in the top-15 potential drugs list were all validated by external databases (e.g., KEGG65, BRENDA66, and DrugBank67).
Similarly, the Yamanishi_08’s dataset (removing DTIs containing CDK 4 and CDK 6) was used to train DTIAM, which is applied to predict the interactions between CDK 4/6 and all drugs in the dataset and 4 approved drugs (Ribociclib, Abemaciclib, Trilaciclib, and Palbociclib) from DrugBank which are used as inhibitors of CDK 4 and CDK 6. Table 2 shows the top-15 potential drugs that potentially interact with CDK 4/6, ranked by the prediction scores of CDK 6. We observed that 4 approved CDK 4/6 inhibitors are successfully predicted by DTIAM. Moreover, Imatinib mesylate, Alvocidib, and Alvocidib hydrochloride were validated to interact with CDK 4/6 by external databases (CTD68 and KEGG65). In addition, the docking studies showed that the five drugs (i.e., Granisetron, Axitinib, Canertinib dihydrochloride, Doramapimod, and Mubritinib) were able to dock to the CDK 6 (Fig. 6B–F). In particular, Granisetron interacted with residue D163(A) and Axitinib interacted with residue V101(A) when docked to CDK 6 (Fig. 6B, C), which were observed similar to Palbociclib (Fig. 6A), the highly selective CDK4/6 inhibitor. All these results indicated that DTIAM can be effectively applied for drug virtual screening and provide a powerful tool to speed up the process of drug development.

A–F The docked poses for the predicted interactions between six potential drugs (i.e., Palbociclib, Granisetron, Axitinib, Canertinib dihydrochloride, Doramapimod, and Mubritinib, where Palbociclib is the reference drug) and the CDK 6. G The performances of DTIAM and baseline models trained on different scales of labeled data (i.e., 20%, 40%, 60%, and 80%) in DTI, DTA, and MoA prediction tasks under the warm start settings.
Ablation study
As mentioned above, DTIAM achieves excellent performance in downstream tasks even with a small amount of labeled data thanks to the pre-trained model. To test the effectiveness of our proposed pre-trained model, we compared DTIAM with the state-of-the-art baseline models with different scales of labeled data on the DTI, DTA, and MoA prediction tasks. We divided 20%, 40%, 60%, and 80% of the samples on six datasets of different tasks for training, and used the remaining samples for validation under the warm start setting.
As can be seen from Fig. 6G, with the increase of the number of training samples, the predictive performance of all methods improves by different degrees. DTIAM achieves the best performance under all data partitions and significantly outperforms other models, especially with less training data (20% and 40%). It is worth noting that DTIAM can outperform other models trained with 60% or even 80% of the samples using only 20% of the samples for supervised training. All the results show that our proposed pre-training model can extract accurate features from massive unlabeled data and can be effectively applied to downstream prediction tasks, even with a small amount of unlabeled data.
To analyze the impact of using different combinations of self-supervised tasks for molecular representation learning pre-training, we conducted ablation experiments on seven combinations of three self-supervised tasks for our proposed molecular pre-training model. The three self-supervised tasks are: (1) Masked Language Modeling (MLM); (2) Molecular Descriptor Prediction (MDP); and (3) Molecular Functional Group Prediction (MFGP). Details about each self-supervised task can be found in the “Self-supervised molecular representation learning” section.
As shown in Table 3, we find that MFGP task has the highest impact on the performance, especially in the drug cold start scenario, with a significant leading margin of 50% in terms of AUPR when using the MFGP task alone (AUPR = 0.305) versus using the MDP task alone (AUPR = 0.203). This demonstrates that the local functional group information of the molecule is very helpful for DTI prediction. At the same time, our molecular representation learning model can accurately capture the key functional group information. Among the combinations of the two self-supervised tasks, the molecular model trained on both MLM and MFGP performed best. Although the best performing model is trained on the combination of MLM, MDP, and MFGP, the additive gain from the MDP task is relatively minor.
Discussion
Accurately predicting DTIs can provide a huge advantage for drug discovery and development. Most existing methods only focus on the DTI binary classification or the DTA regression prediction, neglecting the pharmaceutical MoA information. The MoA prediction can help in understanding modes of drug action and provide new insights into drug discovery. In this study, we developed a unified framework, called DTIAM, to predict DTI, DTA, and MoA by combining the drug and target pre-training models and AutoML techniques. The pre-training models extract the substructure and contextual information from massive unlabeled data via self-supervised learning and can be transferred to various prediction tasks including MoA, DTI, DTA, etc. The high extendibility and generalization ability of the pre-training models have been extensively validated on different types of prediction tasks. Comprehensive comparison tests showed that DTIAM achieved superior performance and significantly outperformed other state-of-the-art machine learning methods on different types of datasets under three cross-validation settings. Further practical application of drug screening demonstrates the reliability of DTIAM predictions and the candidate inhibitor of TMEM16A predicted by DTIAM was verified by wet-lab experiments. Besides, we also validated the applicability of DTIAM for drug virtual screening on EGFR and CDK 4/6 targets, the results showed that the top-15 predicted potential drugs were mostly validated by external databases and literature. All of these results demonstrate that DTIAM can be effectively used for a variety of drug-target prediction tasks and provides a powerful tool for drug development.
DTIAM uses only molecular SMILES and protein sequences as input, and it effectively improves the performance of downstream prediction tasks by using massive amounts of label-free data for self-supervised pre-training. However, the sensitivity of a protein target to a specific compound is influenced by many factors beyond the protein sequences information. These factors, including the protein dynamics, protein mutations, cellular and membrane environment, etc., can alter the compound’s binding efficiency and efficacy. In future work, we will consider the dynamics of proteins in the model to improve the robustness and interpretability of the model, and integrate protein mutation information to improve the sensitivity of the model at key mutation sites.
Methods
The workflow of DTIAM
DTIAM consists of three main components: (1) Self-supervised molecular representation Learning for drug pre-training; (2) Unsupervised protein representation Learning for target pre-training; (3) The representation integration and downstream drug-target inference tasks via automated machine learning (AutoML).
Self-supervised molecular representation learning
We adopt the BERT-style69 method for drug pre-training and develop a molecular representation learning model called BERMol, which stands for Bidirectional Encoder Representations of Molecular. BERMol learns vector representations of molecular substructures from large-scale unlabeled data with the language model and domain-relevant auxiliary tasks. The proposed model is pre-trained on the GuacaMol dataset70 which contains 1.6 million compounds collected from the ChEMBL71 database. To apply the language model to molecular, we define the substructures of molecules as “words” and molecules as “sentences”. We then use the Morgan algorithm72 to extract all substructures of radius 1 for each molecule. After generating the corpus of compounds, the Transformer38 architecture is applied to learn the low-rank representations for all substructures of molecules. Specifically, a molecule can be abstracted as a sentence S = (x1, …, xn), where xi is the i-th word and n is the sentence length, each word in the sentence is then embedded into a d-dimensional vector space \(Z=({\overrightarrow{z}}_{1},\ldots,{\overrightarrow{z}}_{n})\), where \({\overrightarrow{z}}_{i}\in {{\mathbb{R}}}^{d}\) is the d-dimensional embedding of the i-th word. In the encoding step using the transformer architecture, we transform all embeddings into three matrices (Q, K, V) representing queries, keys, and values, respectively, and then compute the self-attention weights between words as follows:
where \(\sqrt{d}\) is the scaling factor used to smooth the gradient of the softmax function, and the output of the attention mechanism is a matrix representing the global relationship between different words. To integrate information from different representation subspaces, multi-head attention is performed with different linear projections, the final output matrix can be written as,
where \({W}_{i}^{Q},{W}_{i}^{K},{W}_{i}^{V}\) are the projection matrices of i-th head. The complete encoder is a stack of multiple blocks combined with a multi-head self-attention mechanism and a fully connected feed-forward network.
In order to learn flexible and high-quality molecular representations, we combine three self-supervised tasks for pre-training: (1) Masked Language Modeling (MLM); (2) Molecular Descriptor Prediction (MDP); (3) Molecular Functional Group Prediction (MFGP). The MLM task was proposed by BERT, whereby the model randomly masks a portion of the tokens and is trained to predict the true identity of the masked tokens. In this task, the final representations of the masked tokes are fed into a neural network model for multiclassification prediction. The task is optimized using the cross-entropy loss as follows:
where Nmask is the number of the masked tokens, V is the size of the vocabulary, i.e., the size of the set of substructures, yij is a one-hot vector representing the true distribution over the vocabulary for the i-th masked token, and pij is the predicted probability of the j-th token in the vocabulary being the correct replacement for the i-th masked word. In a word, the training loss of the MLM task is the sum of the mean masked language modeling likelihood. The goal of the MDP task is to predict a set of real-valued descriptors of chemical characteristics, which is a regression task. The molecular descriptor encodes many physicochemical properties and can be easily calculated by RDKit73. In this task, the final representation of the first token incorporates the global features of the entire molecule and is fed into a neural network model to predict the normalized set of descriptors. The task is optimized using the mean squared error over all predicted values as follows:
where Ndesc is the number of the molecular descriptors used in this task, yi is the normalized value of the i-th descriptors, and \({\hat{y}}_{i}\) is the predicted value of the i-th descriptors. The MFGP task can be formulated as a multi-label classification, which aims to predict the functional groups within the input molecule. The functional group contains rich domain knowledge of molecules and also can be easily detected by RDKit. The final representation of the first token is also fed into a neural network model for multi-label classification. This task is optimized using the cross-entropy loss as follows:
where Nfun is the number of the molecular functional groups used in this task, yi is the true label indicating whether the molecule contains the i-th functional group, and \({\hat{y}}_{i}\) is the predicted probability of the i-th functional group. The final training loss of the self-supervised molecular representation learning model is given by the weighted sum of all individual task losses as follows:
where α and β are two weighting factors. The training objective is to minimize the loss and use backpropagation to optimize the model and update the representations.
Unsupervised protein representation learning
In the target protein representation learning step, we employ ESM-274, a family of large-scale protein language models at scales from 8 million parameters up to 15 billion parameters, to extract the embeddings of target proteins. The ESM-2 language models also use the BERT-style69 encoder with transformer38 architecture to train the masked language modeling objective, which aims to predict the original identity of randomly masked amino acids in a protein sequence based on their context. The UniRef75 protein sequence database is used for the training of ESM-2 models, including ~138 million UniRef90 sequences and ~65 million unique sequences.
The pre-trained ESM-2 models can directly predict the residue-residue contact map of the protein extracted from the Transformer self-attention patterns. Specifically, given a model with L layers, K heads, let cij be a binary random variable, indicating whether the amino acids i, j are in contact. Then the probability of contact between positions i and j is defined as a logistic regression:
where \({a}_{ij}^{kl}\) is attention score between amino acids i and j from the k-th attention head in the l-th layer of the transformer.
And the ESM-2 language models are also enabled to generate high-resolution protein three-dimensional structure predictions from the protein sequence (ESMFold). In this work, we employ one of the ESM-2 models with 33 layers and 650 million parameters and use its hidden states of the last layer as the representations of target proteins.
Downstream drug-target prediction
The last step is to integrate the drug and target representations and make various downstream predictions via AutoGluon76. AutoGluon is an AutoML framework for structured data that automatically utilize state-of-the-art techniques without the need for frequent manual intervention to achieve strong predictive performance in many applications. Unlike prior AutoML frameworks that primarily focus on the task of Combined Algorithm Selection and Hyperparameter optimization (CASH) to find the best model from a sea of possibilities, AutoGluon performs advanced data processing and powerful multi-layer model ensembling to train highly accurate machine learning models. AutoGluon integrates various types of models (such as neural networks, LightGBM boosted trees and Random Forests), and ensembles these models based on novel combinations of multi-layer stacking and repeated k-fold bagging.
In multi-layer stacking, the first layer has multiple base models, whose inputs are the original data features, and outputs are concatenated with data features and then fed into the next layer. And the last stacking layer leverages ensemble selection to aggregate the predictions of the stacker model in a weighted fashion. In the repeated k-fold bagging, the training data is randomly divided into k disjoint chunks, each chunk is used as a test set to produce out-of-fold (OOF) predictions and the remaining chunks are used as a training set to train a model. To minor overfitting in OOF predictions, AutoGluon repeats the k-fold bagging process on n different random partitions of the training data, and all OOF predictions are averaged over the repeated bags. More specifically, the training data (X, Y) is first randomly split into k chunks \({\{{X}_{i}^{j},{Y}_{i}^{j}\}}_{j=1}^{k}\) in the i-th repetition, then train a model on (\({X}_{i}^{-j},{Y}_{i}^{-j}\)) and make predictions \({\hat{Y}}_{m,i}^{j}\) on OOF data \({X}_{i}^{j}\) for each model type m in the family of models M. The outputs of model type m in the stacking layer l are computed by averaging all OOF predictions over the repeated bags, that is,
which are concatenated with the inputs and then fed into the next stacking layer, that is,
The final predictions are the aggregation of the stacker models’ predictions in a weighted manner.
The framework, in this work, is highly adaptable and can be utilized for various drug-target prediction tasks, including DTI, DTA, and MoA. This framework employs pre-trained drug and target representation learning models that can be shared across different tasks, and are then fine-tuned using distinct labeled datasets in a supervised learning manner. The pre-training phase enables the models to learn precise representations from a vast amount of unlabeled data, leading to an exceptional performance on downstream tasks.
Benchmark datasets
In this study, six benchmark datasets for three prediction tasks (i.e., DTI, DTA, and MoA), namely Yamanishi_08, Hetionet, Davis, KIBA, Activation, and Inhibition, were used to comprehensively evaluate the performance and ability of our model.
The Yamanishi_08 dataset and Hetionet are DTI datasets, in which the labels are binary interactions between drugs and targets. The Yamanishi_08’s dataset is originally introduced by Yamanishi et al.16 and consists of four sub-datasets: G-Protein Coupled Receptors (GPCR), Ion Channels (IC), Nuclear Receptors (NR), and Enzymes (E) obtained from KEGG BRITE, BRENDA, SuperTarget, and DrugBank databases65,66,67,77. In this study, we use the combined dataset of the four sub-datasets constructed by Ye et al.9. In total, the dataset contains 791 drugs, 989 targets, and 5127 known DTIs (i.e., positive samples). The Hetionet dataset is constructed by Himmelstein et al.78, which integrated the biomedical data from 29 public resources. It comprises 1384 drugs, 5763 targets, and 49,942 DTIs.
Davis and KIBA are two DTA datasets and are popular standard datasets used in previous work for DTA prediction evaluation36,79. The Davis dataset contains binding intensities of the kinase protein family and the relevant inhibitors measured using dissociation constant (Kd) values. It consists of 68 drugs and 442 targets and was constructed by Davis et al.80. KIBA is a large-scale kinase inhibitor bioactivity dataset constructed by Tang et al.81. It combined different measurement types such as Ki, Kd, and IC50, and contains 2111 drugs and 229 targets.
Activation and Inhibition are two MoA datasets that were obtained from the Therapeutic Target Database (TTD)82. We selected those MOAs that are explicitly defined as activation (e.g., “activator”, “agonist”) or inhibition (e.g., “inhibitor”, “antagonist”). In total, we obtained 1913 activation MoAs between 1426 drugs and 281 targets for the Activation dataset, and 21,055 inhibition MoAs between 14,049 drugs and 1088 targets for the Inhibition dataset.
Baselines
In this work, three types of baseline models are used in the performance comparison for the DTI, DTA, and MoA prediction tasks, including CPI_GNN49, TransformerCPI50, MPNN_CNN51, and KGE_NFM9 for DTI prediction, and DeepDTA36, MONN41, BACPI40, and GraphDTA34 for DTA prediction, and AI-DTI48 for MoA prediction. CPI_GNN, TransformerCPI, MPNN_CNN, DeepDTA, BACPI, and GraphDTA as well as DTIAM require only SMILES strings of compounds and primary sequences of proteins as input. KGE_NFM requires the heterogeneous information extracted from multi-omics data to build a knowledge graph and used the Morgan fingerprints of drugs and CTD descriptors of targets as additional information. MONN requires not only SMILES strings and protein sequences, but also pairwise non-covalent interactions between atoms of compounds and residues of proteins as extra supervision information. In this work, since the non-covalent interactions labels of Davis and KIBA datasets were unavailable, we did not provide the extra supervision information for MONN (denoted as MONNsingle in the original paper). AI-DTI needs SMILES strings of compounds and genetically perturbed transcriptome data of target genes as input. Note that, the MPNN_CNN and DeepDTA models were constructed with DeepPurpose83.
Experimental settings
DTIAM and these baseline methods are evaluated under three different settings of cross-validation, i.e., the warm start setting, the drug cold start setting and the target cold start setting. To explain these settings, we denote the training and test drug sets by Dtrain and Dtest, and training and test drug sets by Ttrain and Ttest, respectively, and use (di, tj) to represent the drug-target pair between the i-th drug and j-th target.
In the warm start setting, for a drug-target pair (di, tj) from the test set (di ∈ Dtest and tj ∈ Ttest), both di and tj are encountered in the training set (di ∈ Dtrain and tj ∈ Ttrain). That is, the test and training sets share common drugs and targets. This scenario is suitable for identifying potential interactions between known drugs and targets.
In the drug cold start setting, for a drug-target pair (di, tj) from the test set, the drug di is unseen in the training phase (di ∈ Dtest, di ∉ Dtrain), while the target tj is present in both training and test sets (tj ∈ Ttest, tj ∈ Ttrain). This experimental setting is relevant if we need to discover potential candidate targets for new drugs.
In the target cold start setting, for a drug-target pair (di, tj) from the test set, we have seen the drug di (di ∈ Dtrain), but the target tj is unseen in the training phase (tj ∉ Ttrain). This scenario is often applied in virtual drug screening of new targets.
Note that the DTI prediction task is evaluated under 10-fold cross-validation, and the DTA and MoA prediction tasks are evaluated under 5-fold cross-validation. In addition, for the KGE_NFM model, the train-test split schemes of the drug/target cold start setting only focus on the drugs/proteins existing in the knowledge graph but without any known DTI relations.
Evaluation metrics
In this study, we use the average scores of the area under the receiver operating characteristics curve (AUROC) and the area under the precision-recall curve (AUPR) to evaluate the performance of each method in the DTI and MoA prediction tasks. In this work, we set the ratio between the positive and negative samples to 1:10 because this is more in line with real-world scenarios. Here, we adopt AUPR as the main evaluation metric, since it gives a more accurate evaluation of a method’s performance in the unbalanced dataset. In the DTA prediction task, the performance of each method was evaluated by the mean squared error (MSE) and the Pearson correlation.
Inhibitor identification of TMEM16A
Cell culture and transfection
HEK293T cells were maintained in Dulbecco’s modified eagle medium (DMEM) (Gibco), supplemented with 10% fetal calf serum (FBS) (Gibco), and 1% penicillin-streptomycin solution (100 U/ml penicillin and 100 μg/ml streptomycin) in a humidified incubator at 37 °C with 5% CO2. The cells were transiently transfected with a cDNA for mouse TMEM16A (mTMEM16A), using Lipofectamine 3000 Transfection Reagent (Thermo Fisher Scientific) with a ratio of 1:3. The mouse cDNA clone mTMEM16A (ANO1, Accession Number: NM_178642.5) was a kind gift from Prof. Young Duk Yang (Seoul National University, Korea) and was subcloned into expression vector pEGFPN1. Following transfection, the cells were maintained at 37 °C for 24 h before patch clamp recording. The UniProt ID of TMEM16A is A0A0A0MQF2. LA795 cells were cultured using RPMI 1640 medium (Gibco) with 10% fetal bovine serum (FBS) (Gibco) and 1% penicillin-streptomycin solution at 37 °C and 5% CO2. LA795 cell is a mouse lung adenocarcinoma cell line with high endogenous expression of TMEM16A.
Electrophysiology
Currents were recorded using an EPC10 amplifier and Pulse software with a Digi LIH1600 interface (HEKA, Lambrecht, Germany). The borosilicate glass patch pipettes with a pipette resistance of 3–5 MΩ (Sutter Instruments, Novato, USA) were drawn by a P-97 puller (Sutter Instruments, Novato, USA). The series resistance is less than 20 MΩ, with a compensation rate ranging from 60 to 80% in the whole-cell patch-clamp. The liquid junction potentials were not corrected and its magnitude was about 10 mV. The glass pipette has been fire-polished. The data was sampled at 10 kHz after being low pass filtered at 2.9 kHz. The stimulation procedures included 150 ms voltage steps from a holding potential of 0 mV and membrane voltage (mV) clamping in 20 mV increments from −80 to +80 mV, with a −80 mV hold at room temperature (22–25 °C). The bath solution used to record TMEM16A currents contained the following (in mM): NaCl 150 mM, MgCl2 ⋅ 6H2O 1 mM, HEPES 10 mM, glucose 10 mM, and mannitol 10 mM adjusted to pH 7.4 with NaOH. The pipette solution contained the following (in mM): CsCl 130 mM, EGTA 10 mM, MgATP 1 mM, MgCl2 ⋅ 6H2O 1 mM, and HEPES 10 mM adjusted to pH 7.3 with CsOH. The 600 nM free Ca2+ bath solution was prepared by adding standard CaCl2 solution (1 M, Sigma-Aldrich, St. Louis, MO, USA) to a final concentration of 8.69 mM, and adjusted to pH 7.3 with CsOH. The osmotic pressure was determined using the OM815 osmometer. The osmotic pressure of the pipette and the bath solutions was in the range of 290–300 mOsm/L and 300–310 mOsm/L, respectively. Outside-out membrane patches are formed by gradually retreating the patch pipette from the cell after a whole-cell recording has been established. This has the effect of drawing out a strand of membrane that eventually breaks and often reseals such that its extracellular surface faces into the recording chamber. The membrane attached to the pipette follows the retreating pipette, and a giga-ohm seal should form again when the membrane breaks and an outside-out patch is excised. The single channel current of TMEM16A is recorded when the drug is administered through the perfusion system. The bath and pipette solutions of the outside-out patch-clamp were the same as those in the whole-cell patch-clamp recording. Different concentrations of dehydrocostus lactone were prepared in the bath solution and administered through the perfusion system. Specifically, whole-cell patch clamp and outside-out patch-clamp were employed to study the inhibitory effect of dehydrocostus lactone on TMEM16A.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The source data of six datasets used to train and evaluate the model is provided at https://github.com/CSUBioGroup/DTIAM/tree/main/data. The MoA datasets were constructed from the Therapeutic Target Database (https://db.idrblab.net/ttd/). The source data of GuacaMol dataset used to pre-train our molecular representation learning model is available at https://github.com/BenevolentAI/guacamol. The targets mentioned in our cases, including ADRA1A, ADRA1B, ADRA1D, CYP3A43, CYP3A5, CYP3A7, DRD1, HTR1B, HTR1D, HTR2A, TMEM16A, EGFR, CDK4 and CDK6 are available in the Uniprot repository (https://www.uniprot.org/) under their accession codes. The virtual screening results are available in the Supplementary data. Source data are provided with this paper through Figshare https://doi.org/10.6084/m9.figshare.27896790.
Code availability
The source data and codes of DTIAM are available on GitHub at https://github.com/CSUBioGroup/DTIAM, which has also been deposited in the Zenodo under accession code https://zenodo.org/records/14847966. ESM-2 v1.0.3 (https://github.com/facebookresearch/esm) is used for calculating target protein representations. AutoGluon v0.5.2 (https://github.com/autogluon/autogluon) is used for constructing predictive models. Data are analyzed using numpy v1.21.2 (https://numpy.org/), pandas v1.3.5 (https://pandas.pydata.org/), and Matplotlib v3.5.1 (https://matplotlib.org/). Structures are visualized by Pymol v2.5.2 (https://www.pymol.org/).
References
Chen, X. et al. Drug–target interaction prediction: databases, web servers and computational models. Brief. Bioinforma. 17, 696–712 (2016).
Wen, M. et al. Deep-learning-based drug–target interaction prediction. J. Proteome Res. 16, 1401–1409 (2017).
DiMasi, J. A., Hansen, R. W. & Grabowski, H. G. The price of innovation: new estimates of drug development costs. J. Health Econ. 22, 151–185 (2003).
Paul, S. M. et al. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 9, 203–214 (2010).
Pushpakom, S. et al. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 18, 41–58 (2019).
Zhang, P., Wei, Z., Che, C. & Jin, B. DeepMGT-DTI: transformer network incorporating multilayer graph information for drug–target interaction prediction. Comput. Biol. Med. 142, 105214 (2022).
Shin, B., Park, S., Kang, K. & Ho, J. C. Self-attention based molecule representation for predicting drug-target interaction. In Machine Learning for Healthcare Conference 230–248 (PMLR, 2019).
Zhu, H., Zhou, R., Cao, D., Tang, J. & Li, M. A pharmacophore-guided deep learning approach for bioactive molecular generation. Nat. Commun. 14, 6234 (2023).
Ye, Q. et al. A unified drug–target interaction prediction framework based on knowledge graph and recommendation system. Nat. Commun. 12, 1–12 (2021).
Batool, M., Ahmad, B. & Choi, S. A structure-based drug discovery paradigm. Int. J. Mol. Sci. 20, 2783 (2019).
Yang, D. et al. G protein-coupled receptors: structure-and function-based drug discovery. Signal Transduct. Target. Ther. 6, 7 (2021).
Saikia, S. & Bordoloi, M. Molecular docking: challenges, advances and its use in drug discovery perspective. Curr. Drug Targets 20, 501–521 (2019).
Salo-Ahen, O. M. et al. Molecular dynamics simulations in drug discovery and pharmaceutical development. Processes 9, 71 (2020).
Schaller, D. et al. Next generation 3D pharmacophore modeling. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 10, 1468 (2020).
Pagadala, N. S., Syed, K. & Tuszynski, J. Software for molecular docking: a review. Biophys. Rev. 9, 91–102 (2017).
Yamanishi, Y., Araki, M., Gutteridge, A., Honda, W. & Kanehisa, M. Prediction of drug–target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 24, 232–240 (2008).
Cichonska, A. et al. Computational-experimental approach to drug-target interaction mapping: a case study on kinase inhibitors. PLoS Comput. Biol. 13, 1005678 (2017).
Mei, J.-P., Kwoh, C.-K., Yang, P., Li, X.-L. & Zheng, J. Drug–target interaction prediction by learning from local information and neighbors. Bioinformatics 29, 238–245 (2013).
Olayan, R. S., Ashoor, H. & Bajic, V. B. DDR: efficient computational method to predict drug–target interactions using graph mining and machine learning approaches. Bioinformatics 34, 1164–1173 (2018).
Cheng, F. et al. Prediction of drug-target interactions and drug repositioning via network-based inference. PLoS Comput. Biol. 8, 1002503 (2012).
Fakhraei, S., Huang, B., Raschid, L. & Getoor, L. Network-based drug-target interaction prediction with probabilistic soft logic. IEEE/ACM Trans. Comput. Biol. Bioinforma. 11, 775–787 (2014).
Wu, Y., Gao, M., Zeng, M., Zhang, J. & Li, M. Bridgedpi: a novel graph neural network for predicting drug–protein interactions. Bioinformatics 38, 2571–2578 (2022).
Wu, Z., Li, W., Liu, G. & Tang, Y. Network-based methods for prediction of drug-target interactions. Front. Pharmacol. 9, 1134 (2018).
Lu, Z., Wang, Y., Zeng, M. & Li, M. Hnedti: prediction of drug-target interaction based on heterogeneous network embedding. In 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), 211–214 (IEEE, 2019).
Luo, Y. et al. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat. Commun. 8, 1–13 (2017).
Bagherian, M. et al. Machine learning approaches and databases for prediction of drug–target interaction: a survey paper. Brief. Bioinforma. 22, 247–269 (2021).
Ding, H., Takigawa, I., Mamitsuka, H. & Zhu, S. Similarity-based machine learning methods for predicting drug–target interactions: a brief review. Brief. Bioinforma. 15, 734–747 (2014).
Chen, R., Liu, X., Jin, S., Lin, J. & Liu, J. Machine learning for drug-target interaction prediction. Molecules 23, 2208 (2018).
D’Souza, S., Prema, K. & Balaji, S. Machine learning models for drug–target interactions: current knowledge and future directions. Drug Discov. Today 25, 748–756 (2020).
Liu, H., Sun, J., Guan, J., Zheng, J. & Zhou, S. Improving compound–protein interaction prediction by building up highly credible negative samples. Bioinformatics 31, 221–229 (2015).
Shi, H. et al. Predicting drug-target interactions using lasso with random forest based on evolutionary information and chemical structure. Genomics 111, 1839–1852 (2019).
Pahikkala, T. et al. Toward more realistic drug–target interaction predictions. Brief. Bioinforma. 16, 325–337 (2015).
Wang, K. & Li, M. Fusion-based deep learning architecture for detecting drug-target binding affinity using target and drug sequence and structure. IEEE J. Biomed. Health Inform. 27, 6112–6120 (2023).
Nguyen, T. et al. GraphDTA: predicting drug–target binding affinity with graph neural networks. Bioinformatics 37, 1140–1147 (2021).
Jiang, M. et al. Drug–target affinity prediction using graph neural network and contact maps. RSC Adv. 10, 20701–20712 (2020).
Öztürk, H., Özgür, A. & Ozkirimli, E. DeepDTA: deep drug–target binding affinity prediction. Bioinformatics 34, 821–829 (2018).
Karimi, M., Wu, D., Wang, Z. & Shen, Y. DeepAffinity: interpretable deep learning of compound–protein affinity through unified recurrent and convolutional neural networks. Bioinformatics 35, 3329–3338 (2019).
Vaswani, A. et al. Attention is all you need. In Advances in Neural Information Processing Systems 30 (2017).
Gao, K. Y. et al. Interpretable drug target prediction using deep neural representation. In IJCAI Vol. 2018, 3371–3377 (2018).
Li, M., Lu, Z., Wu, Y. & Li, Y. BACPI: a bi-directional attention neural network for compound–protein interaction and binding affinity prediction. Bioinformatics 38, 1995–2002 (2022).
Li, S. et al. Monn: a multi-objective neural network for predicting compound-protein interactions and affinities. Cell Syst. 10, 308–322 (2020).
Zhang, S. et al. Drugai: a multi-view deep learning model for predicting drug–target activating/inhibiting mechanisms. Brief. Bioinforma. 24, 526 (2023).
Schenone, M., Dančík, V., Wagner, B. K. & Clemons, P. A. Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 9, 232–240 (2013).
Gibbs, J. B. Mechanism-based target identification and drug discovery in cancer research. Science 287, 1969–1973 (2000).
Sawada, R., Iwata, M., Tabei, Y., Yamato, H. & Yamanishi, Y. Predicting inhibitory and activatory drug targets by chemically and genetically perturbed transcriptome signatures. Sci. Rep. 8, 1–9 (2018).
Wikberg, J. et al. Melanocortin receptors: ligands and proteochemometrics modeling. Ann. N. Y. Acad. Sci. 994, 21–26 (2003).
Mervin, L. H., Afzal, A. M., Brive, L., Engkvist, O. & Bender, A. Extending in silico protein target prediction models to include functional effects. Front. Pharmacol. 9, 613 (2018).
Lee, W.-Y., Lee, C.-Y. & Kim, C.-E. Predicting activatory and inhibitory drug–target interactions based on structural compound representations and genetically perturbed transcriptomes. PLoS One 18, e0282042 (2023).
Tsubaki, M., Tomii, K. & Sese, J. Compound–protein interaction prediction with end-to-end learning of neural networks for graphs and sequences. Bioinformatics 35, 309–318 (2019).
Chen, L. et al. TransformerCPI: improving compound–protein interaction prediction by sequence-based deep learning with self-attention mechanism and label reversal experiments. Bioinformatics 36, 4406–4414 (2020).
Gilmer, J., Schoenholz, S. S., Riley, P. F., Vinyals, O. & Dahl, G. E. Neural message passing for quantum chemistry. In International Conference on Machine Learning 1263–1272 (PMLR, 2017).
Kee, V. R. Hemodynamic pharmacology of intravenous vasopressors. Crit. Care Nurse 23, 79–82 (2003).
Yono, M. et al. Doxazosin-induced up-regulation of α1A-adrenoceptor mRNA in the rat lower urinary tract. Can. J. Physiol. Pharmacol. 82, 872–878 (2004).
Corena-McLeod, M. Comparative pharmacology of risperidone and paliperidone. Drugs RD 15, 163–174 (2015).
Wang, H. et al. Cell-specific mechanisms of TMEM16A Ca2+-activated chloride channel in cancer. Mol. Cancer 16, 1–17 (2017).
Liu, Y., Liu, Z. & Wang, K. The Ca2+-activated chloride channel ANO1/TMEM16A: an emerging therapeutic target for epithelium-originated diseases? Acta Pharm. Sin. B 11, 1412–1433 (2021).
Bai, W., Liu, M. & Xiao, Q. The diverse roles of TMEM16A Ca2+-activated Cl- channels in inflammation. J. Adv. Res. 33, 53–68 (2021).
Al-Hosni, R., Ilkan, Z., Agostinelli, E. & Tammaro, P. The pharmacology of the TMEM16A channel: therapeutic opportunities. Trends Pharmacol. Sci. 43, 712–725 (2022).
Wang, X. et al. Inhibitory effect of daidzein on the calcium-activated chloride channel TMEM16A and its anti-lung adenocarcinoma activity. Int. J. Biol. Macromol. 253, 127261 (2023).
Lam, A. K., Rutz, S. & Dutzler, R. Inhibition mechanism of the chloride channel TMEM16A by the pore blocker 1PBC. Nat. Commun. 13, 2798 (2022).
Miner, K. et al. Drug repurposing: the anthelmintics niclosamide and nitazoxanide are potent TMEM16A antagonists that fully bronchodilate airways. Front. Pharmacol. 10, 51 (2019).
Huang, F. et al. Calcium-activated chloride channel TMEM16A modulates mucin secretion and airway smooth muscle contraction. Proc. Natl Acad. Sci. USA 109, 16354–16359 (2012).
Tradtrantip, L., Namkung, W. & Verkman, A. S. Crofelemer, an antisecretory antidiarrheal proanthocyanidin oligomer extracted from croton lechleri, targets two distinct intestinal chloride channels. Mol. Pharmacol. 77, 69–78 (2010).
De La Fuente, R., Namkung, W., Mills, A. & Verkman, A. S. Small-molecule screen identifies inhibitors of a human intestinal calcium-activated chloride channel. Mol. Pharmacol. 73, 758–768 (2008).
Kanehisa, M. et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 34, 354–357 (2006).
Schomburg, I. et al. BRENDA, the enzyme database: updates and major new developments. Nucleic Acids Res. 32, 431–433 (2004).
Wishart, D. S. et al. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 36, 901–906 (2008).
Davis, A. P. et al. Comparative toxicogenomics database (CTD): update 2021. Nucleic Acids Res. 49, 1138–1143 (2021).
Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. Bert: Pre-training of deep bidirectional transformers for language understanding. In Proc. of the 2019 conference of the North American chapter of the association for computational linguistics: human language technologies, Vol. 1, 4171–4186 (2019).
Brown, N., Fiscato, M., Segler, M. H. & Vaucher, A. C. GuacaMol: benchmarking models for de novo molecular design. J. Chem. Inf. Model. 59, 1096–1108 (2019).
Gaulton, A. et al. The ChEMBL database in 2017. Nucleic Acids Res. 45, 945–954 (2017).
Rogers, D. & Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 50, 742–754 (2010).
Landrum, G. et al. Rdkit: Open-source Cheminformatics (2006).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Suzek, B. E. et al. Uniref clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932 (2015).
Erickson, N. et al. Autogluon-tabular: Robust and accurate automl for structured data. Preprint at https://arxiv.org/abs/2003.06505 (2020).
Günther, S. et al. Supertarget and matador: resources for exploring drug-target relationships. Nucleic Acids Res. 36, 919–922 (2007).
Himmelstein, D. S. et al. Systematic integration of biomedical knowledge prioritizes drugs for repurposing. Elife 6, 26726 (2017).
He, T., Heidemeyer, M., Ban, F., Cherkasov, A. & Ester, M. SimBoost: a read-across approach for predicting drug–target binding affinities using gradient boosting machines. J. Cheminform. 9, 1–14 (2017).
Davis, M. I. et al. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051 (2011).
Tang, J. et al. Making sense of large-scale kinase inhibitor bioactivity data sets: a comparative and integrative analysis. J. Chem. Inf. Model. 54, 735–743 (2014).
Zhou, Y. et al. Therapeutic target database update 2022: facilitating drug discovery with enriched comparative data of targeted agents. Nucleic Acids Res. 50, 1398–1407 (2022).
Huang, K. et al. DeepPurpose: a deep learning library for drug–target interaction prediction. Bioinformatics 36, 5545–5547 (2020).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 62225209 to M.L.), the Project of Xiangjiang Laboratory (No. 24XJJCYJ01003 to M.L.), the Academy of Finland (No. 359752 to J.T.), and Hunan Postgraduate Research and Innovation Project (No. CX20230260 to Z.L.). We acknowledge the High Performance Computing Center of Central South University for support.
Author information
Authors and Affiliations
Contributions
M.L. supervised the research. Z.L. and M.L. conceived the initial idea. Z.L. designed the model and carried out the test experiments. G.S. and Y.C. performed the wet-lab experiments. L.Q., K.W., and C.L. helped with the manuscript preparation. Z.L., G.S., H.Z., X.S., J.T., and M.L. wrote and revised the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lu, Z., Song, G., Zhu, H. et al. DTIAM: a unified framework for predicting drug-target interactions, binding affinities and drug mechanisms. Nat Commun 16, 2548 (2025). https://doi.org/10.1038/s41467-025-57828-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57828-0
This article is cited by
-
Evidential deep learning-based drug-target interaction prediction
Nature Communications (2025)
-
DeepDTAGen: a multitask deep learning framework for drug-target affinity prediction and target-aware drugs generation
Nature Communications (2025)
