
Abstract
Late-stage deuteration of aryl halides with deuterium oxide is a highly desirable but challenging transformation, primarily due to the difficulty of activating inert carbon-halogen bonds and the umpolung of deuterium oxide in the presence of various functional groups. To achieve this transformation, efforts have been made to develop photo-chemical, electro-chemical, or mechano-chemical strategies. However, these approaches often require specialized setups or activated substrates. Despite the well-known functional group tolerance of palladium catalysis, which makes it valuable in late-stage functionalization, a palladium-catalyzed deuteration of aryl halides with deuterium oxide has remained elusive. Herein, a deuteration reaction of aryl bromides, chlorides, and triflates with deuterium oxide has been developed, through palladium catalysis. Chemical equivalent amount of D2O is required for inert substrates like aryl chlorides. The reaction features high functional group tolerance, making it suitable for late-stage deuteration.
Similar content being viewed by others
Introduction
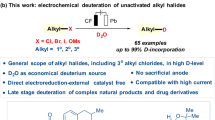
In 2024, the FDA approved an additional deuterated drug for medical use1, with at least ten other deuterated drugs currently in clinical or preclinical trials2. Deuterium on the drug molecules can significantly lower metabolism rates and reduce the dosing frequency because of kinetic isotope effect3,4,5. Consequently, the deuterium labeling technology has been recognized as a crucial tool to enhance the pharmacokinetic and pharmacodynamics properties of drug molecules6,7. As a result, the development of late-stage deuteration methodologies8,9,10,11 is important in drug discovery area. Catalytic deuteration of aryl (pseudo)halides is a highly desirable transformation because various aryl (pseudo)halides are easy to prepare from pharmaceuticals12,13,14,15, and thousands of aryl (pseudo)halides drug intermediates are commercially available. According to stoichiometric calculations, catalytic deuteration of aryl halide requires a deuteride donor. Deuterium sources8,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31 successfully applied to the deuteration of aryl halides include NaBD416, DCOOK16,17, D218,19,PrOD-d820,21, MeOD-d422, CD3CN23, D2O24,25,26,27, etc. Among these, D2O is the most cost-effective commercially available deuterium source (Fig. 1a)8. However, current late-stage deuteration reaction of aryl halides with D2O still require special reaction setups for reduction, or high deuterium source loading24,25,26,27. Herein, a general palladium-catalyzed deuteration reaction of aryl halides with deuterium oxide has been developed, where deuterium oxide effectively serves as the deuteride source under cross-coupling conditions.

a Comparison of the costs of deuterium sources. b Photochemical and electrochemical strategies. c Mechanochemical strategy. d This work: Palladium catalysis strategy.
Traditional deuteration strategies of aryl halides require the preparation of stoichiometric organometallic carbanion reagents, such as organolithium or Grignard reagents24. These organometallic carbanion reagents, when quenched with D2O, yield deuterated products. However, the presence of various functional groups leads to side reactions with the stoichiometric carbanion reagents, complicating the application of the strategy in late-stage functionalization32,33,34,35,36. To address the limitation, the Liu group developed a KOMe/Me3SiSiMe3 system, which may form transient carbanions from aryl halides and then trapped by the large excess of CD3CN to provide the desired deuterated product23. To further investigate the use of D2O as a deuterium source for late-stage aryl halide deuteration, the Gong group explored a photochemical approach, while the Lei group developed an electrochemical strategy. However, both methods require D2O to be used in large excess or as a co-solvent (Fig. 1b)25,26. The Lian group developed a mechanochemical strategy to achieve deuteration reaction of aryl iodides with 2–4 equivalents of D2O27. However, more inert aryl bromides or aryl chlorides substrates have not been successfully applied in the strategy (Fig. 1c). Surprisingly, despite decades of extensive development of palladium-catalyzed cross-coupling reactions, which are known for their high reactivity, excellent functional group tolerance, and broad substrate scope37,38,39,40,41,42, a palladium-catalyzed deuteration of aryl halides with deuterium oxide has not yet been achieved43,44. Herein, a palladium-catalyzed deuteration reaction of various aryl (pseudo)halides with D2O has been developed (Fig. 1d). The reaction exhibits chemical equivalent amount of D2O loading and broad substrate scope. The high functional group tolerance of the transformation enables the late-stage functionalization of several drug molecules.
Results
Optimization of reaction conditions
We propose that the success of a palladium-catalyzed deuteration of aryl halides with D2O lies in an efficient catalytic umpolung reaction of deuterium oxide, followed by a cross-coupling between aryl halide and the in-situ generated deuteride source. The Zhu group has reported that the combination of diboron and water can be used as a hydride donor in a reductive Heck reaction45. Inspired by the work, we propose that, in the presence of diboron, a palladium catalyst may facilitate an efficient umpolung reaction of deuterium oxide to generate the deuteride source, rather than engaging in direct transmetallation with diboron to produce the borylation product. The unusual selectivity for the transmetallation step may provide us a chance to achieve the challenging deuteration reaction of aryl halides with heavy water.
The deuteration reaction of 4-fluorobromobenzene (1a), with deuterium oxide as the deuterium source, was selected as the model reaction, because the yield and deuterated ratio of fluorobenzene (2a) could be easily determined with 19F NMR. The selectivity of deuteration and borylation is controlled by the structures of aryl palladium intermediate and diboron. Traditional basic cross-coupling reaction conditions can produce Ar-[Pd]-OR intermediates that coordinate with the Lewis acidic boron center of diboron, leading to borylation product (Table 1, entry 1–2). However, for the cationic palladium Ar-[Pd]+, it is a different mechanism. The coordination of the cationic species with the Lewis acidic boron center is difficult, while facilitating the hydride coordination, even the in-situ generated hydride could be in low concentration. Generation of the Ar-[Pd]+ intermediate from the Ar-[Pd]-Br intermediate necessitates a halide scavenger46,47. However, traditional silver salt halide scavenger48,49 could oxidize diboron reagents and are incompatible with the palladium-hydride intermediate (Table 1, entry 3). In reported methods for halide-scavenger-mediated deuteration reaction, zinc salt was found to be the privileged additive to promote the transmetallation step without side reactions46,47. A variety of zinc salts have been tested, with zinc oxide showing the highest efficiency (Table 1, entry 4–7). Screening of diboron additives shows that diboron reagents with less steric hindrance such as B2eg2 or B2cat2 are more effective for the deuteration reaction (Table 1, entry 8–10). Commercially available palladium catalysts Pd(tBu3P)2 and SPhos Pd G3 exhibit high efficiency with aryl halide substrates50,51,52. Various other palladium catalysts have been tested but demonstrated much lower yields (Table 1, entry 11–14, Supplementary Information Table S1). Reducing the temperature to room temperature but elongation of the reaction time shows similar results (Table 1, entry 15). Although the reactions of aryl bromide with diboron in the presence of a palladium catalyst are typically considered as borylation conditions, a deuteration product 2a was obtained in 92% yield and 93% deuterated ratio when halide-scavenger-mediated conditions were applied.
Substrate scope
With the optimized reaction conditions in hand, the substrate scope of the deuteration reaction was explored (Fig. 2). The reaction exhibits a broad substrate scope and good functional group tolerance under the optimized reaction conditions. Heteroatom-containing rings such as dibenzothiophene, morpholine, carbazole, and thiophene rings are well-tolerated (2c, 2d, 2e, 2h, and 2u). Coordinative N-heterocycles, such as pyrimidine, also show compatibility (3h). Substrates with ketone, trifluoromethyl, halogens, ester, aldehyde, sulfone, amide, imide, cyanide, and carbamate groups are all successfully deuterated (2k, 2l, 2m, 2o, 2q, 2r, 2s, and 2w), which further broaden the substrate scope of the deuteration reactions. Late-stage modification of pharmaceutical compounds containing C–X bonds leads to the analogs of many drug molecules (2t-2w, 3j). Aryl chlorides, as well as heteroaryl chlorides (3a-3j), are all suitable substrates for the deuteration reactions when pre-catalyst AdBrettPhos Pd G353 and B2eg2 were applied (Supplementary Information Table S1). The results are consistent with a facile oxidative addition even for inert C–Cl bonds. Aryl triflates are also suitable substrates for the deuteration reactions (4a-4j). The oxidative addition of palladium(0) to aryl triflate leads to Ar-[Pd]-OTf, which does not involve coordinating halide species, eliminating the need for a halide scavenger such as ZnO (Supplementary Information Table S2). In comparison to other deuteration reactions of aryl (pseudo)halides with D₂O, the deuteration of aryl triflate substrates has not been reported with previous transition-metal-free methods. The results emphasize the broad substrate scope associated with palladium catalysis, distinguishing it from other strategies.

aReaction conditions: aryl (pseudo)halide (0.3–0.9 mmol, 1 equiv.), SPhos Pd G3 (5 mol%), ZnO (2.0 equiv.), B2cat2 (2.0 equiv.), D2O (2.0–4.0 equiv.), THF (0.2 M), N2 atmosphere, 70 °C, 12 h, isolated yields are reported, and deuterated ratios are reported in parentheses. bYield and deuterated ratio were determined by 19F NMR. cZnO and B2cat2 (4.0 equiv.).dD2O (8.0 equiv.) eB2eg2 (2.0 equiv.). fAdBrettPhos Pd G3 (5–15 mol%). gPd(tBu3P)2 (5–20 mol%), w/o ZnO, B2hex2 (2.0 equiv.).
To further demonstrate the utility of the deuteration strategy, several deuterium-labeled drug molecules were synthesized using current methodology (Fig. 3). Drug molecules, such as naproamide and ipriflavon, could be easily halogenated in the presence of NBS or NCS12,13,14,15. Under standard conditions, the halogenated derivatives could be successfully transformed to the deuterated product [2H]-napropamide (2x), [2H]-aniracetam (2 y), [2H]-clotrimazole (2z), and [2H]-ipriflavone (3k) in high yields and high deuterated ratios. Many aromatic drug molecules metabolize to their phenolic forms because cytochrome P450 enzymes (CYPs) catalyze electrophilic hydroxylation of aromatic compounds during phase one metabolism54,55. Our strategy offers a way to recover these drug metabolites, converting them back into metabolic hotspot-blocked compounds. For example, when the flurbiprofen metabolite56 methyl ester was treated with triflic anhydride, the resulting aryl triflate served as a substrate for the deuteration reaction, leading to the conversion of the drug metabolite into a more metabolically stable compound, [2H]-flurbiprofen methyl ester (4k). Overall, the approach showcases high reactivity in the presence of a variety of functional groups, making it a versatile method for incorporating deuterium into various drug molecules through late-stage modifications.

aReaction conditions: aryl (pseudo)halide (0.3 mmol, 1 equiv.), SPhos Pd G3 (5 mol%), ZnO (2.0 equiv.), B2eg2 (2.0 equiv.), D2O (4.0 equiv.), THF (0.2 M), N2 atmosphere, 70 °C, 12 h, isolated yields are reported, and deuterated ratios are reported in parentheses. bAdBrettPhos Pd G3 (5 mol%). cPd(tBu3P)2 (5 mol%), w/o ZnO, B2hex2 (2.0 equiv.).
Preliminary mechanistic studies
Control experiments with 4-phenylphenyl triflate as substrate show that the deuteration reaction proceeds smoothly, yielding the deuteration product in 69% isolated yield with a 95% deuterated ratio in the presence of D2O and B2pin2 (Fig. 4a). However, when only B2pin2 is used without D2O, the reaction shows <5% conversion under standard conditions. The results are consistent with the mechanism in which the in-situ generated D– equivalent from D₂O plays a key role in enabling efficient transmetallation with aryl palladium. Control experiments with aryl bromide and aryl chloride substrates show a similarly important role of D₂O for high conversion (Supplementary Information Pages S54–S55). When a typical deuteride donor D-Bpin was applied instead of diboron and D2O, an efficient hydride transfer reaction occurred, producing 4a in 41% isolated yield with a 70% deuterated ratio. The results are consistent with the facile transmetallation between Ar-[Pd]-OTf and deuteride donor under reaction conditions. The palladium-catalyzed umpolung reaction of water in the presence of diboron has been reported45,57,58, and our control experiments are consistent with a mechanism in which the aryl palladium undergoes transmetallation with the in-situ generated deuteride donor.

a Control experiment. b Plausible mechanism for the palladium-catalyzed deuteration reaction of aryl halides with deuterium oxide.
Based on the preliminary mechanistic study results, we propose a catalytic cycle (Fig. 4b). First, palladium(0) undergoes oxidative addition with diboron to form an intermediate A. Subsequently, deuterated water could coordinate to the Lewis acidic boron atom in intermediate A to form an tetracoordinated boron species, which then eliminates DO-B(OR)2 to furnish palladium deuteride species intermediate B. Intermediate B exists in equilibrium with the deuteride-boron species and palladium(0). Simultaneously, the palladium(0) can undergo oxidative addition with aryl halides, producing an Ar-[Pd]-X species C. In the presence of a halide scavenger such as zinc salts, a cationic Ar-[Pd]+ species D is generated46,47. The cationic palladium D favors the transmetallation with the in-situ generated deuteride species, rather than diboron, leading to Ar-[Pd]-D intermediate E. Finally, a reductive elimination process of E produces the deuterated product and regenerates palladium(0).
Discussion
In conclusion, we have developed an efficient palladium-catalyzed deuteration reaction of aryl (pseudo)halides with D2O as deuterium source. The reaction is enabled by the merger of a D2O umpolung reaction with a cross-coupling reaction via a cationic aryl palladium intermediate. The efficient method for incorporating deuterium into drug molecules with D2O makes it a practical approach for late-stage deuteration. Further investigation into the deuteration reactions of other electrophiles via the cationic palladium intermediates is in progress in our lab.
Methods
General procedure for deuteration reactions of aryl bromides
Under a nitrogen atmosphere, SPhos Pd G3 (11.7 mg, 15 μmol, 5.0 mol%), ZnO (48.6 mg, 0.600 mmol, 2.00 equiv.), B2cat2 (142.8 mg, 0.600 mmol, 2.00 equiv.), and aryl bromide (0.300 mmol, 1.00 equiv.) were added to a 4 mL vial containing a magnetic stir bar. Subsequently, D2O (24.3 mg, 1.20 mmol, 4.00 equiv.) in THF (1.5 mL, c = 0.2 M) was added into the tube. Subsequently, the reaction mixture was stirred vigorously at 70 °C for 12 h in a heating block. After that, the reaction vessel was opened to air, and the resulting mixture was concentrated by rotary evaporation. The residue was purified by chromatography on silica gel to obtain the pure product.
General procedure for deuteration reactions of aryl chlorides
Under a nitrogen atmosphere, AdBrettPhos Pd G3 (15.2 mg, 15.0 μmol, 5.0 mol%), ZnO (48.6 mg, 0.600 mmol, 2.00 equiv.), B2eg2 (84.6 mg, 0.600 mmol, 2.00 equiv.), and aryl chloride (0.300 mmol, 1.00 equiv.) were added to a 4 mL vial containing a magnetic stir bar. Subsequently, D2O (24.3 mg, 1.20 mmol, 4.00 equiv.) in THF (1.5 mL, c = 0.2 M) was added into the tube. Subsequently, the reaction mixture was stirred vigorously at 70 °C for 12 h in a heating block. After that, the reaction vessel was opened to air, and the resulting mixture was concentrated by rotary evaporation. The residue was purified by chromatography on silica gel to obtain the pure product.
General procedure for deuteration reactions of aryl triflates
Under a nitrogen atmosphere, Pd(tBu3P)2 (7.8 mg, 15 μmol, 5.0 mol%), B2hex2 (152.4 mg, 0.600 mmol, 2.00 equiv.), and aryl triflate (0.300 mmol, 1.00 equiv.) were added to a 4-mL vial containing a magnetic stir bar. Subsequently, D2O (24.3 mg, 1.20 mmol, 4.00 equiv.) in THF (1.5 mL, c = 0.2 M) was added into the tube. Subsequently, the reaction mixture was stirred vigorously at 70 °C for 12 h in a heating block. After that, the reaction vessel was opened to air, and the resulting mixture was concentrated by rotary evaporation. The residue was purified by chromatography on silica gel to obtain the pure product.
Data availability
The data supporting the findings of this study are available within the article and its Supplementary Information file. All other data were available from the corresponding author upon request.
References
Javed, T. FDA approval of deuruxolitinib: a new treatment option for severe alopecia areata. Clin. Res. 5, 23 (2024).
Di Martino, R. M. C., Maxwell, B. D. & Pirali, T. Deuterium in drug discovery: progress, opportunities and challenges. Nat. Rev. Drug Discov. 22, 562–584 (2023).
Pirali, T., Serafini, M., Cargnin, S. & Genazzani, A. A. Applications of deuterium in medicinal chemistry. J. Med. Chem. 62, 5276–5297 (2019).
Mullard, A. Deuterated drugs draw heavier backing. Nat. Rev. Drug Discov. 15, 219–222 (2016).
Atzrodt, J., Derdau, V., Kerr, W. J. & Reid, M. Deuterium‐and tritium‐labelled compounds: applications in the life sciences. Angew. Chem. Int. Ed. 57, 1758–1784 (2018).
Morgan, A. J. et al. Design and synthesis of deuterated boceprevir analogs with enhanced pharmacokinetic properties. J. Label. Compd. Radiopharm. 54, 613–624 (2011).
Sharma, R. et al. Deuterium isotope effects on drug pharmacokinetics. I. System-dependent effects of specific deuteration with aldehyde oxidase cleared drugs. Drug Metab. Dispos. 40, 625–634 (2012).
Kopf, S. et al. Recent developments for the deuterium and tritium labeling of organic molecules. Chem. Rev. 122, 6634–6718 (2022).
Li, N., Li, Y., Wu, X., Zhu, C. & Xie, J. Radical deuteration. Chem. Soc. Rev. 51, 6291–6306 (2022).
Li, H., Shabbir, M., Li, W. & Lei, A. Recent advances in deuteration reactions. Chin. J. Chem. 42, 1145–1156 (2024).
Li, P., Kou, G. S., Qi, L. P. & Qiu, Y. A. Recent advance in electrochemical dehalogenative deuteration. J. Electrochem. 30, 2313005 (2024).
Song, S. et al. DMSO-catalysed late-stage chlorination of (hetero)arenes. Nat. Catal. 3, 107–115 (2020).
Varenikov, A., Shapiro, E. & Gandelman, M. Decarboxylative halogenation of organic compounds. Chem. Rev. 121, 412–484 (2020).
Mondal, H. et al. Late‐stage halogenation of peptides, drugs and (hetero) aromatic compounds with a nucleophilic hydrazide catalyst. Angew. Chem. Int. Ed. 62, e202312597 (2023).
Berger, F. et al. Site-selective and versatile aromatic C−H functionalization by thianthrenation. Nature 567, 223–228 (2019).
Zhang, H. H., Bonnesen, P. V. & Hong, K. Palladium-catalyzed Br/D exchange of arenes: selective deuterium incorporation with versatile functional group tolerance and high efficiency. Org. Chem. Front. 2, 1071–1075 (2015).
Kameo, H. et al. Palladium–borane cooperation: Evidence for an anionic pathway and its application to catalytic hydro‐/deutero‐dechlorination. Angew. Chem. Int. Ed. 58, 18783–18787 (2019).
Zhao, D., Petzold, R., Yan, J., Muri, D. & Ritter, T. Tritiation of aryl thianthrenium salts with a molecular palladium catalyst. Nature 600, 444–449 (2021).
Li, J. et al. Homogenous palladium-catalyzed dehalogenative deuteration and tritiation of aryl halides with D2/T2 Gas. J. Am. Chem. Soc. 146, 31497–31506 (2024).
Zhou, Z. Z., Zhao, J. H., Gou, X. Y., Chen, X. M. & Liang, Y. M. Visible-light-mediated hydrodehalogenation and Br/D exchange of inactivated aryl and alkyl halides with a palladium complex. Org. Chem. Front. 6, 1649–1654 (2019).
Wang, M., Pang, W. H., Yuen, O. Y., Ng, S. S. & So, C. M. Palladium-catalyzed deuterodehalogenation of halogenated aryl triflates using isopropanol-d8 as the deuterium source. Org. Lett. 25, 8429–8433 (2023).
Lin, Z. H., Yao, Y. F. & Zhang, C. P. Deuteration of arylthianthren-5-ium salts in CD3OD. Org. Lett. 24, 8417–8422 (2022).
Wang, X. et al. General and practical potassium methoxide/disilane-mediated dehalogenative deuteration of (hetero) arylhalides. J. Am. Chem. Soc. 140, 10970–10974 (2018).
Caldwell, R. A. Quantitative deuteration of a Grignard reagent. Preparation of 2-butene-2-d1. J. Org. Chem. 35, 1193–1194 (1970).
Li, Y. et al. Organophotocatalytic selective deuterodehalogenation of aryl or alkyl chlorides. Nat. Commun. 12, 2894 (2021).
Lu, L., Li, H., Zheng, Y., Bu, F. & Lei, A. Facile and economical electrochemical dehalogenative deuteration of (hetero) aryl halides. CCS Chem 3, 2669–2675 (2021).
Qu, R. et al. Mechanical‐force‐induced non‐spontaneous dehalogenative deuteration of aromatic iodides enabled by using piezoelectric materials as a redox catalyst. Angew. Chem. Int. Ed. 63, e202400645 (2024).
Mutsumi, T., Iwata, H., Maruhashi, K., Monguchi, Y. & Sajiki, H. Halogen–deuterium exchange reaction mediated by tributyltin hydride using THF-d8 as the deuterium source. Tetrahedron 67, 1158–1165 (2011).
Dewanji, A., Mück‐Lichtenfeld, C. & Studer, A. Radical hydrodeiodination of aryl, alkenyl, alkynyl, and alkyl iodides with an alcoholate as organic chain reductant through electron catalysis. Angew. Chem. Int. Ed. 55, 6749–6752 (2016).
Sun, K. et al. Energy-transfer-enabled photocatalytic transformations of aryl thianthrenium salts. Nat. Commun. 15, 9693 (2024).
Wang, B. et al. Photoinduced catalyst-free deuterodefunctionalization of aryltriazenes with CDCl3. Org. Lett. 26, 4329–4334 (2024).
Wencel-Delord, J. & Glorius, F. C–H bond activation enables the rapid construction and late-stage diversification of functional molecules. Nat. Chem. 5, 369–375 (2013).
Cernak, T., Dykstra, K. D., Tyagarajan, S., Vachal, P. & Krska, S. W. The medicinal chemist’s toolbox for late-stage functionalization of drug-like molecules. Chem. Soc. Rev. 45, 546–576 (2016).
Börgel, J. & Ritter, T. Late-stage functionalization. Chem 6, 1877–1887 (2020).
Guillemard, L., Kaplaneris, N., Ackermann, L. & Johansson, M. J. Late-stage C–H functionalization offers new opportunities in drug discovery. Nat. Rev. Chem. 5, 522–545 (2021).
Zhang, L. & Ritter, T. A perspective on late-stage aromatic C–H bond functionalization. J. Am. Chem. Soc. 144, 2399–2414 (2022).
Diederich, F. & Stang, P. J. (eds.) Metal-Catalyzed Cross-Coupling Reactions (Wiley, 1998).
Lei, A. (ed.) Transition Metal Catalyzed Oxidative Cross-Coupling Reactions (Springer, 2019).
Nicolaou, K. C., Bulger, P. G. & Sarlah, D. Palladium‐catalyzed cross‐coupling reactions in total synthesis. Angew. Chem. Int. Ed. 44, 4442–4489 (2005).
Hartwig, J. F. Evolution of a fourth generation catalyst for the amination and thioetherification of aryl halides. Acc. Chem. Res. 41, 1534–1544 (2008).
Ruiz-Castillo, P. & Buchwald, S. L. Applications of palladium-catalyzed C–N cross-coupling reactions. Chem. Rev. 116, 12564–12649 (2016).
Johansson Seechurn, C. C., Kitching, M. O., Colacot, T. J. & Snieckus, V. Palladium‐catalyzed cross‐coupling: a historical contextual perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. 51, 5062–5085 (2012).
Farizyan, M., Mondal, A., Mal, S., Deufel, F. & van Gemmeren, M. Palladium-catalyzed nondirected late-stage C–H deuteration of arenes. J. Am. Chem. Soc. 143, 16370–16376 (2021).
Dey, J., Kaltenberger, S. & van Gemmeren, M. Palladium(II)‐catalyzed nondirected late‐stage C(sp2)−H deuteration of heteroarenes enabled through a multi‐substrate screening approach. Angew. Chem. Int. Ed. 63, e202404421 (2024).
Kong, W., Wang, Q. & Zhu, J. Water as a hydride source in palladium‐catalyzed enantioselective reductive Heck reactions. Angew. Chem. Int. Ed. 56, 3987–3991 (2017).
Niwa, T. et al. Lewis acid-mediated Suzuki–Miyaura cross-coupling reaction. Nat. Catal. 4, 1080–1088 (2021).
Niwa, T., Takimoto, T., Sakata, Y. & Hosoya, T. Palladium-catalyzed ipso-borylation of aryl halides promoted by Lewis acid-mediated electrophilic activation of aryl (halo)palladium (II) complex. Org. Lett. 25, 8173–8177 (2023).
Stambuli, J. P., Incarvito, C. D., Bühl, M. & Hartwig, J. F. Synthesis, structure, theoretical studies, and ligand exchange reactions of monomeric, T-shaped arylpalladium (II) halide complexes with an additional, weak agostic interaction. J. Am. Chem. Soc. 126, 1184–1194 (2004).
Abelman, M. M., Oh, T. & Overman, L. E. Intramolecular alkene arylations for rapid assembly of polycyclic systems containing quaternary centers. A new synthesis of spirooxindoles and other fused and bridged ring systems. J. Org. Chem. 52, 4130–4133 (1987).
Fu, G. C. The development of versatile methods for palladium-catalyzed coupling reactions of aryl electrophiles through the use of P(t-Bu)3 and PCy3 as ligands. Acc. Chem. Res. 41, 1555–1564 (2008).
Proutiere, F. & Schoenebeck, F. Solvent effect on palladium‐catalyzed cross‐coupling reactions and implications on the active catalytic species. Angew. Chem. Int. Ed. 50, 8192–8195 (2011).
Martin, R. & Buchwald, S. L. Palladium-catalyzed Suzuki−Miyaura cross-coupling reactions employing dialkylbiaryl phosphine ligands. Acc. Chem. Res. 41, 1461–1473 (2008).
Cheung, C. W., Surry, D. S. & Buchwald, S. L. Mild and highly selective palladium-catalyzed monoarylation of ammonia enabled by the use of bulky biarylphosphine ligands and palladacycle precatalysts. Org. Lett. 15, 3734–3737 (2013).
Trager, W. F. Principles of drug metabolism 1: redox reactions, in ADME-Tox Approaches, in Comprehensive Medicinal Chemistry, 2nd Edn., Triggle, D. J. & Taylor, J. (eds.) 87-132 (Elsevier, 2007).
Spatzenegger, M. & Jaeger, W. Clinical importance of hepatic cytochrome P450 in drug metabolism. Drug Metab. Rev. 27, 397–417 (1995).
Adams, W. J., Bothwell, B. E., Bothwell, W. M., VanGiessen, G. J. & Kaiser, D. G. Simultaneous determination of flurbiprofen and its major metabolite in physiological fluids using liquid chromatography with fluorescence detection. Anal. Chem. 59, 1504–1509 (1987).
Cummings, S. P., Le, T. N., Fernandez, G. E., Quiambao, L. G. & Stokes, B. J. Tetrahydroxydiboron-mediated palladium-catalyzed transfer hydrogenation and deuteriation of alkenes and alkynes using water as the stoichiometric H or D atom donor. J. Am. Chem. Soc. 138, 6107–6110 (2016).
Yaghoubi, M., Reyes, I. C. & Stokes, B. J. Pd-catalyzed transfer hydrogenation of alkenes using tetrahydroxydiboron as the sole hydrogen donor. SynOpen 8, 169–172 (2024).
Acknowledgements
The National Natural Science Foundation of China (22401209), Jiangsu Province Basic Research Special Fund (BK20240760), Jiangsu Specially Appointed Professors Plan, and the Start Funding of Soochow University are acknowledged for financial support. We thank Ms. S. Cong, and Ms. J. Shi for the preparation of aryl triflate substrates. We thank Prof. L. Wu for HRMS and Prof. Q. Sun for GC-MS. We thank Prof. L. Jiao, Prof. C. Zhu, Prof. C. Liu, and Prof. J. Li for helpful discussions.
Author information
Authors and Affiliations
Contributions
L.Z. conceived the project. Y.C., R.Y. developed the deuteration reaction. R.Y., Y.C. explored the substrate scope. T.Z., Q.G. prepared the substrates. Y.C. investigated the mechanism with input from Y.Y. L.Z. wrote the manuscript with the input from all authors. L.Z. directed the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, Y., Yuan, R., Zheng, T. et al. Pd-catalyzed deuteration of aryl halides with deuterium oxide. Nat Commun 16, 2584 (2025). https://doi.org/10.1038/s41467-025-57855-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57855-x
This article is cited by
-
Scalable reductive deuteration of (Hetero)Aryl chlorides with D2O
Nature Communications (2025)
