
Abstract
Inward membrane budding, i.e., the bending of membrane towards the cytosol, is essential for forming and maintaining eukaryotic organelles. In eukaryotes, Arf GTPases initiate this inward budding. Our research shows that Asgard archaea genomes encode putative Arf proteins (AArfs). AArfs possess structural elements characteristic of their eukaryotic counterparts. When expressed in yeast and mammalian cells, some AArfs displayed GTP-dependent membrane targeting. In vitro, AArf associated with both eukaryotic and archaeal membranes. In yeast, AArfs interacted with and were regulated by key organelle biogenesis players. Expressing an AArf led to a massive proliferation of endomembrane organelles including the endoplasmic reticulum and Golgi. This AArf interacted with Sec23, a COPII vesicle coat component, in a GTP-dependent manner. These findings suggest certain AArfs are membrane-associating molecular switches with the functional potential to initiate organelle biogenesis, and the evolution of a functional coat could be the next critical step towards establishing eukaryotic cell architecture.
Similar content being viewed by others


Introduction
The emergence of eukaryotic cells in the Proterozoic Eon represents a major leap of organizational complexity in the history of life1. The advantage afforded by such complexity is exemplified by the quick radiation of the last eukaryotic common ancestor (LECA) into multiple lineages, leading to diverse and sophisticated life forms that now dominate Earth. LECA was already a highly complex cell containing multiple organelles. It has been proposed that the duplication and neo-functionalization of a prototypic organelle formation machinery led to the establishment of various non-endosymbiotic organelles in LECA2,3. However, the precise composition of this prototypic organelle biogenesis machinery remains unclear.
The need to form membrane-based organelles necessitates that ancestral eukaryotic cells possess the ability to alter membrane morphology. In particular, the budding or protruding of membrane towards the cytosol, either from the plasma membrane or from organellar surfaces, is essential for organelle formation (Fig. 1a). Such inward budding can produce vesicular transport carriers such as COPI, COPII, and AP/clathrin-coated vesicles4,5. Vesicular transportation interconnects non-endosymbiotic organelles and supplies proteins and membrane as organelle building blocks. Inward budding can also produce organelle precursors, which become functional organelles upon the loading of additional components, as seen during the biogenesis of peroxisomes6. Conversely, outward budding, i.e., budding away from the cytosol, is primarily employed for the disposal of materials into the extracellular space or the lumenal space of endocytic compartments7(Fig. 1a). Examples include exosomes, enveloped viruses, and lumenal vesicles of endosomes. Outward budding can also support the construction of extracellular structures. However, outward budding alone does not support the formation of subcellular organelles.

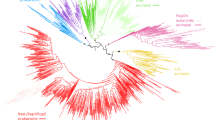
a The role of inward budding in the establishment of eukaryotic endomembrane system. Inward membrane budding refers to the bending of membrane towards the cytosol, either from the surface of organelles or from the plasma membrane. Inward budding supports endomembrane organelle biogenesis either by forming organelle precursors, such as in peroxisome biogenesis, or by forming transport vesicles that supply organelle building blocks. Outward budding refers to the projecting of membrane away from the cytosol, either into the extracellular space or into the lumen of organelles. Outward budding is often employed for disposal of materials to the outside or into the lumen of endocytic/lytic organelles. b Phylogeny of AArfs in relation to other small GTPases. Maximum likelihood phylogenetic tree of small GTPases based on aligned amino acid sequences (179 positions) using IQ-TREE with the LG + R10 model. Branch support values were assessed with the Ultrafast bootstrap algorithm and the SH-aLRT test (both 1000 replicates). c Structural similarity between AArfs and Arf. Structures of AArfs were generated using AlphaFold2. An alignment of yeast Arf1 (PDB: 2K5U) and two AArfs is shown on top. d The G-box motifs are conserved in AArfs. Shown are sequence logos of G-boxes in eukaryotic Arfs, AArfs, and eukaryotic Rabs. e Switch regions are conserved in AArfs. Shown are sequence logos of switch I, II and inter-switch regions in eukaryotic Arfs, AArfs, and eukaryotic Rabs. f Key surface regions, including the guanine nucleotide binding pocket and the effector interacting surface (denoted by yellow circle), are conserved in AArfs. Surface fill models of AArf and eukaryotic Arf are colored by the level of sequence conservation.
Asgard archaea are considered the closest prokaryotic relatives of eukaryotes8,9,10,11,12,13. Experimental evidence has revealed that they contain an actin-based cytoskeleton, and a likely complete ESCRT protein machinery, which may function in outward budding or cytokinesis14,15,16,17. Certain cultured Asgard cells have been observed with membrane tubules protruding away from their cell bodies14,18, consistent with the presence of molecular machinery mediating outward membrane budding. However, the evolutionary status of Asgard archaea with respect to the development of inward membrane budding capacity and the acquisition of a prototypic organelle formation machinery remains unresolved.
In eukaryotes, Arf family GTPases function as molecular switches that initiate the inward budding of coated vesicles19,20,21. An Arf protein can exist in a stable GTP-loaded active state or a GDP-loaded inactive state, unless triggered by regulators to switch between these states. In the active state, an Arf protein specifically binds to certain cellular membrane and recruits coat proteins, which in turn deform the target membrane to protrude towards the cytosol.
Here, through a combination of in silico, biochemical, and cell biology analyses, we demonstrate that Asgard archaea possess ancestral Arf proteins with the functional potential to drive organelle biogenesis, and are poised to take the next critical step in eukaryogenesis to acquire inward budding capacity.
Results
Asgard genomes encode Arf-like proteins
Using a stringent criterion (see Materials and Methods for details), we identified 210 Asgard Arf-like proteins (hereafter referred to as AArfs) from 214 Asgard genomes (Supplementary Dataset D1, D2, D3). AArfs are widely present in Gerdarchaeales, Heimdallarchaeales, and Hodarchaeales, with each genome containing on average five, and up to thirteen AArfs (Supplementary Fig. S1A, B). Phylogenetic analysis placed AArfs in a monophyletic clade clustered with eukaryotic Arf proteins (Fig. 1b, Supplementary Dataset D4). The AArf clade is clearly separated from other Ras superfamily clades, such as eukaryotic Rab, Ran, Rho, Ras, Rag, and prokaryotic MglA. In our analysis, a group of Asgard proteins previously proposed to be Arf-like (denoted as Arfl)22 reside close to Rag and Rup2 clades, and are excluded from the Arf clade.
To take tertiary structure into consideration, we predicted structures of AArfs using AlphaFold2 (Fig. 1c, Supplementary Dataset D5)23, and then used Foldseek server to find AArfs’ closest structural homologues in the PDB database24. We also used Foldseek to query deposited AArf crystal structures (8OUM and 8OUL)25. In both cases, the structural homologues returned by Foldseek were all Arf proteins (Supplementary Dataset D6).
Because proteins can adopt new functions during evolution despite maintaining overall structural similarity, we examined the primary sequences and predicted structures in closer detail to assess the potential of AArfs to function as Arf GTPases. Most P-loop GTPases, including those of the Ras superfamily, contain signature residues in five G-box motifs centered on loop regions26. The residues are important for nucleotide binding and hydrolysis. We found that the G-box motifs are well conserved in AArfs (Fig. 1d). In particular, the [NT]KxD motif of G4 and the SA motif of G5 responsible for guanine selectivity are retained, unlike lineages that switched nucleotide specificity during evolution such as myosin and kinesin26. Arf family-specific features are also present27, such as the leucine and aspartate residues at the second and third positions of G1 motif GxxxGK[ST], and the glycine residue at the third position of G3 motif DxxG.
As molecular switches, small GTPases alter confirmations of a loop region (switch 1) and a helix region (switch 2) during GTP/GDP cycles28. Arf GTPases have two additional structural elements that are sensitive to GTP/GDP binding, a N-terminal amphipathic helix and an inter-switch beta-hairpin that connects switch 1 and switch 2 (Fig. 1c)28,29. The amphipathic helix mediates the association of Arfs with membrane. In the GTP-bond state, the inter-switch hairpin slides out and prevents tight association of N-terminal helix with the GTPase core. We found that key residues in both switch regions are conserved in AArfs (Fig. 1e). Their inter-switch regions are of typical length (Supplementary Dataset D7). At the tertiary structural level, residues from the switches and adjacent elements form a hydrophobic surface on Arf proteins, which mediates interaction with Arf regulators and downstream effectors28,30. This surface is present on AArfs, and contain conserved amino acids (Fig. 1f), suggesting that this surface is functionally important and may assume related functions.
Across the AArf clade, the N-termini are structurally diverse, some of which constitute additional domains (such as longin) (Supplementary Dataset D8). Presence of additional domains have also been observed in other Asgard GTPases31. This is different from eukaryotic Arfs, which primarily contain amphipathic helixes at their N-termini for membrane binding, and implies that AArfs are functionally diverse and many do not act on a membrane. However, AArfs remain clustered with Arfs when we used only the core GTPase domain for phylogenetic analysis (Supplementary Fig. S1C, Supplementary Dataset D9). A group of AArfs (19 out of 210) contain N-terminal helixes that are both amphipathic and comparable in length to their eukaryotic counterparts (Supplementary Dataset D10, Supplementary Fig. S2).
Collectively, our in silico analysis suggests that AArfs constitute a distinct clade of Arf family, which now spans to archaea domain. They not only assume Arf-like structural fold, but also possess structural elements essential for functioning as GTP-dependent molecular switches. Furthermore, structural features suggest that some of them may act on membranes.
A subgroup of AArfs with GTP-dependent membrane targeting
Next, we performed experiments to see whether AArfs can associate with cellular membranes. In the absence of a suitable Asgard experimental system, we opted to express AArfs in eukaryotic models, including yeast and human cells. To refer to individual AArfs, we used the following scheme: first 3–4 letters from the name of the class or order, followed by a code name of the genome assembly, then a number following a dash to denote a particular member of AArfs encoded by that genome (Supplementary Dataset D1). Thus Heim12F4_9–5 refers to No. 5 of the AArfs encoded in genome 12F4_9 of Heimdallarchaeales.
An initial survey of four AArfs with GFP tagged at the C-termini revealed that Heim12F4_9–5 displayed preferential association to the yeast plasma membrane with some polarization towards the bud (Fig. 2a). Association with internal membrane structures was likely present, as its signal within cells was uneven. The other three AArfs displayed diffuse cytosolic signal (Supplementary Fig. S3A). The plasma membrane targeting of Heim12F4_9–5 was dependent on its GTP loading, as the pattern was retained in the GTP-bound mutant, but lost in the GDP-bound mutant (Fig. 2a). These amino acid substitutions followed established paradigms in the G protein field, with Q68L (equivalent to Q61L in Ras) aimed at reducing GTP hydrolysis32,33, and T28N (equivalent to S17N in Ras) for strong preference of GDP binding34,35. The membrane association was also dependent on the presence of its N-terminal amphipathic helix (Fig. 2a).

a Heim12F4_9-5 displayed membrane targeting when expressed in yeast, and the targeting was dependent on GTP binding and its N-terminal helixe. Representative live cell fluorescent microscopy images are shown. DIC, differential interference contrast. b Four other AArfs displayed GTP dependent membrane targeting in yeast. Representative live cell fluorescent microscopy images are shown. c Heim12F4_9-5 and GerdYT_6_3-5 displayed GTP dependent membrane targeting in Hela cells. Cells were mildly fixed. Representative confocal microcopy images are shown. d Principle of liposome floatation assay. Liposomes float together with associated proteins to the top in a density gradient upon centrifugation. Proteins alone do not float. e, f Heim12F4_9-5-His-MBP floated to top fractions with liposomes of both archaeal and eukaryotic compositions. Heim12F4_9-5-His-MBP was purified from E. coli. Archaeal lipid was extracted from cultured Haloferax volcanii. Eukaryotic composition consisted of synthetic phospholipids and sterol. e representative immunoblots. f quantification of protein levels. Mean ± standard deviation, n = 3. a.u., arbitrary unit. g–k Heim12F4_9-5 association with liposomes was dependent on GTP loading, and independent of myristoylation. Purified full length Heim12F4_9-5, Heim12F4_9-5 lacking amphipathic helix (Heim12F4_9-5ΔN), Sar1, and Arf6 were subject to liposome floatation assay using liposomes of eukaryotic composition. g representative silver stained gels. h–k, quantification of protein levels. Mean ± standard deviation, n = 3. a.u., arbitrary unit. White scale bar, 2 μm. Yellow scale bar, 20 μm. Source data are provided in the Source Data.
We expanded our analysis to additional twenty-two AArfs to examine if such membrane association property is linked to particular archaea species or particular AArf lineages. Among them, four more AArfs displayed membrane association (Fig. 2b and Supplementary Fig. S3): GerdFT2–1, GerdMP5_1-6, GerdYT_6_3-5, and HodYT1_057-4. Testing their corresponding point mutations revealed that similar to Heim12F4_9-5, the membrane targeting of these four AArfs was also GTP dependent (Fig. 2b). The GTP/GDP locking mutations did not affect protein stability, as indicated by their comparable protein levels in comparison to the wild type control (Supplementary Fig. S3B). To ensure that membrane association of AArfs is not restricted to a single eukaryotic model, we further examined the subcellular localization of Heim12F4_9-5 and GerdYT_6_3-5 in human Hela cells, and found that the two AArfs also displayed GTP dependent preferential plasma membrane association (Fig. 2c). These five membrane-targeted AArfs are from different genomes, but located within a small branch in the AArf tree (Fig. 3). In contrast, other AArfs from the same genomes, such as GerdYT_6_3-4 and HodYT1_057-1, displayed diffuse cytosolic signal (Supplementary Fig. S3A). The data indicate that a distinct clade of AArfs found across various Heimdallarchaeia species possesses the capacity to bind to cellular membranes in a GTP dependent manner.

A maximum likelihood phylogenetic tree is shown at the center. The tree was inferred in IQ-TREE with LG + R5 model automatically selected. Brach support was evaluated by ultrafast bootstrap algorithm (1000 replicates). Each AArf protein is also denoted by a bar surrounding the tree, with bar length proportional to protein length, colored by species lineage. Predicted structures of select AArfs are shown close to the corresponding bars. Cyan, core Arf GTPase structure; gray, additional non-Arf domains present in certain AArf lineages. Stars denote AArfs experimentally tested in yeast; red stars, those that displayed prominent membrane association; green starts, the rest.
To further characterize the membrane binding property of AArfs, we performed an in vitro liposome co-floatation assay (Fig. 2d). When a protein-liposome mixture is laid at the bottom of a density gradient, liposomes will float to the top upon centrifugation, bringing up membrane-associated proteins. We tried to express Heim12F4_9-5 and GerdYT_6_3-5 in bacteria and were able to obtain soluble Heim12F4_9-5. To test whether AArf membrane association differs between eukaryotic and archaeal lipid compositions36, we prepared two types of liposomes, one using synthetic lipids mimicking eukaryotic membrane, one using lipids extracted from a cultured archaea, Haloferax volcanii. In both cases, Heim12F4_9-5 loaded with GMP-PNP, a non-hydrolysable GTP analogue, co-floated to the top fractions upon centrifugation (Fig. 2e, f), demonstrating that this AArf is capable of binding to both eukaryotic and archaeal membranes. The binding of Heim12F4_9-5 to liposomes was substantially reduced when it was loaded with GDP (Fig. 2g–i). The difference between GMP-PNP loaded and GDP loaded forms was less substantial when its N-terminal amphipathic helix was truncated, suggesting that the N-terminal helix is integral to the nucleotide controlled switch-like behavior.
For many but not all eukaryotic Arfs, their membrane association depends on myristoylation of their N-terminal helixes. We used three tools to evaluate the potential presence of myristoylation on AArfs: myristoylator, “NMT - the MYR Predictor”, and GPS lipid37,38,39, and found that AArfs do not contain recognizable myristoylation sites (Supplementary Dataset D11). Myristoylation often occurs on the second glycine of the N-terminal helix. When we mutated this residue on Heim12F4_9-5, its membrane targeting in yeast cell remained (Supplementary Fig. S3C). This is consistent with our in vitro floatation assay result, in which proteins were expressed in E. coli in the absence of a myristoylation pathway. We further included two Arfs as controls. Sar1 is an Arf family member that is not myristoylated, whereas Arf6 is one that rely on myristoylation. In our floatation assay, both Heim12F4_9-5 and Sar1 were able to bind liposomes, whereas Arf6 was not (Fig. 2g, j, k). These data indicate that AArfs do not rely on myristoylation for membrane association.
AArfs as hubs in a protein network controlling organelle dynamics
Next, we evaluated the potential of AArfs to function like eukaryotic Arfs. We considered two aspects: 1, whether AArfs can be regulated like eukaryotic Arfs, and 2, whether their interactomes imply a role for them in organelle dynamics.
To initiate membrane deformation at the right time and location, eukaryotic Arfs are tightly regulated by guanine nucleotide exchange factors (GEFs), GTPase-activating proteins (GAPs), and other members of Arf family20,21,29. To test whether AArfs can be similarly regulated, we examined the subcellular localization of AArfs upon overexpression of Arf GEFs and GAPs. We opted for overexpression instead of loss of function mutants because many Arfs and their regulators are essential for survival, while functional redundancy exists among others. To avoid saturating potential regulators, here we expressed AArfs at a lower level using the ADH3 promoter. Except for an overall weaker signal resulting from the promoter choice, both Heim12F4_9-5 and GerdYT_6_3-5 displayed polarized plasma membrane association prior to galactose-induced overexpression of GEFs/GAPs (Fig. 4a and Supplementary Fig. S4). Upon induction, seven out of eight tested Arf GEFs altered the localization of AArfs (Fig. 4a). Sec12 and Sed4 are GEFs for Sar1, which functions at the endoplasmic reticulum (ER). Enrichment of AArfs on structures resembling the ER or spots associated with the ER were observed upon their overexpression. Gea1, Gea2, and Syt1 are GEFs for Arf1 and Arl1, which function in Golgi related trafficking. Overexpression of Gea1 and Gea2 led to enrichment of AArfs on intracellular spots. Syt1 abolished AArf polarization, resulting in AArfs concentrating on discontinuous segments along the plasma membrane. Snf1 and Yel1 are GEFs for Arf3, which functions in endocytosis. Overexpression of Snf1 and Yel1 led to enrichment of AArfs on round intracellular structures. Overall, the target of AArf relocation upon GEF overexpression appeared to be related to the endogenous target of the GEFs. To further confirm that AArfs were targeted to the same subcellular location as the GEFs, we constructed a second set of GEF constructs tagged with mCherry. The changes in AArf subcellular distribution upon their expression were overall similar to what we observed with the GST tagged constructs (Supplementary Fig. S5). The colocalization between GFP tagged AArfs and mCherry tagged GEFs implies that the GEFs were directly responsible for the subcellular targeting of AArfs. Among eight Arf GAPs tested, four changed AArf localization (Supplementary Fig. S4). In general, GAP overexpression led to relocation patterns that were different from those of GEF overexpression, potentially reflecting the complex roles of Arf GAPs and of GTP hydrolysis20,40. These data demonstrate that AArfs expressed in eukaryotic host can be regulated by Arf GEFs/GAPs.

a AArfs can be regulated by Arf GEFs. Overexpression of several Arf GEFs altered the subcellular distribution of AArfs. Shown are representative live cell fluorescent microscopy images of yeast cells expressing Heim12F4_9-5-GFP or GerdYT_6_3-5-GFP before and 4 h after galactose-induced GEF overexpression. Fluorescent intensity profiles of cropped areas are presented at the side, with positions of plasma membrane marked by red arrows. Schematic AArf subcellular distribution patterns are shown at the bottom. b, c AArfs can interact with a protein network regulating organelle dynamics. b GO term enrichment in GerdYT_6_3-5 interactome in yeast. Membrane and organelle related terms are colored blue. Data analyzed by two-sided t-test with no adjustment for multiple comparisons. Significance threshold (s0), 0; false discovery rate (FDR), 0.05. c A sub-network of GerdYT_6_3-5 interacting proteins, consisting of those mediating intracellular protein transport, organelle function, and lipid metabolism. Lines and dashed lines denote known protein-protein interactions in STRNG. White scale bar, 2 μm.
The function of eukaryotic Arfs depends on their ability to interact with a network of proteins mediating organelle dynamics. We surveyed the interatomes of AArfs in yeast via affinity purification coupled with mass-spectrometry (Supplementary Fig. S6A, B). Note that because our goal was to identify the interactome of a target protein, not the proteome of a membrane structure, detergent was present in our affinity purification buffer to solubilize membranes. For both Heim12F4_9-5 and GerdYT_6_3-5, we found that their interactomes were enriched in proteins with organelle related functions (Supplementary Fig. S6C and Fig. 4B). These include Arf members (Arl1, Arf1), Arf GEFs (Sed4, Sec7), other classes of organelle dynamics regulators such as Rab GTPases (Ypt31, Sec4), and proteins involved in membrane lipid metabolism (Supplementary Fig. S6D, Fig. 4c, and Supplementary Dataset D12). Notably, the interactome of GerdYT_6_3-5 included Sec24, Sec26, and Sec27, which are components of vesicle coats that function downstream of Arfs to mediate membrane deformation (Fig. 4c)20,40. These data imply that AArfs have the potential to act as central nodes within a protein interaction network that regulates organelle structure and function.
AArf can regulate eukaryotic endomembrane system
We then examined if AArf can indeed act on an endomembrane system. We performed transmission electron microscopy on yeast cells expressing either the wild-type or GDP-bound form of the five membrane enriched AArfs. Strikingly, we found that cells expressing GerdYT_6_3-5 displayed a massive expansion of an ER-like membrane network (Fig. 5a, b and Supplementary Fig. S7A, B). Normally, the yeast ER consists of a peripheral reticulum attached to the plasma membrane via membrane contacts, the nuclear envelope (also known as the nuclear ER), and structures interconnecting the nuclear envelope with the peripheral reticulum. Expression of wild-type GerdYT_6_3-5, but not the GDP-bound form, led to the appearance of an extensive endomembrane network (Fig. 5a, b). Such alteration was visible in 51 ± 7% of the cell sections (mean ± standard deviation, three independent repeats, at least 100 cell sections per repeat). The network was interconnected with the nuclear envelope. It frequently formed additional membrane linings beneath the plasma membrane. Unlike the string-like interconnecting structures that spanned the cytoplasm of wild-type cells, the cytoplasmic portion of this network contained “soap bubble” like structures.

a, b Expression of GerdYT_6_3-5 in yeast triggered the emergence of a massively expanded endomembrane network. a representative transmission electron micrographs of yeast cells expressing wild-type or GDP-bound mutant of GerdYT_6_3-5. b schematic representation of yeast endoplasmic reticulum morphology. Cyan lines denote the endoplasmic reticulum. c, d Expression of GerdYT_6_3-5 in yeast triggered the proliferation of endomembrane organelles. Yeast cells expressing red fluorescent protein tagged organelle markers in combination with GFP alone or GFP tagged wild type or GDP-bound mutant of GerdYT_6_3-5 were observed by live cell fluorescent microscopy. c representative microscopy images of cells carrying ER and Golgi markers. See Fig. S7C for other organelle markers. d quantification of the number of punctate organelles per cell. Mean ± standard deviation, n = 3 independent repeats (>50 cells quantified in each sample of each repeat). Analyzed by two-way ANOVA with Tukey test. Exact P values (from left to right): <0.01, 0.95, <0.01, 0.99, <0.01, 0.52, 0.07, 0.92, <0.01, 0.54. e GerdYT_6_3-5 could interact with Sec23, a component of COPII coat, in a GTP dependent manner. GTP (Q87L) or GDP-bound (T47N) variants of GerdYT_6_3-5-GTP were co-expressed with Sec23-8V5 in yeast. The interaction between GerdYT_6_3-5 and Sec23 was evaluated by co-immunoprecipitation. Representative immunoblots are shown. f, g Structural comparison between Sec23 and Asgard Sec23-like proteins. f Alignment of Sec23 (PDB: 1M2O) and predicted structures of Asgard Sec23-like proteins. g Asgard Sec23-like proteins lack a gelsolin-domain critical for Sar1 interaction and catalytic activity. Sec23-Sar1 complex structure rendered from PDB 1M2O. h Status of Asgard archaea in the evolution of inward membrane budding capacity. Asgard archaea possess AArfs that function as molecular switches in membrane related processes. Sec23-like coat ancestor proteins are present, which possess all but one of the Sec23 constituent domains. Subsequent emergence of a functional coat in a single species would complete the evolution of a prototypic inward membrane budding machinery, promoting endomembrane organelle biogenesis. White scale bar, 2 μm. Source data are provided in the Source Data.
To verify the identity of the expansive membrane network and to examine the status of other organelles that are difficult to discern under transmission electron microscopy, we turned to live cell fluorescent microscopy and examined the signal of red fluorescent organelle markers (Fig. 5c, d and Supplementary Fig. S7C)41. Consistent with our interpretation of the electron micrographs, mCherry-HDEL and Elo3-mCherry both revealed the presence of an expanded ER (Fig. 5c, d). mCherry-HDEL is a marker of ER lumen, and Elo3-mCherry is a marker of ER membrane. Besides altered ER structures, we also observed increased numbers of early Golgi (Anp1), late Golgi/early endosomes (Chs5, Sec7), late endosomes (Vps4), and peroxisomes (Pex3). As expected, all these changes were absent when the GDP-bound mutant of GerdYT_6_3-5 was expressed. These data demonstrate that GerdYT_6_3-5 functioned as a GTP-dependent molecular switch to drive the proliferation of yeast endomembrane organelles.
Considering that we identified components of vesicle coats in the interactome of GerdYT_6_3-5 (Fig. 4c), we further explored whether GerdYT_6_3-5 may engage vesicle coats as effectors. We used co-immuno-precipitation to examine the potential interactions and found that many components of COPI and COPII coat could be co-precipitated by GerdYT_6_3-5 (Supplementary Fig. S8A, B). Intriguingly, interaction between Sec23 and GerdYT_6_3-5 was only detected for the GTP locked variant, but not for the GDP locked variant (Fig. 5e). During the assembly of COPII coat in eukaryotes, Sec23 is the subunit that directly interacts with Sar14,20.Our data indicate that Sec23 behaved as an effector of GerdYT_6_3-5, and might have contributed to GerdYT_6_3-5 driven endomembrane proliferation.
Asgard archaea in evolution of inward membrane deformation
Finally, we evaluated the potential of Asgard proteome to form a functional coat. Based on our interactome data, we focused our analysis on potential ancestors of COPI and COPII coats. We used InterProScan to analyze Asgard proteins for the presence of domains in the coat proteins, and found sporadic existence of proteins resembling ε-COP (Sec28 in yeast) and Sec23/Sec24 (Fig. 5f, Supplementary Fig. S1D, S8C, D and Supplementary Dataset D13, D14, D15-D19). COPI is a complex coat constructed from seven subunits: α-, β-, β’-, γ-, δ-, ε-, and ζ-COP. In the absence of other COPI subunits, especially γ- and β-COP that directly bind Arf14,40, we conclude that COPI evolution is at an extremely primitive stage in Asgard.
COPII is functionally and structurally organized into two layers, with Sec23-Sec24 heterodimer forming the inner coat directly in touch with membrane, and Sec13-Sec31 forming the outer coat assembling into cage like structures4. Sec23 and Sec24 are structurally related, and evolve from a single ancestor42. Sec23 is comprised of five domains, including the β-barrel, Zn-finger, trunk, helical, and gelsolin-like domains. The Sec23–Sar1 interaction interface involves the trunk, helical and gelsolin-like domains (Supplementary Fig. S8D)43. As noted previously, Sec23 acts not only as a component of COPII coat, but also as the GAP of Sar1. Its gelsolin domain contributes a key catalytic residue to fulfill the GAP function. We found that all Asgard Sec23-like proteins lack the gelsolin domain (Supplementary Dataset D14, Fig. 5f, g, and Supplementary Dataset D17-D18). They also lack key surface residues important for interaction with Sar1 (Supplementary Fig. S8E). We experimentally examined if Sec23-like proteins could interact with GerdYT_6_3-5 in yeast. In all three cases, the amounts of co-precipitated Sec23-like proteins were low, and did not display sensitivity to nucleotide loading status (Supplementary Fig. S8F). These data indicate that the Asgard Sec23-like proteins are yet to function like their eukaryotic counterparts.
On the flip side, we consider the Asgard Sec23-like proteins true ancestors of eukaryotic Sec23. Their predicted structures suggest that they already contain most of the Sec23 building blocks in similar spatial arrangements (Fig. 5f, g, Supplementary Dataset D18). Using Foldseek to search for their structural homologues in PDB consistently returned eukaryotic Sec23 (Supplementary Dataset D20). We also noticed the presence of gelsolin domains in many other Asgard proteins (Supplementary Dataset D19, D21, D22). Thus the potential transition from a Sec23-like protein to a functional Sec23 essentially entail two aspects: one is the acquisition of a gelsolin domain to complete the overall Sec23 structure, the other the population of key surface residues enabling Sar1 interaction and GAP catalytic activity. Further considering that the COPII outer coat is functionally dispensable in certain cases44,45,46, we envision that the prototypic inward membrane deformation machinery may consist of a simpler design with just an AArf and an inner coat. In this case, the evolution of a functional Sec23 would be the next critical step towards the emergence of endomembrane structures (Fig. 5h).
Discussion
In this work, we identified a group of archaeal proteins that phylogenetically locate within the Arf GTPase family. In silico analysis suggest that these AArfs share with their eukaryotic relatives key structural elements that enable them to function similarly. A small branch of AArfs possess the ability to associate with cellular membrane in a GTP dependent manner when expressed in yeast or mammalian cells. In vitro, AArf-membrane association occurs with membranes of both eukaryotic and archaeal compositions. In yeast, AArfs can interact with, and can be regulated by key players controlling endomembrane organelle dynamics. Functionally, an AArf can engage vesicle coat protein as effector, and impose dramatic changes on endomembrane organelles. These data suggest that AArfs are membrane-associated molecular switches with the potential to become initiators of organelle biogenesis.
The primary purpose of this work is to evaluate the status of Asgard archaea with respect to the evolution of a prototypic endomembrane organelle formation machinery. For this reason, the protein network mediating organelle formation in extant eukaryotes is the reference point to which we evaluate the Asgard proteins against. Eukaryotic Arfs are hubs in this protein network20,40. They translate upstream signals to membrane-association at the right time and location; and they initiate inward membrane deformation by recruitment of downstream effectors. By these criteria, our data indicate that certain AArfs are already well equipped to function in the same manner. In the presence of upstream regulators and downstream effectors, an AArf can bind to membrane and trigger endomembrane proliferation (Fig. 5a–d, Supplementary Fig. S7B).
Our data indicate that AArfs can be regulated by eukaryotic Arf GEFs and GAPs (Fig. 4a, Supplementary Fig. S4, S5). To see if AArfs may utilize GEFs/GAPs with similar structures in their native environment, we first used esmfold to generate 185,575 predicted structures for 52 Asgard genomes containing AArfs47. We then collected known Arf GEF/GAP structures from PDB and used Foldseek to query our Asgard proteome for potential candidates against either the whole proteins or constituent GEF/GAP domains (Supplementary Dataset D23).
Arf GEF structures we collected include: Sec12 and several GEFs based on Sec7 domain (Supplementary Fig. S9A). Foldseek did not return candidates for Sec7 domain. Closer examination of returned Sec12 candidates revealed that they are all WD40/β-propeller proteins (Supplementary Dataset D24). This is a very common domain present in proteins of diverse functions. Compared with Sec12, the Asgard proteins contain a loop that preclude interaction with Sar1 in a similar spatial orientation (Supplementary Fig. S9B, C, Supplementary Dataset D25). Furthermore, using these Asgard proteins as query for a reverse search via Foldseek returned various non-Sec12 proteins (Supplementary Dataset D26). These results indicate these Asgard β-propeller proteins do not function as Sec12. Besides Sec23, additional Arf GAP structures we collected include: GAPs based on ArfGAP domain, RP2, and C9orf72-SMCR8-WDR41 complex (Supplementary Fig. S10A, Supplementary Dataset D23). As mentioned previously, Asgard Sec23-like proteins lacks essential structural elements for GAP function (Fig. 5f–g, Supplementary Fig. S8D, E). Foldseek did not return results for ArfGAP and RP2 domains. Two candidates for the C9orf72–SMCR8–WDR41 complex were identified (Supplementary Dataset D27). However, alignment of their structures with that of C9orf72–SMCR8–WDR41 complex revealed that the Asgard proteins lack the Arf interacting regions (Supplementary Fig. S10B).
The most important functions of GEF and GAP proteins are to switch a G protein on and off by dictating its bound nucleotide, which in turn determines the conformation of a G protein. Our liposome floatation data on purified Heim12F4_9-5 demonstrate that the nucleotide loaded could control its membrane affinity, confirming the presence of nucleotide dependent conformational change (Fig. 2g, h). We also verified that the intrinsic GTP hydrolysis activity of Heim12F4_9-5 was very low and comparable to that of a eukaryotic G protein in the absence of its cognate GAP (Supplementary Fig. S10C). These in vitro data strongly imply that AArfs are nucleotide dependent molecular switches, which is consistent with data in yeast and human cells. Unlike Arf proteins, which are derived from a common ancestor, Arf GEFs and GAPs are structurally diverse and originate from different protein families48. Thus it is highly plausible that native AArf GEFs/GAPs are structurally distinct from their eukaryotic counterparts, and therefore evaded our structure homology based analysis. At present, the identities of AArf regulators and the nucleotide dependent conformational switching in native archaea cells remain to be established.
At the downstream level, the best characterized Arf effectors are the vesicle coat proteins4,20,40. Our analysis of Sec23-like proteins in Asgard proteomes suggest that these proteins are the precursors of COPII inner coat. Compared to eukaryotic Sec23, Asgard Sec23-like proteins are in a promising state, with most of the constituent domains in place, and only a limited number of steps remaining to become a functional Sec23. Considering that Asgard archaea already possess AArfs, we propose that the subsequent emergence of a functional Sec23 in an AArf containing host, either evolved within a lineage or acquired via horizontal gene transfer, would lead to the completion of a basic inward membrane budding machinery. This event would then ignite the formation of endomembrane organelles, a fundamental transition of cellular architecture during eukaryogensis.
Methods
Asgard genomes collection and annotation
All Asgard genomes were downloaded from GenBank and eLMSG (https://www.biosino.org/elmsg/) at the time of analyses, Sep. 1st, 2021. The quality of these genomes was reassessed using CheckM (v1.0.12)49 to remove low-quality genomes (completeness below 50% and contamination above 10%). The complete list of Asgard genomes used in this study is in Supplementary Dataset D2. To ensure annotation homogeneity, Prodigal (v2.6.1)50 was used to predict protein sequences for these selected genomes. All predicted proteins were then annotated using InterProScan (v5.47-82.0)51 with default settings.
Identification of Asgard Arf like proteins and phylogenetic analysis
Only those sequences simultaneously annotated as the Arf family by Pfam (PF00025), CDD (cd00878), and IPRscan (IPR006689) databases were considered as potential Arf homologues, and those shorter than 160 amino acids were omitted. MAFFT (v7.471) was used to perform multiple sequence alignment with other GTPases under a high accuracy mode (-maxiterate 1000 -localpair) 52. The final alignment was trimmed using trimAl (v1.4.rev15)53 with a gap threshold of 50%. Phylogenetic analyses were conducted by IQ-TREE (v2.0.6)54 using MFP mode. Support values were calculated using ultrafast bootstraps and SH-aLRT test with 1000 replicates. Sequence logos were generated using WebLogo 3 online55.
Protein structure prediction and analysis
Protein structure prediction was performed on AlphaFold 2 server23, and structures with rank 1 were used for subsequent analysis. Docking of GTP molecule into GTPase structures was performed in Schrodinger Maestro Elements 4.6.
To color a protein structure based on sequence conservation level, first a multi-sequence alignment was generated with Clustal Omega56,57. The alignment file and a protein structure were supplied to ConSurf server to generate a PyMol script58. The script was then loaded in PyMol 2.5.2.
The potential presence of amphipathic sequences was assessed on HeliQuest server59. Based on the property of eukaryotic Arf proteins, the following parameters were used: hydrophobicity (H), 0.201–0.629; hydrophobic moment (uH), 0.236–0.636; net charge (Z), −5 ~ 5. The search was restricted to helixes of 18 amino acids or longer. Only helixes with at least five hydrophobic residues uninterrupted on one side were qualified.
To classify AArfs based on protein structure, predicted AArf structures were first simplified by removing N- and C-terminal extensions and switch regions in PyMol to retain only the core GTPase regions. The result was then feed to Foldseek server to search the PDB database (20240101)24.
To find structural homologues of Arf GEFs/GAPs, proteome structures of 52 Asgard genomes containing AArfs were predicted by ESMFold47. Foldseek (v6.29e2557) was used to query the collection of predicted structures for Arf GEF/GAP homologues under parameters: --exhaustive-search -e 1e-5.
Prediction of post-translation modifications
Three tools, Expasy Myristoylator (https://web.expasy.org/myristoylator/), NMT - The MYR Predictor (https://mendel.imp.ac.at/myristate/SUPLpredictor.htm), and GPS-Lipid (http://lipid.biocuckoo.org/webserver.php) were used to predict N-terminal myristoylation of Asgard Arf proteins37,38,39. Only proteins predicted as N-terminally myristoylated by at least two tools were considered as candidates.
Plasmid construction
Plasmids used in this work were constructed using conventional molecular cloning (Supplementary Dataset D28). ORF for AArfs were codon optimized and synthesized. Amplification primers are listed in Supplementary Dataset D29. DNA sequences of the plasmid backbones and inserts are listed in Supplementary Dataset D30. The following substitutions were introduced into AArfs to generate GDP-bound and GTP-bound mutants: Heim12F4_9-5: T28N, Q68L; GerdMP5_1-6: T28N, Q68L; GerdFT2-1: T28N, Q68L; GerdYT_6_3-5: T47N, Q87L; HodYT1_057-4: T28N, Q68L. To generate Heim12F4_9-5 mutant lacking N-terminal amphipathic helix, amino acid 2-12 were removed.
Yeast strain construction, culturing, and microscopy
Transformation of yeast was performed using the common lithium acetate method. Prior to transformation, linear DNA fragments were obtained through restriction digestion. BY4741 (MATa his3∆1 leu2∆0 met15∆0 ura3∆0) was used for characterization of AArf localization. TN124 (MATa leu2 ura3 trp1 pho8Δ60 pho13Δ::LEU2) was used for organelle morphology analysis.
The following media were used for culturing of yeast cells. YPD: 1% yeast extract (Oxoid, LP0021B), 2% peptone (Gibco, 211677), 2% glucose (Sangon Biotech, A501991). YPR: 1% yeast extract, 2% peptone, 2% raffinose (Sangon Biotech, A610503). SMD: 2% glucose, 0.67% yeast nitrogen base without amino acids (Sangon Biotech, A610507), 30 mg/L adenine (Sangon Biotech, A600013), 30 mg/L lysine (Sangon Biotech, A602759), 30 mg/L methionine (Sangon Biotech, A610346), 20 mg/L histidine (Sangon Biotech, A604351), 20 mg/L uracil (Sangon Biotech, A610564), 50 mg/L tryptophan (Sangon Biotech, A601911), 50 mg/L leucine (Sangon Biotech, A600922). SMD + CA: SMD supplemented with 0.5% casaminoacid (Sangon Biotech, A100851); SD-Ura: SMD with uracil opted out.
For general fluorescent microscopy, yeast cells were cultured in YPD to mid-log phase. Yeast cells were then collected by centrifugation, and re-suspended in SMD medium for imaging. Image stacks (z-stepping 0.5 um) were collected on an inverted fluorescent microscope (Olympus IX83) with a 100x oil immersion objective (UPLXAPO100XO, Olympus).
To test the effects of GEF/GAP overexpression, overexpression plasmids encoding GST tagged proteins were extracted from a yeast library60. The overexpression plasmids were then transformed into BY4741 based strains carrying AArf constructs. Transformants were fist cultured in SD-Ura medium overnight. For colocalization analysis, plasmids encoding mCherry tagged proteins (Supplementary Dataset D28) were used, and transformants were cultured in SD-Leu instead. Yeast cells were then shifted to YPR medium with a starting cell density of 0.2 OD600. When cell density reached 0.5 OD600, galactose was added to a final concentration of 2% to induce the expression of GEF/GAPs. Prior to or 4 h after galactose addition, cells were collected and imaged in SMG + CA medium on an inverted fluorescent microscope (Olympus IX83). Image stacks were de-convolved in Sparse Deconvolution (v1.0.3) to reduce background noise61. The deconvolution parameters were: sparse iteration, 100; t/z axial continuity, 0.1; image fidelity, 200; sparsity, 50–5, depending on image; deconvolution times, 5; background, weak; algorithm, Lucy-Richardson; oversampling, no.
For transmission electron microscopy (TEM), samples were prepared as described previously62, using a potassium permanganate-based chemical fixation method. Briefly, yeast cells were grown in YPD medium at 30 °C until they reached OD600 of 0.8. Cells were then collected by centrifugation. Fixation was carried out twice using a 2% v/v KMnO4 (Shanghai Taitan Scientific, 7722-64-7)/water solution. Cells were collected washed in purified water for five times. Following this, the cells were harvested and dehydrated through a series of incubations in 10%, 30%, 50%, 70%, 90%, 95%, and 100% v/v acetone/water at room temperature. For embedding, the resin mixture was prepared using a ratio of 49.9% v/v Eponate 12 resin (glycerol polyglycidyl ether, Ted Pella, 18005), 12.4% v/v DDSA (dodecenyl succinic anhydride, Ted Pella, 18022), 37.7% v/v NMA (methyl nadic anhydride, Ted Pella, 18032), and 1.7% v/v DMP-30 phenol (2,4,6,-tri (dimethylaminomethyl) phenol, Ted Pella, 18042). Cells were then transferred sequentially to 33%, 66%, and 100% v/v resin/acetone at room temperature. Finally, the tubes were incubated at 60 °C for 48 h to polymerize the resin.
Mammalian cell culturing, transfection, and microscopy
Hela cells (ATCC, CCL-2) were cultured in Dulbecco modified Eagle medium (DMEM) (Gibco, C11995500) supplemented with 10% fetal bovine serum (FBS) (HyClone, SV30087.03) and 1% penicillin-streptomycin (BBI Life Sciences Corporation, E607011), at 37 °C with 5% CO2. Plasmids expressing GFP-tagged archaeal proteins were transfected using Lipofectamine 2000 (Thermo Fisher Scientific, 11668019). Culturing media were replaced 9 h after transfection. 24 h after transfection, cells were detached by treatment with 0.25% trypsin-EDTA (Gibco, 2323363). Harvested cells were then allowed to attach to cover glass (NEST, 801008) for 2 h. Finally, culturing media were discarded and cells were fixed with 4% polyformaldehyde (Sangon Biotech, A500684) for 30 min at room temperature. Fixed cells attached to cover glass were imaged on a spinning disk confocal microscope (Olympus, IXplore SpinSR).
Affinity-purification and mass spectrometry to identify AArf interactors
Yeast cells expressing an AArf were grown in YPD to mid-log phase and collected. Cells were re-suspended in IP buffer (0.02 M PIPES/KOH (Sangon Biotech, A600719, A610441) pH = 6.8, 0.3 M KCl (Sangon Biotech, A501159), 1 % v/v glycerol (Sangon Biotech, A600232), 1 mM EDTA (Sangon Biotech, A500838), 1 mM PMSF (Sangon Biotech, A430281), 1 mM DTT (Sangon Biotech, A620058), 0.02% v/v NP-40 (Sangon Biotech, A500109), aprotinin (Sangon Biotech, A422278) 5 μg/ml, leupeptin (Sangon Biotech, A600580) 1 μg/ml, pepstatin A (Sangon Biotech, A421667) 1 μg/ml), and lysed by glass beads. Cell lysate was subject to 120,000 g centrifugation at 4 °C for 10 min. Supernatant was incubated with GFP or V5 affinity beads (Abcam AB206566, ThermoFisher 10004D) at 4 °C for 2 h. The affinity beads were collected and washed twice in IP buffer, once in IP buffer without NP-40. Samples were Trypsin digested and analyzed on an Easy-nLC1200/Q Exactive plus system (ThermoFisher). Mass spectrometry data were analyzed in MaxQuant v2.0.3.1 to obtain peptide abundance63. Data from three independent repeats were then analyzed in Perseus v2.0.3.164 following procedures described in ref. 65. GO term enrichment was analyzed on DAVID server66. Graphic illustration was generated using ggbreak in R67. See Supplementary Note for additional technical details of mass spectrometry.
Preparation of archaeal lipids
Haloferax volcanii was grown in 10 L batch culture in basal medium supplemented with yeast extract, peptone, and casamino acid, pH 7.568. After culturing at 37 °C for 72 h, cells were collected by centrifugation. Lipids were extracted by the modified Bligh-Dyer method69. Briefly, extraction was carried out four times using dichloromethane (Sinopharm Chemical Reagent, 800473191): methanol (Sinopharm Chemical Reagent, 10014128): aqueous buffer = 1:2:0.8, with two extractions using phosphate buffered saline (pH 7.4) and two more using 5% trichloroacetic acid (Sinopharm Chemical Reagent, T104257). The supernatants were combined, and dichloromethane and water were added to reach a dichloromethane: methanol: aqueous buffer ratio of 1:1:0.9. The organic phase was isolated. The remaining aqueous phase underwent three more extractions using dichloromethane to combine with the initial extraction.
Purification of AArf from bacteria
Proteins on pET vector were expressed in Escherichia coli BL21 (DE3) cells for 18 h at 16 °C and induced by addition of isopropyl-β-D-thiogalactoside (IPTG) (Inalco, 1758-1400) at a final concentration of 0.25 mM at OD600 between 0.6 and 0.8. The His6-tagged proteins were purified using Ni2+–nitrilotriacetic acid agarose affinity chromatography (Cytiva) followed by Superdex-200 26/60 size exclusion chromatography in the buffer containing 50 mM Tris pH 8.0 (Sangon Biotech, A600194), 100 mM NaCl (SCR, 10019360), 2 mM MgCl2 (SCR, 10012818), and 2 mM β-mercaptoethanol (J&K Scientific, 249096).
In vitro assay assessing membrane association of purified AArf protein
Liposome floatation assay was performed as previously described with some modification70.
For synthetic lipids mimicking eukaryotic composition, 1 mM lipids mixture containing 80% phospholipids (66%DOPC, 21%DOPE, 8%DOPS, and 5%DOPA) (Avanti Polar Lipids, 850375 P, 850725 P, 840035 P, 840875 P) and 20% ergosterol (Sigma, 45480) was used in each liposome preparation. For archaeal lipids, 1/6 of extracted lipids were used. Lipids were dried, and lipid films were re-suspended in 1 ml proteoliposome buffer (20 mM HEPES (Sangon Biotech, E607018), pH 7.5, 150 mM NaCl (Sangon Biotech, A501218), 1 mM TCEP (tris(2-carboxyethyl)phosphine) (Millipore, 580560), 0.5 mM MgCl2 (Sangon Biotech, A601336)). The re-suspended lipid films were frozen and thawed for six times, then extruded through a 100 nm polycarbonate filter with an extruder for at least 21 times.
Prior to the floatation assay, 285 μL of purified G protein was mixed with 200 μl liposome or proteoliposome buffer in the presence of 5 mM EDTA and 500 μM GMP-PNP in a total volume of 500 μl. The mixture was incubated at 4 °C with gentle inversion for 1 h. Concentration of MgCl2 (Sigma, 68475) was then brought to 15 mM with the addition of 2 M stock solution, and incubated at 4 °C with gentle inversion for 20 min.
Sucrose density gradient was prepared in MSB buffer (25 mM HEPES, 1 mM magnesium acetate (Sangon Biotech, A501341)). 500 μl liposomes were gently mixed with 50% (w/v) sucrose (Sigma, V900116) and MSB buffer to achieve a final sucrose concentration of 35% (w/v), and placed at the bottom of an Ultra-Clear tube. It was overlaid with 4 ml 25% (w/v) sucrose, and then 0.8 ml MSB buffer. Samples were centrifuged at 280,000 g using a SW41Ti rotor (Beckman) for 4 h at 4 °C. After centrifugation, six fractions were collected from top to bottom with volumes of 0.8 ml, 0.8 ml, 1.5 ml, 1.5 ml, 3.5 ml, and 3.5 ml, respectively. For protein detection by immunoblotting, each fraction was treated with a final concentration of 10% trichloroacetic acid (TCA) to precipitate proteins. The pellets were washed with acetone twice and dried. Samples were then solubilized in SDS-PAG loading buffer and processed for SDS-PAGE and immunoblotting. For protein detection by silver staining, fractions were directly mixed with SDS-PAGE loading buffer without TCA precipitation and acetone washes.
Protein co-immunoprecipitation
50 ml of log-phase yeast culture was collected by centrifugation. Samples were resuspended in 1 ml of pre-chilled IP buffer (20 mM PIPES-KOH at pH 6.8, 150 mM KCl, 1 mM EDTA, 1 mM DTT, 10% v/v glycerol, 0.2% v/v NP-40, 1 mM PMSF, 5 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin A) with the addition of 500ul of 0.5 mm glass beads, then mechanically lysed in a bead mill, using a routine of 3 min of beating and 3 min of chill on ice for six rounds. After centrifugation at 13,000×g for 15 min at 4 °C, supernatant was collected.
Prior to use, affinity resin (Dynabeads, Thermo 10004D) were washed three times and resuspended in IP buffer. A mixture of 20ul affinity resin, 3 μl antibody (anti-GFP, Roche 11814460001), and 800 μl of cell lysate was incubated at 4 °C with gentle inversion for 1 h. The affinity resin were then washed three times, each time with 1 ml of IP buffer for 10 min each. Afterwards, 50 μl of SDS-PAGE sample buffer was added to the affinity resin. Samples were then subject to SDS-PAGE and immunoblotting.
GTP hydrolysis assay
Purified G protein was loaded with GTP (Sangon Biotech, A620250) by incubating 100 μM of protein with a 10-fold molar excess of GTP at 30 °C for 30 min in 50 mM Tris-HCl (pH 8.0), 100 mM NaCl, 2 mM β-mercaptoethanol, 2 mM MgCl2, 4 mM EDTA (Sangon Biotech, A500838), and 0.1 mg/ml brain extract total lipid (Avanti, 131101 P). Afterwards, 8 mM MgCl2 was added to system to stop nucleotide loading. Free nucleotide was removed with PD10 (Cytiva, 17085101) pre-equilibrated with buffer-A: 50 mM Tris-HCl (pH 8.0), 100 mM NaCl, 2 mM MgCl2, and 2 mM β-mercaptoethanol. For control, RhoA (aa3-181, F25N) was purified by size-exclusion chromatography Superdex-200 26/60 (Cytiva, 28-9893-36). After removing the Trx-tag, the resulting proteins were further purified by another round of size-exclusion chromatography in buffer-A. GTP hydrolysis assay was performed using the Enzchek Phosphate Assay Kit (Invitrogen, 2563700). GTP-loaded G proteins were mixed with solutions containing the assay reagents, EDTA and GTP and then dispensed into 96-well microplates (Corning). The absorbance at 360 nm was monitored using a Synergy NEO (Agilent).
Statistics and reproducibility
Unless otherwise noted, all biological experiments, including microscopy work and protein work, were repeated independently for at least three times. For microscopy images, images presented in figures represent the most common localization or morphology patterns observed among all the cells or sections.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the iProX partner repository with the dataset identifier PXD053002. Graphical presentations of Alphafold2 results for Supplementary Dataset D5, D16, D18, D22, D25, D27 are available at figshare (https://figshare.com/s/364b1bc2d0c37c3ff080). Previously reported PDB accession codes for [8OUM], [8OUL], [2K5U], [1M2O], [3MKR], [6×90], [1R8Q], [3LVQ], [3BH6], [7MGE] were used for structural analysis. All other data supporting the findings of this study are available within the paper and its Supplementary Information. Source data are provided with this paper.
Code availability
This work did not develop new codes or algorithms. All analyses were performed using publicly available software or web services as described in Methods.
References
Dacks, J. B. et al. The changing view of eukaryogenesis - fossils, cells, lineages and how they all come together. J. Cell Sci. 129, 3695–3703 (2016).
Rout, M. P. & Field, M. C. The evolution of organellar coat complexes and organization of the eukaryotic cell. Annu Rev. Biochem. 86, 637–657 (2017).
Vosseberg J. et al. Timing the origin of eukaryotic cellular complexity with ancient duplications. Nat. Ecol. Evol. 5, 92–100 (2021).
Bethune, J. & Wieland, F. T. Assembly of COPI and COPII vesicular coat proteins on membranes. Annu. Rev. Biophys. 47, 63–83 (2018).
Sanger A., Hirst J., Davies A. K., Robinson M. S. Adaptor protein complexes and disease at a glance. J. Cell Sci. 132, jcs222992 (2019).
Farre J. C., Mahalingam S. S., Proietto M., Subramani S. Peroxisome biogenesis, membrane contact sites, and quality control. Embo Rep. 20, e46864 (2019).
Schoneberg, J., Lee, I. H., Iwasa, J. H. & Hurley, J. H. Reverse-topology membrane scission by the ESCRT proteins. Nat. Rev. Mol. Cell Biol. 18, 5–17 (2017).
Spang, A. et al. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 521, 173-+ (2015).
Gabaldon, T. Origin and early evolution of the eukaryotic cell. Annu Rev. Microbiol. 75, 631–647 (2021).
Xie, R. et al. Expanding Asgard members in the domain of Archaea sheds new light on the origin of eukaryotes. Sci. China Life Sci. 65, 818–829 (2022).
Eme, L. et al. Inference and reconstruction of the heimdallarchaeial ancestry of eukaryotes. Nature 618, 992-+ (2023).
Liu, Y. et al. Expanded diversity of Asgard archaea and their relationships with eukaryotes. Nature 593, 553-+ (2021).
Zaremba-Niedzwiedzka, K. et al. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature 541, 353-+ (2017).
Rodrigues-Oliveira, T. et al. Actin cytoskeleton and complex cell architecture in an Asgard archaeon. Nature 613, 332-+ (2023).
Hatano T. et al. Asgard archaea shed light on the evolutionary origins of the eukaryotic ubiquitin-ESCRT machinery. Nat. Commun. 13, 3398 (2022).
Lu Z. Y. et al. Coevolution of Eukaryote-like Vps4 and ESCRT-III Subunits in the Asgard Archaea. Mbio 11, 10–1128 (2020).
Akil, C. & Robinson, R. C. Genomes of Asgard archaea encode profilins that regulate actin. Nature 562, 439-+ (2018).
Imachi, H. et al. Isolation of an archaeon at the prokaryote-eukaryote interface. Nature 577, 519-+ (2020).
Vargova R. et al. A eukaryote-wide perspective on the diversity and evolution of the ARF GTPase protein family. Genome Biol. Evol. 13, evab157 (2021).
van der Verren, S. E. & Zanetti, G. The small GTPase Sar1, control centre of COPII trafficking. Febs Lett. 597, 865–882 (2023).
Nawrotek, A., Dubois, P., Zeghouf, M. & Cherfils, J. Molecular principles of bidirectional signalling between membranes and small GTPases. Febs Lett. 597, 778–793 (2023).
Klinger, C. M., Spang, A., Dacks, J. B. & Ettema, T. J. G. Tracing the archaeal origins of eukaryotic membrane-trafficking system building blocks. Mol. Biol. Evol. 33, 1528–1541 (2016).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
van Kempen, M. et al. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 42, 243–246 (2024).
Vargova R, et al. The Asgard archaeal origins of Arf family GTPases involved in eukaryotic organelle dynamics. Nat Microbiol 10, 495–508 (2025).
Leipe, D. D., Wolf, Y. I., Koonin, E. V. & Aravind, L. Classification and evolution of P-loop GTPases and related ATPases. J. Mol. Biol. 317, 41–72 (2002).
Rojas, A. M., Fuentes, G., Rausell, A. & Valencia, A. The Ras protein superfamily: evolutionary tree and role of conserved amino acids. J. Cell Biol. 196, 189–201 (2012).
Khan, A. R. & Menetrey, J. Structural Biology of Arf and Rab GTPases’ Effector Recruitment and Specificity. Structure 21, 1284–1297 (2013).
Nawrotek, A., Zeghouf, M. & Cherfils, J. Allosteric regulation of Arf GTPases and their GEFs at the membrane interface. Small GTPases 7, 283–296 (2016).
Cherfils, J. Arf GTPases and their effectors: assembling multivalent membrane-binding platforms. Curr. Opin. Struct. Biol. 29, 67–76 (2014).
Tran, L. T., Akil, C., Senju, Y. & Robinson, R. C. The eukaryotic-like characteristics of small GTPase, roadblock and TRAPPC3 proteins from Asgard archaea. Commun. Biol. 7, 273 (2024).
Pai, E. F. et al. Refined crystal structure of the triphosphate conformation of H-ras p21 at 1.35 A resolution: implications for the mechanism of GTP hydrolysis. EMBO J. 9, 2351–2359 (1990).
Drivas, G. T., Shih, A., Coutavas, E., Rush, M. G. & D’Eustachio, P. Characterization of four novel ras-like genes expressed in a human teratocarcinoma cell line. Mol. Cell Biol. 10, 1793–1798 (1990).
Farnsworth, C. L. & Feig, L. A. Dominant inhibitory mutations in the Mg(2+)-binding site of RasH prevent its activation by GTP. Mol. Cell Biol. 11, 4822–4829 (1991).
Feig, L. A. & Cooper, G. M. Inhibition of NIH 3T3 cell proliferation by a mutant ras protein with preferential affinity for GDP. Mol. Cell Biol. 8, 3235–3243 (1988).
Rezanka, T., Kyselova, L. & Murphy, D. J. Archaeal lipids. Prog. Lipid Res. 91, 101237 (2023).
Xie, Y. et al. GPS-Lipid: a robust tool for the prediction of multiple lipid modification sites. Sci. Rep. 6, 28249 (2016).
Bologna, G., Yvon, C., Duvaud, S. & Veuthey, A. L. N-Terminal myristoylation predictions by ensembles of neural networks. Proteomics 4, 1626–1632 (2004).
Maurer-Stroh, S., Eisenhaber, B. & Eisenhaber, F. N-terminal N-myristoylation of proteins: prediction of substrate proteins from amino acid sequence. J. Mol. Biol. 317, 541–557 (2002).
Taylor, R. J., Tagiltsev, G. & Briggs, J. A. G. The structure of COPI vesicles and regulation of vesicle turnover. Febs Lett. 597, 819–835 (2023).
Zhu J. et al. A validated set of fluorescent-protein-based markers for major organelles in yeast (Saccharomyces cerevisiae). Mbio 10, 10–1128 (2019).
Schlacht, A. & Dacks, J. B. Unexpected ancient paralogs and an evolutionary model for the COPII coat complex. Genome Biol. Evol. 7, 1098–1109 (2015).
Bi, X., Corpina, R. A. & Goldberg, J. Structure of the Sec23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature 419, 271–277 (2002).
Copic, A., Latham, C. F., Horlbeck, M. A., D’Arcangelo, J. G. & Miller, E. A. ER cargo properties specify a requirement for COPII coat rigidity mediated by Sec13p. Science 335, 1359–1362 (2012).
Townley, A. K. et al. Efficient coupling of Sec23-Sec24 to Sec13-Sec31 drives COPII-dependent collagen secretion and is essential for normal craniofacial development. J. Cell Sci. 121, 3025–3034 (2008).
Ishihara, N. et al. Autophagosome requires specific early Sec proteins for its formation and NSF/SNARE for vacuolar fusion. Mol. Biol. Cell 12, 3690–3702 (2001).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Jackson, C. L., Menetrey, J., Sivia, M., Dacks, J. B. & Elias, M. An evolutionary perspective on Arf family GTPases. Curr. Opin. Cell Biol. 85, 102268 (2023).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11, 119 (2010).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Capella-Gutierrez, S., Silla-Martinez, J. M. & Gabaldon, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Crooks, G. E., Hon, G., Chandonia, J. M. & Brenner, S. E. WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 (2004).
Madeira F. et al. The EMBL-EBI Job Dispatcher sequence analysis tools framework in 2024. Nucleic Acids Res. 52, W521-W525 (2024).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Ashkenazy, H. et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350 (2016).
Gautier, R., Douguet, D., Antonny, B. & Drin, G. HELIQUEST: a web server to screen sequences with specific alpha-helical properties. Bioinformatics 24, 2101–2102 (2008).
Zhu, H. et al. Global analysis of protein activities using proteome chips. Science 293, 2101–2105 (2001).
Zhao, W. et al. Sparse deconvolution improves the resolution of live-cell super-resolution fluorescence microscopy. Nat. Biotechnol. 40, 606–617 (2022).
He, C. W. et al. Membrane recruitment of Atg8 by Hfl1 facilitates turnover of vacuolar membrane proteins in yeast cells approaching stationary phase. BMC Biol. 19, 117 (2021).
Tyanova, S., Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 (2016).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016).
Goldman, A. R. et al. The primary effect on the proteome of ARID1A-mutated ovarian clear cell carcinoma is downregulation of the mevalonate pathway at the post-transcriptional level. Mol. Cell Proteom. 15, 3348–3360 (2016).
Sherman, B. T. et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221 (2022).
Xu, S. et al. Use ggbreak to effectively utilize plotting space to deal with large datasets and outliers. Front. Genet. 12, 774846 (2021).
de Silva R. T. et al. Improved growth and morphological plasticity of Haloferax volcanii. Microbiology 167, 001012 (2021).
Sturt, H. F., Summons, R. E., Smith, K., Elvert, M. & Hinrichs, K. U. Intact polar membrane lipids in prokaryotes and sediments deciphered by high-performance liquid chromatography/electrospray ionization multistage mass spectrometry–new biomarkers for biogeochemistry and microbial ecology. Rapid Commun. Mass Spectrom. 18, 617–628 (2004).
Liu X. et al. Functional synergy between the Munc13 C-terminal C1 and C2 domains. Elife 5, e13696 (2016).
Acknowledgements
The authors would like to thank Drs. Qing Zhong, Han-Mai Chen, and Min-Dan Tong (Shanghai Jiao Tong Univ. Sch. of Medicine) for technical assistance, and thank Drs. Kun-Rong Mei (Tianjin Univ.), Yu-Song Guo (Hong Kong Univ. of Sci. & Tech.), Xiao-Wei Chen (Peking Univ.), and Yin-Zhao Wang (Shanghai Jiao Tong Univ.) for helpful discussions. This work was supported by National Natural Science Foundation of China (32270796, 91754110) and Shanghai Municipal Science and Technology Commission (22ZR1433800).
Author information
Authors and Affiliations
Contributions
Conceptualization, J. Zhu, R. Xie, Q. Gong, J-W. Zhu, F. Wang, Z. Xie; Investigation, J. Zhu, R. Xie, Q. Ren, J. Zhou, C. Chen, M.-X. Xie, Y. Zhou, Y. Zhang, N. Liu, J. Wang, Z. Zhang, X. Liu; Writing – Original Draft, R. Xie, Z. Xie; Supervision, W. Yan, Q. Gong, L. Dong, J-W. Zhu, F. Wang, Z. Xie.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Robert Robinson and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, J., Xie, R., Ren, Q. et al. Asgard Arf GTPases can act as membrane-associating molecular switches with the potential to function in organelle biogenesis. Nat Commun 16, 2622 (2025). https://doi.org/10.1038/s41467-025-57902-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57902-7
This article is cited by
-
A dual enzyme-mimicking COF sonosensitizer potentiates cancer sonodynamic therapy via cascade oxygenation and ROS storm
Science China Materials (2026)
