
Abstract
Electro/photocatalytic C-N coupling acts as a key build-block to the next generation of chemicals like amides for wide applications in energy, pharmaceuticals and chemical industries. However, the uncontrolled intermediates coupling challenges the efficient amide production regarding yield or selectivity. Here we propose a photocatalytic radical addition route, where the fundamental active species, including oxygen and photogenerated electron-hole pairs, are regulated for selective intermediates generation and efficient acetamide synthesis from mild co-oxidation of CH3CH2OH and NH3. Sufficient CH3CH2OH is provided to accumulate the stable intermediate (CH3CHO). Meanwhile, the limited NH3 concentration ensures the controllable generation and fast addition of the transient radical (●NH2) on CH3CHO. Through the directed coupling of stable-transient intermediates, the acetamide synthesis rate is pushed forward to a hundred-mmol level (105.61 ± 4.86 mmol·gcat−1·h−1) with a selectivity of 99.17% ± 0.39%, reaching a gram-scale yield (1.82 g) of acetamide. These results illuminate valuable opportunities for the photocatalysis-driven synthetic industry.
Similar content being viewed by others
Introduction
Amides and their derivatives are some of the most important essential chemicals in the food, pharmacy, and chemical industries1,2,3. The efficient and economical synthesis of amides contributes greatly to the sustainable development of human society. However, industrial amide synthesis still relies on coupling the C- and N-containing small molecules under harsh conditions. Taking acetamide synthesis as a typical case, CH3COOH and NH3 are fed for coupling with the assistance of dehydrators and catalysts at a high temperature of 150–180 °C, which consumes massive fossil fuels and leads to huge carbon emissions4,5,6. Hence, substantial research efforts have been devoted to developing alternative routes for amide synthesis under ambient conditions. Among them, the electrochemical method for co-reducing CO2/CO and N2/NOx−, has recently attracted increased interest, in which obvious advances have been achieved for electrochemical amide synthesis from C-N coupling7,8,9,10,11. However, the corresponding synthesis rate and selectivity (or Faraday efficiency, FE) are still restrained to low levels12,13,14. Specifically, under lower bias voltage, the activation of the reactant molecules is insufficient for their catalytic transformation, resulting in a limited synthesis rate15,16,17. On the contrary, the separate deep reduction of C- and N-substance is inevitable when the electric energy supply is excess, making it rather difficult to form the C-N bond selectively, thus failing to increase the selectivity of amides18,19,20. Hence, a mild redox ability, contributed by heterogeneous photocatalysis, may be an alternative approach for the efficient synthesis of amides from selective C-N coupling21,22,23,24.
To this end, the precise regulation and selective generation of the key intermediates for C-N coupling, including carbonyl (-C=O) and amino (-NH2) species, are imperative. However, these species are located in their intermediated valence states, respectively. Therefore, a precisely tailored mild redox reaction route should be constructed for their delicate preparation25,26,27. Otherwise, side reactions of the per-oxidation/per-reduction are unavoidable under a rough redox driving force, which results in decreased selectivity for amide synthesis. Moreover, it is well-acknowledged that a low reaction rate is expected by starting the coupling reactions with two stable intermediates due to their low chemical activity28,29,30. In contrast, the coupling possibility between two transient intermediates, such as reactive radicals, is severely limited since multiple disordered side reactions proceed in their short lifetimes29,31,32. That is, a complementary couple of stable-transient intermediates is required to endow the C-N coupling reaction with both optimum production rate and selectivity synergistically. By making use of the mild redox ability of photocatalysis and optimizing the coupled intermediates, it is anticipated that a meticulously designed photocatalytic reaction system can be developed for efficient synthesis of amides.
Herein, we propose a photocatalytic radical addition route for acetamide synthesis (Fig. 1), using CH3CH2OH and NH3 as the C- and N-source, respectively. Under the mild photocatalytic redox driving force, the fundamental active species, including O2 and electron (e−)-hole (h+) pairs, are regulated for selective co-oxidation of CH3CH2OH and NH3. Specifically, sufficient CH3CH2OH is provided to accumulate CH3CHO as the stable intermediate. Meanwhile, the limited provision of NH3 ensures the controllable generation of ●NH2 as the transient intermediate, in which the fast addition of ●NH2 radical into CH3CHO is subsequently achieved. The oxidative ability of this reaction system is tailored by quantified O2 activation, which guarantees considerable CH3CONH2 selectivity and impedes the peroxidation of CH3CHO and ●NH2 intermediates. The acetamide synthesis rate is pushed forward to a hundred-mmol level (105.61 ± 4.86 mmol gcat−1 h−1) with superior selectivity (99.17% ± 0.39%), significantly exceeding most of the other catalytic routes. The photocatalysis system can be stably and continuously operated for 300 h, during which the acetamide yield reaches the gram scale (1.82 g). A comprehensive mechanism investigation is conducted to reveal the actual coupling coordinates and key intermediates, in which direct evidence has been provided based on the iso-type (D and 15N) labeled in situ EPR, HR-MS, NMR, and in situ ATR-FTIR. It is clarified that the efficient coupling of the stable and transient intermediates is the decisive factor for achieving optimum catalytic activity. We anticipate the current demonstration to be a starting point for realizing the efficient production of amide compounds with a simple but practical heterogeneous photocatalysis scheme, thus increasing the possibility of the science and industry revolution driven by solar light.

Illustration for the coupling of stable and transient intermediates for acetamide photosynthesis.
Results and discussion
Reaction system design and efficiency evaluation
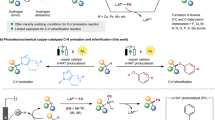
The commercial TiO2 (P25) is applied as a representative photocatalyst to develop the photoredox route for C-N coupling, which only absorbs the photon and forms the active e− and h+ in this system, excluding the specificity of other modified photocatalysts and establishes a general pattern for the superiority of the photocatalytic amides synthesis scheme. Other common photocatalysts are also investigated in the C-N coupling reaction, which processes similar properties, showing the universality of photocatalysts in C-N coupling (Supplementary Fig. 4 and Supplementary Note 3). At first, various photocatalytic routes, including co-oxidation, co-reduction, and combined redox reactions, are screened to examine the practicability of amide synthesis (Table 1). It is identified that the amide bonds can be constructed through the coupling of various C1/C2 and N1 substances, in which significant promotion of the synthesis efficiency is observed in the reaction route of CH3CH2OH and NH3 co-oxidation for CH3CONH2 production (109.24 μmol h−1), remarkably exceeding the other photocatalysis routes. Therefore, CH3CH2OH and NH3 reactants are assigned as the respective C- and N-source for the comprehensive efficiency evaluation and mechanism investigations.
After the optimization of the reaction parameters, including NH3 concentration, CH3CH2OH proportion, O2 proportion (in Ar), and catalyst dosage (Supplementary Figs. 5–9 and Supplementary Note 16), a high CH3CONH2 production rate is achieved at 105.61 ± 4.86 mmol gcat−1 h−1 (Fig. 2a) from the co-oxidation of CH3CH2OH and NH3, which is a significant progression as it reaches the hundred-mmol level for photocatalytic C-N coupling under ambient conditions. Then, as the complete redox reaction proceeds in a heterogeneous photocatalysis process, it is deduced that the co-oxidation reaction is contributed by the light-generated h+ and O2-enabled active species, in which the detailed mechanism requires further investigation. Meanwhile, the hydrogen gas (H2) is yielded by the cooperative e−-driven reduction reaction with the dehydrogenated hydrogen from CH3CH2OH and NH3 (Supplementary Note 13 and Supplementary Figs. 10, 11), in which a minor amount of H2 is detected due to competing O2 activation and reduction for e− consumption.

a catalyst dosage-dependent unit production rate; CH3CONH2 selectivity evaluation regarding the N-(b) and C-sources (c), respectively; d Efficiency comparison between different catalytic routes for photocatalytic C-N coupling, including the targets of production rate and selectivity, the corresponding research works are listed and cited in Supplementary Information (Supplementary Table 3)54,55,56,57,58,59,60; e Long-term stability test. f XRD pattern and the image (inset) of the collected CH3CONH2 sample with rotary evaporation after the long-term stability test. The respective standard curves for detecting the reaction species using ion chromatography (IC, NH4+, NO2−, NO3−, CH3COOH, and NH2OH, Supplementary Figs. 31–35) are provided in Supplementary Information. The error bars in (a–c) were drawn based on the calculated standard error of two parallel tests.
Subsequently, the CH3CONH2 selectivity is evaluated, corresponding to NH3 (N) and CH3CH2OH (C), respectively (Supplementary Note 15). As depicted in Fig. 2b and Supplementary Figs. 12–20, the concentration of CH3CONH2 gradually increases along with the consumption of NH3. It is noted that trace N2 byproduct is generated at the first 30 min, which originates from the NH3 peroxidation. However, as the reaction proceeds, the accumulation of N2 is impeded due to the decrease of the NH3 concentration, which illustrates that the controllable provision of the initial NH3 concentration is vital to determine the oxidative pathways of the system. The concentrations of the other byproducts are exceedingly low, resulting in data points in Fig. 2b that are so closely aligned they appear to overlap, making individual distinctions hard to discern. Under the limited provision of NH3 (100.00 mg L−1), a near-complete conversion of NH3 (97.67% ± 1.67%) for CH3CONH2 production is accomplished under the 60 mins’ light irradiation, presenting a superior CH3CONH2 selectivity (N) of 99.17 ± 0.39%. In addition, it is observed that the synthesis rate decreases after 45 min, which is reasonable due to the remaining NH3 concentration being low after rapid consumption. Based on these results, the recovery of the reaction rate and maintaining of selectivity are expected by conducting cycled experiments through periodical input of limited NH3.
Then, the CH3CONH2 selectivity regarding CH3CH2OH is also evaluated by comprehensively detecting the oxidative products in both the liquid and gaseous phases (Fig. 2c and Supplementary Figs. 21–25). The corresponding selectivity is ~100% at the first 30 min. Along with the consumption of NH3, the generation of the trace byproduct (CH3COOH) is observed due to the peroxidation of CH3CH2OH under low NH3 concentration, which again verifies the importance of limited NH3 provision to guarantee the superior selectivity. After the photocatalysis reaction for 1 h, the CH3CONH2 selectivity (C) is maintained at as high as 95.60 ± 0.22%. The reaction parameters of NH3 concentration for the CH3CONH2 selectivity test regarding NH3 and CH3CH2OH are set differentially. Specifically, a lower concentration of NH3 (100.00 mg L−1) is provided for its efficient conversion to evaluate the selectivity (N). While more NH3 (800.00 mg L−1) is included to impede the peroxidation of CH3CH2OH after the rapid NH3 consumption, where objective evaluations can be established for the selective oxidation of NH3 and CH3CH2OH for CH3CONH2 synthesis, respectively (Supplementary Note 17). These selectivity results imply that the key C- and N-intermediate must be clarified and optimized for the selective C-N coupling, which will, in turn, impede the individual peroxidation of CH3CH2OH and NH3. Moreover, when compared with the conventional industrial synthesis route (Supplementary Table 2), the photocatalytic co-oxidation route for CH3CONH2 synthesis demonstrates significant progress in terms of synthesis conditions, raw material price, and synthesis efficiency.
After systematically optimizing the reactants and reaction parameters, the establishment of the highly effective and selective co-oxidation route of CH3CH2OH and NH3 lays the theoretical foundation for developing the photocatalytic CH3CONH2 synthesis route. In comparison with the other reported routes for photocatalytic C-N coupling (Fig. 2d and Supplementary Table 3), it is significant that a high efficiency is accomplished by applying the photocatalytic co-oxidation routes, including the important indexes of synthesis rate of the targeted coupling products and the corresponding selectivity. Moreover, the as-constructed reaction system has been continuously operated with sufficient CH3CH2OH provision for 300 h (Supplementary Table 1), in which periodic NH3 conversion for stable CH3CONH2 production is maintained, delivering a gram scale yield of CH3CONH2 (1.82 g, Fig. 2e). The collected solid sample, with rotary evaporation for its recovery, is verified as CH3CONH2 by the XRD and nuclear magnetic resonance (1H NMR) technologies, consistent with the crystallography open database (COD #9007523, Fig. 2f) and chemical shift of the hydrogen atom (Supplementary Fig. 26) respectively. No deactivation of the P25 is observed based on the characterization results of the sample before and after the long-term stability test (Supplementary Figs. 27–30 and Supplementary Note 18).
Reaction coordinates for the selective oxidation of CH3CH2OH and NH3
To identify the critical step of the co-oxidation of CH3CH2OH and NH3. The variate-controlled in situ attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR, Supplementary Fig. 36) investigation is conducted by comparing the IR signals for individual ethanol oxidation (EOR), ammonia oxidation (AOR) and their co-oxidation reactions. As the individual EOR proceeds (Fig. 3a, the spectra for the adsorption equilibrium process in the dark are displayed in Supplementary Fig. 37 and Supplementary Table 4), the gradual consumption of CH3CH2OH (C-OH at 1603 and 1364 cm−1) is observed33, which generates the corresponding oxidative intermediates (C-O at 1657 and C = O at 1519 cm−1)34,35. It is found that the peroxidative product (COO− at 1420 cm−1)36 is produced by the excess oxidative ability in the individual EOR. Similarly, the consumption of NH3 (N-H at 3099 cm−1)37 yields the intermediated products of -NH2 (1090 cm−1)38 and NH2OH (1268 cm−1)8 in the individual AOR (Fig. 3b, the spectra for the adsorption equilibrium process in the dark are displayed in Supplementary Fig. 38 and Supplementary Table 5), in which the peroxidative products are also observed (N-O at 1755 cm−1, NO2− at 1530 cm−1 and NO3− at 1626 cm−1)23,39. Since NH2OH species are not detected during the reaction process (Fig. 2b, Supplementary Note 14, and Supplementary Fig. 20), it is clarified that NH2OH is not desorbed from the catalyst surface but is instead converted to other intermediates and products.

Time-dependent IR signals for the individual ethanol oxidation reaction (EOR, a), ammonia oxidation reaction (AOR, b), and combined EOR and AOR (c), respectively; Normalized results of the IR signals of COO− (d), NO3− (e), C=O (f), and -NH2 (g), respectively. The full spectra of the adsorption and photocatalysis process are provided in Supplementary Information for individual EOR (Supplementary Fig. 37), individual AOR (Supplementary Fig. 38), and combined EOR and AOR (Supplementary Fig. 39), respectively.
Most importantly, as illustrated in the combined co-oxidation reactions of EOR and AOR (Fig. 3c, the spectra for the adsorption equilibrium process in the dark are displayed in Supplementary Fig. 39 and Supplementary Table 6), as the co-oxidation of CH3CH2OH (C-OH at 1364 cm−1) and NH3 (N-H at 3087 cm−1) proceeds, the interaction of the C- (C=O at 1544 cm−1) and N-intermediates (-NH2 at 1159 and 1090 cm−1) leads to the generation of the coupling products as indicated by the C-N bond formation at 1416 cm−1 40. Therefore, it is identified that the amide bond (-CONH2) can be effectively generated through the coupling of C=O and -NH2-based intermediates. However, their exact structures require further investigation.
Then, the signals for the peroxidative byproducts (Fig. 3d, e) and critical coupling intermediates (Fig. 3f, g) are normalized for comparison between the individual and combined oxidation reactions. The detailed methods for normalization of the FTIR signals are presented in Supplementary Note 8. It is observed that more peroxidative byproducts, including COO− from EOR (Fig. 3d) and NO3− from AOR (Fig. 3e), are accumulated in the respective individual oxidation reactions, which hinders the selective generation of key intermediates for C-N coupling. By the combination of EOR and AOR, the intensified signals for the intermediates of C=O (Fig. 3f) and -NH2 (Fig. 3g) are noted, leading to the directed coupling rather than the individual peroxidation. Furthermore, since the CH3CH2OH is sufficiently provided, the continuous accumulation of C=O species is reasonable, which provides sufficient C-intermediate for coupling. On the contrary, the limited provision of NH3 yields -NH2 at the first 20 min. Then -NH2 is consumed along with the decrease of NH3 concentration, which follows the efficiency test result (Fig. 2b). Hence, it is deduced that the -NH2-based species can be identified as the key N-intermediate which possesses higher reactive activity than that of the C=O species, due to its gradual accumulation and rapid consumption. It is again verified that continuous and effective C-N coupling can be accomplished by providing sufficient CH3CH2OH and periodically supplying limited NH3. This approach not only generates C- and N-intermediates for coupling but also avoids their peroxidation.
Generation and coupling mechanism of the key intermediates
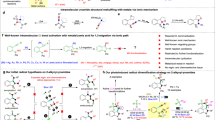
Based on the in situ ATR-FTIR investigation, it is acknowledged that the directed coupling of the C- and N-intermediates is vital to selectively producing CH3CONH2. Then, the generation pathways and coupling mechanism of these key intermediates should be revealed. The O2-proportion-dependent CH3CONH2 synthesis efficiency is first evaluated to identify the oxidative driving force for the generation of these intermediates (Fig. 4a and Supplementary Figs. 40,41). It is observed that the CH3CONH2 yield increases along with the elevation of O2 proportion (in Ar) from 0% to 75%, reaching a yield of 209.77 ± 7.68 μmol h−1. However, a rapid decrease of CH3CONH2 yield (68.82 ± 1.56 μmol h−1) is noted under excess O2 provision (100%), which directly confirms that the O2 molecules, as fundamental active species, require to be quantificationally provided for mild O2 activation, which enables the precise generation of the coupling intermediates. In addition, the production of e−-driven H2 is elevated at the O2 proportion of 0% than that of 75%, which again confirms that the O2 activation and reduction work as competing reactions with H2 evolution (Supplementary Fig. 42).

a O2 proportion-dependent CH3CONH2 yield; DMPO-trapping (with D-labeling, images above) and TEMP-trapping (images below) in situ EPR experiments under the O2 proportion of 0% (b), 75% (c), and 100% (d) respectively. The error bars in (a) were drawn based on the calculated standard error of two parallel tests.
Then, a comprehensive iso-type (deuterium, D) labeled in situ electron paramagnetic resonance (EPR) experiment is conducted to dynamically track the selective EOR pathways and corresponding intermediates, by applying the 5,5-dimethyl-1-pyrroline N-oxide (DMPO) and 2,2,6,6-Tetramethylpiperidinooxy (TEMPO) as the respective trapping agents under different O2 proportion (Fig. 4b–d)41,42,43. The CD3CD2OD, rather than the other common reagents such as CH3OH or H2O, is applied as the only solvent for the D-labeled in situ EPR tests to precisely reproduce the selective EOR for acetamide synthesis. Under the 0% of O2 proportion, it is found that the EOR proceeds under the oxidative driving force of h+, as evidenced by the successful detection of the oxidative intermediate of the DMPO trapped alkyl radical (DMPO-CD3●CDOD) and the absence of the O2-enabled active species (Fig. 4b and Supplementary Figs. 43–45). Interestingly, DMPO trapped alkoxy radical (DMPO-CD3CD2O●) and trace DMPO-CD3●CDOD are both detected under the condition of an O2 proportion of 75%, which follows the optimum O2 proportion parameter for acetamide synthesis (Fig. 4c, Supplementary Fig. 46, 47, and Supplementary Note 19), illustrating the direct relationship between CD3CD2O● generation and acetamide yield promotion. There is a tautomerization transformation from CD3●CDOD to CD3CD2O●, which is driven by the mild oxidative ability of singlet oxygen (1O2, Fig. 4c and Supplementary Fig. 48). The 1O2 is generated from either the activation of the triplet oxygen (3O2) or the h+-triggered rapid oxidation of superoxide radical (●O2−)44,45. Since 3O2 and ●O2− are difficult to distinguish in the current in situ EPR experiments, it is reasonable to infer that both of these two reaction pathways are involved for the generation of 1O2. Moreover, the generation of CD3CD2O●, which is an O-centered active radical, implies that the C-OD bond in the CD3CD2OD molecule can be effectively activated by dehydrogenation, which yields the precursor of -C=O species for C-N coupling. As the concentration of O2 increases, the signal intensity of ●O2− radical gradually increases while the signal intensity of 1O2 gradually decreases. Finally, the excess provision of O2 (100%) leads to the accumulation of the strong oxidative ●O2− radicals (Fig. 4d, Supplementary Fig. 49, and Supplementary Note 20), in which no signals of the 1O2 are detected (Fig. 4d and Supplementary Figs. 50, 51). As the ●O2− radicals bind to the reactants to generate peroxidative products, 1O2 species are no longer generated. Hence, the ●O2−-induced peroxidation is inevitable under such a high O2 concentration, resulting in the decreased coupling efficiency.
By the quantified O2 activation at the proportion of 75%, it is confirmed that the peroxidation of CD3CD2O● is avoided. Therefore, it is reasonably deduced that stable C-intermediate of acetaldehyde (CH3CHO), which possesses the critical -C=O group, can be generated through the hydrogen abstraction from CD3CD2O●. The corresponding control experiment is thereby conducted by replacing CH3CH2OH with CH3CHO as the initial reactant for C-N coupling (Fig. 5a, Supplementary Note 10, Supplementary Fig. 52, and Supplementary Table 7). It is noted that almost complete recovery of the CH3CONH2 yield is accomplished via the NH3-CH3CHO condensation (204.67 ± 6.58 μmol h−1), consistent with that of the co-oxidation of NH3 and CH3CH2OH (209.77 ± 7.68 μmol h−1), which identifies the CH3CHO as the stable C-intermediate, where the generation of CH3CHO requires precise regulation to avoid the decrease of the acetamide. Furthermore, the key role of CH3CHO is illustrated by the gas chromatography (GC, Fig. 5b) and corresponding mass spectra (MS, Supplementary Figs. 53, 54 and Supplementary Note 21) detection results. The provision of CH3CH2OH/CD3CD2OD feedstock results in the generation of H/D labeled acetaldehyde and acetamide, respectively, presenting a selective EOR along the route of CH3CH2OH → CH3●CHOH ↔ CH3CH2O● → CH3CHO → CH3CONH2. Also, it is clarified by the 1H NMR (Fig. 5c, Supplementary Figs. 55 and 56 and Supplementary Note 22) results that the CD3CONH2 is detected with a deuterated ratio of 98.33%, which verifies the reliability and practicability of these designed D-labeled in situ experiments.

a Control experiments for CH3CONH2 synthesis by replacing CH3CH2OH with CH3CHO as the C-source; GC (b) and 1H NMR (c) results with D- (images right) and H-labeling (images left) respectively; d DMPO-trapping in situ EPR experiments with 14N- (image above) and 15N-labeling (image below) respectively; e in situ EPR spectra for e−-induced TEMPO consumption with (image below) and without (image above) NH3 provision respectively; f HR-MS results for CH3CONH2 detection with 14N- (image above) and 15N-labeling (image below) respectively; g Proposed reaction pathways of CH3CH2OH and NH3 co-oxidation for intermediates coupling and CH3CONH2 synthesis. The error bars in (e) were drawn based on the calculated standard error of two parallel tests.
After successfully identifying the EOR for accumulating the stable C-intermediate of CH3CHO, the cooperative AOR mechanism is revealed by a series of 15N-/14N-labeled tracking experiments. The DMPO-●14NH2 and DMPO-●15NH2 are detected without O2 provision, respectively (Fig. 5d, Supplementary Figs. 57, 58, and Supplementary Note 23)46,47, manifesting that h+ is the dominant driving force for the mild oxidation of NH3 for ●NH2 generation. By combining the in situ ATR FTIR and in situ EPR spectra (Fig. 3c, g, 4h), it is indicated that the absorbed -NH2 is oxidized by h+ to produce ●NH2, which is involved in subsequent C-N coupling processes. Besides, further in situ EPR experiments are applied by using TEMPO as the indicator. Under the condition of individual TEMPO provision (Fig. 5e and Supplementary Fig. 59), a slight decrease of TEMPO signals is recorded due to the TEMPO reduction driven by e− under light irradiation for 2 min On the contrary, near-complete TEMPO consumption is observed when adding NH3 into the test solution (Fig. 5e and Supplementary Fig. 60). Since no other oxidative driving force is included due to the absence of O2 (proportion at 0%), it is again confirmed that the selective AOR is dominantly driven by h+ for ●NH2 generation, which consumes more h+ and in turn provides extra e− for accelerating TEMPO reduction.
By the combination of the efficiency test (Figs. 2b, 4a), the in situ ATR-FTIR results (Fig. 3c), as well as the 15N-labeled in situ EPR investigations for ●NH2 detection (Fig. 4h, i), it is acquired that no peroxidative N-containing intermediates or products are generated in the combined EOR and AOR under quantified O2 activation, which leads to the ~100% selectivity (N) for CH3CONH2 synthesis. Hence, it is concluded that the ●NH2 is identified as the transient N-intermediate for C-N coupling. Since the reaction rate coefficient of NH3-to-NH2 oxidation is an order of magnitude faster than that of the NH2-to-NH oxidation, the selective oxidation of NH3 primarily yields NH2-based intermediates or radicals than that of the NH48,49. Moreover, the fast formation (~1 ps) and relatively long lifetime (>1 ns) endow the ●NH2 radicals with a high possibility to participate in the coupling reactions, in comparison with the other N-centered radicals possessing short lifetimes, such as ●NH4 (13 ps)50,51, which again verifies that ●NH2 can act as the transient N-intermediate for C-N coupling. The illustration for ●NH2 detection parameters, especially the O2 proportion, is listed in Supplementary Note 24, which explains that the absence of the O2 provision is indispensable for the detection of ●NH2. Based on these results, it is clarified that the controllable generation and fast addition of transient ●NH2 radicals on stable CH3CHO intermediate should contribute directly to the efficient and selective C-N coupling for CH3CONH2 synthesis. The AOR pathways, along with the route of NH3 → ●NH2 → CH3CONH2, are then verified by the 14N/15N labeled high-resolution MS (HR-MS, Fig. 5f, Supplementary Figs. 61,62, and Supplementary Note 25) results, in which both CH3CO14NH2 and CH3CO15NH2 are detected, by introducing 14NH3 and 15NH3 (in CH3CH2OH) as the feedstocks respectively.
After these comprehensive mechanism investigations of the reaction pathways and coupling intermediates, the heterogeneous photocatalytic scheme for CH3CONH2 synthesis is proposed (Fig. 5g). Under the sufficient provision of CH3CH2OH and quantified O2 activation (75% in Ar), the CH3CHO, from selective oxidation of CH3CH2OH, is generated and accumulated as the stable C-intermediate for coupling. Meanwhile, the limited and periodic provision of NH3 leads to the controllable generation and fast addition of transient N-intermediate (●NH2) on stable CH3CHO intermediate, in which the rapid consumption of ●NH2 for C-N coupling avoids its peroxidation for side product generation. The coupling of long-lived C-intermediate and short-lived N-intermediate ensures a high selectivity and yield rate of CH3CONH2. The C-N coupling selectivity can be effectively maintained by the tailored oxidative ability and oxidative species, including h+ and e−-driven O2-enabled active species (1O2 and ●O2−), in which the excess e− contributes to the H2 evolution to construct a complete photocatalytic redox reaction. For the overall photocatalytic synthesis pathway of CH3CONH2, optimizing the interaction between the initial reactive species and the photogenerated charge carriers is of crucial importance to achieve carrier-driven radical generation. By fully leveraging the radical characteristics of lifetime and migration distance, radical-mediated photocatalytic reactions can proceed effectively, which ensures the high efficiency of mass transfer and the sufficient utilization of light energy, thereby achieving a significant enhancement of the redox efficiency.
Here, we have demonstrated a compelling solar-driven photocatalytic synthesis system. By combining mild co-oxidation of CH3CH2OH and NH3, efficient and selective access to CH3CONH2 synthesis is achieved with heterogeneous photocatalysis technology. Through the precise regulation of the O2 activation and e−-h+ carriers, the CH3CONH2 yield reaches the gram scale with satisfying production rate, selectivity, and stability. Comprehensive mechanism investigation results indicate that the controllable generation and fast addition of the transient ●NH2 radical on stable CH3CHO intermediate contributes essentially to this catalytic performance in acetamide synthesis. The C-N coupling from stable and transient intermediates could provide scientific and technical feasibility for the solar light-driven synthetic fields, which, with further promotion, might be industrially and economically practical, thereby illuminating valuable opportunities for the mild-photocatalysis-driven synthetic industry.
Methods
Photocatalytic synthesis route and efficiency evaluation
Different N-source (NH3, KNO2, or KNO3) was added into 50.00 mL of absolute methanol (CH3OH), ethanol (CH3CH2OH), formic acid (HCOOH), or acetic acid (CH3OOH) to screen the C-N coupling routes (Supplementary Note 1). After the construction of the route of CH3CH2OH and NH3 co-oxidation for CH3CONH2 synthesis, the parameters of NH3 concentration, CH3CH2OH volume, O2 proportion, and catalyst dosage were optimized for elevating the synthesis efficiency (Supplementary Note 2). The corresponding parameters of the selectivity test regarding N and C were also optimized (Supplementary Notes 4,5). The reactants, intermediates, and products, in liquid and gaseous phases, were detected and quantified by high-performance liquid chromatography (HPLC, Shimadzu Essentia LC-16i/MSD), ion chromatography (IC, Shimadzu IC-16) and infrared flue gas analyzer (Bruker MATRIX-MG5), respectively (Supplementary Note 26).
After the long-term stability test, the collected catalyst sample was characterized by the X-ray diffraction technology (XRD, Shimadzu XRD-6100) for its crystal stability, scanning electron microscopy (SEM, FEG ESEM XL30) and transmission electron microscopy (TEM, FEI Talos F200S) for its geometric stability respectively. The obtained reaction mixture was centrifuged for the separation of the solution and catalyst. Then, a rotary evaporator was applied to remove the solvent and potential side products from the solution. The collected product sample was characterized by the XRD (Supplementary Notes 6, 7).
In situ ATR-FTIR investigation
An INVENIO R FTIR (Bruker) spectrometer equipped with a mercury cadmium telluride (MCT) detector was utilized for the measurements. Before the test, 100.0 uL of catalyst ink (a mixture of 10.00 mg of P25, 25.0 uL of Nafion, and 1.00 mL of CH3CH2OH) was deposited onto the Si crystal and then dried in air. The reaction chamber was filled with a total of 10.00 mL of the reaction solution, and 75% of O2 (in Ar) was continuously injected into the system. An Xe lamp (Bobei BBZM-1) was applied as the light source, and detection was performed during light irradiation52.
The pristine IR signals were normalized to evaluate the species’ evolution directly. Between the columns of data, the highest value was set to be 1, and the lowest value was set to be 0. The rest were correspondingly normalized from 0 to 1. The resulting normalized data were thus described as a function of the IR scanning time53.
In situ EPR investigation by H/D labeling
All the in situ EPR measurements were conducted on the equipment of Bruker EMX Nano. 1000.0 μL of CH3CH2OH (or CD3CD2OD), 40.0 μL of NH3·H2O, 50.0 μL of the well-mixed P25 suspension (1000.00 mg L−1 in CH3CH2OH or CD3CD2OD), 10.0 μL of DMPO were added into the reactor under the O2 proportion (in Ar) of 0, 75, and 100%, respectively. The EPR signals were recorded at 1, 3, 5, and 10 min under the dark or light irradiation conditions, respectively, collected by a capillary (Supplementary Notes 9).
In situ EPR investigation by 14N/15N labeling
About 550.0 μL of 14NH3·H2O (10.00 g L−1) or 15NH4Cl (10.00 g L−1 of 15NH4+), 390.0 μL of DI and 50.0 μL of the well-mixed P25 suspension (in CH3CH2OH, 1000.00 mg L−1) were added into the reactor under the Ar (99.999%) gas injection. The EPR signals were recorded at 1, 3, 5, and 10 min under the dark or light irradiation condition respectively, collected by a capillary (Supplementary Notes 11, 12).
The other experimental details are provided in the Supplementary Information.
Data availability
All source data generated in this study are provided in the Supplementary Information and Source Data files. Source data are provided with this paper.
References
Pattabiraman, V. R. & Bode, J. W. Rethinking amide bond synthesis. Nature 480, 471–479 (2011).
Tao, Z., Rooney, C. L., Liang, Y. & Wang, H. Accessing organonitrogen compounds via C-N coupling in electrocatalytic CO2 reduction. J. Am. Chem. Soc. 143, 19630–19642 (2021).
Beyazay, T., Martin, W. F. & Tüysüz, H. Direct synthesis of formamide from CO2 and H2O with nickel-iron nitride heterostructures under mild hydrothermal conditions. J. Am. Chem. Soc. 145, 19768–19779 (2023).
Kakumoto, T., Saito, K. & Imamura, A. Thermal decomposition of formamide: shock tube experiments and ab initio calculations. J. Phys. Chem. 89, 2286–2291 (1985).
Comer, B. M. et al. Prospects and challenges for solar fertilizers. Joule 3, 1578–1605 (2019).
Shindell, D. & Smith, C. J. Climate and air-quality benefits of a realistic phase-out of fossil fuels. Nature 573, 408–411 (2019).
Wu, Y., Jiang, Z., Lin, Z., Liang, Y. & Wang, H. Direct electrosynthesis of methylamine from carbon dioxide and nitrate. Nat. Sustain. 4, 725–730 (2021).
Li, M. et al. Electrosynthesis of amino acids from NO and α-keto acids using two decoupled flow reactors. Nat. Catal. 6, 906–915 (2023).
Chen, C. et al. Coupling N2 and CO2 in H2O to synthesize urea under ambient conditions. Nat. Chem. 12, 717–724 (2020).
Meng, N. et al. Electrosynthesis of formamide from methanol and ammonia under ambient conditions. Nat. Commun. 13, 5452 (2022).
Yuan, M. et al. Unveiling electrochemical urea synthesis by co‐activation of CO2 and N2 with Mott–Schottky heterostructure catalysts. Angew. Chem. Int. Ed. 60, 10910–10918 (2021).
Li, J., Zhang, Y., Kuruvinashetti, K. & Kornienko, N. Construction of C-N bonds from small-molecule precursors through heterogeneous electrocatalysis. Nat. Rev. Chem. 6, 303–319 (2022).
Wan, Y., Zheng, M., Yan, W., Zhang, J. & Lv, R. Fundamentals and rational design of heterogeneous C‐N coupling electrocatalysts for urea synthesis at ambient conditions. Adv. Energy Mater. 14, 2303588 (2024).
Wu, Q., Zhu, F., Wallace, G., Yao, X. & Chen, J. Electrocatalysis of nitrogen pollution: transforming nitrogen waste into high-value chemicals. Chem. Soc. Rev. 53, 557–565 (2024).
Song, H., Chipoco Haro, D. A., Huang, P.-W., Barrera, L. & Hatzell, M. C. Progress in photochemical and electrochemical C-N bond formation for urea synthesis. Acc. Chem. Res. 56, 2944–2953 (2023).
Peng, X. et al. Electrochemical C-N coupling of CO2 and nitrogenous small molecules for the electrosynthesis of organonitrogen compounds. Chem. Soc. Rev. 52, 2193–2237 (2023).
Kim, J. E. et al. Electrochemical synthesis of glycine from oxalic acid and nitrate. Angew. Chem. Int. Ed. 60, 21943–21951 (2021).
Li, L. et al. Tandem dual-site PbCu electrocatalyst for high-rate and selective glycine synthesis at industrial current densities. Nano Lett. 24, 2392–2399 (2024).
Hu, Q. et al. Pulsed co-electrolysis of carbon dioxide and nitrate for sustainable urea synthesis. Nat. Sustain. 7, 442–451 (2024).
Zhang, X. et al. Identifying and tailoring C-N coupling site for efficient urea synthesis over diatomic Fe-Ni catalyst. Nat. Commun. 13, 5337 (2022).
Tan, H. et al. Photocatalysis of water into hydrogen peroxide over an atomic Ga-N5 site. Nat. Synth. 2, 557–563 (2023).
Pi, S. et al. Solar-driven waste-to-chemical conversion by wastewater-derived semiconductor biohybrids. Nat. Sustain. 6, 1673–1684 (2023).
Li, J. et al. Subnanometric alkaline-earth oxide clusters for sustainable nitrate to ammonia photosynthesis. Nat. Commun. 13, 1098 (2022).
Li, J. et al. Dynamic in situ formation of Cu2O sub‐nanoclusters through photoinduced pseudo‐Fehling’s reaction for selective and efficient nitrate‐to‐ammonia photosynthesis. Angew. Chem. Int. Ed. 63, e202317575 (2024).
Xia, P. et al. Designing a redox heterojunction for photocatalytic “overall nitrogen fixation” under mild conditions. Adv. Mater. 34, 2200563 (2022).
Chen, S. et al. Direct electroconversion of air to nitric acid under mild conditions. Nat. Synth. 3, 76–84 (2023).
Meng, X. et al. Direct methane conversion under mild condition by thermo-, electro-, or photocatalysis. Chem 5, 2296–2325 (2019).
Lee, S., Bae, H. S. & Choi, W. Selective control and characteristics of water oxidation and dioxygen reduction in environmental photo(electro)catalytic systems. Acc. Chem. Res. 56, 867–877 (2023).
Jiang, Y. et al. Enabling specific photocatalytic methane oxidation by controlling free radical type. J. Am. Chem. Soc. 145, 2698–2707 (2023).
Jiang, W. et al. Pd-modified ZnO-Au enabling alkoxy intermediates formation and dehydrogenation for photocatalytic conversion of methane to ethylene. J. Am. Chem. Soc. 143, 269–278 (2021).
Zhang, L. et al. Photoelectrocatalytic arene C-H amination. Nat. Catal. 2, 266–373 (2019).
Nosaka, Y. & Nosaka, A. Y. Generation and detection of reactive oxygen species in photocatalysis. Chem. Rev. 117, 11302–11336 (2017).
Li, J. N. & Kornienko, N. Electrochemically driven C-N bond formation from CO2 and ammonia at the triple-phase boundary. Chem. Sci. 13, 3957–3964 (2021).
Liu, Q., Lin, J., Cheng, H., Wei, L. & Wang, F. Simultaneous co-photocatalytic CO2 reduction and ethanol oxidation towards synergistic acetaldehyde synthesis. Angew. Chem. Int. Ed. 62, e202218720 (2023).
Kuang, S. et al. Acetamide electrosynthesis from CO2 and nitrite in water. Angew. Chem. Int. Ed. 63, e202316772 (2024).
Meng, N., Huang, Y., Liu, Y., Yu, Y. & Zhang, B. Electrosynthesis of urea from nitrite and CO2 over oxygen vacancy-rich ZnO porous nanosheets. Cell Rep. Phys. Sci. 2, 100378 (2021).
Wang, J. et al. Regulating the ammonia oxidation selectivity via the quantified provision of molecular oxygen. ACS Catal. 13, 8783–8791 (2023).
Dong, X. A. et al. Insights into dynamic surface bromide sites in Bi4O5Br2 for sustainable N2 photofixation. Angew. Chem. Int. Ed. 61, e202200937 (2022).
Hu, Q. et al. Ammonia electrosynthesis from nitrate using a ruthenium-copper cocatalyst system: a full concentration range study. J. Am. Chem. Soc. 146, 668–676 (2023).
Li, Y. et al. Sequential co-reduction of nitrate and carbon dioxide enables selective urea electrosynthesis. Nat. Commun. 15, 176 (2024).
Finkelstein, E., Rosen, G. M. & Rauckman, E. J. Spin trapping of superoxide and hydroxyl radical: practical aspects. Arch. Biochem. Biophys. 200, 1–16 (1980).
Attwood, A. L., Edwards, J. L., Rowlands, C. C. & Murphy, D. M. Identification of a surface alkylperoxy radical in the photocatalytic oxidation of acetone/O2 over TiO2. J. Phys. Chem. A 107, 1779–1782 (2003).
Tasdemir, H. U., Sayin, U., Turkkan, E. & Ozmen, A. EPR investigation of gamma irradiated single crystal guaifenesin: a combined experimental and computational study. Radiat. Phys. Chem. 121, 61–68 (2016).
Nonell, S. & Flors, C. Singlet Oxygen: Applications in Biosciences and Nanosciences (Royal Society of Chemistry, 2016).
Kearns, D. R. Physical and chemical properties of singletmolecular oxygen. Chem. Rev. 71, 395–427 (1971).
Burgess, V. A. & Easton, C. J. An EPR study of polar effects in radical reactions of N-acylamino acid derivatives. Spectrosc. Lett. 24, 1059–1070 (1991).
Eloranta, A. et al. EPR, ENDOR and TRIPLE resonance of amino-substituted 9,10-anthraquinone radicals and the rotation of the amino groups in the solution phase. Magn. Reson. Chem. 34, 903–907 (1996).
Elishav, O. et al. Progress and prospective of nitrogen-based alternative fuels. Chem. Rev. 120, 5352–5436 (2020).
Dove, J. E. & Nip, W. S. A shock-tube study of ammonia pyrolysis. Can. J. Chem. 57, 689–701 (1979).
Fuke, K. & Takasu, R. Ultrafast photochemistry of ammonia clusters: formation and decay of hypervalent molecular clusters containing the NH4 radical. Bull. Chem. Soc. Jpn. 68, 3309–3318 (2006).
Savee, J. D., Mann, J. E. & Continetti, R. E. Stability of the ground and low-lying vibrational states of the ammonium radical. J. Phys. Chem. Lett. 4, 3683–3686 (2013).
Chen, R. et al. Promoting the efficiency and selectivity of NO3-to-NH3 reduction on Cu-O-Ti active sites via preferential glycol oxidation with holes. Proc. Natl Acad. Sci. USA 120, e2312550120 (2023).
Li, J. et al. The spatially oriented charge flow and photocatalysis mechanism on internal van der Waals heterostructures enhanced g-C3N4. ACS Catal. 8, 8376–8385 (2018).
Yang, S. et al. Photocatalytic Co‐reduction of N2 and CO2 with CeO2 catalyst for urea synthesis. Angew. Chem. Int. Ed. 62, e202312076 (2023).
Shi, C. et al. Nitric acid‐mediated artificial urea photo‐synthesis with N2 And CO2. Adv. Energy Mater. 14, 2400201 (2024).
Yuan, M. et al. Host–guest molecular interaction promoted urea electrosynthesis over a precisely designed conductive metal–organic framework. Energy Environ. Sci. 15, 2084–2095 (2022).
Yang, W., Xiao, L., Dai, W., Mou, S. & Dong, F. Efficient solar driven upgrading of N2 to urea through photoredox reactions on Pt cluster/TiO2. Adv. Energy Mater. 14, 2303806 (2024).
Yang, W. et al. Photocatalytic formamide synthesis via coupling of electrophilic and nucleophilic radicals over atomically dispersed Bi sites. Angew. Chem. Int. Ed. 63, e202408379 (2024).
Li, P., Zhao, W., Wang, K., Wang, T. & Zhang, B. Photocatalytic synthesis of glycine from methanol and nitrate. Angew. Chem. Int. Ed. 63, e202405370 (2024).
Li, W. et al. Atomic ruthenium‐promoted cadmium sulfide for photocatalytic production of amino acids from biomass derivatives. Angew. Chem. Int. Ed. 63, e202320014 (2024).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Nos. 22225606, 22422607, and 22276029), the Young Elite Scientists Sponsorship Program by CAST (2023QNRC001) and the Natural Science Foundation of Sichuan Province (2025NSFTD0003).
Author information
Authors and Affiliations
Contributions
F.D. and J.L. conceived the idea and designed this study. X.L. conducted characterizations, photocatalytic tests, and mechanism investigations. W.Y. helped develop the photocatalytic C-N coupling reaction routes. J.Y. conducted the HR-MS and NMR detection experiments. S.S. helped screen the reaction routes from various C- and N-sources. R.C. and J.W. helped design the reactors and test the parameters for photocatalytic reactions and in situ characterizations. H.D. helped conduct the long-term stability test. D.Y. provided suggestions for conducting the mechanism investigation of intermediate coupling. X.L., J.L., and F.D. wrote the paper. All authors discussed the results and edited the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zhou Chen, Zupeng Chen and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, X., Yang, W., Yue, J. et al. Photocatalytic C-N coupling from stable and transient intermediates for gram-scale acetamide synthesis. Nat Commun 16, 3590 (2025). https://doi.org/10.1038/s41467-025-58840-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58840-0
This article is cited by
-
Revisiting Pt foil catalysts for formamide electrosynthesis achieved at industrial-level current densities
Nature Communications (2025)
