
Abstract
Charge separation of particulate photocatalysts has been considered as the rate-determining step in artificial photocatalysis since the finding of Honda-Fujishima effect, whose efficiency is generally much lower than that of natural photosynthesis. To approach its upper limit, it requires the photoexcited electrons and holes be efficiently transferred to the spatially separated redox reaction sites over a single photocatalyst particle. Herein, it is demonstrated the spatial charge separation among facets of BiVO4:Mo can be notably promoted by creating an electron transfer layer. It not only favors electrons to transfer to its surface, but also promotes the built-in electric field intensity of the inter-facet junction by over 10 times. Consequently, the charge separation efficiency of the modified BiVO4:Mo with loading of CoFeOx oxidation cocatalyst exceeds 90% at 420 nm, comparable to that of the natural photosynthesis system, over which notably enhanced photocatalytic activities are achieved. Our findings demonstrate the effectiveness of electron transfer layer in intensifying charge separation of particulate photocatalysts.
Similar content being viewed by others


Introduction
Charge separation is crucial for the achievement of efficient artificial photosynthetic reactions over particulate photocatalysts, such as photocatalytic overall water splitting (OWS)1,2,3,4, CO2 reduction5,6,7, and CH4 conversion8,9,10, etc. The separation and transfer of the photogenerated electrons and holes are generally relevant to the intrinsic electronic structure, crystallinity, morphology, and defects of the semiconductors. The appearance of even one disadvantageous factor derived from the above-mentioned aspects can result in poor charge separation, which is one of the key reasons responsible for the current low efficiency of photocatalysts11,12,13,14,15,16. Hence, the exploration of effective strategies to promote charge separation is highly desirable for the development of efficient artificial photocatalysts.
Construction of different surface and interface junction structures is an effective way to promote charge separation by creating an energy level difference in or between the surface or the interface, such as heterojunctions (i.e., CdTe/V-In2S3, InGaN/GaN)17,18,19, phase junctions (i.e., α/β Ga2O3, anatase/rutile)20,21, and inter-facet junctions (i.e., BiVO4 {110}/{010} facet and SrTiO3 {110}/{100} facet)22,23,24. For example, Li et al. have demonstrated that there is an obvious potential difference between {010} and {110} facets of the monoclinic BiVO4. The conduction band minimum (CBM) and valence band maximum (VBM) of the {010} facet are lower than those of the {110} facet, which facilitates the migration of electrons to the {010} facet and the accumulation of holes on the {110} facet. As a result, the spatial separation of photogenerated electrons and holes can be achieved. However, the charge separation of photocatalysts that aim to achieve visible-light driven photocatalytic overall reactions is much lower than that of the natural photosynthesis system25,26. To further promote the charge separation, more effective strategies need to be explored based on it. Different from the fields of photoelectrocatalysis27 and photovoltaics28 that highly efficient spatial charge separation in cascade can be realized by rationally integrating multiple cooperative strategies in the electrode devices, it is rather challenging for the case of particulate photocatalysts. It is mainly because each photocatalyst particle can be regarded as one integral reaction system, where charge transfer is generally disordered and difficult to be manipulated for the subsequent redox reactions, resulting in serious charge recombination. To achieve comparable charge separation efficiency of particulate artificial photocatalysts as the natural photosynthesis, the key issue should be addressed based on how to render the photoexcited electrons and holes to be efficiently transferred and accumulated at the spatially separated redox reaction sites over a single photocatalyst particle.
To address this issue, a visible-light-responsive oxide photocatalyst of the decahedral Mo-doped BiVO4 (denoted as BiVO4:Mo) with spatial charge separation among facets is selected as a model in this work. It is demonstrated that its charge separation efficiency can be greatly enhanced by constructing an electron transfer layer (ETL) on the {010} surface (an electron accumulation facet) via a simple NaOH etching treatment. Detailed analysis shows that the complex defect (VO2 vacancy with the V site occupied by the Na, denoted as Na(VO2)) is formed in the etching layer, and induces the downward shifting of band edges with respect to the pristine {010} surface, leading to the formation of an external built-in electric field perpendicular to the {010} direction. What’s more important, due to such downshift of the band edges of the etching layer on the {010} surface, the potential difference between {110} facet (a hole accumulation facet) and the treated {010} facet is further enlarged with the electric field intensity enhanced by 12 times, facilitating the notably promoted spatial charge separation among facets. After loading of CoFeOx cocatalyst, the charge separation efficiency of over 90% at 420 nm is achieved for the modified BiVO4:Mo photocatalyst, making a breakthrough in visible-light-responsive oxide photocatalysts with comparable value as that of the natural photosynthesis system. This work demonstrates the developed ETL strategy can effectively cooperate with the inter-facet junction strategy to remarkably intensify the spatial charge separation among facets of BiVO4:Mo, which is expected to be widely extended for efficient photocatalytic applications.
Results
Preparation and structural characterizations of BiVO4:Mo-derived samples
BiVO4:Mo was synthesized by a hydrothermal method29. Subsequently, it was further etched in NaOH aqueous solution to prepare the surface modified sample (denoted as BiVO4:Mo(NaOH), see the catalyst preparation part for details)30. The NaOH etching causes the dissolution of V atoms, while Bi atoms are rarely dissolved. In addition, Na atoms are found to be incorporated in the BiVO4:Mo(NaOH) sample (Supplementary Table 1). X-ray diffraction (XRD) patterns and ultraviolet-visible diffuse reflectance spectra (UV-Vis DRS) results show that both samples exhibit the monoclinic phase (Supplementary Fig. 1a) and similar absorption edge of 520 nm (Supplementary Fig. 1b)31. Further information of the Rietveld refinement for their XRD patterns indicate that such alkali etching treatment can cause partial lattice distortion in BiVO4:Mo crystal structure (see Supplementary Fig. 2 and the detailed analysis in Supplementary Table 2), in which the lengths of V-O and Bi-O bonds become shorter, and the asymmetric degree of V-O bond turns greater (Supplementary Fig. 3)32. Scanning electron microscopy (SEM) images and energy dispersive spectrometer (EDS) mapping images further show that both BiVO4:Mo and BiVO4:Mo(NaOH) samples have the similar particle size and decahedral morphology, where the top and side surfaces are {010} and {110} facets, respectively (Supplementary Fig. 4)33. It should be noted that spatial charge separation among facets can be similarly observed on both samples (Supplementary Figs. 5, 6), in accordance with our previous report24. In addition, similar specific surface areas are also confirmed for both samples (Supplementary Table 3). Therefore, it can be briefly concluded that the macrostructure properties of BiVO4:Mo are well-maintained except that a little distortion in the main crystal structure is induced after the NaOH etching treatment.
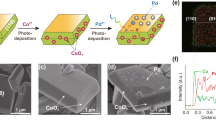
The location of surface atoms after etching was verified by annular dark field scanning transmission electron microscopy (ADF-STEM). As shown in the atomic resolution ADF-STEM images of BiVO4:Mo and BiVO4:Mo(NaOH) along the [001] direction in Fig. 1a, b, both Bi and V atoms are imaged as bright spots, while O atoms are not visible. Correspondingly, the brighter atoms are assigned to Bi atoms, and the darker ones are V atoms. The atomic arrangement matches well with the crystal structure of {010} facet of BiVO4 in Fig. 1c. It can be observed that two Bi atoms and two V atoms are arranged alternately on the (040) facet of BiVO4:Mo (Fig. 1a), while V atoms are selectively etched with Bi atoms are kept for the BiVO4:Mo(NaOH) sample (Fig. 1b). In addition, a reduction in the atomic spacing of BiVO4:Mo(NaOH) [001] orientation along the a-axis and c-axis is found (Supplementary Fig. 7), but the main crystal skeleton is maintained. This may result from the fact that the incorporated Na occupies the V vacancy, which keeps the crystal structure from being seriously reconstructed. It should be pointed out that it is difficult to distinguish these two different atoms on the {110} facet via the ADF-STEM characterization due to the overlap of Bi and V atoms in top view (Supplementary Fig. 8).

Atomic-resolution ADF-STEM images of {010} facet of (a) BiVO4:Mo and (b) BiVO4:Mo(NaOH) samples. c Atomic arrangement of the {010} facet of BiVO4. STEM images of (d, e) BiVO4:Mo and (g, h) BiVO4:Mo(NaOH) particles with the probing path of EELS shown by an arrow (this illustration is a simulated probe position). f, i The corresponding EELS of V L2,3 edge.
To further check the change of surface composition of the {110} facet, electron energy loss spectroscopy (EELS) was thus carried out. Meanwhile, it was performed along the {110} to {010} facet direction (see the arrows in Fig. 1e, h) to analyze the valence states of V and O elements by probing V L2,3 edge and O K edge (Fig. 1d–i and Supplementary Fig. 9). As shown in Fig. 1f and Supplementary Fig. 9a, there is no shift for the peaks of the V L2,3 and O K edges along the {110} to {010} facet direction for the BiVO4:Mo sample. After etching with NaOH, the peaks of the V L2,3 and O K edges on the {110} facet are the same as the case in BiVO4:Mo sample. However, both peaks are obviously decreased on the {010} facet (Fig. 1i and Supplementary Fig. 9b). These results indicate that the valence states of V and O elements on the {110} facet of the samples with and without etching are the same, while the valences on the {010} facet become a little lower after the etching. Therefore, it is reasonable to deduce that the NaOH etching treatment will dissolve a part of V and O atoms on the {010} facet, while it does not obviously affect the composition of {110} facet of the BiVO4:Mo sample. This may be due to the fact that the surface hydrogenation energies of both {010} and {110} facets with distinct surface coordination structure are strongly dependent on the pH value, and the hydrogenated {010} facet tends to become more unstable than the hydrogenated {110} facet with increasing the pH value34.
To further analyze the thickness and composition of the etching layer on the {010} surface, Ar+ sputtering was applied to controllably peel the surface of the sample, and then X-ray photoelectron spectroscopy (XPS) characterization was carried out to detect the peeled sample (denoted as: Ar+ sputtering X nm, X = 1–5). As shown in the high-resolution V 2p XPS of BiVO4:Mo in Fig. 2a, two characteristic peaks observed at 517.0 and 524.6 eV are ascribed to V5+ 2p3/2 and V5+ 2p1/2, respectively35. These two peaks with lower binding energies of ca. 0.3 eV are also observed in BiVO4:Mo(NaOH) than those of BiVO4:Mo sample, revealing the reduced valence state of V after the etching. With increasing the thickness of the peeled layer over the BiVO4:Mo(NaOH), the binding energies of V 2p peaks will be gradually increased. When the thickness of the peeled layer reaches ca. 4 nm, the binding energy of V over the etched sample is consistent with that in BiVO4:Mo. Similar results are also found for the Bi3+ 4f7/2, Bi3+ 4f5/2 and O2– 1 s peaks in Fig. 2b, c36,37,38. In addition, it needs to be noted that the intensity of the Na+ 1 s peak at 1070.8 eV decreases with increasing the sputtering depth, and disappears until the depth is 4 nm (Fig. 2d). These results integrally indicate that the etched thickness is ca. 4 nm. Detailed surface atom ratios of Na:Bi:V:O for those samples were calculated and summarized in Supplementary Table 4. In the whole etching layer, one V vacancy is approximately accompanied by two O vacancies and one Na impurity (see the details in Supplementary Fig. 10). In this case, both defects of Vac(VO2) and NaV are proposed to form, which can be assigned to p-type defect in BiVO4 and are not conducive to the electron transfer39. When the Vac(VO2) and NaV share the same V occupation site, two different p-type defects can be coupled thermodynamically to form a neutral complex defect of Na(VO2) in the etching layer (Fig. 2e and Supplementary Fig. 11, and see the details in Supplementary Table 5, denoted as ion-substituted layer).

XPS (a) V 2p, (b) Bi 4 f, (c) O 1 s, and (d) Na 1 s of series BiVO4:Mo(NaOH) samples with different etched depth. e Schematic diagram of the ion-substituted layer in BiVO4:Mo(NaOH). Herein, 1, Ar+ sputtering 1 nm, 2, Ar+ sputtering 2 nm, 3, Ar+ sputtering 3 nm, 4, Ar+ sputtering 4 nm, 5, Ar+ sputtering 5 nm.
Effect of the ion-substituted layer on the spatial charge separation among facets of BiVO4
To reveal the influence of the ion-substituted layer on the charge separation, firstly, the structures and the corresponding total density of states (TDOS) between the pristine {010} surface and the ion-substituted {010} surface were compared and analyzed. As shown in Fig. 3a–c, one Vac(VO2) causes one electron loss of the surface, and therefore the VBM of BiVO4 {010} surface with Vac(VO2) defect is obviously upshifted relative to the VBM of the perfect BiVO4 {010} surface, further confirming the p-type character of Vac(VO2) defect. When V is replaced by Na in the finite surface model, both the CBM and VBM are upshifted relative to those of the pristine surface, further illustrating the p-type character of NaV defect. However, when Vac(VO2) and NaV defects are coupled to form Na(VO2), the lost one electron is compensated by the addition of Na. The coupled neutral defect of Na(VO2) makes the CBM and VBM downshift relative to those of the perfect surface, thereby enabling the ion-substituted layer to function as an ETL that facilitates electron transfer from the perfect {010} surface to the ion-substituted {010} surface (Fig. 3d).

a Optimized atomic models, (b) TDOS, and (c) band alignment of different BiVO4 {010} surfaces. d A schematic illustration of the band structure of BiVO4 and ion-substituted layer. Herein, 1, BiVO4 {010} surface, 2, BiVO4 {010} surface-Vac(VO2), 3, BiVO4 {010} surface-NaV, 4, BiVO4 {010} surface-Na(VO2). CBM conduction band minimum, VBM valance band maximum.
The effect of the ETL on the charge separation of BiVO4:Mo(NaOH) in the scale of single particle was further analyzed according to our home-made light-assisted Kelvin probe force microscopy (KPFM) characterization. Herein, Atomic Force Microscopy (AFM) scanning was used to obtain 3D topographic images to identify BiVO4:Mo and BiVO4:Mo(NaOH) particles (Fig. 4a, b), and KPFM was applied to measure the contact potential difference (CPD) of typical samples under both dark and illuminated states (Supplementary Fig. 12a, b). The work functions of {010} and {110} facets (denoted as Wf(sample-010) and Wf(sample-110), respectively) can be calculated with the measured CPD under the darkness (see the details in Supplementary Fig. 12). It is worth noting that the Wf(BiVO4:Mo(NaOH)-110) is close to the Wf(BiVO4:Mo-110). However, the Wf(BiVO4:Mo(NaOH)-010) increases from 5.43 to 5.51 eV due to the existence of the ETL, implying the formation of an external built-in electric field can facilitate the electron transfer from the bulk to the treated {010} surface (Supplementary Fig. 12c, d).

a1, b1 3D AFM topographic images of BiVO4:Mo and BiVO4:Mo(NaOH) samples. 3D KPFM potential images under (a2, b2) dark and (a3, b3) illuminated conditions. c, d Cross-sectional SPV of BiVO4:Mo and BiVO4:Mo(NaOH) samples acquired from 3D KPFM potential images under dark and illuminated conditions. e, f Analysis of electrical field calculated from the results in (c, d). SPV: surface photovoltage.
The degree of band bending and the driving force of charge separation were further identified by the value of surface photovoltage (SPV, SPV = CPDillumination – CPDdark) and ΔSPV (ΔSPV = SPV{110} – SPV{010}), respectively (see the details in Supplementary Fig. 13)40. It is striking to notice that the ΔSPV (ca. 80–100 mV) of BiVO4:Mo(NaOH) can be promoted by ca. 5 times with respect to the BiVO4:Mo (ca. 10–20 mV) (Fig. 4c, d). Based on the present SPV data, the profile diagram showing the distribution of linear electric field intensity can be calculated41. As shown in Figs. 4e, f, the maximum electric field intensity of BiVO4:Mo(NaOH) can reach ca. 2000 kV m–1, which is ca. 12 times higher than that of BiVO4:Mo (ca. 150 kV m–1). This means that the ion-substitution-induced built-in electric field can be superimposed on the pristine built-in electric field among facets, cooperatively promoting the charge separation.
The charge separation efficiency at 420 nm was quantitatively evaluated via one photochemical reaction using dual scavengers to eliminate the influence of surface catalytic conversion processes, in which the charge injection efficiency is considered to be almost 100% (see the detail in Supplementary Fig. 14). In this case, the obtained apparent quantum efficiency (AQE) can be approximated as the charge separation efficiency. Specifically, the Ag+ ions and methanol were employed as electron and hole sacrificial agents, respectively, and the amount of photodeposited Ag was quantified to insight the AQE. As shown in Supplementary Fig. 14, the photodeposition rates of Ag on BiVO4:Mo, BiVO4:Mo(NaOH), and CoFeOx-BiVO4:Mo(NaOH) samples can be calculated as 0.0106, 0.0204, and 0.0249 mg s–1, respectively. It means that the photogenerated charge separation efficiency of BiVO4:Mo(NaOH) is doubled to that of the pristine BiVO4:Mo, which is even comparable to the highest quantum efficiency of the reported BiVO4 photocatalyst42. After loading CoFeOx oxidation cocatalyst on the {110} facet, a Schottky junction is formed between CoFeOx and BiVO4:Mo, which facilitates the hole transfer and ultimately enables the charge separation efficiency of CoFeOx-BiVO4:Mo(NaOH) exceeding 90% (Table 1).
Photocatalytic performance
As encouraged by the significant promotion of charge separation, the BiVO4:Mo(NaOH) modified with cocatalysts was thus investigated for water oxidation in the presence of [Fe(CN)6]3– ions (Supplementary Figs. 15, 16)43. The photocatalytic water oxidation activity of 270 μmol h–1 is achieved over the optimized ion-substituted photocatalyst, which is 1.5 times higher than that of the pristine BiVO4:Mo photocatalyst (Supplementary Figs. 17, 18). It is worth noting that similar promotion effect of NaOH etching is observed for the BiVO4 photocatalyst without Mo doping (Supplementary Fig. 18), which demonstrates that Mo doping does not remarkably affect the construction of the ETL for the promoted charge separation. As shown in Supplementary Fig. 19, good reproducibility of the photocatalytic O2 evolution rates is observed on the BiVO4:Mo(NaOH) samples in different batches. Considering the state of the deposited co-catalysts and the absorption edges of BiVO4:Mo and BiVO4:Mo(NaOH) samples are similar, the main difference in activity should be ascribed to the distinct charge separation efficiency (Supplementary Figs. 20, 21). In addition, another photocatalytic oxygen evolution reaction in the presence of Fe3+ ions and photocatalytic degradation of tetracycline were also evaluated, where the similar promotion effect was observed (Supplementary Fig. 22).
Finally, a photocatalytic Z-scheme OWS system was assembled by employing Au/CoFeOx-BiVO4:Mo(NaOH), RhyCr2–yO3-ZrO2/TaON and [Fe(CN)6]3–/4– couple as an O2-evolving photocatalyst (OEP), an H2-evolving photocatalyst (HEP) and a redox mediator, respectively (Fig. 5a). The HEP was synthesized according to the previous report44 (see the catalyst preparation part for details), and the basic structural characterization results of HEP were shown in Supplementary Figs. 23–25. RhyCr2–yO3 was applied as a reduction cocatalyst over the ZrO2/TaON photocatalyst, in which the optimal contents of Rh and Cr were 1.0 and 1.1 wt%, respectively (Supplementary Figs. 26, 27). Under the optimized reaction condition (Supplementary Figs. 28–30), the H2 and O2 evolution rates of 270 and 135 μmol h–1 from the OWS reaction are achieved under visible light irradiation (Fig. 5b), and the multiple consecutive cycles test shows the good stability of the assembled photocatalytic system (Fig. 5c). On this basis, an AQE of 22.8% at 420 nm is achieved (Supplementary Table 6), surpassing the previously reported values of redox-mediated Z-scheme OWS systems (Supplementary Table 7). It should be pointed out that Na+ ions remained within the BiVO4:Mo lattice are not obviously leached out (Supplementary Table 8), implying the robustness of the ETL during the reaction.

a Schematic demonstration of photocatalytic Z-scheme OWS over RhyCr2–yO3-ZrO2/TaON and Au/CoFeOx-BiVO4:Mo(NaOH). b Time courses and (c) multiple cycles of curve of Z-scheme OWS. Reaction conditions: 75 mg of Au/CoFeOx-BiVO4:Mo(NaOH) and 125 mg of RhyCr2–yO3-ZrO2/TaON in the 150 mL of sodium phosphate buffer solution (pH = 6.0, 25 mM) containing K4[Fe(CN)6] (15 mM), 300 W xenon lamp (λ > 420 nm), temperature: 283 K, Pyrex top-irradiation type.
Discussion
In summary, the ion-substitution-induced ETL is demonstrated to be effective in cooperation with the inter-facet junction for notably promoted spatial charge separation of BiVO4:Mo photocatalyst, leading to a high charge separation efficiency of over 90% at 420 nm for CoFeOx-BiVO4:Mo(NaOH), marking a breakthrough in artificial photosynthesis comparable to that of the natural photosynthesis system. Detailed analysis shows that the neutral defect of Na(VO2) in the layer induces the downshift of the band edges of the {010} surface. Consequently, an external built-in electric field perpendicular to the {010} direction is generated, leading to the enlarged potential difference between {110} facet and the treated {010} facet, which not only promotes the electron transfer from the bulk to the ion-substituted {010} surface, but also facilitates the spatial charge separation between {110} and the ion-substituted {010} facets. As benefited from the notably promoted charge separation, such photocatalytic systems with remarkably promoted efficiencies as reactions including water oxidation, Z-scheme OWS, and tetracycline degradation can be fabricated. Specifically, one redox-mediated Z-scheme OWS system with a benchmarking AQE of 22.8% at 420 nm is assembled. This work highlights the importance and effectiveness of ETL in promoting the spatial charge separation of particulate photocatalysts, especially when it is cooperated with other “junction”-based strategies, as may open a venue to develop highly efficient photocatalytic systems.
Methods
Materials and reagents
For the preparation of ZrO2/TaON and BiVO4:Mo samples, Ta2O5 (99.9%, High Purity Chemicals), ZrO(NO3)2·2H2O (ZrO2 45.0%, Guangfu Chemical Reagent), Bi(NO3)3·5H2O (99.0%, Sinopharm Chemical), NH4VO4 (99%, Rhawn Chemical Reagent), Na2MoO4 (99.0%, Sinopharm Chemical), NaOH (98%, Alfa Aesar), HNO3 (97%, Jiangtian Chemical Technology Co., LTD), NH3·H2O (25%–28%, Aladdin Chemical Reagent) and (CH2OH)2 (99%, Aladdin Chemical Reagent) were used. NaH2PO4 (99.9%, Macklin Chemical Reagent) and Na2HPO4 (99%, Macklin Chemical Reagent) were used to prepare phosphoric acid buffer solution. Na3RhCl6·nH2O (Rh ≥ 17%, Alfa Aesar), K2CrO4 (99.5%, Tianjin Kemiou Chemical Reagent Co., LTD), HAuCl4 (Au 48%–50%, Sinopharm Chemical), and CoSO4 (99.5%, Rhawn Chemical Reagent) were used for the deposition of cocatalysts. K4[Fe(CN)6] (99.5%, Sinopharm Chemical) or methanol (99.9%, Aladdin Chemical Reagent) was used as a hole acceptor, while K3[Fe(CN)6] (99.5%, Sinopharm Chemical), Fe(NO3)3·9H2O (99.9%, Aladdin Chemical Reagent) or AgNO3 (99%, Aladdin Chemical Reagent) was used as an electron acceptor. H2SO4 (98%, Tianjin Bohua Chemical Reagent Co., LTD) was used to mediate the pH value of the solution. Tetracycline (96%, Aladdin Chemical Reagent) was used as a model pollutant. All chemicals were used as purchased without further purification.
Catalyst preparation
Synthesis of ZrO2/TaON
Typically, Ta2O5 powder was mixed with ZrO(NO3)2 in methanol, then the obtained mixture was dried and calcined at 1073 K for 2 h in air to yield ZrO2/Ta2O5 composite. The composite was nitrided with an NH3 flow (20 mL min–1) at 1123 K for 15 h to synthesize ZrO2/TaON (Zr/Ta = 0.15 by mol)44.
Synthesis of BiVO4:Mo
BiVO4:Mo (Mo/(Mo+V) = 0.04 mol%) was synthesized by a hydrothermal method29. Typically, Bi(NO3)3·5H2O and NH4VO3 were dissolved into HNO3 solution (2 M) and NaOH solution (2 M), respectively. A certain amount of NaMoO4 was added to the NaOH solution for Mo doping. Then Bi(NO3)3 solution was added dropwise to the NH4VO3 solution, and an orange precipitate was formed. Subsequently, (CH2OH)2 was added to the mixed solution, and the pH value of the solution was adjusted to 2.1 with NH3·H2O. After stirring the solution for 0.5 h, the suspension was transferred to a Teflon-lined stainless-steel autoclave and maintained at 473 K for 24 h.
Synthesis of BiVO4:Mo(NaOH)
Typically, the as-prepared BiVO4:Mo powder was dispersed in NaOH aqueous solution (0.01 M) and stirred for 6 min at room temperature. Then the powder was filtered and dried. The obtained powder is denoted as BiVO4:Mo(NaOH).
Cocatalyst deposition
Preparation of RhyCr2–yO3-ZrO2/TaON
The samples were prepared by an in situ photodeposition method. Typically, 50 mg of ZrO2/TaON was dispersed in 100 mL of methanol solution (20 vol%) containing a calculated amount of Na3RhCl6 and K2CrO4. The solution was vacuumed to remove air and then irradiated for 6 h under a 300 W xenon lamp without any cut-off filter. After the reaction, the as-obtained powder sample was collected by filtration and denoted as RhyCr2–yO3-ZrO2/TaON.
Preparation of Au/CoFeOx-BiVO4:Mo and Au/CoFeOx-BiVO4:Mo(NaOH)
Cocatalysts of Au and CoFeOx were loaded by an in situ photodeposition method. Typically, 50 mg of BiVO4:Mo or BiVO4:Mo(NaOH) powder was dispersed in 25 mM of phosphate buffer solution (pH = 6, 100 mL) containing calculated amounts of HAuCl4, CoSO4, and K3[Fe(CN)6] (0.5 mM). Before the irradiation, the reaction system was thoroughly vacuumed for the removal of the air. Then a 300 W xenon lamp without any cut-off filter was introduced to irradiate the dispersed solution for 2 h. After the reaction, the as-loaded powder was collected by filtration and dried naturally, which was denoted as Au/CoFeOx-BiVO4:Mo or Au/CoFeOx-BiVO4:Mo(NaOH).
Preparation of CoFeOx-BiVO4:Mo(NaOH)
CoFeOx cocatalyst was loaded by an in-situ photodeposition method. Typically, 50 mg of BiVO4:Mo was dispersed in 25 mM of phosphate buffer solution (pH = 6, 100 mL) containing the calculated amounts of CoSO4 (0.2 wt%), K4[Fe(CN)6] (0.8 mM), and NaIO3 (20 mM). The reaction system was thoroughly evacuated, and then the dispersion solution was irradiated with a 300 W xenon lamp without any cutoff filter for 2 h. After the reaction, the as-obtained powder sample was collected by filtration and denoted as CoFeOx-BiVO4:Mo(NaOH).
Characterizations
Field emission SEM (JEOL, JSM-7800F) was used to analyze the morphology of the catalysts. Atomic-resolution TEM (JEOL, JEM-ARM200F) was equipped with a cold field emission gun, an aberration corrector, and an EELS (Enfina, Gatan) to analyze the atomic arrangement of the catalyst surface and valence states of elements. TEM (JEOL, JEM-2800) was used to analyze the elemental distribution and lattice fringe. The XRD (Rigaku, MiniFlex 600) with Cu Kα (λ = 0.154178 nm) radiation and Raman spectroscopy (TEO, SR-500I-A) were employed to verify structural characteristics of the catalysts. The absorbance of the samples was measured by UV-Vis DRS (Thermo Fisher, Evolution 220). ICP-OES (Agilent 5110) was used to analyze the content of Na, Bi, V, and Ag elements. The surface area was calculated based on the BET model using a Micromeritics ASAP 2460 at 77 K. XPS was operated on a Thermo Scientific ESCALAB 250Xi with a monochromatic Al Kα source (150 W, 1486.6 eV, 500 μm, the C 1 s positioned at 284.8 eV was applied as the internal standard for the calibration of binding energies) was employed to study the chemical state. Depth profiling was accomplished using Ar+. The Ar+ was operated at 3 kV and 10 mA with a 1 × 1 mm2 raster size at an angle of 40° to the surface normal with an etching speed of 0.2 nm s–1. Therefore, each sputtering of Ar+ for every 5 s is considered to etch a depth of 1 nm.
The CPD images of samples were measured by KPFM (Bruker Dimension FastScan) in the amplitude-modulated mode under ambient atmosphere. A lift mode with a lifting height of 75 nm was applied for the SPV test. The Kelvin tip used was made of Sb-doped Si tip coated with Pt/Ir (SCM-PIT-V2). CPD under light was measured using a blue LED (λ = 405 nm, 4 mW cm–2) almost parallel to the substrate. SPV refers to the CPD difference with or without light illumination, which is defined as SPV = CPDillumination – CPDdark45.
Density functional theory calculations and models
First-principles calculations in this work were performed employing the plane-wave pseudopotential method within density functional theory (DFT), using the Perdew-Burke-Ernzerhof (PBE) method as exchange-correlation functional, and implemented through the Vienna Ab-initio Simulation Package (VASP) code46. The interaction of electron-ion was described using the projector augmented wave (PAW) pseudopotential method47. 5d106s26p3 for Bi, 3d43s1 for V, 2s22p4 for O and 2p63s1 for Na atoms were adopted as valence electrons. Considering the Mo dopant with a low doping proportion is recognized as uniformly distributed in the bulk, the role of the Mo dopant in the ETL is quite minor. The stoichiometric BiVO4 {010} surface was adopted to represent the BiVO4:Mo {010} facet. The BiVO4 {010} surface was represented by a slab model repeated periodically with a vacuum region of 15 Å between repeated slabs. The {010} slab was constructed by 32 Bi atoms in 8 Bi layers, 32 V atoms in 8 V layers, and 128 O atoms in 16 O layers. The top 4 Bi and 4 V layers and top 8 O layers are fully relaxed (see the details in Supplementary Data 1). The 3 × 3 × 1 Monkhorst-Pack-type k-points samplings were used for (010) 2 × 2 surface unit cells. To ensure that the residual forces converged to less than 0.02 eV Å–1, the conjugate gradient method was employed to optimize the surface structure48. The electronic wavefunction was expanded using a kinetic energy cutoff of 520 eV. All of the eigenvalues of the slabs with different defects are aligned by the Bi 1 s core state of the same fixed Bi atom. For defects in the surface model, their formation Gibbs free energies can be calculated by using the well-established formalism39,49,50. The detailed defect calculation results can be found in Supplementary Table 5 and Supplementary Fig. 11.
Measurement of the charge separation efficiency
The charge separation efficiency was measured using a 300 W Xe lamp (Beijing Perfectlight, PLS-SXE 300D) with a band-pass filter (Beijing Perfectlight, λ = 420 nm, full width at half maximum (FWHM): 15 nm) to illuminate a Pyrex top-irradiation-type reaction vessel that was connected to a closed glass circulation system (Beijing Perfectlight, Labsolar-6A). 75 mg of photocatalyst was added into the 100 mL of methanol solution (20 vol%) containing AgNO3 (50 mM). The reaction solution was fully vacuumed to remove the air before the irradiation, and the system was maintained at 283 K using cooling water. The charge separation efficiency was calculated based on the following Eq. (1):
where A is a coefficient, which is 1 for Ag photodeposition. R is the photodeposition rate of Ag (mol s–1), Na is the Avogadro constant, and I is the number of photons measured at a specific wavelength. The number of photons reaching the solution was measured by a calibrated silicon photodiode (HAMAMATSU, S2281-04). As measured, the total number of the incident photons at the wavelength of 420 nm was 1.54 × 1017 photons s–1 (see the details in Supplementary Fig. 31, Supplementary Tables 9, 10). The photodeposition rates of Ag over BiVO4:Mo, BiVO4:Mo(NaOH), and CoFeOx-BiVO4:Mo(NaOH) were quantified by ICP-OES.
Evaluation of hydrogen evolution and water oxidation
Photocatalytic hydrogen evolution and water oxidation reactions were conducted in a Pyrex vessel that was connected to a closed glass circulation system (Beijing Perfectlight, Labsolar-6A) under top irradiation from a 300 W xenon lamp (Beijing Perfectlight, PLS-SXE 300D) with a cutoff filter (λ > 420 nm). For the test of hydrogen evolution, 50 mg of photocatalyst was added into the 100 mL of phosphoric acid buffer solution (25 mM, pH = 6.0) containing 10 mM of K4[Fe(CN)6]. For the test of water oxidation with [Fe(CN)6]3– ions, 50 mg of photocatalyst was added into the 100 mL of phosphoric acid buffer solution (25 mM, pH = 6.0) containing 10 mM of K3[Fe(CN)6]. For the evaluation of water oxidation with Fe3+ ions, 50 mg of photocatalyst was added into the 100 mL of H2O containing 10 mM of Fe(NO3)3, which H2SO4 was added to the solution to mediate the pH value to 2.5. The reaction was carried out under vacuum conditions and maintained at 283 K. Cooling water was used to maintain the reaction system at 283 K. The evolved gases were analyzed by an on-line gas chromatography (Shimadzu, GC-2014C, 5 Å molecular sieve columns, thermal conductivity detector, Ar carrier gas).
Evaluation of photocatalytic degradation of tetracycline
Photocatalytic degradation of tetracycline was conducted in a Pyrex vessel, which was implemented under top irradiation from a 300 W xenon lamp with a cutoff filter (λ > 420 nm). 50 mg of photocatalyst was added into 50 mL of H2O containing tetracycline (20 mg L–1). Before the irradiation, the suspension was vigorously stirred for 1 h in the darkness to reach a balance and was maintained at 298 K. The concentration of tetracycline was determined via the high-performance liquid chromatography (Agilent, 1260 Infinity II) with a UV detector at 355 nm.
Evaluation of Z-scheme overall water splitting
Photocatalytic Z-scheme OWS reaction was conducted in the same glass circulation system with photocatalytic hydrogen evolution and water oxidation reactions under top irradiation from a 300 W xenon lamp with a cutoff filter (λ > 420 nm). 75 mg of OEP and 125 mg of HEP were added into the 150 mL of phosphoric acid buffer solution (25 mM, pH = 6.0) containing 15 mM of K4[Fe(CN)6]. The reaction system occurred under vacuum conditions and was maintained at 283 K. The evolved gases were analyzed by an on-line gas chromatography.
Measurement of apparent quantum efficiency
An AQE was measured using a 300 W Xe lamp with a band-pass filter (λ = 420 nm, FWHM: 15 nm) to illuminate a Pyrex top-irradiation-type reaction vessel. The AQE was calculated based on the following Eq. (2):
where A is a coefficient, which is 4 for hydrogen evolution and 8 for oxygen evolution. R is the rate of hydrogen or oxygen evolution (mol s–1), Na is the Avogadro constant, and I is the number of photons measured at a specific wavelength.
Data availability
The source data generated in this study are provided in the Source Data file. Source data are provided with this paper.
References
Nishiyama, H. et al. Photocatalytic solar hydrogen production from water on a 100-m2 scale. Nature 598, 304–307 (2021).
Nishioka, S. et al. Photocatalytic water splitting. Nat. Rev. Methods Prim. 3, 42 (2023).
Lin, L. et al. Molecular-level insights on the reactive facet of carbon nitride single crystals photocatalysing overall water splitting. Nat. Catal. 3, 649–655 (2020).
Xiao, M. et al. Hollow nanostructures for photocatalysis: advantages and challenges. Adv. Mater. 20, 1801369 (2019).
Wang, Q. et al. Molecularly engineered photocatalyst sheet for scalable solar formate production from carbon dioxide and water. Nat. Energy 5, 703–710 (2020).
Yoshino, S. et al. Photocatalytic CO2 reduction using water as an electron donor under visible light irradiation by Z-scheme and photoelectrochemical systems over (CuGa)0.5ZnS2 in the presence of basic additives. J. Am. Chem. Soc. 144, 2323–2332 (2022).
Wang, Y. et al. Direct and indirect Z-scheme heterostructure-coupled photosystem enabling cooperation of CO2 reduction and H2O oxidation. Nat. Commun. 11, 3043 (2020).
Feng, N. et al. Efficient and selective photocatalytic CH4 conversion to CH3OH with O2 by controlling overoxidation on TiO2. Nat. Commun. 12, 4652 (2021).
Luo, L. et al. Nearly 100% selective and visible-light-driven methane conversion to formaldehyde via. single-atom Cu and Wδ+. Nat. Commun. 14, 2690 (2023).
Fan, Y. et al. Selective photocatalytic oxidation of methane by quantum-sized bismuth vanadate. Nat. Sustain. 4, 509–515 (2021).
Tao, C. et al. Surface reconstruction and passivation of BiVO4 photoanodes depending on the “structure breaker” Cs+. JACS Au 3, 1851–1863 (2023).
Kim, D., Sakimoto, K., Hong, D. & Yang, P. Artificial photosynthesis for sustainable fuel and chemical production. Angew. Chem. Int. Ed. 54, 3259–3266 (2015).
Xue, Z., Luan, D., Zhang, H. & Lou, X. Single-atom catalysts for photocatalytic energy conversion. Joule 6, 92–133 (2022).
Pan, L. et al. Boosting the performance of Cu2O photocathodes for unassisted solar water splitting devices. Nat. Catal. 1, 412–420 (2018).
Li, Z., Luo, W., Zhang, M., Feng, J. & Zou, Z. Photoelectrochemical cells for solar hydrogen production: current state of promising photoelectrodes, methods to improve their properties, and outlook. Energy Environ. Sci. 6, 347–370 (2013).
Li, X., Tung, C. & Wu, L. Semiconducting quantum dots for artificial photosynthesis. Nat. Rev. Chem. 2, 160–173 (2018).
Zhou, P. et al. Solar-to-hydrogen efficiency of more than 9% in photocatalytic water splitting. Nature 613, 66–70 (2023).
Zhao, D. et al. Boron-doped nitrogen-deficient carbon nitride-based Z-scheme heterostructures for photocatalytic overall water splitting. Nat. Energy 6, 388–397 (2021).
Zhang, Y. et al. Internal quantum efficiency higher than 100% achieved by combining doping and quantum effects for photocatalytic overall water splitting. Nat. Energy 8, 504–514 (2023).
Wang, X. et al. Photocatalytic overall water splitting promoted by an α–β phase junction on Ga2O3. Angew. Chem. Int. Ed. 51, 13089–13092 (2012).
Gao, Y. et al. Directly probing charge separation at interface of TiO2 phase junction. J. Phys. Chem. Lett. 8, 1419–1423 (2017).
Takata, T. et al. Photocatalytic water splitting with a quantum efficiency of almost unity. Nature 581, 411–414 (2020).
Mao, X. & Chen, P. Inter-facet junction effects on particulate photoelectrodes. Nat. Mater. 21, 331–337 (2022).
Li, R. et al. Spatial separation of photogenerated electrons and holes among {010} and {110} crystal facets of BiVO4. Nat. Commun. 4, 1432 (2013).
Wang, L., Zhang, Y., Chen, L., Xu, H. & Xiong, Y. 2D polymers as emerging materials for photocatalytic overall water splitting. Adv. Mater. 30, 1801955 (2018).
Wang, Z., Li, C. & Domen, K. Recent developments in heterogeneous photocatalysts for solar-driven overall water splitting. Chem. Soc. Rev. 48, 2109–2125 (2019).
Kim, T. & Choi, K. Nanoporous BiVO4 photoanodes with dual-layer oxygen evolution catalysts for solar water splitting. Science 343, 990–994 (2014).
Wang, J. et al. Halide homogenization for low energy loss in 2-eV-bandgap perovskites and increased efficiency in all-perovskite triple-junction solar cells. Nat. Energy 9, 70–80 (2024).
Zhang, B. et al. Facet-oriented assembly of Mo:BiVO4 and Rh:SrTiO3 particles: integration of p-n conjugated photo-electrochemical system in a particle applied to photocatalytic overall water splitting. ACS Catal. 12, 2415–2425 (2022).
Lee, D. et al. The impact of surface composition on the interfacial energetics and photoelectrochemical properties of BiVO4. Nat. Energy 6, 287–294 (2021).
Rao, P. et al. Simultaneously efficient light absorption and charge separation in WO3/BiVO4 core/shell nanowire photoanode for photoelectrochemical water oxidation. Nano Lett. 14, 1099–1105 (2014).
Dutta, A. et al. A tandem (Bi2O3 → Bimet) catalyst for highly efficient ec-CO2 conversion into formate: operando raman spectroscopic evidence for a reaction pathway change. ACS Catal. 11, 4988–5003 (2021).
Zhen, C. et al. Liquid metal-embraced photoactive films for artificial photosynthesis. Nat. Commun. 15, 16728 (2024).
Zhao, Y. et al. Surface etching to tune the behaviours of photogenerated charges on a decahedron BiVO4 crystal for efficient photocatalysis. Environ. Sci.: Adv. 2, 1189–1195 (2023).
Zhang, B. et al. Nitrogen-incorporation activates NiFeOx catalysts for efficiently boosting oxygen evolution activity and stability of BiVO4 photoanodes. Nat. Commun. 12, 6969 (2021).
Wang, S. et al. In situ formation of oxygen vacancies achieving near-complete charge separation in planar BiVO4 photoanodes. Adv. Mater. 32, 2001385 (2020).
Qiu, W. et al. Freeing the polarons to facilitate charge transport in BiVO4 from oxygen vacancies with an oxidative 2D precursor. Angew. Chem. Int. Ed. 58, 19087–19095 (2019).
Pei, H. et al. Gradient-tuned VO4 vacancies in BiVO4 photoanode for boosting bulk hole transport and oxygen evolution reaction performance. Adv. Funct. Mater. 34, 2401122 (2024).
Yin, W., Wei, S., Al-Jassim, M., Turner, M. & Yan, Y. Doping properties of monoclinic BiVO4 studied by first-principles density-functional theory. Phys. Rev. B 83, 155102 (2011).
Talebian-Kiakalaieh, A. et al. In situ characterizations revealing ruthenium-atom-induced raise of photocatalytic performance. Adv. Energy Mater. 13, 2301594 (2023).
Guo, Y., Zhu, B., Tang, C., Zhou, Q. & Zhu, Y. Photogenerated outer electric field induced electrophoresis of organic nanocrystals for effective solid–solid photocatalysis. Nat. Commun. 15, 428 (2024).
Zhao, Y. et al. A hydrogen farm strategy for scalable solar hydrogen production with particulate photocatalysts. Angew. Chem. Int. Ed. 59, 9653–9658 (2020).
Qi, Y. et al. Redox-based visible-light-driven Z-scheme overall water splitting with apparent quantum efficiency exceeding 10%. Joule 2, 2393–2402 (2018).
Maeda, K., Higashi, M., Lu, D., Abe, R. & Domen, K. Efficient nonsacrificial water splitting through two-step photoexcitation by visible light using a modified oxynitride as a hydrogen evolution photocatalyst. J. Am. Chem. Soc. 132, 5858–5868 (2010).
Zhao, J. & Osterloh, F. Photochemical charge separation in nanocrystal photocatalyst films: insights from surface photovoltage spectroscopy. J. Phys. Chem. Lett. 5, 782–786 (2014).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for Ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Gonze, X. First-principles responses of solids to atomic displacements and homogeneous electric fields: implementation of a conjugate-gradient algorithm. Phys. Rev. B 55, 10337 (1997).
Kang, J., Zhang, X. & Wei, S. Advances and challenges in DFT-based energy materials design. Chin. Phys. B 31, 107105 (2022).
Zhang, X., Kang, J. & Wei, S. Defect modeling and control in structurally and compositionally complex materials. Nat. Comput. Sci. 3, 210 (2023).
Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (No.22272082 to S.C., No.21925206 to F.Z., No.22332005 to F.Z., No.12104181 to J.Y., No.11991060 to S.-H.W. and No.12088101 to S.-H.W.), the Fundamental Research Funds for the Central Universities, Nankai University (No. 63213098 to S.C.), and the Foundation from Hebei Provincial Department of Science and Technology (No. 226Z4307G to S.C.).
Author information
Authors and Affiliations
Contributions
S.W. carried out the sample synthesis, characterizations, and photocatalysis measurements. C.L., Y.Q. and N.W. performed the Z-scheme OWS. J.Z., M.L., B.Z. and G.M. helped with the SPV measurements and analysis. X.C., S.-H.W. and J.Y. conducted the DFT calculations. H.Z. discussed the result. S.C. and F.Z. conceived the idea and supervised the project. All authors discussed the results and approved the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Mohamad Mohsen Momeni, Nageswara Rao Peela, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, S., Li, C., Qi, Y. et al. Etched BiVO4 photocatalyst with charge separation efficiency exceeding 90%. Nat Commun 16, 3776 (2025). https://doi.org/10.1038/s41467-025-59076-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59076-8
This article is cited by
-
Bi2WO6-cellulose acetate hybrid films as green photocatalysts for wastewater treatment
Colloid and Polymer Science (2026)
-
Mg powder mediated flux-assisted nitridation synthesis of BaTaO2N for efficient photocatalytic water splitting
Science China Chemistry (2025)
