
Abstract
Recent advances have shown that single-nucleus RNA sequencing (snRNA-seq) can be applied to formalin-fixed, paraffin-embedded (FFPE) tissues, opening avenues for transcriptomic analysis of archived specimens. Yet, isolating intact nuclei remains difficult due to RNA cross-linking. Here, we introduce a cryogenic enzymatic dissociation (CED) strategy for rapid, high-yield and fidelity nuclei extraction from FFPE samples and validate its utility with snRandom-seq (snCED-seq) using male C57/BL6 mice. Compared with conventional approaches, CED delivers a tenfold increase in nuclei yield with significantly reduced hands-on time, while minimizing secondary RNA degradation and preserving intranuclear transcripts. snCED-seq enhances gene detection sensitivity, lowers mitochondrial and ribosomal contamination, and increases overall gene expression quantification. In Alzheimer’s disease studies, it distinguished two astrocyte subpopulations, microglia, and oligodendrocytes, revealing cellular heterogeneity. Additionally, snCED-seq identify major cell types in a single 50 μm FFPE human lung section. Our results demonstrate that snCED-seq is robust for FFPE specimens and poised to enable multi-omics analyses of clinical samples.
Similar content being viewed by others
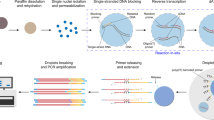

Introduction
High-throughput single-cell/nuclei RNA sequencing (scRNA/snRNA-seq) methods have revolutionized the entire-field of biomedical research1,2,3. scRNA/snRNA-seq has been highly successful at disease mechanisms, discovering biomarkers to help stratify patients, and identifying novel therapeutic targets as well as determining the impact of drugs. However, fresh/frozen specimen procurement is not a standard clinical and diagnostic practice in most institutions, and fresh/frozen samples cannot be obtained for certain sample types. Routine formalin-fixed paraffin-embedded (FFPE) tissues are the most common archivable specimens, constituting a vast and valuable patient material bank for clinical history4. Inevitably, the irreversible modifications caused by formalin fixation on macromolecules in FFPE samples always make it challenging for molecular biology applications. The studies have made great progress in transcription profiling in FFPE samples by optimal RNA extraction methods5,6 or spatial in situ profiling7. What’s more, the combinations of scRNA-seq and spatial technologies have been applied to FFPE tissues. Currently, three methods have been posted, snPATHO-Seq8, snFFPE-seq9, and snRandom-seq10, provided optimized methods to isolate single intact nuclei from FFPE tissues to perform snRNA-seq, which demonstrates the feasibility of snRNA-Seq in FFPE tissues and unlocking possibilities for these challenging samples. Accurate transcriptomic characterization of each cell in clinical FFPE specimens is believed to provide a better understanding of cell heterogeneity and population dynamics, thereby improving accurate diagnosis, treatment, and prognosis of human disease. With the development of snRNA-seq techniques for FFPE samples, there is growing interest in the use of the vast archives of samples for diagnostic purposes.
The application of snRNA-seq in FFPE samples is premised on obtaining superior nuclei. However, isolation of intact and high-quality nuclei remains challenging due to RNA crosslinking, modification, and degradation caused by formaldehyde fixation. The strategies of nuclei preparation for FFPE tissues are longstanding and can date back to the last century, but previous applications are only limited to DNA content11, fluorescence in situ hybridization12,13, genome-wide association studies14,15, and chromatin accessibility profiling16. Specifically, nuclei were dissociated by hyperthermia of biological tissue sections in protease solution, a technique that is sensitive to heating time and easily destroys the nuclear membrane, resulting in the loss of nuclear morphology. Moreover, prolonged exposure to enzyme buffers may increase the permeability of the nuclear membrane, resulting in RNA molecule leakage and adversely affecting snRNA-seq experiments conducted in droplets. The current state-of-the-art snRNA-seq for FFPE samples uses a mechanical homogenization method that is suitable for frozen samples before, and combined with a hyperthermic enzyme dissociation approach for nuclei preparation8,9,10. However, the homogenization of formaldehyde-fixed tissue poses challenges, leading to the presence of debris in the resulting nuclei suspension, which necessitates multiple filtration steps. This, in turn, affects the yield of nuclei and may result in the loss of smaller nuclei. The presence of tissue debris remains a challenge, introducing a higher amount of ribosomal RNA (rRNA), which can affect sequencing data quality. Therefore, the acquisition of high-quality nuclei from PFA-fixed or FFPE samples will be an important basis for transcriptome study of clinical samples.
To address these limitations, here we develop the cryogenic enzymatic dissociation (CED) method—an efficient, high‑fidelity protocol for extracting nuclei from FFPE tissues—and apply it to full-length and total RNA single-nucleus sequencing of post-fixed brain samples. We perform a comprehensive side-by-side comparison of nuclei isolated by CED versus conventional methods, and across fresh-frozen, PFA-fixed, and FFPE tissues, to demonstrate snCED-seq’s robustness in FFPE samples. Despite optimizing traditional protocols, CED-derived nuclei consistently outperformed conventional preparations in RNA integrity, nuclei yield, genes and UMIs per nucleus, and overall gene expression richness. We then apply snCED-seq to over 60,000 single nuclei from the AD hippocampus, providing an in-depth analysis of cellular heterogeneity in AD mice at single cell resolution. Additionally, we profile all major cell types in a human lung micro-tissue. Our findings establish snCED-seq as a reliable platform for transcriptomic analysis of FFPE tissues from neurodegenerative diseases and suggest its broader potential for unlocking valuable archived pathology specimens for clinical applications.
Results
Overview of the cryogenic enzymatic dissociation of nuclei for post-fixed tissues
The acquisition of high-fidelity nuclei is a prerequisite for the research and application of snRNA-seq for FFPE samples, and also a key factor for its full mining of transcriptional information. Since the last century, nuclear preparation of FFPE tissues requires enzymatic dissociation at high temperatures (HED) worldwide and are limited to non-transcriptomic applications. We converted the idea of traditional protocols of preparing nuclei from FFPE tissues, the factors that we deemed pertinent to affect transcriptome analysis, such as dissociation temperature, reagents and time. We established a method of nucleus preparation for post-fixed (paraformaldehyde fixed (PFA-fixed) and FFPE) tissues, CED strategy. For this method, sarcosyl was used instead of sodium N-dodecyl sulfate (SDS) or Triton X-100 as an anionic surfactant to participate in the nuclei preparation, which was more friendly to the nuclear membrane than the cell membrane, and became the preferred component for nucleus isolation in CED method. Moreover, proteinase K (PK) was used to digest proteins of tissue to minimize background contamination. Our CED method eliminates the need for ultracentrifugation through a sucrose cushion, nor any filtration procedure, maximizing product retention, increasing nucleation rates, and preventing nuclear membranes and cytoplasmic contamination. Most importantly, the entire nucleus preparation process was carried out at low temperature, which protects the nuclear membrane and maximally retains the RNA molecules within nucleus, providing high-fidelity nucleus for snRNA-seq research of FFPE samples. In addition, by adjusting the experimental parameters, CED method is not only suitable for tissue slides, but also has good compatibility with FFPE blocks, which is more in line with the application needs of snRNA-seq in disease research. Next, the full-length total RNAs within nuclei from frozen, PFA-fixed and FFPE brains were captured by random primers for snRNA-seq (snCED-seq), and the main workflow of snCED-seq was shown in Fig. 1.

The workflow of snCED-seq for post-fixed tissues includes single nuclei isolation by CED and HED method with the snRandom-seq method used in this study. The steps from nucleus extraction to targeted sequencing are shown. In contrast to HED, the nuclei prepared with CED were morphologically intact without leakage of RNA molecular. Created in BioRender. Tu, D. (2025) https://BioRender.com/uq32z5z.
The nuclei derived from FFPE brains prepared by CED method had intact morphology, good dispersion, high purity without agglomeration (Fig. 2a). Confirmation of the integrity and dispersion of nuclear morphology were also verified using epifluorescence microscopy (Fig. 2b). Representative images of nuclei isolated from the hippocampus of three biological replicates showed much less debris and a size distribution was centered around 6–8 µm (Supplementary Fig. 1a), slightly smaller than normal frozen brain nuclei17, presumably due to the tissue being fixed. Perhaps, CED method without cumbersome filtering procedures, tiny nuclei could be preserved. Statistics showed that at least a million levels of nuclei were obtained from each pair of hippocampi (Supplementary Fig. 1a, bottom). The recent snRNA-seq techniques for FFPE tissues based on random primer capture10 or gene probe capture8 require the input of nearly one million nuclei to ensure the output of about 10,000 nuclei. Our CED method effectively circumvents the shortcomings of the current two mainstream nuclear preparation strategies, and can export nuclei stably without introducing more impurities and destroying the nuclear membrane.

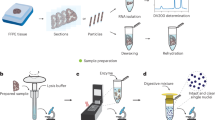
a, b Image of trypan blue-stained (a) and DAPI-stained (b) nuclei isolated from FFPE mo°use brain by CED before cell encapsulation, respectively. Scale bar, 50 μm. c Nuclei yield at different proteinase K concentrations. d, e Bar plots showing the relationship between the nuclei numbers of the PFA-fixed hippocampus with the dissociation time was isolated by HED (c) and CED (d) methods. f Nuclear yield per gram of hippocampal tissue, using CED and mechanical homogenization, respectively. p < 0.0001, two-sided Welch’s t-test. g, h RNA integrity number (RIN) (g) and total RNA yield (h) of RNA extracted from nuclei. i Representative peak values of amplified cDNA in different groups. n = 3 biological duplication, and bars show mean ± standard deviation (SD) (c–i). n = 3 times the experiment was repeated with similar results (a, b). Source data are provided as a Source Data file (c–i).
We optimized the proteinase K (PK) concentration in the nuclear dissociation system as measured by morphology and count of nuclei, and found that the optimal concentration for HED was 0.4 mg/mL, while a higher was required in the CED for mouse brain, which was due to the reduced enzyme activity at low temperature (Fig. 2c). The nuclei count gradually decreased with the extension of dissociation time at 37 °C (Fig. 2d), but was not observed in our method (Fig. 2e) and with the intact morphology throughout. Since the release and disappearance of nuclei occur simultaneously during enzyme dissociation, the traditional HED method will force the preferentially obtained nuclei to digest in enzyme solution, or damage the nuclear membrane, affecting nuclear yield, and this method was very sensitive to reaction time, increasing the burden on the experimenter. In addition, since molecular cross-linking makes it difficult to adequately homogenize the tissue, a lot of impurities in the dissociative system that cannot be removed, which must be removed by additional density gradient methods combined with filtration, thereby reducing the nuclear yield. The result reflected CED method could obtain more than 100,000 nuclei per gram of hippocampal tissue, which was more than 10 times that of commercially nuclear extraction kits based on mechanical methods (Fig. 2f and Supplementary Fig. 2j). Finaly, given the clinical demand for snRNA-seq in a variety of organs, we also dissociated the nuclei of multiple organs (Soybean size) by CED method, including heart, liver, spleen, lung, stomach, intestines, kidney and pancreas (Supplementary Fig. 2). We observed strong applicability of this approach to multiple organs, except to the heart and lung. Especially in spleen, intestines and kidney, with the tens of millions of nuclei numbers, and despite their abundance and dense arrangement, they remained independent, intact, and unaggregated (Supplementary Fig. 2c, f and g). Despite the lower fitness of CED method in the heart and lung, it still outperformed the mechanical homogenization (Supplementary Fig. 2h). Subsequently, the nuclei were dissociated from 50 μm paraffin scroll of human organs (organoid brain, lung, liver, kidney and pancreas) (Supplementary Fig. 3). Due to the small size of the brain organoids, we fixed them with PFA followed by OCT embedding and ice cutting (Supplementary Fig. 3c). Although the diameter of the brain organoid section was only about 2 mm, more than 30,000 nuclei could be dissociated from two sections (Supplementary Fig. 3b, c), which was enough to prove the superiority of CED method in the nuclei yield. Moreover, conventional wisdom suggests that nuclei are not freeze-thaw friendly, forced to improve the experimenter’s awareness of time control. Notably, nuclear envelope rupture and aggregation did not occur in nuclei isolated by CED even after one month of dry ice or storage at −80 °C. This property breaks the restriction that the nucleus cannot be cryopreserved.
Without damage of RNA molecules in the nucleus from CED method
The morphology of the nucleus ensures the independence of single nucleus data, while the quality of RNA molecules in the nucleus can ensure the high-quality output of snRNA-seq, which is also one of most important factors affecting its application in transcriptome research. PFA fixation of cells induces cross-linking between nucleic acids and proteins, whereas the preparation of FFPE samples requires hours of high temperature wax immersion, both result in RNA damage. How to avoid secondary damage to RNAs during nuclear preparation is crucial for snRNA-seq. We extracted RNA from nuclei to verify the harmlessness of CED method on RNA molecules. We first investigated how to extract RNA molecules from the nuclei of fixed brains. The conventional commercial RNA extraction kits are obviously not suitable for the nuclei of PFA-fixed and FFPE tissues. We combined two lysis systems suitable for fresh or frozen tissue to extract RNA from cross-linked nuclei. The effects of different heating conditions and proteinase K concentrations on RNA integrity (RIN) and RNA yield were tested using Drop-seq buffer and commercial RNA extraction kits. We found that PFA cross-linking was effectively reversed by incubation at 56 °C for 15 min in standard Drop-seq lysis buffer (Supplementary Fig. 1b), significantly shortening the heating time compared to the reported18,19,20. PK has been reported to increase RNA yield18,19, but our results showed that the PK concentration has little effect on RIN and RNA yield (Supplementary Fig. 1c). In addition, we performed the same experimental exploration on lysis systems of other high-throughput sequencing platforms, although comparable amount of RNA could also be obtained, RIN values were low (2–4). This means that the standard Drop-seq lysis buffer can be used directly as lysate for FFPE nuclei at 56 °C.
The results showed that the CED method had almost no damage to the RNA molecules compared with the HED method. The RIN values of nuclei were basically consistent with the RIN values of PFA-fixed tissues [PFA(4 C) vs PFA-fixed section] (Supplementary Fig. 1d), but far higher than those of the nuclei prepared by HED method [PFA(37 C)], and even the RIN values of FFPE [FFPE(4 C)] nuclei were higher than that of PFA(37 C) (Fig. 2f). Then, the cDNA libraries were generated from multiple tissues to truly reflect the quality of polyA_RNA. The major peak size of cDNA for both PFA(4 C) and FF(0 h) was above 1200 bp, while around 800 bp for PFA(37 C), which was even lower than that of FFPE(4 C) (Fig. 2h), which again confirmed that CED method was less damaging to RNA molecules in the nucleus than the HED method. Notably, the RNA yield of nuclei isolated by CED method was consistent with that of freshly frozen (FF) samples, but much higher than that of traditional methods (Fig. 2g), which might be a key reason for limiting the application of snRNA-seq for post-fixed samples. We found that free RNA penetrates into the enzyme solution during high-temperature dissociation process, resulting in the reduction of the amount of RNA in the nucleus (Supplementary Fig. 1e), which we concluded to be a fatal shortcoming of the conventional method. It has been reported that 3× or 5× SSC proved to be a good medium for the prevention of cellular RNA degradation, but we found that that the two types of buffers had almost the same effect on RNA molecules (Supplementary Fig. 1f). In conclusion, CED method can minimize the damage to the nuclear morphology and RNA molecules of post-fixed brains.
Validation the nuclei quality derived from CED method by snRNA-seq
We employed droplet-based snRNA-seq technology to capture total RNA (M20 Genomics) and poly(A) RNA (10× Genomics). snRNA-seq was performed on mouse hippocampus samples with three treatment conditions: (1) fresh frozen tissue-the nuclei were extracted by mechanical homogenization, capture by poly(T) [FF (10×)] and random primers [FF(M20)]; (2) PFA-fixed tissue—the nuclei were prepared by HED [PFA(37 C)] and CED method [and PFA(4 C)] for snRandom-seq; (3) The nuclei of FFPE tissue were dissociated by CED method and capture by random primers [FFPE(4 C)/FFPE]. With snRNA-seq data of frozen tissue for reference, the influence of high and low temperature dissociation on sequencing data was evaluated to verify the fidelity of nuclei obtained by the CED method, and also to evaluate the applicability of CDE method in FFPE samples. Before microfluidic encapsulation, the nuclei were imaged to confirm single nucleus morphology and counted, and the results returned that the nuclei numbers of all samples were about million level. snRandom-seq requires a substantial amount of input material, millions of nuclei fully meet the requirement of nuclear detection rate of 10,000. After barcoding and amplification, the fragment size of the cDNA from FF(M20) main peaked about at 700 bp (Supplementary Fig. 4a). While the main peak of cDNA from PFA(4 C) between 300 and 1000 bps, and longer than PFA(37 C), which potentially due to RNA degradation (Supplementary Fig. 4a). In addition, the next-generation sequencing (NGS) library with equal input of cDNA showed the lowest library for PFA(37 C) (Supplementary Fig. 4b). The amount of cDNA and NGS library from PFA(4 C) was slightly higher than that in FFPE(4 C) and FF(M20), but much higher than PFA(37 C), indicating that CED method effectively blocked the leakage of nuclear RNA and almost maintained the true level RNA molecules within the nucleus (Supplementary Fig. 4b). In fact, we have optimized the HED method to work well for snRNA-seq, which has improved its suitability for transcriptomics, but the results are still unsatisfactory. In short, the nuclei prepared by the CED method from fixed or paraffin-embedded samples are more suitable for the research of snRNA-seq.
Performance of nuclei from FFPE tissues in snRNA-seq
We identified 150,507 high-quality unique nucleus barcodes using the barcode-gene rank plot, with clear separation of nuclei from background noise, and an average of more than 10,000 nuclei were identified in every sample (Fig. 3a). Gene and UMI count distribution showed that the total UMIs and genes in FF(M20) and PFA(4 C) were significantly (p < 0.05) higher than in PFA(37 C) (Supplementary Fig. 5a, b), which confirmed that CED method could maximize the retention of RNA molecules. But the total number of genes detected in all samples were comparable, all above 20,000 (Supplementary Fig. 5c). In addition, snRNA-seq captured a mean of 1835, 2725, 1347, 2013, 1847 genes and 4189, 11100, 4996, 8759, 7072 UMIs in single nucleus by sequencing average ~27 k, 19 k, 13 k, 17 k, 12 k reads per nucleus for FF(10×), FF(M20), PFA(37 C), PFA(4 C) and FFPE(4 C) samples, respectively (Fig. 3b, c). The number of genes and UMIs in FF(M20) was higher than FF(10×), which benefit from the principle of capturing full-length and transcripts by random primers. Moreover, the gene and UMI counts per nucleus in PFA(4 C) were slightly lower than FF(M20), but about 1.5 to 1.75 times higher than PFA(37 C), and even about 2–2.5 times in individual samples, and their numbers in FFPE(4 C) were also higher than that in FF(10×) and PFA(37 C) (Fig. 3b, c). The saturation analysis showed that FF(M20) and PFA(4 C) had the highest sensitivity, followed by FFPE(4 C), with 4000 to 5000 detected genes per nucleus, respectively, at a sequencing depth of 30,000 trimmed reads per nucleus, and both exhibited a higher gene detection rate than PFA(37 C) (Fig. 3g). We next compared our data with other reported results (Fig. 3d and Supplementary Fig. 5d). The genes detected per nucleus in snCED-seq datasets of brains comparable with other parenchymatous organs, despite inherently lower RNA abundance in brains (Supplementary Fig. 5d). The number of genes detected in snRNA-seq reached saturation between 100 k and 150 k uniquely aligned reads per nucleus (Supplementary Fig. 5d). Beyond that, lower rates of mitochondrial and ribosomal genes in PFA(4 C) and FFPE samples than others, almost 0, indicating that the CED nuclei were pure without cytosolic contamination (Fig. 3e, f). Unlike FF(10×), samples captured by random primers exhibited homogeneous coverage across the body of protein-coding, but with a slight bias toward the 3′-end due to the extra addition of oligo(dT) primer in reverse transcription (Supplementary Fig. 5e). However, PFA(37 C) had more 3′ bias, possibly related to the greater fragmentation of RNA within nuclei (Supplementary Fig. 5e).

a–c Number of nuclei (a), UMIs per nucleus (b) and genes (c) per nucleus detected in fresh frozen, PFA-fixed and FFPE samples. d Gene detection per nucleus comparison of our data (> 10,000 nuclei) with mouse tissues (5795 (kidney), 4287 (liver), 6732 (heart) and 3774 (testis) nuelci) by snRandom-seq10, mouse brain (7031) by snFFPE-seq9 and breast (5721) by snPATHO-seq8. Data in the box plot correspond to the first (lower hinges) quartiles, third quartiles (upper hinges), and median (center). The upper whisker extends from the hinge to the maxima no further than 1.5 * IQR from the hinge. The lower whisker extends from the hinge to the minima at most 1.5 * IQR of the hinge. e, f Percentage of mitochondrial (e) and ribosomal (f) genes. g Saturation analysis of snCED-seq based on the different samples. h Percentage of reads mapped to different genomic regions under different conditions. i Counts of different RNA biotypes detected in FFPE brains. j The Pearson’s correlation coefficient (R) of the normalized gene expressions between technical replication samples and post-fixed/fresh samples (All p values = 0). n = 3 biological duplication, and bars show mean ± SD (a–c, e, f and i). a–c, ns p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001 (Tukey’s multiple comparisons test), and the specific p values are in the source data. Source data are provided as a Source Data file (a–j).
In our snRNA sequencing experiment of PFA-fixed and FFPE brains, less than 10% uniquely aligned reads were mapped to exons and intergenic regions, and with more reads mapped to introns (Fig. 3h). In contrast, frozen samples had a higher proportion of exons (FF(M20): ~13% and FF(10×): ~21%) (Fig. 3h). We suspected that the nuclei of frozen samples were prepared by the homogenization method, which was more susceptible to the cytoplasm of pollution. By comparison, higher coverage of intronic regions in the post-fixed groups, especially in PFA(4 C) and FFPE(4 C) (Fig. 3h), suggesting that our nuclei had little cytosolic contamination, with higher fidelity. The higher proportion of introns might lead to more accurate RNA velocity measurements across differentiation trajectories21. A broad spectrum of RNA biotypes was detected, and protein-coding genes were the most highly detected biotype across all groups, but also other biotypes (Supplementary Fig. 6a). Unexpectedly, a substantial amount of full transcripts was detected in all group samples, especially in FF(10×) (Supplementary Fig. 6a), used 10× Chromium Single Cell 3′ Solution, which is consistent with our previous bulk RNA-seq analysis22. Contrary to our previous knowledge, we speculated that perhaps there is a wider range of A-capped non-coding RNA molecules within the nucleus. However, at least our data show that extensive and ought to exist non-coding genes can be detected in the FFPE nuclei prepared by CED method (Fig. 3i).
Next, gene expression correlation analysis was performed on our data. To prove the repeatability of our method, duplicate samples were sequenced independently, and a high correlation (Pearson R: 0.99, p < 2.2e-16) of gene expression profiles across random batches was seen in PFA(37 C), PFA(4 C) and FFPE(4 C) groups (Fig. 3j), indicating the robustness of nuclei from fixed/FFPE samples. We then analyzed the correlation of gene expression between fixed/FFPE and frozen samples. Consistently, the total RNA profiles of fixed/FFPE and FF(M20) samples displayed a good correlation (PearsonR: > 0.9, p < 2.2e-16), more genes were underexpressed in PFA(37 C) group, but not observed in PFA(4 C) and FFPE(4 C) samples (Fig. 3j). A poor correlation between fixed/FFPE and FF(10×) (PearsonR: ~0.7, p < 2.2e-16) (Supplementary Fig. 5f), and higher gene expression in fixed samples, which likely stem from differences in technique. In addition, the correlation between FFPE(4 C) and PFA(4 C) was as high as 0.99, reaching the within-group level. Compared with PFA(37 C), it was only 0.95, and the gene expression was higher in FFPE(4 C) samples (Supplementary Fig. 5f). These results suggest that nuclei from the CED method behave more similarly to frozen samples.
Cell heterogeneity analysis in PFA-fixed and FFPE tissues
We next compared the cell types identified in all group samples at single-cell resolution. Unsupervised clustering of the above filtered high-quality single brain nucleus profiles, by merging the data from PFA-fixed, FFPE samples and frozen samples. By merging the data from all batch of samples, we obtained a robust cell clustering by Uniform Manifold Approximation and Projection (UMAP), and the low similarity cellular landscapes between FF and fixed/FFPE samples before batch (Supplementary Fig. 7a). Batch-based processing resulted in integrated UAMP profiles revealed over 21 distinct clusters (Fig. 4a and Supplementary Fig. 7b). All clusters could be further annotated based on classical known cell-type markers (Fig. 4b), and 11 major cell types were identified with cell-specific genes reliably mapped on the corresponding clusters (Fig. 4b and Supplementary Fig. 7c). Most of the recommended terms in mouse hippocampus samples were identified, including excitatory neuron (Ex1–8), inhibitory neuron (Inh1-4), Interneuron (Inter_N), astrocytes (AST), Oligodendrocytes (Oligo), Oligodendrocyte progenitor cells (OPC), Microglial (Micro) and Cajal Retzius cells (CRC) (Fig. 4a). Besides the known cell types, we also annotated choroid plexus cells (CPC) markered by Prlr, which are rarely detected in reported data (Fig. 4a), and we suspect that our nuclei were more abundant and contained more cell types. The choroid plexus is a relatively small structure in the brain ventricles, due to their low abundance, CPC will be masked by other cell types such as neurons, glial cells in snRNA-seq. Understanding the role of CPC can provide valuable insights into both normal brain physiology and pathological conditions. The identification of CPC is a breakthrough in understanding rare cell types in the brain, not only contributes to a better understanding of the molecular landscape of brain tissue but also paves the way for future investigations into the role of CPC in brain function and disease.

a Cell map of mouse hippocampus. UMAP of 150,507 single-nucleus RNA profiles from hippocampi of fresh frozen, PFA-fixed and FFPE samples. colored by cluster. b Dot plot of the average expressions of top two markers in each of the 21 clusters. The color bars indicate the gene expression level, the bluer the color, the higher the expression level. The bubble diameter indicates the proportion of expression of the gene in that cell cluster, and the larger the diameter, the stronger the specific expression. c Number of nuclei (Left) and proportion (Right) of annotated cell types of all samples by snCED-seq. d The number of differentially expressed genes (DEGs) between each comparison group; Red and blue indicate upregulation and downregulation, respectively. Source data are provided as a Source Data file (a–d).
Subsequently, we analyzed the proportion of cell types across all groups. As expected, the proportion of cells differed between 3’ and random primer capture techniques, mainly in AST, Oligo, and Endo cells (Fig. 4c and Supplementary Fig. 7d). However, a similar population of cells between frozen and post-fixed samples in our datasets was seen (Fig. 4c). We surmise that we shortened the time of enzymatic dissociation of the samples at high temperatures, thereby retaining most of the cell types in PFA(37 C) group. However, the experimentalists are required to be experienced, otherwise resulting in poor batch. Despite this, CPC cells were severely lost in PFA(37 C) samples, but a considerable number of cells were detected in both PFA(4 C) and FFPE samples (Supplementary Fig. 7d), indicating that CED detected more scarce cells than conventional methods. In addition, higher number of cell clusters was obtained at a resolution of 0.1 than other reported results23,24,25. Therefore, we counted the cluster numbers under different resolutions and found that absolute advantage in PFA(4 C) samples, and even a higher cluster number of FFPE samples than PFA(37 C) group (Supplementary Fig. 7e). But the number of clusters reached a comparable level in all samples when resolution at 1.0 (Supplementary Fig. 7e). We confirm our previous inference that the nucleus prepared by CED method may bring more cellular heterogeneity information and has the potential to recognize more cell types.
Systematic comparison of the CED with mechanical homogenization methods
To verify whether the CED technique maintains its established nuclear yield and genetic detection advantages, we conducted a direct comparison with nuclei prepared by mechanical homogenization dissociation (MHD) methods of two previously reported and a commercial kit by using snRandom-seq, and named FFPE(CED), FFPE(MHD1)10, FFPE(MHD2)26, and FFPE(MHD3), respectively. Among them, MHD3 could only obtain tens of thousands of nuclei in the hippocampus, which could not be used for subsequent snRNA-seq, while MHD1 and MDH2 with unclean background compared with CED nuclei, and there were still larger or smaller fragmented tissues (Supplementary Fig. 8). The cDNA and library structures of FFPE(MHD) were more complex and the main peak fragment distribution was smaller than that of FFPE(CED), which might be related to the nuclear purity (Supplementary Figs. 3 and 8a, b). In contrast, fewer nuclei and significantly (p < 0.05) reduced mapping rates were observed in FFPE(MHD) (Supplementary Fig. 9a, c). Despite the higher sequencing depth with significantly increased (p < 0.05) sequencing saturation in FFPE(MHD) (Supplementary Fig. 9b, c), the number of UMIs and genes per nuclei were significantly (p < 0.05) lower than that in FFPE(CED), only about one-third (Supplementary Fig. 9d, e). In addition, the proportion of ribosomal genes and mitochondrial genes increased significantly (p < 0.05) in FFPE (MHD), which shown again hat the purity of nuclei was insufficient (Supplementary Fig. 9f, g). Saturation analysis showed that the number of genes detected in FFPE(CED) was significantly better than that in FFPE(MHD) samples at any sequencing depth, and FFPE(MHD) were reached saturation point by 5–10 K uniquely aligned reads per nucleus, but FFPE(CED) had not yet reached saturation point at 20 k (Supplementary Fig. 9j). Although theoretically, mechanical homogenization tissues at low temperature had little effect on the integrity and yield of RNA (such as FF(M20)), the current application of this strategy in FFPE samples requires the help of high-temperature enzyme dissociation for about 1 h, which will lead to RNA molecule leakage, and might be the important reason for the reduction of gene number. Compared with the enzymatic dissociation method, the genes within nuclei from MHD were distributed with higher proportion of exons and lower introns, both in frozen and formalin-fixed tissues (Supplementary Fig. 9h). We speculate that it might be related to purity, due to more immature RNA within nuclei than in the cytosol, and theoretically more introns. Moreover, the exons proportion in the nuclei of FFPE samples based on MHD dissociation was higher than that of frozen tissues (Supplementary Fig. 9h), which also confirmed that the post-fixed samples were difficult to homogenize thoroughly, resulting in more debris. Considerable amount of total RNA in all samples by using snRNA-seq technology based on random primer (Supplementary Fig. 9i). Next, we performed a correlation analysis of gene expression between FFPE and frozen nuclei. Naturally formed two modules with FFPE(CED)_FF(MM20) and FFPE(MHD1) _FFPE(MHD2) as matrices. Lower repetition within group of FFPE(MHD1), and weaker correlated with FFPE(CED) and FF(M20) gene expression than FFPE(MHD2) (Supplementary Fig. 10e). In addition, a higher degree of processing-related artefacts was observed in the nuclei from other samples compared to FFPE(CED), including the stress response pathway, which could introduce bias in the interpretation of these pathways (Supplementary Fig. 10f).
Although the nuclei numbers were similar before quality control (Supplementary Fig. 9a), the cell sacrifice rates reached a staggering 37.2% and 72.7% in FFPE(MHD1) and FFPE(MHD2) after screening with the same criteria in all samples, respectively (Supplementary Fig. 10a, b). FFPE(CED) with only 9.9% cell screening rate proved that the quality of nuclei prepared by CED method was higher and more compatible with snRNA-seq technology. Consistently, we found that an average of 34.5% and 63.9% of reads were distributed in rRNA in the two FFPE(MHD) samples, compared to almost zero in our data (Supplementary Fig. 10c). Many reads were distributed in rRNA, which might be the reason for the lower UMI and gene numbers, and also the factor that more cells were filtered. Subsequently, cluster analysis was performed after removing batch effect to construct a merged UMAP map with 22 cell clusters (Supplementary Fig. 10d). While both samples identified a consistent number of cell types, their proportions differed significantly (Supplementary Fig. 11a–c). Both in frozen and FFPE samples, the nuclei obtained by MHD technologies contained more neuronal cells, especially in FFPE(MHD2), which captured 95.9% of neuronal cells (Supplementary Fig. 11c, d). Whereas the nuclei prepared by CED identified more non-neurons, such as Ast, CPC, CRC, Micro, and OPC, which might be related to the nuclei preparation strategies, and the homogenized nuclear suspension requires multiple decontamination processes, resulting in the loss of scarce cells or cells of small size, leading to cell proportion artifacts. In addition, the cluster numbers under different resolutions were counted, and FFPE(MHD2) showed the worst cell heterogeneity, while FFPE(CED) was comparable to FF(M20), and even at low resolution, it was more advantageous for cell heterogeneity analysis (Supplementary Fig. 11e).
Consistently, more genes under-expressed in FFPE samples compared to FF (M20) (Fig. 4d and Supplementary Fig. 11f). However, gene expression perturbations varied widely between nuclei prepared by different strategies. Compared with FF(M20), 547 downregulated DEGs in FFPE(CED) (Fig. 4d), while 4960 and 4447 genes were significantly (p < 0.05) under-expressed in FFPE(MHD1) and FFPE(MHD2), respectively. Moreover, 82% and 96% of DEGs were upregulated in FFPE(CED) compared to FFPE(MHD) (Supplementary Fig. 11f). The differences in gene expression between nuclei prepared by the two MHD methods were also remarkable, with 4314 genes downregulated in FFPE(MHD1) (Supplementary Fig. 11f). In summary, the effect of nuclear preparation strategy on gene expression should not be ignored, which is crucial for accurate disease mechanism study. In addition to the high yield and no RNA damage within nuclei, CED method has higher gene expression similarity and richer gene expression between dissociated nuclei and frozen samples, which is more suitable for the application of clinical samples.
snCED-seq revealed cell diversity and heterogeneity in FFPE hippocampal from AD mice
To validate the promise of our nuclei for the research of brain diseases, we applied snCED-seq on the FFPE hippocampus of AD and matched wild type (WT) mice (Fig. 5a) to explore the specific-cell state changes of AD samples. After nuclei with over or under expression of genes were filtered out, snCED-seq identified 62,000 true nuclei in the FFPE brains, and with approximately zero mitochondrial and ribosomal genes in all samples (Supplementary Fig. 12a). Unsupervised clustering of the single nuclei revealed 19 distinct clusters at a resolution of 0.1 (Supplementary Fig. 12b). The main cell types of AD and WT hippocampus could be identified based on the known cell-type markers, including Ex1-6 (Hs6st3, Pdzrn3), Inh1-4 (Gad1, Gad2), AST (Slc1a2), Oligo (Mbp, Mobp), Micro (Dock2), OPC (Vcan), Endo (Flt1, Mecom), Smooth muscle cell (SMC, Ebf1). Olfactory ensheathing glia (OEG, Bnc2) and CRC (Cdh4, Reln) with minimal number of cells were also identified (Supplementary Fig. 12c).Abundant cell types could be identified in our data, but differences in cell proportions compared with previous data from frozen hippocampal of 7-month-old mice [2], such as an increased proportion of most neuronal cells in the AD model, but AD-related cells (Ast, Micro, Oligo, OPC) and vascular-related cells (Endo, SMC) were absent (Supplementary Fig. 12f). The AST proportion gradually increased with the increase of age of AD25, and a decrease in the proportion of AST of AD mice has also been reported23. We speculate that it might be due to inconsistent methods of nuclear preparation or age of AD mice.

a Overview of the experimental strategy. Created in BioRender. Tu, D. (2025) https://BioRender.com/hrx13a. b Cell map of mouse hippocampus in WT and AD by supervised clustering with ref. 25. UMAP of 62,000 single-nucleus RNA profiles from hippocampi of Then, Nuclei were collected and resuspended 5-month-old male mice, three WT and three 5 × FAD (AD); colored by cluster. c Heat map showing expression of specific markers in all cell types, identifying each cluster in B. Expression level (color scale) of marker genes across clusters and the percentage of cells expressing them (dot size). d The frequency of each cluster in every sample. e The percentage of cell types in AD and WT. AD1 sample was screened. Data in the box plot correspond to the first (lower hinges) quartiles, third quartiles (upper hinges), and median (center). The upper whisker extends from the hinge to the maxima no further than 1.5 * IQR from the hinge. The lower whisker extends from the hinge to the minima at most 1.5 * IQR of the hinge. f DEG counts for each cell type The intensity of the blue colour was proportional to entry values. g The odds ratios of DEGs and AD-disease genes in every cluster. The dot size expresses cells association with the AD disease. Empty dot indicated statistical significance and crossed dot indicated non-significance (fisher’s exact test). and the specific p values are in the source data. n = 3 biological duplication, and bars show mean ± SD (e). Source data are provided as a Source Data file (b–g).
To identify cell types more accurately and precisely, a reference of the published snRNA-seq data of frozen AD hippocampal25 was used for supervised clustering using Approximate Nearest Neighbors Oh Yeah (Annoy) (Fig. 5b and Supplementary Fig. 13). 90.7% of the FFPE data and the reference data were predicted to be high predicted scores, and only a minority of the cells had a score less than 0.8 (Supplementary Fig. 12d, e). However, we could also infer cell types from the distribution of low-predicted cells in the UMAP map, such as Ast and ExN cells (Supplementary Fig. 22a). The gene and UMI numbers in all cell types from the high predicted score were higher, and the other quality control data was also better (Supplementary Fig. 22b). In addition, the ExN.IEGs cells from the reference data were successfully detected in our data, but in a small proportion, and overlapped with ExN.CA1.1 cells, so we included them in the ExN.CA1.1 (Supplementary Fig. 22c). Finally, we determined the atlas of supervised clustering, and identified 22 clusters covering 11 cell types, including 9 Ex cells (ExN, ExN.CA1.1–1.3, ExN.CA3.1–3.3, ExN.DG, and ExN.sub), 4 Inh cells (GABAergic.1-4, GABA1-4), and 9 non-neuronal cells (Fig. 5b, c). We observed a disproportion of cells in the AD1 that did not conform to conventional wisdom (Fig. 5d), but the proportion of diseased cells before and after removal of the AD1 sample was barely affected (Fig. 5e and Supplementary Fig. 12g). Overall, the cell types in the reference data were all detectable in our FFPE nuclei, with excitatory cells accounted for the largest proportion (52%), followed by Ast (21%), Oligo (11%), GABA (6%), OPC (4%), and Micro (3%), and the other cells accounted for about 1% respectively (Supplementary Fig. 22d).
The proportions of cells obtained by both clustering methods were similar (Supplementary Fig. 7f, g). We observed that most cells coincided with unsupervised clustering by marker gene comparison, with ExN.DG corresponding to Ex.neuron6 and GABA4 corresponding to Inh.neuron2 (Fig. 5c and Supplementary Fig. 12c). However, the annotation of some cells changed. For example, we merged Ast1-2 in the reference data into AST, and labeled SMCs and OEGs in unsupervised clustering as pericytes_Per and Fibroblasts_Fib, respectively (Fig. 5c and Supplementary Fig. 14c). OECs are a glial cell between Schwann cells and oligo, which have the functions of neurotrophic, inhibition of gliosis, scar formation and sheath formation, and can provide a suitable microenvironment for axon growth and strong migration characteristics. It has been reported that OEC transplantation reduced amyloid burden in amyloid precursor protein transgenic mouse model27. OECs injected into the hippocampus of AD mice can improve the learning and memory ability and increase the activity of mitochondrial cytochrome oxidase in the hippocampal CA1 region, which has an obvious therapeutic effect on AD. This is consistent with our results that OECs in AD undergo loss (Supplementary Fig. 12f). Notably, CR cells are only present in our data other than reference data (Supplementary Fig. 12d). The number of CR cells decreases with brain development, and a handful of CR cells can still be detected in the hippocampus of old mice28. Since the dominant advantage of the CED strategy to maximize the retention of nuclei of FFPE tissues, relatively few CR cells distributed in the hippocampus were efficiently enriched, and detected by snRNA-seq. We then observed that the proportion of all glial and vascular-associated and other nonneuronal cells were reduced in AD, compared with WT (Fig. 5e). Although the proliferation of Ast and Micro cells is deemed to be the cellular changes of AD disease, but the frozen hippocampal snRNA-seq data reported by Regev, except for Micro and Endo cells, the remaining proportion changes of non-neural cells are consistent with ours25. Also, in the cortical data, Ast was in a status of missing, Micro was the only cell type that increased in AD, and the rest were in a stable state. But in the hippocampus, Ast, OPC and vascular cells were reduced in AD, the proportion of Micro increased, and Oligo remained almost unchanged23. The similar results further demonstrate the reliability of our data.
In brief, more abundant cell types can be detected in our data, and provide the superior nuclei for omics research in brain diseases. We next used these cells to characterize the heterogeneity of AD-disease cells and the perturbing nature of perturbation of gene expression.
Multidimensional identification of AD disease-specific cells
To reveal AD-associated cells, we compared levels of gene expression in nuclei isolated from AD versus WT individuals by cell type, and identified 4225 unique differentially expressed genes (DEGs) that implicated all major cell types, and 95.6% of DEGs were overexpressed genes (Fig. 5f). Except for Ex, the remaining cells showed a strong signature (log2FC > 0.25, p < 0.01) of activation, especially in GABA, Micro and Oligo cells (Fig. 5f), where the activation of GABA.2 sub-cells was dominant (Supplementary Fig. 15a). More DEGs were upregulated in Ex neurons than underexpressed genes, possibly originating from the neural activation of Ex.CA3.1 and Ex.CA3.2 sub-cells. We then analyzed the DEGs with multiple methods, and the results were consistent, especially in neurons and glial cells (Supplementary Fig. 15b). Moreover, both up- and downregulated DEGs were highly cell type specific, 62% of DEGs in neurons, whereas DEGs in non-neuronal populations were substantially smaller, probably owing to reduced power in lower-abundance cell types29. Furthermore, vascular cells (Endo and Epend) also showed no less differential changes than glial cells (Fig. 5f, Supplementary Fig. 15a, and Supplementary Data 1). These contrasting observations on the number and dominant directionality of DEGs reveal a heterogeneous response to AD across cell types-a recurrent theme that will be observed throughout the study.
The vast majority of DEGs (50%) were perturbed only in a single cell type, which indicates that these perturbations are strongly cell-type specific (Supplementary Fig. 15c). But a small number of genes was highly expressed in 82 % of cell types, such as Magi2, Cadm2, Grm7, Adgrl3, Ctnna2, Ctnnd2, Camta1, Dgki, Drc1, Lsamp, Mbd5, Nrg3, Ppfia2, and Prkce (Supplementary Data 2). Rn18s-rs5 and Malat1 genes were under-expressed in most cells. Among them, Magi2 has been reported to be associated with AD phenotypes30 and is considered a potential candidate locus in the etiology of divergent AD, which was involved in the regulation of protein degradation, apoptosis, neuron loss, and neurodevelopment31. We speculate that these genes preferentially undergo perturb changes in expression in AD pathology, which may be therapeutic targets associated with AD disease. Overall, these results of our snRNA-seq for FFPE brains indicate that all major cell types are affected at the transcriptional level by AD pathology. Finally, we evaluated whether AD-associated variants are enriched in genomic regions with genes whose expression pattern is cell-type-specific. Fisher’s test enrichment scores of each cell type-specific DEGs and AD risk genes were calculated, and AD risk variants were found to be associated with genes from Micro, OPC, Ast, and Oligo cells, and they were also significantly (p < 0.05) enriched in GABA1 and GABA2 (Fig. 5g).
The multi-dimensional analyses results showed that nuclei prepared by CED method could identify disease-specific cell types comparable to those in frozen samples by snRNA-seq, and vascular cells, which have been less studied in AD, also surfaced. Next, the traditional AD-associated cells (Micor, Ast, Oligo) were used for further analysis firstly.
Microglial heterogeneity analysis associated with AD-related traits
Using the single-cell resolution feature, we sub-clustered Micro cells of AD and WT mice, and identified four subpopulations (Micro0-3) (Supplementary Fig. 16a). The micro2 was representative cells only in 5xFAD mouse, but micro0 and micro1 were mainly distributed in WT (Supplementary Fig. 16b). A scanty of 15 micro3 cells were distributed independently in the UMAP atlas, and with the 1 to 2 ratio in AD and WT (Supplementary Fig. 16b), indicating that Micro cells are significantly affected by AD pathology and the disease-induced differences result in two nonoverlapping cellular states. We observed that the top 10 upregulated DEGs in AD were highly expressed only in Micro2 and Micro3, implying specific disease changes for these cells (Supplementary Fig. 16c). Micro’s DEGs overlapped with the AD disease gene set, and 11 genes were identified (Supplementary Fig. 16d). These genes were mainly expressed in Micro2 and Micro3, and individual genes were upregulated in Micro1 (Supplementary Fig. 16e). Thus, we determined Micro2 is a diseases-associated microglial (DAM). Although Micro3 cells accounted for less than 1% of the total, disease genes were specifically highly expressed in them, such as the β-amyloid precursor protein related gene (App) (Supplementary Fig. 16e). We found that the expression of these genes was higher in Micro3 of AD mice (Supplementary Fig. 16f), implying that five Micro3 cells in AD were also DAM cells, indicating that our nuclei are highly cellular heterogeneous and more suitable for the application of transcriptomics in diseases.
To demonstrate the accuracy of the DAM cells we identified, we employed multi-channel data for validation. The expression of DAM genes in human cerebral cortex was verified in our data (Supplementary Fig. 16g). The proton divalent cation transporter Slc11a1, which regulates ion homeostasis and has pleiotropic effects on proinflammatory responses, was expressed only in Micro3. The zinc efflux transporter gene Tmem163 and immune cell adaptor gene Skap1 were expressed only in Micro2, which were specific for AD brain (Supplementary Fig. 16g). SKAP1 is an immune-cell adaptor that couples T-cell receptors to the “inside-out” signaling pathway of LFA-1-mediated T-cell adhesion. Studies have reported that Skap1-deficient mice are highly resistant to collagen-induced arthritis, which is a potential target for therapeutic intervention of autoimmune and inflammatory diseases32. Thus, the high expression of Skap1 in Micro2, lose its anti-inflammatory resistance, which is promising to be a checkpoint for studying the mechanism of AD disease. Then, DAM genes in cortical Micro of 7-month-old 5XFAD mice were also used to verify the accuracy of our DAM identification23. The results showed that Axl, Lgf1, and Csf1, which were upregulated in AD cortex, were all highly expressed in AD hippocampus compared with WT, but the homeostatic genes, such as P2ry12, Crybb1, Tmem119, and Cx3cr1, were under expressed in AD (Supplementary Fig. 16h, Left). However, Trem2 was under-expression in AD of our data, whereas Csf1, a gene reported Trem2-dependent upregulation, does not appear to be affected by Trem2 deficiency in the AD hippocampus. Similar results were obtained, the reported DAM genes were mainly highly expressed in Micro2 and Micro3, while most of the downregulated genes were more prominently expressed in Micro1 (Supplementary Fig. 16h, Right). The expression profiles of reported disease-pathway genes associated with AD in microglia subclusters were also analyzed. The antioxidant defense system is essential for cell survival in the central nervous system, and oxidative stress dysfunction is associated with neurodegenerative diseases33. Therefore, we first analyzed the genes involved in the regulatory pathway of oxidative stress-induced neuronal death. We found that these genes were highly expressed in AD mice such as the amyloid gene (App), the ubiquitin protein ligase gene (Prkn), and the oxidation resistance 1 gene (Oxr1) (Supplementary Figs. 16i and 17). Analogous results were presented again, that all the genes related to this pathway were significantly (p < 0.05) highly expressed in Micro2 and Micro3 (Supplementary Fig. 16j). In addition, genes involved in the B-cell receptor signaling pathway, regulation of neuronal apoptotic processes, stress-activated protein kinase signaling cascade, regulation of GTPase activity, and immune response-activation signaling pathways were all significantly overexpressed in the AD hippocampus (Supplementary Fig. 16k).
Our snRNA-seq data from FFPE hippocampal nuclei prepared by CED method identified two DAM cells, which Micro2 was the proliferating DAM in AD, and DAM-signature gene expression was independent of Trem2 expression. Moreover, Micro3 is also affected by AD development and exists in AD independent of Micro2. In addition, the combined analysis of multiple datasets indicated a strong robustness of our nuclei, which will be further validated in subsequent analyses.
Similar disease-related transcriptional changes occur in astrocytes and oligodendrocytes
Next, we identified five unique AST transcriptomically defined clusters characterized by high expression of Luzp2 (Ast0), Rgs6 (Ast1), Kcnip4 (Ast2), Cdh4 (Ast3), and Rnf213 (Ast4) (Supplementary Fig. 18a, b). The DEGs of AST subclusters between AD and WT were evaluated, and most DEGs were upregulated in AD (Supplementary Fig. 18c). We identified DEGs that were unique to single or combinations of AST subclusters (Supplementary Fig. 19a) and evaluated these gene sets by Gene Ontology (GO) analysis (Supplementary Fig. 19b). We observed that the specific DEG numbers (Supplementary Fig. 19a) and their enriched GO terms (Supplementary Fig. 19b) were largest in AST2, and the greatest change (log2FC) in specific expression (Supplementary Fig. 18c). In addition, when comparing the top-10 up/downregulated DEGs by cluster and disease state, few conserved/common transcriptomic changes were found across all AST subpopulations but instead found highly cluster-specific transcriptomic changes based on disease state (Supplementary Fig. 18d). The perturbation of gene expression changes in Ast2 and Ast4 was the most prominent (Supplementary Fig. 18d), and was provisionally defined as DAA (Disease-associated astrocytes). The DEGs that were significantly upregulated were exactly the marker genes of Ast2 (Supplementary Figs. 18b–d and 11c). Despite the overall absence of Ast cells in AD, Ast2 was highly enriched in 5xFAD mice, and Ast4 also exhibited slight cellular proliferation (Supplementary Fig. 18e), consistent with the pathological features of astrogliosis in AD. DAA genes, Kcnip4, Erc2, Nrg3, Nrxn3, and Csmd1 were notably highly expressed in Ast2 of AD mice compared with other subclusters (Supplementary Fig. 18g). We suspected that these DAA genes were primed activated in the hippocampus, and were preferentially activated during disease induction to dominate cell state changes.
DAA cells of Gfap-high state in the report25, its upregulated genes were highly and unique expression in Ast4, such as Gfap, Aqp4, C4b, and the gene encoding a serine protease inhibitor linked to increased amyloid accumulation (Serpina3n) and encoding a lysosomal cysteine protease involved in proteolytic processing of amyloid precursor protein, Ctsb (Cathepsin B) (Supplementary Fig. 18f). Moreover, a set of genes including those involved in endocytosis (Vim), complement cascade (Osmr) and senescence (Ggta1) were also overexpression in AST4, confirming our AST4 as a DAA (Supplementary Fig. 18f). Gsk3b (glycogen synthase kinase 3β gene), Psen1 (presenile factor gene), Bdnf (brain-derived neurotrophic factor), and AD risk gene Sorl1 (encoding endosomal recycling receptor gene) and App, associated with AD pathological pathways, were also highly expressed in our two DAA cells (Supplementary Fig. 18h, i). Then, we examined the expression levels of RNA signatures from bulk datasets, only ischemic related genes (Mcao) and inflammation related genes (Lps) were overexpressed in Ast2, but downregulated in Ast4 (Supplementary Figs. 19d and 20 top).
Following the approach of AST, we also characterized the Oligo subpopulations (Supplementary Fig. 22a). Six Oligo subclusters were identified, and Oligo2 characterized by Kcnip4, Nrg3, Csmd1, and Grin2a, which was consistent with Ast2 (Supplementary Fig. 22b). Moreover, the top upregulated genes in AD were similarly distributed in the Oligo2 (Supplementary Fig. 22d), and its unique DEG and GO term numbers were higher than those in other subclusters (Supplementary Figs. 22c and 23a), while the downregulated DEGs were mainly derived from Oligo5 (Supplementary Fig. 22c). Unlike Miro and AST, comparable cell proportions of Oligo subclusters between AD and WT (Supplementary Fig. 22e), which was consistent with conventional cognition. We also observed that AD-related genes were overexpressed in Oligo2 (Supplementary Fig. 22g, h). Strikingly, marker genes of Oligo5, such as Gpc5, Ntm, Rora, and Nrxn1, were also highly expressed in Ast4, which was specifically expressed by pathologically (Supplementary Fig. 22i). Among them, Gpc5 (Glypican 5) was the susceptibility gene for inflammatory demyelinating diseases34. Ntm was involved in the negative regulation of neuronal projection development and acts upstream or within cell adhesion. Downregulation of Rora inhibits glioma proliferation through NF-κB signaling pathway35, and its regulatory effect was lost when Ast4 and Oligo5 are upregulated. Nrxn1 (Neurophin 1) was a cell adhesion molecule that plays a key role in establishing and maintaining synaptic connections, and its abnormal expression has been implicated in schizophrenia36. Next, we analyzed the expression of marker genes from AD-pathology-associated Oligo29 in Oligo0-5, and most of genes were more significantly expressed in the Oligo2 and Oligo5 (Supplementary Fig. 22j). In particular, Qdpr, Nlgn1, Lama2, and Fchsd2, closely related to AD-pathology genes reported, were highly expressed in Oligo5 (Supplementary Figs. 22j and 20 bottom).
Finally, to further confirm the accuracy of DAA and Disease-associated Oligodendrocytes (DAO) identification, we collected functional terms associated with AD pathology of previously reported, and analyzed the enrichment of these disease functions in Ast and Oligo subpopulations (Supplementary Fig. 21 and Supplementary Data 3). The results showed that almost all functions were enriched in the Ast2, including lipids, glial cell regeneration, endocytosis, NFκB, endothelial cell differentiation and cognition (Supplementary Fig. 21a). Ast4 cells, however, were enriched with relatively independent functional sets, including functions in the regulation and regulation of growth, response to oxygen levels, and autophagy (Supplementary Fig. 21a). Moreover, GO terms enriched in Ast2 were also enriched in Oligo2 with stronger significance and enrichment index (Supplementary Fig. 21b). And the DEGs of Oligo2 were also enriched in autophagy, apoptosis, mRNA regulation and myelination (Supplementary Fig. 21b). In addition, AD-related genes from DisGeNet database and the reported DAA, DAM, and DAO genes were mainly enriched in Ast, Micro and Oligo, followed by Ex.CA3.1 and GAB.2 cells, which was consistent with the results of DEGs analysis (Supplementary Fig. 21c).
In a nutshell, the characteristics of DAA and DAO were similar in our snRNA-seq data of FFPE samples. Oligo2 and Ast2 had the same specific expression genes and transcription differences, while Ast4 and Oligo5 have similar transcription characteristics. We conjectured that a group of disease-susceptible gene sets caused similar transcriptional changes in different cell types, which, in turn, affected the occurrence and progression of AD.
Integration of astrocytes and oligodendrocytes from multiple datasets
Given the abundance of high-quality, well-powered AD sample AST and Oligo snRNA-seq datasets in the literature, we next sought to determine whether we could resolve the same transcriptomic differences previously reported, and in turn demonstrate the availability of our nucleus. We evaluated AST and Oligo subtypes in each individual dataset and compared them with ours. We compared five AST clusters (G0–G4) in the Grubman dataset37, four AST clusters (M0–M3) in the Mathys dataset29, and 7 AST clusters (Z0–Z9) in the Zhou dataset23, and 9 AST clusters (L0–L8) in the Liddelow dataset38 were integrated with our AST0-4 for analysis, separately (Supplementary Fig. 19e). A similar analysis was performed on Oligo subtypes (Supplementary Fig. 23d). Using our AST and Oligo subpopulation profiles as a reference, we identified sub-cells that were also recognizable in the individual datasets. Although a complete one-to-one correspondence was not possible, we still observed that AST and Oligo subtypes were analyzed in the individual data, and disease-associated cells (Ast2, Ast4, Oligo2, and Oligo5) were clearly identified in all datasets, especially in Mathys and Liddelow and Multi-datasets. For example, AST2 was highly correlated with G0, G3, M3, M4, L3-6 cells, while AST4 was more correlated with G1, G4, M0, L7, L8 (Supplementary Fig. 19e). In contrast, AST0, Oligo0, and Oligo1 showed poor agreement in these datasets. In conclusion, the results of multi-channel data integration analysis of our AST and Oligo subclusters confirmed the aforementioned argument that the diversity of distribution detected in multiple frozen samples could be detected in our data, again demonstrating that our nucleus has cellular diversity, which lays the foundation for the study of disease heterogeneity.
Transcriptional similarities in different disease-specific cell types
Although there are minimal transcriptional changes in neurons and other cells in AD cortex23, and a recent work also focused only on astrocytes and oligodendrocytes39. But the reported data of AD shows that all major cell types are affected by AD pathology at the transcriptional level29, which was consistent with our results. In our snRNA-seq data of FFPE tissues, the Micro, GABA1, OPCs, AST, Ex.Neu, Oligo, Ex.CA3.1, GABA.2 cells were more perturbed by AD (Supplementary Fig. 24a). Moreover, more than 400 upregulated DEGs were identified in two vascular related cells, Epend and Endo, which even exceeded AST (Supplementary Fig. 24a). To test the previous conjecture that there is a disease susceptibility gene-set with consistent transcriptional differences in different cell types. We performed differential analysis of gene expression for all cell types perturbed by AD to explore the transcriptional similarities of disease-related cell types. (Supplementary Fig. 24a).
We observed that only the DEGs of Micro were cell-specific, while the most significant DEGs of the remaining glial cells and vascular cells were highly heterogeneous, and the top DEGs of neuronal cells also overlapped strongly (Supplementary Fig. 24b and Supplementary Data 4). Since the Log2FC of two vascular cells were too large to annihilating the information of the other cells, we present them independently (Supplementary Fig. 24c). The positional candidate or therapeutic marker genes, including the immune-related hub genes (Fgf1340 and Etl441), the anti-inflammatory gene (Myo1e)42, the multichannel transmembrane tonic transporter gene (Ank), and the cadherin-related protein gene (Ctnna3)43 showed the greatest transcriptional changes only in micro cells (Supplementary Fig. 24b). However, Kcnip4, Grin2a, and Lrp1b were among the most differentially transcribed in other nonneuronal cells. The gene encoding Kv channel interacting protein 4 (Kcnip4) was a candidate gene for attention deficit hyperactivity disorder44. The inability of the Kcnip4 isoform to interact with the secretase complex leads to increased secretion of beta-amyloid enriched in the more toxic Aβ-42 species45. And it also has been reported that Kcnip4 interacts with presenilin, and the presenilin gene is associated with early-onset familial AD46. Sleep deprivation (SD) could increases the risk of AD, and N-methyl-D-aspartate receptors (NMDAR) is an important cognitive regulator. Specific knockdown of hippocampal astrocytic Grin2a (the gene encoding the NMDAR subunit GluN2A) aggravated SD-induced cognitive decline, elevated Aβ, and attenuated the SD-induced increase in autophagy flux47. Most of these conclusions were based on the results of immunofluorescence staining, while our snRNA-seq data showed exactly the opposite, the Grin2a gene was not only highly expressed in Ast, but also positively expressed in most of the cell types associated with AD (Supplementary Fig. 24b). The low-density lipoprotein receptor-associated protein 1B (LRP1B) can interact with APP and regulate its processing to Aβ48. In summary, the transcriptional profiling of all cell types closely associated with AD reconfirmed our previous hypothesis that a single disease-susceptible gene set causes similar transcriptional changes in different cell types.
Performance of snCED-seq in human lung FFPE micro-samples
For archived clinical human FFPE samples, we successfully isolated approximately 150,000 intact and pure nuclei from one 50 μm scroll of paracancerous tissue using the CED method (Supplementary Figs. 3b and 25a). These nuclei were then subjected to snCED-seq, and the fragment size of library peaked about 317 bps (Supplementary Fig. 25b). Unlike FFPE(MHD) data, only 0.8% of the sequencing reads were distributed in rRNA, while the majority of reads were distributed in introns in FFPE(Lung) (Supplementary Fig. 25c), which might be due to the higher proportion of unspliced RNA present within nucleus, or the preservation conditions and processing methods of FFPE samples, which may affect RNA splicing efficiency. In the FFPE(Lung) dataset with a sequencing depth of 9886k per nuclei, 1830 UMIs and 770 genes were detected per nucleus, respectively (Supplementary Fig. 25d). Compared with scRNA-seq of frozen lung tissues, our sequencing depth was only one-tenth of that, but the number of detected genes was only reduced by half, and the proportion of mitochondrial and ribosomal genes significantly lower than the reported49 (Supplementary Fig. 25d). Unsupervised clustering of the above filtered high-quality 4271 single lung nucleus profile revealed ten distinct clusters (Fig. 6a), and further annotated based on classical known cell-type markers49,50 (Fig. 6b). Immune cells (B/T cells, mast and macrophages), alveolar epithelial type cells (alveolar epithelial type cells_AT1/2 and lung epithelial cells_ epithelial), vascular cells (endothelial) and interstitial cells (fibroblasts) were successfully detected, covering almost all major cell types of the lung (Fig. 6a). Gene expressions of classical known cell-type marker genes, such as chloride intracellular channel 5 (CLIC5) and caveolin-1 (CAV1) were highly expressed in AT1, surfactant protein B (SFTPB), ATP-binding cassette subfamily A member 3 (ABCA3), and surfactant protein C (SFTPC) were highly expressed in AT2, were reliably mapped on the corresponding clusters ((Fig. 6b, c). Besides the known top markers of cell types, such as such as SNX30, TMEM164, and CACNA2D2, which are typical of non-lung cells, were highly expressed in AT2 cells (Supplementary Fig. 26). In addition, the dataset contained a larger fraction of unclassified cells (cluster 3) with nonspecific gene expression, which was annotated AT2 and immune cells (T cells and macrophages) by top DEGs. Compared with scRNA-seq, our results were consistent with the undefined cells in snRNA-seq data of lung tissue51. Next, we compared the proportion of cells in FFPE(Lung) with that in frozen lung samples that had been reported from healthy donors50, and found that most of the cell proportion were similar to that of healthy frozen samples (Fig. 6e). For example, the proportion of AT cells was approximately 50%, indicating that they had not been replaced by cancer cells, and the status of immune cells remained stable.

a UMAP of 4271 single-nucleus RNA profiles of human lung tissue. colored by cluster. b Expression of selected cell-type markers in the UMAPs of FFPE human lung. Gene expression levels are indicated by shades of bule. c Dot plot of the average expressions of top five markers in each of the 10 cell types. d Counts of different RNA biotypes detected in FFPE(lung). Expression level (color scale) of marker genes across clusters and the percentage of cells expressing them (dot size). e Proportion of annotated cell types of FFPE(lung) by snCED-seq. Source data are provided as a Source Data file (a–e).
In short, the number of nuclei sufficient for FFPE snRNA-seq platform can be obtained from such a micro-tissue, and all major cell types in lung organ can be detected, which provides assistance for transcriptome studies of clinical puncture samples.
Discussion
In this study, we developed a strategy for high-quality nuclear preparation from FFPE tissues by enzymatic dissociation of archived samples at low temperature without the need for tedious filtration step, which therefore provides a critical advance to profile single nuclei transcriptome from low-quality biological samples of PFA-fixed or FFPE tissues of. Meanwhile, we performed snRNA-seq on frozen, PFA-fixed and FFPE brains using 10× Genome and snRandom-seq technologies, and performed head-to-head comparison. To prove our method, we performed validation and obtained promising results. snRNA-seq was conducted on frozen samples using 10× Genome and snRandom-seq technologies to eliminate platform differences, and the data of frozen samples was used as the gold standard for reference. We used snRandom-seq to perform snRNA-seq on PFA-fixed samples to compare the nucleus preparation strategies, and explored the applicability of snRNA-seq on FFPE samples as well as its application performance in brain diseases. The CED method and snCED-seq represent a significant advance in single-nuclei sequencing, enabling researchers to retrospectively select samples from a large paraffin sample bank, and facilitating mechanistic studies of brain disease samples that are difficult to obtain clinically.
Molecular biological application of FFPE tissues has always been challenging due to the chemical cross-linking and low-quality RNA. Although researchers have gradually become aware of the potential for obtaining expression profiles of individual cells or nuclei FFPE tissues, the approaches of extracting or isolating high-quality nuclei remains challenging. The acquisition of nuclei is a crucial step for snRNA-seq of FFPE samples, and its quality directly determines transcriptome analysis. The preparation methods of FFPE sample nuclei are longstanding, and are mainly divided into two categories, hyperthermal enzymatic dissociation strategies and mechanical extraction strategies. Enzymatically obtained nuclei are unhea1rd of in transcriptomics studies. In fact, prolonged high-temperature treatment resulted in secondary RNA degradation of FFPE samples, and prolonged exposure of nuclei to the enzyme buffer may increase the permeability of the nuclear membrane, leading to RNA molecules leakage and adversely affecting snRNA-seq experiments performed in droplets. The mechanical homogenization strategy was less damaging to RNA molecules within nucleus. However, tissue homogenization for fixed and FFPE tissues, becomes increasingly challenging due to molecular cross-linking within the nuclei. Firstly, effectively removing excessive tissue debris poses difficulties and leads to severe contamination of the snRNA-seq data9; Moreover, the high proportion of rRNA requires additional removal processes when employing total RNA protocols21,52. In our early experiments, the large amount of tissue debris interfered with accurate identification of nuclei due to the molecular cross-linking introduced by formaldehyde fixation, which requires a complicated debris removal process, thus affecting the yield of nuclei and losing smaller nuclei. Currently, high-throughput snRNA-seq platforms employ a combination of enzyme dissociation and homogenate10. Despite the optimization of nuclear suspension and RNA quality within the nucleus, their own shortcomings have not been dismissed. Furthermore, all current methods for preparing nuclei from FFPE samples focus on tissue sections (5–100 μm), while disease research often involves the tissue blocks.
Due to the characteristics of dissociation and digestion of nuclei in the preparation process of enzyme dissociation, the traditional high-temperature method makes the nuclei prepared first be digested in the dissociation solution, or the nuclear membrane is damaged, which affects the nuclear yield and is very sensitive to the reaction time, increasing the burden on the experimenter. Our nuclei were obtained by enzymatic hydrolysis of molecularly cross-linked tissues with a single step at low temperature, and without ultracentrifugation through a sucrose cushion and any filtration procedures, thereby maximizing product retention and nucleation rates. Taking a pair of mouse hippocampus as an example, the number of nuclei prepared by CED method was about 10 times that of the traditional method, and CED method can better enrich the small diameter nuclei missed by the traditional method. Most importantly, our CED method can effectively protect the nuclear membrane and maximally retains the nuclear molecules, providing high-fidelity nuclei for snRNA-seq research. For the latest snRNA-seq technology based on random primer capture10 or gene probe capture8, it is necessary to input nearly one million nuclei on the premise of ensuring the output of about 10,000 nuclei. Our CED method effectively avoids the current two major nucleus preparation strategies, and can export the nucleus stably without introducing more impurities and damaging the nuclear membrane. The nuclei prepared by our CED method could be successfully preserved or transported on dry ice, which we speculated might be due to the fact that the permeability of the nuclear membrane was not damaged. In addition, our CED method has good applicability to a variety of organs, such as brain, liver, kidney, pancreas, spleen tissues, but slightly poor compatibility with heart and lung, although the yield of nuclei was still higher than that of the mechanical method. The heart has a complex cellular composition, mainly including myocardial tissue, nerve tissue, Purkinje fiber, connective tissue, epithelial tissue, etc. Similarly, the main connective component of the lung is composed of connective tissue, which is rich in collagen fibers, elastic fibers, reticular fibers. Connective tissue and cell relatively dense structure greatly increases the difficulty of the heart and lung tissue, and the choice of operating conditions and enzymes need further adjustment.
The excellent performance of the CED method was maintained for both PFA-fixed and FFPE tissues in our benchmarking effort. Compared with the HED high-throughput snRNA-Seq database of the PFA-fixed samples, snCED-seq outperforms well in various perspectives, supported by the genes and transcripts per nucleus, the percentage of mitochondrial and ribosomal genes, gene detection sensitivity, gene expression correlation with frozen samples, especially in gene expression richness. High-quality and high-sensitivity snRNA-seq data from post-fixed (PFA-fixed and FFPE) specimens by snCED-seq allows for the identification of rare cell populations. We further provide a detailed map of cell-type-specific expression of FFPE samples from AD and WT mice, which highlights the predominance of gene expression richness in our nuclei. Multiple disease-related subpopulations have been successfully identified, and the DAM has transcriptional independence, while the transcriptional similarity between DAA and DAO subpopulations. There is even a population of genes (Kcnip4, Grin2a, Lrp1b, etc.) that are in the waiting state of activation, priority in different cells by the interference of the disease. In short, nuclei from CED method excels in revealing cellular heterogeneity, which contributes to the precision diagnosis and treatment to human disease.
Overall, this study proposes a method for the preparation of high-fidelity nuclei from post-fixed samples, which outperforms traditional methods in various aspects, and demonstrates good compatibility with a variety of FFPE organs. The application of FFPE samples in AD was also investigated and found that our nuclei have potential for uncovering disease cellular heterogeneity. The simple experimental protocols and comprehensive transcriptomic information from the FFPE tissues described in this study are expected to enable large-scale snCED-seq applications in both basic and clinical research in the future. Our nuclear preparation strategy lays the foundation for revealing transcriptomic and even multi-omic information from FFPE samples.
Methods
Ethical statement
The study was approved by the animal ethical and welfare committee of Zhongda Hospital Southeast University (approval numbers: 20200104005). All procedures were conducted following the guidelines of the animal ethical and welfare committee of SEU. The collection of human samples and research conducted in this study was approved by the Research Ethics Committee of the Affiliated Hospital of Yangzhou University (approval numbers: 2025-YKL02-001). Written informed consent was obtained from all individual participants. All applicable institutional and/or national guidelines for the care and use of animals were followed.
Experimental model
Male wildtype (WT) C57/BL6 mice (8 weeks of age) were ordered from Qinglongshan Animal Farm, Nanjing, China. Different human tissues were derived from the Affiliated Hospital of Yangzhou University. AD and their control mice were purchased from Jiangsu Huachuang sinoPharmaTechCo., Ltd, Taizhou, China. Five-month-old heterozygous 5xFAD transgenic mice (on a C57/BL6 background) co-overexpress mutant forms of human amyloid precursor protein associated with familial AD, the Swedish mutation (K670N/M671L), the Florida mutation (I716V), the London mutation (V717I) and carry two FAD mutations (M146L and L286V) people PSEN1. The expression of both transgenes is regulated by the mouse neurospecific regulatory element Thy1 promoter to drive transgene overexpression in the brain. Throughout the study, all mice in each experiment were nontransgenic littermates from the same mouse colony. All mouse samples were from male animals. The samples in this study were used to validate the feasibility of the method, with no specific gender requirements, so sex and age information of human tissue donors were not collected in this study.
All the mice were single-housed under standard laboratory conditions, including a 12 h light/darkcycle, temperatures of 25 °C with 40% humidity, with free access to mouse diet and water. The animals were anesthetized with 500 mg/kg tribromoethanol (Sigma, Saint Louis, MO, USA) and were killed by cervical dislocation. After the animals were sacrificed, hippocampi were isolated. Fresh frozen (FF) tissues were obtained by quickly frozen in liquid nitrogen; PFA-fixed (PFA) tissues were prepared by adding PFA (4%) to the hippocampus and fixed for 20 h at 4 °C; FFPE samples were prepared by dehydration the fixed hippocampus twice in 70%, 90%, and 100% ethanol, respectively, and then clearing with xylene solution for 15 min, twice, followed by paraffin embedding for 2 h (62 °C). Frozen samples and PFA-fixed samples were stored in a −80 °C, and FFPE samples were stored at 4 °C. FFPE tissues were prepared from human organ tissues using a standardized protocol and subsequently sectioned at 50 μm. The brain organoids were embedded using OCT after fixation.
Single nuclei isolation from PFA-fixed and FFPE tissues
For snap-frozen samples
The nuclei prepared by the homogenization method by Singleron Biological Tech Co. for 10× Genomics snRNA-seq and M20 Genomics for snRandom-seq.
For PFA-fixed samples
-
(1)
Cryogenic enzymatic dissociation (CED) method: The brain tissue was washed three times with 1 mL PBS (1×, pH = 7.4) and cut into 1 mm3 pieces in a 2 mL enzyme-free centrifuge tube, adding 1 mL dissociation buffer (1.5 mg/mL protease K, TE buffer, pH = 8, 0.5 % sarkosyl, SDS or Triton X-100). The centrifuge tube was placed transversely in a closed low-temperature (4 °C) box, and placed on a shaker to sway slightly 6–15 h (The concentration of protease K depends on organ types, and dissociation timing depends on tissue size and organ types) to ensure that the solution dissociation system flowed back and forth between the tube mouth and the tube bottom. The nuclear suspension was transferred to a 1.5 mL test tube and centrifuged, centrifuged at 10,000 rpm for 10 min at 4 °C. Discarded supernatant, and then washed nuclei with 1 mL pre-cold PBS (1×, pH = 7.4) twice, centrifuged at about 10,000 × g for 10 min at 4 °C. Finally, the nuclei were resuspended in 200–400 uL of nuclear store buffer (NSB, 1× PBS, 0.2 U/mL RNase Inhibitor) and stored at −80 °C.
-
(2)
High temperatures enzymatic dissociation (HED) method: We optimized and improved the previously described method of enzymatic dissociation nuclei at 37 °C overnight13,15,53,54. These methods are mainly applied to paraffin sections or scrolls, and the cell suspension after digestion and the undigested bulk tissue are separated by the nylon pores or 25-G needles. Considering that most of the research samples of snRNA-seq technology are tissue blocks, we extended the optimization method to tissue blocks. In brief, the brain tissue was washed three times with 1 mL PBS (1×, pH = 7.4) and cut into 1 mm3 pieces in a 2 mL enzyme-free centrifuge tube, adding 1 mL dissociation buffer (0.6 mg/mL protease K, TE buffer, pH = 8, 0.5% SDS/Triton X-100), and shaken slightly for 5 and 6 h. Then, Nuclei were collected and resuspended in NSB. In order to ensure the yield and morphology integrity of the nuclei, the nuclei of dissociation process for DAPI staining and detection under a microscope. Insufficient dissociation time affects the number of nuclei, but the nuclear membrane will be damaged or even the nuclei will be digested if the dissociation time is too long.
For FFPE samples
-
(1)
CED method: The target tissue was trimmed out of the FFPE blocks with a sterilized scalpel and placed in a 2 mL tube, and washed thrice with 1.5 mL xylene for 2 h at 4 °C to remove the paraffin. The samples were gently rehydrated by immersing the samples in a graded series of ethanol solutions, starting with pure 100% ethanol and ending with 50% ethanol (100% × 2, 95%, 70%, 50% × 1) for 1 h, then washed twice with pre-cold water. The steps of nuclei prepared was same as PFA-fixed tissue. An aliquot of nuclei was stained with DAPI (4′,6-diamidino-2-phenylindole) staining solution, loaded on a hemocytometer and observed under an inverted fluorescence microscope. Finally, morphologically intact, free of impurities, and abundant (in millions) nuclei were stored on dry ice and sent to M20 Genomics.
-
(2)
Mechanical homogenization dissociation method: FFPE brain nuclei were extracted entirely with the use of three mechanical homogenization strategies without any modification, including two reported methods (snPATHO-seq26 and snRandom-seq10) and a commercial kit. (1) MHD1: FFPE sections were cut from the paraffin block and were washed with xylene to remove paraffin and rehydrated via a sequence of ethanol baths, and then rinsed with RPMI1640. Then, tissue disruption physically using a pestle in 100 μL digestion mix (1 mg/mL Liberase TM, 1 mg/mL Collagenase D, and 1 U/μL of RNAse inhibitor in RPMI1640), and then filled up to 1 mL and subjected to digestion at 37 °C for 60 min. The pre-digested tissue was treated with 1× Nuclei Ez lysis buffer with 2% BSA, and the separated nuclei were filtered through a 70 μm mesh, double-rinsed with 1× PBS + 1% BSA, and once with a 0.5× PBS + 0.02% BSA blend. They were then resuspended in this blend and re-filtered via a 40 μm mesh. (2) MHD2: FFPE sections were cut from the paraffin block and were washed with xylene to remove paraffin and rehydrated via a sequence of ethanol baths as reported10. The samples were then washed twice with pre-cold wash buffer and homogenized with Dounce homogenizer with the presence of pre-cold lysis buffer (1 × PBS, 0.1% Triton X-100, 1 U/μL RNase Inhibitor) on ice, then 1 mL lysis buffer to rinse the douncer, and 100 μL of 10 mg/mL proteinase K was added into the lysis buffer, incubating at 37 °C for 5 min. Then, the isolated nuclei were filtered through a 20 μm cellstrainer and washed twice with wash buffer. (3) Nuclei Isolation Kit for FFPE Tissue_MHD3 (52301-10): The 50 μm sections was placed in a 1.5 mL centrifuge tube after deparaffinized with xylene and hydrated with ethanol, then added 200 μL of lysate and fully ground, another 400 μL of lysate was added and placed on ice for 5 min. Subsequently, 150 μL of digestive enzyme lysate was added and shaken at 500 rpm for 10 min at 50 °C. The mixture was filtered using a 40 μm cell sieve, the supernatant was discarded, and 500 μL of debris removal solution was added, centrifuged at 2000 × g for 5 min at 4 °C, and discarded supernatant. The nuclei were then resuspended in 500 μL of the wash buffer and collected and stored nuclei in the wash buffer. The single nuclei were processed to snRNA-seq according to the reported protocol10. All FFPE sections of mouse were derived from the whole mouse hippocampus.
Nuclear quality assessment
Morphology and counting of nuclei
Trypan Blue and DAPI (20 μg/mL) were mixed with nuclear suspension 1:1, the quality control step was performed by viewing the nuclei under the microscope on a hemocytometer to check nuclei shape and count. Among them, the nuclei morphology and counting results of different organs (heart, liver, spleen, lung, kidney, pancreas, etc.), snPATHO-seq and snRandom-seq protocols were performed by M20 Genomics. The nuclei from Nuclei Isolation Kit were detected by the company. The other nuclear quality inspection data were obtained from the method in our study.
Extraction RNA of nuclear
We modified the previously described of RNA extracted18. Bulk nuclei were first suspended in 100 uL PBS (1×, 7.4), and with 1-to-1 volume ratio lysis buffer (200 mM Tris pH 7.5, 6% Ficoll type 400, 0.2% Sarkosyl, 20 mM EDTA, 50 mM DTT and proteinase K), incubated at 56 °C for different time, then RNA was extracted using the RNeasy Plus Mini Kit (Qiagen, Hilden, Germany). The specific process according to the manufacturer’s protocol. Proteinase K concentration (0–1.5 mg/mL) was optimized to improve RNA yield and decross-linking time (15–60 min) was optimized to reduce RNA degradation. Finally, RNA quality was evaluated by the Qubit 4.0 and the RNA 6000 Nano Kit (Agilent, Santa Clara, CA, USA) with Agilent 4150 Bioanalyzer.
Construction of cDNA
The cDNA was prepared using the Smart-Seq2 protocol with the following modifications. Prior to cDNA synthesis, two master mixes were prepared on ice: Mix1 (1 ng RNA, 10 μM Oligo dT (AAGCAGTGGTATCAACGCAGAGTACT30VN) and 10 mM dNTP), Mix2 (5× First-Strand Buffer, 25 mM MgCl2, 40 U/μL RNase Inhibitor, 100 μM DTT, 10 μM TSO adaptor (AAGCAGTGGTATCAACGCAGAGTACAT/rG//rG//iXNA_G/), 5 M Betaine, and 100 U/μL Reverse Transcriptase). All prepared solutions were kept on ice for subsequent use. Mix1 was denatured at 72 °C for 3 min (lid temperature: 105 °C), immediately chilled on ice. Subsequently, Mix1 and Mix2 were combined, reverse transcription (RT) was performed at 42 °C (90 min), followed by 10 cycles (50 °C_2 min and 42 °C_2min), 70 °C (15 min), and 4 °C hold. Following RT and template switching, cDNA was amplified using KAPA HotStart HIFI 1× ReadyMix and PCR primer (AAGCAGTGGTATCAACGCAGAGT). The PCR conditions were as follows: 98 °C (3 min), followed by 20 cycles (98 °C_20 s, 67 °C_15 s and 72 °C_6 min), 72 °C (5 min), and 4 °C hold. The PCR products were purified using 0.8× AMPure XP beads, and cDNA/library quality was assessed using a Bioanalyzer 4150 (Agilent, Santa Clara, CA, USA) with a High Sensitivity DNA chip.
Library construction and sequencing
For frozen samples
Isolated nuclei were subjected to droplet-based 3′ end massively parallel sequencing using Chromium Single Cell 3′ Reagent Kits per the manufacturer’s instructions (10× Genomics) and droplet-based snRNA sequencing technology by capturing full-length total RNAs with random primers (M20 Genomics).
For PFA-fixed and FFPE samples
In the single-cell transcriptome sequencing experiments of this study, we utilized the VITAcruizer single-cell preparation instrument DP400 (Cat #E20000131, M20 Genomics) to achieve droplet generation, single-cell partition and encapsulation, and nucleic acid capture. The VITApilote high-throughput FFPE single-cell transcriptome kit (Cat #R20121124, M20 Genomics) was employed for pre-library sample processing, single-cell library construction, and purification. Experimental procedures were conducted following the respective kit and instrument manuals. The main workflow is outlined below.
Nuclei were removed from dry ice and thawed at 4 °C, and the qualified single nuclei were subjected to snRNA-seq processing according to the snRandom-seq protocol10. In brief, the counted nuclei were used for in situ DNA blocking using the DNA Polymerase kit (M20 Genomics), then quickly put on ice. Prepare the in situ reverse transcription reaction (ISRT) mix (above nuclei,10 µM random primer, 10 µM oligo(dT) primer, Reverse Transcriptase, 5× reverse transcription buffer, 100 mM dNTP and 200 U/µL RNase Inhibitor), and the ISRT was performed by 12 cycles (8 °C_10 s, 15 °C_15 s, 20 °C_15 s, 30 °C_30 s, and 42 °C_1 min), 42 °C (30 min) and 4 °C hold. Then, added the dA tailing by the TdT reaction kit. Subsequently, the reverse-transcribed nuclei, 2× DNA extension reaction mixed and barcoded beads containing cell barcodes and UMIs were encapsulated into droplets using the microfluidic platform of M20 Genomics, and the emulsions were incubated at 37 °C (1 h), 50 °C (30 min), 60 °C (30 min), and 75 °C (20 min). After the barcoding reaction, droplets were broken by mixing with PFO buffer (M20 Genomics). The aqueous phase was taken out and purified by Ampure XP beads. PCR was performed to amplify the purified product with PCR Primer1 (GGAGTTGGAGTGAGTGGATGAGTGATG) and Primer2 (GTG AGT GAT GGT TGA GGA TGT GTG GAG ATA) at 95 °C (1 min), followed by 20 cycles (95 °C_30 s, 60 °C_30 s and 72 °C_2 min), 72 °C (5 min) and 4 ̊C hold. The amplified product was purified by Ampure XP beads and quantified by Qubit. VAHTS Universal DNA Library Prep Kit for Illumina V3 was used to construct library. Library sequencing was performed using the NovaSeq 6000 and S4 Reagent Kit with paired end reads of 150.
Data analysis for C57/BL6 mice
Preprocessing of snRNA-seq data
Mus_musculus. GRCm39.109 reference genome was downloaded from ensemble database. Then we used STARsolo module in STAR (2.7.10a) with default parameters to generate the gene expression matrix and filter the valid nuclei. The Seurat v4.2 was applied for the major downstream analysis. Before we started downstream analysis, there are some filtering metrics to guarantee the reliability of each data.
detected in fewer than 3 cells were filtered to avoid cellular stochastic events. We deleted mitochondrial genes after the quality control, the left genes used for downstream analysis. For the cell part, we set different filter standards for each dataset according to the UMI and gene numbers distribution to filter low quality cells. Finally, we got 23248 genes and 142661 cells as the expression matrix to do downstream analysis in the method comparison part.
Clustering and cell annotation
After quality control, unsupervised clustering was performed using Seurat v4.2. A series of preprocessing procedures including normalization, variance stabilization and scaling data, were performed in an R function ‘SCTransform’based on regularized negative binomial regression. Then, we selected 2000 highly variable genes to integrate all sequencing libraries using ‘FindIntegrationAnchors’ and ‘IntegrateData’ functions, followed by the regression of technical noise. Principal component analysis (PCA) was performed using integrated output matrix, and the reasonable principal component (PC) numbers was chosen using the ‘JackStraw’ function. And we chose the top 30 significant PCs for downstream cluster identification and visualization. Clusters were defined based on ‘FindClusters’ function with resolution from 0.1 to 1 with 0.1 as seperation. UMAP was used for the final dimension reduction and visualization. Based on the cluster results with resolution equal to 0.2, we next used ‘FindAllMarkers’ function with MAST algorithm. We ranked the marker genes according to the p-value and log2 fold change (log2 FC) within each cluster and searched top genes in Cell Marker database55 and Panglao DB56 databases to annotate cell types of clusters.
Differential expression analysis
Within each cluster, we calculated differentially expressed genes (DEGs) between 2 different conditions by using ‘FindMarkers’ function. we used ‘MAST’ setting as well and the Benjamini–Hochberg procedure to adjust p value. Then we set threshold q_adjust <0.05, absolute value of log2 FC > 0 to filter DEGs. The DEGs functional enrichment analysis based on GO and Kyoto Encyclopedia of Genes and Genomes (KEGG) was applied by an R package ClusterProfile v4.10.0 using a hypergeometric test and corrected for multiple hypothesis by FDR.
Data analysis for AD and WT mice
Preprocessing of snRNA-seq data
Mus_musculus. GRCm39.109 reference genome was downloaded from ensemble database. Then we used STARsolo module in STAR (2.7.10a) with default parameters to generate the gene expression matrix and filter the valid nuclei. =Seurat v4.4.0 was applied for the major downstream analysis. Before starting the downstream analysis, we used four filtering metrics to ensure the reliability of the data. (1) Gene detected in fewer than 3 cells were filtered to avoid cell random events; (2) Remove nuclei with mitochondrial gene expression percentage >10% to exclude apoptotic cells; (3) Remove UMI > 30000 cells; (4) Remove cells outside the range of 300 to 5000 genes. After filtering cells and genes based on the above metrics, we further use Doublet Finder V2.0 with default parameters to predict and remove potential doublet in each sample. Only cells that have passed a rigorous multi-step quality control regimen are considered for downstream analysis. Thus, 23,248 genes and 63,789 nuclei were retained in AD part.
Clustering and cell annotation
After quality control, we aligned the data from different batches using the SCTransform57 integration workflow in Seurat with default settings. We identified high-resolution clusters (resolution = 0.1) using the Seurat functions FindNeighbors and FindClusters (Leiden clustering algorithm) based on the first 30 principal components. To annotate cell types within this dataset, we employed two distinct approaches: (1) manual annotation using previously published databases Cellmarker 2.0 and and Panglao DB databases, and (2) projecting annotations onto the cells analyzed in this study by integrating the clustering results with the dataset from Habib et al.25 using the Seurat functions FindTransferAnchors and TransferData. Combining these methods yielded detailed and reliable cell cluster information. We also excluded certain less reliable cell types (e.g., Ex.neuron2 and Ex.IEG). During the subsequent cell proportion analysis, we observed a significant imbalance in the cell proportions of the AD1 sample, with neurons comprising the majority and non-neuronal cells only accounting for 4%. Thus, we deemed this sample unreliable and excluded it from further analysis. Ultimately, we retained 23,248 genes and 52,569 cells.
For the subcluster analysis of astrocytes, oligodendrocytes, and microglia, we similarly utilized the Seurat functions FindNeighbors and FindClusters on the top 30 principal components, setting the resolution to 0.1.
Differential expression analysis
Within each cluster, we used the “FindMarkers” function in the Seurat package to detect DEGs between AD and WT conditions. We applied the “MAST” setting and controlled the false discovery rate (FDR) using the Benjamini–Hochberg procedure. We set thresholds of |avg_log2FC| > 0.25 and p_val_adj <0.05 to filter DEGs, identifying both upregulated and downregulated genes in AD relative to WT for each cluster.
Enrichment analysis
The functional enrichment analysis of Differentially Expressed Genes (DEGs) based on GO biological processes and the KEGG was conducted using the R package ClusterProfile v4.11.1. The analysis employed a hypergeometric test and corrected for multiple hypotheses using the False Discovery Rate (FDR). For enrichment between gene sets, the testGeneOverlap function from the R package GeneOverlap v1.34.0 was utilized to perform Fisher’s exact test, identifying overlaps among different gene sets.
Comparison with external data sets
To compare our data with external datasets, we collected single-cell data from studies by Grubman, Mathys, Zhou, Liddelow, and others. Specifically, we gathered the top markers for astrocytes, oligodendrocytes, and microglia from these studies. We then analyzed the relative expression levels of these markers in our own dataset to identify corresponding AD-related cell subtypes mentioned in these publications. This comparative approach allowed us to validate our findings and highlight specific cell types associated with Alzheimer’s disease in our study.
Data analysis for human tissues
Quality control and filtering
Potential doublets were identified and removed using scDblFinder. For gene filtering, genes detected in fewer than three cells were excluded to minimize the impact of stochastic cellular events. Following quality control, mitochondrial genes (with the percent of mitochondrial gene > 10%) were removed, and the remaining genes were retained for downstream analysis. For cell filtering, dataset-specific thresholds were applied based on UMI (retaining cells with <20,000 UMIs) and gene counts (retaining cells with <6000 detected genes) to eliminate low-quality cells. After these filtering steps, the final expression matrix consisted of 29,849 genes and 4271 cells for subsequent analyses.
Clustering and cell annotation
After quality control, unsupervised clustering was performed using Seurat v5.2. The preprocessing pipeline included normalization, variance stabilization, and data scaling. Since only a single sample was analyzed, batch correction or integration was unnecessary. PCA was conducted using the normalized expression matrix, and significant principal components (PCs) were determined via the JackStraw method. The top 10 significant PCs were selected for downstream clustering and visualization. Clusters were identified using the FindClusters function, testing resolutions ranging from 0.4 to 1.2 (in increments of 0.1). UMAP was applied for dimensionality reduction and visualization.
For cell type annotation, the clustering result at resolution = 0.4 was used to identify marker genes via the FindAllMarkers function (Wilcoxon rank-sum test). Marker genes were ranked based on adjusted p-values and log2 FC. The top marker genes were then cross-referenced with established cell-type databases, including CellMarker2 and PanglaoDB, to assign biological identities to each cluster.
Statistics and reproducibility
Statistical details for each experiment are provided in the figure legends. The FFPE single nuclei isolation experiment, RNA quality comparison experiment and DNA fragments analysis experiment were repeated more than three times independently with similar results. The snRNA-seq experiment was repeated three times independently with similar results. No statistical method was used to predetermine the sample size. No data were excluded from the analyses. The experiments were not randomized. The Investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The snRNA-seq datasets generated in this study have been deposited in the National Center for Biotechnology Information (NCBI) under accession code PRJNA1117576. Source data are provided with this paper.
References
Shalek, A. K. et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 498, 236–240 (2013).
Jaitin, D. A. et al. Massively parallel single-cell rna-seq for marker-free decomposition of tissues into cell types. Science 343, 776–779 (2014).
Rozenblatt-Rosen, O., Stubbington, M. J. T., Regev, A. & Teichmann, S. A. The human cell atlas: from vision to reality. Nature 550, 451 (2017).
Gnanapragasam, V. J. Unlocking the molecular archive: the emerging use of formalin-fixed paraffin-embedded tissue for biomarker research in urological cancer. Bju Int. 105, 274–278 (2010).
Amini, P. et al. An optimised protocol for isolation of RNA from small sections of laser-capture microdissected FFPE tissue amenable for next-generation sequencing. BMC Mol. Biol. 18, 22 (2017).
Maliga, Z. et al. Micro-region transcriptomics of fixed human tissue using Pick-Seq. Preprint at https://biorxiv.org/content/early/2021/03/18/431004 (2021).
Liu, Y. et al. High-plex protein and whole transcriptome co-mapping at cellular resolution with spatial CITE-seq. Nat. Biotechnol. 41, 1405–1409 (2023).
Wang T. P. et al. snPATHO-seq, a versatile FFPE single- nucleus RNA sequencing method to unlock pathology archives. Commun. Biol. 7, 1340 (2024).
Chung, H. et al. SnFFPE-Seq: towards scalable single nucleus RNA-Seq of formalin-fixed paraffin embedded (FFPE) tissue. Preprint at bioRxiv https://doi.org/10.1101/2022.08.25.505257 (2022).
Xu, Z. et al. High-throughput single nucleus total RNA sequencing of formalin-fixed paraffin-embedded tissues by snRandom-seq. Nat. Commun. 14, 2734 (2023).
Hedley, D. Method for analysis of cellular DNA content of paraffin-embedded pathological material using flow cytometry. J. Histochem. Cytochem. 31, 1333–1335 (1983).
Liehr, T., Uwe, C. & Erich, G. Nucleus extraction from single mounted tissue sections. Genet. Anal.-Biomol. E 15, 65–69 (1999).
Paternoster, S. F. et al. A new method to extract nuclei from paraffin-embedded tissue to study lymphomas using interphase fluorescence in situ hybridization. Am. J. Pathol. 160, 1967–1972 (2002).
Schutte, B., Reynders, M. M., Bosman, F. T. & Blijham, G. H. Flow cytometric determination of DNA ploidy level in nuclei isolated from paraffin-embedded tissue. Cytometry 6, 26–30 (1985).
Martelotto, L. G. et al. Whole-genome single-cell copy number profiling from formalin-fixed paraffin-embedded samples. Nat. Med. 23, 376–385 (2017).
Chin, H. G. et al. Universal NicE-seq for high resolution accessible chromatin profiling for formaldehyde fixed and FFPE tissues. Clin. Epigenetics 12, 143 (2020).
Maitra, M. et al. Extraction of nuclei from archived post-mortem tissues for single-nucleus sequencing applications. Nat. Protoc. 16, 2788–2801 (2021).
Phan, H. V. et al. High-throughput RNA sequencing of paraformaldehyde-fixed single cells. Nat. Commun. 12, 5636 (2021).
Thomsen, E. R. et al. Fixed single-cell transcriptomic characterization of human radial glial diversity. Nat. Methods 13, 87–93 (2016).
Alles, J. et al. Cell fixation and preservation for droplet-based single-cell transcriptomics. BMC Biol. 15, 44 (2017).
Salmen, F. et al. High-throughput total RNA sequencing in single cells using VASA-seq. Nat. Biotechnol. 40, 1780–1793 (2022).
Guo, Y. et al. Transcriptomic profiling of nuclei from paraformaldehyde-fixed and formalin-fixed paraffin-embedded brain tissues. Anal. Chim. Acta 1281, 341861 (2023).
Zhou, Y. et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Medthods 26, 131–142 (2020).
Guo, Y. et al. Defining specific cell states of mptp-induced parkinson’s disease by single-nucleus rna sequencing. Int. J. Mol. Sci. 23, 10774–10794 (2022).
Habib, N. et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 23, 701–706 (2020).
Wang, T. et al. snPATHO-seq, a versatile FFPE single-nucleus RNA sequencing method to unlock pathology archives. Commun. Biol. 7, 1340 (2024).
Cao, J. L., Liu, J. Y., Xiao, J., Jiang, X. R. & Huang, H. Y. Olfactory ensheathing cells transplanation reduced amyloid burden in amyloid precursor protein transgenic mouse model. Acta Anatomica Sin. 41, 823–826 (2010).
Fan, W. J., Cheng, W. J., Niu, Y. L., Li, M. S. & Deng, J. B. Normal development of Cajal-Retzius cells in mouse hippocampus and their changes in APPswe transgenic mice. Acta Anatomica Sin. 41, 211–218 (2010).
Mathys, H. et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019).
Shen, L., Thompson, P. M., Potkin, S. G., Bertram, L. & Saykin, A. J. Genetic analysis of quantitative phenotypes in AD and MCI: imaging, cognition and biomarkers. Brain Imaging Behav. 8, 183–207 (2014).
Potkin, S. G. et al. Hippocampal atrophy as a quantitative trait in a genome-wide association study identifying novel susceptibility genes for alzheimer’s disease. PLoS ONE 4, 6501 (2009).
Smith, X., Taylor, A. & Rudd, C. E. T-cell immune adaptor SKAP1 regulates the induction of collagen-induced arthritis in mice - ScienceDirect. Immunol. Lett. 176, 122–127 (2016).
Wu, Y., Davies, K. E. & Oliver, P. L. The antioxidant protein Oxr1 influences aspects of mitochondrial morphology. Free Radic. Biol. Med. 95, 255–267 (2016).
Shin, J.-G. et al. Putative association of GPC5 polymorphism with the risk of inflammatory demyelinating diseases. J. Neurol. Sci. 335, 82–88 (2013).
Jiang, Y. et al. MiR-18a-downregulated RORA inhibits the proliferation and tumorigenesis of glioma using the TNF-α-mediated NF-κB signaling pathway. EBioMedicine 52, 102651 (2020).
Uzunhan, T. A. & Ayaz, A. Homozygous exonic and intragenic NRXN1 deletion presenting as either West syndrome or autism spectrum disorder in two siblings. Clin. Neurol. Neurosur. 214, 107141 (2022).
Grubman, A. et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 22, 2087–2097 (2019).
Liddelow, S. A. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017).
Sadick, J. S. et al. Astrocytes and oligodendrocytes undergo subtype-specific transcriptional changes in Alzheimer’s disease. Neuron 110, 1788–1805 (2022).
Zhao, K. et al. Integrated analysis and identification of hub genes as novel biomarkers for Alzheimer’s disease. Front. Aging Neurosci. 14, 1–14 (2022).
Kamphuis et al. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol. Aging 35, 2746–2760 (2014).
Rangaraju, S. et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease- associated-microglia in alzheimers disease. Mol. Neurodegener. 13, 24–49 (2018).
Busby, V. L., Goossens, S., Nowotny, P. & Lovestone, S. α-t-catenin is expressed in human brain and interacts with the wnt signaling pathway but is not responsible for linkage to chromosome 10 in alzheimer’s disease. Neuromol. Med. 5, 133–146 (2004).
Weissflog, L., Scholz, C.-J., Jacob, C. P., Nguyen, T. T. & Reif, A. KCNIP4 as a candidate gene for personality disorders and adult ADHD. Eur. Neuropsychopharmacol. 23, 436–447 (2012).
Massone, S. et al. RNA polymerase III drives alternative splicing of the potassium channel-interacting protein contributing to brain complexity and neurodegeneration. J. Cell Biol. 193, 851–866 (2011).
Morohashi, Y. Molecular cloning and characterization of calp/kchip4, a novel ef-hand protein interacting with presenilin 2 and voltage-gated potassium channel subunit Kv4. J. Biol. Chem. 277, 14965–14975 (2002).
Ros-Bernal, F. et al. Neuroinflammation and Parkinson’s Disease (Springer New York, 2014).
Cam, J. A. et al. The low density lipoprotein receptor-related protein 1B retains β-amyloid precursor protein at the cell surface and reduces amyloid-β peptide production. J. Biol. Chem. 279, 29639–29646 (2004).
Madissoon, E. et al. scRNA-seq assessment of the human lung, spleen, and esophagus tissue stability after cold preservation. Genome Biol. 21, 1 (2019).
Chen, H. et al. snRNA-Seq analysis reveals ten hub genes associated with alveolar epithelial cell injury during pulmonary acute respiratory distress syndrome. Heliyon 9, 17160 (2023).
Renaut, S. et al. Single-cell and single-nucleus RNA-sequencing from paired normal-adenocarcinoma lung samples provide both common and discordant biological insights. PLoS Genet. 20, 26 (2024).
Sang, G. et al. High throughput detection of variation in single-cell whole transcriptome through streamlined scFAST-seq. Preprint at bioRxiv. https://doi.org/10.1101/2023.03.19.533382 (2023).
Rossi, E. et al. High-level detection of gene amplification and chromosome aneuploidy in extracted nuclei from paraffin-embedded tissue of human cancer using FISH: a new approach for retrospective studies. Eur. J. Histochem. 49, 53–58 (2005).
Hedley, D., Friedlander, M., Taylor, I., Rugg, C. & Musgrove, E. Method for analysis of cellular DNA content of paraffin-embedded pathological material using flow cytometry. J. Histochem. Cytochem. 31, 1333–1335 (1983).
Zhang, X. et al. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 47, 721–728 (2018).
Oscar, F., Li-Ming, G. & Johan, B. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. J. Biol. Data 2019, 1–9 (2019).
Hafemeister, C. & Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Cold Spring Harb. Lab. 20, 296 (2019).
Acknowledgements
The project was supported by the National Natural Science Foundation of China (81827901, 82361138570, to X.Z.). We thank all members of X.Z., G.W. and J.Z. lab for their help and suggestions, and the team of M20 Genomics for their help with snRNA-seq experiments. I would like to thank J.M. and R.Q. for their support.
Author information
Authors and Affiliations
Contributions
X.Z. and Y.G. designed the project. Y.G. conceived and developed CED method and snCED-seq, and conducted all the experiments. Y.G. visualized the results, prepared the figures and wrote the paper. G.W., J.M., and R.Q. performed and analyzed the snCED-seq data. R.M. and J.Z. contributed to the FFPE human tissues and analyzed data. J.X. contributed to the mouse brain tissue sampling. X.Y., K.Y., Y.H., and X.Y. revised the manuscript. X.Z., G.W., and J.Z. supervised the project. M20 Genomics prepared libraries and performed next-generation sequencing. All authors have revised and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guo, Y., Ma, J., Qi, R. et al. snCED-seq: high-fidelity cryogenic enzymatic dissociation of nuclei for single-nucleus RNA-seq of FFPE tissues. Nat Commun 16, 4101 (2025). https://doi.org/10.1038/s41467-025-59464-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59464-0
